Summary
Clinical characteristics.
SCN9A neuropathic pain syndromes (SCN9A-NPS) comprise SCN9A erythromelalgia (EM), SCN9A paroxysmal extreme pain disorder (PEPD), and SCN9A small fiber neuropathy (SFN).
- SCN9A-EM is characterized by recurrent episodes of bilateral intense, burning pain, and redness, warmth, and occasionally swelling. While the feet are more commonly affected than the hands, in severely affected individuals the legs, arms, face, and/or ears may be involved.
- SCN9A-PEPD is characterized by neonatal or infantile onset of autonomic manifestations that can include skin flushing, harlequin (patchy or asymmetric) color change, tonic non-epileptic attacks (stiffening), and syncope with bradycardia. Later manifestations are episodes of excruciating deep burning rectal, ocular, or submandibular pain accompanied by flushing (erythematous skin changes).
- SCN9A-SFN is characterized by adult-onset neuropathic pain in a stocking and glove distribution, often with a burning quality; autonomic manifestations such as dry eyes, mouth, orthostatic dizziness, palpitations, bowel or bladder disturbances; and preservation of large nerve fiber functions (normal strength, tendon reflexes, and vibration sense).
Diagnosis/testing.
The diagnosis of SCN9A-NPS is established in a proband with a heterozygous pathogenic variant in SCN9A identified by molecular genetic testing.
Management.
Treatment of manifestations: Most affected individuals are treated in dermatology clinics, neurology clinics, or pain clinics, or by anesthesiologists specializing in the management of chronic pain.
- SCN9A-EM. Cooling the extremities reduces pain; note that use of a fan is preferable to prolonged immersion in cold water, which can result in skin maceration, infection, and gangrene. Medications to consider are nonselective sodium channel blockers (e.g., carbamazepine, lidocaine infusion, or oral mexiletine).
- SCN9A-PEPD. Use of stool softeners and passing stool slowly to reduce the likelihood of triggering an attack. Carbamazepine is the most effective (albeit not completely effective) treatment in reducing the number and severity of attacks. Other anti-seizure medications with varying effectiveness include lamotrigine, topiramate, tiagabine, and sodium valproate.
- SCN9A-SFN. Lacosamide is associated with reduced pain ratings, improved general well-being and sleep quality, but not with changed overall quality of life or autonomic manifestations.
Surveillance: Follow up with a neurologist or neuromuscular specialist to assess for disease progression. Routine monitoring for side effects of medications used in treatment (such as Stevens-Johnson syndrome, liver toxicity, neutropenia seen with carbamazepine).
Agents/circumstances to avoid: Triggers including warmth, standing, alcohol, and spicy foods (SCN9A-EM); defecation, cold wind, eating, and emotion (SCN9A-PEPD); diabetes mellitus, alcohol, and chemotherapy (SCN9A SFN).
Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently asymptomatic older and younger relatives of an affected individual at risk for SCN9A-NPS in order to identify as early as possible those who would benefit from avoidance of activities that are known to trigger onset of pain.
Pregnancy management: Potential teratogenic effects of medications used for treatment of SCN9A-NPS should be discussed with affected women of childbearing age, ideally prior to conception.
Genetic counseling.
SCN9A neuropathic pain syndromes are inherited in an autosomal dominant manner. Each child of an individual with an NPS-causing variant in SCN9A has a 50% chance of inheriting the variant. Once the SCN9A pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
GeneReview Scope
Table
SCN9A erythromelalgia (SCN9A-EM) SCN9A paroxysmal extreme pain disorder (SCN9A-PEPD)
Diagnosis
Suggestive Findings
An SCN9A neuropathic pain syndrome (SCN9A-NPS) should be suspected in individuals with one of the following three neuropathic pain disorders.
Erythromelalgia (EM), characterized by:
- Recurrent episodes of bilateral intense, burning pain;
- Redness, warmth, and occasionally swelling of the distal extremities;
- Feet more commonly affected than the hands; although in severely affected individuals, the legs, arms, face, and/or ears may be involved.
Note: If manifestations are intermittent, photographs of the affected extremities during a flare can help with diagnosis (see Figure 1).
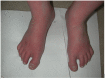
Figure 1.
Red and swollen feet of an individual with SCN9A erythromelalgia
Paroxysmal extreme pain disorder (PEPD), characterized by:
- Neonatal or infantile onset of autonomic manifestations that can include skin flushing, harlequin (patchy or asymmetric) color change, tonic non-epileptic attacks (stiffening), and syncope with bradycardia;
- Later, episodes of excruciating deep burning rectal, ocular, or submandibular pain accompanied by flushing (erythematous skin changes).
- Attacks are precipitated by defecation or perineal wiping (rectal attacks), eating (jaw attacks), or cold wind, temperature change, or crying (ocular attacks).
- The pain typically is localized at the beginning of an episode, but spreads (e.g., from the rectum to the abdomen).
- Attacks usually begin abruptly and range in duration from seconds to as long as two hours.
Between episodes, constipation is common (from a reluctance to pass stool, thereby precipitating a painful attack).
Inherited small fiber neuropathy (I-SFN), characterized by [Faber et al 2012]:
- Adult-onset neuropathic pain in a stocking and glove distribution, often with a burning quality;
- Autonomic manifestations such as dry eyes, mouth, orthostatic dizziness, palpitations, and bowel or bladder disturbances;
- Preservation of large nerve fiber functions (normal strength, tendon reflexes, and vibration sense);
- Normal nerve conduction studies;
- Reduced intraepidermal nerve fiber density and/or abnormal quantitative sensory testing.
Note: Exogenous and systemic causes of small fiber neuropathy such as diabetes mellitus, HIV, and neurotoxic drugs (chemotherapy) must be excluded.
Establishing the Diagnosis
The diagnosis of SCN9A-NPS is established in a proband with a heterozygous pathogenic variant in SCN9A by molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing and multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of SCN9A-NPS is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of SCN9A-NPS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SCN9A is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Depending on the sequencing method used, (multi)exon or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is typically to perform gene-targeted deletion/duplication analysis to detect an exon or whole-gene deletion or duplication; however, because SCN9A-NPS is caused by gain-of-function variants, intragenic deletions or duplications are not expected to be causative, and none have been reported.
A peripheral neuropathy multigene panel that includes SCN9A and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of SCN9A-NPS, some panels for erythromelalgia and/or paroxysmal extreme pain disorder may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in SCN9A Neuropathic Pain Syndromes
Clinical Characteristics
Clinical Description
Pain is the predominant symptom of SCN9A neuropathic pain syndromes. Triggers for episodes of pain vary with the specific syndrome. Pain may be accompanied by signs of redness, warmth, or autonomic dysfunction. Erythromelalgia (EM) and paroxysmal extreme pain disorder (PEPD) typically begin in infancy or childhood, whereas small fiber neuropathy (SFN) typically begins in adulthood, and the prevalence increases with age. All three disorders have been reported with intrafamilial variability in age of onset and severity.
Erythromelalgia (EM) is characterized by recurrent attacks of intense pain, redness, warmth, and swelling involving the feet and, less frequently, the hands [Dib-Hajj et al 2013, McDonnell et al 2016]. Warmth is an essential part of the syndrome. During the attacks, the extremities appear red or purple and may be swollen. Commonly, the attacks occur in the evening or at night and so may not be observed by a physician. The individual may seek medical advice for painful extremities but neglect to mention the characteristic warmth or redness (especially if limited to the soles of the feet). The symptoms are usually bilateral and symmetric. Within a family, the manifestations of the disorder may vary considerably.
Onset of SCN9A-EM is usually in childhood or adolescence; however, it has been recognized in infants in families who are familiar with the disorder. While rare, later onset (age >20 years) has been reported in some individuals and families [Choi et al 2010].
Initially, the symptoms involve the soles of the feet and the hands; with age, the lower legs and the arms may become involved. In individuals with advanced disease, symptoms may occur many times a day and last hours, especially at night, or become constant and unremitting.
At the onset, the episodes are triggered by exposure to warmth. A pathognomonic feature is triggering of episodes by warm or hot ambient temperature and relief with cooling of the extremities.
Less consistent precipitating factors include exercise, tight shoes, wearing socks, alcohol, spicy foods, and other vasodilating agents.
Some individuals have allodynia (pain evoked by a normally innocuous stimulus) and hyperalgesia (increased sensitivity to a painful stimulus).
The episodes may be disabling, interfere with sleep, and severely limit normal activities such as walking, participation in sports, wearing socks and shoes, and attending school or going to work. Individuals tend to limit their activities in warm weather and to stay in air-conditioned environments. Some individuals move from hot, humid climates to cooler climates.
Affected individuals prefer to wear open-toed shoes and to sleep with their feet uncovered. Swimming can be helpful because it keeps the limbs cool during exercise.
Neurologic examination is typically normal, although reduced ankle reflexes and decreased distal sensation can be seen.
Histopathologic examination of skin biopsy in individuals with erythromelalgia shows nonspecific thickening of blood vessel basement membrane, perivascular edema and mononuclear infiltrate, and reduced density of the autonomic nerve plexuses.
Paroxysmal extreme pain disorder (PEPD) is characterized by episodes of rectal, ocular, or submandibular pain accompanied by erythema. The onset is in early infancy, and may occur prenatally [Fertleman et al 2007]. Infants may appear stiff and red at the time of delivery, but recover within minutes. Attacks are triggered by defecation, wiping the perineum, taking rectal temperature, or feeding. Pain is inferred from the infant's grimacing and crying.
Older children and adults describe the pain as excruciating, and burning or sharp. The attacks are always accompanied by erythematous flushing of the skin, which usually corresponds to the affected region. In severe attacks, the pain spreads and become generalized.
The number of rectal attacks can decrease with age, whereas the ocular and jaw attacks can increase with age [Fertleman et al 2007].
In PEPD, biopsies of rectum or colon have been reported as normal or only nonspecific findings [Fertleman et al 2007]
Inherited small fiber neuropathy (SFN) is characterized by symmetric, distal burning pain, numbness, and paresthesias. Autonomic manifestations such as dry eyes or mouth, orthostatic dizziness, change in sweating, and bowel or bladder dysfunction may be present. The prevalence of SFN increases with age.
Pathogenic missense variants in SCN9A were reported in eight of 28 adults (age ≥18 years) with inherited small fiber neuropathy (I-SFN) in whom other known causes for small fiber neuropathy (e.g., diabetes mellitus, impaired glucose tolerance, hyperlipidemia) were excluded, and who met strict diagnostic criteria for I-SFN with reduced intraepidermal nerve fiber density on skin biopsy or abnormal quantitative sensory testing [Faber et al 2012].
Of note, in SFN, the intraepidermal nerve fiber density (IENFD) in a skin biopsy is quantitatively reduced compared with age- and sex-adjusted normative values [Faber et al 2012], although I-SFN can occur in persons with normal IENFD [Eijkenboom et al 2019].
Genotype-Phenotype Correlations
Disease severity varies within families. In one family, an infant with SCN9A-PEPD had a father with only ocular attacks [Fertleman et al 2007].
Although SCN9A variants resulting in greater hyperpolarizing shifts in Nav1.7 sodium channels correlated with onset of manifestations at younger ages (P = .016), unknown inter-individual differences also influence age of onset: in two families in which individuals with erythromelalgia had the same variant (p.Ser241Thr), the age of onset was in the first decade in one family and the second decade in the other [Geha et al 2016].
Penetrance
The penetrance in families with SCN9A erythromelalgia and paroxysmal extreme pain disorder reported to date is 100%; individuals with SCN9A small fiber neuropathy show incomplete penetrance.
Of note, in 2008 the Erythromelalgia Association conducted a survey of its members regarding diagnosis, symptoms, and response to medications (see www.erythromelalgia.org). In this heterogeneous group of 427 participants (of which an unknown subset has SCN9A-EM), only 5% reported a relative with a diagnosis of erythromelalgia – fewer than would be expected for an autosomal dominant disorder with high penetrance; however, a pattern of autosomal dominant inheritance with 100% penetrance was always observed in multi-generation families with SCN9A-EM.
Nomenclature
Erythromelalgia. The term "erythromelalgia" was coined by the American neurologist S Weir Mitchell, MD, and is derived from the Greek words: erythro (red), melos (extremity), and algos (pain).
Some authors prefer "erythromelalgia" for both primary EM (OMIM 133020) and acquired forms of the disease (secondary EM); others use inherited EM as a synonym for primary EM.
Primary EM may also be referred to as "erythermalgia."
Paroxysmal extreme pain disorder was previously referred to as familial rectal pain [Fertleman et al 2007].
Prevalence
No accurate data on the worldwide prevalence of SCN9A-NPS are available. Molecular genetic data in the medical literature are available for between 100 and 200 individuals with SCN9A erythromelalgia and SCN9A paroxysmal extreme pain disorder.
It is likely that SCN9A neurogenic pain syndromes are underdiagnosed or often misdiagnosed.
Genetically Related (Allelic) Disorders
Disorders caused by germline pathogenic variants in SCN9A that are not associated with neuropathic pain are summarized in Table 2.
Table 2.
SCN9A Allelic Disorders
Differential Diagnosis
Erythromelalgia
The differential diagnosis of SCN9A erythromelalgia (SCN9A-EM) includes secondary EM resulting from an underlying organic disease, medication, or toxin; neuropathies; other conditions with some overlapping features; and inherited erythromelalgia in which no SCN9A pathogenic variant is identified See Table 3.
Table 3.
Differential Diagnosis of SCN9A Erythromelalgia
Paroxysmal Extreme Pain Disorder
Table 4.
Differential Diagnosis of SCN9A Paroxysmal Extreme Pain Disorder
SCN9A Small Fiber Neuropathy (SFN)
Table 5.
Conditions in the Differential Diagnosis of SCN9A Small Fiber Neuropathy
In a cohort of 921 patients with SFN, diagnostic evaluations identified immunologic conditions (19%), sodium channel gene variants (16.7%), diabetes mellitus (7.7%), vitamin B12 deficiency (4.7%), alcohol abuse (3%), chemotherapy side effects (2.2%), monoclonal gammopathy of undetermined significance (1.4%), and hemochromatosis (0.3%). No cause was found in 53% of patients [de Greef et al 2018].
In one study, 58 of 1139 (5%) of individuals with SFN of unknown cause were found to have a heterozygous SCN9A variant [Eijkenboom et al 2019].
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with an SCN9A neuropathic pain syndrome (SCN9A-NPS), the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Neurologic examination, including a quantitative sensory test and pain assessment
- Clinical assessment of SCN9A-NPS that may include questionnaires such as the Small Fibre Neuropathy and Symptoms Inventory Questionnaire [Terkelsen et al 2017] or the Neuropathic Pain Scale [Galer & Jensen 1997]
- Assessment of the pain management strategies used (see Treatment of Manifestations)
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Most affected individuals are treated in dermatology, neurology, or pain clinics; or by anesthesiologists specializing in the management of chronic pain.
SCN9A Erythromelalgia (SCN9A-EM)
The pain is often refractory to treatment.
Cooling the extremities reduces pain in a symptomatic person. Cooling with a fan is generally safer than immersion in water; complications from prolonged immersion in ice baths include skin maceration, infection, and gangrene. Amputation has occasionally been necessary to treat these complications.
Medications. The individual's treating physician should determine treatment based on factors including other medical conditions, known medication allergies, and potential for drug-drug interactions.
- Nonselective sodium channel blockers including carbamazepine, lidocaine infusion, or oral mexiletine have been used successfully in SCN9A-EM [Choi et al 2009, Fischer et al 2009, Cregg et al 2014].
- Carbamazepine. While it is generally less effective in treating EM than PEPD, a family with the SCN9A pathogenic variant p.Val400Met reported significant improvement of EM symptoms while using carbamazepine [Fischer et al 2009]. Since then, patients with the p.Ser241Thr and p.Ile234Thr variants have reported pain relief from carbamazepine [Geha et al 2016, Yang et al 2018].
- A combination of gabapentin and carbamazepine has been reported effective in at least one individual with the SCN9A variant p.Ile848Thr [Natkunarajah et al 2009].
Other
- See Tham & Giles [2018] for a review of additional combinations of medical, surgical, and alternative treatments used with varying success in individuals with erythromelalgia (not limited to SCN9A-EM).
- Use of oxcarbazepine in treating EM has not been reported.
SCN9A Paroxysmal Extreme Pain Disorder (SCN9A-PEPD)
Use of stool softeners and passing stool slowly can reduce the likelihood of triggering an attack.
Medications. The most effective (albeit not completely effective) treatment in reducing the number and severity of attacks in PEPD is carbamazepine [Fertleman et al 2007]. While a correlation may exist between individual pathogenic variants and response to drugs that block the sodium channel [Drenth & Waxman 2007, Dib-Hajj et al 2013], no current comprehensive review addresses this issue.
Additional anti-seizure medications including lamotrigine, topiramate, tiagabine, and sodium valproate have been reported to have varying effectiveness. See Supplemental Table E3 in Fertleman et al [2007].
Analgesics and opiates are ineffective in the treatment of PEPD.
SCN9A Small Fiber Neuropathy (SCN9A-SFN)
A small randomized placebo-controlled double-blind crossover-design clinical trial of the anti-seizure drug lacosamide in 24 individuals with SCN9A-SFN reported reduction in pain ratings, improvement in general well-being and sleep quality, but no change in overall quality of life or autonomic manifestations compared with 23 individuals who received placebo [de Greef et al 2019].
Surveillance
There are no published guidelines for surveillance for SCN9A-NPS.
Follow up with a neurologist or neuromuscular specialist to assess for progression of the disease.
It is important to monitor for known side effects of treatment of medications (e.g., Stevens-Johnson syndrome, liver toxicity, and neutropenia – associated with carbamazepine treatment).
Agents/Circumstances to Avoid
SCN9A erythromelalgia. Symptoms are triggered by warmth and standing and, in some individuals, by alcohol and spicy foods including chili peppers or garlic.
In some individuals, exercise can trigger symptoms. However, for many individuals, the benefits of mild exercise outweigh the disadvantages. Swimming is a preferred exercise because the extremities remain cool.
SCN9A paroxysmal extreme pain disorder is often triggered by defecation, cold wind, eating, and emotion.
SCN9A small fiber neuropathy. Avoid additional risk factors for small fiber neuropathy such as diabetes mellitus, alcohol, and chemotherapy.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger relatives of an affected individual at risk for SCN9A-NPS in order to identify as early as possible those who would benefit from avoidance of activities that are known to trigger onset of pain.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Potential teratogenic effects of medications given for treatment of SCN9A-NPS should be discussed with affected women of childbearing age, ideally prior to conception.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
SCN9A neuropathic pain syndromes (SCN9A-NPS) are inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Many individuals diagnosed with an SCN9A-NPS have an affected parent.
- A proband with an SCN9A-NPS may have the disorder as the result of a de novo pathogenic variant. The proportion of cases caused by a de novo pathogenic variant is unknown because the evaluation of family members for milder symptoms has been incomplete and molecular genetic data are insufficient.
- Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent [Han et al 2006].Note: Misattributed parentage can also be explored as an alternative explanation for an apparent de novo pathogenic variant.
- The family history of some individuals diagnosed with an SCN9A-NPS may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has been performed on the parents of the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- Penetrance of SCN9A erythromelalgia and SCN9A paroxysmal extreme pain disorder is complete; SCN9A small fiber neuropathy is associated with incomplete penetrance.
- A sib heterozygous for the same SCN9A pathogenic variant may be more or less severely affected than the proband (see Genotype-Phenotype Correlations).
- If the proband has a known SCN9A pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism. Two affected sibs with an unaffected mosaic father have been reported [Han et al 2006].
- If the parents have not been tested for the SCN9A pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk because of the possibility of germline mosaicism or, in SCN9A-SFN, reduced penetrance in a heterozygous parent.
Offspring of a proband. Each child of an individual with an NPS-causing variant in SCN9A has a 50% chance of inheriting the variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected or has a pathogenic variant, his or her family members are at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the SCN9A pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider decisions regarding prenatal testing to be the choice of the parents, discussion of these issues is appropriate.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Erythromelalgia Association200 Old Castle LaneWallingford PA 19086Phone: 610-566-0797
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
SCN9A Neuropathic Pain Syndromes: Genes and Databases
Table B.
OMIM Entries for SCN9A Neuropathic Pain Syndromes (View All in OMIM)
Molecular Pathogenesis
The normal product of SCN9A is the sodium channel protein type 9 alpha subunit (voltage-gated sodium channel Nav1.7). Nav1.7 comprises 1,977 amino acids organized into four domains, each with six transmembrane segments (S1-6), similar to members of the voltage-gated sodium and calcium ion channels [Dib-Hajj et al 2013]. The channel produces a fast inactivating sodium current that is sensitive to nanomolar concentrations of the neurotoxin tetrodotoxin (TTX-S). A number of reviews describe the molecular pathogenesis of SCN9A-related disorders [Emery et al 2015, Dib-Hajj & Waxman 2019].
The Nav1.7 sodium channel is preferentially expressed within dorsal root ganglia and sympathetic ganglion neurons. Disease-associated variants alter the biophysical properties of the channel (see Mechanism of disease causation) [Dib-Hajj et al 2013].
Mechanism of disease causation. The heterozygous SCN9A variants that cause the autosomal dominant disorders that comprise SCN9A neuropathic pain syndromes (erythromelalgia, PEPD, small fiber neuropathy) and epilepsy (see Table 2) cause disease by gain of function of the NaV1.7 sodium channel [Dib-Hajj et al 2013].
Voltage-clamp recordings have shown that these gain-of-function attributes include:
- A hyperpolarizing shift in activation of the channel (making it easier to open the channel;
- Increased amplitude of ramp current (an enhanced response to slow, small depolarizations);
- Slower deactivation (slower closing of the channel when the stimulus is removed);
- Depolarizing shifts in fast and slow inactivation (allowing more channels to be available to open).
Individual pathogenic variants may manifest one or more of these attributes, but rarely all of them.
When abnormal Nav1.7 channels are expressed in dorsal root ganglion (DRG) sensory neurons, they increase the excitability of these neurons (making it easier to fire) and increased firing frequency; these are the cellular underpinnings of the pain that patients experience.
In contrast, SCN9A variants that cause loss of function of the NaV1.7 sodium channel cause the autosomal recessive disorders HSAN2D and congenital insensitivity to pain (see Table 2) [Cox et al 2006, Yuan et al 2013].
Table 6.
Notable SCN9A Pathogenic Variants
References
Literature Cited
- Choi JS, Cheng X, Foster E, Leffler A, Tyrrell L, Te Morsche RH, Eastman EM, Jansen HJ, Huehne K, Nau C, Dib-Hajj SD, Drenth JP, Waxman SG. Alternative splicing may contribute to time-dependent manifestation of inherited erythromelalgia. Brain. 2010;133:1823–35. [PubMed: 20478850]
- Choi JS, Zhang L, Dib-Hajj SD, Han C, Tyrrell L, Lin Z, Wang X, Yang Y, Waxman SG. Mexiletine-responsive erythromelalgia due to a new Nav1.7 mutation showing use-dependent current fall-off. Exp Neurol. 2009;216:383–9. [PubMed: 19162012]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al-Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, McHale DP, Wood JN, Gribble FM, Woods CG. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444:894–8. [PMC free article: PMC7212082] [PubMed: 17167479]
- Cregg R, Cox JJ, Wood JN, Werdehausen R. Mexilitine as a treatment for primary erythromelalgia: normalization of biophysical properties of mutant L858F Nav1.7 sodium channels. Br J Pharmacol. 2014;171:4455–63. [PMC free article: PMC4209151] [PubMed: 24866741]
- de Greef BTA, Hoeijmakers JGJ, Geerts M, Oakes M, Church TJE, Waxman SG, Dib-Hajj SD, Faber CG, Merkies ISJ. Lacosamide in patients with Nav1.7 mutations-related small fibre neuropathy: a randomized controlled trial. Brain. 2019;142:263–75. [PubMed: 30649227]
- de Greef BTA, Hoeijmakers JGJ, Gorissen-Brouwers CML, Geerts M, Faber CG, Merkies ISJ. Associated conditions in small fiber neuropathy - a large cohort study and review of the literature. Eur J Neurol. 2018;25:348–55. [PMC free article: PMC5814938] [PubMed: 29112785]
- Dib-Hajj SD, Waxman SG. Sodium channels in human pain disorders: genetics and pharmacogenomics. Annu Rev Neurosci. 2019;42:87–106. [PubMed: 30702961]
- Dib-Hajj SD, Yang Y, Black JA, Waxman SG. The NaV1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62. [PubMed: 23232607]
- Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117:3603–9. [PMC free article: PMC2096434] [PubMed: 18060017]
- Eijkenboom I, Sopacua M, Hoeijmaker JGJ, De Greef BTA, Lindsey P, Almomani R, Marchi M, Vanoevelen J, Smeets HJM, Waxman SG, Lauria G, Merkies ISJ, Faber CG, Gerrits MM. Yield of peripheral sodium channels gene screening in pure small fibre neuropathy. J Neurol Neurosurg Psychiatry. 2019;90:342–52. [PubMed: 30554136]
- Emery EC, Habib AM, Cox JJ, Nicholas AK, Gribble FM, Woods CG, Reiman F. Novel SCN9A mutations underlying extreme pain phenotypes: unexpected electrophysiological and clinical correlations. J Neurosci. 2015;35:7674–81. [PMC free article: PMC4438121] [PubMed: 25995458]
- Faber CG, Hoeijmakers JGJ, Ahn H-S, Cheng X, Han C, Choi J-S, Estacion M, Lauria G, Vanhoutte EK, Gerrits MM, Dib-Hajj S, Drenth JPH, Waxman SG, Merkles IJS. Gain of function Nav1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012;71:26–39. [PubMed: 21698661]
- Fertleman CR, Ferrie CD, Aicardi J, Bednarek NA, Eeg-Olofsson O, Elmslie FV, Griesemer DA, Goutieres F, Kirkpatrick M, Malmros IN, Pollitzer M, Rossiter M, Roulet-Perez E, Schubert R, Smith VV, Testard H, Wong V, Stephenson JB. Paroxysmal extreme pain disorder (previously familial rectal pain syndrome). Neurology. 2007;69:586–95. [PubMed: 17679678]
- Fischer TZ, Gilmore ES, Estacion M, Eastman E, Taylor S, Melanson M, Dib-Hajj SD, Waxman SG. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol. 2009;65:733–41. [PMC free article: PMC4103031] [PubMed: 19557861]
- Galer BS, Jensen MP. Development and preliminary validation of a pain measure specific to neuropathic pain: the Neuropathic Pain Scale. Neurology. 1997;48:332–8. [PubMed: 9040716]
- Geha P, Yang Y, Estacion M, Schulman BR, Tokuno H, Apkarian AV, Dib-Hajj SD, Waxman SG. Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol. 2016;73:659–67. [PubMed: 27088781]
- Han C, Rush AM, Dib-Hajj SD, Li S, Xu Z, Wang Y, Tyrrell L, Wang X, Yang Y, Waxman SG. Sporadic onset of erythermalgia: A gain-of-function mutation in Na(v)1.7. Ann Neurol. 2006;59:553–8. [PubMed: 16392115]
- Huang J, Mis MA, Tanaka B, Adi T, Estacion M, Liu S, Walker S, Dib-Hajj SD, Waxman SG. Atypical changes in DRG neuron excitability and complex pain phenotype associated with a Nav1.7 mutation that massively hyperpolarizes activation. Sci Rep. 2018;8:1811. [PMC free article: PMC5788866] [PubMed: 29379075]
- McDonnell A, Schulman B, Ali Z, Dib-Hajj SD, Brock F, Cobain S, Mainka T, Vollert J, Tarabar S, Waxman SG. Inherited erythromelalgia due to mutations in SCN9A: natural history, clinical phenotype and somatosensory profile. Brain. 2016;139:1052–65. [PubMed: 26920677]
- Natkunarajah J, Atherton D, Elmslie F, Mansour S, Mortimer P. Treatment with carbamazepine and gabapentin of a patient with primary erythermalgia (erythromelalgia) identified to have a mutation in the SCN9A gene, encoding a voltage-gated sodium channel. Clin Exp Dermatol. 2009;34:e640–2. [PubMed: 19549232]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. The Human Gene Mutation Database: towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 2017;136:665–77. [PMC free article: PMC5429360] [PubMed: 28349240]
- Terkelsen AJ, Karlsson P, Lauria G, Freeman R, Finnerup NB, Jensen TS. The diagnostic challenge of small fibre neuropathy: clinical presentations, evaluations, and causes. Lancet Neurol. 2017;16:934–44. [PubMed: 29029847]
- Tham SW, Giles M. Current pain management strategies for patients with erythromelalgia: a critical review. J Pain Res. 2018;11:1689–98. [PMC free article: PMC6121769] [PubMed: 30214279]
- Yang Y, Adi T, Effraim P, Chen L, Dib-Hajj SD, Waxman SG. Reverse pharmacogenomics: carbamazepine normalizes activation and attenuates thermal-induced hyperexcitability of sensory neurons due to Nav1.7 mutation I234T. Br J Pharmacol. 2018;175:2261–71. [PMC free article: PMC5980548] [PubMed: 28658526]
- Yuan J, Matsuura E, Higuchi Y, Hashiguchi A, Nakamura T, Nozuma S, Sakiyama Y, Yoshimura A, Izumo S, Takashima H. Hereditary sensory and autonomic neuropathy type IID caused by an SCN9A mutation. Neurology. 2013;80:1641–9. [PubMed: 23596073]
Chapter Notes
Acknowledgments
This work is supported in part by a gift from the Erythromelalgia Association.
Revision History
- 23 January 2020 (bp) Comprehensive update posted live
- 15 August 2013 (me) Comprehensive update posted live
- 25 September 2008 (cd) Revision: prenatal testing available
- 26 August 2008 (cg) Comprehensive update posted live
- 28 March 2007 (fmh) Revision: sequence analysis for SCN9A clinically available
- 18 January 2007 (cd) Revision: mutations in SCN9A resulting in loss of function of sodium channel protein type 9 subunit alpha cause channelopathy-associated insensitivity to pain.
- 5 May 2006 (me) Review posted live
- 2 August 2005 (fmh) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: May 6, 2006; Last Update: January 23, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Hisama FM, Dib-Hajj SD, Waxman SG. SCN9A Neuropathic Pain Syndromes. 2006 May 6 [Updated 2020 Jan 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
