Clinical Description
Neurofibromatosis 1 (NF1) is an extremely variable multisystem disease; the progression and severity may differ throughout life in an affected individual as well as in affected family members with the same NF1 pathogenic variant [Gutmann et al 2017, Monroe et al 2017, Gianluca et al 2020]. The cardinal clinical manifestations of NF1 include multiple café au lait macules, intertriginous freckling, multiple cutaneous neurofibromas, subcutaneous or deep nodular neurofibromas, plexiform neurofibromas, and characteristic ocular signs. Problems with learning, behavior, and social adaptation are unusually common among people with NF1. Optic or non-optic gliomas occur much more often than expected, but most of these tumors exhibit a benign course. Individuals with NF1, especially those with plexiform or deep nodular neurofibromas in large numbers or of large size, are at high risk of developing malignant peripheral nerve sheath tumors, which tend to occur at a much younger age and have a worse prognosis than in the general population. Women with NF1 are at increased risk of developing breast cancer and have complications of pregnancy more often than expected. Hypertension is frequent in people with NF1, and NF1 vasculopathy may cause stroke or other cardiovascular complications in affected children and young adults. Vertebral or tibial dysplasia can cause major disability in some individuals, and NF1-associated gastrointestinal, endocrine, or pulmonary disease – although less frequent – may be quite serious.
Table 2.
Neurofibromatosis 1: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature 1 | Typical Age of Onset | Comment |
|---|
| Café au lait macules | >99% | Infancy & childhood | ↑ in number & size during 1st few yrs of life; macules fade in older persons. |
| Intertrigenous freckling | 85% | Infancy & early childhood | Frequency ↑ w/age during childhood. |
| Lisch nodules | >95% | Early childhood | Frequency ↑ w/age during childhood. |
| Choroidal abnormalities | 82%-98% | Early childhood | ↑ in number & size during childhood |
| Optic pathway glioma | 15%-20% | Birth - 6 yrs | Frequency lower in adults |
| Non-optic glioma | 2%-5% | Any age | Frequency lower in children than adults |
Cutaneous
neurofibromas | 99% | Adolescence-adulthood | Infrequent in childhood; variably ↑ in size & number throughout life |
| Nodular neurofibromas (subcutaneous or deep) | ~15% | Adolescence | Frequency shown is on clinical exam; frequency is 2-3x higher on whole-body MRI. |
Plexiform
neurofibroma(s) | ~30% | Infancy (sometimes congenital) or childhood | Frequency shown is on clinical exam; frequency is ~50% on whole-body MRI. |
Malignant peripheral
nerve sheath tumor | 8%-13% | Adolescence - adulthood | Cross-sectional prevalence 2%-5% after mid-childhood |
| Intellectual disability | 4%-8% | Childhood | Persists throughout life |
| Learning difficulties | 50%-60% | Childhood | Persist throughout life |
| Behavior issues | 30%-67% | Childhood |
| Seizures | 6%-7% | Any age | |
| Long bone dysplasia | 2% | Infancy (congenital) | |
| Dystrophic scoliosis | 5% | 6-10 yrs | Rapidly progressive scoliosis due to vertebral dysplasia |
| Nondystrophic scoliosis | 5% | Adolescence | Milder scoliosis w/o vertebral anomalies |
| Osteoporosis | ~20% | Mid-adulthood | Osteopenia is frequent at all ages; osteoporosis occurs earlier than in general population but is rare in children & uncommon in young adults |
| Hypertension | ≥15%-20% | Any age | Prevalence greater in adults than children |
- 1.
Many of the features listed in this table have different frequencies at different ages. The table gives life-time cumulative incidence figures that may be higher, and sometimes much higher, than the prevalence at any given age. Most frequencies in this table are from Ferner & Gutmann [2013] or DeBella et al [2000]. References for more recently defined features are given in the discussion of individual features below.
Cutaneous Features
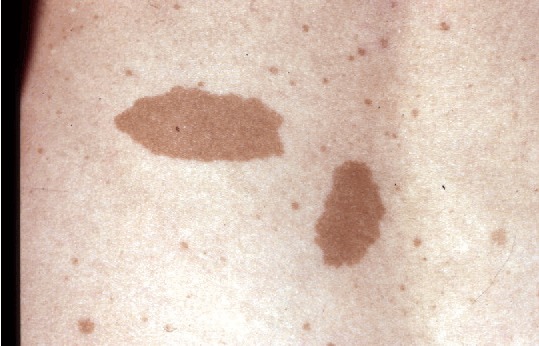
Café au lait macules (CALMs). Typically, CALMs in individuals with NF1 are ovoid in shape with well-defined borders, uniform in color (a little darker than the background pigmentation of the individual's skin), and about 1-3 cm in size; however, they may be smaller or much larger, lighter or darker, or irregular in shape [Ozarslan et al 2021, Albaghdadi et al 2022]. The pigmentation may also be irregular, with freckling or a more deeply pigmented smaller CALM within a larger, more typically colored lesion. They usually appear in infancy and early childhood, and once established remain stable in number and size (except for growth of the skin itself). They may be difficult to see in older adults with NF1 because of the wrinkling and diffuse pigmentation that often occur with aging.
CALMs are flat and flush with the surrounding skin; if the skin of the lesion is raised or has an unusually soft or irregular texture in comparison to the surrounding skin, an underlying plexiform neurofibroma is likely. The darker pigmentation of CALMs may be difficult to see in people with very fair skin or very dark skin, where the color of the lesions is similar to that of the rest of the skin. A Wood's light is useful in such individuals to demonstrate the pigmented macules. CALMs are not seen on the palms or soles in people with NF1 but can occur almost anywhere else on the body.
Freckling. Clusters of freckles are frequent in sun-exposed areas and may also be seen diffusely over the trunk, proximal extremities, and neck in people with NF1. Individuals with NF1 also develop freckles in areas where skin rubs against skin – in the axilla, groin, and under the breasts in women [Ozarslan et al 2021, Albaghdadi et al 2022].
Neurofibromas. See Other Tumors.
Other skin findings. Juvenile xanthogranuloma and nevus anemicus are more common than expected in people with NF1 and may be useful in supporting the diagnosis in young children who do not meet the standard diagnostic criteria [Miraglia et al 2020, Ozarslan et al 2021]. Juvenile xanthogranulomas are small, tan- or orange-colored papules that may occur in clusters. Nevus anemicus is an irregularly shaped macule that is paler than surrounding skin and does not get red when rubbed, as the skin surrounding it does.
Ocular Findings
Lisch nodules are innocuous iris hamartomas that can be demonstrated on slit lamp examination in almost all adults with NF1, but in fewer than half of children with NF1 younger than age five years. They can be distinguished from iris freckles by their three-dimensional nodular appearance.
Choroidal freckling cannot be seen on standard ophthalmologic examination but can be visualized by scanning laser ophthalmoscopy with infrared or near-infrared light, infrared reflectance imaging, or optical coherence tomography [Vagge et al 2016, Moramarco et al 2018]. The lesions, which are Schwann cell proliferations arrayed in concentric rings around an axon, occur in the majority of people with NF1 of all ages and increase in prevalence, number, and size with age during childhood [Touzé et al 2021].
Other findings frequently seen on ophthalmologic examination of individuals with NF1 include microvascular abnormalities of the retina [Parrozzani et al 2018, Moramarco et al 2019] and hyperpigmented spots of the fundus [Moramarco et al 2021].
Optic pathway gliomas in individuals with NF1 are usually asymptomatic and remain so throughout life [Di Nicola & Viola 2020, Shofty et al 2020]. The clinical course in individuals with optic pathway gliomas tends to be milder in individuals with NF1 than in those who do not have NF1. Optic pathway gliomas in NF1 are frequently stable for many years or only very slowly progressive [Sellmer et al 2018, Kinori et al 2021]. In addition, the majority of optic pathway gliomas appear to regress spontaneously – their prevalence declines from approximately 20% in young children to less than 5% in older adults with NF1 [Sellmer et al 2018]. Symptomatic optic pathway gliomas in individuals with NF1 usually present before age six years with loss of visual acuity, proptosis, or strabismus, but these tumors may not become symptomatic until later in childhood or adulthood [Friedrich & Nuding 2016, Kinori et al 2021].
Increased tortuosity of the optic nerve can be seen on brain MRI in children with NF1, but optic nerve tortuosity is not associated with the occurrence of optic pathway glioma among individuals with NF1 [Ji et al 2013].
Other Tumors
Neurofibromas are benign Schwann cell tumors that can affect virtually any nerve in the body [Brena et al 2020, Serra et al 2020, Ozarslan et al 2021]. Cutaneous neurofibromas are discrete, well-circumscribed masses, usually ranging in size from 1-2 mm to a few centimeters. Their consistency varies from soft to rubbery to firm. They may be sessile or pedunculated, and the involved skin may be the same color and tone as adjacent uninvolved skin or may be pinker or browner or bluer. Most are asymptomatic, but they may itch or be tender to touch. Cutaneous neurofibromas are rare in children but present in almost all adults with NF1.
Subcutaneous neurofibromas lie under the skin (the skin can be moved over them). Most feel rubbery and are nodular, but they may be diffuse, with indistinct borders, and of soft or heterogeneous consistency. The skin overlying a superficial diffuse neurofibroma may exhibit unusual pigmentation or hair patterning. Subcutaneous neurofibromas may be isolated or occur in clusters or continuously like beads on a string along a nerve. Most are small, but subcutaneous neurofibromas can grow to be 5 cm in diameter or more. They may be tender and are sometimes painful. Subcutaneous neurofibromas are uncommon in children but present in about 15% of adults with NF1 on clinical examination.
Cutaneous and subcutaneous neurofibromas continue to develop throughout life, although the rate of appearance may vary greatly from year to year. The total number of neurofibromas seen on clinical examination in adults with NF1 varies from a few to hundreds or even thousands. Some women experience a rapid increase in the number and size of neurofibromas during pregnancy, but this does not appear to produce a persistent increase in the tumor burden in comparison to those of child-bearing age with NF1 who have not been pregnant [Well et al 2020].
Plexiform neurofibromas. About half of people with NF1 have plexiform neurofibromas. Most of these tumors are internal, and thus not apparent on clinical examination. They can, however, be seen on MRI (see Imaging). Plexiform neurofibromas tend to grow in childhood and adolescence and then remain stable throughout adulthood [Nguyen et al 2012]. Although most plexiform neurofibromas are asymptomatic, they may cause pain, grow to cause disfigurement, produce overgrowth or erosion of adjacent tissue, or impinge on the function of nerves and other structures.
Superficial diffuse plexiform neurofibromas are soft and irregular; they are often associated with thickening, hypertrophy, and/or hyperpigmentation of the associated skin. More extensive diffuse plexiform neurofibromas may have a characteristic “bag of worms” feel on palpation, indicating involvement of multiple nerves and branches. Plexiform neurofibromas may also be firm and nodular, occurring singly or extending for some distance or even along the entire extent of a nerve, producing a “beads on a string” feel on palpation. Deeper plexiform neurofibromas that are not apparent on clinical examination may also be diffuse or nodular, and singular or clustered on any nerve, nerve root, or nerve plexus.
Malignant peripheral nerve sheath tumors (MPNST) are the most common malignant neoplasms associated with NF1. In comparison to the general population, MPNST tend to occur at a younger age and be associated with a poorer prognosis in people with NF1 [Martin et al 2020, Sharma et al 2021]. Most, if not all, MPNST arise in preexisting diffuse or nodular plexiform neurofibromas. The most frequent clinical sign of malignant change is persistent pain, either as a new symptom or as exacerbation of existing pain. This pain may be accompanied by rapid growth or change in texture of the tumor clinically or on MRI.
Individuals with NF1 who have a type 1 whole-gene deletion or benign subcutaneous neurofibromas, or whose burden of benign internal plexiform neurofibromas is high, appear to be at greater risk of developing MPNST than people with NF1 who do not have these features [Nguyen et al 2014]. Individuals with atypical neurofibromas also appear to be at unusually high risk of developing MPNST [Higham et al 2018].
Brain tumors. Non-optic gliomas in people with NF1 are usually asymptomatic, and most are discovered as incidental findings on head MRI done as a routine screen or for other indications. These are usually low-grade tumors that grow slowly or not at all over many years [Sellmer et al 2017], although symptomatic and/or high-grade brain tumors are seen occasionally [Byrne et al 2017, Glombova et al 2019].
At least 20% of people with NF1 who have one non-optic glioma have two or more of these tumors [Sellmer et al 2017, Glombova et al 2019]. Second central nervous system (CNS) gliomas occur in 17%-20% of individuals with NF1 who have optic pathway gliomas [Sharif et al 2006, Sellmer et al 2018].
Breast cancer. Women with NF1 are at substantially increased risk of developing breast cancer before age 50 years [Uusitalo et al 2016] and of dying of breast cancer [Evans et al 2020]. The cumulative risk of developing contralateral breast cancer is greater than expected among women with NF1 [Evans et al 2020]. Breast cancers in women with NF1 are more likely to be HER2-positive and to have other unfavorable tumor markers [Evans et al 2020].
Hematologic malignancies. Although still rare, juvenile myelomonocytic leukemia (JMML) is hundreds of times more frequent in children with NF1 than in other children [Niemeyer & Flotho 2019]. The usual clinical features at presentation are splenomegaly, hepatomegaly, and leukemic infiltrates of the lung in association with a peripheral blood smear that shows myelocytes, meta-myelocytes, and sometimes nucleated red cells. Juvenile xanthogranulomas may be seen in children with NF1 and JMML but do not appear to be more frequent than expected in other children with NF1 [Liy-Wong et al 2017]. It is not clear whether lymphoreticular malignancies occur more frequently than expected in adults with NF1 [Bergqvist et al 2021].
Additional tumors. A variety of other tumors may also be seen more often than expected in individuals with NF1, including rhabdomyosarcomas [Crucis et al 2015], pheochromocytomas [Gruber et al 2017], paragangliomas [Gruber et al 2017], gastrointestinal stromal tumors [Nishida et al 2016], and glomus tumors [Kumar et al 2014]. People with NF1 may also be at increased risk for some other cancers [Seminog & Goldacre 2013, Varan et al 2016, Landry et al 2021].
Other Neurologic Manifestations
Motor function. Hypotonia and impairments in coordination, balance, and fine motor function are frequent in children with NF1 [Iannuzzi et al 2016, Haas-Lude et al 2018, Pardej et al 2022]. Children with NF1 also have less muscle strength than unaffected children of the same age, sex, and weight [Summers et al 2015].
Intellectual and learning disabilities. Deficits in visual-spatial performance are most common in individuals with NF1, but specific learning disorders and problems with executive function, memory, and language are also frequent [Vogel et al 2017]. The average IQ of people with NF1 is ~1 SD lower than individuals in the general population, and frank intellectual disability (IQ <70) occurs in 4%-8% of individuals with NF1, a frequency about twice that in the general population [Vogel et al 2017, Al-Farsi et al 2022]. Intellectual disability is more frequent among individuals whose NF1 is caused by a whole-gene deletion.
Behavioral issues include problems with social competence and attention. Both children and adults with NF1 report increased social difficulties such as isolation and reduced peer acceptance and exhibit fewer social skills and prosocial behaviors [Chisholm et al 2018, Payne et al 2020]. Attention-deficit/hyperactivity disorder is present in 30%-50% of children and adolescents with NF1 [Vogel et al 2017, Chisholm et al 2018, Domon-Archambault et al 2018] and may persist into adulthood [Mautner et al 2015]. Symptoms of autism are frequent in children with NF1, and 25% of children with NF1 meet standard diagnostic criteria for autism spectrum disorder [Vogel et al 2017, Chisholm et al 2018, Domon-Archambault et al 2018, Payne et al 2020, Chisholm et al 2022]. Autistic symptoms appear to be less common among adults than children with NF1 [Morris et al 2016]. Sleep disturbance is frequent in individuals with NF1 at all ages [Domon-Archambault et al 2018, Fjermestad et al 2018]. Psychiatric diseases such as mood disorders, anxiety disorders, and emerging personality disorders may also occur more often than expected among adults with NF1 [Domon-Archambault et al 2018, Kenborg et al 2021].
Polyneuropathy. A few percent of people with NF1 develop a diffuse polyneuropathy, often (though not always) in association with multiple nerve root tumors [Barnett et al 2019, Bayat & Bayat 2020]. NF1 polyneuropathy may be asymptomatic or produce sensory deficits, pain, or itching. The risk for MPNST appears to be higher in individuals who have polyneuropathy than in those who do not.
Seizures occur in about 5% of individuals with NF1, with a slightly higher prevalence in adults than in children [Bernardo et al 2020, Sorrentino et al 2021]. The seizures may be generalized but more often are focal, occurring in association with a brain tumor, area of infarction, or mesial temporal sclerosis [Pecoraro et al 2017, Bernardo et al 2020, Sorrentino et al 2021]. Neurodevelopmental abnormalities are more common in individuals with NF1 and epilepsy [Sorrentino et al 2021].
The approach to treating epilepsy in individuals with NF1 is similar to that used in those who do not have NF1 [Bernardo et al 2020, Sorrentino et al 2021]. Control of focal seizures may require the use of more than one anti-seizure drug or surgical removal of the affected part of the brain.
Headaches occur in at least half of individuals with NF1 and are more frequent in adults than children [Fjermestad et al 2018, Hirabaru & Matsuo 2018, Kongkriangkai et al 2019]. Migraine headaches are most common in NF1, but other kinds of headaches may occur. NF1-associated lesions are often seen on head MRI in people with headaches, but the frequency is similar to that found in those without headaches [Afridi et al 2015].
Neuroimaging. Hyperintense lesions (unidentified bright objects [UBOs] or focal areas of high signal intensity [FASI]) are seen on T2-weighted brain MRI in more than half of children with NF1 [Sellmer et al 2018]. These may occur in the optic tracts, basal ganglia, brain stem, cerebellum, or cortex, and they usually show no evidence of a mass effect. Typical UBOs are not seen on T1-weighted MRI imaging or on CT scan. UBOs correspond pathologically to areas of spongiform myelinopathy [DiPaolo et al 1995]. They peak in number and size at about age seven years and then tend to regress, but some persist into adulthood [Sellmer et al 2018, Calvez et al 2020]. The presence of UBOs does not appear to be related to the occurrence of seizures in children with NF1 [Hsieh et al 2011]. Some studies have suggested that the presence, number, volume, location, or disappearance of UBOs over time correlates with learning disabilities or behavioral abnormalities in children with NF1, but findings have not been consistent across investigations [Payne et al 2014, Roy et al 2015, Parmeggiani et al 2018, Eby et al 2019, Baudou et al 2020].
Enlargement of the corpus callosum is seen in some children with NF1 and has been associated with learning disabilities [Pride et al 2010, Aydin et al 2016].
MRI is the imaging method of choice for demonstrating optic gliomas, brain tumors, and neurofibromas of cranial, spinal, or peripheral nerves, as well as diffuse or nodular neurofibromas anywhere in the body. Positron emission tomography is useful in recognizing MPNST [Nishida et al 2021], and high-resolution ultrasound examination can be used to characterize dermal and superficial plexiform neurofibromas [Winter et al 2020].
Musculoskeletal Features
Long bone, sphenoid wing, or vertebral dysplasia. Osseous dysplasia may occur as a primary abnormality in individuals with NF1 (typical of long-bone dysplasia), or in association with an adjacent plexiform neurofibroma or dural ectasia (vertebral or sphenoid wing dysplasia). Healing of fractured or defective bone in any of these focal lesions is often unsatisfactory [Elefteriou et al 2009]. Surgical treatment of osseous dysplasia and its associated deformities of the long bones, craniofacies, and spine is frequently difficult and best accomplished by experienced specialists [Mladenov et al 2020].
Dysplasia of the long bones, most often the tibia and fibula, is an infrequent but characteristic feature of NF1 [Elefteriou et al 2009]. The lesion is congenital and almost always unilateral. It usually presents in infancy with anteriolateral bowing of the lower leg. Early recognition of tibial dysplasia permits bracing, which may prevent fracture. The initial radiographic changes are narrowing of the medullary canal with cortical thickening at the apex of the bowing [Stevenson et al 2007].
Sphenoid wing dysplasia typically presents with asymmetry of the orbits, but it is sometimes found incidentally on cranial imaging. Sphenoid wing dysplasia is often static but may be progressive, occasionally disrupting the integrity of the orbit and producing pulsating enophthalmos [Chauvel-Picard et al 2020].
Vertebral dysplasia usually presents as dystrophic scoliosis, which typically develops between ages six and eight years, much earlier than common adolescent scoliosis. Dystrophic scoliosis, which is characterized by acute angulation of the spine over a short segment, may be rapidly progressive within a few months after becoming apparent [Kaspiris et al 2022].
Nondystrophic scoliosis, which is not usually associated with vertebral abnormalities in individuals with NF1, resembles common adolescent scoliosis in its age at onset and more benign course [Elefteriou et al 2009].
Osteoporosis. Generalized osteopenia is more common than expected in people with NF1 [Rodari et al 2018, Filopanti et al 2019, Jalabert et al 2021, Kaspiris et al 2022], and fractures occur more often than expected [Heervä et al 2012]. Adults with NF1 develop osteoporosis more frequently and at a younger age than in the general population [Filopanti et al 2019, Kaspiris et al 2022]. The pathogenesis of these bony changes is not fully understood, but individuals with NF1 often have lower-than-expected serum 25-hydroxy vitamin D concentrations, elevated serum parathyroid hormone levels, and evidence of increased bone resorption [Riccardi et al 2020, Tezol et al 2021, Kaspiris et al 2022]. The function of both osteoblasts and osteoclasts appears to be abnormal in bone from people with NF1 [Riccardi et al 2020, Kaspiris et al 2022].
Vascular Involvement
Arterial hypertension occurs in at least 15%-20% of individuals with NF1 [Dubov et al 2016, Sivasubramanian & Meyers 2021]. It may develop at any age but is more frequent in adults than children. Often no specific cause is found, but hypertension may be caused by renal artery stenosis or mid-aortic syndrome, usually as a manifestation of NF1 vasculopathy, especially in children [Celik et al 2021, Sivasubramanian & Meyers 2021]. Hydronephrosis and other structural abnormalities of the urinary tract are frequent in individuals with NF1-related hypertension but are also seen in those without hypertension [Dubov et al 2016, Celik et al 2021].
Although much more frequent in people with NF1 than in the general population, pheochromocytomas or paragangliomas are found in fewer than 1% of adults with NF1. These tumors are usually asymptomatic, but they can cause arterial hypertension [Al-Sharefi et al 2019].
Pulmonary hypertension is a rare but very serious complication of NF1 in older adults [Jutant et al 2018, Jutant et al 2020]. It is usually associated with parenchymal lung disease, which itself may be a manifestation of NF1 vasculopathy affecting vessels in the lungs [Jutant et al 2018].
Stroke is more common and often occurs at a younger age among people with NF1 than in the general population [Terry et al 2016]. Anatomically variant stenotic or ectatic cerebral arteries and intracranial aneurysms occur more frequently in individuals with NF1 than in the general population [Bekiesińska-Figatowska et al 2014, D'Arco et al 2014, Barreto-Duarte et al 2021]. The internal carotid, middle cerebral, or anterior cerebral artery are most often affected. Small telangiectatic vessels form around the stenotic area and appear as a "puff of smoke" (moya moya) on cerebral angiography. Moya moya vasculopathy develops about three times more often than expected in children with NF1 after cranial irradiation for primary brain tumor [Murphy et al 2015].
Cardiac Issues
Congenital anomalies of the circulatory system were observed 3.35 times (95% confidence interval 1.64-6.83x) more often than expected among children with NF1 in a study performed through Finnish population-based registries [Leppävirta et al 2018]. Pulmonary valve stenosis and mitral valve anomalies are the most frequent cardiac defects seen in individuals with NF1 [Lin et al 2000, Pinna et al 2019]. Congenital heart defects and hypertrophic cardiomyopathy may be especially frequent among persons with NF1 whole-gene deletions [Nguyen et al 2013, Pinna et al 2019]. Intracardiac neurofibromas may also occur [Nguyen et al 2013].
Pulmonary Disease
NF1-associated diffuse lung disease occurs in 10%-20% of adults [Jutant et al 2018, Alves Júnior et al 2019]. Symptoms are usually nonspecific and may include dyspnea on exertion, shortness of breath, chronic cough, or chest pain. Symptoms do not usually appear until the third or fourth decade of life, although characteristic signs may be found on imaging studies in children with NF1 [Spinnato et al 2019]. Chest CT is the method of choice for identifying NF1-associated diffuse lung disease, which typically is characterized by upper-lobe cystic and bullous disease and basilar interstitial disease. NF1 pulmonary disease is often poorly responsive to available medical treatments.
Growth
Individuals with NF1 tend to be below average in height and above average in head circumference for age [Zessis et al 2018]. However, very few individuals with NF1 have height more than 3 SD below the mean or head circumference more than 4 SD above the mean.
In contrast, individuals with whole-gene NF1 deletions have overgrowth (especially in height) between ages two and six years [Ning et al 2016, Kehrer-Sawatzki et al 2020]. The clinical features in some of these individuals resemble those of Weaver syndrome (see Genotype-Phenotype Correlations).
Pubertal development is usually normal, although decreased pubertal growth velocity occurs in both girls and boys [Zessis et al 2018]. However, delayed puberty is common [Virdis et al 2003], and precocious puberty and/or growth hormone excess may also occur in children with NF1, especially in those with tumors of the optic chiasm [Cambiaso et al 2017, Hannah-Shmouni & Stratakis 2019].
Quality of Life
Quality of life assessments are lower in both children and adults with NF1 than in comparison groups [Vranceanu et al 2015]. Cosmetic, medical, social, and behavioral features of NF1 all may compromise the quality of life in people with NF1, and clinical depression may impair their ability to function effectively [Domon-Archambault et al 2018]. A population-based Danish registry study found that people with NF1 were more than twice as likely to be hospitalized and to have more frequent and longer hospitalizations at all ages in comparison to the general population [Kenborg et al 2020].
NF1 Phenotypic Variants
Mosaic NF1 (i.e., somatic mosaicism for an NF1 pathogenic variant) may present as clinical features of NF1 localized to one or more segments of the body or as typical (generalized) NF1 [Ejerskov et al 2021]. Mosaic NF1 is usually milder than typical NF1 involving the same pathogenic variant, and some adults with mosaic NF1 have no clinical features of NF1 [Kluwe et al 2020, Yang et al 2020]. Mosaic NF1 involving just a single body segment is sometimes called "segmental NF1," but the term "localized mosaic NF1" is preferred because it is more informative and because non-mosaic NF1 may sometimes involve just one part of the body by chance, especially in young children. Adults with mosaic NF1 may have children with typical (i.e., non-mosaic) NF1 [Legius & Brems 2020] (see Genetic Counseling).
NF1-Noonan syndrome phenotype occurs in approximately 12% of individuals with NF1. The features may include ocular hypertelorism, downslanted palpebral fissures, ptosis, low-set ears, webbed neck, pectus anomaly, and pulmonic stenosis. Affected family members with NF1 may or may not have concomitant features of Noonan syndrome [Chen et al 2014, Ekvall et al 2014]. The NF1-Noonan syndrome phenotype is genetically heterogeneous. Some individuals have disease-causing variants of both NF1 and PTPN11 (the gene most often involved in Noonan syndrome) [D'Amico et al 2021]. In other individuals, only an NF1 variant is identified [De Luca et al 2005]. Watson syndrome, an overlapping phenotype characterized by pulmonary valvular stenosis, CALMs, short stature, and mild intellectual disability, is caused by NF1 variants [Allanson et al 1991].
Familial spinal neurofibromatosis is characterized by neurofibromas of every spinal nerve root but few (if any) cutaneous manifestations of NF1 [Bettegowda et al 2021]. Despite the name, this phenotype may also occur in simplex cases [Ruggieri et al 2015].
Multiple spinal ganglioneuromas (rather than neurofibromas) and multiple subcutaneous tumors were reported in an adult with an NF1 pathogenic variant whose clinical features were not diagnostic of NF1 [Bacci et al 2010].
Multiple lipomas have been observed in association with a pathogenic variant of NF1 but no other clinical features of NF1 [Ramirez et al 2021].
Symptomatic optic glioma was reported in a male with an NF1 pathogenic variant and no other diagnostic features of NF1 at age 21 years [Buske et al 1999].
Encephalocraniocutaneous lipomatosis was reported in one child with more than five CALMs and an NF1 pathogenic variant [Legius et al 1995].