Summary
Clinical characteristics.
Mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS) is part of a continuum of progressive neurodegenerative disorders caused by abnormalities of mitochondrial energy generation, which includes the overlapping phenotypes mtDNA-associated Leigh syndrome and mtDNA-associated Leigh-like syndrome.
Mitochondrial DNA-LSS is characterized by onset of manifestations typically between ages three and 12 months, often following an intercurrent illness (usually viral) or metabolic challenge (vaccinations, surgery, prolonged fasting). Decompensation (often with elevated lactate levels in blood and/or cerebrospinal fluid) is typically associated with developmental delay and/or regression. Neurologic features include hypotonia, spasticity, seizures, movement disorders, cerebellar ataxia, and peripheral neuropathy. Brain stem dysfunction may manifest with respiratory symptoms, swallowing difficulties, ophthalmoparesis, and abnormalities in thermoregulation. Extraneurologic manifestations may include poor weight gain, cardiomyopathy, and conduction defects. Up to 50% of individuals die by age three years, most often from respiratory or cardiac failure.
Diagnosis/testing.
The diagnosis of mtDNA-LSS is established in a proband fulfilling clinical diagnostic criteria for LSS by identification of a heteroplasmic or homoplasmic pathogenic variant in one of the 15 mtDNA genes known to be involved in mtDNA-LSS.
Management.
Treatment of manifestations: Treatment is supportive. Sodium bicarbonate or sodium citrate for significant acidosis (THAM may be used if there is hypernatraemia); anti-seizure medication for seizures; dystonia therapy with benzhexol, baclofen, tetrabenazine, or gabapentin alone or in combination, or by botulinum toxin injection; treatment of respiratory compromise per pulmonologist; caloric and nutritional supplementation and feeding therapy as needed; developmental and educational support; physical therapy and occupational therapy; standard treatment of eye movement disorders; medical management of cardiomyopathy; treatment of constipation as needed; treatment of liver failure per hepatologist; treatment of electrolyte abnormalities per nephrologist; hearing aids or cochlear implants for sensorineural hearing loss; speech therapy and hearing support services as needed; management of diabetes mellitus and adrenal insufficiency per endocrinologist; standard treatments for anxiety and/or depression; psychological support and care coordination for the affected individual and family.
Surveillance: Affected individuals should be followed at regular intervals to monitor for progression of disease and associated complications. Neurologic assessment for ataxia and seizures at each visit along with an assessment of pulmonary issues, growth, nutrition, and gastrointestinal manifestations. Development, educational, and cognitive assessment and assessment of mobility and self-help skills at least annually. Ophthalmology assessment every six to 12 months or as advised by ophthalmologist. Annual blood pressure, EKG, and echocardiogram or as advised by cardiologist. Liver function tests, urinalysis, urine albumin-to-creatinine ratio, urine amino acids, serum electrolytes, blood urea nitrogen, creatinine, complete blood count, and fasting glucose annually. Annual audiology assessment. Assessment of care coordination and family psychosocial needs at each visit.
Agents/circumstances to avoid: Sodium valproate, medications that cause acidosis, and dichloroacetate should be avoided or used with caution; administration of anesthesia requires careful consideration to avoid aggravation of respiratory symptoms and precipitation of respiratory failure.
Genetic counseling.
Mitochondrial DNA-LSS is transmitted by maternal inheritance. The mother of a proband may have the mtDNA pathogenic variant and may exhibit mild clinical manifestations of mtDNA-LSS. Many affected individuals have no known family history of mtDNA-LSS or other mitochondrial disorder. The explanation for apparently simplex cases may be absence of a comprehensive and/or reliable family history, a maternal heteroplasmy level below the disease threshold (i.e., the minimum level of heteroplasmy expected to result in mitochondrial disease), or a de novo mtDNA pathogenic variant in the proband. If the mother of the proband has the mtDNA pathogenic variant identified in the proband, all sibs of the proband are at risk of inheriting the pathogenic variant. Sibs may inherit the pathogenic variant at varying heteroplasmy levels due to the bottleneck effect and variant-specific segregation patterns. The risk to a sib of developing clinical manifestations is difficult to determine and depends on heteroplasmy level, the variation in heteroplasmy levels among different tissues, and the disease threshold for the specific variant. Recurrence risk assessment and prenatal testing for disorders caused by pathogenic variants in mtDNA is challenging due to the intricacies of mtDNA transmission and the inherent challenge in using prenatal genetic test results to predict clinical outcome. Reproductive options for the family members of a proband with an mtDNA pathogenic variant may include prenatal testing, preimplantation genetic testing, and oocyte donation.
Diagnosis
Diagnostic criteria for Leigh syndrome spectrum (LSS) have been published [McCormick et al 2023].
Suggestive Findings
Mitochondrial DNA (mtDNA)-associated LSS (mtDNA-LSS) should be suspected in probands with several of the following clinical, laboratory, and/or imaging findings.
Clinical findings
- Progressive neurologic disease with developmental delay and neurodevelopmental regression
- Manifestations of brain stem and/or basal ganglia disease (e.g., respiratory abnormalities, nystagmus, ophthalmoparesis, optic atrophy, ataxia, and dystonia)
- Seizures
- Psychiatric disturbance
Laboratory findings
- Elevated blood lactateElevation tends to be more marked in postprandial samples.
- Elevated cerebrospinal fluid (CSF) lactateNote: While elevated blood and/or CSF lactate is suggestive of a mitochondrial disorder, a normal lactate does not exclude mtDNA-LSS.
- Plasma amino acids may show increased alanine concentration, reflecting persistent hyperlactatemia. Decreased plasma citrulline concentration and/or elevated 3-hydroxy-isovalerylcarnitine (C5-OH) levels has been reported in individuals with MT-ATP6 pathogenic variants [Balasubramaniam et al 2017, Larson et al 2019, Peretz et al 2021,Tise et al 2023].
- Urine organic acid analysis results are often nonspecific and may show lactic aciduria, increased Krebs cycle intermediates, and increased dicarboxylic acids.Note: 3-methylglutaconic aciduria may be observed in some nuclear gene-encoded causes of LSS, notably 3-methylglutaconic aciduria with deafness-dystonia, [hepatopathy], encephalomyopathy, and Leigh-like syndrome (MEGD[H]EL syndrome caused by SERAC1 deficiency), and ethylmalonic aciduria is a feature of ethylmalonic encephalopathy caused by biallelic variants in ETHE1. Identification of increased methylmalonic acid or propionic acid is suggestive of other genetic causes of LSS or organic acidemias (e.g., SUCLG1- or SUCLA2-related mtDNA depletion syndrome, methylmalonic acidemia, propionic acidemia) (see Causes of Nuclear Gene-Encoded Leigh Syndrome Spectrum and Differential Diagnosis).
Brain imaging findings
- Characteristic findings include bilateral symmetric hyperintense signal abnormality in the brain stem and/or basal ganglia on T2-weighted MRI or bilateral symmetric hypodensities in the basal ganglia on CT that may progress/fluctuate with time [Bonfante et al 2016, Alves et al 2020]. Corresponding restricted diffusion may be evident on diffusion-weighted imaging [Alves et al 2020].
- Additional findings can include cerebral atrophy, bilateral hyperintense signal abnormality in the thalamus, cerebellum, cerebral white matter, and/or spinal cord on T2-weighted MRI [Bonfante et al 2016, Alves et al 2020, Hong et al 2020, Stenton et al 2022].
- Magnetic resonance spectroscopy (MRS) lactate peak (in the absence of acute seizures)Note: Characteristic patterns of brain lesions have been described for specific causes of nuclear gene-encoded LSS. For example, specific brain lesions affecting the mammillothalamic tracts, substantia nigra, medial lemniscus, medial longitudinal fasciculus, spinothalamic tracts, and cerebellum appear to be characteristic of NDUFAF2-related LSS. SERAC1 deficiency is associated with a distinctive brain MRI pattern affecting the basal ganglia, especially the putamen. Initially there are T2-weighted signal changes of the pallidum, and later swelling of the putamen and caudate nucleus with an "eye" representing early sparing of the dorsal putamen, followed by progressive involvement of the putamina (see Causes of Nuclear Gene-Encoded Leigh Syndrome Spectrum).
Histopathology of muscle tissue may be normal or may show nonspecific changes such as accumulation of intracytoplasmic neutral lipid droplets or predominance of type II muscle fibers. Ragged red fibers and/or cytochrome c oxidase-negative fibers may occasionally be observed.
Since the advent of next-generation sequencing, muscle biopsy is performed less frequently (especially in the pediatric population), as obtaining a muscle biopsy is invasive and often requires general anesthesia, which carries a risk of metabolic decompensation. Muscle biopsy is now generally reserved for critically unwell individuals or (occasionally) for functional validation of genetic findings.
Note: (1) Although muscle histopathology is only occasionally abnormal, when it is abnormal it can be as much of a contributor to diagnostic certainty as respiratory chain enzymes or molecular testing. (2) If muscle biopsy is obtained for respiratory chain enzyme studies, histopathology should also be performed.
Respiratory chain enzyme studies. Biochemical analysis of tissue biopsies or cultured cells often detects deficient activity (<30% of control mean values) of one or more of the respiratory chain enzyme complexes. Isolated defects of complex I or complex IV are the most common enzyme abnormalities observed in individuals with LSS and can help guide subsequent molecular genetic testing of mtDNA or nuclear genes [Sofou et al 2018, Hong et al 2020, Ogawa et al 2020, Ardissone et al 2021]. Combined respiratory chain enzyme defects can also be observed, particularly in disorders of mitochondrial gene expression. Normal respiratory chain enzyme analysis does not exclude mtDNA-LSS and can be seen in individuals with mtDNA pathogenic variants affecting complex V subunits such as the MT-ATP6 pathogenic variants m.8993T>G, m.8993T>C, and m.9176T>C.
Note: (1) Skeletal muscle (vastus lateralis) is usually the tissue of choice for respiratory chain enzyme studies. (2) Skin fibroblasts can be used, but only about 50% of respiratory chain enzyme defects identified in skeletal muscle are also identified in skin fibroblasts. (3) Approximately 10%-20% of individuals with normal skeletal muscle respiratory chain enzyme analysis may have an enzyme defect detected in liver or cardiac muscle, particularly if those tissues are involved clinically [Frazier et al 2020].
Establishing the Diagnosis
The diagnosis of mtDNA-LSS is established in a proband fulfilling criteria for LSS [McCormick et al 2023] in whom a heteroplasmic or homoplasmic pathogenic variant in one of the genes listed in Table 1 has been identified.
Clinical Criteria for mtDNA-LSS
Typical neuroradiologic findings. Bilateral, typically symmetric hyperintensities on T2-weighted MRI or hypodensities on CT in the brain stem and/or basal ganglia with or without bilateral, T2-weighted hyperintensities on MRI or hypodensities on CT in the thalamus, cerebellum, subcortical white matter, and/or spinal cord
AND ≥1 of the following characteristic neurologic clinical features:
- Developmental regression
- Developmental delay
- Psychiatric features
AND ≥1 of the following biochemical and/or mitochondrial abnormalities:
- Elevated lactate in plasma and/or CSF
- MRS lactate peak (in absence of acute seizures)
- Respiratory chain enzyme activity deficiency (<30% enzyme activity) in affected tissues (muscle, liver, fibroblasts)
Note: Historically, the diagnosis of LSS was achieved by demonstrating mitochondrial dysfunction through relevant tissue biopsy to inform a targeted molecular genetic approach. However, the introduction of comprehensive genomic sequencing has removed the need for tissue biopsy in most scenarios.
Molecular Genetic Testing
Molecular genetic testing approaches for mtDNA-LSS can include comprehensive genomic testing (genome sequencing or mtDNA sequencing and exome sequencing) or targeted single-gene testing. Choice of approach may be dependent on clinical suspicion, age of presentation, and availability of genomic testing.
Option 1
Comprehensive genomic testing includes sequencing of mtDNA and nuclear DNA. Comprehensive genomic testing does not require a phenotype-driven hypothesis and may provide or suggest a diagnosis not previously considered (e.g., variant in a different gene[s] that results in a similar clinical presentation).
- Genome sequencing is preferred (if available), as it allows analysis of the mitochondrial and nuclear genome, and typically has the highest diagnostic rate.
- If genome sequencing is not available, mtDNA sequencing is recommended. Exome sequencing should be considered in conjunction with mtDNA sequencing, especially in the pediatric population, given that approximately 70% of individuals will have nuclear gene-encoded LSS (see Differential Diagnosis). For children, a parent-child trio should be considered to assist in the identification of rare autosomal recessive or de novo pathogenic variants. It may be possible to extract mtDNA sequence data from some exome kits, but coverage varies and may not be of clinical quality, hence stand-alone mtDNA sequencing is recommended.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Option 2
Targeted analysis of common pathogenic variants may be considered if there is a high clinical suspicion based on clinical or biochemical findings or if comprehensive testing is not available (see Table 1).
Note: (1) Most mtDNA pathogenic variants associated with mtDNA-LSS are "heteroplasmic" (i.e., abnormal mtDNA coexists with wild type mtDNA) and for some pathogenic variants, the heteroplasmy level may vary among different tissues and may increase or decrease with age. (2) Mitochondrial DNA pathogenic variants may be lost from the leukocyte population with increasing age [Schon et al 2021, Davis et al 2022]; therefore, in adults with milder manifestations and asymptomatic older maternal relatives, the pathogenic variant may only be detected in hair follicles, urine sediment cells, or skeletal muscle, which is the most reliable source of mtDNA [Davis et al 2022]. (3) Leukocyte testing is recommended in children, particularly when using next-generation sequencing methods, which allow detection of very low heteroplasmy levels. (4) Long-range PCR can be performed to identify large mtDNA rearrangements. (5) Mitochondrial DNA deletions are not usually detectable in leukocyte DNA in adults; in this age group, skeletal muscle (or urinary sediment cells) is the tissue of choice for analysis [Schon et al 2021, Davis et al 2022]. (6) Deletions/duplications of mtDNA are an extremely rare cause of LSS in adults. (7) The authors are not aware of any published reports of children with mtDNA-LSS when the pathogenic variant was present in another tissue but not detected in blood.
Table 1.
Molecular Genetics of Mitochondrial DNA-Associated Leigh Syndrome Spectrum
Clinical Characteristics
Clinical Description
Mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS) is a continuum of progressive neurodegenerative disorders, with typical onset in infancy or early childhood of sudden neurodevelopmental regression. Individuals with LSS may have variable extraneurologic manifestations including gastrointestinal, cardiac, hepatic, renal, and endocrine disease. The clinical features of mtDNA-LSS are similar to those observed in individuals with nuclear gene-encoded Leigh syndrome spectrum (LSS). Clinically, it is difficult to distinguish mtDNA-LSS from nuclear gene-encoded LSS, and there are relatively few publications describing the full range of clinical features in mtDNA-LSS. The following description of the phenotypic features summarizes the more robust data on features of all individuals with LSS; specific information and references for mtDNA-LSS are included if available.
Table 2.
Mitochondrial DNA-Associated Leigh Syndrome Spectrum: Frequency of Select Features
Onset of symptoms is typically between age three and 12 months, often following an intercurrent illness (usually viral) or metabolic challenge (vaccinations, surgery, prolonged fasting). However, disease onset can vary, ranging from prenatal onset to adulthood. Early onset of symptoms within the first 24 months of life has been reported in approximately 75% of individuals with LSS. Later onset (age >2 years) includes presentation in childhood, adolescence, or adulthood and is generally associated with slower progression of disease.
Initial features may be nonspecific and vary with age of onset:
- Prenatally, intrauterine growth restriction, cardiomegaly, and oligohydramnios have been reported in LSS [Sofou et al 2014, Hong et al 2020, Stendel et al 2020, Ardissone et al 2021].
- Early-onset LSS (age <2 years) may present with nonspecific features such as feeding difficulties, poor weight gain, hypotonia, developmental delay, and persistent vomiting.
- Late-onset LSS (age >2 years) may manifest with predominant muscle weakness and movement disorders including ataxia and dystonia.
- Onset in adulthood may manifest with ataxia, peripheral neuropathy, and psychiatric disturbance [Wei et al 2018, Hong et al 2020].
Decompensation (often with increased blood and/or cerebrospinal fluid [CSF] lactate concentrations) during an intercurrent illness is typically associated with sudden loss of developmental skills. A period of recovery may follow the initial decompensation, but the individual rarely returns to the developmental status achieved prior to the presenting illness. Most individuals with LSS will experience at least one acute decompensation that may require hospitalization.
Neurologic features include developmental delay/regression, hypotonia, spasticity, dystonia, muscle weakness, hypo- or hyperreflexia, seizures (myoclonic, generalized tonic-clonic, infantile spasms), movement disorders (including chorea), cerebellar ataxia, and peripheral neuropathy. Adolescents and adults with LSS may exhibit psychiatric disturbance.
Brain stem lesions may cause respiratory difficulty (apnea, central hypo- or hyperventilation, or irregular respiration), bulbar problems (e.g., abnormal swallowing and speech), persistent vomiting, and abnormalities of thermoregulation (hypo- and hyperthermia). Sensorineural hearing loss may be present in approximately 15% of individuals, originating from the cochlea or auditory nerves.
Ophthalmologic findings include optic atrophy, retinitis pigmentosa, strabismus, ptosis, nystagmus, and ophthalmoparesis. Pigmentary retinopathy was reported in 12 out of 13 individuals with an MT-ATP6 m.8993T>G pathogenic variant in one retrospective cohort [Ng et al 2019].
Gastrointestinal manifestations include poor weight gain, vomiting, dysphagia, gastroparesis, and intestinal dysmotility. One natural history study suggested that gastrointestinal symptoms were the most common extraneurologic manifestation in individuals with LSS, with 25% exhibiting gut dysmotility and nearly half of the cohort requiring gastrostomy or nasogastric tube feeding at follow up [Lim et al 2022].
Cardiac manifestations including hypertrophic or dilated cardiomyopathy and conduction defects (e.g., Wolff-Parkinson-White syndrome and paroxysmal supraventricular tachycardia) are reported in approximately 15% of individuals with LSS. Cardiomyopathy has been reported in up to 70% of individuals with an MT-ND5 pathogenic variant [Stenton et al 2022, Kistol et al 2023].
Hepatic manifestations including elevated liver transaminases, hepatomegaly, or liver failure have been reported in approximately 10% of individuals with LSS [Naess et al 2009, Van Hove et al 2010, Sofou et al 2014, Duff et al 2015, Alves et al 2020].
Renal manifestations including renal tubulopathy or diffuse glomerulocystic kidney damage have been reported in approximately 5% of individuals with LSS [López et al 2006, Naess et al 2009, Sofou et al 2018, Alves et al 2020, Hong et al 2020, Stenton et al 2022].
Endocrine manifestations including diabetes mellitus and adrenal insufficiency have been reported in a small proportion of individuals with LSS [Alves et al 2020, Ardissone et al 2023].
Prognosis in individuals with LSS is generally poor. Most affected individuals experience episodic deterioration of motor and/or cognitive function interspersed with plateaus in development. Up to 50% of individuals with LSS will die before age three years, most often from respiratory or cardiac failure, neurologic deterioration, or sepsis. However, disease progression can be variable. Some individuals have slower progression of disease, with stable periods lasting years in between decompensations, even into adulthood. In previously undiagnosed individuals, death may appear to be sudden and unexpected.
Poor prognosis has been associated with early-onset disease [Sofou et al 2014, Lee et al 2016, Hong et al 2020, Ogawa et al 2020]. Individuals with pathogenic variants in MT-ND5 [Ogawa et al 2020] and those with MT-ATP6 pathogenic variant m.8993T>G [Stendel et al 2020, Na & Lee 2022] have been reported to be more likely to have a severe disease phenotype.
Intermediate phenotypes. Maternal relatives of individuals with mtDNA-LSS can have any one or a combination of clinical manifestations of mtDNA-LSS, NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa), or other mitochondrial disorders. These include mild learning difficulties, ataxia, muscle weakness, night blindness, deafness, diabetes mellitus, migraine, or sudden unexpected death (see Genetically Related Disorders).
Genotype-Phenotype Correlations
For most mtDNA pathogenic variants, it is difficult to distinguish a correlation between genotype and phenotype because clinical expression of mtDNA pathogenic variants is influenced by pathogenicity of the variant, relative amount of abnormal and wild type mtDNA (heteroplasmy level), the variation in heteroplasmy level in different tissues, and the energy requirements of brain and other tissues, which may vary with age. Intrafamilial and interfamilial clinical variability are seen in individuals with the same genotype [Stendel et al 2020].
Several studies have tried to describe genotype-phenotype correlations in mtDNA-LSS; however, these are generally inconsistent between cohorts, limiting their translation into clinical practice.
MT-ATP6 pathogenic variants, specifically m.8993T>G and m.8993T>C, probably show the strongest genotype-phenotype correlation of any mtDNA pathogenic variants. They show very little tissue-dependent [Ng et al 2019] or age-dependent variation in the proportion of abnormal mtDNA [White et al 1999b] as well as a strong correlation between the heteroplasmy level and disease severity. Logistic regression models were developed that predict the probability of a severe outcome in an individual based on the measured heteroplasmy level of m.8993T>G and m.8993T>C (see Figure 1) [White et al 1999a]. Across all MT-ATP6 variants, heteroplasmy levels have been shown to have a significant negative association with age of onset of disease [Ganetzky et al 2019]. These findings are supported by Ng et al [2019], who generated logistic regression models to predict the risk of disease manifestation based on the blood heteroplasmy level of MT-ATP6 pathogenic variants m.8993T>C, m.8993T>G, m.9176T>C, and m.9185T>C (see Figure 2). Note, however, that in such retrospective studies it is not possible to completely avoid ascertainment bias, and the data should be regarded as broadly indicative rather than precise.
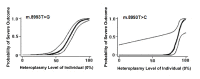

MT-ATP6 pathogenic variants m.8993T>G and m.8993T>C tend to be associated with early-onset and severe clinical disease, with m.8993T>G causing a more severe phenotype [Sofou et al 2018, Ganetzky et al 2019, Stendel et al 2020]. Although the cause of this difference is unknown, it is postulated to result from the instability of the abnormal protein.
- m.8993T>G. Individuals with a heteroplasmy level below 60% are usually asymptomatic or have only mild pigmentary retinopathy or migraine headaches; however, asymptomatic adults with up to 75% abnormal mtDNA have been reported [Ganetzky et al 2019, Ng et al 2019, Stendel et al 2020]. Individuals with moderate heteroplasmy levels (70%-90%) of pathogenic variant m.8993T>G may present with the milder phenotype of NARP, while those with more than 90% abnormal mtDNA typically present with LSS [Ng et al 2019, Stendel et al 2020, Stenton et al 2022].
- m.8993T>C is a less severe variant than m.8993T>G, and generally all symptomatic individuals with m.8993T>C have a heteroplasmy level greater than 90%. However, some asymptomatic individuals with m.8993T>C have also been reported to have a heteroplasmy level greater than 90% [Stendel et al 2020, Stenton et al 2022].Note: Overlap in MT-ATP6 pathogenic variant heteroplasmy level is observed between some asymptomatic individuals and others with NARP or mild neurologic manifestations, particularly for variants other than m.8993T>G. There can also be overlap in heteroplasmy levels between some individuals with NARP and others with LSS [Claeys et al 2016, Ganetzky et al 2019, Ng et al 2019, Stendel et al 2020].
Genotype-phenotype correlations are much weaker for other mtDNA pathogenic variants (e.g., m.3243A>G in MT-TL1, m.8344A>G in MT-TK, m.9176T>C in MT-ATP6, m.14459G>A and m.14487T>C in MT-ND6, m.10158T>C and m.10191T>C in MT-ND3, and m.13513G>A in MT-ND5). It is not possible to use heteroplasmy level to predict outcome (e.g., in asymptomatic family members or in prenatal diagnosis) unless the value is near 0% or near 100%. Some individuals with MT-ND3 and MT-ND5 pathogenic variants have been reported to have relatively severe phenotypes at low heteroplasmy levels [Wei et al 2018, Ogawa et al 2020, Stenton et al 2022, Na & Lee 2023].
Other genotype-phenotype correlations have been suggested for mtDNA pathogenic variants affecting respiratory chain enzyme complex I, although confirmation of these findings requires further systematic analysis of larger cohorts. Individuals with complex I deficiency are more commonly reported to have cardiac and visual involvement [Sofou et al 2018], with pathogenic variants in MT-ND5 strongly associated with cardiomyopathy [Stenton et al 2022, Kistol et al 2023]. A retrospective study of neuroimaging in children with mtDNA-LSS suggested that cerebral cortical and white matter involvement is more common in MT-ND5- and MT-ND3-related LSS [Alves et al 2020]; however, this finding was not supported by a subsequent study [Stenton et al 2022]. Poor prognosis has been associated with MT-ND5 pathogenic variants in addition to MT-ATP6 pathogenic variant m.8993T>G [Ogawa et al 2020, Stenton et al 2022].
Individuals with mtDNA-LSS may exhibit overlapping phenotypes with other mitochondrial phenotypes, including MELAS. MT-ND pathogenic variants (most commonly involving MT-ND5 and MT-ND3) have been described in multiple individuals with an LSS/MELAS overlapping phenotype in which individuals display clinical and radiologic features of both syndromes [Wei et al 2021].
Penetrance
Penetrance is reduced (see Genotype-Phenotype Correlations).
Nomenclature
Leigh syndrome was originally described in an affected infant as "subacute necrotizing encephalomyelopathy" [Leigh 1951]. Leigh syndrome was historically defined by characteristic neuropathologic features including multiple focal symmetric necrotic lesions in the basal ganglia, thalamus, brain stem, dentate nuclei, and optic nerves that histologically demonstrate a spongiform appearance and are characterized by demyelination, gliosis, and vascular proliferation. The advent of MRI and next-generation sequencing has made it possible to establish the diagnosis without postmortem examination. Individuals with mtDNA-LSS are often referred to as having "maternally inherited Leigh syndrome" (MILS) [Ciafaloni et al 1993]. This term can cause confusion given perhaps one quarter of probands with mtDNA-LSS have de novo pathogenic variants (see Genetic Counseling).
The term Leigh-like syndrome was used previously when clinical and other features were strongly suggestive of Leigh syndrome but did not fulfill the stringent diagnostic criteria because of atypical neuropathology (i.e., variation in the distribution or character of lesions or the presence of unusual features such as extensive cortical destruction), neuroimaging inconsistent with Leigh syndrome, normal blood and CSF lactate levels, and/or incomplete evaluation.
Leigh syndrome spectrum encompasses both Leigh syndrome and Leigh-like syndrome. Leigh syndrome and Leigh-like syndrome represent a continuum of overlapping clinical and biochemical features and result from pathogenic variants in the same group of mitochondrial and nuclear genes.
Prevalence
LSS is the most frequently observed phenotype of pediatric-onset mitochondrial disorders. Mitochondrial DNA-associated LSS accounts for approximately 30% of LSS.
The following prevalence data are for all genetic causes of LSS. In southeastern Australia, Leigh syndrome occurs in 1:77,000 infants, and the combined birth prevalence of LSS was estimated to be at least 1:40,000 [Rahman et al 1996]. In western Sweden, the prevalence of Leigh syndrome in preschool children was 1:34,000 [Darin et al 2001]. Thus, the prevalence of LSS is likely to be 1:30,000-40,000.
Analyses of 67 individuals with LSS reported by Rahman et al [1996] identified mtDNA pathogenic variants in 34% [Authors, unpublished data]. Hence, the prevalence of mtDNA-LSS is likely to be 1:100,000-140,000.
Genetically Related Disorders
Pathogenic variants in mtDNA genes known to be associated with mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS) can also be associated with a variety of other mitochondrial disorders. For example, the pathogenic variants most often identified in individuals with Leber hereditary optic neuropathy (m.11778A>G in MT-ND4 and m.14484T>C in MT-ND6) have also been reported in individuals with mtDNA-LSS [Lim et al 2022, Stenton et al 2022]. See Table 3 and Primary Mitochondrial Disorders Overview.
Table 3.
Selected Allelic Disorders
Differential Diagnosis
Leigh syndrome spectrum (LSS) is most often caused by a pathogenic variant in a nuclear gene. Pathogenic variants in 98 nuclear genes have been associated with autosomal recessive, autosomal dominant, and X-linked nuclear gene-encoded LSS [McCormick et al 2023] (see Causes of Nuclear Gene-Encoded Leigh Syndrome Spectrum).
Other monogenic disorders that cause or resemble LSS are listed in Table 4.
Table 4.
Other Monogenic Disorders That Cause or Resemble Leigh Syndrome Spectrum
Acquired conditions that cause or resemble LSS with similar clinical features and/or similar neuroimaging findings include the following:
- Acute necrotizing encephalopathy (may be triggered by viral infections)
- Viral encephalopathies
- Hypoxic-ischemic encephalopathy
- Wernicke encephalopathy (thiamine deficiency)
Management
Evaluation Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS), the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Mitochondrial DNA-Associated Leigh Syndrome Spectrum: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
As for most mitochondrial disorders, no specific curative treatment for mtDNA-LSS exists [Pitceathly et al 2021], and currently there are no licensed or approved treatments for mtDNA-LSS. Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 6).
Table 6.
Mitochondrial DNA-Associated Leigh Syndrome Spectrum: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 7 are recommended.
Table 7.
Mitochondrial DNA-Associated Leigh Syndrome Spectrum: Recommended Surveillance
Agents/Circumstances to Avoid
An international Delphi-based consensus on the safety of medication use in individuals with primary mitochondrial disorders was published in 2020 to assist clinical decision making. However, it is important to tailor medication recommendations to the individual [De Vries et al 2020].
Sodium valproate should be avoided if possible, because of its inhibitory effect on respiratory chain enzymes.
Medications that can cause acidosis should be avoided or, if necessary, prescribed with caution, with frequent monitoring of blood acid-base levels.
Anesthesia can potentially aggravate respiratory symptoms and precipitate respiratory failure, so careful consideration should be given to its use and to monitoring of the individual prior to, during, and after anesthetic procedures [Parikh et al 2017]. Catabolism should be prevented by minimizing preoperative fasting and considering intravenous glucose perioperatively during prolonged anesthesia (unless the individual is on a ketogenic diet). Prolonged propofol use during maintenance anesthesia may increase the risk of lactic acidosis. Neuromuscular blocking drugs should be avoided in individuals with muscle disease, or if necessary, under strict monitoring. Avoid lactate-containing agents (e.g., Ringer lactate) in those with risk of lactic acidosis [Parikh et al 2017, De Vries et al 2020].
Dichloroacetate (DCA) reduces blood lactate by activating the pyruvate dehydrogenase complex. Mixed clinical outcomes have been observed following the use of DCA in individuals with mitochondrial disorders. DCA was reported to worsen peripheral neuropathy in a randomized clinical trial in individuals with MELAS [Kaufmann et al 2006]. It is therefore recommended that DCA should be used with caution in individuals with LSS, particularly in individuals with established peripheral neuropathy or with a genetic cause associated with increased risk of peripheral neuropathy.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
A number of vitamins, cofactors, and other compounds, including riboflavin, thiamine, creatine, biotin, coenzyme Q10, carnitine, and alpha-lipoic acid, have been prescribed in individuals with mtDNA-LSS. To date, there is no consistency in prescribing practice between international centers. Despite the theoretic potential, there is no established evidence base for any of these compounds in mtDNA-LSS [Pfeffer et al 2012b]. Complex phenotypic and genetic heterogeneity, as well as a lack of robust clinical outcome measures, are some of the obstacles to developing effective therapies for mtDNA-LSS. Evidence for some of these therapies has been shown in specific vitamin/cofactor metabolism and transporter defects in nuclear gene-encoded Leigh syndrome spectrum. In recent years, there has been an increase in clinical trials for pharmacologic and non-pharmacologic therapies for mtDNA-LSS.
Antioxidants such as coenzyme Q10 and its analogs have demonstrated potential antioxidant activities against toxic metabolites and accumulated reactive oxygen species. Apart from disorders of coenzyme Q10 biosynthesis, coenzyme Q10 has limited evidence for efficacy in the treatment of LSS.
Vatiquinone (PTC-743; previously known as EPI-743) is a structurally modified variant of coenzyme Q10 that may help to replenish intracellular glutathione levels [Shrader et al 2011]. An open-label clinical trial in an end-of-life setting in 14 individuals with primary mitochondrial disorders (including mtDNA-LSS) showed clinical improvement in 11 of the 14 individuals, which corresponded to increased regional and whole-brain technetium-99m hexamethylpropylene oxime uptake on single-photon emission computed tomography [Enns et al 2012]. An open-label Phase IIA single-center clinical trial of vatiquinone therapy showed stabilization of disease progression in ten children with mtDNA-LSS or nuclear gene-encoded LSS [Martinelli et al 2012]. The unpredictable natural history of mtDNA-LSS causes difficulty in interpretation of open-label studies. A Phase IIB randomized placebo-controlled double-blind crossover clinical trial of vatiquinone in children with mtDNA-LSS or nuclear gene-encoded LSS was completed in May 2015 (NCT01721733), and a long-term safety and efficacy evaluation of vatiquinone in children with mtDNA-LSS or nuclear gene-encoded LSS was completed in late 2023 (NCT02352896). The findings of these studies have not yet been published. A recent clinical trial of vatiquinone in individuals with mitochondrial disorders with associated epilepsy phenotype (NCT04378075) failed to meet its primary end point (see PTC Therapeutics press release).
KH176 (sonlicromanol) is a redox-modulating agent that targets the thioredoxin/peroxiredoxin system to reduce cellular reactive oxygen species. Promising preclinical studies in a mouse model for LSS [de Haas et al 2017] led to a Phase I study, which reported that KH176 was tolerated but at higher doses was associated with changes in cardiac electrophysiology (QTc prolongation and T wave morphologic changes) [Koene et al 2017]. A Phase II randomized, double-blind, placebo-controlled, parallel group clinical trial in children with genetically confirmed primary mitochondrial disorders with motor symptoms is currently recruiting (NCT04846036).
Gene therapy. Gene-editing techniques with mitochondrial targeted restriction endonucleases (mitoREs) or programmable nucleases, such as mitochondrial targeted zinc finger nucleases (mtZFN) and transcription activator-like effector nucleases (mito-TALEN), are postulated to shift heteroplasmy toward wild type mtDNA by inducing mtDNA double-stranded breaks, leading to rapid degradation of mutated mtDNA. Gene-editing techniques that introduce specific de novo nucleotide changes in mtDNA include bacterial cytidine deaminases. However, to date, gene therapy has not progressed to clinical trials in humans.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Other
Ketogenic diet (KD) has been proposed as a therapy for mitochondrial disorders, in particular complex I deficiency [Paleologou et al 2017]. Although this high-fat, low-carbohydrate diet has proven efficacy for refractory epilepsies, high-quality evidence for its use in the treatment of primary mitochondrial disorders, including mtDNA-LSS, is currently lacking. Studies in patient-derived fibroblasts have postulated that KD may be beneficial in mitochondrial disorders [Santra et al 2004]. A systematic review on the use of KD in mitochondrial disorders highlighted the paucity of high-quality studies but suggested that KD can be considered in individuals with mitochondrial disorders (including mtDNA-LSS) and therapy-refractory epilepsy (except mtDNA deletion-related myopathy and pyruvate carboxylase deficiency) [Zweers et al 2021]. A prospective controlled study in individuals with mitochondrial disorders (including one individual with LSS) found that KD reduced seizure frequency in 76%, with a high efficacy in individuals with MELAS [Huang et al 2022]. Further clinical trials are required to determine the efficacy of KD in mtDNA-LSS. Another proposed option is supplementation with the fatty acid decanoic acid, which is thought to be the active component of the KD and appears to stimulate mitochondrial biogenesis in cell models [Hughes et al 2014], including fibroblasts from individuals with complex I-deficient nuclear gene-encoded LSS [Kanabus et al 2016]. A prospective open-label feasibility study of K.Vita, a medical food containing a unique ratio of decanoic acid to octanoic acid, had a beneficial effect on frequency of seizures, including in individuals with primary mitochondrial disorders [Schoeler et al 2021]. Further clinical trials are needed.
Mitochondrial biogenesis is the process in which cells increase the mitochondrial mass. Several studies have investigated whether upregulation of mitochondrial biogenesis may provide an effective therapeutic approach for mitochondrial respiratory chain disorders. This approach involves using agonists such as bezafibrate or resveratrol to stimulate the peroxisome proliferator-activated receptor gamma (PPARgamma) coactivator alpha (PGC-1a) pathway [Bastin et al 2008, Steele et al 2020]. Alternatively, nicotinamide analogs such as nicotinamide riboside or nicotinamide mononucleotide have been used to boost reduced nicotinamide adenine dinucleotide (NAD+) levels and induce mitochondrial biogenesis via the same PGC-1a pathway [Cerutti et al 2014, Khan et al 2014, Felici et al 2015, Long et al 2015]. KL1333, a novel NAD+ modulator, interacts with NAD(P)H:dehydrogenase 1 (NQO1) to increase intracellular NAD+ levels via NADH oxidation [Seo et al 2018]. Results of a Phase Ia/Ib trial assessing the safety and tolerability in individuals with primary mitochondrial disorders are yet to be published (NCT03888716). Further studies are needed to investigate the safety and efficacy of these therapies in mtDNA-LSS.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS) is transmitted by mitochondrial (maternal) inheritance.
Risk to Family Members
Parents of a proband
- The father of a proband is not at risk of having the mtDNA pathogenic variant.
- The mother of a proband may have the mtDNA pathogenic variant and may exhibit mild clinical manifestations of mtDNA-LSS.
- In most instances, the mother has a much lower heteroplasmy level (i.e., proportion of abnormal mtDNA) than the proband and usually remains asymptomatic or develops only mild manifestations such as gait disturbance or retinopathy.
- Occasionally the mother has a substantial heteroplasmy level and develops severe manifestations of mtDNA-LSS in adulthood [de Vries et al 1993, Wei et al 2018].
- Many affected individuals have no known family history of mtDNA-LSS or other mitochondrial disorders. The explanation for apparently simplex cases may be absence of a comprehensive and/or reliable family history, a maternal heteroplasmy level below the threshold level that causes disease, or a de novo mtDNA pathogenic variant in the proband. A study of 105 probands with childhood-onset mitochondrial disease, including mtDNA-LSS, suggested that approximately 25% of individuals had a de novo pathogenic variant [Sallevelt et al 2017].
- Molecular genetic testing of the mother for the mtDNA pathogenic variant identified in the proband is recommended. Assessment in at least two different maternal tissues (e.g., blood, urine sediment cells, buccal cells) is suggested [Mavraki et al 2023]. Testing of maternal relatives can be considered to provide reassurance or appropriate surveillance and genetic counselling. (Note: Mitochondrial DNA pathogenic variants may be lost from the leukocyte population with increasing age [Schon et al 2021, Davis et al 2022]; therefore, in adults with milder manifestations and for asymptomatic older maternal relatives, the pathogenic variant may only be detected in tissues such as hair follicles, urine sediment cells, or skeletal muscle.)
- If the proband represents a simplex case (i.e., the only family member known to be affected) and the mtDNA pathogenic variant identified in the proband cannot be identified in maternal tissues, possibilities to consider include a de novo pathogenic variant in the proband and germline (gonadal) mosaicism in the mother. Care should be used when determining if a pathogenic variant is de novo. For some pathogenic variants, homoplasmic (or apparently homoplasmic levels) in an affected child may result from very low heteroplasmy levels in the mother.
Sibs of a proband. The risk to the sibs depends on the genetic status of the mother.
- If the mother of the proband has the mtDNA pathogenic variant identified in the proband, all sibs are at risk of inheriting the pathogenic variant. Sibs may inherit the pathogenic variant at varying heteroplasmy levels due to the bottleneck effect and variant-specific segregation patterns.
- For the MT-ATP6 pathogenic variants m.8993T>G and m.8993T>C, a strong positive relationship exists between the mother's heteroplasmy level and the predicted recurrence risk [White et al 1999a] (see Figure 3). However, the 95% confidence interval of the risk estimates are wide, and these data may be of limited use for genetic counseling.
- Due to positive selection, the m.8993T>G pathogenic variant exhibits skewed segregation [Otten et al 2018], with the heteroplasmy levels in oocytes/embryos generally displaying a distribution to the extremes, either 0% or approximately 100%.
- The risk to a sib of developing clinical manifestations is difficult to determine and depends on heteroplasmy level, the variation in heteroplasmy levels among different tissues, and the disease threshold (i.e., the minimum level of heteroplasmy expected to result in mitochondrial disease) for the specific variant (see Genotype-Phenotype Correlations).
- In mtDNA-LSS, the MT-ATP6 pathogenic variants m.8993T>G and m.8993T>C probably exhibit the strongest genotype-phenotype correlation of any mtDNA pathogenic variants [Ng et al 2019]; they show very little tissue-dependent or age-dependent variation in heteroplasmy level as well as a strong correlation between heteroplasmy level and clinical outcome.
- It is difficult to use heteroplasmy level to predict clinical outcome (e.g., in asymptomatic family members or in prenatal diagnosis) for other mtDNA pathogenic variants detected in individuals known to be associated with mtDNA-LSS unless the value is near 0% or near 100%. Retrospective studies for some of the more common mtDNA pathogenic variants can be used to indicate an approximate (empiric) recurrence risk. See Chinnery et al [1998] and Pickett et al [2019] for MT-TL1 pathogenic variant m.3243A>G and MT-TK pathogenic variant m.8344A>G and Sallevelt et al [2017], and Steffann et al [2021], and Vachin et al [2018] for other mtDNA pathogenic variants.
- Variation in clinical manifestations is seen among family members with the same genotype [Stendel et al 2020].
- If the proband is presumed to have a de novo mtDNA pathogenic variant (i.e., the pathogenic variant is absent from the blood and at least one other sample from the mother and any maternal relatives who were tested), sibs are at low risk [Sallevelt et al 2017]. However, the possibility of maternal germline (gonadal) mosaicism cannot be excluded and therefore the recurrence risk is not 0%.

Offspring of a proband
- Offspring of a male proband with an mtDNA pathogenic variant are not at risk.
- All offspring of a female proband with an mtDNA pathogenic variant are at risk of inheriting the pathogenic variant. Offspring may inherit the pathogenic variant at varying heteroplasmy levels due to the bottleneck effect and variant-specific segregation patterns.
- The risk to offspring of a female proband of developing clinical manifestations is difficult to determine and depends on the maternal heteroplasmy level, the variation in heteroplasmy levels among different tissues in the proband, and the specific mtDNA pathogenic variant. Retrospective studies for some of the most common mtDNA pathogenic variants can be used to indicate an approximate (empiric) recurrence risk for the offspring of females. See White et al [1999a] for the MT-ATP6 pathogenic variants m.8993T>G and m.8993T>C, Chinnery et al [1998] and Pickett et al [2019] for the MT-TL1 pathogenic variant m.3243A>G and MT-TK pathogenic variant m.8344A>G, and Sallevelt et al [2017] and Steffann et al [2021] for other mtDNA pathogenic variants.
Other family members. The risk to other family members depends on the genetic status of the proband's mother: if the proband's mother has an mtDNA pathogenic variant, her sibs and mother are also at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy. Similarly, decisions regarding testing to determine the genetic status of at-risk asymptomatic family members are best made before pregnancy.
- It is appropriate to offer genetic counseling (including general discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk. It should be emphasized in genetic counseling for mtDNA-LSS that it is not possible to make specific predictions about clinical outcome in individuals or their offspring.
Phenotypic variability. The phenotype of an individual with an mtDNA pathogenic variant results from a combination of factors, including the severity of the pathogenic variant, the percentage of abnormal mitochondria (mtDNA heteroplasmy), and the organs and tissues in which the pathogenic variant is found (tissue distribution). Family members often inherit different percentages of abnormal mtDNA and therefore can have a wide range of clinical manifestations.
Testing in asymptomatic at-risk family members. Testing to evaluate the genetic status of an apparently asymptomatic family member is technically possible once the mtDNA pathogenic variant has been identified in the proband. However, interpretation of test results of asymptomatic at-risk family members is extremely difficult. Prediction of phenotype based on test results is not possible. Furthermore, absence of the mtDNA pathogenic variant in one tissue (e.g., blood) does not guarantee that the pathogenic variant is absent in other tissues.
Prenatal Testing and Preimplantation Genetic Testing
Recurrence risk assessment and prenatal testing for disorders caused by pathogenic variants in mtDNA are challenging due to the intricacies of mtDNA transmission, such as the mtDNA bottleneck effect (resulting in variation in heteroplasmy levels between generations), pathogenic variant-specific selection, the threshold effect (the heteroplasmy level associated with clinical manifestations of mitochondrial disease), and the inherent challenge in using prenatal genetic test results to predict clinical outcome.
The European Neuromuscular Disease Centre International Consensus Workshops have been instrumental in developing guidelines for the counseling of reproductive options for families with mitochondrial disease [Poulton & Turnbull 2000, Poulton & Bredenoord 2010, Poulton et al 2019].
Reproductive options for the family members of a proband with an mtDNA pathogenic variant may include prenatal testing, preimplantation genetic testing (PGT), and oocyte donation. Prenatal testing appears to be the preferred reproductive testing option for mtDNA pathogenic variants that appear to have occurred de novo in the proband and are associated with low recurrence risk [Sallevelt et al 2017], as there is a high likelihood that the pathogenic variant will not be present in prenatal samples. Prenatal testing may also be suitable for women thought to have low recurrence risk due to having non-zero but low levels of the pathogenic mtDNA variant, such as <20% heteroplasmy. PGT is currently considered an appropriate reproductive option for females with familial heteroplasmic mtDNA pathogenic variants and is likely the best option available, at present, for those with moderate recurrence risk. However, PGT may not be successful for women at moderate risk and is not recommended for females containing homoplasmic or near homoplasmic levels of an mtDNA pathogenic variant [Poulton et al 2019]. For families in which prenatal testing and/or PGT are unlikely to successfully reduce recurrence risk, oocyte donation may be considered.
Prenatal testing involves quantification of the heteroplasmy level in fetal tissue obtained by chorionic villus biopsy (CVS) (usually performed between 11-12 weeks' gestation) or amniocentesis (usually performed at 15-18 weeks' gestation).
- Available evidence suggests that the proportion of abnormal mtDNA in extraembryonic and embryonic tissues is similar and does not change substantially during pregnancy. For a number of mtDNA pathogenic variants, including m.8993T>G, m.8993T>C, and m.9176T>C in MT-ATP6, m.3243A>G in MT-TL1, m.10191T>C in MT-ND3, and m.13513G>A in MT-ND5, the proportion of abnormal mtDNA has been reported to remain relatively stable throughout embryofetal development and in postnatal tissue [White et al 1999a, Dahl et al 2000, Thorburn & Dahl 2001, Jacobs et al 2005, Steffann et al 2007, Monnot et al 2011, Steffann et al 2021]. However, variation in mtDNA heteroplasmy levels between the placenta and embryonic/postnatal tissue or between multiple placental samples has also been reported [Marchington et al 2006, Monnot et al 2011, Vachin et al 2018, Steffann et al 2021].
- There is no consensus between centers regarding the preference for CVS or amniocentesis for prenatal testing for mtDNA disease, although some studies support the use of amniocentesis given reports of intraplacental variation with CVS [Steffann et al 2021].
- Analysis should be done on direct CVS/amniocytes, not on cultured cells, as the heteroplasmy level of mtDNA pathogenic variants can change during cell culture.
The major limitation of prenatal testing is the potential difficulty in predicting clinical outcome based on fetal genetic testing results. For prediction of clinical outcome to be reliable, there must be a close correlation between the heteroplasmy level and disease severity; uniform distribution of heteroplasmy levels in all tissues; and stability of the heteroplasmy level over time [Poulton & Turnbull 2000]. See Genotype-Phenotype Correlations for pathogenic variant-specific information.
Clinical practice varies in determining which thresholds are used to determine the likelihood a fetus will be affected or unaffected based on prenatal test results. A systematic review suggested a generic threshold of 18% after it was found that individuals with muscle heteroplasmy levels below approximately 18% had a 95% chance or higher of being unaffected, independent of the mtDNA variant [Hellebrekers et al 2012]. Prenatal heteroplasmy risk groups of low (<30%), intermediate (30%-60%) and high (>60%) have been applied in different studies [Nesbitt et al 2014, Vachin et al 2018, Steffann et al 2021]. An intermediate proportion of abnormal DNA would represent a "gray zone" in which interpretation is difficult or impossible. For some pathogenic variants, using this risk stratification is feasible; however, it may not be appropriate for other pathogenic variants, such as m.3243A>G in MT-TL1 [Hellebrekers et al 2012].
- While the MT-ATP6 pathogenic variant m.8993T>G is known to exhibit skewed segregation in embryos or oocytes (either 0% or approximately 100%) [Blok et al 1997, Otten et al 2018], prenatal testing is still considered a suitable option for females with low heteroplasmy levels given that a reliable prediction is possible for most heteroplasmy levels [Smeets et al 2015].
- A model has been developed for the MT-TL1 m.3243A>G pathogenic variant to facilitate predictions of fetal heteroplasmy (below a specified threshold of 18%) based on maternal heteroplasmy levels [Pickett et al 2019].
- For other mtDNA pathogenic variants, prenatal diagnosis may be offered to females with a low proportion of abnormal mtDNA. However, weaker genotype-phenotype correlation or lack of data would mean that accurate prediction of the phenotype may not possible. Couples would require careful counseling before embarking on these procedures.
Preimplantation genetic testing (PGT) involves quantification of the heteroplasmic variant load to facilitate selection of the most appropriate embryo for transfer. PGT is traditionally performed by sampling one or two blastomeres on day three or four (cleavage stage) after fertilization. At the cleavage stage, there are robust data supporting the correlation between the heteroplasmic variant load in individual blastomeres and the whole embryo [Steffann et al 2006, Monnot et al 2011, Sallevelt et al 2013]. Most centers offering PGT for inherited diseases now use trophectoderm biopsy rather than cleavage stage biopsy. There are limited data using trophectoderm biopsy for mtDNA PGT, with one study suggesting it may be less suitable for estimating heteroplasmy in the embryo [Mitalipov et al 2014], although this remains unclear [Poulton et al 2019].
Embryos should only be regarded as suitable for implantation if they have a very low proportion of abnormal mtDNA, preferably 0%, although threshold levels of 18% may imply low recurrence risk [Hellebrekers et al 2012, Smeets et al 2015]. Nevertheless, as disease thresholds and patterns of transmission vary with different mtDNA pathogenic variants, it is important to assess this on an individual basis, exploring the couple's own views on what level of residual risk they are willing to accept. Heteroplasmy levels of clinically affected and unaffected individuals within a family should also be considered [Poulton et al 2019].
In some females, particularly those with a high proportion of abnormal mtDNA in blood or urine sediment cells, a large proportion of oocytes may have a high level of abnormal mtDNA, and multiple cycles of ovarian stimulation may not result in an embryo suitable for implantation. However, PGT for mtDNA pathogenic variants may provide valuable information for future reproductive planning even if a successful unaffected conception is not achieved.
- If most of the embryos tested have a substantial proportion of abnormal mtDNA, oocyte donation is likely to be the only current option for ensuring an unaffected embryo.
- In contrast, if most of the embryos tested have undetectable abnormal mtDNA, the parents may opt for prenatal testing in subsequent unassisted (natural) pregnancies.
Other Reproductive Options
Oocyte donation accompanied by IVF using the partner's sperm. Use of a maternal relative as the oocyte donor should be avoided, since the relative may have oocytes with a high proportion of abnormal mtDNA even though her leukocytes may lack detectable abnormal mtDNA.
Mitochondrial replacement therapy (MRT) involves the transfer of a nucleus removed from either a zygote (pronuclear transfer) or an oocyte (maternal spindle transfer) into an enucleated cell at the same embryonic stage from an unaffected donor. Clinical trials are currently under way to investigate whether this reproductive option would be suitable for females with pathogenic mtDNA variants where PGT is not likely to be (or has not been) an effective option (e.g., homoplasmic variants and/or heteroplasmic variants at high heteroplasmy levels).
The first child born after maternal spindle transfer was reported in 2017, born to a female with MT-ATP6 pathogenic variant m.8993T>G [Zhang et al 2017]. The boy was reported to be well at age seven months but long-term neurodevelopmental follow up has yet to be published.
After extensive scientific, ethical, and public consultation, in 2016, the UK government gave permission for MRT to prevent the transmission of severe mitochondrial disease caused by specific mtDNA pathogenic variants [Herbert & Turnbull 2017]. The use of MRT in the UK is regulated by the Human Fertilisation and Embryology Authority (HFEA) and is limited to families who are at substantial risk for transmitting severe mtDNA disease in a situation in which PGT is deemed inappropriate or likely to be unsuccessful. A five-year clinical trial is being carried out at the Newcastle Fertility Clinic, and outcomes are yet to be published. In 2022, the Australian government passed the Mitochondrial Donation Law Reform Bill (Maeve's Law) regarding the clinical implementation of MRT. Under the bill, MRT may be introduced in a staged manner, with current legislation allowing for the commencement of a clinical trial of the use of MRT techniques (see National Health and Medical Research Council).
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- United Mitochondrial Disease FoundationPhone: 888-317-UMDF (8633)Email: info@umdf.org
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Mito FoundationAustraliaPhone: 61-1-300-977-180Email: info@mito.org.au
- Mitochondrial Disease Registry and Tissue BankMassachusetts General HospitalPhone: 617-726-5718Fax: 617-724-9620Email: nslate@partners.org
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321Email: ResourceCenter@mdausa.org
- People Against Leigh Syndrome (PALS)Phone: 713-248-8782
- Retina InternationalIreland
- The Lily FoundationUnited KingdomEmail: liz@thelilyfoundation.org.uk
- RDCRN Patient Contact Registry: North American Mitochondrial Disease Consortium
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Mitochondrial DNA-Associated Leigh Syndrome Spectrum: Genes and Databases
Table B.
OMIM Entries for Mitochondrial DNA-Associated Leigh Syndrome Spectrum (View All in OMIM)
Molecular Pathogenesis
Human mitochondrial DNA (mtDNA) encodes 37 genes, including 13 genes encoding protein subunits of the mitochondrial respiratory chain and oxidative phosphorylation system, 22 transfer RNA (tRNA) genes, and two ribosomal RNA (rRNA) genes. All mtDNA genes lack introns and are transcribed as large polycistronic transcripts that are processed into monocistronic mRNAs. Protein-coding genes are then translated by the mitochondrial-specific translational machinery. The mitochondrial-specific translational machinery is required because translation of mtDNA-encoded genes is physically separated from the cytosolic translational machinery and because the mtDNA genetic code differs from the universal genetic code in several codons.
For some mtDNA pathogenic variants associated with Leigh syndrome spectrum (LSS), a strong correlation exists between the proportion of abnormal-to-wild type mtDNA and severity of the biochemical phenotype in cultured cells. For some variants, such as MT-ATP6 pathogenic variants m.8993T>G and m.8993T>C, a strong correlation also exists between the proportion of abnormal-to-wild type mtDNA and clinical severity. However, the mechanism by which some mtDNA pathogenic variants cause a phenotype such as LSS, while others cause myopathy, deafness, or diabetes mellitus, is unknown.
Pathogenic mtDNA variants causing LSS fall into two major classes, namely, those in tRNA genes and those in protein-coding genes.
- Transfer RNA pathogenic variants cause decreased mitochondrial protein synthesis by abnormalities in base modification and aminoacylation of the tRNA.
- Pathogenic variants in protein-coding mtDNA genes typically cause decreased activity of the respiratory chain complex of which that subunit is a part.
Mechanism of disease causation. Loss of function
Table 8.
Pathogenic Variants Referenced in This GeneReview by Gene
Chapter Notes
Author Notes
Prof Rahman, Prof Thorburn, and Dr Ball are actively involved in clinical research regarding individuals with mitochondrial DNA-associated Leigh syndrome spectrum (mtDNA-LSS). Prof Thorburn is a member of the mitoHOPE Program team performing an Australian clinical trial of mitochondrial replacement therapy.
The authors would be happy to communicate with persons who have any questions regarding diagnosis of mtDNA-LSS or other considerations.
Acknowledgments
The authors would like to acknowledge the support of clinicians, researchers, and patient advocacy groups for mtDNA-LSS. They would also like to acknowledge all the patients and families who have generously given their time and shared their experiences to advance the understanding of mtDNA-LSS.
Author History
Megan Ball, MD (2024-present)
Joyeeta Rahman, BSc; UCL Great Ormond Street Institute of Child Health (2017-2024)
Shamima Rahman, PhD, FRCP, FRCPCH (2003-present)
David R Thorburn, PhD, FHGSA, FFSc(RCPA) (2003-present)
Revision History
- 9 May 2024 (sw) Comprehensive update posted live
- 28 September 2017 (sw) Comprehensive update posted live
- 17 April 2014 (me) Comprehensive update posted live
- 8 February 2011 (me) Comprehensive update posted live
- 3 February 2006 (me) Comprehensive update posted live
- 30 October 2003 (me) Review posted live
- 3 July 2003 (dt) Original submission
References
Published Guidelines / Consensus Statements
- Diodato D, Schiff M, Cohen BH, Bertini E, Rahman S; Workshop participants. 258th ENMC international workshop Leigh syndrome spectrum: genetic causes, natural history and preparing for clinical trials 25-27 March 2022, Hoofddorp, Amsterdam, The Netherlands. Neuromuscul Disord. 2023;33:700-9. [PubMed]
- McCormick EM, Keller K, Taylor JP, Coffey AJ, Shen L, Krotoski D, Harding B; NICHD ClinGen U24 Mitochondrial Disease Gene Curation Expert Panel; Gai X, Falk MJ, Zolkipli-Cunningham Z, Rahman S. Expert panel curation of 113 primary mitochondrial disease genes for the Leigh syndrome spectrum. Ann Neurol. 2023;94:696-712. [PubMed]
- Parikh S, Goldstein A, Karaa A, Koenig MK, Anselm I, Brunel-Guitton C, Christodoulou J, Cohen BH, Dimmock D, Enns GM, Falk MJ, Feigenbaum A, Frye RE, Ganesh J, Griesemer D, Haas R, Horvath R, Korson M, Kruer MC, Mancuso M, McCormack S, Raboisson MJ, Reimschisel T, Salvarinova R, Saneto RP, Scaglia F, Shoffner J, Stacpoole PW, Sue CM, Tarnopolsky M, Van Karnebeek C, Wolfe LA, Cunningham ZZ, Rahman S, Chinnery PF. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19:10.1038/gim.2017.107. [PubMed]
- Poulton J, Turnbull DM. 74th European Neuromuscular Centre International Consensus Workshop on genetic counseling and prenatal diagnosis of mitochondrial DNA disorders. 19-20 November 1999, Naarden, The Netherlands. Available online. 2000. Accessed 4-23-24.
Literature Cited
- Alves C, Teixeira SR, Martin-Saavedra JS, Guimaraes Goncalves F, Lo Russo F, Muraresku C, McCormick EM, Falk MJ, Zolkipli-Cunningham Z, Ganetzky R, Vossough A, Goldstein A, Zuccoli G. Pediatric Leigh syndrome: neuroimaging features and genetic correlations. Ann Neurol. 2020;88:218-32. [PubMed: 32445240]
- Ardissone A, Bruno C, Diodato D, Donati A, Ghezzi D, Lamantea E, Lamperti C, Mancuso M, Martinelli D, Primiano G, Procopio E, Rubegni A, Santorelli F, Schiaffino MC, Servidei S, Tubili F, Bertini E, Moroni I. Clinical, imaging, biochemical and molecular features in Leigh syndrome: a study from the Italian network of mitochondrial diseases. Orphanet J Rare Dis. 2021;16:413. [PMC free article: PMC8501644] [PubMed: 34627336]
- Ardissone A, Ferrera G, Lamperti C, Tiranti V, Ghezzi D, Moroni I, Lamantea E. Phenotyping mitochondrial DNA-related diseases in childhood: a cohort study of 150 patients. Eur J Neurol. 2023;30:2079-91. [PubMed: 37038312]
- Balasubramaniam S, Lewis B, Mock DM, Said HM, Tarailo-Graovac M, Mattman A, van Karnebeek CD, Thorburn DR, Rodenburg RJ, Christodoulou J. Leigh-like syndrome due to homoplasmic m.8993T>G variant with hypocitrullinemia and unusual biochemical features suggestive of multiple carboxylase deficiency (MCD). JIMD Rep. 2017;33:99-107. [PMC free article: PMC5413447] [PubMed: 27450367]
- Baldo MS, Nogueira C, Pereira C, Janeiro P, Ferreira S, Lourenco CM, Bandeira A, Martins E, Magalhaes M, Rodrigues E, Santos H, Ferreira AC, Vilarinho L. Leigh syndrome spectrum: a Portuguese population cohort in an evolutionary genetic era. Genes (Basel). 2023;14:27. [PMC free article: PMC10454233] [PubMed: 37628588]
- Bastin J, Aubey F, Rötig A, Munnich A, Djouadi F. Activation of peroxisome proliferator-activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients' cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433-41. [PubMed: 18211970]
- Blok RB, Gook DA, Thorburn DR, Dahl HH. Skewed segregation of the mtDNA nt 8993 (T-->G) mutation in human oocytes. Am J Hum Genet. 1997;60:1495-501. [PMC free article: PMC1716104] [PubMed: 9199572]
- Bonfante E, Koenig MK, Adejumo RB, Perinjelil V, Riascos RF. The neuroimaging of Leigh syndrome: case series and review of the literature. Pediatr Radiol. 2016;46:443-51. [PubMed: 26739140]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, Viscomi C, Zeviani M. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014;19:1042-9. [PMC free article: PMC4051987] [PubMed: 24814483]
- Chinnery PF, Howell N, Lightowlers RN, Turnbull DM. MELAS and MERRF. The relationship between maternal mutation load and the frequency of clinically affected offspring. Brain. 1998;121:1889-94. [PubMed: 9798744]
- Ciafaloni E, Santorelli FM, Shanske S, Deonna T, Roulet E, Janzer C, Pescia G, DiMauro S. Maternally inherited Leigh syndrome. J Pediatr. 1993;122:419-22. [PubMed: 8095070]
- Claeys KG, Abicht A, Hausler M, Kleinle S, Wiesmann M, Schulz JB, Horvath R, Weis J. Novel genetic and neuropathological insights in neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP). Muscle Nerve. 2016;54:328-33. [PubMed: 27015314]
- Dahl HH, Thorburn DR, White SL. Towards reliable prenatal diagnosis of mtDNA point mutations: studies of nt8993 mutations in oocytes, fetal tissues, children and adults. Hum Reprod. 2000;15 Suppl 2:246-55. [PubMed: 11041530]
- Darin N, Oldfors A, Moslemi AR, Holme E, Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001;49:377-83. [PubMed: 11261513]
- Davis RL, Kumar KR, Puttick C, Liang C, Ahmad KE, Edema-Hildebrand F, Park JS, Minoche AE, Gayevskiy V, Mallawaarachchi AC, Christodoulou J, Schofield D, Dinger ME, Cowley MJ, Sue CM. Use of whole-genome sequencing for mitochondrial disease diagnosis. Neurology. 2022;99:e730-e742. [PMC free article: PMC9484606] [PubMed: 35641312]
- de Haas R, Das D, Garanto A, Renkema HG, Greupink R, van den Broek P, Pertijs J, Collin RWJ, Willems P, Beyrath J, Heerschap A, Russel FG, Smeitink JA. Therapeutic effects of the mitochondrial ROS-redox modulator KH176 in a mammalian model of Leigh Disease. Sci Rep. 2017;7:11733. [PMC free article: PMC5601915] [PubMed: 28916769]
- de Vries DD, van Engelen BG, Gabreëls FJ, Ruitenbeek W, van Oost BA. A second missense mutation in the mitochondrial ATPase 6 gene in Leigh's syndrome. Ann Neurol. 1993;34:410-2. [PubMed: 8395787]
- De Vries MC, Brown DA, Allen ME, Bindoff L, Gorman GS, Karaa A, Keshavan N, Lamperti C, McFarland R, Ng YS, O'Callaghan M, Pitceathly RDS, Rahman S, Russel FGM, Varhaug KN, Schirris TJJ, Mancuso M. Safety of drug use in patients with a primary mitochondrial disease: An international Delphi-based consensus. J Inherit Metab Dis. 2020;43:800-18. [PMC free article: PMC7383489] [PubMed: 32030781]
- Duff RM, Shearwood AM, Ermer J, Rossetti G, Gooding R, Richman TR, Balasubramaniam S, Thorburn DR, Rackham O, Lamont PJ, Filipovska A. A mutation in MT-TW causes a tRNA processing defect and reduced mitochondrial function in a family with Leigh syndrome. Mitochondrion. 2015;25:113-9. [PubMed: 26524491]
- Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, Amagata A, Barnes A, Kheifets V, Shrader WD, Thoolen M, Blankenberg F, Miller G. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105:91-102. [PubMed: 22115768]
- Felici R, Lapucci A, Cavone L, Pratesi S, Berlinguer-Palmini R, Chiarugi A. Pharmacological NAD-boosting strategies improve mitochondrial homeostasis in human complex I-mutant fibroblasts. Mol Pharmacol. 2015;87:965-71. [PubMed: 25788480]
- Frazier AE, Vincent AE, Turnbull DM, Thorburn DR, Taylor RW. Assessment of mitochondrial respiratory chain enzymes in cells and tissues. Methods Cell Biol. 2020;155:121-156. [PubMed: 32183956]
- Ganetzky RD, Stendel C, McCormick EM, Zolkipli-Cunningham Z, Goldstein AC, Klopstock T, Falk MJ. MT-ATP6 mitochondrial disease variants: phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases. Hum Mutat. 2019;40:499-515. [PMC free article: PMC6506718] [PubMed: 30763462]
- Hellebrekers DM, Wolfe R, Hendrickx AT, de Coo IF, de Die CE, Geraedts JP, Chinnery PF, Smeets HJ. PGD and heteroplasmic mitochondrial DNA point mutations: a systematic review estimating the chance of healthy offspring. Hum Reprod Update. 2012;18:341-9. [PubMed: 22456975]
- Herbert M, Turnbull D. Mitochondrial donation — clearing the final regulatory hurdle in the United Kingdom. N Engl J Med. 2017;376:171-3. [PubMed: 28030773]
- Hong CM, Na JH, Park S, Lee YM. Clinical characteristics of early-onset and late-onset Leigh syndrome. Front Neurol. 2020;11:267. [PMC free article: PMC7174756] [PubMed: 32351444]
- Huang L, Li H, Zhong J, Yang L, Chen G, Wang D, Zheng G, Han H, Han X, Long Y, Wang X, Liang J, Yu M, Shen X, Fan M, Fang F, Liao J, Sun D. Efficacy and safety of the ketogenic diet for mitochondrial disease with epilepsy: a prospective, open-labeled, controlled study. Front Neurol. 2022;13:880944. [PMC free article: PMC9377015] [PubMed: 35979062]
- Hughes SD, Kanabus M, Anderson G, Hargreaves IP, Rutherford T, O'Donnell M, Cross JH, Rahman S, Eaton S, Heales SJ. The ketogenic diet component decanoic acid increases mitochondrial citrate synthase and complex I activity in neuronal cells. J Neurochem. 2014;129:426-33. [PubMed: 24383952]
- Jacobs LJ, de Coo IF, Nijland JG, Galjaard RJ, Los FJ, Schoonderwoerd K, Niermeijer MF, Geraedts JP, Scholte HR, Smeets HJ. Transmission and prenatal diagnosis of the T9176C mitochondrial DNA mutation. Mol Hum Reprod. 2005;11:223-8. [PubMed: 15709156]
- Kanabus M, Fassone E, Hughes SD, Bilooei SF, Rutherford T, Donnell MO, Heales SJR, Rahman S. The pleiotropic effects of decanoic acid treatment on mitochondrial function in fibroblasts from patients with complex I deficient Leigh syndrome. J Inherit Metab Dis. 2016;39:415-26. [PMC free article: PMC4851692] [PubMed: 27080638]
- Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66:324-30. [PubMed: 16476929]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, Suomalainen A. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721-31. [PMC free article: PMC4203351] [PubMed: 24711540]
- Kistol D, Tsygankova P, Krylova T, Bychkov I, Itkis Y, Nikolaeva E, Mikhailova S, Sumina M, Pechatnikova N, Kurbatov S, Bostanova F, Migiaev O, Zakharova E. Leigh syndrome: spectrum of molecular defects and clinical features in Russia. Int J Mol Sci. 2023;24:1597. [PMC free article: PMC9865855] [PubMed: 36675121]
- Koene S, Spaans E, Van Bortel L, Van Lancker G, Delafontaine B, Badilini F, Beyrath J, Smeitink J. KH176 under development for rare mitochondrial disease: a first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J Rare Dis. 2017;12:163. [PMC free article: PMC5644106] [PubMed: 29037240]
- Larson AA, Balasubramaniam S, Christodoulou J, Burrage LC, Marom R, Graham BH, Diaz GA, Glamuzina E, Hauser N, Heese B, Horvath G, Mattman A, van Karnebeek C, Lane Rutledge S, Williamson A, Estrella L, Van Hove JKL, Weisfeld-Adams JD. Biochemical signatures mimicking multiple carboxylase deficiency in children with mutations in MT-ATP6. Mitochondrion. 2019;44:58-64. [PMC free article: PMC10201920] [PubMed: 29307858]
- Lee JS, Kim H, Lim BC, Hwang H, Choi J, Kim KJ, Hwang YS, Chae JH. Leigh syndrome in childhood: neurologic progression and functional outcome. J Clin Neurol. 2016;12:181-7. [PMC free article: PMC4828564] [PubMed: 27074294]
- Lee JS, Yoo T, Lee M, Lee Y, Jeon E, Kim SY, Lim BC, Kim KJ, Choi M, Chae JH. Genetic heterogeneity in Leigh syndrome: highlighting treatable and novel genetic causes. Clin Genet. 2020;97:586-94. [PubMed: 32020600]
- Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry. 1951;14:216-21. [PMC free article: PMC499520] [PubMed: 14874135]
- Lim AZ, Ng YS, Blain A, Jiminez-Moreno C, Alston CL, Nesbitt V, Simmons L, Santra S, Wassmer E, Blakely EL, Turnbull DM, Taylor RW, Gorman GS, McFarland R. Natural history of Leigh syndrome: a study of disease burden and progression. Ann Neurol. 2022;91:117-30. [PMC free article: PMC9534328] [PubMed: 34716721]
- Lin Y, Xu X, Zhao D, Liu F, Luo Y, Du J, Wang D, Ji K, Zhao Y, Yan C. A novel m.11406 T > A mutation in mitochondrial ND4 gene causes MELAS syndrome. Mitochondrion. 2020;54:57-64. [PubMed: 32659360]
- Long AN, Owens K, Schlappal AE, Kristian T, Fishman PS, Schuh RA. Effect of nicotinamide mononucleotide on brain mitochondrial respiratory deficits in an Alzheimer's disease-relevant murine model. BMC Neurol. 2015;15:19. [PMC free article: PMC4358858] [PubMed: 25884176]
- López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet. 2006;79:1125-9. [PMC free article: PMC1698707] [PubMed: 17186472]
- Marchington DR, Scott-Brown M, Barlow DH, Poulton J. Mosaicism for mitochondrial DNA polymorphic variants in placenta has implications for the feasibility of prenatal diagnosis in mtDNA diseases. Eur J Hum Genet. 2006;14:816-23. [PubMed: 16670690]
- Martinelli D, Catteruccia M, Piemonte F, Pastore A, Tozzi G, Dionisi-Vici C, Pontrelli G, Corsetti T, Livadiotti S, Kheifets V, Hinman A, Shrader WD, Thoolen M, Klein MB, Bertini E, Miller G. EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol Genet Metab. 2012;107:383-8. [PubMed: 23010433]
- Mavraki E, Labrum R, Sergeant K, Alston CL, Woodward C, Smith C, Knowles CVY, Patel Y, Hodsdon P, Baines JP, Blakely EL, Polke J, Taylor RW, Fratter C. Genetic testing for mitochondrial disease: the United Kingdom best practice guidelines. Eur J Hum Genet. 2023;31:148-63. [PMC free article: PMC9905091] [PubMed: 36513735]
- McCormick EM, Keller K, Taylor JP, Coffey AJ, Shen L, Krotoski D, Harding B, Panel NCUMDGCE, Alves C, Ardissone A, Bai R, de Barcelos IP, Bertini E, Bluske K, Christodoulou J, Clause AR, Copeland WC, Diaz GA, Diodato D, Dulik MC, Enns G, Feigenbaum A, Fratter C, Ghezzi D, Goldstein A, Gropman A, Haas R, Karaa A, Koenig MK, Monteleone B, Parikh S, Duenas BP, Rajkumar R, Saada A, Saneto RP, Sergeant K, Shoffner J, Smith C, Stanley C, Thiffault I, Thorburn D, Walker M, Wallace D, Wong LJ, Gai X, Falk MJ, Zolkipli-Cunningham Z, Rahman S. Expert panel curation of 113 primary mitochondrial disease genes for the Leigh syndrome spectrum. Ann Neurol. 2023;94:696-712. [PMC free article: PMC10763625] [PubMed: 37255483]
- Mezuki S, Fukuda K, Matsushita T, Fukushima Y, Matsuo R, Goto YI, Yasukawa T, Uchiumi T, Kang D, Kitazono T, Ago T. Isolated and repeated stroke-like episodes in a middle-aged man with a mitochondrial ND3 T10158C mutation: a case report. BMC Neurol. 2017;17:217. [PMC free article: PMC5729248] [PubMed: 29237403]
- Mitalipov S, Amato P, Parry S, Falk MJ. Limitations of preimplantation genetic diagnosis for mitochondrial DNA diseases. Cell Rep. 2014;7:935-7. [PMC free article: PMC4349563] [PubMed: 24856294]
- Monnot S, Gigarel N, Samuels DC, Burlet P, Hesters L, Frydman N, Frydman R, Kerbrat V, Funalot B, Martinovic J, Benachi A, Feingold J, Munnich A, Bonnefont J-P, Steffann J. Segregation of mtDNA throughout human embryofetal development: m.3243A>G as a model system. Hum Mutat. 2011;32:116-25. [PMC free article: PMC3058134] [PubMed: 21120938]
- Na JH, Lee YM. Genotype-phenotype analysis of MT-ATP6-associated Leigh syndrome. Acta Neurol Scand. 2022;145:414-22. [PubMed: 34877647]
- Na JH, Lee YM. Heteroplasmic mutant load differences in mitochondrial DNA-associated Leigh syndrome. Pediatr Neurol. 2023;138:27-32. [PubMed: 36335839]
- Naess K, Freyer C, Bruhn H, Wibom R, Malm G, Nennesmo I, von Dobeln U, Larsson NG. MtDNA mutations are a common cause of severe disease phenotypes in children with Leigh syndrome. Biochim Biophys Acta. 2009;1787:484-90. [PubMed: 19103152]
- Nesbitt V, Alston CL, Blakely EL, Fratter C, Feeney CL, Poulton J, Brown GK, Turnbull DM, Taylor RW, McFarland R. A national perspective on prenatal testing for mitochondrial disease. Eur J Hum Genet. 2014;22:1255-9. [PMC free article: PMC4200441] [PubMed: 24642831]
- Ng YS, Martikainen MH, Gorman GS, Blain A, Bugiardini E, Bunting A, Schaefer AM, Alston CL, Blakely EL, Sharma S, Hughes I, Lim A, de Goede C, McEntagart M, Spinty S, Horrocks I, Roberts M, Woodward CE, Chinnery PF, Horvath R, Nesbitt V, Fratter C, Poulton J, Hanna MG, Pitceathly RDS, Taylor RW, Turnbull DM, McFarland R. Pathogenic variants in MT-ATP6: a United Kingdom-based mitochondrial disease cohort study. Ann Neurol. 2019;86:310-5. [PMC free article: PMC6771528] [PubMed: 31187502]
- Ogawa E, Fushimi T, Ogawa-Tominaga M, Shimura M, Tajika M, Ichimoto K, Matsunaga A, Tsuruoka T, Ishige M, Fuchigami T, Yamazaki T, Kishita Y, Kohda M, Imai-Okazaki A, Okazaki Y, Morioka I, Ohtake A, Murayama K. Mortality of Japanese patients with Leigh syndrome: effects of age at onset and genetic diagnosis. J Inherit Metab Dis. 2020;43:819-26. [PMC free article: PMC7383885] [PubMed: 31967322]
- Otten ABC, Sallevelt S, Carling PJ, Dreesen J, Drusedau M, Spierts S, Paulussen ADC, de Die-Smulders CEM, Herbert M, Chinnery PF, Samuels DC, Lindsey P, Smeets HJM. Mutation-specific effects in germline transmission of pathogenic mtDNA variants. Hum Reprod. 2018;33:1331-41. [PMC free article: PMC6551225] [PubMed: 29850888]
- Paleologou E, Ismayilova N, Kinali M. Use of the ketogenic diet to treat intractable epilepsy in mitochondrial disorders. J Clin Med. 2017;6:56. [PMC free article: PMC5483866] [PubMed: 28587136]
- Parikh S, Goldstein A, Karaa A, Koenig MK, Anselm I, Brunel-Guitton C, Christodoulou J, Cohen BH, Dimmock D, Enns GM, Falk MJ, Feigenbaum A, Frye RE, Ganesh J, Griesemer D, Haas R, Horvath R, Korson M, Kruer MC, Mancuso M, McCormack S, Raboisson MJ, Reimschisel T, Salvarinova R, Saneto RP, Scaglia F, Shoffner J, Stacpoole PW, Sue CM, Tarnopolsky M, Van Karnebeek C, Wolfe LA, Cunningham ZZ, Rahman S, Chinnery PF. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19:1380-97. [PMC free article: PMC7804217] [PubMed: 28749475]
- Peretz RH, Ah Mew N, Vernon HJ, Ganetzky RD. Prospective diagnosis of MT-ATP6-related mitochondrial disease by newborn screening. Mol Genet Metab. 2021;134:37-42. [PMC free article: PMC8578202] [PubMed: 34176718]
- Pfeffer G, Blakely EL, Alston CL, Hassani A, Boggild M, Horvath R, Samuels DC, Taylor RW, Chinnery PF. Adult-onset spinocerebellar ataxia syndromes due to MTATP6 mutations. J Neurol Neurosurg Psychiatry. 2012a;83:883-6. [PMC free article: PMC4034166] [PubMed: 22577227]
- Pfeffer G, Majamaa K, Turnbull DM, Thorburn D, Chinnery PF. Treatment for mitochondrial disorders. Cochrane Database Syst Rev. 2012b;2012:CD004426. [PMC free article: PMC7201312] [PubMed: 22513923]
- Pickett SJ, Blain A, Ng YS, Wilson IJ, Taylor RW, McFarland R, Turnbull DM, Gorman GS. Mitochondrial donation - which women could benefit? N Engl J Med. 2019;380:1971-2. [PubMed: 31091381]
- Pitceathly RD, Murphy SM, Cottenie E, Chalasani A, Sweeney MG, Woodward C, Mudanohwo EE, Hargreaves I, Heales S, Land J, Holton JL, Houlden H, Blake J, Champion M, Flinter F, Robb SA, Page R, Rose M, Palace J, Crowe C, Longman C, Lunn MP, Rahman S, Reilly MM, Hanna MG. Genetic dysfunction of MT-ATP6 causes axonal Charcot-Marie-Tooth disease. Neurology. 2012;79:1145-54. [PMC free article: PMC3525307] [PubMed: 22933740]
- Pitceathly RDS, Keshavan N, Rahman J, Rahman S. Moving towards clinical trials for mitochondrial diseases. J Inherit Metab Dis. 2021;44:22-41. [PMC free article: PMC8432143] [PubMed: 32618366]
- Poulton J, Bredenoord AL. 174th ENMC international workshop: Applying pre-implantation genetic diagnosis to mtDNA diseases: implications of scientific advances 19-21 March 2010, Naarden, The Netherlands. Neuromuscul Disord. 2010;20:559-63. [PubMed: 20627569]
- Poulton J, Steffann J, Burgstaller J, McFarland R, workshop p. 243rd ENMC international workshop: developing guidelines for management of reproductive options for families with maternally inherited mtDNA disease, Amsterdam, the Netherlands, 22-24 March 2019. Neuromuscul Disord. 2019;29:725-33. [PubMed: 31501000]
- Poulton J, Turnbull DM. 74th ENMC International Workshop: Mitochondrial Diseases 19-20 November 1999, Naarden, The Netherlands. Neuromuscul Disord. 2000;10:460-2. [PubMed: 10899455]
- Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, Christodoulou J, Thorburn DR. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343-51. [PubMed: 8602753]
- Sallevelt SC, de Die-Smulders CE, Hendrickx AT, Hellebrekers DM, de Coo IF, Alston CL, Knowles C, Taylor RW, McFarland R, Smeets HJ. De novo mtDNA point mutations are common and have a low recurrence risk. J Med Genet. 2017;54:73-83. [PMC free article: PMC5502310] [PubMed: 27450679]
- Sallevelt SC, Dreesen JC, Drusedau M, Spierts S, Coonen E, van Tienen FH, van Golde RJ, de Coo IF, Geraedts JP, de Die-Smulders CE, Smeets HJ. Preimplantation genetic diagnosis in mitochondrial DNA disorders: challenge and success. J Med Genet. 2013;50:125-32. [PubMed: 23339111]
- Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56:662-9. [PubMed: 15389892]
- Schoeler NE, Orford M, Vivekananda U, Simpson Z, Van de Bor B, Smith H, Balestrini S, Rutherford T, Brennan E, McKenna J, Lambert B, Barker T, Jackson R, Williams RSB, Sisodiya SM, Eaton S, Heales SJR, Cross JH, Walker MC, Group KVS. K.Vita: a feasibility study of a blend of medium chain triglycerides to manage drug-resistant epilepsy. Brain Commun [Internet]. 2021;3. Available online. [PMC free article: PMC8557697] [PubMed: 34729477]
- Schon KR, Horvath R, Wei W, Calabrese C, Ibanez K, Ratnaike T, Pitceathly RDS, Bugiardini E, Quinlivan R, Hanna MG, Clement E, Ashton E, Sayer JA, Brennan P, Josifova D, Izatt L, Fratter C, Nesbitt V, Barrett T, McMullen DJ, Smith A, Deshpande C, Smithson SF, Festenstein R, Canham N, Caulfield M, Houlden H, Rahman S, Chinnery PF, Ambrose JC, Arumugam P, Bevers R, Bleda M, Boardman-Pretty F, Boustred CR, Brittain H, Caulfield MJ, Chan GC, Elgar G, Fowler T, Giess A, Hamblin A, Henderson S, Hubbard TJP, Jackson R, Jones LJ, Kasperaviciute D, Kayikci M, Kousathanas A, Lahnstein L, Leigh SEA, Leong IUS, Lopez FJ, Maleady-Crowe F, McEntegart M, Minneci F, Moutsianas L, Mueller M, Murugaesu N, Need AC, O'Donovan P, Odhams CA, Patch C, Pereira MB, Perez-Gil D, Pullinger J, Rahim T, Rendon A, Rogers T, Savage K, Sawant K, Scott RH, Siddiq A, Sieghart A, Smith SC, Sosinsky A, Stuckey A, Tanguy M, Taylor Tavares AL, Thomas ERA, Thompson SR, Tucci A, Welland MJ, Williams E, Witkowska KA, Wood SM. Use of whole genome sequencing to determine genetic basis of suspected mitochondrial disorders: cohort study. BMJ. 2021;375:e066288. [PMC free article: PMC8565085] [PubMed: 34732400]
- Seo KS, Kim JH, Min KN, Moon JA, Roh TC, Lee MJ, Lee KW, Min JE, Lee YM. KL1333, a novel NAD(+) modulator, improves energy metabolism and mitochondrial dysfunction in MELAS fibroblasts. Front Neurol. 2018;9:552. [PMC free article: PMC6041391] [PubMed: 30026729]
- Shrader WD, Amagata A, Barnes A, Enns GM, Hinman A, Jankowski O, Kheifets V, Komatsuzaki R, Lee E, Mollard P, Murase K, Sadun AA, Thoolen M, Wesson K, Miller G. Alpha-tocotrienol quinone modulates oxidative stress response and the biochemistry of aging. Bioorg Med Chem Lett. 2011;21:3693-8. [PubMed: 21600768]
- Smeets HJ, Sallevelt SC, Dreesen JC, de Die-Smulders CE, de Coo IF. Preventing the transmission of mitochondrial DNA disorders using prenatal or preimplantation genetic diagnosis. Ann N Y Acad Sci. 2015;1350:29-36. [PubMed: 26312584]
- Sofou K, De Coo IFM, Isohanni P, Ostergaard E, Naess K, De Meirleir L, Tzoulis C, Uusimaa J, De Angst IB, Lönnqvist T, Pihko H, Mankinen K, Bindoff LA, Tulinius M, Darin N. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet Journal of Rare Diseases. 2014;9:52. [PMC free article: PMC4021638] [PubMed: 24731534]
- Sofou K, de Coo IFM, Ostergaard E, Isohanni P, Naess K, De Meirleir L, Tzoulis C, Uusimaa J, Lonnqvist T, Bindoff LA, Tulinius M, Darin N. Phenotype-genotype correlations in Leigh syndrome: new insights from a multicentre study of 96 patients. J Med Genet. 2018;55:21-7. [PubMed: 29101127]
- Steele H, Gomez-Duran A, Pyle A, Hopton S, Newman J, Stefanetti RJ, Charman SJ, Parikh JD, He L, Viscomi C, Jakovljevic DG, Hollingsworth KG, Robinson AJ, Taylor RW, Bottolo L, Horvath R, Chinnery PF. Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol Med. 2020;12:e11589. [PMC free article: PMC7059007] [PubMed: 32107855]
- Steffann J, Frydman N, Gigarel N, Burlet P, Ray PF, Fanchin R, Feyereisen E, Kerbrat V, Tachdjian G, Bonnefont JP, Frydman R, Munnich A. Analysis of mtDNA variant segregation during early human embryonic development: a tool for successful NARP preimplantation diagnosis. J Med Genet. 2006;43:244-7. [PMC free article: PMC2563237] [PubMed: 16155197]
- Steffann J, Gigarel N, Corcos J, Bonniere M, Encha-Razavi F, Sinico M, Prevot S, Dumez Y, Yamgnane A, Frydman R, Munnich A, Bonnefont JP. Stability of the m.8993T->G mtDNA mutation load during human embryofetal development has implications for the feasibility of prenatal diagnosis in NARP syndrome. J Med Genet. 2007;44:664-9. [PMC free article: PMC2597968] [PubMed: 17545557]
- Steffann J, Monnot S, Magen M, Assouline Z, Gigarel N, Ville Y, Salomon L, Bessiere B, Martinovic J, Rotig A, Bengoa J, Borghese R, Munnich A, Barcia G, Bonnefont JP. A retrospective study on the efficacy of prenatal diagnosis for pregnancies at risk of mitochondrial DNA disorders. Genet Med. 2021;23:720-31. [PubMed: 33303968]
- Stendel C, Neuhofer C, Floride E, Yuqing S, Ganetzky RD, Park J, Freisinger P, Kornblum C, Kleinle S, Schols L, Distelmaier F, Stettner GM, Buchner B, Falk MJ, Mayr JA, Synofzik M, Abicht A, Haack TB, Prokisch H, Wortmann SB, Murayama K, Fang F, Klopstock T. Delineating MT-ATP6-associated disease: from isolated neuropathy to early onset neurodegeneration. Neurol Genet. 2020;6:e393. [PMC free article: PMC6975175] [PubMed: 32042921]
- Stenton SL, Zou Y, Cheng H, Liu Z, Wang J, Shen D, Jin H, Ding C, Tang X, Sun S, Han H, Ma Y, Zhang W, Jin R, Wang H, Sun D, Lv JL, Prokisch H, Fang F. Leigh syndrome: a study of 209 patients at the Beijing Children's Hospital. Ann Neurol. 2022;91:466-82. [PubMed: 35094435]
- Thorburn DR, Dahl HH. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet. 2001;106:102-14. [PubMed: 11579429]
- Tinker RJ, Falk MJ, Goldstein A, George-Sankoh I, Xiao R, Adang L, Ganetzky R. Early developmental delay in Leigh syndrome spectrum disorders is associated with poor clinical prognosis. Mol Genet Metab. 2022;135:342-9. [PMC free article: PMC8965798] [PubMed: 35216885]
- Tise CG, Verscaj CP, Mendelsohn BA, Woods J, Lee CU, Enns GM, Stander Z, Hall PL, Cowan TM, Cusmano-Ozog KP. MT-ATP6 mitochondrial disease identified by newborn screening reveals a distinct biochemical phenotype. Am J Med Genet A. 2023;191:1492-501. [PubMed: 36883293]
- Vachin P, Adda-Herzog E, Chalouhi G, Elie C, Rio M, Rondeau S, Gigarel N, Jabot Hanin F, Monnot S, Borghese R, Bengoa J, Ville Y, Rotig A, Munnich A, Bonnefont JP, Steffann J. Segregation of mitochondrial DNA mutations in the human placenta: implication for prenatal diagnosis of mtDNA disorders. J Med Genet. 2018;55:131-6. [PubMed: 28754700]
- Van Hove JL, Saenz MS, Thomas JA, Gallagher RC, Lovell MA, Fenton LZ, Shanske S, Myers SM, Wanders RJ, Ruiter J, Turkenburg M, Waterham HR. Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res. 2010;68:159-64. [PMC free article: PMC2928220] [PubMed: 20453710]
- Wei Y, Cui L, Peng B. Mitochondrial DNA mutations in late-onset Leigh syndrome. J Neurol. 2018;265:2388-95. [PubMed: 30128709]
- Wei Y, Huang Y, Yang Y, Qian M. MELAS/LS overlap syndrome associated with mitochondrial DNA mutations: clinical, genetic, and radiological studies. Front Neurol. 2021;12:648740. [PMC free article: PMC8137909] [PubMed: 34025555]
- White SL, Collins VR, Wolfe R, Cleary MA, Shanske S, DiMauro S, Dahl HH, Thorburn DR. Genetic counseling and prenatal diagnosis for the mitochondrial DNA mutations at nucleotide 8993. Am J Hum Genet. 1999a;65:474-82. [PMC free article: PMC1377946] [PubMed: 10417290]
- White SL, Shanske S, McGill JJ, Mountain H, Geraghty MT, DiMauro S, Dahl HH, Thorburn DR. Mitochondrial DNA mutations at nucleotide 8993 show a lack of tissue- or age-related variation. J Inherit Metab Dis. 1999b;22:899-914. [PubMed: 10604142]
- Yamashita S, Nishino I, Nonaka I, Goto Y. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. J Hum Genet. 2008;53:598–606. [PubMed: 18414780]
- Yu XL, Yan CZ, Ji KQ, Lin PF, Xu XB, Dai TJ, Li W, Zhao YY. Clinical, neuroimaging, and pathological analyses of 13 Chinese Leigh syndrome patients with mitochondrial DNA mutations. Chin Med J. 2018;131:2705-12. [PMC free article: PMC6247594] [PubMed: 30425197]
- Zhang J, Liu H, Luo S, Lu Z, Chavez-Badiola A, Liu Z, Yang M, Merhi Z, Silber SJ, Munne S, Konstantinidis M, Wells D, Tang JJ, Huang T. Live birth derived from oocyte spindle transfer to prevent mitochondrial disease. Reprod Biomed Online. 2017;34:361-8. [PubMed: 28385334]
- Zweers H, van Wegberg AMJ, Janssen MCH, Wortmann SB. Ketogenic diet for mitochondrial disease: a systematic review on efficacy and safety. Orphanet J Rare Dis. 2021;16:295. [PMC free article: PMC8254320] [PubMed: 34217336]
Publication Details
Author Information and Affiliations
Victoria, Australia
Victoria, Australia
Victoria, Australia
University of Melbourne
Melbourne, Australia
Victoria, Australia
UCL Great Ormond Street Institute of Child Health
London, United Kingdom
Publication History
Initial Posting: October 30, 2003; Last Update: May 9, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Ball M, Thorburn DR, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome Spectrum. 2003 Oct 30 [Updated 2024 May 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
%20for%20an%20individual%20with%20MT-ATP6%20pathogenic%20variants%20m.&p=BOOKS&id=1173_narp-Image001.jpg "Click on image to zoom")
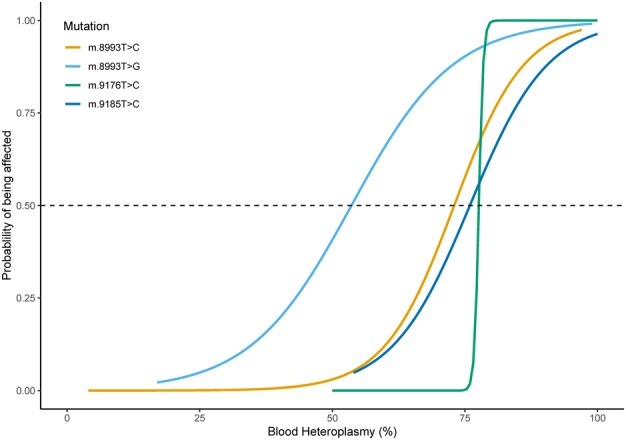
%20to%20develop%20clinical%20manifestations%20of%20Leigh%20syndrome%20spectrum%20due%20to%20MT-ATP6%20pathogenic%20variants%20m.&p=BOOKS&id=1173_narp-Image003.jpg "Click on image to zoom")