Summary
Clinical characteristics.
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) is characterized by fractures (resulting from radiologically demonstrable polycystic osseous lesions), frontal lobe syndrome, and progressive presenile dementia beginning in the fourth decade. The clinical course of PLOSL can be divided into four stages:
- 1.
The latent stage is characterized by normal early development.
- 2.
The osseous stage (3rd decade of life) is characterized by pain and tenderness, mostly in ankles and feet, usually following strain or injury. Fractures are typically diagnosed several years later, most commonly in the bones of the extremities.
- 3.
In the early neurologic stage (4th decade of life), a change of personality begins to develop insidiously. Affected individuals show a frontal lobe syndrome (loss of judgment, euphoria, loss of social inhibitions, disturbance of concentration, and lack of insight, libido, and motor persistence) leading to serious social issues.
- 4.
The late neurologic stage is characterized by progressive dementia and loss of mobility.
Death usually occurs before age 50 years.
Diagnosis/testing.
The diagnosis of PLOSL can be established in a proband with radiologically demonstrable polycystic osseous lesions, frontal lobe syndrome, and progressive presenile dementia beginning in the fourth decade. Identification of biallelic pathogenic variants in TYROBP or TREM2 can confirm the diagnosis if radiographic and clinical features are inconclusive.
Management.
Treatment of manifestations: Treatment is symptomatic. Orthopedic surgery and/or devices may be of value in individual cases. Anti-seizure medication can be used to prevent epileptic seizures and secondary worsening of the condition.
Prevention of secondary complications: Some of the social consequences of PLOSL may be avoided if family members are informed early about the nature of the disorder.
Surveillance: Intervals for follow up of bone lesions and neurologic and psychiatric manifestations must be determined on an individual basis.
Genetic counseling.
PLOSL is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a TREM2 or TYROBP pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives and prenatal testing for a pregnancy at increased risk are possible if the pathogenic variants in the family have been identified.
Diagnosis
No consensus clinical diagnostic criteria for polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) have been published.
Suggestive Findings
PLOSL should be suspected in individuals with the following features:
- Radiologically demonstrable polycystic osseous lesions and fractures of the wrists and ankles after minor trauma at the mean age of 27 years (range 18-33 years) [Paloneva et al 2001]. Cyst-like lesions and loss of bone trabeculae are most conspicuous in the fingers and in the carpal and tarsal bones (see Figure 1 and Figure 2) [Mäkelä et al 1982, Nwawka et al 2014]. See also Figures 2 and 3 in Nwawka et al [2014]. If an individual has multiple lytic or cyst-like lesions on radiograph primarily in the distal extremities, CT and MRI are useful in making an accurate diagnosis [Nwawka et al 2014].
- Frontal lobe syndrome in the fourth decade manifested by euphoria and loss of judgment and social inhibitions
- Progressive presenile dementia beginning in the fourth decade. Dementia is mild at the onset of neurologic symptoms. The disease culminates in severe dementia; affected individuals typically die before age 50 years.

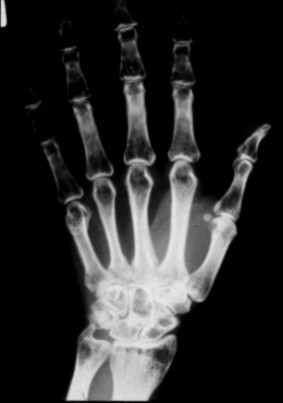
Figure 1.
A radiograph of the hand of a person with PLOSL demonstrates multiple cyst-like lesions and loss of bone trabeculae.

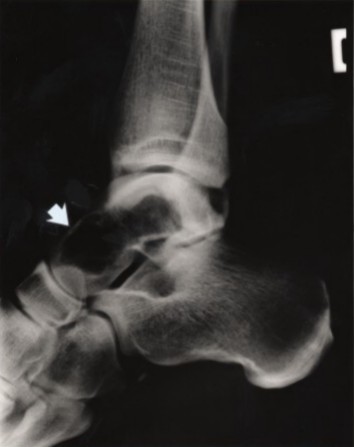
Figure 2.
A radiograph shows a well-demarcated cyst-like lesion (arrow) in the talus of a person with PLOSL, age 28 years. Paloneva et al [2001]; reprinted with permission from Neurology
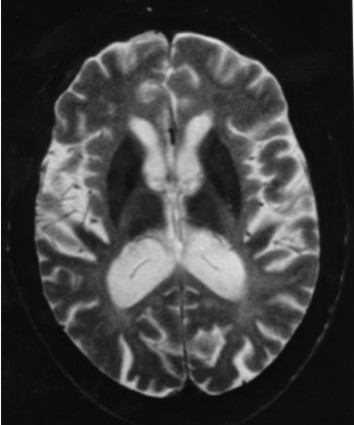
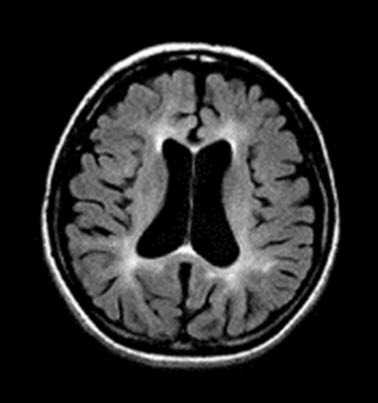
Neuroradiologic findings (Figure 3, Figure 4, Figure 5)


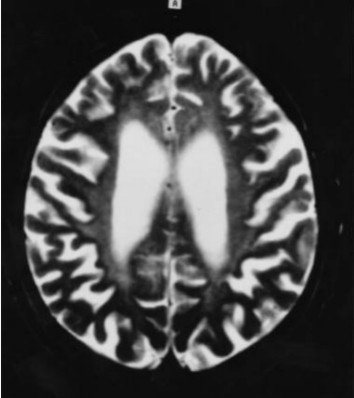
Figure 5.
Brain MRI. High-intensity lesions in the bilateral periventricular white matter on an axial FLAIR image (TR: 8000 ms, TE: 120 ms) Kuroda et al [2007]; reprinted with permission from Elsevier
- Cerebral atrophy. Dilated ventricles, atrophy of the basal ganglia and thalamus, prominent sulci, or thin corpus callosum is a constant finding on CT and MRI and is evident before the appearance of neuropsychiatric symptoms. Cerebellar atrophy may also be present [Paloneva et al 2001, Klünemann et al 2005, Solje et al 2014].
- Bilateral calcifications of the basal ganglia are a common finding on CT. The basal ganglia and thalamus, particularly the putamina, may show very low signal intensities on T2-weighted MRI [Klünemann et al 2005]. Calcifications may occur before central nervous system (CNS) symptoms [Paloneva et al 2001, Solje et al 2014].
- Increased signal intensities of the cerebral white matter are usually found on T2-weighted images after the appearance of clinical CNS symptoms. These white matter changes are diffuse and have no region of predilection, apart from the frontal lobes. The changes are usually centrally located (periventricular white matter, centrum semiovale, internal capsules). As the disease progresses, the high periventricular signal intensity spreads toward the periphery sparing most of the arcuate fibers. In some instances, the white matter changes also extend to the cortex. However, the white matter may look normal in some individuals with CNS symptoms [Paloneva et al 2001].
- SPECT and PET findings are variable. Hypoperfusion of the cortical areas, thalamus, and basal ganglia have been reported [Klünemann et al 2005, Takeshita et al 2005].
Electroencephalogram is normal early in the disease. With advancing disease, individuals show accentuation of theta and delta activity. Initially, theta is typically rhythmic, 6-8 Hz, dominating in the centrotemporal areas; later, diffuse slowing becomes evident. In the late stage of the disease, irritative activity usually appears on EEG [Paloneva et al 2001].
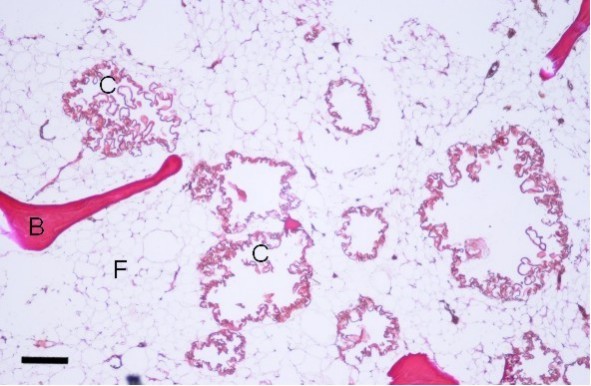
Bone biopsy is not required to establish the diagnosis. The cyst-like bone lesions are filled with lipid material that microscopically consists of characteristic 1-2 µm thick lipid membranes and amorphous lipid substance.
Establishing the Diagnosis
The clinical diagnosis of PLOSL can be established in a proband based on clinical diagnostic criteria [Paloneva et al 2001], or the molecular diagnosis can be established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in TREM2 or TYROBP identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic TREM2 or TYROBP variants of uncertain significance (or of one known pathogenic variant and one variant of uncertain significance) does not establish or rule out the diagnosis.
Clinical diagnosis. The combination of radiologically demonstrable polycystic osseous lesions, frontal lobe syndrome, and progressive presenile dementia beginning in the fourth decade is diagnostic [Paloneva et al 2001].
Molecular diagnosis. Molecular testing approaches can include a combination of gene targeted testing (serial single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of PLOSL has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Serial single-gene testing. The order of testing is based on the individual's ethnicity. In individuals outside of Finland and Japan, TREM2 pathogenic variants appear to be more frequent than those in TYROBP [Klünemann et al 2005].
- 1.
Sequence analysis of each gene is performed first.
- 2.
If only one or no pathogenic variant is found, deletion/duplication analysis is performed next.
A multigene panel that includes TREM2 and TYROBP and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the diagnosis of PLOSL has not been considered because an individual has atypical phenotypic features, comprehensive genomic testing, which does not require the clinician to determine which gene is likely involved, is an option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
Table 1.
Molecular Genetic Testing Used in Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL)
Clinical Characteristics
Clinical Description
The clinical course of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) can be divided into four stages: latent, osseous, early neurologic, and late neurologic [Klünemann et al 2005].
Latent stage. Early development is normal.
Osseous stage (3rd decade of life). The first symptoms of PLOSL appear in early adulthood as pain and tenderness, mostly in the ankles and feet, usually following strain or a minor accident. Fractures are typically diagnosed several years later, most commonly in the bones of the extremities [Paloneva et al 2001]. The first fractures usually occur shortly before age 30 years; however, affected individuals may have had pain and swelling of the ankles and wrists after strain for years. The fractures heal well. It is important to note that some individuals may present with neurologic symptoms without any preceding osseous manifestations [Paloneva et al 2001, Bock et al 2013].
Early neurologic stage (4th decade of life). Personality changes begin insidiously in the fourth decade. Affected individuals show progressive loss of judgment, leading to serious social consequences, including divorce, unemployment, and financial trouble [Paloneva et al 2001, Ilonen et al 2012]. Some individuals may attempt suicide. The full-blown picture of frontal lobe syndrome subsequently appears: loss of judgment; euphoria; lack of social inhibitions, including Witzelsucht; disturbance of concentration; and lack of insight, libido, and motor persistence.
Progressive signs of upper motor neuron involvement (spasticity, extensor plantar reflexes) are noticed. With advancing disease, lack of initiative and activity conceal the aforementioned symptoms [Paloneva et al 2001].
Memory disturbances begin at approximately the same age as the personality changes, and are best detectable by psychometric tests [Vanhanen et al 2013]. The memory disturbance is less severe than the personality change, and affected individuals retain basic personal information until the last stage of the disease.
Other disturbances of higher cortical function, such as motor aphasia, agraphia, acalculia, and apraxia, appear only at the last stage of the disease.
Affected individuals may develop postural dyspraxia: they walk or sit in peculiar skewed postures. Involuntary athetotic or choreatic movements or myoclonic twitches are common. Individuals who reach their mid-thirties frequently experience epileptic seizures. In some individuals, impotence or lack of libido and urinary incontinence are among the first symptoms [Paloneva et al 2001].
Late neurologic stage. In the last stage of the disease, individuals lose their ability to walk and progress to a vegetative state. Primitive reflexes, such as visual and tactile grasp and mouth-opening reflexes, as well as the sucking reflex, may become noticeable. Affected individuals typically die before age 50 years [Paloneva et al 2001].
Bone pathology. The cyst-like bone lesions are filled with lipid material that microscopically consists of characteristic 1-2 µm thick lipid membranes and amorphous lipid substance [Kitajima et al 1989]. See Figure 6.

Neuropathology. Generalized cerebral gyral atrophy with frontal accentuation is observed at autopsy. The corpus callosum is abnormally thin. The central white matter is severely reduced in amount, grayish, and tough. The basal ganglia, particularly the caudate nuclei, are variably reduced in size [Paloneva et al 2001]. All affected individuals show marked hydrocephalus ex vacuo.
Histologic examination reveals scattered neurons showing features of central chromatolysis. Intraneuronal or glial pathologic inclusions have not been observed [Paloneva et al 2001]. Neuronal loss as well as astrocytic proliferation and hypertrophy are observed in the caudate nuclei. In addition, scattered calcospherites are seen, particularly in the putamina and globi pallidi [Kalimo et al 1994, Paloneva et al 2001]. Thalamic degeneration may occur [Kobayashi et al 2000]. Affected individuals show advanced loss of axons and myelin and a pronounced astrocytic reaction in the centrum semiovale, accentuated in the frontal and temporal lobes, with moderate involvement of the gyral white matter. In addition, widespread activation of microglia in the cerebral white matter is seen [Paloneva et al 2001]. Scattered small arterioles and capillaries in the deep frontal and temporal white matter show concentric thickening of the vascular wall with multiple thickened basement membranes and narrowing or obliteration of the lumen [Paloneva et al 2001]. The cerebral cortices are less severely affected [Aoki et al 2011].
Pathologic findings in other organs. Characteristic lipomembranous changes have been described in systemic adipose tissue [Nasu et al 1973]. Pathologic manifestations in organs other than the central nervous system (CNS) and the skeletal system have been insufficiently characterized.
Genotype-Phenotype Correlations
Individuals with homozygous pathogenic variants in TYROBP or TREM2 develop similar CNS manifestations [Paloneva et al 2002, Klünemann et al 2005].
Some TREM2 pathogenic variants have been reported to cause a dementia syndrome resembling PLOSL without evident osseous manifestations (see Genetically Related Disorders).
Nomenclature
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy may be abbreviated as PLOSL.
The first affected individuals were described in the 1960s independently by Järvi and Hakola in Finland and Nasu in Japan.
In the early literature, PLOSL was also known as membranous lipodystrophy. This term is outdated and should not be used.
Prevalence
The prevalence of PLOSL is highest in Finland due to a TYROBP founder variant, deletion of exons 1-4 (c.-2897_277-1227del5265), with an estimated prevalence of 1:1,000,000 to 2:1,000,000 [Pekkarinen et al 1998]. The prevalence of PLOSL in other countries is lower; no detailed data on the prevalence elsewhere are available. Most affected individuals have been diagnosed in Japan (>100 cases) [Pekkarinen et al 1998]. Single families with PLOSL have been diagnosed worldwide [Klünemann et al 2005].
Genetically Related (Allelic) Disorders
No disorders other than those discussed in this GeneReview are known to be caused by mutation of TYROBP. Other phenotypes associated with germline pathogenic variants in TREM2 are summarized in Table 2.
Table 2.
TREM2 Allelic Disorders
Differential Diagnosis
The combination of frontal-type dementia beginning in the fourth decade and radiologically demonstrable polycystic osseous lesions makes it easy to clinically distinguish polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) from the established forms of familial and nonfamilial frontotemporal dementia (e.g., Pick disease, nonspecific frontal lobe degeneration, CSF1R adult-onset leukoencephalopathy with axonal spheroids and pigmented glia, and the various entities of frontotemporal dementia and parkinsonism linked to pathogenic variants in MAPT).
Management
No clinical practice guidelines for polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with PLOSL, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy
Treatment of Manifestations
Only symptomatic treatment is available.
Table 4.
Treatment of Manifestations in Individuals with Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy
Surveillance
The interval of surveillance for bone lesions, neurologic, and psychiatric manifestations must be determined individually.
Table 5.
Recommended Surveillance for Individuals with Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Calcium substitution alone has been shown to be ineffective in preventing the development of the osseous manifestations. The effect of bisphosphonates has not been studied.
It has been speculated that nonsteroidal anti-inflammatory drugs could slow the progression of PLOSL; however, clinical trials have not been performed.
A single individual with PLOSL improved temporarily when taking donepezil [D Hemelsoet, personal observation]. Clinical trials in a series of individuals with PLOSL have not been reported.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., presumed to be carriers of one TREM2 or TYROBP pathogenic variant based on family history).
- If a molecular diagnosis has been established in the proband, molecular genetic testing of the parents is recommended to confirm that both parents are heterozygous for a PLOSL-related pathogenic variant and to allow reliable recurrence risk assessment. (De novo variants are known to occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017].)
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a PLOSL-related pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.Note: Sibs who are younger than age 40 years are at risk for PLOSL. Polycystic osseous lesions in radiographs of the hands and feet of an at-risk adult suggest the diagnosis.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband
- Unless an affected individual's reproductive partner also has PLOSL or is a carrier, offspring will be obligate heterozygotes for a TREM2 or TYROBP pathogenic variant.
- Because of the low carrier rate in the general population, the risk that an affected individual would have children with a carrier is very low except in genetic isolates (see Prevalence).
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a TREM2 or TYROBP pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the PLOSL-related pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being affected or carriers.
Predictive testing (i.e., testing of asymptomatic at-risk individuals)
- Predictive testing for at-risk relatives is possible once the PLOSL-related pathogenic variants have been identified in an affected family member.
- Potential consequences of such testing (including but not limited to socioeconomic changes and the need for long-term follow up and evaluation arrangements for individuals with a positive test result) as well as the capabilities and limitations of predictive testing should be discussed in the context of formal genetic counseling prior to testing.
Predictive testing in minors (i.e., testing of asymptomatic at-risk individuals younger than age 18 years)
- For asymptomatic minors at risk for adult-onset conditions for which early treatment would have no beneficial effect on disease morbidity and mortality, predictive genetic testing is considered inappropriate, primarily because it negates the autonomy of the child with no compelling benefit. Further, concern exists regarding the potential unhealthy adverse effects that such information may have on family dynamics, the risk of discrimination and stigmatization in the future, and the anxiety that such information may cause.
- For more information, see the National Society of Genetic Counselors position statement on genetic testing of minors for adult-onset conditions and the American Academy of Pediatrics and American College of Medical Genetics and Genomics policy statement: ethical and policy issues in genetic testing and screening of children.
In a family with an established diagnosis of PLOSL, it is appropriate to consider testing of symptomatic individuals regardless of age.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the TREM2 or TYROBP pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for PLOSL are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Library of Medicine Genetics Home Reference
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy: Genes and Databases
Table B.
OMIM Entries for Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (View All in OMIM)
Molecular Pathogenesis
TYROBP is a transmembrane adaptor protein that mediates the activation of a wide variety of cells of myeloid and lymphoid origin. On the cell plasma membrane, TYROBP is expressed as a disulfide-bonded homodimer linked to the associated cell surface receptors. Numerous TYROBP-associated cell surface receptors have been reported [Lanier 2009]. The cytoplasmic domain of TYROBP contains an immunoreceptor tyrosine-based activation motif which on receptor engagement becomes phosphorylated and binds the cytoplasmic protein tyrosine kinases SYK and ZAP70.
The protein encoded by TREM2 is an activating cell-surface receptor that forms a complex with the transmembrane adaptor protein TYROBP. The TREM2-TYROBP protein complex regulates the differentiation and function of osteoclasts, the bone-resorbing cells. The differentiation of osteoclasts in TYROBP-deficient individuals is impaired, and the osteoclasts show a reduced bone resorption capability in vitro [Cella et al 2003, Paloneva et al 2003].
TREM2 is expressed by a variety of cells of myeloid origin. TREM2 also activates monocyte-derived dendritic cells and is expressed by macrophages [Thrash et al 2009]. In the central nervous system (CNS), TREM2 and TYROBP are expressed by microglial cells. The exact function of TREM2 in the CNS is unknown [Painter et al 2015]. TREM2/DAP12-mediated signaling has been postulated to promote proliferation, phagocytosis, and migration of microglia by induction and maintenance of microglial activation in the CNS [Konishi & Kiyama 2018].
The intracellular responses underlying the pathogenic mechanisms of PLOSL are poorly understood.
Mechanism of disease causation. Loss of function
Table 6.
Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy: Notable Pathogenic Variants by Gene
Chapter Notes
Revision History
- 10 December 2020 (sw) Comprehensive update posted live
- 12 March 2015 (me) Comprehensive update posted live
- 26 August 2010 (me) Comprehensive update posted live
- 16 April 2009 (jp) Revision: sequence analysis available on a clinical basis for TYROBP and TREM2
- 1 May 2006 (me) Comprehensive update posted live
- 15 March 2004 (me) Comprehensive update posted live
- 24 January 2002 (me) Review posted live
- 31 October 2001 (jp) Original submission
References
Published Guidelines / Consensus Statements
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 12-30-22.
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset conditions. Available online. 2017. Accessed 12-30-22.
Literature Cited
- Aoki N, Tsuchiya K, Togo T, Kobayashi Z, Uchikado H, Katsuse O, Suzuki K, Fujishiro H, Arai T, Iseki E, Anno M, Kosaka K, Akiyama H, Hirayasu Y. Gray matter lesions in Nasu-Hakola disease: a report on three autopsy cases. Neuropathology. 2011;31:135–43. [PubMed: 20880319]
- Arıkan M, Yıldırım A, Togral G, Ekmekçi AB. Extremity manifestations and surgical treatment for Nasu Hakola disease. Case Rep Orthop. 2014;2014:458728. [PMC free article: PMC3965938] [PubMed: 24711942]
- Bock V, Botturi A, Gaviani P, Lamperti E, Maccagnano C, Piccio L, Silvani A, Salmaggi A. Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy (PLOSL): a new report of an Italian woman and review of the literature. J Neurol Sci. 2013;326:115–9. [PubMed: 23399524]
- Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer's disease and other neurodegenerative disorders. Lancet Neurology. 2018;17:721–30. [PubMed: 30033062]
- Cella M, Buonsanti C, Strader C, Kondo T, Salmaggi A, Colonna M. Impaired differentiation of osteoclasts in TREM-2-deficient individuals. J Exp Med. 2003;198:645–51. [PMC free article: PMC2194167] [PubMed: 12913093]
- Chouery E, Delague V, Bergougnoux A, Koussa S, Serre JL, Mégarbané A. Mutations in TREM2 lead to pure early-onset dementia without bone cysts. Hum Mutat. 2008;29:E194–E204. [PubMed: 18546367]
- Dardiotis E, Siokas V, Pantazi E, Dardioti M, Rikos D, Xiromerisiou G, Markou A, Papadimitriou D, Speletas M, Hadjigeorgiou GM. A novel mutation in TREM2 gene causing Nasu-Hakola disease and review of the literature. Neurobiol Aging. 2017;53:194.e13–194.e22. [PubMed: 28214109]
- Guerreiro RJ, Lohmann E, Brás JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H, Emre M, Singleton A, Hardy J. Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia-like syndrome without bone involvement. JAMA Neurol. 2013;70:78–84. [PMC free article: PMC4001789] [PubMed: 23318515]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Ilonen T, Hakola P, Vanhanen M, Tiihonen J. Rorschach assessment of personality functioning in patients with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Acta Neuropsychiatr. 2012;24:236–44. [PubMed: 25286817]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Kalimo H, Sourander P, Järvi O, Hakola P. Vascular changes and blood-brain barrier damage in the pathogenesis of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (membranous lipodystrophy). Acta Neurol Scand. 1994;89:353–61. [PubMed: 8085433]
- Kitajima I, Kuriyama M, Usuki F, Izumo S, Osame M, Suganuma T, Murata F, Nagamatsu K. Nasu-Hakola disease (membranous lipodystrophy). Clinical, histopathological and biochemical studies of three cases. J Neurol Sci. 1989;91:35–52. [PubMed: 2746291]
- Klünemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE, Peltonen L, Paloneva J. The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology. 2005;64:1502–7. [PubMed: 15883308]
- Kobayashi K, Kobayashi E, Miyazu K, Muramori F, Hiramatsu S, Aoki T, Nakamura I, Koshino Y. Hypothalamic haemorrhage and thalamus degeneration in a case of Nasu-Hakola disease with hallucinatory symptoms and central hypothermia. Neuropathol Appl Neurobiol. 2000;26:98–101. [PubMed: 10787346]
- Kondo T, Takahashi K, Kohara N, Takahashi Y, Hayashi S, Takahashi H, Matsuo H, Yamazaki M, Inoue K, Miyamoto K, Yamamura T. Heterogeneity of presenile dementia with bone cysts (Nasu-Hakola disease): three genetic forms. Neurology. 2002;59:1105–7. [PubMed: 12370476]
- Konishi H, Kiyama H. Microglial TREM2/DAP12 signaling: a double-edged sword in neural diseases. Front Cell Neurosci. 2018;12:206. [PMC free article: PMC6087757] [PubMed: 30127720]
- Kuroda R, Satoh J, Yamamura T, Anezaki T, Terada T, Yamazaki K, Obi T, Mizoguchi K. A novel compound heterozygous mutation in the DAP12 gene in a patient with Nasu-Hakola disease. J Neurol Sci. 2007;252:88–91. [PubMed: 17125796]
- Lanier LL. DAP10- and DAP12-associated receptors in innate immunity. Immunol Rev. 2009;227:150–60. [PMC free article: PMC2794881] [PubMed: 19120482]
- Le Ber I, De Septenville A, Guerreiro R, Bras J, Camuzat A, Caroppo P, Lattante S, Couarch P, Kabashi E, Bouya-Ahmed K, Dubois B, Brice A. Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging. 2014;35:2419.e23–5. [PMC free article: PMC4208293] [PubMed: 24910390]
- Mäkelä P, Järví O, Hakola P, Virtama P. Radiologic bone changes of polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Skeletal Radiol. 1982;8:51–4. [PubMed: 7079784]
- Nasu T, Tsukahara Y, Terayama K. A lipid metabolic disease-"membranous lipodystrophy"-an autopsy case demonstrating numerous peculiar membrane-structures composed of compound lipid in bone and bone marrow and various adipose tissues. Acta Pathol Jpn. 1973;23:539–58. [PubMed: 4800725]
- Nwawka OK, Schneider R, Bansal M, Mintz DN, Lane J. Membranous lipodystrophy: skeletal findings on CT and MRI. Skeletal Radiol. 2014;43:1449–55. [PubMed: 24777445]
- Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, Bu G. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener. 2015;10:43. [PMC free article: PMC4560063] [PubMed: 26337043]
- Paloneva J, Autti T, Raininko R, Partanen J, Salonen O, Puranen M, Hakola P, Haltia M. CNS manifestations of Nasu-Hakola disease: a frontal dementia with bone cysts. Neurology. 2001;56:1552–8. [PubMed: 11402114]
- Paloneva J, Kestilä M, Wu J, Salminen A, Böhling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH, Pekkarinen P, Lanier LL, Timonen T, Peltonen L. Loss-of-function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet. 2000;25:357–61. [PubMed: 10888890]
- Paloneva J, Mandelin J, Kiialainen A, Bohling T, Prudlo J, Hakola P, Haltia M, Konttinen YT, Peltonen L. DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med. 2003;198:669–75. [PMC free article: PMC2194176] [PubMed: 12925681]
- Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, Bianchin M, Bird T, Miranda R, Salmaggi A, Tranebjaerg L, Konttinen Y, Peltonen L. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet. 2002;71:656–62. [PMC free article: PMC379202] [PubMed: 12080485]
- Pekkarinen P, Hovatta I, Hakola P, Järvi O, Kestilä M, Lenkkeri U, Adolfsson R, Holmgren G, Nylander PO, Tranebjaerg L, Terwilliger JD, Lönnqvist J, Peltonen L. Assignment of the locus for PLO-SL, a frontal-lobe dementia with bone cysts, to 19q13. Am J Hum Genet. 1998;62:362–72. [PMC free article: PMC1376898] [PubMed: 9463329]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Satoh JI, Yanaizu M, Tosaki Y, Sakai K, Kino Y. Targeted sequencing approach to identify genetic mutations in Nasu-Hakola disease. Intractable Rare Dis Res. 2016;5:269–274. [PMC free article: PMC5116862] [PubMed: 27904822]
- Solje E, Hartikainen P, Valori M, Vanninen R, Tiihonen J, Hakola P, Tienari P, Remes A. The C9ORF72 expansion does not affect the phenotype in Nasu-Hkola disease with the DAP12 mutation. Neurobiology of aging. 2014;35:1780.e13–1780.e17. [PubMed: 24612676]
- Takeshita T, Kaminaga T, Tatsumi T, Hatanaka Y, Furui S. Regional cerebral blood flow in a patient with Nasu-Hakola disease. Ann Nucl Med. 2005;19:309–12. [PubMed: 16097640]
- Thrash JC, Torbett BE, Carson MJ. Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu-Hakola disease. Neurochem Res. 2009;34:38–45. [PMC free article: PMC2655126] [PubMed: 18404378]
- Tranebjaerg L, Schrader H, Paloneva J. Polycystic lipomembranous osteodysplasia. Tidsskr Nor Laegeforen. 2000;120:3196. [Article in Norwegian] [PubMed: 11109371]
- Vanhanen M, Hakola P, Ilonen T, Tiihonen J. Word list learning in patients with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy. Dement Geriatr Cogn Dis Extra. 2013;3:10–5. [PMC free article: PMC3618099] [PubMed: 23569454]
- Williamson JC, Larner AJ. Behavioral variant frontotemporal dementia-like syndrome with novel heterozygous TREM2 frameshift mutation. Alzheimer Dis Assoc Disord. 2019;33:75–76. [PubMed: 30106757]
- Yamazaki K, Yoshino Y, Mori Y, Ochi S, Yoshida T, Ishimaru T, Ueno S. A case of Nasu-Hakola disease without fractures or consanguinity diagnosed using exome sequencing and treated with sodium valproate. Clin Psychopharmacol Neurosci. 2015;13:324–6. [PMC free article: PMC4662179] [PubMed: 26598595]
Publication Details
Author Information and Affiliations
Niuvanniemi Hospital
University of Kuopio
Kuopio, Finland
Haartman Institute
University of Helsinki
Helsinki, Finland
Publication History
Initial Posting: January 24, 2002; Last Update: December 10, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Paloneva J, Autti T, Hakola P, et al. Polycystic Lipomembranous Osteodysplasia with Sclerosing Leukoencephalopathy. 2002 Jan 24 [Updated 2020 Dec 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.