Molecular Pathogenesis
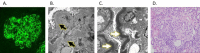
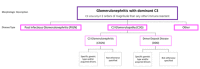
The complement system, composed of the classic pathway and the alternate pathway, is a component of the immune system that enhances the function of antibodies and phagocytes. C3 glomerulopathy (C3G) is caused by uncontrolled activation of the complement alternative pathway.
With the exception of DGKE, all the genes discussed in association with C3G encode proteins in the complement system. C3 and CFB are integral to complement activation and together form C3bBb, a C3 convertase that amplifies the initial complement response, and C3bBbC3b, a C5 convertase that cleaves C5 into C5a and C5b to trigger the terminal pathway. CD46, CFH, CFHR1, CFHR5, and CFI are complement regulators. The role of DGKE in complement activation, although minimal, is believed to be essential in normal podocyte function.
Familial cases of C3G are uncommon and when identified are most often highly penetrant heterozygous copy number variants involving the CFHR1-5 genes (see ), homozygous variants that lead to CFH deficiency, or heterozygous gain-of-function variants in C3. Common to all of these variants is an impact on the regulation of the AP in the fluid phase [Noris & Remuzzi 2017].
Since C3G is rarely inherited in a simple mendelian fashion, the study of rare variants and haplotypes associated with disease is important.
Several studies have shown that some common variants in complement genes are also associated with C3G and increase the odds ratio of developing disease [Abrera-Abeleda et al 2011, Kobayashi et al 2017]. While the identification of common variants that are C3G "risk alleles" cannot be used to direct clinical care, the identification of rare variants in complement genes does affect patient care, especially in the context of a comprehensive assessment of complement function, which includes plasma levels of complement proteins and their split products, assays for autoantibodies, and tests of overall complement activity. For more detailed information, see Osborne et al [2018].
C3
Gene structure.
C3 comprises 41 exons that encode complement C3, which has a molecular weight of 176 kd. The mature protein forms a beta chain and an alpha chain. For a detailed summary of gene and protein information, see Table A.
Benign variants. Several C3 variants (some of which are common in the population) are associated with C3G and define an at-risk haplotype [Hageman et al 2005, Smith et al 2007, Fremeaux-Bacchi et al 2008, Abrera-Abeleda et al 2011, Iatropoulos et al 2016, Riedl et al 2017, Osborne et al 2018].
Pathogenic variants. Pathogenic variants and their location are shown in Table 4.
Table 4.
C3 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Affected Domain | Reference Sequences |
|---|
| c.443G>A | p.Arg148Gln | C3β chain |
NM_000064.3
NP_000055.2
|
| c.1855G>A | p.Val619Met | Linker |
| c.3125G>A | p.Arg1042Gln | TED |
| c.3908G>A | p.Arg1303His | CUBf |
| c.3959G>A | p.Arg1320Gln | CUBf |
| c.4552T>C | p.Cys1518Arg | C345C |
| c.4873T>C | p.Asp1625His | C345C |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
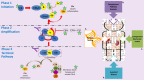
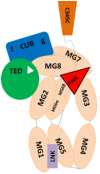
C3 encodes complement component C3, the central activating protein of the AP. This 1,663-amino-acid protein has 15 domains, including the anaphylatoxin (ANA), αNT, CUB (C1r/C1s, Uegf, Bmp1), C345C, thioester (TED), linker (LNK), and anchor domains and eight macroglobulin (MG) domains (see ). C3 is mainly synthesized in the liver, and it is the central protein of the complement system. Spontaneous or proteolytic cleavage of C3 generates the anaphylatoxin C3a (inflammatory effector cells) and the C3b fragment that deposits on cell surfaces triggering the complement cascade activation.
Schematic representation of complement component C3 The structural organization of C3 contains eight macroglobulins, anaphylatoxin (ANA), αNT, CUB (C1r/C1s, Uegf, Bmp1), C345C, linker, and thioester (TED) domains.
Abnormal gene product. Most pathogenic variants listed in Table 4 affect proper cleavage of C3 protein by affecting recognition sites for the binding of CFH and CFI, two regulators of complement activation. Pathogenic variants may also produce reduced quantities of C3 protein (i.e., truncating variants or frameshifts that lead to premature stop variants) [Martínez-Barricarte et al 2010].
CD46
Gene structure.
CD46 (cluster differentiation 46) has 14 exons that encode the 43.7-kd membrane cofactor protein (MCP).
Benign variants. Several variants in CD46 have been associated with C3G and define an at-risk haplotype [Servais et al 2007, Fang et al 2008, Servais et al 2012, Osborne et al 2018].
Normal gene product.
CD46 encodes MCP, a complement regulatory protein of 392 amino acids that is highly expressed in the kidney. It is a transmembrane protein and a member of the regulators of complement activation (RCA), and has eight domains: four short consensus repeat (SCRs 1-4-sushi) domains, which contain ligand-binding domains for decay and cofactor activity; an O-linked-glycosylation site, rich in serine, threonine, and proline (STP) domain, which can be alternately spliced; a helical transmembrane domain; and two cytoplasmic topological domains. Its major role in controlling complement activity is to inactivate C3b and C4b by functioning as a cofactor for factor I [Servais et al 2012, Liszewski & Atkinson 2015].
Abnormal gene product. Pathogenic variants in CD46 typically lead to reduced surface expression of MCP, which contributes to defective surface regulation of complement [Servais et al 2012].
CFB
Gene structure.
CFB comprises 18 exons that encode complement factor B, which has a molecular weight of 93 kd. For a detailed summary of gene and protein information, see Table A.
Benign variants. Benign variants in CFB associated with complement regulation have been described [Gold et al 2006].
Pathogenic variants. The p.Ser367Arg pathogenic variant is in the von Willebrand factor A (VWFa) domain, the catalytic unit of Bb, and is a gain-of-function variant that contributes to AP dysregulation [Imamura et al 2015].
Table 5.
CFB Pathogenic Variants Discussed in This GeneReview
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff has not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
CFB is composed of 764 amino acids and has five domains: three complement component protein (sushi) domains; a VWFa domain; and a peptidase S1 (serine protease) domain. Factor B (fB) is a component of the AP. It binds to C3b and is then cleaved by fD into Ba, a non-catalytic fragment that is released, and Bb, a catalytic subunit that remains bound to C3b to form C3 convertase (C3bBb). The fB cleavage site is near the N-terminus of the VWFa domain.
Abnormal gene product. Pathogenic variants in CFB may lead to gain-of-function properties that contribute to the dysregulation of the complement cascade [Alberts et al 2002, Imamura et al 2015].
CFH
Gene structure.
CFH has 23 exons that encode complement factor H, a protein of 1,231 amino acids. For a detailed summary of gene and protein information, see Table A, Gene.
Benign variants. Several variants of CFH (some of which are common in the population) define an at-risk haplotype that has been associated with C3G [Hageman et al 2005, Servais et al 2007, Smith et al 2007, Zhang et al 2012, Johnson et al 2014, Xiao et al 2014, Merinero et al 2018, Riedl et al 2017, Osborne et al 2018].
Pathogenic variants.
CFH pathogenic variants in C3G occur in heterozygous, homozygous, and compound heterozygous states. CFH has been implicated in C3G by the following:
A report of two sisters with DDD who were homozygous for the
c.670_672delAAG (p.Lys224del) pathogenic variant in
CFH [
Licht et al 2006,
Zipfel et al 2006]. Factor H (fH) carrying this pathogenic variant is present in the serum at normal concentrations but is nonfunctional [
Zipfel et al 2006]. The normal N-terminal activities of fH (C3b binding and complement regulation) are defective, indicating that dysfunctional fH is associated with the development of DDD [
Licht et al 2006].
Studies of skin fibroblasts from a child with fH deficiency and chronic hypocomplementemic renal disease and abnormal fH localization. One copy of a
p.Cys536Arg missense variant and one copy of a
p.Cys959Tyr missense variant were detected in
CFH. Both pathogenic variants affect conserved cysteine residues characteristic of the short consensus-repeat (SCR) modules of fH and therefore predict profound changes in the higher-order structure of the 155-kd protein [
Ault et al 1997].
Two brothers with MPGN were reportedly homozygous for a
p.Arg127Leu amino acid change in
CFH [
Dragon-Durey et al 2004]. Homozygosity for this missense variant is associated with absence of fH in the serum, suggesting that this variant results in sequestration of the protein in the endoplasmic reticulum.
Table 6.
CFH Pathogenic Variants Discussed in This GeneReview
View in own window
DNA Nucleotide Change
(Alias 1) | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.380G>T | p.Arg127Leu |
NM_000186.3
NP_000177.2
|
| c.670_672delAAG | p.Lys224del
(ΔLys224) |
c.1606T>C
(1679T>C) | p.Cys536Arg
(Cys518Arg) |
| c.2655del | p.Arg885SerfsTer13 |
c.2876G>A
(2949G>A) | p.Cys959Tyr
(Cys991Tyr) |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
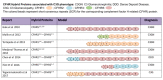
Normal gene product. Complement factor H (fH) protein is the key regulator of the alternative pathway of complement and is composed of 1,231 amino acids. Its structural organization is based on 20 homologous repeat domains (short consensus repeats [SCRs] or sushi domains). Each SCR has 60 amino acids. The first four SCRs (1-4) at the N-terminus are essential for fluid-phase complement regulation, binding of fH to C3b, decay acceleration activity of C3bBb, and fI cofactor activity in mediating the cleavage of C3b to iCb3. The last two SCRs (19-20) at the C terminus bind to cell surfaces to regulate C3 convertase in that microenvironment. See .
Schematic representation of factor H The 20 bead-like homologous short consensus repeats (SCRs) each have 60 amino acids. The first four SCRs (1-4) (red beads) regulate activation in the fluid phase, binding to C3b, decay acceleration activity, and fI (more...)
Abnormal gene product.
CFH pathogenic variants associated with C3G are more frequently found in the N-terminal short consensus repeats (SCRs 1-4), a region essential for fluid-phase complement control. In contrast, in atypical hemolytic uremic syndrome (aHUS), pathogenic variants are more frequently located in the carboxy terminus (SCRs 18-20), a region involved in cell surface regulation.
CFHR1
Gene structure.
CFHR1 comprises six exons that encode complement factor H-related protein 1. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. A duplication of exons 2 through 5 results in an abnormal fusion protein (). The duplication alters the complex oligomerization of the FHR proteins, leading to increased FHR deregulation likely due to increased binding to C3b, iC3b, and C3dg, with decreased fH binding and decreased AP control (see ) [Tortajada et al 2013].
Table 7.
CFHR1 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| Dup exons 1-4 | Duplication of all amino acids in SCR 1-4 of protein |
NM_002113.2
NP_002104.2
|
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
CFHR1 encodes complement factor H-related 1 (FHR1), a protein of 330 amino acids and a member of the regulators of complement activation (RCA) family. Its structural organization is highly homologous to fH, although FHR1 has only five SCRs. The last two SCRs (SCRs 4 and 5) share 96%-100% homology to SCRs 19 and 20 of fH. FHR1 homodimerizes through its first two SCRs and also forms heterodimers with FHR2 and FHR5.
Abnormal gene product. Fusion proteins of FHR1 lead to the formation of complex multimers with FHR1, FHR2, and FHR5 that alter complement regulation by outcompeting fH, which results in reduced convertase regulation (see ) [Xiao et al 2016].
CFHR5
Gene structure.
CFHR5 has ten exons that encode complement factor H-related 5 (FHR5), a protein of 551 amino acids organized into nine SCRs. For a detailed summary of gene and protein information, see Table A, Gene.
Benign variants. Several CFHR5 variants (some of which are common in the population) are associated with C3G and define an at-risk haplotype [Abrera-Abeleda et al 2006, Zipfel et al 2015, Osborne et al 2018].
Pathogenic variants. Pathogenic variants are more frequently associated with C3GN than with DDD (CFHR5 nephropathy is a type of C3GN) [Goicoechea de Jorge et al 2009].
Normal gene product. The normal gene product encoded by CFHR5 is complement factor H-related protein 5 (FHR5), a plasma protein organized like fH in repetitive SCRs. FHR5 has nine SCRs and possesses fI-dependent cofactor activity that leads to inactivation of C3b [McRae et al 2001, Rodríguez de Córdoba et al 2004, McRae et al 2005]. In 92 renal biopsies from patients with different glomerular diseases, FHR5 was present in all complement-containing glomerular immune deposits [Murphy et al 2002], suggesting that FHR5 plays an important role in protecting the glomerulus from complement activation. The precise role of FHR5 in the physiopathology of C3G remains to be determined.
Abnormal gene product. Several abnormal gene products of CFHR5 have been reported in association with C3G. These products usually arise as a consequence of nonallelic homologous recombination, which results in hybrid gene formation (see ).
The presence of CFHR5 pathogenic variants in C3G is consistent with the hypothesis that FHRs play an important role in the complement regulation and disease pathogenesis [Abrera-Abeleda et al 2006, Zhang et al 2013].
CFI
Gene structure.
CFI comprises 13 exons that encode complement factor I (fI), which has a molecular weight of 33 kd. For a detailed summary of gene and protein information, see Table A.
Benign variants. Several CFI variants (some of which are common in the population) are associated with C3G and define an at-risk haplotype [Fremeaux-Bacchi et al 2013, Imamura et al 2015, Chauvet et al 2017, Osborne et al 2018].
Pathogenic variants
c.Ala240Gly – in LDL receptor class A 1 and associated with C3G, aHUS, and AMD. The variant results in partial reduction in secretion [
Nilsson et al 2010].
Table 8.
CFI Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.719C>G | p.Ala240Gly
(Ala222Gly) |
NM_000204.3
NP_000195.2
|
| c.-4C>T | |
| c.170G>A | p.Gly57Asp |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
Normal gene product.
CFI encodes complement fI, a protein of 583 amino acids synthesized in the liver. The fI protein cleaves fluid-phase and cell-bound C3b and C4b, inhibiting their activity in the complement cascade. Factor H and several other RCA proteins are obligatory cofactors for fI activity.
Abnormal gene product. Abnormal fI may lead to a partial reduction in secreted fI, thus compromising its cofactor activity with fH [Servais et al 2012]. Deficiency in fI can lead to complement dysregulation and consequently low serum levels of other complement proteins.
DGKE
Gene structure.
DGKE comprises 11 exons that encode diacylglycerol kinase epsilon, which has a molecular weight of 64 kd. For a detailed summary of gene and protein information, see Table A.
Benign variants. Several DGKE variants (some of which are common in the population) are associated with C3G and define an at-risk haplotype [Osborne et al 2018].
Pathogenic variants. Several loss-of-function variants have been associated with C3G including the following:
Table 9.
DGKE Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.127C>T | p.Gln43Ter |
NM_003647.2
NP_003638.1
|
| c.301A>T | p.Lys101Ter |
| c.610delA | p.Thr204GlnfsTer6 |
| c.966G>A | p.Trp322Ter |
| c.1050G>A | p.Trp350Ter |
Note on variant classification: Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
Note on nomenclature: GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
DGKE is unrelated to the complement pathway. DGKE encodes diacylglycerol kinase epsilon (DGKE), a lipid kinase of 567 amino acids expressed in podocytes, glomerular capillary endothelial cells, and platelets. DGKE plays a key role in signal transduction by affecting the balance between diacylglycerol kinase and phosphatidic acid, thus controlling DAG levels within the cell [Lemaire et al 2013]. The role of DGKE in complement activation is minimal; however, it is believed to play an essential role in normal podocyte function [Ozaltin et al 2013].
In the kidney, DGKE is ubiquitously expressed in podocytes and endothelial cells. Although Dgke
null mice do not have spontaneous clinical signs of kidney disease and have normal serum creatinine and urinary albumin, they develop subclinical microscopic anomalies of the glomerular endothelium that worsen with age, as well as glomerular capillary occlusion when exposed to nephrotoxic serum [Zhu et al 2016].
Abnormal gene product. Abnormal DGKE may lead to prothrombosis through sustained signaling by arachidonic acid-containing diacylglycerol (AA-DAG) [Lemaire et al 2013].