|
AMER1
|
Osteopathia striata with cranial sclerosis
| XL | Longitudinal striations of sclerotic long bones in combination w/osteosclerosis of cranial & facial bones | Short stature, delayed closure of anterior fontanelle, micrognathia, linear striations in long bones of females |
|
FLNA
| Frontometaphyseal dysplasia type 1 (See Otopalatodigital Spectrum Disorders.) | XL | Skeletal findings are frontal bone hyperostosis & metaphyseal dysplasia (similar to those seen in Pyle disease). | Urogenital defects, contractures in hands, elbows, knees, & ankles |
|
GJA1
| Autosomal recessive craniometaphyseal dysplasia (AR-CMD) (OMIM 218400) | AR | Hyperostosis of cranial base & cranial vault w/metaphyseal flaring similar to AD-CMD | Skeletal phenotype may be less severe than in typical AD-CMD. |
|
LRP5
| Autosomal dominant osteopetrosis type 1 (OMIM 607634) | AD | Cranial sclerosis & high bone mass w/o ↑ fragility | Diffuse osteosclerosis, no metaphyseal flaring |
|
SFRP4
| Pyle disease (OMIM 265900) | AR | Metaphyseal dysplasia | Little or no involvement of cranial bones in Pyle disease |
|
SOST
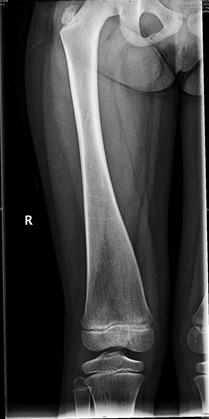
| Craniodiaphyseal dysplasia (CDD) (OMIM 218300) | AD | Progressive overgrowth of craniofacial bones w/deafness, facial palsy, & visual disturbance due to nerve entrapment Choanal stenosis is a clinically significant complication. Radiologically, cranial & facial bones are hyperostotic while diaphyses of limb bones are expanded, w/thin cortices.
| Cranial & facial thickening are generally more severe in CDD than in CMD. |
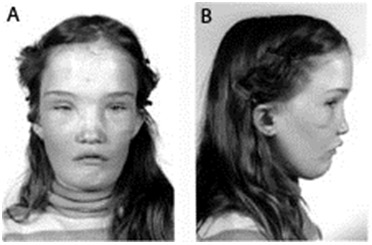
| Sclerosteosis (See SOST Sclerosing Bone Dysplasias.) | AR | Progressive skeletal overgrowth (most pronounced in skull & mandible) & variable syndactyly Facial distortion due to bossing of forehead & mandibular overgrowth becomes apparent in early childhood w/progression into adulthood. Hyperostosis of skull → narrowing of foramina & entrapment of 7th cranial nerve (→ facial palsy) w/other, less common nerve entrapment syndromes. Hyperostosis of calvarium ↓ intracranial volume, ↑ risk for potentially lethal elevation of intracranial pressure. Survival into old age is unusual but not unprecedented.
| Sclerosis in spine & pelvis, 2-3 finger syndactyly, nail dysplasia, no metaphyseal flaring, gigantism |
| Van Buchem disease (See SOST Sclerosing Bone Dysplasias.) | AR | Progressive skeletal overgrowth Van Buchem disease is generally milder than sclerosteosis; no syndactyly. Life span appears normal.
| Osteosclerosis incl clavicles & ribs; hyperphosphatasemia. |
|
TGFB1
| Progressive diaphyseal dysplasia | AD | Hyperostosis of skull results in narrowing of foramina, causing facial palsy & deafness. | Diaphyseal hyperostosis of long bones is pronounced. |