Summary
Clinical characteristics.
Aceruloplasminemia is characterized by iron accumulation in the brain and viscera. The clinical triad of retinal degeneration, diabetes mellitus (DM), and neurologic disease is seen in individuals ranging from age 30 years to older than 70 years. The neurologic findings of movement disorder (blepharospasm, grimacing, facial and neck dystonia, tremors, chorea) and ataxia (gait ataxia, dysarthria) correspond to regions of iron deposition in the brain. Individuals with aceruloplasminemia often present with anemia prior to onset of DM or obvious neurologic problems. Cognitive dysfunction including apathy and forgetfulness occurs in more than half of individuals with this condition.
Diagnosis/testing.
Aceruloplasminemia, a disorder of iron metabolism caused by the complete absence of ceruloplasmin ferroxidase activity, is associated with very low to absent serum ceruloplasmin and some combination of the following:
Low serum copper concentration
Low serum iron concentration
High serum ferritin concentration
Increased hepatic iron concentration
The diagnosis of aceruloplasminemia is established in a proband with typical clinical findings and the identification of biallelic pathogenic variants in CP by molecular genetic testing.
Management.
Treatment of manifestations: Iron chelating agents (i.e., desferrioxamine, deferiprone, or deferasirox) to decrease serum ferritin concentration, decrease brain and liver iron stores, and prevent progression of neurologic signs/symptoms in symptomatic individuals with blood hemoglobin concentration higher than 9 g/dL; combined IV desferrioxamine and fresh-frozen human plasma (FFP) is effective in decreasing iron content in the liver; repetitive FFP treatment can improve neurologic signs/symptoms; antioxidants such as vitamin E may be used along with a chelator or oral administration of zinc to prevent tissue damage, particularly to the liver and pancreas.
Surveillance: Annual glucose tolerance test starting at age 15 years to evaluate for the onset of diabetes mellitus; ECG evaluation early in the course of the disease; evaluation of thyroid and liver function and complete blood count annually starting at the time of diagnosis.
Agents/circumstances to avoid: Iron supplements.
Evaluation of relatives at risk: If the pathogenic variants in the family are known, molecular genetic testing of asymptomatic sibs of a proband allows for early diagnosis and initiation of surveillance and treatment. If pathogenic variants are unknown, monitoring of serum concentrations of hemoglobin and hemoglobin A1c in asymptomatic sibs is recommended.
Genetic counseling.
Aceruloplasminemia is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible if the CP pathogenic variants in the family have been identified.
Diagnosis
Aceruloplasminemia is characterized by iron accumulation in the brain and viscera.
Suggestive Findings
Aceruloplasminemia should be suspected in individuals with characteristic MRI findings, more than one of the following clinical findings, and typical results on laboratory testing.
MRI. Abnormal low intensities in the liver as well as the striatum, thalamus, and dentate nucleus of the brain on T1- and T2-weighted images are consistent with iron deposition and support a diagnosis of aceruloplasminemia (see ).
Clinical findings
Laboratory test results
Serum ceruloplasmin <2 mg/dL (normal serum concentration: 21-36 mg/dL)
Serum copper concentration <20 µg/dL (normal range: 70-125 µg/dL)
Serum iron concentration <45 µg/dL (normal range: male 60-180 µg/dL; female 60-140 µg/dL)
Serum ferritin concentration >400 ng/mL (normal range: male 45-200 ng/mL; female 30-100 ng/mL)
Plasma ceruloplasmin ferroxidase activity that is not detectable using the method described by
Erel [1998] (normal activity: 500-680 U/L)
Establishing the Diagnosis
The diagnosis of aceruloplasminemia is established in a proband with typical clinical findings and identification of biallelic pathogenic variants in CP by molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, concurrent or serial single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of aceruloplasminemia is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with atypical clinical findings or in whom the diagnosis of aceruloplasminemia has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of aceruloplasminemia, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
Single-gene testing. Sequence analysis of CP detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If only one or no pathogenic variant is found perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
A
multigene panel that includes
CP and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the diagnosis of aceruloplasminemia is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Aceruloplasminemia
View in own window
| Gene 1 | Method | Proportion of Pathogenic Variants 2 Detectable by Method |
|---|
|
CP
| Sequence analysis 3 | >94% 4 |
| Gene-targeted deletion/duplication analysis 5 | Unknown 6 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
Individuals of Japanese heritage [Miyajima et al 1999]. Sequence analysis identifies at least one pathogenic variant in all individuals with abnormal low-intensity areas in both the basal ganglia and liver on MRI [Kono & Miyajima 2015].
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
Rare single-exon deletions have been reported [Pelucchi et al 2018]. No data on detection rate of gene-targeted deletion/duplication analysis are available.
Clinical Characteristics
Clinical Description
The clinical manifestations of aceruloplasminemia are retinal degeneration, diabetes mellitus (DM), and neurologic signs/symptoms [Miyajima 2003]. Individuals with aceruloplasminemia often present with iron-restricted microcytic anemia prior to onset of DM or neurologic signs/symptoms. Phenotypic expression varies even within families.
A summary of clinical manifestations and age of onset in 71 Japanese individuals is shown in Table 2. The manifestations (in order of frequency) are anemia, retinal degeneration, diabetes mellitus, and neurologic signs/symptoms. The neurologic signs/symptoms correspond to regions of brain iron accumulation and include ataxia, involuntary movement, parkinsonism, and cognitive dysfunction [Miyajima et al 2003, Kono 2012, Kono & Miyajima 2015].
Table 2.
Clinical Manifestations / Age at Onset in 71 Individuals with Aceruloplasminemia
View in own window
| Clinical Manifestations | Age at Onset of Manifestation |
|---|
| Iron-restricted microcytic anemia (80%) | <20 yrs |
| Diabetes mellitus (70%) | <30 yrs: 18%
30-39 yrs: 35%
40-49 yrs: 31%
>50 yrs: 16% |
| Retinal degeneration (76%) | At least >20 yrs |
| Neurologic signs/ symptoms (68%) | Ataxia (71%) incl dysarthria, gait ataxia, limb ataxia, nystagmus | <40 yrs: 7%
40-49 yrs: 38%
50-59 yrs: 42%
>60 yrs: 13% |
| Involuntary movement (64%) incl dystonia (blepharospasm, grimacing, neck dystonia), tremors, chorea |
| Parkinsonism (20%) incl rigidity, akinesia |
| Cognitive dysfunction (60%) incl apathy, forgetfulness |
Additional Findings
Laboratory findings
Undetectable serum ceruloplasmin
Elevated serum ferritin
Decreased serum iron
Iron refractory microcytic anemia
Low serum copper and normal urinary copper levels
MRI (magnetic resonance imaging) reveals low intensity on both T1- and T2-weighted MRI in the liver and the basal ganglia, including the caudate nucleus, putamen and pallidum, and thalamus.
Liver biopsy results
Excess iron accumulation (>1,200 µg/gram dry weight) within hepatocytes and reticuloendothelial cells
Normal hepatic architecture and histology without cirrhosis or fibrosis
Normal copper accumulation
Pathologic findings. Visceral organs, especially the liver, pancreas, and heart, have iron deposition:
The liver shows no cirrhotic changes. The iron content in the liver is greater than the iron content in the brain. The hepatic iron concentration (HIC) is determined in µmol/g of dry weight. The hepatic iron index (HII) is then calculated by dividing the hepatic iron concentration by the age (in years) of the individual. Normal individuals have an HII of 1.1 or less; more than 80% of individuals with aceruloplasminemia have an HII greater than 1.3. (HIC [µg/g dry weight] 56 = HIC [µmol/g dry weight], HIC [µmol/g dry weight]/age [years] = HII).
Islet beta cells demonstrate iron deposition, which results in diabetes mellitus.
The distribution in order of iron level in the brain is globus pallidus > putamen > cerebral cortex > cerebellar cortex. Severe iron overload and extensive neuronal loss are observed in the basal ganglia, while iron deposition and neuronal cell loss are trivial in the frontal cortices. The cerebellar cortex shows marked loss of Purkinje cells. Iron deposition is more prominent in the astrocytes than in the neurons. Astrocytic deformity and globular structures are characteristic features in brains of individuals with aceruloplasminemia. The globular structures in the astrocytes are seen in proportion to the degree of iron deposition [
Kaneko et al 2002,
Miyajima 2003,
Oide et al 2006].
Genotype-Phenotype Correlations
No clear genotype-phenotype correlation exists for aceruloplasminemia.
Nomenclature
Aceruloplasminemia was originally called familial apoceruloplasmin deficiency [Miyajima et al 1987].
Prevalence
The serum ceruloplasmin concentrations of about 5,000 adults undergoing medical examination were screened (Table 3). The prevalence of aceruloplasminemia was estimated at one in 2,000,000 in nonconsanguineous marriages in Japan [Miyajima et al 1999].
There is no data on the prevalence of this disorder outside of Japan are available.
Table 3.
Frequencies of Aceruloplasminemia
View in own window
| Parental Relatedness | Homozygotes | Heterozygotes |
|---|
| Nonconsanguineous | 4.90 x 10-7 | 1.40 x 10-3 |
| Consanguineous | 3.29 x 10-6 | 1.39 x 10-3 |
Heterozygotes for the ceruloplasmin gene have been estimated to account for 0.1% of individuals with diabetes in Japan [Daimon et al 1997].
Differential Diagnosis
Table 4.
Disorders to Consider in the Differential Diagnosis of Aceruloplasminemia
View in own window
| Disorder | Gene(s) | MOI | Clinical Features |
|---|
| Overlapping | Distinguishing (in aceruloplasminemia) |
|---|
|
Other types of slowly progressive
NBIA
with later onset 1
|
| Atypical pantothenate kinase-associated neurodegeneration (PKAN) |
PANK2
| AR | Radiographic evidence of focal iron accumulation in brain, usually basal ganglia |
|
|
Neuroferritinopathy
|
FTL
| AD |
|
Copper metabolic disorder
|
| Wilson disease 2, 3 |
ATP7B
| AR | Ceruloplasmin deficiency | Radiographic evidence of iron accumulation in basal ganglia, especially thalamus Iron accumulation in several visceral organs as well as brain
|
|
Iron metabolic disorder
|
| HFE-associated hereditary hemochromatosis 3, 4 |
HFE
| AR | Iron accumulation in visceral tissues |
|
|
Other
|
|
Huntington disease
|
HTT
| AD | Neurologic manifestations | Radiographic evidence of iron accumulation in basal ganglia, especially thalamus Iron accumulation in several visceral organs as well as brain
|
|
DRPLA
|
ATN1
| AD |
| Dystonia (See Hereditary Dystonia Overview.) | >25 genes | AD
AR
XL 5 |
| Hereditary spinocerebellar ataxias (See Hereditary Ataxia Overview.) | >50 genes | AD
AR
XL |
| Drug effects or toxicity | No history of drug use or toxicity Radiographic evidence of iron accumulation in basal ganglia, especially thalamus Iron accumulation in several visceral organs as well as brain
|
AD = autosomal dominant; AR = autosomal recessive; DRPLA = dentatorubral-pallidoluysian atrophy; MOI = mode of inheritance; NBIA = neurodegeneration with brain iron accumulation; XL = X-linked
- 1.
Aceruloplasminemia is a type of NBIA. NBIA is a group of inherited neurologic disorders in which iron accumulates in the basal ganglia resulting in progressive dystonia, spasticity, parkinsonism, neuropsychiatric abnormalities, and optic atrophy or retinal degeneration. Ten types and their associated genes are recognized (PANK2, PLA2G6, C19orf12, FA2H, ATP13A2, WDR45, COASY, FTL, CP, and DCAF17 (see NBIA Disorders Overview). The age of onset ranges from infancy to late adulthood; the rate of progression varies.
- 2.
In Wilson disease, an inability to transfer copper into the ceruloplasmin precursor protein, apoceruloplasmin, and a decrease in biliary copper excretion results in serum ceruloplasmin deficiency and excess copper accumulation.
- 3.
Because aceruloplasminemia has features of Wilson disease and HFE-associated hereditary hemochromatosis, it could be incorrectly diagnosed as mild hemochromatosis with hypoceruloplasminemia or mild Wilson disease with hemosiderosis.
- 4.
HFE-associated hereditary hemochromatosis is the most common iron metabolic disorder.
- 5.
While hereditary, isolated, and combined dystonias are usually inherited in an autosomal dominant manner, complex dystonias are often inherited in an autosomal recessive or mitochondrial manner. In general, X-linked forms are rare.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with aceruloplasminemia, the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Iron deposition. Serum ferritin concentration; brain and abdomen MRI findings
Neurologic findings. Brain MRI
Diabetes mellitus. Glucose tolerance test; blood concentrations of insulin and HbA1c
Retinal degeneration. Examination of the optic fundi and fluorescein angiography
Anemia. Complete blood count
Other. Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Note: Individual case reports indicate the effectiveness of treatment in individuals with aceruloplasminemia; however, no large series of symptomatic persons treated with iron chelators and zinc is available and there is no universally accepted treatment regimen. A systematic review/analysis of studies designed to evaluate the clinical effectiveness of desferrioxamine, deferiprone, deferasirox, and zinc as monotherapy for the initial treatment of various clinical presentations of aceruloplasminemia is needed.
Desferrioxamine. Treatment with iron chelating agents (i.e., desferrioxamine) can be considered for symptomatic individuals whose blood hemoglobin concentration is higher than 9 g/dL. Treatment can decrease serum ferritin concentration as well as brain and liver iron stores, and can prevent progression of the neurologic signs/symptoms [Miyajima et al 1997].
Intravenous infusions of 500 mg of desferrioxamine (desferoxamine mesylate) dissolved in 100 mL of isotonic saline solution are given over one hour. Desferrioxamine is infused twice a week for six to ten months.
In the
Miyajima et al [1997] study, head MRI evaluations were performed before and after treatment to evaluate the effect of treatment on iron storage in the brain. Serum concentrations of iron, ferritin, copper, hemoglobin, and hemoglobin A1c, as well as C-peptide immunoreactivity, were measured before and after treatment. Lipid peroxidation in plasma samples was also measured by the thiobarbituric acid method. T
2-weighted MRI showed an increase in the signal intensity of the basal ganglia. Serum ferritin concentration was markedly reduced and hepatic iron concentration was decreased, whereas serum iron concentration was elevated and anemia and DM were ameliorated.
In the
Mariani et al [2004] report, the brain MRI did not change after more than one year of desferoxamine treatment, whereas excess iron in the liver was removed.
Pan et al [2011] reported an affected individual age 52 years who was treated with desferrioxamine (500 mg) by intravenous infusion in a 5% glucose solution once a week for four years. After four years of treatment, brain MRI evaluation demonstrated improvement in low-intensity areas in the basal ganglia, suggesting that iron chelation can reduce abnormal iron deposition in the central nervous system.
Deferasirox. Iron chelation therapy with deferasirox, an oral iron chelating agent, led to a mild improvement in clinical symptoms, including cognitive performance, gait, and balance, in an individual with aceruloplasminemia who had no response to deferoxamine or fresh-frozen plasma therapy [Skidmore et al 2008]. Oral administration of deferasirox may prevent tissue damage, particularly to the liver and pancreas [Finkenstedt et al 2010].
Deferiprone has a lower molecular weight and more lipophilic properties. Deferiprone therapy had no beneficial effects in an individual in a previous report [Mariani et al 2004]; however, it has been shown to protect against retinal degeneration and neurodegeneration and to increase the life span if initiated early in mice exhibiting knockout for ceruloplasmin and hephaestin [Hadziahmetovic et al 2011].
Fresh-frozen human plasma (FFP). After the intravenous administration of FFP containing ceruloplasmin, serum iron content increases for several hours because of ferroxidase activity of ceruloplasmin. Iron content in the liver decreases more with the combined intravenous administration of FFP and desferrioxamine than with FFP administration alone. Neurologic signs/symptoms can improve following repetitive FFP treatment [Yonekawa et al 1999].
Antioxidants such as vitamin E may be used along with a chelator or oral administration of zinc to prevent tissue damage, particularly to the liver and pancreas [Kuhn et al 2007].
Complications of aceruloplasminemia. Diabetes is treated in the standard manner. Transfusion is not needed for anemia, and there is no specific treatment for retinal disease.
Prevention of Primary Manifestations
Zinc concentrations in affected individuals were decreased in the brain and visceral organs, and zinc showed opposing distributions to those for iron. Because zinc has antioxidant activity, treatment with an iron chelator accompanied by zinc may be useful in individuals with aceruloplasminemia to diminish iron accumulation in the brain and body and to prevent or ameliorate systemic and neurologic symptoms [Miyajima 2015].
Surveillance
Marked accumulation of iron in parenchymal tissues including the liver, pancreas, heart, and thyroid can result in diabetes mellitus, cardiac failure, and hypothyroidism.
All affected individuals should have an annual glucose tolerance test starting at age 15 years to evaluate for the onset of diabetes mellitus.
Cardiac evaluation should be performed early in the course of the disease and repeated every year.
Evaluation of thyroid and liver function and complete blood count are indicated annually starting at the time of diagnosis.
Agents/Circumstances to Avoid
Iron supplements. Individuals with aceruloplasminemia erroneously diagnosed as having iron deficiency anemia and treated with iron supplements had accelerated iron accumulation.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger sibs of a proband (starting at age 15 years) in order to identify as early as possible those who would benefit from surveillance and initiation of treatment. However, the proper preventive approach for asymptomatic sibs is unknown. Evaluations can include:
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Aceruloplasminemia is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
The parents of an individual with aceruloplasminemia are obligate heterozygotes (i.e., carriers of one CP pathogenic variant).
Clinical disease is not known to occur in heterozygotes, although data are not adequate to exclude the possibility in older individuals.
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) with clinical signs/symptoms have not been reported, although data are not adequate to exclude the possibility in older individuals.
Offspring of a proband. The offspring of an individual with aceruloplasminemia are obligate heterozygotes (carriers) for a pathogenic variant in CP.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a CP pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the CP pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the CP pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for aceruloplasminemia are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
NBIA Disorders Association
NBIAcure
Center of Excellence for NBIA Clinical Care and Research
International Registry for NBIA and Related Disorders
Oregon Health & Science University
Email: info@nbiacure.org
Treat Iron-Related Childhood Onset Neurodegeneration (TIRCON)
Germany
Email: TIRCON@med.uni-muenchen.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Aceruloplasminemia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Gene structure.
CP is approximately 4.4 kb in a total of 19 exons; it encodes the ceruloplasmin precursor. For a detailed summary of gene and protein information, see Table A, Gene.
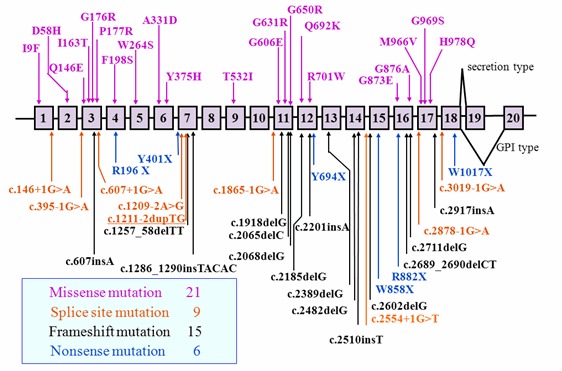
Pathogenic variants. More than 70 pathogenic variants in CP have been identified in more than 60 affected families across different populations [Yoshida et al 1995, Yazaki et al 1998, Miyajima et al 1999, Daimon et al 2000, Hellman et al 2000, Kohno et al 2000, Bosio et al 2002, Hellman et al 2002, Loréal et al 2002, Hatanaka et al 2003, Mariani et al 2004, Kuhn et al 2005, Pérez-Aguilar et al 2005, Kono et al 2006, Shang et al 2006, Kono 2012]. See . No hot spots for pathogenic variants in CP have been observed.
More than half of the pathogenic variants in CP are predicted to be loss-of-function variants.
Table 5.
CP Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.2962G>A | p.Gly988Ser
(Gly969Ser) |
NM_000096.3
NP_000087.1
|
| c.2991T>G | p.His997Gln
(His978Gln) |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions; in this instance the alias refers to the length of the mature peptide.
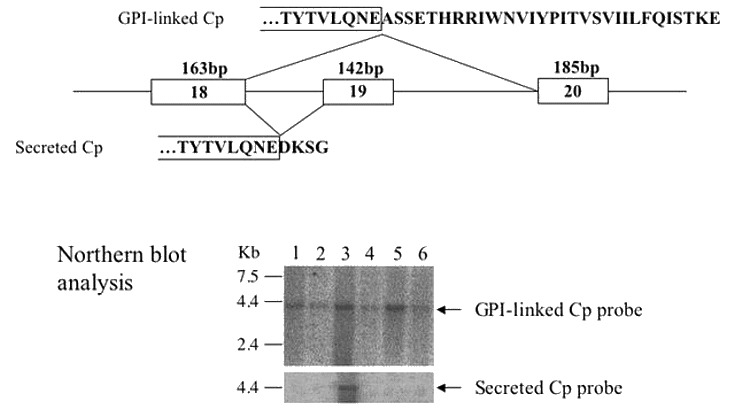
Normal gene product. The product of CP, ceruloplasmin, is a blue copper oxidase that carries more than 95% of the plasma copper content in vertebrates.
Ceruloplasmin has two forms: (1) a secreted form (1,040 amino acids) mainly produced and secreted by hepatocytes, and (2) a glycosylphosphatidylinositol (GPI)-anchored form (1,065 amino acids) mainly expressed in astrocytes as well as visceral organs (see ) [Patel et al 2000]:
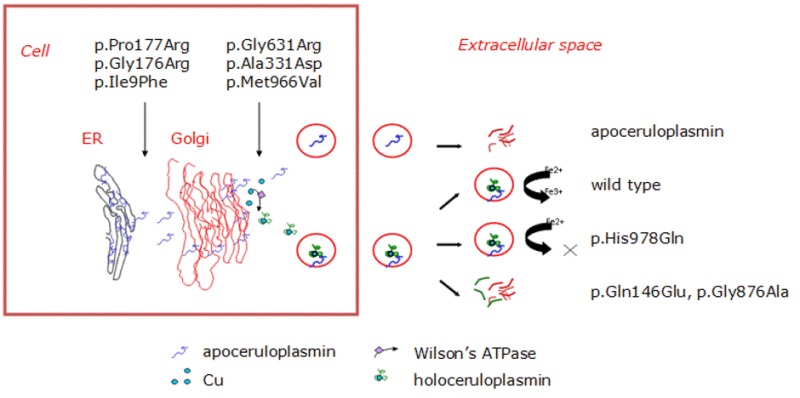
Abnormal gene product. Molecular analysis of the pathogenic missense variants has shown several different mechanisms by which pathogenic variants in the ceruloplasmin gene can result in the lack of enzymatic activity (see ). Abnormal gene products are usually degraded immediately after release from the hepatocytes. With some CP pathogenic nonsense variants, abnormal ceruloplasmin is retained within the endoplasmic reticulum (early secretory pathway); with other pathogenic variants, abnormal ceruloplasmin results in structural abnormalities in the protein that prevent the incorporation of copper into ceruloplasmin in the Golgi apparatus (late secretory pathway) [Hellman et al 2002, Kono & Miyajima 2006, Kono et al 2006].
References
Literature Cited
Bosio S, De Gobbi M, Roetto A, Zecchina G, Leonardo E, Rizzetto M, Lucetti C, Petrozzi L, Bonuccelli U, Camaschella C. Anemia and iron overload due to compound heterozygosity for novel ceruloplasmin mutations.
Blood. 2002;100:2246–8. [
PubMed: 12200392]
Daimon M, Susa S, Ohizumi T, Moriai S, Kawanami T, Hirata A, Yamaguchi H, Ohnuma H, Igarashi M, Kato T. A novel mutation of the ceruloplasmin gene in a patient with heteroallelic ceruloplasmin gene mutation (HypoCPGM).
Tohoku J Exp Med. 2000;191:119–25. [
PubMed: 10997552]
Daimon M, Yamatani K, Tominaga M, Manaka H, Kato T, Sasaki H. NIDDM with a ceruloplasmin gene mutation.
Diabetes Care. 1997;20:678. [
PubMed: 9097006]
Erel O. Automated measurement of serum ferroxidase activity.
Clin Chem. 1998;44:2313–9. [
PubMed: 9799759]
Finkenstedt A, Wolf E, Hofner E, Gasser BI, Bosch S, Bakry R, Creus M, Kremser C, Schocke M, Theurl M, Moser P, Schranz M, Bonn G, Poewe W, Vogel W, Janecke AR, Zoller H. Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation.
J Hepatol. 2010;53:1101–7. [
PMC free article: PMC2987498] [
PubMed: 20801540]
Hadziahmetovic M, Song Y, Wolkow N, Lacovelli J, Grieco S, Lee J, Lyubarsky A, Pratico D, Connelly J, Spino M, Harris ZL, Dunaief JL. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration.
Invest Ophthalmol Vis Sci. 2011;52:959–68. [
PMC free article: PMC4183363] [
PubMed: 21051716]
Hatanaka Y, Okano T, Oda K, Yamamoto K, Yoshida K. Aceruloplasminemia with juvenile-onset diabetes mellitus caused by exon skipping in the ceruloplasmin gene.
Intern Med. 2003;42:599–604. [
PubMed: 12879954]
Hellman NE, Kono S, Miyajima H, Gitlin JD. Biochemical analysis of a missense mutation in aceruloplasminemia.
J Biol Chem. 2002;277:1375–80. [
PubMed: 11689569]
Jeong SY, David S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system.
J Biol Chem. 2003;278:27144–8. [
PubMed: 12743117]
Kaneko K, Yoshida K, Arima K, Ohara S, Miyajima H, Kato T, Ohta M, Ikeda SI. Astrocytic deformity and globular structures are characteristic of the brains of patients with aceruloplasminemia.
J Neuropathol Exp Neurol. 2002;61:1069–77. [
PubMed: 12484569]
Kohno S, Miyajima H, Takahashi Y, Inoue Y. Aceruloplasminemia with a novel mutation associated with parkinsonism.
Neurogenetics. 2000;2:237–8. [
PubMed: 10983721]
Kono S. Aceruloplasminemia.
Curr Drug Targets. 2012;13:1190–9. [
PubMed: 22515740]
Kono S, Miyajima H. Molecular and pathological basis of aceruloplasminemia.
Biol Res. 2006;39:15–23. [
PubMed: 16629161]
Kono S, Miyajima H. Aceruloplasminemia. In: Rosenberg RN, Pascual JM, eds. Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease. 5 ed. Elsevier, Academic Press. 2015:495-506.
Kono S, Suzuki H, Oda T, Miyajima H, Takahashi Y, Shirakawa K, Ishikawa K, Kitagawa M. Biochemical features of ceruloplasmin gene mutations linked to aceruloplasminemia.
Neuromolecular Med. 2006;8:361–74. [
PubMed: 16775387]
Kuhn J, Bewermeyer H, Miyajima H, Takahashi Y, Kuhn KF, Hoogenraad TU. Treatment of symptomatic heterozygous aceruloplasminemia with oral zinc sulphate.
Brain Dev. 2007;29:450–3. [
PubMed: 17307325]
Kuhn J, Miyajima H, Takahashi Y, Kunath B, Hartmann-Klosterkoetter U, Cooper-Mahkorn D, Schaefer M, Bewermeyer H. Extrapyramidal and cerebellar movement disorder in association with heterozygous ceruloplasmin gene mutation.
J Neurol. 2005;252:111–3. [
PubMed: 15654567]
Loréal O, Turlin B, Pigeon C, Moisan A, Ropert M, Morice P, Gandon Y, Jouanolle AM, Verin M, Hider RC, Yoshida K, Brissot P. Aceruloplasminemia: new clinical, pathophysiological and therapeutic insights.
J Hepatol. 2002;36:851–6. [
PubMed: 12044538]
Mariani R, Arosio C, Pelucchi S, Grisoli M, Piga A, Trombini P, Piperno A. Iron chelation therapy in aceruloplasminaemia: study of a patient with a novel missense mutation.
Gut. 2004;53:756–8. [
PMC free article: PMC1774045] [
PubMed: 15082597]
Miyajima H. Aceruloplasminemia, an iron metabolic disorder.
Neuropathology. 2003;23:345–50. [
PubMed: 14719552]
Miyajima H. Investigated and available therapeutic options for treating aceruloplasminemia. Expert Opin Orphan Drugs. 2015;3:1011–20.
Miyajima H, Kohno S, Takahashi Y, Yonekawa O, Kanno T. Estimation of the gene frequency of aceruloplasminemia in Japan.
Neurology. 1999;53:617–9. [
PubMed: 10449129]
Miyajima H, Nishimura Y, Mizoguchi K, Sakamoto M, Shimizu T, Honda N. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration.
Neurology. 1987;37:761–7. [
PubMed: 3574673]
Miyajima H, Takahashi Y, Kamata T, Shimizu H, Sakai N, Gitlin JD. Use of desferrioxamine in the treatment of aceruloplasminemia.
Ann Neurol. 1997;41:404–7. [
PubMed: 9066364]
Miyajima H, Takahashi Y, Kono S. Aceruloplasminemia, an inherited disorder of iron metabolism.
Biometals. 2003;16:205–13. [
PubMed: 12572680]
Oide T, Yoshida K, Kaneko K, Ohta M, Arima K. Iron overload and antioxidative role of perivascular astrocytes in aceruloplasminemia.
Neuropathol Appl Neurobiol. 2006;32:170–6. [
PubMed: 16599945]
Pan PL, Tang HH, Chen Q, Song W, Shang HF. Desferrioxamine treatment of aceruloplasminemia: Long-term follow-up.
Mov Disord. 2011;26:2142–4. [
PubMed: 21594898]
Patel BN, Dunn RJ, David S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain.
J Biol Chem. 2000;275:4305–10. [
PubMed: 10660599]
Pelucchi S, Mariani R, Ravasi G, Pelloni I, Marano M, Tremolizzo L, Alessio M, Piperno A. Phenotypic heterogeneity in seven Italian cases of aceruloplasminemia.
Parkinsonism Relat Disord. 2018;51:36–42. [
PubMed: 29503155]
Pérez-Aguilar F, Burguera JA, Benlloch S, Berenguer M, Rayon JM. Aceruloplasminemia in an asymptomatic patient with a new mutation. Diagnosis and family genetic analysis.
J Hepatol. 2005;42:947–9. [
PubMed: 15885371]
Shang HF, Jiang XF, Burgunder JM, Chen Q, Zhou D. Novel mutation in the ceruloplasmin gene causing a cognitive and movement disorder with diabetes mellitus.
Mov Disord. 2006;21:2217–20. [
PubMed: 17013908]
Skidmore FM, Drago V, Foster P, Schmalfuss IM, Heilman KM, Streiff RR. Aceruloplasminaemia with progressive atrophy without brain iron overload: treatment with oral chelation.
J Neurol Neurosurg Psychiatry. 2008;79:467–70. [
PubMed: 17911185]
Yamaguchi K, Takahashi S, Kawanami T, Kato T, Sasaki H. Retinal degeneration in hereditary ceruloplasmin deficiency.
Ophthalmologica. 1998;212:11–4. [
PubMed: 9438577]
Yazaki M, Yoshida K, Nakamura A, Furihata K, Yonekawa M, Okabe T, Yamashita N, Ohta M, Ikeda S. A novel splicing mutation in the ceruloplasmin gene responsible for hereditary ceruloplasmin deficiency with hemosiderosis.
J Neurol Sci. 1998;156:30–4. [
PubMed: 9559983]
Yonekawa M, Okabe T, Asamoto Y, Ohta M. A case of hereditary ceruloplasmin deficiency with iron deposition in the brain associated with chorea, dementia, diabetes mellitus and retinal pigmentation: administration of fresh-frozen human plasma.
Eur Neurol. 1999;42:157–62. [
PubMed: 10529542]
Yoshida K, Furihata K, Takeda S, Nakamura A, Yamamoto K, Morita H, Hiyamuta S, Ikeda S, Shimizu N, Yanagisawa N. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans.
Nat Genet. 1995;9:267–72. [
PubMed: 7539672]
Chapter Notes
Acknowledgments
Aceruloplasminemia research is funded in part by a Grant-in-Aid of Science from the Ministry of Education, Science, Culture, Sports, and Technology, Japan.
Revision History
27 September 2018 (ha) Comprehensive update posted live
5 November 2015 (me) Comprehensive update posted live
18 April 2013 (me) Comprehensive update posted live
17 February 2011 (me) Comprehensive update posted live
29 April 2008 (me) Comprehensive update posted live
15 August 2005 (me) Comprehensive update posted live
12 August 2003 (me) Review posted live
23 June 2003 (hm) Original submission