Summary
Clinical characteristics.
Alkaptonuria is caused by deficiency of homogentisate 1,2-dioxygenase, an enzyme that converts homogentisic acid (HGA) to maleylacetoacetic acid in the tyrosine degradation pathway. The three major features of alkaptonuria are dark urine or urine that turns dark on standing, ochronosis (bluish-black pigmentation in connective tissue), and arthritis of the spine and larger joints. Ochronosis generally occurs after age 30 years; arthritis often begins in the third decade. Other manifestations can include pigment in the sclera, ear cartilage, and skin of the hands; aortic or mitral valve calcification or regurgitation and occasionally aortic dilatation; renal stones; prostate stones; and hypothyroidism.
Diagnosis/testing.
The biochemical diagnosis of alkaptonuria in a proband is based on the detection of a significant amount of HGA in the urine (usually 1 to 8 grams per day).
The molecular diagnosis (needed to provide genetic counseling to family members) is based on identification of biallelic pathogenic variants in HGD.
Management.
Treatment of manifestations: Symptomatic management of joint pain is tailored to the individual; physical and occupational therapy help promote optimal muscle strength and flexibility; knee, hip, and shoulder replacements are options when needed; surgical intervention for prostate stones and renal stones as needed; aortic stenosis may necessitate valve replacement; thyroid hormone replacement.
Treatment with nitisinone, which has been shown to slow the progression of symptoms, is approved for use in treatment of alkaptonuria in Europe but not the United States.
Surveillance: In individuals older than age 40 years, echocardiography to detect aortic dilatation, aortic or mitral valve calcification, and stenosis. In individuals with suggestive symptoms, consider CT imaging to detect coronary artery calcification. Assess thyroid function at the time of initial diagnosis, and monitor every 1-2 years thereafter.
Agents/circumstances to avoid: Physical stress to the spine and large joints (including heavy manual labor or high-impact sports) to try to reduce progression of severe arthritis.
Evaluation of relatives at risk: It is appropriate to evaluate apparently asymptomatic older and younger sibs of an affected individual in order to identify as early as possible those who would benefit from preventive measures to help preserve overall joint mobility and function.
Genetic counseling.
Alkaptonuria is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an HGD pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. When both HGD pathogenic variants in the family are known, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
Diagnosis
No consensus clinical diagnostic criteria for alkaptonuria have been published.
Suggestive Findings
Alkaptonuria should be suspected in individuals with the following clinical findings and family history.
Clinical findings
- Dark urine or urine that turns dark on standing. Oxidation of homogentisic acid (HGA) excreted in the urine produces a melanin-like product and causes the urine to turn dark on standing or exposure to an alkaline agent. However, darkening may not occur for several hours after voiding and many individuals never observe any abnormal color to their urine.
- Ochronosis (bluish-black pigmentation of connective tissue). Accumulation of HGA and its oxidation products (e.g., benzoquinone acetic acid) in connective tissue leads to ochronosis.
- Brown pigmentation of the sclera is observed midway between the cornea and the outer and inner canthi at the insertion of the recti muscles. Pigment deposition may also be seen in the conjunctiva and cornea (Figure 1A). The pigmentation does not affect vision [Chévez Barrios & Font 2004].
- Ear cartilage pigmentation is seen in the concha and antihelix (Figure 1B). The cartilage is slate blue or gray and feels irregular or thickened. Calcification of the ear cartilage may be observed on radiographs.
- Pigment also appears in cerumen and in perspiration, causing discoloration of clothing.
- A deep purple or black discoloration may be seen on the skin of the hands, corresponding to the underlying tendons, or in the web between the thumb and index finger.
- Arthritis, often beginning in the spine and resembling ankylosing spondylitis in its large-joint distribution. Radiographs of the spine showing flattened and calcified intervertebral discs are pathognomonic (Figure 1C). Findings include degeneration of the intervertebral discs followed by disc calcification and eventually fusion of the vertebral bodies. Osteophyte formation and calcification of the intervertebral ligaments also occur. Radiographs of the large joints may show joint space narrowing, subchondral cysts, and osteophyte formation. Enthesopathy can be seen at the muscle insertions [Mannoni et al 2004].Note: In some individuals, the diagnosis of alkaptonuria is identified only after the individual seeks medical attention for chronic joint pain or after black articular cartilage is noted during orthopedic surgery.
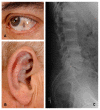
Figure 1
A. Ochronosis of the sclera of the eye B. Ochronosis of the antihelix and concha
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The biochemical diagnosis of alkaptonuria is established in a proband with suggestive clinical findings and a significant amount of homogentisic acid (HGA) in a urine sample detected by gas chromatography-mass spectrometry analysis or liquid chromatography tandem mass spectrometry [Phornphutkul et al 2002; Hughes et al 2014]. The amount of HGA excreted in the urine per day in individuals with alkaptonuria is usually between one and eight grams; a control 24-hour urine sample contains 20-30 mg of HGA. (Note: Elevated HGA can also be detected on a random urine sample.)
The molecular diagnosis of alkaptonuria is established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in HGD identified by molecular genetic testing (see Table 1). While molecular genetic testing is not required to establish the diagnosis of alkaptonuria in a proband, it is required to provide genetic counseling to family members (see Genetic Counseling).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic HGD variants of uncertain significance (or of one known HGD pathogenic variant and one HGD variant of uncertain significance) does not establish or rule out the diagnosis.
Single-gene testing is used when laboratory findings have established the diagnosis of alkaptonuria. Sequence analysis of HGD is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Table 1.
Molecular Genetic Testing Used in Alkaptonuria
Clinical Characteristics
Clinical Description
The clinical findings of alkaptonuria include connective tissue ochronosis and arthritis of the spine and larger joints. Urinary excretion of homogentisic acid (HGA) and disease severity can vary significantly within the same family. Alkaptonuria does not cause developmental delay or cognitive impairment and does not generally reduce the life span of affected individuals.
Connective Tissue
In general, pigmentary changes are observed after age 30 years. Tendon-related findings, including a thickened Achilles tendon, tendonitis, and rupture, have also been observed clinically [Phornphutkul et al 2002] and are demonstrable by MRI.
Joints
Ochronotic arthritis is a regular manifestation of longstanding alkaptonuria. Involvement of the spine usually appears in the third decade. In one large series, low back pain was observed prior to age 30 years in 49% of individuals and prior to age 40 years in 94% [Phornphutkul et al 2002].
Lumbar and thoracic spine symptoms precede cervical spine symptoms. The sacroiliac region is usually spared. Limitation of spine flexion directly correlates with degree of disability. Individuals with decreased forward flexion demonstrate impaired function and increased fatigue [Perry et al 2006]. In a review of case reports of symptomatic myelopathy requiring surgical intervention, the most frequent to least frequent causes were cervical, thoracic, and lumbar myelopathy [Donaldson et al 2019].
Joint disease appears to start earlier and progress more rapidly in males than in females. Knees, hips, and shoulders are frequently affected. Fifty percent of individuals require at least one joint replacement by age 55 years [Phornphutkul et al 2002]. Small joint involvement is less common.
Because the kidneys are responsible for secreting massive quantities of HGA, impaired renal function can accelerate the development of ochronosis and joint destruction [Introne et al 2002].
Other Organ Involvement
Heart. Pigment deposition in the heart valves and blood vessels leads to aortic or mitral valve calcification with stenosis or regurgitation and occasionally aortic dilatation. Aortic valve stenosis occurs at a high frequency in the sixth and seventh decades of life. Unlike cardiac valve disease that occurs in the general population, correlation with standard cardiovascular risk factors is not observed. Aortic stenosis may necessitate aortic valve replacement. Coronary artery calcification has been demonstrated on chest CT [Hannoush et al 2012].
Renal stones. By age 64 years, 50% of individuals with alkaptonuria have a history of renal stones.
Renal function is critical for individuals with alkaptonuria as the kidney actively secretes HGA. Kidney injury, either acute or chronic, may impair the elimination of HGA and cause HGA to accumulate in the blood and tissues. This may lead to acidosis, hemolysis, and methemoglobinemia, which may be fatal [Davison et al 2016, Freeman & Wills 2018, Hugar et al 2019].
Prostate stones. Black prostate stones occur relatively frequently in individuals with alkaptonuria. In one series, eight of 27 men age 31-60 years had prostate stones. Prostate stones may contribute to recurrent infection or urinary obstruction and require surgical removal.
Thyroid. Hypothyroidism occurs at an increased frequency in alkaptonuria. In a single-center study, the prevalence of hypothyroidism was 16% compared to a prevalence of 3.7% in the general population [Avadhanula et al 2020].
Ochronotic pigment in the eye. In one individual with alkaptonuria, the ochronotic pigment in the eye was misdiagnosed as melanosarcoma, resulting in enucleation of the eye [Skinsnes 1948].
Genotype-Phenotype Correlations
No correlation is observed between the type of HGD pathogenic variant and amount of HGA excreted or disease severity.
While analysis of HGD variants from 172 individuals with alkaptonuria revealed that residual HGA activity ranged from 1% to more than 30%, there was no observed difference in serum HGA, urinary excretion of HGA, or clinical manifestations [Ascher et al 2019].
Nomenclature
Occasionally alkaptonuria is referred to collectively (and incorrectly) as ochronosis.
Prevalence
At least 1000 affected individuals have been described in the literature; this is likely an underestimate. The incidence of alkaptonuria in the US is estimated at 1:250,000 to 1:1,000,000 live births.
Alkaptonuria occurs worldwide.
The prevalence of alkaptonuria in Slovakia is estimated at 1:19,000 [Zatkova et al 2003]. A high prevalence has been observed in northwestern Slovakia, likely as the result of a founder effect. Four pathogenic variants (c.457dup, c.481G>A, c.808G>A, and c.1111dup) represent Slovak founder variants, accounting for 80% of all pathogenic variants found in the Slovak population (see Table 2).
A high prevalence has been observed in the Dominican Republic due to the founder variant c.360T>G [Goicoechea De Jorge et al 2002].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with biallelic pathogenic variants in HGD.
Differential Diagnosis
Ochronosis. Ochronosis resulting from alkaptonuria may be confused with acquired, reversible pigmentary changes following prolonged use of carbolic acid dressings for chronic cutaneous ulcers [La Du 2001].
Chemically induced ochronosis has also been described following long-term use of either the antimalarial agent Atabrine® [Ludwig et al 1963], the skin-lightening agent hydroquinone, or the antibiotic minocycline [Suwannarat et al 2004, Stichman & West 2016].
A thorough history combined with lack of excessive HGA excretion in the urine should eliminate false positive diagnoses.
Arthritis. While the arthritis of alkaptonuria resembles ankylosing spondylitis in its damage to the spine and large joints, it differs in sparing the sacroiliac joint and in its radiographic appearance. Radiographic findings of the spine also differentiate alkaptonuria from rheumatoid arthritis and osteoarthritis.
Management
No clinical practice guidelines for alkaptonuria have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with alkaptonuria, the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Complete history and physical examination with particular attention to range of motion in the spine and large joints
- Physical medicine and rehabilitation evaluation if limited range of motion or joint pain occurs
- Electrocardiogram and echocardiogram in individuals older than age 40 years
- Renal ultrasound examination or helical abdominal CT to evaluate for the presence of renal calculi
- Measurement of TSH and free thyroxine to evaluate for primary hypothyroidism
- Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature, mode of inheritance, and implications of alkaptonuria in order to facilitate medical and personal decision making
Treatment of Manifestations
In the US, treatment of alkaptonuria remains symptomatic. While the European Medicines Agency has authorized marketing of nitisinone for treatment of alkaptonuria, to date the US Food and Drug Administration has not approved use of nitisinone for treatment of alkaptonuria.
Symptomatic Management
Joint pain is substantial in individuals with alkaptonuria; close attention to pain control is necessary. Many different pain management options may be considered including nonsteroidal anti-inflammatory drugs, medications for neuropathic pain, and in some cases opiates. Alternative pain management options including physical therapy, a transcutaneous electrical nerve stimulation unit, and nerve blocks may also be considered. Each individual is different and may have contraindications to some classes of medications. Optimal pain management should be tailored to the individual with close follow up and long-term management. Consultation with specialists in pain management may also be considered.
Physical and occupational therapy are important to promote optimal muscle strength and flexibility. Maintaining joint range of motion through moderate non-weight-bearing exercise such as swimming may have beneficial effects.
Knee, hip, and shoulder replacement surgeries are options for managing significant arthritis. In general, the goal of joint replacement is pain relief rather than increased range of motion. Joint replacement in individuals with alkaptonuria is associated with prosthetic survival comparable to that found in individuals with osteoarthritis. In individuals with alkaptonuria total joint replacement significantly improves function compared with preoperative disability [Rajkumar et al 2020].
Aortic stenosis may necessitate valve replacement.
Prostate stones and renal stones may require surgical intervention.
Hypothyroidism should be treated with thyroid hormone supplementation.
Nitisinone
Nitisinone, an inhibitor of 4-hydroxyphenylpyruvate dioxygenase (the enzyme that produces HGA), is approved for the treatment of tyrosinemia type I in the US and Europe.
Nitisinone therapeutic trials in alkaptonuria. In a three-year therapeutic trial, 2 mg of nitisinone taken daily reduced urine and plasma HGA by 95% throughout the study duration [Introne et al 2011]. Without dietary restriction, plasma tyrosine averaged 800 μmol/L, an approximate tenfold elevation. Side effects were minimal: one individual developed corneal crystals that required discontinuation of nitisinone; another had elevated liver transaminases. While statistically significant improvements in hip range of motion and measurements of musculoskeletal function were not observed in the treatment group compared to the control group, there was a positive trend (which was not statistically significant) in the slowing of the rate of progression of aortic stenosis.
A follow-up multi-center randomized controlled study (ClinicalTrials.gov; NCT01916382) using 10 mg of nitisinone daily reduced urinary HGA by 98%-99% [Ranganath et al 2020]. The primary outcome of this study was sustained reduction of urinary HGA at 12 months, with similar reductions in serum concentration of HGA. Using the Alkaptonuria Severity Score Index as a measure of disease severity, the study showed a decreased rate of progression in individuals receiving nitisinone compared to controls receiving no treatment. Adverse events included corneal keratopathy (due to elevated plasma tyrosine concentration) in 13% of treated individuals and an increased rate of infections (the cause of which was unclear).
Ineffective Treatments
Ineffective treatments include the following:
- Studies of dietary restriction of phenylalanine and tyrosine to reduce the production of HGA have not shown efficacy; furthermore, severe restriction of these amino acids is not practical in the long term and may be dangerous.
- Although high-dose vitamin C decreases urinary benzoquinone acetic acid (a derivative of HGA), it has no effect on HGA excretion [Wolff et al 1989]. Furthermore, no credible studies have demonstrated clinical efficacy of ascorbic acid [La Du 2001].
Surveillance
Cardiac. Surveillance for cardiac complications every one to two years is advisable after age 40 years,]; surveillance should include echocardiography to detect aortic dilatation and aortic or mitral valve calcification and stenosis.
Individuals with symptoms suggestive of coronary artery disease may be candidates for CT imaging, depending on the recommendation of a medical provider.
Urology. Urologic complications become more prevalent after age 40 years:
- Routine surveillance is not recommended, but awareness of this potential complication is advised.
- Ochronotic prostate stones appear on radiography; kidney stones can be identified by ultrasonography and helical abdominal CT.
Thyroid. Assess thyroid function (using TSH and free thyroxine) at the time of initial diagnosis, and monitor for primary hypothyroidism every one to two years thereafter.
Agents/Circumstances to Avoid
Avoidance of physical stress to the spine and large joints, including heavy manual labor or high-impact sports, may reduce the progression of severe arthritis.
Younger individuals with alkaptonuria should be directed toward non-contact and lower-impact sports.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger sibs of an affected individual in order to identify as early as possible those who would benefit from preventive measures. Evaluations can include:
- Biochemical testing for the presence of elevated urinary homogentisic acid (HGA);
- Molecular genetic testing if the HGD pathogenic variants in the family are known.
Those found to have alkaptonuria should be counseled to avoid high-impact and contact sports. Career considerations include avoidance of occupations involving heavy physical labor. Instruction on joint strengthening and flexibility exercises, in conjunction with appropriate physical activity, can help preserve overall joint mobility and function.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Alkaptonuria is inherited in an autosomal recessive manner.
Note: The molecular diagnosis of alkaptonuria must be established in a proband (i.e., both HGD pathogenic variants identified) in order to provide reliable recurrence risk assessment for family members.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one HGD pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an HGD pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an HGD pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Urinary excretion of homogentisic acid and disease severity can vary significantly between sibs with the same HGD pathogenic variants.
- Heterozygotes are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with alkaptonuria are obligate heterozygotes (carriers) for a pathogenic variant in HGD.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an HGD pathogenic variant.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the HGD pathogenic variants in the family.
Biochemical testing. Biochemical genetic testing is not reliable as a method of carrier detection.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the HGD pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing for alkaptonuria are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- AKU Society of North America
- Alkaptonuria Society66 Devonshire RoadCambridge CB1 2BLUnited KingdomPhone: +44 1223 322897Email: info@akusociety.org
- Alkaptonuria: A Fact Sheet for PatientsNational Institutes of HealthPhone: 800-411-1222 (toll-free)Email: prpl@cc.nih.gov
- MedlinePlus
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- ApreciseKUre Registry
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Alkaptonuria: Genes and Databases
Table B.
OMIM Entries for Alkaptonuria (View All in OMIM)
Molecular Pathogenesis
Alkaptonuria is caused by a deficiency of the enzyme homogentisate 1,2-dioxygenase (HGD), the third enzyme of the tyrosine degradation pathway (Figure 2). Biallelic pathogenic variants in HGD lead to significantly decreased enzyme function. Deficiency of HGD causes accumulation of homogentisic acid (HGA). HGA is subsequently oxidized to benzoquinones, forming melanin-like pigments that bind to cartilage and connective tissue [Ranganath et al 2019].
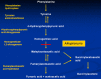
Figure 2.
The tyrosine degradation pathway. Alkaptonuria is characterized by deficiency of homogentisate 1,2-dioxygenase, which converts homogentisic acid (HGA) to maleylacetoacetic acid.
Mechanism of disease causation. Loss of function
Table 2.
Notable HGD Pathogenic Variants
Chapter Notes
Author Notes
Dr Introne is a pediatrician, clinical geneticist, and biochemical geneticist who has cared for more than 170 patients with alkaptonuria since 2000.
Dr Perry is a physiatrist who has cared for more than 170 patients with alkaptonuria.
Dr Chen is a cardiologist who specializes in cardiovascular CT and MRI, and has imaged more than 150 patients with alkaptonuria.
Acknowledgments
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland. The authors would like to thank Dr William Gahl for his previous authorship on this review and his commitment to patients with alkaptonuria. We would also like to thank the AKU Society and all of the member chapters who have supported our research as well as patients, families, and health care providers.
Author History
Marcus Chen, MD (2021-present)
William A Gahl, MD, PhD; National Human Genome Research Institute (2003-2021)
Wendy J Introne, MD (2003-present)
Michael A Kayser, DO; Saint Francis Hospital, Tulsa (2007-2013)
Monique Perry, MD (2021-present)
Chanika Phornphutkul, MD; Brown University (2003-2007)
Pim Suwannarat, MD; Mahidol University (2003-2007)
Revision History
- 10 June 2021 (bp) Comprehensive update posted live
- 12 May 2016 (sw) Comprehensive update posted live
- 22 August 2013 (me) Comprehensive update posted live
- 10 March 2011 (me) Comprehensive update posted live
- 2 July 2009 (cd) Revision: sequence analysis available clinically
- 4 December 2007 (me) Comprehensive update posted live
- 26 June 2006 (ca) Revision: targeted mutation analysis available for eight mutations
- 24 May 2005 (me) Comprehensive update posted live
- 9 May 2003 (me) Review posted live
- 4 March 2003 (ps) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Alkaptonuria" is in the public domain in the United States of America.
References
Literature Cited
- Ascher DB, Spiga O, Sekelska M, Pires DEV, Bernini A, Tiezzi M, Kralovicova J, Borovska I, Soltysova A, Olsson B, Galderisi S, Cicaloni V, Ranganath L, Santucci A, Zatkova A. Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU. Eur J Hum Genet. 2019;27:888–902. [PMC free article: PMC6777518] [PubMed: 30737480]
- Avadhanula S, Introne WJ, Auh S, Soldin SJ, Stolze B, Regier D, Ciccone C, Hannah-Shmouni F, Filie AC, Burman KD, Klubo-Gwiezdzinska J. Assessment of thyroid function in patients with alkaptonuria. JAMA Netw Open. 2020;3:e201357. [PMC free article: PMC7090965] [PubMed: 32202644]
- Beltrán-Valero de Bernabé D, Granadino B, Chiarelli I, Porfirio B, Mayatepek E, Aquaron R, Moore MM, Festen JJ, Sanmartí R, Peñalva MA, de Córdoba SR. Mutation and polymorphism analysis of the human homogentisate 1, 2-dioxygenase gene in alkaptonuria patients. Am J Hum Genet. 1998;62:776–84. [PMC free article: PMC1377044] [PubMed: 9529363]
- Beltrán-Valero de Bernabé D, Peterson P, Luopajarvi K, Matintalo P, Alho A, Konttinen Y, Krohn K, Rodriguez de Cordoba S, Ranki A. Mutational analysis of the HGO gene in Finnish alkaptonuria patients. J Med Genet. 1999;36:922–3. [PMC free article: PMC1734273] [PubMed: 10594001]
- Chévez Barrios P, Font RL. Pigmented conjunctival lesions as initial manifestation of ochronosis. Arch Ophthalmol. 2004;122:1060–3. [PubMed: 15249376]
- Davison AS, Milan AM, Gallagher JA, Ranganath LR. Acute fatal metabolic complications in alkaptonuria. J Inherit Metab Dis. 2016;39:203–10. [PubMed: 26596578]
- Donaldson CJ, Mitchell SL, Riley LH 3rd, Kebaish KM. "As black as ink": a case of alkaptonuria-associated myelopathy and a review of the literature. Spine (Phila Pa 1976). 2019;44:E53–E59. [PubMed: 29933333]
- Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, Renedo M, Fernández-Ruiz E, Peñalva MA, Rodríguez de Córdoba S. The molecular basis of alkaptonuria. Nat Genet. 1996;14:19–24. [PubMed: 8782815]
- Freeman AR, Wills SM. Fatal methemoglobinemia complicating alkaptonuria (ochronosis): a rare presentation. Forensic Sci Med Pathol. 2018;14:236–40. [PubMed: 29572623]
- Gehrig A, Schmidt SR, Muller CR, Srsen S, Srsnova K, Kress W. Molecular defects in alkaptonuria. Cytogenet Cell Genet. 1997;76:14–6. [PubMed: 9154114]
- Goicoechea De Jorge E, Lorda I, Gallardo ME, Pérez B, Peréz De Ferrán C, Mendoza H, Rodríguez De Córdoba S. Alkaptonuria in the Dominican Republic: identification of the founder AKU mutation and further evidence of mutation hot spots in the HGO gene. J Med Genet. 2002;39:E40. [PMC free article: PMC1735184] [PubMed: 12114497]
- Hannoush H, Introne WJ, Chen MY, Lee S-J, O'Brien K, Suwannarat P, Kayser MA, Gahl WA, Sachdev V. Aortic stenosis and vascular calcifications in alkaptonuria. Mol Genet Metab. 2012;105:198–202. [PMC free article: PMC3276068] [PubMed: 22100375]
- Hugar SB, Shulman J, Yanta J, Nine J. Ochronosis presenting as methemoglobinemia. J Forensic Sci. 2019;64:913–6. [PubMed: 30229904]
- Hughes AT, Milan AM, Christensen P, Ross G, Davison AS, Gallagher JA, Dutton JJ, Ranganath LR. Urine homogentisic acid and tyrosine: simultaneous analysis by liquid chromatography tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;963:106–12. [PubMed: 24952314]
- Introne WJ, Perry MB, Troendle J, Tsilou E, Kayser MA, Suwannarat P, O'Brien KE, Bryant J, Sachdev V, Reynolds JC, Moylan E, Bernardini I, Gahl WA. A 3-year randomized therapeutic trial of nitisinone in alkaptonuria. Mol Genet Metab. 2011;103:307–14. [PMC free article: PMC3148330] [PubMed: 21620748]
- Introne WJ, Phornphutkul C, Bernardini I, McLaughlin K, Fitzpatrick D, Gahl WA. Exacerbation of the ochronosis of alkaptonuria due to renal insufficiency and improvement after renal transplantation. Mol Genet Metab. 2002;77:136–42. [PubMed: 12359141]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- La Du BN. Alkaptonuria. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York, NY: McGraw-Hill; 2001:2109-23.
- Ludwig GD, Toole JF, Wood JC. Ochronosis from quinacrine (atabrine). Ann Intern Med. 1963;59:378–84. [PubMed: 14065956]
- Mannoni A, Selvi E, Lorenzini S, Giorgi M, Airo P, Cammelli D, Andreotti L, Marcolongo R, Porfirio B. Alkaptonuria, ochronosis, and ochronotic arthropathy. Semin Arthritis Rheum. 2004;33:239–48. [PubMed: 14978662]
- Müller CR, Fregin A, Srsen S, Srsnova K, Halliger-Keller B, Felbor U, Seemanova E, Kress W. Allelic heterogeneity of alkaptonuria in Central Europe. Eur J Hum Genet. 1999;7:645–51. [PubMed: 10482952]
- Perry MB, Suwannarat P, Furst GP, Gahl WA, Gerber LH. Musculoskeletal findings and disability in alkaptonuria. J Rheumatol. 2006;33:2280–5. [PubMed: 16981292]
- Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y, Gerber LH, Gahl WA. Natural history of alkaptonuria. N Engl J Med. 2002;347:2111–21. [PubMed: 12501223]
- Rajkumar N, Soundarrajan D, Dhanasekararaja P, Rajasekaran S. Clinical and radiological outcomes of total joint arthroplasty in patients with ochronotic arthropathy. Eur J Orthop Surg Traumatol. 2020;30:923–9. [PubMed: 32172376]
- Ranganath LR, Norman BP, Gallagher JA. Ochronotic pigmentation is caused by homogentisic acid and is the key event in alkaptonuria leading to the destructive consequences of the disease-A review. J Inherit Metab Dis. 2019;42:776–92. [PubMed: 31282009]
- Ranganath LR, Psarelli EE, Arnoux JB, Braconi D, Briggs M, Bröijersén A, Loftus N, Bygott H, Cox TF, Davison AS, Dillon JP, Fisher M, FitzGerald R, Genovese F, Glasova H, Hall AK, Hughes AT, Hughes JH, Imrich R, Jarvis JC, Khedr M, Laan D, Le Quan Sang KH, Luangrath E, Lukáčová O, Milan AM, Mistry A, Mlynáriková V, Norman BP, Olsson B, Rhodes NP, Rovenský J, Rudebeck M, Santucci A, Shweihdi E, Scott C, Sedláková J, Sireau N, Stančík R, Szamosi J, Taylor S, van Kan C, Vinjamuri S, Vrtíková E, Webb C, West E, Záňová E, Zatkova A, Gallagher JA. Efficacy and safety of once-daily nitisinone for patients with alkaptonuria (SONIA 2): an international, multicentre, open-label, randomised controlled trial. Lancet Diabetes Endocrinol. 2020;8:762–72. [PubMed: 32822600]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Skinsnes OK. Generalized ochronosis; report of an instance in which it was misdiagnosed as melanosarcoma, with resultant enucleation of an eye. Arch Pathol (Chic). 1948;45:552–8. [PubMed: 18891026]
- Stichman JR, West SG. Minocycline-induced cartilage hyperpigmentation mimicking alkaptonuria in a patient with knee pain. J Rheumatol. 2016;43:825. [PubMed: 27037242]
- Suwannarat P, Phornphutkul C, Bernardini I, Turner M, Gahl WA. Minocycline-induced hyperpigmentation masquerading as alkaptonuria in individuals with joint pain. Arthritis Rheum. 2004;50:3698–701. [PubMed: 15529343]
- Vilboux T, Kayser M, Introne W, Suwannarat P, Bernardini I, Fischer R, O’Brien K, Kleta R, Huizing M, Gahl WA. Mutation spectrum of homogentisic acid oxidase (HGD) in alkaptonuria. Hum Mutat. 2009;30:1611–9. [PMC free article: PMC2830005] [PubMed: 19862842]
- Wolff JA, Barshop B, Nyhan WL, Leslie J, Seegmiller JE, Gruber H, Garst M, Winter S, Michals K, Matalon R. Effects of ascorbic acid in alkaptonuria: alterations in benzoquinone acetic acid and an ontogenic effect in infancy. Pediatr Res. 1989;26:140–4. [PubMed: 2771520]
- Zatkova A, Chmelikova A, Polakova H, Ferakova E, Kadasi L. Rapid detection methods for five HGO gene mutations causing alkaptonuria. Clin Genet. 2003;63:145–9. [PubMed: 12630963]
- Zatková A, de Bernabé DB, Poláková H, Zvarík M, Feráková E, Bosák V, Ferák V, Kádasi L, de Córdoba SR. High frequency of alkaptonuria in Slovakia: evidence for the appearance of multiple mutations in HGO involving different mutational hot spots. Am J Hum Genet. 2000;67:1333–9. [PMC free article: PMC1288576] [PubMed: 11017803]
Publication Details
Author Information and Affiliations
National Human Genome Research Institute
National Institutes of Health
Bethesda, Maryland
Clinical Center
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
Publication History
Initial Posting: May 9, 2003; Last Update: June 10, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Introne WJ, Perry M, Chen M. Alkaptonuria. 2003 May 9 [Updated 2021 Jun 10]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

