

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Primary generalized glucocorticoid resistance syndrome is a rare genetic disorder characterized by resistance of entire tissues to glucocorticoids. Affected subjects demonstrate elevation of serum cortisol without Cushingoid manifestations, as the hypothalamic-pituitary-adrenal (HPA) axis is upregulated to compensate for the reduced action of this steroid in local tissues. Instead, these patients develop hypertension and/or signs of hyperandrogenism, because hyper-secreted adrenocorticotropic hormone (ACTH) stimulates production of adrenal mineralocorticoids and/or androgens in addition to the glucocorticoid cortisol. At the molecular level, this syndrome is caused by inactivating mutations in the NR3C1 gene that encodes the human glucocorticoid receptor (hGR) protein. Biochemical, molecular and structural exploration on pathologic mutant receptors revealed a variety of functional defects, such as reduced affinity to glucocorticoids or target DNA, inability to transactivate glucocorticoid-responsive genes, and slowing of the cytoplasmic to nuclear translocation. The clinical spectrum of this syndrome is thus broad, ranging from asymptomatic to severe cases of mineralocorticoid and/or androgen excess depending on the severity of genetic defects and resulting dysfunction of the mutated receptors. When this syndrome is suspected, a detailed personal and family history should be obtained. Physical examination should include an assessment for signs of mineralocorticoid and/or androgen excess. In neonates and young children, severe hypoglycemia and loss of consciousness due to reduced actions of glucocorticoids in the liver may be present as initial manifestations in addition to hypertension and/or genital abnormalities. Suspected subjects should undergo a detailed endocrinologic evaluation with particular emphasis on the measurement of diurnal serum cortisol and plasma ACTH concentrations and determination of the 24-hour urinary free cortisol excretion to identify upregulation of the HPA axis with preservation of the normal circadian rhythmicity. The diagnosis of this syndrome should be confirmed by sequencing of the NR3C1 gene including exon/intron junctions and subsequent validation of functional defects of the mutated receptors. Treatment involves administration of high doses of mineralocorticoid activity-sparing pure glucocorticoids like dexamethasone, which stimulate the mutant and/or the wild-type hGR, and suppress the endogenous secretion of ACTH and adrenal steroids in the affected subjects. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Organisms are exposed continuously to internal and external stressors, and live through them by maintaining the internal equilibrium called homeostasis (1). In order to respond adequately to such stressors through coordinating various body activities, we humans are equipped with a highly sophisticated stress responsive system, the hypothalamic-pituitary-adrenal (HPA) axis, which consists of the brain hypothalamus, the anterior pituitary gland, and the adrenal cortex, and employs glucocorticoids as its end-effector hormones. Actions of glucocorticoids, which are essential for life, can be determined by a balance between circulating levels of these hormones and local tissue sensitivity (2, 3). Exceeding appropriate ranges of tissue sensitivity to glucocorticoids may present either as glucocorticoid resistance or glucocorticoid hypersensitivity with their specific manifestations (3, 4). Such alterations in tissue glucocorticoid actions can occur in general (that is, throughout the body) or in tissue-specific manner (restricted in some organs and tissues; e.g., immune organs/cells, central nervous system (CNS), liver and fat tissues) (3). They are caused primarily by genetic defects of the molecules involved in the glucocorticoid signaling pathway or secondary through modulation of this pathway by other pathologic conditions, such as infectious, inflammatory and autoimmune diseases, obesity, and insulin resistance/overt diabetes mellitus. One such condition is the primary generalized glucocorticoid resistance syndrome, which is caused by inactivating mutations in the glucocorticoid receptor gene (5). Affected subjects develop partial glucocorticoid resistance observed in entire organs and tissues of the affected subjects (5). In recognition of Professor George P. Chrousos' novel and extensive research work in this field, the term “Chrousos Syndrome” may be used for this syndrome (6, 7).
GLUCOCORTICOIDS
Glucocorticoids (cortisol in humans and corticosterone in rodents) are produced from cholesterol through multiple enzymatic reactions in the zona fasciculata of the adrenal cortex in response to the adrenocorticotropic hormone (ACTH) released from the pituitary gland (1). Glucocorticoids regulate a broad spectrum of physiologic functions essential for life, such as growth, reproduction, immunity, intermediary metabolism, cardiovascular tone, and CNS functions, playing essential and indispensable roles in the maintenance of resting and stress-related homeostasis (1, 7, 8). In addition, glucocorticoids exert potent anti-inflammatory and immunomodulatory effects particularly with their stress-equivalent or pharmacologic doses, thus they are widely used in the treatment of inflammatory, autoimmune, and lymphoproliferative diseases (8).
GLUCOCORTICOID RECEPTOR PROTEINS, ISOFORMS AND ITS ENCODING GENE, NR3C1
Circulating cortisol freely passes through the cytoplasmic membrane and enters into the cytoplasm of its target cells, and binds to an intracellular protein, the glucocorticoid receptor (GR) (9, 10). The human (h) GR is one of the steroid/thyroid/retinoic acid nuclear hormone receptor superfamily proteins, which consist of over 600 members in the animal kingdom (11). Many of them mediate extracellular signals transduced mainly by lipophilic hormones/compounds into the cell nucleus by binding them as ligands and by acting as ligand-dependent transcription factors (12, 13). hGR influences transcription rates of numerous glucocorticoid-responsive genes (up to 3~5% of the entire protein-coding genes) in a positive or a negative fashion by interacting directly or indirectly with promoter/enhancer regions of these genes (14). The hGR gene (NR3C1: nuclear receptor subfamily 3, group C, member 1) consists of 9 exons and is located at chromosome 5q31.3. Exons 2-9 constitute the protein-coding sequence, whereas exon 1 encodes an untranslated region (12, 14, 15). The human NR3C1 gene has multiple exon 1s (see below) that harbor specific promoters containing a respective transcription start site for conferring tissue-specific expression of the receptor protein (15). Alternative splicing of the NR3C1 gene in exon 9s generates two highly homologous receptor isoforms, the hGRα and the hGRβ (16). They share amino (N)-terminal 727 common amino acids, but then diverge, with hGRα having an additional 50 amino acids and hGRβ having an additional, nonhomologous 15 amino acids at their carboxyl (C)-termini (17). hGRα resides primarily in the cytoplasm of cells and represents the classic GR that binds natural and synthetic glucocorticoids and mediates most of the actions of these hormones (15). On the other hand, hGRβ does not bind glucocorticoids, has intrinsic, gene-specific transcriptional activity, and exerts a dominant negative effect on the transcriptional activity of hGRα (18). Although physiologic and pathologic roles of hGRβ are still largely unknown (19, 20), recent studies demonstrated that this isoform is implicated in modulation of the insulin signaling and participates in the pathogenesis of brain gliomas (21-23).
The hGRα mRNA expresses not only the classic, full-length hGRα, but also multiple translational isoforms by using at least eight alternative amino-terminal translation initiation sites (24). All these hGRα isoforms are differentially distributed in the cytoplasm and/or the nucleus in the absence of ligand, have different transcriptional activity, and display distinct transactivating or transrepressing activities on various glucocorticoid-responsive genes (24). Since hGRβ shares with hGRα a common amino-terminal domain that contains the same translation initiation sites, the hGRβ variant mRNA might also be translated through the same translation initiation sites to a similar host of hGRβ isoforms (14).
The human NR3C1 has 11 different promoters with their alternative first exons (1A1, 1A2, 1A3, 1B, 1C, 1D, 1E, 1F, 1H, 1I and 1J) (25, 26). Therefore, it can produce 11 different hGRα mRNA transcripts from different promoters that encode the same hGRα protein, as these transcripts share common exon 2 to exon 9α that contains the same translation initiation codon. 1A1, 1A2, 1A3 and 1I are located in the distal promoter region spanning ~32,000-36,000 bps upstream of the translation initiation site, while 1B, 1C, 1D, 1E, 1F, 1H and 1J position in the proximal promoter region located up to ~5,000 bps upstream of this site (25). Through differential use of these promoters, expression levels of hGRα can vary among tissues in different physiologic and pathologic conditions, as each tissue has specific expression profiles of local transcription factors and epigenetic modification of chromatin-associated molecules bound on these exon 1-associated promoters (25, 27, 28). Again, differential tissue-specific expression of hGRβ through the use of these promoters appears to be present. The above-indicated marked complexity in transcription/translation of the human NR3C1 gene enables target tissues to respond differently to circulating cortisol and accounts for stochastic, but still a highly organized nature of tissue glucocorticoid actions, in order to fulfil specific local needs of glucocorticoid hormonal effects (14). Such complexity of the glucocorticoid signaling at the receptor level also indicates that the proper biologic action of glucocorticoids in every target tissue is extremely important.
The hGRα protein consists of three major domains and one region, namely the N-terminal (NTD), DNA-binding (DBD) and ligand-binding domain (LBD), and the hinge region (HR) (15). Exact amino acid location of these domains/region in the hGRα protein explained below is based on the data retrieved from the Pfam source of the Ensembl database (www.ensembl.org). NTD is encoded by exon 2 and represents the largest domain of the receptor, spanning over amino acids 1 to 401. It contains an unstructured acidic transactivation surface called activation function (AF) -1, which is used as a molecular platform for modulating the transcription of glucocorticoid-responsive genes (10). This domain also undergoes several post-translational modifications particularly at AF-1. DBD is expressed from exons 3 and 4, and lies between amino acids 417 and 494. This domain consists of two 4C (cysteine)-type zinc fingers and support the interaction between the receptor and its target DNA sequences known as glucocorticoid response elements (GREs) (10, 29). LBD is encoded by exons 5-9 and positions at the C-terminal end of the receptor corresponding to amino acids 531 to 777. This domain is structurally formed with 12 α-helices and four β-sheets, and contains two functional structures, the ligand-binding pocket (LBP) and the second transactivation surface called AF-2, as well as several other molecular platforms including the one responsible for nuclear translocation of the receptor (29, 30). Most of the protein surfaces of LBD that mediate these LBD-specific functions are formed upon binding of the receptor to a ligand and following conformational changes of this domain (15). Finally, HR lies between DBD and LBD, is encoded by 5’ part of exon 5, and spans between amino acids 495 and 530. This region provides appropriate structural flexibility to the receptor and allows the dimerized receptors to interact with different classic/alternative tandem GREs with various length of spacing nucleotides (the classic tandem GREs has three spacing nucleotides) (15).
MOLECULAR ACTIONS OF hGRα
Intracellular Shuttling of hGRα and its Regulators
At target cells, hGRα in the absence of glucocorticoids resides primarily in the cytoplasm as part of the hetero-oligomeric complex consisting of chaperone heat shock proteins (HSPs) 90, 70 and 50, immunophilins (e.g., FK506-binding protein (FKBP)), and possibly other proteins (31) (Figure 1). Binding of HSP90 to hGRα induces a conformational change in receptor’s LBD, and confers its ligand-friendly state, exposing the LBP to glucocorticoids and masking two nuclear localization signals (NLS), NL1 and NL2. Upon binding to a ligand, hGRα dissociates from the complex, exposes NL1 and NL2 to their counterpart molecular machinery, and translocates into the nucleus through the nuclear pore. NL1 harbors a classic NLS and is located between the C-terminal portion of DBD and the N-terminal part of HR (32). The function of NL1 is dependent on the importin α, a protein component of the nuclear pore-associated nuclear import system, which transports a liganded GRα as a cargo from the cytoplasm to the nucleus through the nuclear pore in an ATP-dependent fashion (33). NL2 spans over most of the LBD whose molecular mechanism(s) for supporting nuclear translocation of the receptor has(ve) not yet been elucidated (31, 34). Inside the nucleus, ligand-bound hGRα dimerizes and modulates transcription rates of glucocorticoid-responsive genes by associating with promoter/enhancer regions of their encoding genes (15) (Figure 1). The receptor subsequently liberates the ligand and is dissociated from its target genes and slowly translocates back to the cytoplasm with the molecular mechanisms described below (15). The ubiquitin-proteasomal pathway degrades some of the liganded hGRα in the nucleus, facilitating clearance of the receptor from GREs; thus this system negatively regulates the transcriptional activity of hGRα (35).
In addition to translocating into the nucleus, some liganded hGRαs migrate to the cytoplasmic membrane where they modulate the activity of cell surface receptors by associating with their intracellular signaling molecules, such as classic and small GTP-binding (G) proteins, and several serine/threonine and tyrosine kinases (36-38). The ligand-bound hGRα is also known to translocate into the mitochondria and to modulate the activity of this intracellular organelle (39). After modulating transcription rates of glucocorticoid-responsive genes in the nucleus, ligand-liberated hGRα is exported back to the cytoplasm and is re-incorporated into the HSP-containing multiprotein complex to function again as a ligand-binding competent receptor (31, 40) (Figure 1). Several mechanisms are postulated for mediating the GRα export from the nucleus to the cytoplasm. The Ca2+-binding protein calreticulin plays a role in this process, directly binding to DBD of the receptor (41-43). The chromosomal maintenance 1 (CRM1, also known as exportin 1)- and the classic nuclear export signal (NES)-mediated nuclear export machinery does not appear to function directly on hGRα (32, 42). Rather, NES-harboring and phospho-serine/threonine-binding proteins 14-3-3s can bind hGRα, and shift its intracellular localization toward the cytoplasm (44, 45). This action of 14-3-3s on hGRα appears to be independent to the ligand-induced nuclear translocation of the receptor, which is mediated in part by the NL1/importin α-associated nuclear pore complex. Numbers of serine and threonine residues of hGRα are phosphorylated by several serine/threonine kinases at their specific target residues, some of which function as phosphorylation-dependent binding sites of 14-3-3 proteins (46). For example, the v-akt murine thymoma viral oncogene homolog 1 (AKT1) (or the protein kinase B α) phosphorylates serine (S) 134 of the hGRα, and 14-3-3 binds to phosphorylated S134. Binding of 14-3-3 on hGRα at this site shifts subcellular localization of the latter to the cytoplasm and downregulates its transcriptional activity inside the nucleus (45, 47). The misshapen-like kinase 1 (MINK1) and the Rho-associated protein kinase (ROCK) respectively phosphorylate threonine (T) 524 and S617 (48). 14-3-3s bind phosphorylated forms of these residues as a dimer (48), possibly modulating subcellular localization and transcriptional activity of the hGRα.
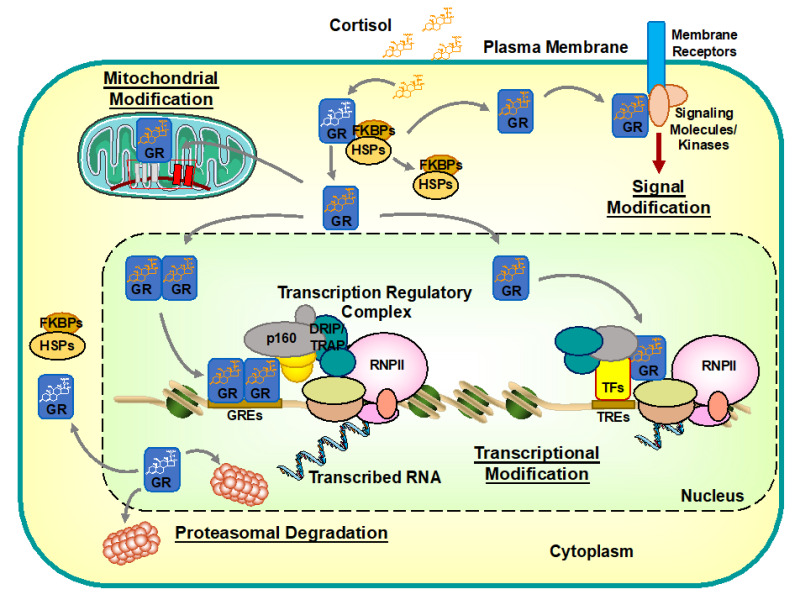
Figure 1:
Intracellular circulation and actions of hGRα. hGRα resides in the cytoplasm in the absence of ligand by forming a heterocomplex with several heat shock proteins (HSPs), immunophilins (e.g., FKBP), and some other proteins. Upon binding to ligand cortisol, hGRα dissociates from the complex and translocates into the nucleus through the nuclear pore. Inside the nucleus, hGRα binds directly to glucocorticoid response elements (GREs) located in promoter/enhancer regions of glucocorticoid-responsive genes. DNA-bound hGRα then stimulates transcription rates of glucocorticoid-responsive genes by attracting the regulatory regions the transcription regulatory complex including the RNA polymerase II (RNPII) and its ancillary components through bridging coactivators, such as p300/CBP and p160 proteins. Promoter/enhancer-bound hGRα also recruits in collaboration with these coactivators various chromatin remodeling molecules, including the DRIP/TRAP complex (DRIP/TRAP), the SWI/SNF chromatin modulator (SWI/SNF), and the Mediator complex (MED). In addition to binding directly to DNA and regulating transcription, hGRα interacts indirectly with regulatory regions of glucocorticoid-responsive genes via protein-protein interaction with other transcription factors (TFs) and/or attracted cofactor molecules, ultimately modulating positively and negatively the transcriptional activity of GRE- and non-GRE-containing glucocorticoid-responsive genes. hGRα then moves back to the cytoplasm to re-form a heterocomplex with HSPs for regaining a ligand-friendly status or is cleared from DNA by proteasomal degradation. Further, hGRα can influence the action of cell surface receptors by associating with their intracellular signaling molecules, such as classic and small G-proteins, and several serine/threonine and tyrosine kinases (known as non-genomic actions of glucocorticoids). Accumulating evidence suggests that liganded hGRα also influences the transcription of mitochondrial genes by translocating into this intracellular organelle. CBP: cAMP-responsive element-binding protein (CREB)-binding protein; DRIP/TRAP: vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein complex; FKBPs: FK506-binding proteins; GREs: glucocorticoid response elements; GR: glucocorticoid receptor; HSPs: heat shock proteins; MED: Mediator complex; p160: p160-type nuclear receptor coactivator; RNPII: RNA polymerase II; SWI/SNF: switching/sucrose non-fermenting complex; TFs: transcription factors; TREs: transcription factor response elements.
Genomic and Non-genomic Actions of hGRα
After binding to glucocorticoids and translocating into the nucleus, hGRα binds as a dimer to a tandem GREs located in promoter/enhancer regions of glucocorticoid-responsive genes, and regulates their mRNA expression positively or negatively, depending on the GRE sequence and the promoter/enhancer context (15, 49, 50) (Figure 1). GRE-bound hGRα stimulates transcription of responsive genes by facilitating formation of the transcription regulatory complex, which includes the RNA polymerase II (RNPII) and its ancillary components (51). Mechanically, hGRα uses its two transactivation domains, AF-1 and AF-2, as protein surfaces for interacting with and attracting nuclear receptor coactivators (51). These proteins then act as bridges between the DNA-bound hGRα and the RNPII-containing transcription initiation complex (52, 53) (Figure 1). In addition, they act in themselves as histone acetyltransferases (HAT) as well as attract other enzymatic proteins, and loosen tightly packed chromatin DNA by chemically modulating specific amino acid residues of histones and other chromatin-associated molecules (54). Representatives of these HAT coactivators include p300 and its homologous cAMP-responsive element-binding protein (CREB)-binding protein (CBP), and the p160 family of nuclear receptor coactivators (NCoAs). The former proteins serve as macromolecular docking “platforms” for many transcription factors, including nuclear hormone receptors, CREB, activator protein-1 (AP-1), nuclear factor-κB (NF-κB), p53, and signal transducers and activators of transcription (STATs), and thus, are called co-integrators (55). On the other hand, the p160 family of nuclear receptor coactivators (NCoAs) is more specific to nuclear hormone receptors including hGRα, and play a central role in the initiation of transcription by hGRα, as they are first attracted to the DNA-bound receptor molecule (55, 56). For physical interaction with hGRα, p160-type coactivators employ the LxxLL motif in which “L” is leucine and “x” is any amino acids. They harbor in their nuclear receptor-binding domain (NRB) multiple LxxLL motifs, each of which have different affinity to respective nuclear hormone receptors (55-58). The LxxLL motif forms the α-helical structure and is deeply buried into the molecular cleft formed by the AF-2 surface of the liganded hGRα (58). Interestingly, p160 family proteins also serve as transcriptional coactivators for some other transcription factors (e.g., NF-κB) (59, 60). In collaboration with these transcriptional coactivators and promoter/enhancer-bound other transcription factors, hGRα interacts with and attracts several distinct chromatin remodeling complexes (e.g., the mating-type switching/sucrose non-fermenting (SWI/SNF) complex, the vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein (DRIP/TRAP) complex, and the Mediator (MED) complex) as well as various enzymatic molecules, scaffold proteins, and long non-coding RNAs (e.g., the steroid receptor RNA coactivator (SRA) and the growth arrest-specific 5 (Gas5)), ultimately forming a huge transcriptional regulatory complex for initiating transcription of the downstream coding sequence though the attracted RNPII (61-64). These newly identified functional oligonucleotides exert their transcriptional regulatory activity in part by modulating the liquid-liquid phase separation among various proteins inside the transcription regulatory complex formed on the DNA-bound hGRα (65).
Similar to the transcription factors incorporated in the transcriptional regulatory complex recruited by GREs-bound hGRα, liganded hGRα is also attracted to the transcription regulatory complex formed by DNA-bound other transcription factors (e.g., AP-1, NF-κB, p53, STATs, and forkhead transcription factors: FOXOs). This incorporation of hGRα can be independent to its physical association with DNA GREs, and the recruited hGRα modulates their transcriptional activity positively or negatively (15, 66) (Figure 1). The interaction between hGRα and these transcription factors are mediated by mutual protein-protein interactions between these proteins or indirectly through bridging coactivators, such as p300/CBP and p160-type coactivators (15). This GRE-independent activity of hGRα may be more important than the GRE-mediated one, given that the mice harboring a mutant GR defective in the dimerization surface, and thus, active in protein-protein interaction but inactive in transactivation via tandem GREs, survive and procreate, in contrast to the mice with Nr3c1 gene knock-out, which die immediately after birth due to respiratory failure (67). Suppression of transactivation of other transcription factors through such protein-protein interactions appears to be important particularly in the suppression of immune functions and inflammation by glucocorticoids (68-70).
Mounting evidence suggests that glucocorticoids also signal within seconds or minutes. These effects are called “non-genomic”, since they do not require the transcriptional activity of hGRα (15). Representative examples of these actions are: (i) the immediate suppression of ACTH release from the anterior pituitary gland by glucocorticoids (71); (ii) the increased frequency of excitatory post-synaptic potentials by glucocorticoids in the brain hippocampus (72); (iii) the cardioprotective role of glucocorticoids through nitric oxide-mediated vasorelaxation (73); and (iv) some immunomodulatory effects of glucocorticoids via inhibition of the T-cell receptor signaling (74). Some of the molecular mechanisms underlying these actions of hGRα have been proposed. For example, ligand-activated hGRα physically interacts with the classic G protein β through its NTD, and may modulate the action of G protein-coupled receptors located at the cytoplasmic membrane (36). Recent studies also demonstrated that hGRα influences the activity of kinase-mediated signaling, such as of the mitogen-activated protein kinase and the phosphatidylinositol 3-kinase through interacting with their key signaling molecules residing under the cytoplasmic membrane or in the cytoplasm (71-75) (Figure 1).
These non-genomic effects of hGRα modulate the action of some intracellular signaling pathways, whereas the latter can influence the activity of hGRα through post-translational modifications (PTMs) of this receptor protein. Such PTMs include phosphorylation, ubiquitination, acetylation, and sumoylation (15). These covalent changes may influence receptor stability, subcellular localization, as well as its interaction with other proteins including transcription factors and transcriptional cofactors/regulators (10). Thus, enzymes catalyzing these PTMs act as molecular effectors of their upstream intracellular signaling pathways for modulating the biologic effects of glucocorticoids by targeting the hGRα protein.
In addition to the above-explained diverse actions, glucocorticoids can modulate expression of the mitochondrial genes by translocating into this cytoplasmic organelle, and by binding to the classic GREs located in some regulatory sites (D-loop) of these genes (76-78) (Figure 1). This action of hGRα in the mitochondria appears to play a role in the glucocorticoid-mediated modulation of apoptosis, a well-known process of the programmed cell death, and may contribute to the therapeutic effects of glucocorticoids on hematologic and other malignancies (79).
PRIMARY GENERALIZED GLUCOCORTICOID RESISTANCE SYNDROME
Pathophysiology and Clinical Manifestations
This syndrome is a condition first described by Chrousos, et.al., as a rare, familial or sporadic, genetic disorder characterized by generalized, partial target tissue insensitivity to glucocorticoids (80). Because of glucocorticoid insensitivity in the central components of the HPA axis, glucocorticoid-mediated negative feedback inhibition on the brain hypothalamus and the anterior pituitary gland is decreased (5, 81) (Figure 2). These changes result in compensatory elevation of the corticotropin-releasing hormone (CRH) and the arginine-vasopressin (AVP) at the hypothalamus and systemic release of the ACTH from the anterior pituitary gland. Excess ACTH secretion then causes bilateral adrenocortical hyperplasia and increased production/secretion of cortisol, which compensates for its reduced actions in target tissues. However, elevated circulating ACTH also stimulates production of other adrenal steroids, such as mineralocorticoids (e.g., deoxycorticosterone (DOC) and corticosterone) and/or adrenal androgens (e.g., androstenedione, dehydroepiandrosterone (DHEA), and DHEA-sulfate (DHEA-S)), leading to the development of excess manifestations of these hormones, because tissue sensitivity to these steroids is not altered. Increased mineralocorticoids may cause hypertension and/or hypokalemic alkalosis, whereas elevated adrenal androgens may develop manifestations (see below) through their direct effects on target tissues and/or indirect actions via modulation of the hypothalamic-pituitary-gonadal axis.
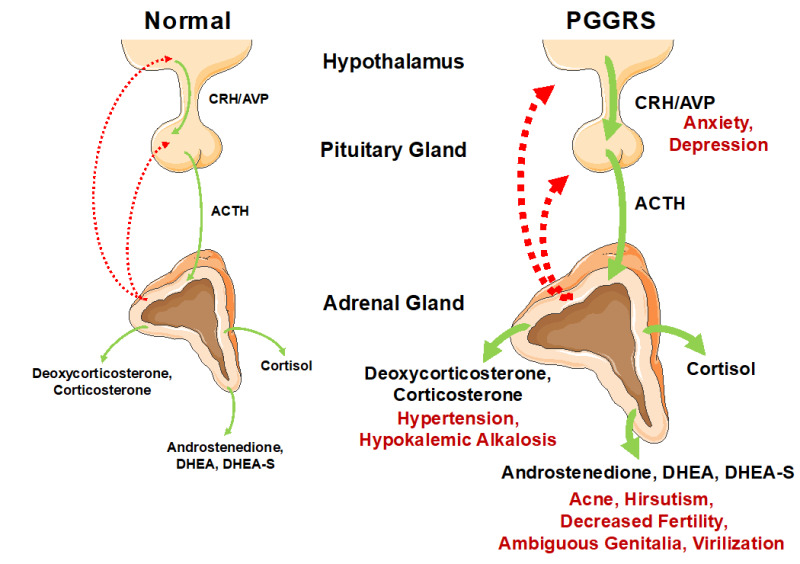
Figure 2.
Pathophysiologic mechanisms and clinical manifestations of primary generalized glucocorticoid resistance syndrome (PGGRS). The HPA axis consists of the brain hypothalamus, the anterior pituitary gland, and the adrenal cortex with their secreting hormones/peptides, CRH/AVP, ACTH and cortisol, respectively. In patients with this syndrome, their HPA axis is re-set to upward with preservation of circadian rhythmicity due to generalized, partial insensitivity to glucocorticoids in entire tissues. Thus, hypothalamic CRH/AVP, pituitary ACTH and adrenal cortisol are all hyper-secreted in order to compensate for the reduced actions of cortisol in both CNS and peripheral tissues. In addition to augmenting production of cortisol in the adrenal glands, elevated ACTH stimulates secretion of mineralocorticoids (e.g., deoxycorticosterone and corticosterone) and androgens (e.g., androstenedione, dehydroepiandrosterone (DHEA) and DHEA-sulfate(S)), which in turn cause a variety of manifestations associated with excess secretion of these hormones. In contrast, manifestations associated with overproduction of cortisol are rare in adult patients but neonates/young children may develop hypoglycemia and associated seizures due to reduced actions of cortisol in the liver. Elevated CRH/AVP in CNS may precipitate anxiety and depression in some patients. Solid lines indicate positive effects, whereas dashed lines show negative effects. Manifestations associated with elevation of the indicated molecules/compounds are shown with red letters. ACTH: adrenocorticotropic hormone; AVP: arginine vasopressin; CNS: central nervous system; CRH: corticotropin-releasing hormone; DHEA: dehydroepiandrosterone; DHEA-S: DHEA-sulfate; PGGRS; primary generalized glucocorticoid resistance syndrome.
Manifestations associated with excess adrenal androgens observed in patients with this syndrome include acne, hirsutism (more common in females), decreased fertility in both sexes, male-pattern hair loss, menstrual irregularities and oligo-anovulation in females, and oligospermia in males. Affected children may develop advanced bone age and subsequent short stature in their adulthood. In female new born babies, clitoromegaly/ambiguous genitalia may be seen (5, 82, 83).
Clinical manifestations of glucocorticoid deficiency are rare in adult patients but are reported in neonates/young children as severe hypoglycemia and associated seizures/coma, because gluconeogenesis depends on the proper action of glucocorticoids in the liver during early childhood (84-86). Some adult patients develop anxiety and/or chronic fatigue, which appear to be caused by elevated hypothalamic CRH and/or AVP (87-92). Increased circulating ACTH may cause bilateral adrenal hyperplasia (5). Some patients harbor adrenal incidentalomas (93, 94). Although this adrenal neoplasm is very common in general population (95), elevated circulating ACTH may facilitate tumor development and/or its growth. Further, one patient with this syndrome harbored an ACTH-producing pituitary adenoma, which might have been caused/facilitated by the elevated CRH/AVP (96).
Finally, the clinical spectrum of this syndrome is broad, ranging from severe to mild forms, and a number of patients may even be asymptomatic, displaying biochemical alterations only (5, 93, 97, 98). This heterogeneity is mainly due to variable impact of the patients’ genetic changes in the receptor protein, but other factors, such as their genetic backgrounds and/or epigenetic and biochemical changes, for example, associated with their ageing and lifestyles, may also contribute to variability of disease expression.
NR3C1 Gene Mutations That Cause Primary Generalized Glucocorticoid Resistance Syndrome
The molecular basis of this syndrome is ascribed to inactivating mutations in the NR3C1 gene, which impair molecular actions of hGRα and hence decrease tissue sensitivity to glucocorticoids. Currently, 36 pathologic mutations that cause this syndrome have been reported (Table 1 and Figure 3). Chrousos, et. al., reported the first family of this syndrome who carried a homozygous miss-sense mutation, which replaces adenine by thymine at nucleotide position 1,922 (80). The NR3C1 gene harboring this mutation expresses the hGRαD641V mutant receptor, which has valine (V) instead of aspartic acid (D) at amino acid position 641 in the LBD (80). Since then, numbers of patients were reported whose pathologic mutations were identified mostly as heterozygous in the coding sequence of LBD (90, 96, 99-102). Most of these patients demonstrated characteristic manifestations, such as those of mineralocorticoid and androgen excess, similar to the original case of Chrousos, et. al., thus they may be considered as “classic cases”. More recently, technological progress in the genome sequencing including the use of capillary or high through-put next generation sequencers enabled clinical researchers to conduct large studies with recruitment of the subjects with conventional/unconventional manifestations, (e.g., obesity and bilateral adrenal incidentalomas, as evident in the French Muta-GR study (ClinicalTrials.gov Identifier: NCT02810496) (97)). Clinicians are now able to obtain much easier and faster than before the data of patients’ genome sequence around the NR3C1 gene. Together with growing acknowledgement of this syndrome among clinicians and clinical researchers, such technological progress appears to have facilitated the discovery of new cases with classic symptoms, as well as those with much milder and/or alternative manifestations or even with biochemical changes only. Further, the identified mutations tend to distribute over the entire NR3C1 gene including coding areas of all three major domains and intronic sequences (Table 1 and Figure3).
Among 36 pathologic NR3C1 mutations, only three are homozygous mutations, while the other 33 are heterozygous (Table 1 and Figure 3). One patient harbors two different NR3C1 mutations each of which are identified in different alleles (thus, compound heterozygous) (86). Among 34 mutations found in the NR3C1 coding sequence, 24 are miss-sense mutations, which replace one amino acid with another (thus, point mutations), five are non-sense mutations, which introduce a stop codon and generate truncated receptor proteins, and another five are frame-shift mutations, which also develop truncated receptors but with additional unrelated amino acids after the mutation point. At the receptor protein level, 22 mutations are located in LBD, two in HR, seven in DBD, and four are in NTD (Figure 3). In addition to these coding sequence mutations, two mutations are identified in the intronic sequence, located in intron F (between exon 5 and 6) and in intron I (between exon 7 and 8), respectively (91, 103).
Table 1.
The NR3C1 Gene Mutations that Cause Primary Generalized Glucocorticoid Resistance Syndrome†.
| Amino Acid Change | Nucleotide Change | Zygosity | Mutation Type | Proband’s Gender and Age | Clinical Manifestations | Molecular Defects | References |
|---|---|---|---|---|---|---|---|
| NTD Mutations | |||||||
| P9R | 26C>G | Heterozygous | Point Mutation | M, 33 | Hypertension | N.D. | (104) |
| Q123X | 367G>T | Heterozygous | Point Mutation | F, 31 | Fatigue, Anxiety, Hirsutism, Irregular menstruation, Infertility | N.D. | (87) |
| E198X | 592G>T | Compound heterozygous with 2141G>A mutation | Point Mutation | F, 3 | Hypoglycemia Hypertension | Also harbors R714Q expressed from a different allele | (86) |
| D401H† | 1201G>T | Heterozygous | Point Mutation | F, 43 | Hypertension Hyperglycemia | Increased transcriptional activity | (105) |
| DBD Mutations | |||||||
| V423A | 1268T>C | Heterozygous | Point Mutation | M, 9 | Fatigue Anxiety Hypertension | Decreased DNA-binding activity | (88) |
| R469X | 1405C>T | Heterozygous | Point Mutation | M, 46 | Adrenal hyperplasia Hypertension Hypokalemia | No GR mRNA and protein expression from the affected allele | (106) |
| R477C | 1429C>T | Heterozygous | Point Mutation | F, 12 | Mild hirsutism Elevated cortisol | N.D. | (92) |
| R477H | 1430G>A | Heterozygous | Point Mutation | F, 41 | Hypertension, Hirsutism, Fatigue | No DNA-binding activity | (107) |
| R477S | 1429C>A | Heterozygous | Point Mutation | F, 30 | Hypertension Elevated serum cortisol | No DNA-binding activity | (93) |
| Y478C | 1433A>G | Heterozygous | Point Mutation | M, 49 | Adrenal incidentaloma No symptoms | Decreased DNA-binding activity | (93) |
| HR Mutations | |||||||
| R491X | 1471C>T | Heterozygous | Point Mutation | M, 44 | Bilateral adrenal hyperplasia Elevation of ACTH and cortisol | Decreased transcriptional activity | (97) |
| Q501H | 1503G>T | Heterozygous | Point Mutation | F, 60 | No symptoms Mild elevation of urinary free cortisol | Decreased transcriptional activity | (97) |
| LBD Mutations | |||||||
| S551Y | 1652C>A | Heterozygous | Point Mutation | M, 14 | Fatigue Hypokalemia Hypertension Polyuria | Decreased affinity to ligand Decreased transcriptional activity | (108) |
| T556I | 1667C>T | Heterozygous | Point Mutation | M, 56 | Adrenal incidentaloma Increased UFC | N.D. | (94) |
| I559N | 1676T>A | Heterozygous | Point Mutation | M, 33 | Hypertension, Oligospermia, Infertility | No ligand-binding activity | (96, 99) |
| V571A | 1724T>C | Heterozygous | Point Mutation | F, 9 | Ambiguous genitalia*, Hypertension, Hypokalemic Alkalosis Hyperandrogenism | Highly decreased ligand-binding activity | (82, 100) |
| V575G | 1724T>G | Heterozygous | Point Mutation | M, 70 | Bilateral adrenal hyperplasia (His daughters have mild hirsutism) | Decreased affinity to ligand Decreased transcriptional activity | (98) |
| H588LfsX5 | 1762-1765insTTAC>G | Heterozygous | Frame Shift | F, 41 | Hirsutism Anxiety Fatigue | N.D. | (92) |
| L595V | 1915C>G | Heterozygous | Point Mutation | F, 16 | No symptoms | Decreased affinity to ligand Decreased transcriptional activity | (98) |
| S612YfsX15 | 1835delC | Heterozygous | Frame Shift | F, 20 | Fatigue Hirsutism | No ligand-binding activity | (109) |
| D641V | 1922A>T | Homozygous | Point Mutation | M, 48 | Hypertension, Hypokalemic alkalosis | Reduced affinity to ligand Reduced transcriptional activity | (80) |
| Y660X | 1992A>T | Heterozygous | Point Mutation | F, 70 | Hypokalemia Hypertension | No transcription activity | (110) |
| L672P | 2015T>C | Heterozygous | Point Mutation | M, 46 | No symptom Mild elevation of urinary free cortisol Adrenal incidentaloma | No ligand-binding activity No transcriptional activity | (93) |
| G679S | 2035G>A | Heterozygous | Point Mutation | F, 19 | Hirsutism Fatigue Hypertension | Decreased affinity to ligand Decreased transcriptional activity | (111) |
| R714Q | 2141G>A | Heterozygous | Point Mutation | F, 2 | Hypertension Mild clitoromegaly Advanced bone age Precocious puberty Hypokalemia | Decreased affinity to ligand Decreased transcriptional activity | (84) |
| R714Q | 2141G>A | Heterozygous | Point Mutation | F, 31 | Unsuccessful attempts for pregnancy for 2.5 years | Decreased affinity to ligand Decreased transcriptional activity | (112) |
| R714Q | 2141G>A | Compound heterozygous with 592G>T mutation | Point Mutation | F, 3 | Hypoglycemia Hypertension | Also harbors E198X expressed from the other allele | (86) |
| H726R | 2177A>G | Heterozygous | Point Mutation | F, 30 | Hirsutism Acne Alopecia Anxiety Fatigue Irregular menstrual cycles | Decreased affinity to ligand Decreased transcriptional activity | (89) |
| V729I | 2185G>A | Homozygous | Point Mutation | M, 6 | Precocious puberty Hyperandrogenism | Reduced affinity to ligand Reduced transcriptional activity | (101) |
| F737L | 2209T>C | Heterozygous | Point Mutation | M, 7 | Hypertension Hypokalemia | Decreased affinity to ligand Decreased transcriptional activity | (7) |
| I747M | 2241T>G | Heterozygous | Point Mutation | F, 18 | Hirsutism Oligo/amenorrhea | Decreased affinity to ligand Decreased transcriptional activity | (102) |
| I757V | 2269A>G | Heterozygous | Point Mutation | F, 23 | No symptoms | Decreased affinity to ligand Decreased transcriptional activity | (97) |
| L773P | 2318T>C | Heterozygous | Point Mutation | F, 29 | Hypertension Hirsutism Fatigue Anxiety | Decreased affinity to ligand Decreased transcriptional activity | (90) |
| L773VfsX25 | 2317-2318delCT | Heterozygous | Frame Shift | M, 27 | Hypoglycemia Fatigability with feeding Hypertension | No ligand-binding activity | (113) |
| F774SfsX24 | 2318-2319delTG | Homozygous | Frame Shift | M, 1 | Hypokalemia Hypoglycemia Hypertension | No ligand-binding activity | (85) |
| Intronic Mutations | |||||||
| NR (No protein expression) | 1891-1894delGAGT | Heterozygous | Destruction of the splice donor site | F, 26 | Hirsutism, Menstrual Irregularities | No GR mRNA and protein expression from the affected allele | (103) |
| N.D. Predicted to generate V675GfsX10 | 2024G > T | Heterozygous | Predicted to skip exon 8 | F, 49 | Hirsutism, Menstrual Irregularities, Anxiety | N.D. | (91) |
- *
: The case also harbors a heterozygous mutation in the 21-hydroxylase gene.
- †
: The 1201G>T D401H mutation causes mild glucocorticoid hypersensitivity.
N.D.; not determined。
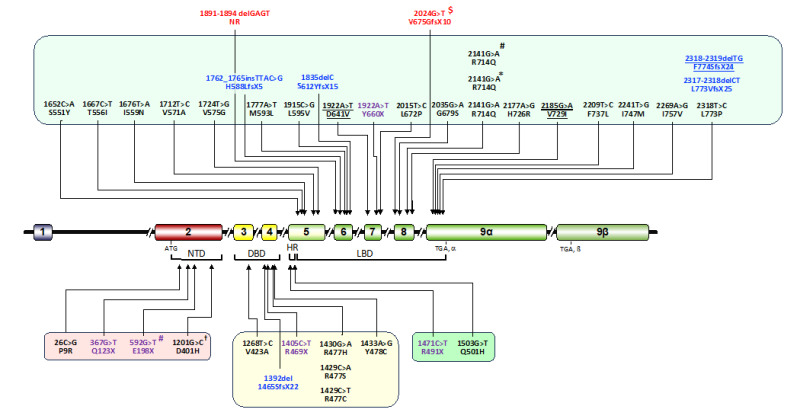
Figure 3.
Location of the NR3C1 gene mutations that cause primary generalized glucocorticoid resistance syndrome†. Currently, 36 independent mutations are reported. The mutations identified in the coding sequence of LBD, HR, DBD and NTD are shown in a light green, green, yellow and red box, respectively. Miss-sense mutations, non-sense mutations and frame-shift mutations are shown with black, purple and blue letters, respectively. Two mutations identified in the intronic sequence are shown with red letters. Homozygous mutations are shown with underlines. †: The 1201G>T D410H mutation causes mild glucocorticoid hypersensitivity; *: The same miss-sense mutation but found in unrelated subjects/families; $: Prediction only (the mutated hGRα protein was not biologically identified); #: These two mutations were found as compound heterozygous in one affected subject. Numbers of nucleotides and amino acids are based on the transcription initiation site and the first methionine of the hGRα protein, respectively. DBD: DNA-binding domain; HR: hinge region; LBD: ligand-binding domain; NTD: N-terminal domain.
Molecular Defects of Pathologic hGRα Mutants
Molecular defects of pathologic mutant receptors have been extensively investigated by focusing on their defects in ligand-association, transactivation of glucocorticoid-responsive genes, cytoplasmic to nuclear translocation, and others (5). Recently, computer-based in silico structural simulation has also been used for estimating the structural impact of mutations to hGRα LBD and DBD (88, 114).
Pathologic mutant receptors generally cause inactivation/reduction of one or some of the receptor functions, whereas they are in most cases heterozygous mutations that enables affected subjects to harbor both mutated and intact hGRα protein in their tissues (5, 6). Thus, affected subjects of this syndrome demonstrate partial loss of glucocorticoid actions in their tissues, consistent with the experimental evidence that genetic knock-out (inactivation) of the Nr3c1 gene in mice (thus, complete abbreviation of the GR protein and its actions) is lethal (115). However, one homozygous case who only expresses a mutant receptor with complete loss of the ligand-binding activity was reported (2318-2319delTG F774SfsX24) (85). Given that the ligand-binding is essential for subsequent receptor activation, this mutant receptor might have residual activities including minimal association to glucocorticoids or other steroids, enabling the patient to survive even though he only expresses this highly damaged receptor.
LBD MUTATIONS
There are 22 pathologic mutations whose amino acid changes are identified in the LBD. Among them, 17 are miss-sense mutations (see Table 1 and Figure 3 for details), one is a non-sense mutation (1992A>T Y660X) (115), and four are frame-shift mutations (1762-1765insTTAC>G H588LfsX5, 1835delC S612YfsX15, 2317-2318delCT L773VfsX25 and 2318-2319delTG F774SfsX24) (85, 92, 109, 113). Since LBD is the domain harboring a majority of receptor functions with established evaluation means (15), molecular defects of these mutant receptors have been most extensively and systemically investigated. These molecular examinations include: i) the affinity of the mutant receptors for the ligand (the synthetic pure glucocorticoid dexamethasone was used in most cases, thus the method is called “dexamethasone binding assay”); ii) the transcriptional activity of the mutant receptors on endogenous glucocorticoid-responsive genes and/or transiently introduced exogenous GRE-driven reporters; iii) the ability of in vitro physical interaction of the mutant receptors with p160-type nuclear receptor coactivators, such as the glucocorticoid receptor-interacting protein 1 (GRIP1 or NCoA2); iv) the subcellular localization of the mutant receptors and their nuclear translocation in response to glucocorticoids (in most cases, dexamethasone was used as a ligand); v) the ability of the mutant receptors to bind endogenous DNA GREs (using the chromatin-immunoprecipitation (ChIP) assay); vi) the structural analysis on the mutant receptors’ LBDs by employing the computer-based in silico three-dimensional (3D) simulation using as a template crystallographic data of the LBD peptide; vii) the motility of the mutant receptors inside the nucleus using the fluorescence recovery after photobleaching (FRAP) analysis.
Molecular defects in two major functions of the hGRα, the ability to bind glucocorticoids and the transactivation of glucocorticoid-responsive genes are summarized in Table 1. Compared with the wild-type receptor, all mutant receptors demonstrate variable reduction in their affinity to dexamethasone, and attenuate their transactivation of GREs-driven genes following exposure to this steroid, with the most severe impairment observed in the cases of I559N, V571A, D641V, L672P, R714Q, I747M, L773P, L773VfsX25 and F774fsX24 mutations (80, 82, 84, 85, 96, 99, 100, 102, 110, 113). In the in silico 3D structural simulation analysis on LBD of the miss-sense point mutant receptors, most of the replaced amino acids are located outside the molecular structures, which directly mediate these two major functions, LBP and the AF-2 surface, respectively (114). The latter is used for physical interaction with the LxxLL motif of p160-type coactivators (58). Further analysis revealed that these point mutations damage and/or alter multiple intramolecular amino acid interactions necessary for maintaining the proper structural conformation of LBD, resulting in the alteration in these two protein surfaces indirectly but simultaneously (114). More detailed structural analysis revealed that the amino acid replacements damage LBP by indirectly reducing the electrostatic interaction between key residues of LBP and those of the dexamethasone molecule (especially, the interaction formed against the carbonyl oxygen of carbon (C) 3 of this steroid) (114). Their impact on the interaction between the AF-2 surface and the LxxLL motif of the p160-type coactivator GRIP1 protein is variable, but tends to damage the ionic interaction (or salt bridge) of non-core leucines of this motif as well as the noncovalent interaction of its core leucine residues formed against key amino acids of the AF-2 surface, ultimately reducing the affinity of this motif to the hydrophobic cleft of the AF-2 surface (114).
The C-terminal portion of the hGRα LBD that follows the α-helix-12 of this domain is one of the hot spots of pathologic hGRα mutations, as evident in the accumulation of three independent mutations to this region (L773P, L773VfsX25 and F774fsX24) (85, 90, 113). Indeed, this molecular area is particularly important for creating the AF-2 surface and for maintaining the ligand-bound LPB conformation through its dramatic intramolecular shift upon binding to a ligand (30). Arginine (R) at amino acid position 714 is another hot spot of the point mutations, as three patients independently harbor this mutation that replaces this amino acid to glutamine (Q) (84, 86, 112). In the structural simulation analysis on the R714Q mutant receptor, substitution of R for Q in LBD causes a rearrangement of the side chains resulting in forming a new salt bridge between R704 and D662 and displacing Q714 (84). This relaxes some constraint on the helix-10 and results in structural changes throughout the LBD, indirectly damaging conformation of both LBP and the AF-2 surface (84). Interestingly, the third case with the R714Q mutation harbors another point mutation (592G>T E198X) in the other allele (compound heterozygous), which generates a truncated receptor at E198 (E198X) (86). Thus, the patient expresses both R714Q and E198X mutant receptors but no intact receptor in her tissues.
The LBD mutant receptors frequently demonstrate delay of their translocation from the cytoplasm to the nucleus compared to the wild-type receptor, consistent with the fact that the ligand-binding “turns on” the nuclear translocation of the receptor by inducing the conformational change that allows the receptor to expose NL1 and NL2 surfaces to their counterpart nuclear import systems (7, 84, 85, 89, 90, 98-100, 102, 116). Although detailed molecular mechanisms underlying this defect have not been examined yet, it is likely that the mutations interrupt proper functions of these domains (32). Some mutant receptors, such as hGRαV729I and hGRαF737L, shift their subcellular localization toward the nucleus in the absence of ligand (7, 100), possibly by their defective intracellular circulation, such as through defective NL1 activity and/or altered interaction with14-3-3 proteins, calreticulin or others.
All LBD mutant receptors tested for their interaction with DNA GREs preserve their ability to bind this recognition sequences, because they have intact DBD, which can function independently to LBD (7, 84, 85, 89, 90, 98-100, 102, 116). Further, many of these mutant receptors demonstrate a dominant negative effect on the transcriptional activity of the wild-type receptor, because they are in most cases partially active mutants, and thus, can interfere with the full activity of the wild-type receptor, such as by competing for the molecules mediating the latter’s transcriptional activity (e.g., by squelching transcriptional cofactors including p160-type coactivators) (5, 6, 102). Finally, the LBD point mutant receptors tested in the FRAP analysis demonstrate dynamic motility defects inside the nucleus of living cells, possibly due to their reduced affinity to ligand and/or inability to interact properly with key cofactors and/or chromatin molecules (117).
Molecular characterization of the LBD mutants explained above have been performed mostly by employing cell-based bioassays. However, Kaziales, et. al., recently performed in vitro biochemical assays on the L773P mutant receptor by employing its purified peptide consisting of DBD, HR, and intact or mutated LBD (118). The “wild-type” receptor peptide (called GRm) employed for their assays harbors multiple amino acid replacements for conferring its peptide stability. Thus, the authors compared GRm and GRmL773P, and found that the latter has altered physical interaction with HSP90 (118). They suggested that this molecular defect underlies the reduced interaction of the receptor peptide to dexamethasone, the LxxLL motif, and further, DNA GREs, although exact molecular evidence and associated mechanisms were not demonstrated.
HR MUTATIONS
Two pathologic mutations were identified in HR (Table 1 and Figure 3). One is a non-sense mutation (1471 C>T R491X) and the other is a miss-sense mutation (1503 G>T Q501H) (97). Both are located in exon 5. The patient harboring R491X developed typical manifestations of Chrousos syndrome, as the mutant receptor lacks the entire LBD (97). On the other hand, the subject harboring Q501H demonstrated biochemical changes only, while the mutant receptor showed weakly reduced transactivation of the exogenous glucocorticoid-responsive gene (97).
DBD MUTATIONS
Currently, seven pathologic mutations were identified in DBD (Table 1 and Figure 3). Among them, five are miss-sense (point) mutations. The other two are a non-sense mutation and a frame-shift mutation. All five point mutant receptors reduce or lose their affinity to DNA GREs (88, 92, 93, 97, 107). In contrast, they retain intact affinity for ligand dexamethasone, because DBD and LBD function independently with each other (88, 92, 93, 97, 107). Among these point mutations, three (1430G>A R477H, 1429C>T R477S and 1429C>T R477C) replace arginine (R) at amino acid position 477 to other amino acids (histidine (H), serine (S) and cysteine (C), respectively), while one targets tyrosine (Y) at position 478 and changes it to cysteine (C) (1433A>G Y478C). Thus, the area around R477 and Y478 appears to be a hot-spot of DBD mutations. These two amino acids are located just C-terminally to the fourth cysteine residue of the second zinc finger of DBD, which participates in holding a zinc ion together with the other three cysteines of this finger motif. R477 is critical for maintaining the ability of the receptor to bind GREs by providing the hydrophobicity required for its interaction with the backbone chain of the GRE DNA. Thus, replacement of either of these two amino acids seems to reduce the affinity of the mutant receptors to the GRE DNA through damaging this local hydrophobicity.
The point mutation 1268T>C V423A replaces valine (V) at amino acid position 423 to alanine (A) (88). V423 is located just N-terminally to the second cysteine of the first zinc finger of DBD. Replacement of this valine to alanine at amino acid position 423 permits water molecules to diffuse into the zinc-binding region of the receptor and indirectly damages the hydrophobicity maintained by R477, leading to the reduction in the affinity of this mutant receptor to the GRE DNA (88).
Interestingly, the mutant receptors V423A, R477S and Y478C demonstrate delayed cytoplasmic to nuclear translocation upon exposure to dexamethasone (88, 93). Molecular defect(s) underlying this impairment have(s) not been elucidated, but these mutations appear to affect indirectly the function of NL1, because this molecular surface spans over the second zinc finger of DBD, while these mutations damage the hydrophobic circumstance around this finger (88, 107). The second zinc finger of the DBD is also critical for receptor homodimerization, which is a prerequisite for the receptor to bind a tandem GREs and subsequent transactivation of glucocorticoid-responsive genes harboring this DNA sequence (119). Thus, defective homodimerization may also contribute to the reduced transcriptional activity of these DBD mutant receptors.
NTD MUTATIONS
Four independent point mutations are reported in NTD. These include 26C>G P9R, 367G>T Q123X, 592G>T E198X and 1201G>T D401H (86, 87, 104, 105). The 367G>T Q123X and the 592G>T E198X are non-sense mutations generating truncated receptors, respectively at amino acid position 123 and 198. Because both receptors appear to be highly damaged as they lack the entire DBD and LBD, the affected subjects demonstrated clear-cut manifestations of Chrousos syndrome (86, 87).
The patient harboring the 26C>G P9R mutation demonstrated mild clinical manifestations with slight increase in ACTH and cortisol secretion (104). Molecular characterization was not performed for this mutant receptor (104), thus there is a possibility that the identified nucleotide change is not pathologic. Indeed, NTD (exon 2) is the domain most harboring single nucleotide polymorphisms (SNPs) among all three major domains throughout the nuclear hormone receptor genes (13), thus this domain can well tolerate to nucleotide replacements and tends to maintain its proper functions compared to the other domains.
The patient harboring the 1201G>T D401H mutation demonstrated mild hypersensitivity to glucocorticoids in contrast to the other pathologic mutations that cause glucocorticoid resistance (105). Compared to the wild-type receptor, the D401H mutant receptor demonstrated ~2-fold stronger transcriptional activity in a reporter assay, which is equivalent to the activity of the N363S mutant receptor in a side-by-side assay. The nucleotide change causing the N363S replacement is a well-known polymorphism associated with mild glucocorticoid hypersensitivity (120-122). Thus, the 1201G>T D401H may be another weakly functional polymorphism causing mild tissue hypersensitivity to glucocorticoids.
The 3-year-old girl with the 592G>T E198X mutation additionally harbors the 2141G>A R714Q mutation in the other allele as explained in the section of “LBD mutations” (86). She developed severe manifestations of glucocorticoid resistance, such as uncontrollable hypertension, brain micro-infarctions, and hypoglycemic coma, because both mutant receptors she harbored are highly damaged. The family study revealed that the 592G>T E198X mutation is maintained among her family, while the 2141G>A R714Q mutation is de novo in the affected girl (86).
INTRONIC MUTATIONS
So far, only two intronic mutations are reported. One is the 1891-1894 delGAGT NR, which deletes four nucleotides (GAGT) at the nucleotide position 1891-1894 (in intron F located between exon 4 and 5) that destroys the intron-acceptor site located 5’ terminally to exon 6 (91, 103). The mutated mRNA expressed from the affected allele loses its biological stability, therefore the mutation functionally “knocks-out” NR3C1 of this allele (103). The amount of patient’s tissue hGRα is thus 50% of the healthy subjects as the receptor protein is only expressed from the intact allele (103). The other mutation is the 2024G>T, which replaces G with T at the position one nucleotide 5’ terminally to exon 8 (thus, located at the 3’-terminal portion of intron I) (91). Although biochemical characterization on the mutant receptor was not performed, the computer-based prediction indicated that the mutation appears to cause a skip of the entire exon 8 and to generate the V675GfsX10 truncated receptor whose molecular function appears to be highly damaged (91). It is also possible that the mutation reduces stability of its mRNA, leading to functional “knock-out” of NR3C1 of the affected allele similar to the 1891-1894 delGAGT NR mutation (103). Thus, biochemical evaluation on the mutated mRNA and hGRα protein is needed.
Clinical Evaluation of Patients with Primary Generalized Glucocorticoid Resistance Syndrome
Key for evaluating patients with this syndrome is to identify the manifestations suggesting upregulation of the HPA axis without Cushingoid features (5) (Table 2). Circadian rhythmicity of circulating ACTH and cortisol should be preserved, in contrast to the patients with Cushing syndrome (5). In addition, any evidence suggesting psychiatric problems (e.g., anxiety and depression), possibly through upregulation of brain CRH and/or AVP may be noted (5).
Physical examination should include an assessment for signs of hypertension and associated metabolic alkalosis caused by elevated levels of adrenal mineralocorticoids (5). Arterial blood pressure should be recorded and should be monitored over a 24-hour period. Signs of hyperandrogenism and/or virilization caused by over-production of the adrenal androgens, such as acne, hirsutism, pubic and axillary hair development, male-pattern hair loss, and clitoromegaly, should be evaluated. Hirsutism should be assessed using the Ferriman-Gallwey score (123), while pubic hair development should be classified according to the Tanner scale (124, 125). All subjects should be screened for signs associated with Cushing syndrome or therapeutic use of high-dose glucocorticoids.
Table 2.
Clinical Manifestations and Diagnostic Evaluation of Primary Generalized Glucocorticoid Resistance Syndrome
| Clinical Presentation |
| Glucocorticoid excess Apparently normal glucocorticoid actions without Cushingoid features (However, hypoglycemia and associated coma/seizures can be observed in affected neonates/young children) Mineralocorticoid excess Hypertension Hypokalemic alkalosis Adrenal androgen excess Children: Ambiguous genitalia at birth*, clitoromegaly, premature adrenarche, gonadotropin-independent precocious puberty Females: Acne, hirsutism, male-pattern hair loss, menstrual irregularities, oligo-anovulation, infertility Males: Acne, hirsutism, oligospermia, adrenal rests in the testes, infertility CRH/AVP excess in brain hypothalamus and elevation of circulating ACTH levels Anxiety Benign pituitary tumors (ACTH-producing) Bilateral adrenal hyperplasia Adrenal adenomas |
| Diagnostic Evaluation |
| Upward shift of the HPA axis activity and responsiveness to high-dose glucocorticoids Elevated plasma ACTH concentrations Elevated serum cortisol concentrations Increased 24-hour urinary free cortisol (UFC) excretion Resistance of the HPA axis to dexamethasone suppression but positive response to its high, grading doses Normal circadian rhythmicity of circulating cortisol and ACTH concentrations Presence of glucocorticoid resistance in patients’ tissues The thymidine incorporation assay using patients’ PBMCs: Reduced dexamethasone-induced suppression of phytohemagglutinin-stimulated thymidine incorporation compared to normal subjects The dexamethasone binding assay using patients’ PBMCs: Decreased affinity to dexamethasone compared to normal subjects Identification of mutation(s) in the NR3C1 gene (mostly in its coding sequence but can be in exon/intron junctions) Identification of molecular defects of mutant receptors in appropriate assay systems * The case demonstrating this manifestation also harbored a heterozygous mutation in the 21-hydroxylase gene. |
Endocrinological Evaluation of Patients with Primary Generalized Glucocorticoid Resistance Syndrome
The aim of the endocrinological evaluation is to demonstrate up-regulation of the HPA axis with preservation of its normal circadian rhythmicity and blunted responsiveness to exogenous glucocorticoids (5). Concentrations of plasma ACTH, renin activity and aldosterone, as well as serum cortisol, corticosterone, deoxycorticosterone, testosterone, androstenedione, DHEA, and DHEA-S should be measured. Determination of 24-hour UFC excretion on 2 or 3 consecutive days is important to access the presence of hypercortisolism. Diurnal fluctuation of plasma ACTH and serum cortisol should be evaluated, for example, by monitoring them both in the morning and in the evening.
Responsiveness of the HPA axis to exogenous glucocorticoids should be examined using the dexamethasone suppression test (5). Increasing doses of dexamethasone (e.g., 0.3, 0.6, 1.0, 1.5, 2.0, 2.5, and 3.0 mg) should be given orally at midnight every other day, and a serum sample should be drawn at 0800h the following morning for determining serum cortisol concentrations. Affected subjects demonstrate resistance of the HPA axis to administered dexamethasone but can respond to higher doses. Concurrent measurement of serum dexamethasone concentrations is recommended in order to exclude the possibility of increased metabolic clearance or decreased absorption of this compound (83). Pituitary and adrenal imaging studies should be performed, because patients with this syndrome frequently harbor hypertrophy of these organs or may develop their benign tumors.
Cellular and Molecular Studies on Patients with Primary Generalized Glucocorticoid Resistance Syndrome
The purpose of cellular studies is to identify the presence of tissue resistance to glucocorticoids in actual tissues of the affected subjects. The thymidine incorporation assay and the dexamethasone binding assay employing subjects’ peripheral blood mononuclear cells (PBMCs) are generally employed (5, 126) (Table 2). In the former assay, dexamethasone administration strongly suppresses phytohemagglutinin-stimulated thymidine incorporation of PBMCs in normal subjects. However, this response is significantly blunted in the affected subjects due to reduced affinity/actions of this steroid in these cells. The dexamethasone binding assay can address reduction in the affinity of patients’ tissue hGRα to dexamethasone, because mutant receptors harboring their defects in LBD almost always show reduced affinity for this steroid.
As part of the molecular examination for verifying pathologic causes and their molecular mechanisms, sequencing of the coding region of the NR3C1 gene including exon/intron junctions should be performed (126). Identification of mutations in the NR3C1 gene is critical for diagnosing this syndrome. Once mutations are identified, the next step is to prove that the identified mutations have biologic impact. Because the NR3C1 gene harbors so many neutral polymorphisms (13), there is always a possibility that the identified nucleotide changes are just coincidental but not pathologic. Population incidence of the identified nucleotide changes is important if available, as pathologic mutations generally have a very low allele frequency. Molecular studies can be started by constructing the mutant hGRα-expressing plasmids. Then, molecular actions of mutant receptors can be examined by transfecting the created plasmids (transiently or stably) to appropriate cell lines (e.g., GR-negative African green monkey kidney CV1 and COS7 cells, and GR-positive human cervical cancer HeLa cells). Using mutant receptor-expressing cultured cells, reporter transactivation assays using the GREs-driven luciferase gene can be performed to address the reduced transcriptional activity of mutant receptors. The dexamethasone binding assay can also be performed in the COS7 cells transiently expressing mutant receptors to evaluate their affinity to dexamethasone in the absence of the wild-type GR. In microscope-based imaging studies on the cells transfected with plasmids expressing mutant receptors, their abnormal subcellular localization and delayed nuclear translocation in response to dexamethasone can be evaluated.
Management of Patients with Primary Generalized Glucocorticoid Resistance Syndrome
The aim of the treatment for patients with this syndrome is to suppress the excess ACTH secretion in order to reduce production of the adrenal steroids with mineralocorticoid and/or androgenic activity to minimize their pathologic effects (5). Treatment involves the administration of high doses of mineralocorticoid activity-sparing pure glucocorticoids (e.g., dexamethasone), which activate mutated and/or wild-type hGRα in the hypothalamus/pituitary gland of the affected subjects and suppress their ACTH secretion. Adequate suppression of the HPA axis is of particular importance, given that the treatment is virtually life-long, thus any side effects of exogenous glucocorticoids should be avoided as much as possible. Long-term dexamethasone treatment should be titrated carefully according to the clinical manifestations and biochemical profiles of the affected subjects.
CONCLUSIVE REMARKS AND FUTURE PERSPECTIVES
Primary generalized glucocorticoid resistance syndrome is characterized by hypercortisolism without Cushingoid features but with manifestations caused by upregulation of the HPA axis, such as hypertension (by mineralocorticoid excess) and signs of hyperandrogenism (by adrenal androgen excess) (81). The pathologic cause of this syndrome is ascribed to mutations in the NR3C1 gene, which decrease the action of its encoding protein hGRα, a ligand-dependent transcription factor (15, 81). In honor to Professor George P. Chrousos who discovered the first case and significantly contributed to the progress of this field, this syndrome may be called “Chrousos syndrome”, particularly for the cases who demonstrate classic and characteristic manifestations of this syndrome (6, 80). Recent progress in genome technology including high through-put sequencing has enabled clinical researchers to handle large patient cohorts and clinicians can get access to the NR3C1 gene sequencing much easier and faster than before. Consequently, 35 cases/families of this syndrome are currently reported world-wide who harbor pathologic mutations in the NR3C1 gene. It is of note that some of the recent cases tend to demonstrate much milder manifestations compared to the classic cases of Chrousos syndrome (97, 110). Further, some of them even lack obvious manifestations but show biochemical or imaging abnormalities only (93, 97). For these cases with very mild or no manifestations, their genetic changes may be considered as rare polymorphisms rather than pathologic mutations. Further discussion is needed for distinguishing pathologic mutations and mildly functional polymorphisms based on their clinical manifestations and allele frequency of the nucleotide changes.
In some reported cases, molecular defects of the mutated receptors were not evaluated. Testing them in tandem with the wild-type receptor is crucial for avoiding false-diagnosis, because the NR3C1 gene harbor substantial numbers of biologically silent polymorphisms (13). On the other hand, there are patients who demonstrate characteristic manifestations of Chrousos syndrome but do not harbor pathologic mutations in the NR3C1 gene. These “mutation-silent” subjects might carry their genetic defects not in NR3C1 but in other genes whose encoding proteins function in the glucocorticoid signaling pathway. For example, there was a boy who demonstrated manifestations compatible with multiple steroid hormone resistance (127). He harbored a small gene segmental deletion around one zinc finger protein (ZNF) gene, and Its encoding protein ZNF764 turned out to function as a coactivator of several steroid hormone receptors including the hGRα (127). As our knowledge of the glucocorticoid signaling pathway increases, including new players like long non-coding RNAs (15, 128, 129), we hope that genetic cause(s) of undiagnosed cases with Chrousos syndrome will soon be identified, by employing classic genetic methods (e.g., the linkage analysis) as well as cutting-edge genome-related methodologies including the whole genome/exome sequencing and sophisticated bioinformatical/statistical analysis tools.
REFERENCES
- 1.
- Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinology. 2009;5(7):374-381. [PubMed: 19488073]
- 2.
- Chrousos GP, Charmandari E, and Kino T. Glucocorticoid action networks -an introduction to systems biology. J Clin Endocrinol Metab. 2004;89(2):563-564. [PubMed: 14764762]
- 3.
- Kino T, De Martino MU, Charmandari E, Mirani M, and Chrousos GP. Tissue glucocorticoid resistance/hypersensitivity syndromes. J Steroid Biochem Mol Biol. 2003;85(2-5):457-467. [PubMed: 12943736]
- 4.
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332(20):1351-1362. [PubMed: 7715646]
- 5.
- Charmandari E, Kino T, Ichijo T, and Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93(5):1563-1572. [PMC free article: PMC2386273] [PubMed: 18319312]
- 6.
- Charmandari E, and Kino T. Chrousos syndrome: a seminal report, a phylogenetic enigma and the clinical implications of glucocorticoid signalling changes. Eur J Clin Invest. 2010;40(10):932-942. [PMC free article: PMC2948853] [PubMed: 20649902]
- 7.
- Charmandari E, Kino T, Ichijo T, Jubiz W, Mejia L, Zachman K, et al. A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2007;92(10):3986-3990. [PubMed: 17635946]
- 8.
- Kino T, and Chrousos GP. In: Steckler T, Kalin NH, and Reul JMHM eds. Handbook on Stress and the Brain. Amsterdam: Elsevier BV; 2005:295-312.
- 9.
- Chrousos GP. The glucocorticoid receptor gene, longevity, and the complex disorders of Western societies. Am J Med. 2004;117(3):204-207. [PubMed: 15300973]
- 10.
- Nicolaides NC, Galata Z, Kino T, Chrousos GP, and Charmandari E. The human glucocorticoid receptor: molecular basis of biologic function. Steroids. 2010;75(1):1-12. [PMC free article: PMC2813911] [PubMed: 19818358]
- 11.
- Nuclear Receptors Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell. 1999;97(2):161-163. [PubMed: 10219237]
- 12.
- Germain P, Staels B, Dacquet C, Spedding M, and Laudet V. Overview of nomenclature of nuclear receptors. Pharmacol Rev. 2006;58(4):685-704. [PubMed: 17132848]
- 13.
- Mackeh R, Marr AK, Dargham SR, Syed N, Fakhro KA, and Kino T. Single-nucleotide variations of the human nuclear hormone receptor genes in 60,000 individuals. J Endocr Soc. 2018;2(1):77-90. [PMC free article: PMC5779106] [PubMed: 29379896]
- 14.
- Chrousos GP, and Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 2005;2005(304):pe48. [PubMed: 16204701]
- 15.
- Nicolaides NC, Chrousos G, and Kino T. Glucocorticoid Receptor. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al. eds. Endotext. South Dartmouth (MA): MDText.com, Inc. Copyright © 2000-2024, MDText.com, Inc.; 2000.
- 16.
- Bamberger CM, Bamberger AM, de Castro M, and Chrousos GP. Glucocorticoid receptor β, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95(6):2435-2441. [PMC free article: PMC295915] [PubMed: 7769088]
- 17.
- Lewis-Tuffin LJ, and Cidlowski JA. The physiology of human glucocorticoid receptor β (hGRβ) and glucocorticoid resistance. Ann N Y Acad Sci. 2006;1069:1-9. [PubMed: 16855130]
- 18.
- Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, and Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor β isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274(39):27857-27866. [PubMed: 10488132]
- 19.
- Kino T, Su YA, and Chrousos GP. Human glucocorticoid receptor isoform β: Recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66(21):3435-3448. [PMC free article: PMC2796272] [PubMed: 19633971]
- 20.
- Ramos-Ramírez P, and Tliba O. Glucocorticoid receptor β (GRβ): Beyond its dominant-negative function. Int J Mol Sci. 2021;22(7);3649. [PMC free article: PMC8036319] [PubMed: 33807481]
- 21.
- Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, and Hinds TD, Jr. Glucocorticoid receptor β stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem. 2014;289(25):17885-17894. [PMC free article: PMC4067219] [PubMed: 24817119]
- 22.
- Yin Y, Zhang X, Li Z, Deng L, Jiao G, Zhang B, et al. Glucocorticoid receptor β regulates injury-mediated astrocyte activation and contributes to glioma pathogenesis via modulation of β-catenin/TCF transcriptional activity. Neurobiol Dis. 2013;59:165-176. [PubMed: 23906498]
- 23.
- Wang Q, Lu PH, Shi ZF, Xu YJ, Xiang J, Wang YX, et al. Glucocorticoid receptor β acts as a co-activator of T-cell factor 4 and enhances glioma cell proliferation. Mol Neurobiol. 2015;52(3):1106-1118. [PubMed: 25301232]
- 24.
- Lu NZ, and Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18(3):331-342. [PubMed: 15866175]
- 25.
- Presul E, Schmidt S, Kofler R, and Helmberg A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J Mol Endocrinol. 2007;38(1-2):79-90. [PubMed: 17242171]
- 26.
- Turner JD, and Muller CP. Structure of the glucocorticoid receptor (NR3C1) gene 5' untranslated region: Identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol. 2005;35(2):283-292. [PubMed: 16216909]
- 27.
- Sinclair D, Fullerton JM, Webster MJ, and Shannon Weickert C. Glucocorticoid receptor 1B and 1C mRNA transcript alterations in schizophrenia and bipolar disorder, and their possible regulation by GR gene variants. PLoS One. 2012;7(3):e31720. [PMC free article: PMC3302776] [PubMed: 22427805]
- 28.
- Sinclair D, Webster MJ, Fullerton JM, and Weickert CS. Glucocorticoid receptor mRNA and protein isoform alterations in the orbitofrontal cortex in schizophrenia and bipolar disorder. BMC Psychiatry. 2012;12:84. [PMC free article: PMC3496870] [PubMed: 22812453]
- 29.
- Frank F, Ortlund EA, and Liu X. Structural insights into glucocorticoid receptor function. Biochem Soc Trans. 2021;49(5):2333-2343. [PMC free article: PMC9274455] [PubMed: 34709368]
- 30.
- Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110(1):93-105. [PubMed: 12151000]
- 31.
- Pratt WB. The role of heat shock proteins in regulating the function, folding, and trafficking of the glucocorticoid receptor. J Biol Chem. 1993;268(29):21455-21458. [PubMed: 8407992]
- 32.
- Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Haché RJ, et al. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol. 1999;19(2):1025-1037. [PMC free article: PMC116033] [PubMed: 9891038]
- 33.
- Hoelz A, Debler EW, and Blobel G. The structure of the nuclear pore complex. Annu Rev Biochem. 2011;80:613-643. [PubMed: 21495847]
- 34.
- Terry LJ, Shows EB, and Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science. 2007;318(5855):1412-1416. [PubMed: 18048681]
- 35.
- Kinyamu HK, Chen J, and Archer TK. Linking the ubiquitin-proteasome pathway to chromatin remodeling/modification by nuclear receptors. J Mol Endocrinol. 2005;34(2):281-297. [PubMed: 15821097]
- 36.
- Kino T, Tiulpakov A, Ichijo T, Chheng L, Kozasa T, and Chrousos GP. G protein β interacts with the glucocorticoid receptor and suppresses its transcriptional activity in the nucleus. J Cell Biol. 2005;169(6):885-896. [PMC free article: PMC2171637] [PubMed: 15955845]
- 37.
- Kino T, Souvatzoglou E, Charmandari E, Ichijo T, Driggers P, Mayers C, et al. Rho family guanine nucleotide exchange factor Brx couples extracellular signals to the glucocorticoid signaling system. J Biol Chem. 2006;281(14):9118-9126. [PMC free article: PMC4152920] [PubMed: 16469733]
- 38.
- Boldizsar F, Szabo M, Kvell K, Czompoly T, Talaber G, Bjorkan J, et al. ZAP-70 tyrosines 315 and 492 transmit non-genomic glucocorticoid (GC) effects in T cells. Mol Immunol. 2013;53(1-2):111-117. [PubMed: 22898186]
- 39.
- Kokkinopoulou I, and Moutsatsou P. Mitochondrial glucocorticoid receptors and their actions. Int J Mol Sci. 2021;22(11):6054. [PMC free article: PMC8200016] [PubMed: 34205227]
- 40.
- Haché RJ, Tse R, Reich T, Savory JG, and Lefebvre YA. Nucleocytoplasmic trafficking of steroid-free glucocorticoid receptor. J Biol Chem. 1999;274(3):1432-1439. [PubMed: 9880517]
- 41.
- Black BE, Holaska JM, Rastinejad F, and Paschal BM. DNA binding domains in diverse nuclear receptors function as nuclear export signals. Curr Biol. 2001;11(22):1749-1758. [PubMed: 11719216]
- 42.
- Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, and Paschal BM. Calreticulin is a receptor for nuclear export. J Cell Biol. 2001;152(1):127-140. [PMC free article: PMC2193655] [PubMed: 11149926]
- 43.
- Holaska JM, Black BE, Rastinejad F, and Paschal BM. Ca2+-dependent nuclear export mediated by calreticulin. Mol Cell Biol. 2002;22(17):6286-6297. [PMC free article: PMC133999] [PubMed: 12167720]
- 44.
- Kino T, Souvatzoglou E, De Martino MU, Tsopanomihalu M, Wan Y, and Chrousos GP. Protein 14-3-3σ interacts with and favors cytoplasmic subcellular localization of the glucocorticoid receptor, acting as a negative regulator of the glucocorticoid signaling pathway. J Biol Chem. 2003;278(28):25651-25656. [PubMed: 12730237]
- 45.
- Habib T, Sadoun A, Nader N, Suzuki S, Liu W, Jithesh PV, et al. AKT1 has dual actions on the glucocorticoid receptor by cooperating with 14-3-3. Mol Cell Endocrinol. 2017;439:431-443. [PubMed: 27717743]
- 46.
- Kino T. GR-regulating serine/threonine kinases: New physiologic and pathologic implications. Trends Endocrinol Metab. 2018;29(4):260-270. [PubMed: 29501228]
- 47.
- Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24(6):766-776. [PMC free article: PMC3878658] [PubMed: 24291004]
- 48.
- Munier CC, De Maria L, Edman K, Gunnarsson A, Longo M, MacKintosh C, et al. Glucocorticoid receptor Thr524 phosphorylation by MINK1 induces interactions with 14-3-3 protein regulators. J Biol Chem. 2021;296:100551. [PMC free article: PMC8080530] [PubMed: 33744286]
- 49.
- Ramamoorthy S, and Cidlowski JA. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev. 2013;24:41-56. [PMC free article: PMC4770453] [PubMed: 23392094]
- 50.
- Schaaf MJ, and Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol. 2002;83(1-5):37-48. [PubMed: 12650700]
- 51.
- Beato M, and Sánchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17(6):587-609. [PubMed: 8969970]
- 52.
- McKenna NJ, and O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108(4):465-474. [PubMed: 11909518]
- 53.
- Osz J, Brélivet Y, Peluso-Iltis C, Cura V, Eiler S, Ruff M, et al. Structural basis for a molecular allosteric control mechanism of cofactor binding to nuclear receptors. Proc Natl Acad Sci U S A. 2012;109(10):E588-594. [PMC free article: PMC3309777] [PubMed: 22355136]
- 54.
- Bulynko YA, and O'Malley BW. Nuclear receptor coactivators: Structural and functional biochemistry. Biochemistry. 2011;50(3):313-328. [PMC free article: PMC3647688] [PubMed: 21141906]
- 55.
- Mahajan MA, and Samuels HH. Nuclear hormone receptor coregulator: Role in hormone action, metabolism, growth, and development. Endocr Rev. 2005;26(4):583-597. [PubMed: 15561801]
- 56.
- York B, and O'Malley BW. Steroid receptor coactivator (SRC) family: Masters of systems biology. J Biol Chem. 2010;285(50):38743-38750. [PMC free article: PMC2998129] [PubMed: 20956538]
- 57.
- Heery DM, Kalkhoven E, Hoare S, and Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387(6634):733-736. [PubMed: 9192902]
- 58.
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, et al. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12(21):3343-3356. [PMC free article: PMC317236] [PubMed: 9808622]
- 59.
- Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. 2012;109(29):11776-11781. [PMC free article: PMC3406827] [PubMed: 22753499]
- 60.
- Sheppard KA, Rose DW, Haque ZK, Kurokawa R, McInerney E, Westin S, et al. Transcriptional activation by NF-κB requires multiple coactivators. Mol Cell Biol. 1999;19(9):6367-6378. [PMC free article: PMC84607] [PubMed: 10454583]
- 61.
- Richter WF, Nayak S, Iwasa J, and Taatjes DJ. The Mediator complex as a master regulator of transcription by RNA polymerase II. Nat Rev Mol Cell Biol. 2022;23(11):732-749. [PMC free article: PMC9207880] [PubMed: 35725906]
- 62.
- Colley SM, and Leedman PJ. SRA and its binding partners: an expanding role for RNA-binding coregulators in nuclear receptor-mediated gene regulation. Crit Rev Biochem Mol Biol. 2009;44(1):25-33. [PubMed: 19280430]
- 63.
- Colley SM, and Leedman PJ. Steroid Receptor RNA Activator - A nuclear receptor coregulator with multiple partners: Insights and challenges. Biochimie. 2011;93(11):1966-1972. [PubMed: 21807064]
- 64.
- Kino T, Hurt DE, Ichijo T, Nader N, and Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3(107):ra8. [PMC free article: PMC2819218] [PubMed: 20124551]
- 65.
- Gao Y, Liu C, Wu T, Liu R, Mao W, Gan X, et al. Current status and perspectives of non-coding RNA and phase separation interactions. Biosci Trends. 2022;16(5):330-345. [PubMed: 36273890]
- 66.
- Johnson TA, Chereji RV, Stavreva DA, Morris SA, Hager GL, and Clark DJ. Conventional and pioneer modes of glucocorticoid receptor interaction with enhancer chromatin in vivo. Nucleic Acids Res. 2018;46(1):203-214. [PMC free article: PMC5758879] [PubMed: 29126175]
- 67.
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93(4):531-541. [PubMed: 9604929]
- 68.
- Reichardt HM, Tuckermann JP, Göttlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20(24):7168-7173. [PMC free article: PMC125338] [PubMed: 11742993]
- 69.
- Cruz-Topete D, and Cidlowski JA. One hormone, two actions: anti- and pro-inflammatory effects of glucocorticoids. Neuroimmunomodulation. 2015;22(1-2):20-32. [PMC free article: PMC4243162] [PubMed: 25227506]
- 70.
- Oakley RH, and Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol. 2013;132(5):1033-1044. [PMC free article: PMC4084612] [PubMed: 24084075]
- 71.
- Hinz B, and Hirschelmann R. Rapid non-genomic feedback effects of glucocorticoids on CRF-induced ACTH secretion in rats. Pharm Res. 2000;17(10):1273-1277. [PubMed: 11145234]
- 72.
- Karst H, Berger S, Turiault M, Tronche F, Schütz G, and Joëls M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A. 2005;102(52):19204-19207. [PMC free article: PMC1323174] [PubMed: 16361444]
- 73.
- Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FP, Plumier JC, Rebsamen MC, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8(5):473-479. [PMC free article: PMC2668717] [PubMed: 11984591]
- 74.
- Löwenberg M, Verhaar AP, Bilderbeek J, Marle J, Buttgereit F, Peppelenbosch MP, et al. Glucocorticoids cause rapid dissociation of a T-cell-receptor-associated protein complex containing LCK and FYN. EMBO Rep. 2006;7(10):1023-1029. [PMC free article: PMC1618362] [PubMed: 16888650]
- 75.
- Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, and Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: Genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012;26(12):4805-4820. [PubMed: 22954589]
- 76.
- Demonacos C, Djordjevic-Markovic R, Tsawdaroglou N, and Sekeris CE. The mitochondrion as a primary site of action of glucocorticoids: the interaction of the glucocorticoid receptor with mitochondrial DNA sequences showing partial similarity to the nuclear glucocorticoid responsive elements. J Steroid Biochem Mol Biol. 1995;55(1):43-55. [PubMed: 7577720]
- 77.
- Demonacos C, Tsawdaroglou NC, Djordjevic-Markovic R, Papalopoulou M, Galanopoulos V, Papadogeorgaki S, et al. Import of the glucocorticoid receptor into rat liver mitochondria in vivo and in vitro. J Steroid Biochem Mol Biol. 1993;46(3):401-413. [PubMed: 9831490]
- 78.
- Psarra AM, and Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta. 2011;1813(10):1814-1821. [PubMed: 21664385]
- 79.
- Lee SR, Kim HK, Song IS, Youm J, Dizon LA, Jeong SH, et al. Glucocorticoids and their receptors: insights into specific roles in mitochondria. Prog Biophys Mol Biol. 2013;112(1-2):44-54. [PubMed: 23603102]
- 80.
- Chrousos GP, Vingerhoeds A, Brandon D, Eil C, Pugeat M, DeVroede M, et al. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest. 1982;69(6):1261-1269. [PMC free article: PMC370198] [PubMed: 6282933]
- 81.
- Chrousos G. Q&A: primary generalized glucocorticoid resistance. BMC Med. 2011;9:27. [PMC free article: PMC3086527] [PubMed: 21429213]
- 82.
- Mendonca BB, Leite MV, de Castro M, Kino T, Elias LL, Bachega TA, et al. Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab. 2002;87(4):1805-1809. [PubMed: 11932321]
- 83.
- Nicolaides NC, and Charmandari E. Chrousos syndrome: from molecular pathogenesis to therapeutic management. Eur J Clin Invest. 2015;45(5):504-514. [PubMed: 25715669]
- 84.
- Nader N, Bachrach BE, Hurt DE, Gajula S, Pittman A, Lescher R, et al. A novel point mutation in helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J Clin Endocrinol Metab. 2010;95(5):2281-2285. [PMC free article: PMC2869551] [PubMed: 20335448]
- 85.
- McMahon SK, Pretorius CJ, Ungerer JP, Salmon NJ, Conwell LS, Pearen MA, et al. Neonatal complete generalized glucocorticoid resistance and growth hormone deficiency caused by a novel homozygous mutation in Helix 12 of the ligand binding domain of the glucocorticoid receptor gene (NR3C1). J Clin Endocrinol Metab. 2010;95(1):297-302. [PubMed: 19933394]
- 86.
- Tatsi C, Xekouki P, Nioti O, Bachrach B, Belyavskaya E, Lyssikatos C, et al. A novel mutation in the glucocorticoid receptor gene as a cause of severe glucocorticoid resistance complicated by hypertensive encephalopathy. J Hypertens. 2019;37(7):1475-1481. [PMC free article: PMC10913058] [PubMed: 31145715]
- 87.
- Paragliola RM, Costella A, Corsello A, Urbani A, and Concolino P. A novel pathogenic variant in the N-terminal domain of the glucocorticoid receptor, causing glucocorticoid resistance. Mol Diagn Ther. 2020;24(4):473-485. [PubMed: 32607951]
- 88.
- Roberts ML, Kino T, Nicolaides NC, Hurt DE, Katsantoni E, Sertedaki A, et al. A novel point mutation in the DNA-binding domain (DBD) of the human glucocorticoid receptor causes primary generalized glucocorticoid resistance by disrupting the hydrophobic structure of its DBD. J Clin Endocrinol Metab. 2013;98(4):E790-795. [PMC free article: PMC3615201] [PubMed: 23426617]
- 89.
- Nicolaides NC, Geer EB, Vlachakis D, Roberts ML, Psarra AM, Moutsatsou P, et al. A novel mutation of the hGR gene causing Chrousos syndrome. Eur J Clin Invest. 2015;45(8):782-791. [PubMed: 26031419]
- 90.
- Charmandari E, Raji A, Kino T, Ichijo T, Tiulpakov A, Zachman K, et al. A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J Clin Endocrinol Metab. 2005;90(6):3696-3705. [PubMed: 15769988]
- 91.
- Mauri S, Nieto-Moragas J, Obón M, and Oriola J. The glucocorticoid resistance syndrome. Two cases of a novel pathogenic variant in the glucocorticoid receptor gene. JCEM Case Rep. 2024;2(1):luad153. [PMC free article: PMC10759794] [PubMed: 38170043]
- 92.
- Velayos T, Grau G, Rica I, Pérez-Nanclares G, and Gaztambide S. Glucocorticoid resistance syndrome caused by two novel mutations in the NR3C1 gene. Endocrinol Nutr. 2016;63(7):369-371. [PubMed: 27211791]
- 93.
- Vitellius G, Fagart J, Delemer B, Amazit L, Ramos N, Bouligand J, et al. Three novel heterozygous point mutations of NR3C1 causing glucocorticoid resistance. Hum Mutat. 2016;37(8):794-803. [PubMed: 27120390]
- 94.
- Zhu HJ, Dai YF, Wang O, Li M, Lu L, Zhao WG, et al. Generalized glucocorticoid resistance accompanied with an adrenocortical adenoma and caused by a novel point mutation of human glucocorticoid receptor gene. Chin Med J (Engl). 2011;124(4):551-555. [PubMed: 21362280]
- 95.
- Sherlock M, Scarsbrook A, Abbas A, Fraser S, Limumpornpetch P, Dineen R, et al. Adrenal Incidentaloma. Endocr Rev. 2020;41(6):775-820. [PMC free article: PMC7431180] [PubMed: 32266384]
- 96.
- Karl M, Lamberts SW, Koper JW, Katz DA, Huizenga NE, Kino T, et al. Cushing's disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians. 1996;108(4):296-307. [PubMed: 8863343]
- 97.
- Vitellius G, Trabado S, Hoeffel C, Bouligand J, Bennet A, Castinetti F, et al. Significant prevalence of NR3C1 mutations in incidentally discovered bilateral adrenal hyperplasia: Results of the French MUTA-GR Study. Eur J Endocrinol. 2018;178(4):411-423. [PubMed: 29444898]
- 98.
- Nicolaides NC, Roberts ML, Kino T, Braatvedt G, Hurt DE, Katsantoni E, et al. A novel point mutation of the human glucocorticoid receptor gene causes primary generalized glucocorticoid resistance through impaired interaction with the LXXLL motif of the p160 coactivators: dissociation of the transactivating and transreppressive activities. J Clin Endocrinol Metab. 2014;99(5):E902-907. [PMC free article: PMC4010692] [PubMed: 24483153]
- 99.
- Kino T, Stauber RH, Resau JH, Pavlakis GN, and Chrousos GP. Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: Importance of the ligand-binding domain for intracellular GR trafficking. J Clin Endocrinol Metab. 2001;86(11):5600-5608. [PubMed: 11701741]
- 100.
- Charmandari E, Kino T, Souvatzoglou E, Vottero A, Bhattacharyya N, and Chrousos GP. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab. 2004;89(4):1939-1949. [PubMed: 15070967]
- 101.
- Malchoff CD, Javier EC, Malchoff DM, Martin T, Rogol A, Brandon D, et al. Primary cortisol resistance presenting as isosexual precocity. J Clin Endocrinol Metab. 1990;70(2):503-507. [PubMed: 2105334]
- 102.
- Vottero A, Kino T, Combe H, Lecomte P, and Chrousos GP. A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab. 2002;87(6):2658-2667. [PubMed: 12050230]
- 103.
- Karl M, Lamberts SW, Detera-Wadleigh SD, Encio IJ, Stratakis CA, Hurley DM, et al. Familial glucocorticoid resistance caused by a splice site deletion in the human glucocorticoid receptor gene. J Clin Endocrinol Metab. 1993;76(3):683-689. [PubMed: 8445027]
- 104.
- Lin L, Wu X, Hou Y, Zheng F, and Xu R. A novel mutation in the glucocorticoid receptor gene causing resistant hypertension: A case report. Am J Hypertens. 2019;32(11):1126-1128. [PubMed: 31414133]
- 105.
- Charmandari E, Ichijo T, Jubiz W, Baid S, Zachman K, Chrousos GP, et al. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR-mediated gene expression. J Clin Endocrinol Metab. 2008;93(12):4963-4968. [PMC free article: PMC2626453] [PubMed: 18827003]
- 106.
- Bouligand J, Delemer B, Hecart AC, Meduri G, Viengchareun S, Amazit L, et al. Familial glucocorticoid receptor haploinsufficiency by non-sense mediated mRNA decay, adrenal hyperplasia and apparent mineralocorticoid excess. PLoS One. 2010;5(10):e13563. [PMC free article: PMC2962642] [PubMed: 21042587]
- 107.
- Ruiz M, Lind U, Gåfvels M, Eggertsen G, Carlstedt-Duke J, Nilsson L, et al. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol (Oxf). 2001;55(3):363-371. [PubMed: 11589680]
- 108.
- Ma L, Tan X, Li J, Long Y, Xiao Z, De J, et al. A novel glucocorticoid receptor mutation in primary generalized glucocorticoid resistance disease. Endocr Pract. 2020;26(6):651-659. [PubMed: 32045292]
- 109.
- Trebble P, Matthews L, Blaikley J, Wayte AW, Black GC, Wilton A, et al. Familial glucocorticoid resistance caused by a novel frameshift glucocorticoid receptor mutation. J Clin Endocrinol Metab. 2010;95(12):E490-499. [PMC free article: PMC4110505] [PubMed: 20861124]
- 110.
- Vitellius G, Delemer B, Caron P, Chabre O, Bouligand J, Pussard E, et al. Impaired 11β-hydroxysteroid dehydrogenase type 2 in glucocorticoid-resistant patients. J Clin Endocrinol Metab. 2019;104(11):5205-5216. [PubMed: 31225872]
- 111.
- Raef H, Baitei EY, Zou M, and Shi Y. Genotype-phenotype correlation in a family with primary cortisol resistance: possible modulating effect of the ER22/23EK polymorphism. Eur J Endocrinol. 2008;158(4):577-582. [PubMed: 18362306]
- 112.
- Molnár Á, Patócs A, Likó I, Nyírő G, Rácz K, Tóth M, et al. An unexpected, mild phenotype of glucocorticoid resistance associated with glucocorticoid receptor gene mutation case report and review of the literature. BMC Med Genet. 2018;19(1):37. [PMC free article: PMC5840839] [PubMed: 29510671]
- 113.
- Donner KM, Hiltunen TP, Jänne OA, Sane T, and Kontula K. Generalized glucocorticoid resistance caused by a novel two-nucleotide deletion in the hormone-binding domain of the glucocorticoid receptor gene NR3C1. Eur J Endocrinol. 2013;168(1):K9-k18. [PubMed: 23076843]
- 114.
- Hurt DE, Suzuki S, Mayama T, Charmandari E, and Kino T. Structural analysis on the pathologic mutant glucocorticoid receptor ligand-binding domains. Mol Endocrinol. 2016;30(2):173-188. [PMC free article: PMC4792232] [PubMed: 26745667]
- 115.
- Cole TJ, Blendy JA, Monaghan AP, Krieglstein K, Schmid W, Aguzzi A, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9(13):1608-1621. [PubMed: 7628695]
- 116.
- Nicolaides NC, and Charmandari E. Novel insights into the molecular mechanisms underlying generalized glucocorticoid resistance and hypersensitivity syndromes. Hormones (Athens). 2017;16(2):124-138. [PubMed: 28742501]
- 117.
- Kino T, Liou SH, Charmandari E, and Chrousos GP. Glucocorticoid receptor mutants demonstrate increased motility inside the nucleus of living cells: time of fluorescence recovery after photobleaching (FRAP) is an integrated measure of receptor function. Mol Med. 2004;10(7-12):80-88. [PMC free article: PMC1431369] [PubMed: 16307173]
- 118.
- Kaziales A, Rührnößl F, and Richter K. Glucocorticoid resistance conferring mutation in the C-terminus of GR alters the receptor conformational dynamics. Sci Rep. 2021;11(1):12515. [PMC free article: PMC8206104] [PubMed: 34131228]
- 119.
- Dahlman-Wright K, Wright A, Gustafsson JA, and Carlstedt-Duke J. Interaction of the glucocorticoid receptor DNA-binding domain with DNA as a dimer is mediated by a short segment of five amino acids. J Biol Chem. 1991;266(5):3107-3112. [PubMed: 1993683]
- 120.
- van Rossum EF, and Lamberts SW. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog Horm Res. 2004;59:333-357. [PubMed: 14749509]
- 121.
- Vitellius G, Trabado S, Bouligand J, Delemer B, and Lombès M. Pathophysiology of glucocorticoid signaling. Ann Endocrinol (Paris). 2018;79(3):98-106. [PubMed: 29685454]
- 122.
- Wester VL, Koper JW, van den Akker EL, Franco OH, Stolk RP, and van Rossum EF. Glucocorticoid receptor haplotype and metabolic syndrome: the Lifelines cohort study. Eur J Endocrinol. 2016;175(6):645-651. [PubMed: 27634941]
- 123.
- Ferriman D, and Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab. 1961;21:1440-1447. [PubMed: 13892577]
- 124.
- Marshall WA, and Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child. 1969;44(235):291-303. [PMC free article: PMC2020314] [PubMed: 5785179]
- 125.
- Marshall WA, and Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child. 1970;45(239):13-23. [PMC free article: PMC2020414] [PubMed: 5440182]
- 126.
- Chrousos GP, Kino T, and Charmandari E. Evaluation of the hypothalamic-pituitary-adrenal axis function in childhood and adolescence. Neuroimmunomodulation. 2009;16(5):272-283. [PMC free article: PMC2790806] [PubMed: 19571588]
- 127.
- Kino T, Pavlatou MG, Moraitis AG, Nemery RL, Raygada M, and Stratakis CA. ZNF764 haploinsufficiency may explain partial glucocorticoid, androgen, and thyroid hormone resistance associated with 16p11.2 microdeletion. J Clin Endocrinol Metab. 2012;97(8):E1557-1566. [PMC free article: PMC3410270] [PubMed: 22577170]
- 128.
- Kadmiel M, and Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34(9):518-530. [PMC free article: PMC3951203] [PubMed: 23953592]
- 129.
- Chrousos GP, and Kino T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress (Amsterdam, Netherlands). 2007;10(2):213-219. [PubMed: 17514590]
- Primary cortisol resistance: a familial syndrome and an animal model.[J Steroid Biochem. 1983]Primary cortisol resistance: a familial syndrome and an animal model.Chrousos GP, Loriaux DL, Brandon D, Tomita M, Vingerhoeds AC, Merriam GR, Johnson EO, Lipsett MB. J Steroid Biochem. 1983 Jul; 19(1B):567-75.
- Glucocorticoid resistance syndrome caused by a novel NR3C1 point mutation.[Endocr J. 2018]Glucocorticoid resistance syndrome caused by a novel NR3C1 point mutation.Al Argan R, Saskin A, Yang JW, D'Agostino MD, Rivera J. Endocr J. 2018 Nov 29; 65(11):1139-1146. Epub 2018 Aug 30.
- Review Novel causes of generalized glucocorticoid resistance.[Horm Metab Res. 2007]Review Novel causes of generalized glucocorticoid resistance.Charmandari E, Kino T. Horm Metab Res. 2007 Jun; 39(6):445-50.
- Review Syndromes of glucocorticoid and mineralocorticoid resistance.[Steroids. 1995]Review Syndromes of glucocorticoid and mineralocorticoid resistance.Arai K, Chrousos GP. Steroids. 1995 Jan; 60(1):173-9.
- A Novel Pathogenic Variant in the N-Terminal Domain of the Glucocorticoid Receptor, Causing Glucocorticoid Resistance.[Mol Diagn Ther. 2020]A Novel Pathogenic Variant in the N-Terminal Domain of the Glucocorticoid Receptor, Causing Glucocorticoid Resistance.Paragliola RM, Costella A, Corsello A, Urbani A, Concolino P. Mol Diagn Ther. 2020 Aug; 24(4):473-485.
- Primary Generalized Glucocorticoid Resistance Syndrome - EndotextPrimary Generalized Glucocorticoid Resistance Syndrome - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...