CLINICAL RECOGNITION
Hypercalcemia can be defined as a serum calcium greater than 2 standard deviations above the normal mean in a reference laboratory. Calcium in the blood is normally transported:
partly bound to plasma proteins (about 45%), notably to albumin; partly bound to small anions such as phosphate and citrate (about 10%); partly in the free or ionized state (about 45%).
Only the ionized calcium is metabolically active i.e. subject to transport into cells, but most laboratories report total serum calcium concentrations. Hypercalcemia is therefore often defined as a total serum calcium (bound plus ionized) greater than 10.6 mg/dl (2.65 mM) or an ionized serum calcium greater than 5.3 mg/dl (1.3 mM) but values may vary between laboratories.
Dehydration, or hemoconcentration during venipuncture, may elevate total serum albumin whereas ionized calcium may remain normal. Consequently, a falsely elevated total serum calcium may be reported. Conversely when serum albumin levels are low, total serum calcium may be falsely low. To correct for an abnormally high or low serum albumin the following formula can be used:
Corrected calcium (mg/dL) = measured total serum calcium (mg/dL) + [4.0-serum albumin (g/dL) X 0.8] or Corrected calcium (mM) = measured total serum Ca (mM) + [40 - serum albumin (g/L) X 0.02]
Changes in blood pH can also alter the equilibrium constant of the albumin-calcium complex: Acidosis reduces binding and alkalosis enhances binding. Consequently, when major shifts in serum protein or pH are present it is prudent to directly measure the ionized calcium level in order to determine the presence of hypercalcemia.
Clinical Manifestations may be due to hypercalcemia or may be due to the causal disorder or may be due to both. Hypercalcemic manifestations will vary depending on whether the hypercalcemia is of acute onset and severe (greater than 12 mg/dL or 3 mM) or whether it is chronic and relatively mild. Patients may also tolerate higher serum calcium levels more readily if the onset is relatively gradual, but at concentrations above 14 mg/dL (3.5 mM) most patients are symptomatic. In both acute and chronic cases, the major manifestations affect gastrointestinal, renal and neuromuscular function (Table 1).
Table 1.
Manifestations of Hypercalcemia
PATHOPHYSIOLOGY
Fluxes of calcium across the skeleton, the gut, and the kidney play a major role in maintaining calcium homeostasis. When the extracellular fluid (ECF) calcium is raised above the normal range, the calcium ion per se, by stimulating the G-protein coupled calcium sensing receptor (CaSR), can inhibit parathyroid hormone (PTH) release. Decreased PTH and CaSR stimulation will both facilitate reduced renal calcium reabsorption, and decreased PTH will result in reduced bone resorption and diminished release of calcium from bone. Decreased PTH and hypercalcemia will also reduce renal production of the active form of vitamin D, 1,25-dihydroxyvitamin D [1,25(OH)2D], and decrease gut absorption of calcium. The net effect of the diminished renal calcium reabsorption, intestinal calcium absorption, and skeletal calcium resorption will be to reduce the elevated ECF calcium to normal. Consequently, decreased levels of PTH and decreased levels of 1,25(OH)2D should accompany hypercalcemia unless the PTH or 1,25(OH)2D is the cause of the hypercalcemia. The converse sequence of events occurs when the ECF calcium is reduced below the normal range.
A genetic relative of PTH, PTH-related peptide (PTHrP), can also resorb bone, when released from certain tumors. Both PTH and PTHrP act on osteoblastic cells to increase production of cytokines, notably receptor activator of nuclear factor kappa B ligand (RANKL) which increases production and activation of multinucleated osteoclasts which then resorb mineralized bone.
DIAGNOSIS AND DIFFERENTIAL (FIGURE 1)
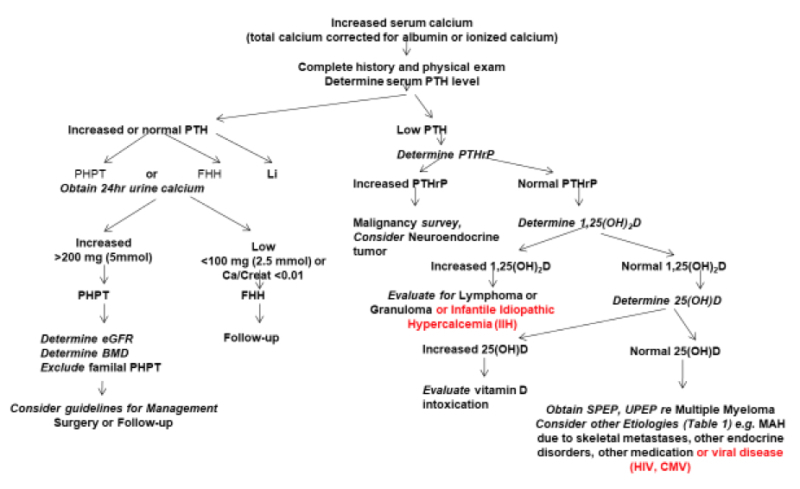
Figure 1
Algorithm for Diagnosing the Cause of Hypercalcemia
Hypercalcemic disorders can be broadly grouped into Endocrine Disorders, Malignant Disorders, Inflammatory Disorders, Medication-Induced Hypercalcemia, and Immobilization as given in Tables 2-8. Primary hyperparathyroidism (HPTH) and malignancy-associated hypercalcemia (MAH) account for the vast majority of hypercalcemic disorders. (For a more complete discussion of hypercalcemic disorders and the underlying pathophysiology, see reference 1)
Table 2.
Endocrine Disorders Associated with Hypercalcemia
Table 3.
Malignancy-Associated Hypercalcemia (MAH)
Table 4.
Granulomatous Disorders Causing Hypercalcemia
Table 5.
Pediatric Syndromes
Table 6.
Viral Syndromes
Table 7.
Medication-Induced
Table 8.
Immobilization
DIAGNOSTIC TESTS NEEDED AND SUGGESTED
Laboratory testing should be guided by the results of a careful history and a detailed physical examination and should be geared toward assessing the extent of the alteration in calcium homeostasis and toward establishing the underlying diagnosis and determining its severity. Most patients with primary HPTH, the most common cause of hypercalcemia in the clinic, present with mild hypercalcemia discovered on a routine biochemical assessment. There may be a history of a recent or remote renal stone. Bone pain and fractures are rare although the patient may carry a diagnosis of osteoporosis based on a previous bone mineral density (BMD) measurement. A history of a documented peptic ulcer is rare in primary sporadic HPTH and should raise concern about MEN1. Although cardiovascular and neuropsychiatric manifestations have been described they appear to require more validation. Documentation of at least two elevated corrected (or ionized) serum calcium levels with concomitant elevated (or at least normal) serum PTH levels is required to establish the diagnosis (Figure 1). Lithium treatment has been associated with hypercalcemia, elevated or normal serum PTH, and increased renal calcium reabsorption. The presence of a family history of hypercalcemia or of kidney stones should raise suspicion of MEN1 or MEN2a (reference 3 and 4). If, in addition to primary HPTH in the proband, one or more first-degree relatives are found to have at least one of the three tumors characterizing MEN1 (parathyroid, pituitary, pancreas) or MEN2a (parathyroid, medullary thyroid carcinoma, pheochromocytoma) then it is highly likely that the disease is familial. The presence of ossifying fibromas of the mandible and maxilla, and renal lesions such as cysts and hamartomas in addition to HPTH would suggest HPTH-jaw tumor syndrome. In all patients with documented primary HPTH, a 24-hour urine calcium and creatinine level should be obtained to exclude familial hypocalciuric hypercalcemia (FHH). If the urine calcium to creatinine ratio is less than 0.01 and if testing serum and urine calcium in three relatives discloses hypercalcemia and relative hypocalciuria in other family members, then this diagnosis is likely and parathyroid surgery is to be avoided. If the urine calcium to creatinine ratio is greater than 0.01 then estimated glomerular filtration rate (eGFR) and a BMD test should be performed and guidelines for treatment of primary HPTH should be considered (see below).
Tertiary hyperparathyroidism with hypercalcemia and elevated PTH has been described in chronic kidney disease patients on hemodialysis, or in patients with hypophosphatemic syndromes (e.g. x-linked hypophosphatemic rickets) receiving long-term oral phosphate therapy without concomitant calcitriol.
If hypercalcemia is associated with very low or suppressed serum PTH levels, then malignancy would be an important consideration, either in association with elevated serum PTHrP or in its absence, in which case it is generally as a result of the production of other cytokines, often with osteolytic metastases. When malignancy-associated hypercalcemia is suspected then an appropriate malignancy screen should be done including skeletal imaging to identify skeletal metastases. As well appropriate general biochemical assessment such as a complete blood count and serum creatinine and specific biochemical assessment such as serum and urine protein electrophoresis to exclude multiple myeloma would be appropriate.
Detection of elevated serum 1,25(OH)2D levels in the absence of elevated serum PTH levels, suggests the need for a search for lymphoma or for non-infectious (e.g. sarcoidosis) or infectious granulomatous disease.
Hypercalcemia may also occur with thyrotoxicosis, pheochromocytoma, VIPoma, and hypoadrenalism. Increased PTHrP may be associated with neuroendocrine tumors. Serum PTH levels are suppressed in these disorders and 1,25(OH)2D levels are not elevated. Although these conditions may be suspected from clinical examination, detailed biochemical evaluation of these non-PTH associated endocrine disorders is required for confirmation.
Detection of elevated serum 25-hydroxyvitamin D [25(OH)D], should lead to a search for vitamin D intoxication. Vitamin A intoxication may also lead to hypercalcemia, but in the absence of elevated serum 25(OH)D, 1,25(OH)2D, or PTH. Hypercalcemia has been reported in association with human immunodeficiency virus (HIV), HTLV-III or cytomegalovirus (CMV) infections of the skeleton, presumably due to direct skeletal resorption. Use of foscarnet as an antiviral agent has also been associated with hypercalcemia. Transient hypercalcemia may accompany thiazide diuretic ingestion, possibly associated with dehydration, but prolonged hypercalcemia with thiazides requires a search for other causes. Hypercalcemia may be seen in patients with advanced breast cancer with skeletal metastases, at the initiation of treatment with tamoxifen. Aminophylline and theophylline used as bronchodilators have (rarely) been reported to be associated with hypercalcemia. The use of aluminum-containing phosphate binders in patients on chronic hemodialysis was associated with hypercalcemia in the past but, with the advent of other modes of therapy, this is rarely seen today. Similarly, the use of absorbable alkali (NaHCO3) along with large quantities of milk for ulcer treatment was a cause of hypercalcemia in the past but this therapy has been superseded today.
In the pediatric age group, hypercalcemia may include Jansens’s Metaphyseal Chondrodysplasia due to an activating mutation of the type 1 PTH/PTHrP receptor; neonatal severe hyperparathyroidism (NSHPTH) which may present with life-threatening hypercalcemia in neonates that are homozygous for inactivating mutations in CaSR; William’s Syndrome, an autosomal dominant disorder with hemizygous submicroscopic deletions of chromosome 7q11.23, characterized phenotypically by multiple congenital abnormalities, and in which hypercalcemia may occur possibly due to aberrant vitamin D metabolism; and idiopathic infantile hypercalcemia (IIH) in which hypercalcemia may be associated with increased 1,25(OH)2D.due to loss-of-function mutations in CYP24A1, the gene encoding the enzyme responsible for the first step in inactivation of 1,25(OH)2D. IIH may also be caused by loss-of-function mutations in SLC34A1, encoding the renal proximal tubular sodium-phosphate cotransporter, Na/Pi-IIa, leading to phosphaturia, phosphate depletion, suppression of the hormone fibroblast growth factor-23 (FGF-23), decreased CYP24A1,and increased 1,25(OH)2D production.
THERAPY
If the patient's serum calcium concentration is less than 12 mg/dL (3 mM) then treatment of the hypercalcemia can be aimed solely at treatment of the underlying disorder. If the patient has symptoms and signs of acute hypercalcemia as described above and serum calcium is greater than 12 mg/dL (3 mM), then a series of urgent measures should be instituted (Table 9). These measures are almost always required with a serum calcium above 14 mg/dL (3.5 mM).
Table 9.
Management of Acute Hypercalcemia
FOLLOW-UP
In the patient with primary sporadic HPTH who presents with kidney stones, fractures, or a low BMD (T-score less than -2.5) surgery would be indicated. In the patient with documented asymptomatic primary HPTH, follow-up should be done annually with measurement of serum calcium and serum creatinine (to determine estimated GFR). BMD should be repeated every one to two years. Guidelines below should be considered for recommending surgery (reference 2). The diagnosis of familial disease raises issues of management of HPTH in the proband and affected family members in view of the fact that familial HPTH generally is generally associated with multigland disease, whereas the sporadic disease is usually due to an adenoma. In HPTH jaw tumor syndrome there should be recognition of the high frequency of parathyroid carcinoma.
Table 10.
Guidelines for Surgery in Primary HPTH
Management of other etiologies of hypercalcemia are generally directed toward the specific entity involved.
REFERENCES
- 1.
- Goltzman D. Approach to Hypercalcemia. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2016 Aug 8.
- 2.
- Bilezikian JP. Primary Hyperparathyroidism. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2017 Jan 15.
- 3.
- Vinik A, Perry RR, Hughes MS, Feliberti E. Multiple Endocrine Neoplasia Type 1. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2017 Oct 7.
- 4.
- Hughes MS, Feliberti E, Perry RR, Vinik A. Multiple Endocrine Neoplasia Type 2A (including Familial Medullary Carcinoma) and Type 2B. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2017 Oct 8.
- 5.
- Kaltsas G, Dimitriadis GK, Androulakis II, Grossman A. Paraneoplastic Syndromes related to Neuroendocrine Tumours. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000- 2017 Feb 16.
Publication Details
Author Information and Affiliations
Publication History
Last Update: August 4, 2019.
Copyright
This electronic version has been made freely available under a Creative Commons (CC-BY-NC-ND) license. A copy of the license can be viewed at http://creativecommons.org/licenses/by-nc-nd/2.0/.
Publisher
MDText.com, Inc., South Dartmouth (MA)
NLM Citation
Goltzman D. Hypercalcemia. [Updated 2019 Aug 4]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.