NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.
ABSTRACT
Aldosterone is crucial for regulating sodium conservation in the kidney, salivary glands, sweat glands, and colon. This adrenal steroid hormone acts via the mineralocorticoid receptor (MR) to promote active transport of sodium and potassium excretion in its target tissues, through activation of specific amiloride-sensitive sodium channels (ENaC) and a Na-K ATP-ase pump. Defective aldosterone biosynthesis or action results in various clinical and laboratory test manifestations, such as hypotension, hyponatremia, hyperkalemia, and acidosis. Primary adrenal insufficiency and congenital adrenal hypoplasia are discussed in other chapters. In this chapter the mechanisms underlying aldosterone-deficient conditions, such as hyporeninemic hypoaldosteronism, primary hypoaldosteronism, including aldosterone synthase deficiency (ASD), acquired forms of the disease, and pseudohypoaldosteronism, an aldosterone resistance syndrome due to insensitivity of target tissues to aldosterone, are reviewed. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Aldosterone is crucial for sodium conservation in the kidney, salivary glands, sweat glands, and colon. Aldosterone is synthesized exclusively in the zona glomerulosa of the adrenal gland. Destruction or dysfunction of the adrenal gland in conditions such as primary adrenal insufficiency, congenital adrenal hypoplasia, isolated mineralocorticoid deficiency, acquired secondary aldosterone deficiency (hyporeninemic hypoaldosteronism), acquired primary aldosterone deficiency, and inherited enzymatic defects in aldosterone biosynthesis cause clinical symptoms and laboratory characteristics owing to aldosterone deficiency. Pseudohypoaldosteronism is an aldosterone resistance syndrome i.e. a condition due to the insensitivity of target tissues to aldosterone. In this chapter, aldosterone-deficiency conditions other than primary adrenal insufficiency and congenital adrenal hypoplasia are reviewed.
ALDOSTERONE BIOSYNSTHESIS
All human steroid hormones are derived from cholesterol. Aldosterone is synthesized in the zona glomerulosa of the adrenal cortex through four enzymes, cholesterol desmolase (CYP11A1), 21-hydroxylase (CYP21A2), aldosterone synthase (CYP11B2), and 3β-hydroxysteroid dehydrogenase (3β-HSD) (Figure 1). CYP11A1, CYP21A2 and CYP11B2 are cytochrome 450 enzymes (CYP), which are membrane-bound, heme-containing enzymes that accept electrons from NADPH through accessory proteins and use molecular oxygen to perform hydroxylation or other oxidative conversions (1). CYP11A1, which is a side-chain cleavage enzyme, cleaves the side chain from C21 of cholesterol, converting cholesterol to pregnenolone in adrenal mitochondria and this is the first step in steroidogenesis. The CYP11A1 gene is located on the long arm of human chromosome 15q24-q25 (2). Pregnenolone is returned to the cytosolic compartment and is converted to progesterone by 3β-HSD. Progesterone is then hydroxylated at C21 by CYP21A2, an enzyme located in the smooth endoplasmic reticulum, to yield deoxycorticosterone (DOC). The CYP21A2 gene is located on the short arm of human chromosome 6 (3). Only CYP21A2 is active in humans, the other, CYP21A1P is a pseudogene (4). CYP11B1, which is a mitochondrial enzyme, catalyzes β-hydroxylation at C11 and converts DOC to corticosterone. The terminal two steps in the conversion of corticosterone to aldosterone (18-hydroxylation and 18-methyloxidation) are catalyzed by CYP11B2 (aldosterone synthase) (5) which was previously named corticosterone 18-hydroxylase/18-methyloxidase (CMO I/CMO II) or 18-hydroxylase/isomerase. These two steps previously proposed to be catalyzed by separate enzyme, CMO 1 and II, are known to involve only one enzyme substrate interaction, aldosterone synthase encoded by CYP11B2 gene (6). The CYP11B1 and CYP11B2 genes are located on the long arm of chromosome 8 and the amino acid sequence of CYP11B2 shares more than 90% homology with that of CYP11B1 (7). In humans, the expression of CYP11B1 and CYP11B2 in the adrenal glands is spatially separated. While expression of CYP11B1 takes place in the zona reticularis/fasciculata, CYP11B2 expression and aldosterone synthesis are restricted to the zona glomerulus (8).
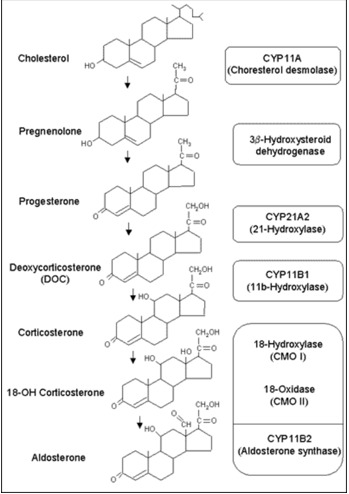
Figure 1.
Aldosterone Biosynthesis. Aldosterone is derived from cholesterol. Biosynthetic pathway of aldosterone and structure of adrenal steroids and their biosynthetic precursors are shown in the figure. The enzymes that catalyze each step are listed in the adjacent box at the right side of the figure.
Epigenetic Regulation Of Cyp11b2 Expression
CYP11B2 gene expression is epigenetically controlled. DNA methylation at CpG dinucleotides alter gene expression by affecting transcription factor binding activity (9). Cyclic AMP responsive element binding protein 1 (CREB 1) /ATF family members and nuclear receptor subfamily 4, group A (NR4A) members bind the CYP11B2 promoter at Ad1 (cAMP response element at -71/-64) and Ad5 (cAMP response element at -129/-114), respectively, leading to activation of transcription. DNA methylation at CpG1 greatly decreased CREB 1 binding to Ad1 in the promoter lesion of CYP11B2 gene (10). In addition, DNA methylation at CpG2 reduced basal binding activities of NR4A1 and NR4A2 with Ad5 by 30% and 50%, respectivly (10). Ang II infusion in the rat decreased the methylation ratio of CYP11B2 gene and increased gene expression in the adrenal gland (10). A low-salt diet induced hypomethylation of rat CYP11B2 and increased CYP11B2 mRNA levels parallel with aldosterone synthesis (10).
REGULATION OF ALDOSTERONE SECRETION
Aldosterone secretion is regulated by multiple factors. The renin-angiotensin system and potassium ion are the major regulators, whereas ACTH and other POMC peptides, sodium ion, vasopressin, dopamine, ANP, β-adrenergic agents, serotonin and somatostatin are minor modulators.
The Renin-Angiotensin System
Renin is a 430 amino acid enzyme that cleaves renin substrate or angiotensinogen, which is a 453 amino acid alpha-globulin product of the liver, to produce the decapeptide, angiotensin I. Angiotensin I is rapidly cleaved by angiotensin-converting enzyme (ACE) in the lung and other tissues to form the octapeptide, angiotensin II. Moreover, angiotensinase cleaves the NH2-terminal Asp residue from angiotensin II and produces the heptapeptide, angiotensin III, then to the hexapeptide angiotensin IV. The circulating levels of angiotensin III are 15 to 25% of those of angiotensin II. Angiotensin II, III and IV stimulate aldosterone secretion and vasoconstriction, while angiotensin II is more potent for vasoconstriction. The angiotensins are inactivated within minutes by tissue and plasma peptidase. The levels of the circulating renin are the rate-limiting factor in this process.
Renin is synthesized by the juxtaglomerular cells in the renal cortex and its secretion is controlled by renal arterial blood pressure, sodium concentrations of tubular fluid sensed by the macula densa, and renal sympathetic nervous activity (11). Factors that decrease renal blood flow, such as hemorrhage, dehydration, salt restriction, upright posture, and renal artery narrowing, increase renin levels. In contrast, factors that increase blood pressure, such as high salt intake, peripheral vasoconstrictors, and supine posture, decrease renin levels. Hypokalemia increases and hyperkalemia decreases renin release.
The effect of angiotensin II and III on the adrenal glomerulosa is initiated by binding to G-protein coupled receptors. The first mechanism of the intracellular signal transduction is activation of phospholipase C, which hydrolyzes PIP2 to IP3, which then releases intracellular calcium ions (12). Interestingly, angiotensin II does not stimulate adenylate cyclase activity. Angiotensin II stimulation leads to increased transfer of cholesterol to the inner mitochondrial membrane and increased conversion of cholesterol to pregnenolone and corticosterone to aldosterone (13).
Potassium
Potassium directly increases aldosterone secretion by the adrenal cortex and aldosterone then lowers serum potassium by stimulating its excretion by the kidney. High dietary potassium intake increases plasma aldosterone and enhances the aldosterone response to a subsequent potassium or angiotensin II infusion (12). The primary action of potassium for stimulating aldosterone secretion is to depolarize the plasma membrane, which activates voltage-dependent calcium channels, that permit influx or efflux of extracellular calcium (12–14), leading to the activation of calmodulin and calmodulin-dependent kinase, subsequently. The activated kinase phosphorylates both activating transcription factor and members of CRE-binding protein family which bind to 5’ flanking promotor regions of the CYP11B2 gene and trigger gene transcription in the zona glomerulosa, followed by increased aldosterone biosynthesis (13,14).
Pituitary Factors
ACTH and possibly other POMC-derived peptides, including α-MSH, α-MSH, β-LPH, and β-END, influence aldosterone secretion, however, the role of ACTH in aldosterone secretion is minor (12). ACTH increases aldosterone secretion by binding to glomerulosa cell-surface melanocortin-2 receptor, by activating adenylate cyclase, and increasing intracellular cAMP (15). Like other agents, ACTH stimulates the same two early and late steps of aldosterone biosynthesis.
Vasopressin has a modest and transient stimulatory effect on aldosterone secretion from zona granulosa cells in vitro. This effect is probably mediated via V2 receptors and phospholipase C generating IP3 and diacylglycerol (16).
Sodium
Sodium intake influences aldosterone secretion by an indirect effect through renin and to a minor extent by direct effects on zona glomerulosa responsiveness to angiotensin II. High sodium intake increases vascular volume, which suppresses renin secretion and angiotensin II generation and decreases the sensitivity of aldosterone response to angiotensin II.
Inhibitory Agents
Dopamine inhibits aldosterone secretion in humans by a mechanism that is independent of the effects of prolactin, ACTH, electrolytes, and the renin-angiotensin system (17,18). This inhibitory effect may involve binding to D2 receptors on glomerulosa cells (19). Atrial natriuretic peptide (ANP) directly inhibits aldosterone secretion and blocks the stimulatory effects of angiotensin II, potassium and ACTH, at least in part, by interfering with extracellular calcium influx (20).
MECHANISMS OF ALDOSTERONE ACTION
Effect of Aldosterone
Aldosterone is crucial for sodium conservation in the kidney, salivary glands, sweat glands, and colon. Aldosterone promotes active sodium transport and excretion of potassium in its major target tissues. It exerts its effects via the mineralocorticoid receptor (MR) and the resultant activation of specific amiloride-sensitive sodium channels (ENaC) and the Na-K ATP-ase pump (21). Aldosterone and the MR may be involved in the regulation of genes coding for the subunits of the amiloride sensitive sodium channel and the Na-K ATP-ase pump, serum and glucocorticoid regulated kinase (SGK), channel-inducing factor, as well as of other proteins (22,23). Activated SGK1 phosphorylates the neural precursor cell-expressed, developmentally down-regulated protein 4-2 (Nedd4-2) which allows binding of 14-3-3 proteins (24). Then, the interaction of Nedd4-2 and ENaC causes an accumulation of ENaC at the plasma membrane and enhances epithelial sodium transport by increasing open probability of ENaC. In a later phase translation and allocation of ENaC, basolateral Na-K ATP-ase and apical K channel (ROMK) are enhanced in its target tissues (25–27).
On the other hand, rapid effects in response to aldosterone but independent of the MR were described as so-called non-genomic or rapid signaling of aldosterone. The G protein-coupled estrogen receptor (GPER) [previously known as G protein-coupled receptor 30 (GPR30)], a member of the seven transmembrane domain family of cell surface receptors, has been reported to be a membrane receptor for aldosterone (28). The expression of GPER is ubiquitous, including in vascular cells (both endothelial cells and smooth muscle cells) and is required for rapid MR-independent effects of aldosterone in vascular smooth muscle cells (28). Aldosterone has both vasodilator and vasoconstrictor effects. The effect of aldosterone on endothelial function would vary depending on the balance between GPER and MR expression. In vascular endothelial cells, aldosterone activation of GPER mediates vasodilation, while activation of endothelial MR has been linked to enhanced vasoconstrictor and/or impaired vasodilator response (28–30).
Mineralocorticoid Receptor
The mineralocorticoid receptor (MR) is found in the cytoplasm and nucleus and the sodium channels are expressed in the apical membrane of epithelial cells of the distal convoluted tubule as well as in cells of other tissues involved with conservation of salt, such as colon, sweat glands, lung, and tongue. MR is a member of the nuclear receptor superfamily. Together with the glucocorticoid, progesterone, and androgen receptors, MR forms the steroid receptor subfamily (30). Steroid receptors display a modular structure comprised of five regions (A-E). The N-terminal A/B region harbors an autonomous activation function. The central C region, corresponding to the DNA-binding domain, is highly conserved and is composed of two zinc fingers involved in DNA binding and receptor dimerization. The D region is a hydrophilic region and it forms a hinge between DNA-binding domain and ligand-binding domain. The E region corresponds to the C-terminal ligand-binding domain and mediates numerous functions, including ligand binding, interaction with heat-shock proteins, dimerization, nuclear targeting, and hormone-dependent activation (31) (Figure 2). The human MR (hMR) and human glucocorticoid receptor (hGR) have almost identical DNA-binding domains (94% homology in the amino acid) and very similar ligand-binding domains (57%), but divergent N-terminal A/B regions (<15%) (32). The hMR gene was mapped on chromosome 4q31.1-31.2 (33,34) and hMR cDNA encodes a 107 kilodalton polypeptide with 984 amino acids (32). The hMR gene consists of 10 exons, including two exons 1 that encode different 5'-untranslated sequences (35). Expression of the two different hMR variants is under the control of two different promoters that contain no obvious TATA element, but multiple GC boxes. Both hMRα and hMRβ mRNAs are expressed at approximately the same level in the mineralocorticoid target tissues (36).
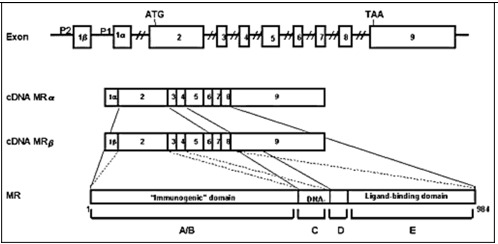
Figure 2.
The linearized structures of the mineralocorticoid receptor gene, mRNAs and protein. The MR gene consists of 10 exons. The MR has two exons 1 (exon 1α and exon 1β), each with an alternative promoter; however, the finally translated MR protein is the same. Exons 1 are untranslated regions, exon 2 codes for the immunogenic domain (A/B), exons 3 and 4 for the DNA-binding domain (C), and exons 5-9 for the hinge region (D) and the ligand-binding domain (E) (37)
Molecular and Cellular Mechanisms of the Aldosterone Action
MRs in its unliganded state is located in the cytoplasm, as part of hetero-oligomeric complexes containing heat shock proteins 90, 70 and 50 (38). Upon binding with their ligand, the receptor-ligand complex dissociates from the heat shock proteins, homo- or heterodimerizes and translocates into the nucleus. Homodimers or heterodimers of the MR interact with hormone-responsive elements (HRE) and/or other transcription factors in the promoter regions of target genes, including the subunits of the ENaC or other proteins related to this channel and sodium transport in general, and modulates the transcription rates of these genes (39) (Figure 3).
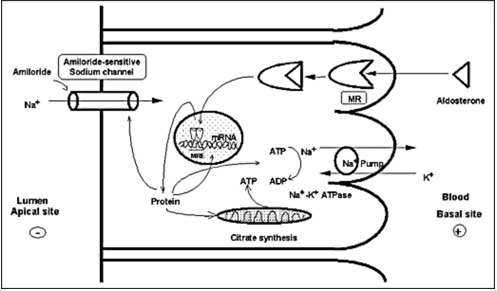
Figure 3.
Mechanism of aldosterone action on sodium reabsorption at the distal convoluted tubule of the nephron. Aldosterone binds to the MR, which is located in the cytoplasm in complex with heat shock proteins 90, 70 and 50. After binding, the receptor-ligand complex translocates into the nucleus, binds to hormone-responsive elements (HRE) of target genes where it modulates their transcription rate. Amiloride-sensitive sodium channel (ENaC) subunits or other related proteins may be targets of such regulation (40).
Pre-Receptor Regulation
Since cortisol circulates at plasma concentrations several orders of magnitude higher than those of aldosterone does, and since it has a high affinity for the MR, it would be expected to overwhelm this receptor in mineralocorticoid target tissues and cause mineralocorticoid excess. A local enzyme, 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2), however, converts active cortisol to inactive cortisone, and protects the MRs from the effects of cortisol (40) 11β-HSD catalyzes the inter-conversion of hormonally active C11-hydroxylated corticosteroids (cortisol in humans or corticosterone in rodents) and their inactive C11-keto metabolites (cortisone in humans or 11-dehydrocorticosterone in rodents). Two isozymes of 11β-HSD have been identified, 11β-HSD type 1 (11β-HSD1) and 11β-HSD2, which differ in their biological properties and tissue distributions. 11β-HSD2, a potent NAD-dependent 11β-hydrogenase, rapidly inactivates glucocorticoids. The human 11β-HSD2 gene encodes 405 amino acids and its molecular weight is approximately 40-kilodalton (41). 11β-HSD2 has a hydrophilic N-terminal domain that is thought to anchor the protein into membranes (42). 11β-HSD2 is localized as a dimer in the nucleus and cytoplasm of cells of the cortical collecting duct and colon (42,43). Prednisolone and prednisone are substrates for both 11β-HSD isozymes (44,45) and dexamethasone is metabolized slightly by 11β-HSD2 (46). Licorice derivatives, such as glycyrrhizic acid, and the hemisuccinate derivative carbenoxolone are inhibitors of 11β-HSD2. Inhibition of 11β-HSD2 with such agents, confers mineralocorticoid potency to physiologic concentrations of endogenous glucocorticoids in the kidney and colon (47). Thus, in normal physiology, 11β-HSD2 protects the MR by converting cortisol to the inactive cortisone and allows aldosterone-selective access to the inherently nonselective MR in mineralocorticoid target tissues.
Amiloride-Sensitive Sodium Channel (Epithelial Sodium Channel; ENaC)
The cDNA of the α-subunit of the ENaC (αENaC) was cloned from the rat colon in 1993 (48) and soon after the cDNAs of the β- and γ-subunits of this channel were cloned for the same species (49). The human α-, β- and γ-subunits of ENaC were also cloned (50,51). In vitro studies demonstrated that the α subunit of the ENaC itself had the majority of Na channel function, while, the β- and γ- subunits alone were not shown to play as major a role in sodium transport (48). However, the β- and γ-subunits enhanced the function of the α-subunit and all subunits are required for full ENaC activity (52). It appears then that this channel consists of the α-, β- and γ-subunits and an amiloride-binding protein (Figure 4). Aldosterone increases transcription of αENaC but not β- and γ-subunits, resulting enhanced channel assembly and transported from endoplasmic reticulum to Golgi (53). In Golgi, furin proteolytically cleaves specific sites in the extracellular domains of α- and γ-ENaC, resulting in channel activation. At the cell surface, Nedd4-2 binds to ENaC, increasing endocytosis and degeneration (54).The proline-rich region of the C-terminal of the αENaC is important for binding to α-spectrin and for stabilization of the sodium channel in the membrane (55). Recently, several studies demonstrated abnormalities of the β- and γ-subunits of the ENaC in patients with Liddle's syndrome, characterized by mineralocorticoid excess (hypertension and hypokalemic alkalosis), and suppressed aldosterone secretion (56–59). The truncation caused by these mutations influenced the PY motif at the N-terminal of the molecule. This motif is responsible for the binding of the channel subunits with NEDD4, a carrier protein facilitating clearance of the channel (60). Moreover, a point mutation of the αENaC gene, located close to the N-terminal of the protein, was reported to cause a decrease of the probability of an open sodium channel, resulting in defective reabsorption (40,61).
The ENaC-Regulatory Complexes in Aldosterone-Mediated Sodium Transport
Aldosterone-induced trans-epithelial Na+ transport via ENaC involves the coordinate functioning of stimulatory signaling proteins such as serum- and glucocorticoid-induce kinase-1 (SGK1) (23,62), glucocorticoid-induced leucine zipper protein-1 (GILZ1) (63) and connector enhancer of kinase suppressor of Ras 3 (CNK3) (64), with inhibitory proteins, such as neural precursor cell expressed, developmentally downregulated protein (Nedd4-2) (24) and extracellular signal-regulated kinase (ERK) 1/2 (23,24,62,65).
SGK1 is an aldosterone-regulated protein kinase that stimulates renal ENaC through many mechanisms. First, SGK1 phosphorylates the E3 ubiquitin ligase and Nedd4-2, and inhibits its actions. Nedd4-2 interacts with the C-terminal tail of ENaC subunits, decrease surface expression of the channel via channel ubiquitinoylation (23,24,62). Second, SGK1 phosphorylates kinase with no lysine (WNK) 4 and prevents ENaC endocytosis (66). Third, SGK1 directly phosphorylates alpha ENaC and transforms silent ENaC channels to active ones (67). Then, SGK1 alters ENaC expression, trafficking and activity, and stimulates Na+ transport in the kidney cortical collecting duct (CCD) (68). However, SGK1 is a short-lived protein. Following synthesis, SGK1 is rapidly targeted to the endoplasmic reticulum (ER), where ER-associated ubiquitin ligases CHIP and HRD1 aid in its ubiquitinoylation and subsequent proteasome-mediated degradation (69). Another aldosterone-induced ENaC-regulator, GILZ, which protects SGK1 from rapid ER-associated degradation by controlling protein-protein interaction (53.6). In kidney CCD, GILZ1 is robustly induced by aldosterone (70). GILZ1 stimulates ENaC cell surface expression and activity at least in part by inhibiting ERK1/2, which abrogates ENaC function (65,71,72).
The recently identified MR target gene CNKSR3 (connector enhancer of kinase suppressor of Ras 3), commonly referred as CNK3, is highly expressed in the connecting tubule (CNT) and the CCD (73). CNK3, like SGK1 and GILZ1, is rapidly induced by physiological concentrations of aldosterone (64). CNK3 acts to assembly various ENaC-regulatory components in close vicinity of the channel and thereby exerts its stimulatory effects on channel function (74).
Epigenetic Control of ENaC Transcription by Aldosterone-Sensitive Dot1A-Af9 Complex
Chromatin regulates gene transcription by the post-translational modification of histone N-terminal tails such as acetylation and methylation. The histone H3 Lys 79 methyltransferase disruptor of telomeric silencing alternative splice variant a (Dot1a) methylates histone H3 Lys79, which resides in the globular domain (75). ALL-1 fused gene from chromatin 9 (Af9), putative transcription factor, physically and functionally interact with Dot1a to form a nuclear repressor complex that directly or indirectly binds specific site of the alpha ENaC promoter. Aldosterone reduces the level of Af9 mRNA and protein. Then, Af9 overexpression induces hypermethylation of histone H3 Lys 79 and repression of alpha ENaC transcription (76). Aldosterone impairs the formation of Dot1a -Af9 complex associated with alpha ENaC promoter by 1) decreasing abundance of Dot1a and Af9; 2) attenuating the interaction between Dot1a and Af9 via Sgk-1-catalyzed phosphorylation of Af9 at Ser 435; 3) counterbalancing the repression through binding to mineralocorticoid receptor (MR) and facilitating its translocation into the cell nucleus, where MR and Dot1a compete for binding to Af9. These are aldosterone-dependent and -independent mechanisms for Dot1a-Af9-mediated repression of alpha ENaC transcription. While aldosterone -independent de-repression achieved through the action of ALL-1 fused gene from chromatin 17 (Af17), Af17 upregulates alpha ENaC transcription by decreasing Af9 binding to Dot1a and relieving Dot1a-Af9-mediated repression of ENaC (77). 4) SGK1 phosphorylates Af9, thus, down-regulating Dot1a-Af9 complex, and relieving the basal repression on alpha ENaC transcription (67,78).
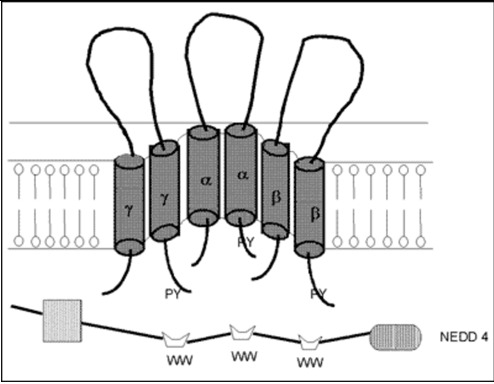
Figure 4.
Model of a putative amiloride-sensitive sodium channel (ENaC). The amiloride-sensitive sodium channel appears to consist of the α-, β- and γ- subunits and an amiloride-binding protein. This channel is located at the apical site of the renal epithelium and plays a role in passive sodium transport, which is mainly regulated by mineralocorticoids (79).
THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM IN NEWBORNS AND INFANTS
Aldosterone secretion rate of newborns and infants was similar to that of older children and adults. Therefore, the aldosterone secretion rate corrected by body surface was much higher in infancy than later in life (80). Urinary aldosterone at birth depends on gestational age and increases progressively, concurrently with the levels of plasma aldosterone. Plasma renin activity, plasma aldosterone and urinary excretion rate of aldosterone decrease with age (81). At birth, human kidneys display tubular immaturity leading to sodium wasting and impaired ability to reabsorb water. Past studies showed that plasma potassium concentrations were significantly higher in newborns than in their respective mothers, while neonatal and maternal plasma sodium concentrations were closely related. Aldosterone and renin levels in newborns differs significantly from the corresponding maternal concentrations (82). The aldosterone-renin ratio significantly increases with gestational age. Thus, neonatal partial aldosterone resistance was previously suggested because of the high urinary sodium loss in the presence of hyperactivity of the renin-angiotensin-aldosterone system (83). Previous study found that the highest aldosterone levels detected in the cord blood originated from de novo synthesis by the fetal adrenal glands (84). In addition, neonatal aldosterone resistance was associated with weak or undetectable renal MR expression at birth. MR mRNA is transiently expressed between 15 and 24 weeks of gestation, but it is undetectable in late gestational age and neonatal kidney (85). 11 beta-hydroxysteroid dehydrogenase type 2 (11 beta HSD2) and alpha ENaC are closely correlated with cyclic MR expression.
CLASSIFICATION OF HYPOALDOSTERONISM
Various syndromes are characterized by or associated with hypoaldosteronism. Hypoaldosteronism is classified in three large categories, defective stimulation of aldosterone secretion, primary defects in adrenal synthesis or secretion of aldosterone, and aldosterone resistance, according to their pathophysiology and summarized in Table 1.
Table 1.
Causes of Hypoaldosteronism and Hormonal Profiles
| Causes of Hypoaldosteronism | Hormonal Profiles |
|---|---|
| DEFECTIVE STIMULATION OF ALDOSTERONE | |
| ❖ Congenital keep tablehyporeninemic hypoaldosteronism ❖ Acquired hyporeninemic hypoaldosteronism Associated with diabetes mellitus Associated with nephropathy Glomerulonephritis Gouty nephritis Pyelonephritis Nephropathy associated with multiple myeloma Nephropathy associated with systemic lupus erythematosa Mixed cryoglobulinemia Nephrolithiasis Analgesic nephropathy Renal amyloidosis Iga nephropathy ❖ Associated with autonomic insufficiency ❖ Associated with liver cirrhosis ❖ Associated with sickle cell anemia ❖ Associated with acquired immune deficiency syndrome ❖ Associated with polyneuropathy, organomegaly, endocrinopathy, m protein and skin changes syndrome ❖ Lead poisning ❖ Excess sodium bicarbonate ❖ Sjogren's syndrome ❖ Drugs interfering with renin production Β-blocker Prostaglandin synthetase inhibitors Non-steroidal anti-inflammatory drugs Calcium channel blocker ❖ Other drugs Cyclosporin a Mitomycin c Cosyntropin | Low plasma renin; Low plasma and urinary aldosterone |
| ❖ Drugs interfering with angiotensin ii production Angiotensin ii converting enzyme inhibitors | High plasma renin; low plasma aldosterone; low angiotensin ii |
| PRIMARY DEFECTS IN ADRENAL SECRETION OF ALDOSTERONE | |
❖ Combined with defective cortisol synthesis
Congenital adrenal hyperplasia -Cholesterol desmolase deficiency (lipoid adrenal hyperplasia) -3β-hydroxysteroid dehydrogenase deficiency -21-hydroxylase deficiency -11β-hydroxylase deficiency Adrenoleukodystrophy, adrenomyeloneuropathy | Low plasma renin; low plasma aldosterone; low plasma cortisol High plasma deoxycorticosteorne |
| b) Acquired causes Autoimmune adrenal destruction -Addison's disease -Multiple autoimmune endocrinopathy Infectious adrenal destruction -Bacterial infection -Fungal infection Infiltration of adrenal glands -Amyloidosis -Hemochromatosis -Sarcoidosis Metastatic or infiltrative malignant disease Bilateral adrenalectomy Drug induced -Mitotane -Aminoglutethimide -Torilostane -Ketoconazole | Low plasma renin; low plasma aldosterone; low plasma cortisol |
❖ Isolated deficiency of aldosterone secretion
-Corticosterone methyloxidase type i (cmo i) deficiency -Corticosterone methyloxidase type ii (cmo ii) deficiency | High plasma renin; low plasma aldosterone Normal plasma 18-hydroxycorticosterone/aldosterone ratio High plasma 18-hydroxycorticosterone/aldosterone ratio |
-Sepsis -Pneumonia -Peritonitis -Cholangitis -Liver failure After removal of mineralocorticoid secreting adrenal tumor Discontinuation of agents with mineralocorticod activity Heparin or chlorbutol administration | Low plasma aldosterone concentration; inappropriate elevated plasma renin |
| DEFECTIVE ALDOSTERONE ACTION | |
| ❖ Pseudohypoaldosteronism (pha) type 1 Renal (autosomal dominant pha) Systemic pha (autosomal recessive pha) ❖ Secondary pseudohypoaldosteronism Associated with urinary tract infection Associated with medication that blocks epithelial sodium channel (enac) -Amiloride -Triamterene -Trimethoprim -Pentamidine Administration of aldosterone antagonists -Spironolactone -Progesterone -17-hydroxyprogesterone -Synthetic progestin Drugs that may lead to aldosterone resistance -Caludinerin inhibitor (cyclosporin a, tacrolimus) | High plasma renin; high plasma and urinary aldosterone |
Defective Stimulation of Aldosterone
The first category of conditions, which is characterized by defective stimulation of aldosterone secretion, includes the syndromes of congenital and acquired hyporeninemic hypoaldosteronism. One of these conditions is due to a defect of renin secretion such as hyporeninemia resulting from β-blockers, prostaglandin synthetase inhibitors, and calcium channel blockers. Another condition is due to decrease in the conversion of angiotensin I to angiotensin II mediated by converting enzyme inhibitor medications and is associated with hyperreninemia.
Primary Defects in Adrenal Biosynthesis or Secretion of Aldosterone
The second category of conditions, which are characterized by primary defects in adrenal synthesis or secretion of aldosterone, includes all causes of primary adrenal insufficiency and primary hypoaldosteronism caused by aldosterone synthase (CYP11B2) deficiency or as an acquired state. Primary adrenal insufficiency causes include congenital adrenal hypoplasia, congenital adrenal hyperplasia, adrenoleukodystrophy/ adrenomyeloneuropathy, acquired adrenal insufficiency due to autoimmune, infectious and infiltrative disease, bilateral adrenalectomy and use of adrenolytic agents and enzyme inhibitors that block cortisol and aldosterone biosynthesis. These conditions are usually combined with defective cortisol synthesis. Aldosterone synthase (CYP11B2) deficiency (ASD) leads to reduced aldosterone production associated with low or high levels of 18-hydroxycorticosterone, referred to as CMO I or CMO II deficiency, respectively. Several conditions may be associated with aldosterone biosynthetic activity. Heparin suppresses aldosterone synthesis. Critically ill patients with persistent hypovolemia and hypotension also have inappropriately low plasma aldosterone concentrations in relation to the activity of the renin-angiotensin system. Isolated primary hypoaldosteronism in occasionally associated with metastatic cancer of the adrenal gland.
Defective Aldosterone Actions
The third category which is characterized by defective aldosterone action includes syndromes of aldosterone resistance such as pseudohypoaldosteronism type 1 and sodium-wasting states resulting from excessive amounts of circulating mineralocorticoid antagonists, such as spironolactone and its analogues, and synthetic progestin or natural agonists, such as progesterone or 17-hydroxyprogesterone. These mineralocorticoid antagonists may antagonize aldosterone at the levels of mineralocorticoid receptor (86) and frequently, these states are compensated for by elevated concentrations of plasma aldosterone.
HYPORENINEMIC HYPOALDOSTERONISM
The most common form of isolated hypoaldosteronism is caused by impaired renin release from the kidney. Hudson et al. first described this syndrome in 1957 (87), however, hyporeninemia was first recognized in 1972 (88) (89). The typical patient is 50 to 70 years old and usually presents with chronic and asymptomatic hyperkalemia and mild to moderate renal insufficiency with a 40-70% decrease in the glomerular filtration rate when compared to that of age matched healthy subjects. Hyperchloremic metabolic acidosis is seen in approximately half of the patients. This acidosis is classified as a renal tubular acidosis type IV (90). The acidosis is a consequence of decreased renal ammonia neogenesis, reduced hydrogen ion-secretory capacity in the distal nephron, and mild reduction in the proximal tubular threshold for bicarbonate reabsorption. Occasionally, muscle weakness or cardiac arrhythmias are present in some patients. More than a half of the patients have diabetes mellitus (91). Other frequently associated states include autonomic neuropathy, hypotension, and various nephropathies such as glomerulonephritis, gouty nephropathy, and pyelonephritis. Also, this syndrome is associated with nephropathies associated with multiple myeloma and systemic lupus erythematosus, mixed cryoglobulinemia, nephrolithiasis, analgesic nephropathy, renal amyloidosis, IgA nephropathy, cirrhosis, sickle cell anemia, acquired immune deficiency syndrome (AIDS), polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes (POEMS) syndrome, lead poisoning, excess sodium bicarbonate, and Sjogren’s syndrome (90,92–101) . Moreover, this syndrome occurs transiently in association with use of non-steroidal anti-inflammatory drugs, cyclosporin A, mitomycin C, cosyntropin, and other agents in susceptible individuals (102–104).
Pathophysiology
Urinary aldosterone excretion is low under basal conditions and fails to increase after sodium restriction. Plasma renin activity is also low and does not increase appropriately during sodium restriction, periods of prolonged upright posture, or diuretic administration (88). Interstitial renal disease and damage to the juxtaglomerular apparatus seems the most likely cause for the primary defect in renin generation or release and secondary deficiency of aldosterone. However, in some patients with this syndrome there is an absent or blunted aldosterone response to angiotensin II (94,104), suggesting a coexisting primary defect in aldosterone secretion or it reflects atrophy of the zona glomerulosa caused by chronic renin deficiency.
There are various mechanisms to be explained for the hyporeninemia. First possible mechanism is the hypervolemia. The expanded extracellular fluid volume due to hypertension may suppress renin. In fact, long-term sodium restriction and diuretic administration increase plasma renin activity in these patients, however, the increments of plasma renin activity are less than those of normal subjects (97). A second possible mechanism is insufficiency of the autonomic nervous system, particularly in patients with diabetic neuropathy. Impaired adrenergic response to postural change may contribute to insufficient renin release. Besides, these patients exhibit decreased sensitivity to β-adrenergic agonists, suggesting defects in both production and action of catecholamines (96). A third proposed mechanism is secretion of abnormal forms of renin, such as a defect in the conversion of prorenin to renin. Insufficiency of autonomic nervous system may be associated with impaired conversion of prorenin to renin. Indeed, patients with diabetes mellitus and autonomic neuropathy have elevated plasma levels of prorenin (105). A fourth possibility is prostaglandin deficiency. Production of prostaglandin I2 (prostacyclin), which mediates renin release, is apparently diminished in patients with hyporeninemic hypoaldosteronism as assessed by measurement of the stable urinary metabolite 6-keto-prostaglandin F1α (95). Furthermore, the prostaglandin I2 in these patients was unresponsive to the potent stimulator’s norepinephrine and calcium. Prostaglandin I2 deficiency may cause hyporeninemic hypoaldosteronism by causing defects in the conversion of prorenin to renin and renin release (106).
Diagnosis
The diagnosis of hyporeninemic hypoaldosteronism must be considered in any patient with unexplained hyperkalemia. Excess potassium intake from food or drugs does not cause sustained hyperkalemia, if renal function is normal. Renal function should be evaluated and drugs that impair renal potassium excretion should be excluded as a cause. The clinical diagnosis is confirmed by low plasma renin activity and low plasma concentrations or urinary aldosterone excretion under conditions that activate the renin-angiotensin-aldosterone axis by maintenance of upright posture and/or furosemide administration. A low random plasma renin concentration associated with a normal ratio of aldosterone to plasma renin activity is also useful for the diagnosis (94).
Therapy
The therapeutic approach should be chosen after taking into consideration the age of the patients and other concurrent disorders. Only monitoring potassium concentrations is enough for patients with moderate hyperkalemia and without electro-cardiographic changes. Drugs that promote hyperkalemia, such as β-adrenergic antagonists, cyclooxygenase inhibitors, angiotensin-converting enzyme inhibitors, heparin, and potassium-sparing diuretics, should be avoided. Dietary potassium intake should be reduced, if possible. Diuretics are the initial treatment for patients who have disorders associated with sodium retention, such as hypertension and congestive heart failure. Mineralocorticoid replacement with fludrocortisone is reserved for patients with severe hyperkalemia without hypertension and congestive heart failure.
PRIMARY HYPOALDOSTERONISM- ALDOSTERONE SYNTHASE DEFICIENCY (ASD)
Congenital hypoaldosteronism is a rare inherited disorder transmitted as either an autosomal recessive or autosomal dominant trait with mixed penetrance. This disorder was previously termed "corticosterone methyloxidase (CMO)” deficiency and subdivided into two types according to the relative levels of aldosterone and its precursors in an affected person. Patients with "corticosterone methyloxidase I (CMO I)" deficiency have elevated serum levels of corticosterone and low levels of 18-hydroxycorticosterone and aldosterone. In contrast, patients with "corticosterone methyloxidase II (CMO II)" deficiency have high levels of 18-hydroxycorticosterone, the immediate precursor of aldosterone (107). With greater understanding of structure-activity relationships in the CYP11B2 enzyme, this disorder may be better considered a spectrum of hormonal deficiencies, depending on the nature of the CYP11B2 gene defect (108). Two steps of aldosterone biosynthesis from corticosterone previously proposed to be catalyzed by separate enzymes, CMO I and II, previously, are known to involve only one enzyme substrate interaction (6). Isolated aldosterone deficiency results from loss of activity of aldosterone synthase encoded by CYP11B2 gene (109–118). Therefore, the term aldosterone synthase deficiency type 1 (ASD1) and type 2 (ASD2) reflects more appropriately the molecular basis of this disease. In both ASD1 and 2, glomerulosa zone corticosterone is increased and aldosterone decreased, but 18-hydroxycorticosterone is increased in ASD2 (108). ASD1 is associated with loss of both 18-hydroxilation and 18-oxidation enzyme activities. In ASD2, the ability to convert corticosterone (B) to 18-hydorxytetrahydro11-dehydrocorticosterone (18-OH-B) is preserved with failure of further oxidation of 18-hhdroxicorticosrerone to aldosterone (119). The deficiency of aldosterone is much more severe in ASD1. In contrast, aldosterone may reach normal levels under intense stimulation of renin-angiotensin system in ASD2 (108). The clinical presentations of these deficiencies are otherwise similar.
Clinical Presentation
The clinical presentation is typical of aldosterone deficiency, including electrolyte abnormalities such as a variable degree of hyponatremia, hyperkalemia and metabolic acidosis, with poor growth in childhood, but there are usually no symptoms in adults (107,120). Miao et al. reviewed 45 ASD patients (20 of ASD1, 12 of ASD2, 13 of undefined subtype) (121). From their review, 95% of the patients having ASD1 and all of having ASD2 and an undefined subtype had hyponatremia, while 89% showed hyperkalemia. In infants, it is characterized by recurrent dehydration, salt wasting and failure to thrive. These symptoms are present generally within the first 3 months of life, and most often after the first 5 days of life. A modest uremia with a normal creatinine level reflects dehydration in the presence of intrinsically normal renal function. Plasma renin activity might vary, while elevated plasma renin activity levels were more likely to be found in the ASD1 (121).
Diagnosis and Therapy
The diagnosis can be established by measuring the appropriate corticosteroids or their major metabolic products, such as 11-deoxycorticosterone (DOC), corticosterone, 18-hydroxycorticosterone, 18-hydroxy-DOC, and aldosterone levels in plasma. The ratio of plasma 18-hydroxycorticosterone to plasma aldosterone differentiates the two disorders; it is less than 10 in ASD1 (CMO I deficiency) and more than 100 in ASD2 (CMO II deficiency) (121,122). Patients with ASD2 (CMO II deficiency) tend to have increased plasma cortisol levels that may result from increased adrenal sensitivity to ACTH induced by the increased plasma angiotensin II levels in response to sodium depletion (123).
Both forms of the syndrome are treated by replacement of mineralocorticoid with the usual dosage of fludrocortisone (0.1-0.3 mg/ day). Almost infants and children require oral sodium supplementation (2 g/day as NaCl alone or in combination with NaHCO3), although some infants with severe symptom need intravenous fluids. Oral sodium supplementation may be discontinued once plasma rennin activity has decreased to normal, but mineralocorticoid replacement is usually maintained through childhood.
Molecular Mechanism of CYP11B2 Deficiency
ASD has been identified in Jews of European, North American, and Iranian descent (119). In Asians, it was reported in the Thai (124), Indian (124), Japanese (125) and Chinese populations (120,126).
To date, approximately 40 mutations, such as missense and nonsense mutations, splicing mutations, small insertions/deletions, gross deletions, and complex rearrangements, in the CYP11B2 have been reported in cases of ASD; the most common mutations were missense and nonsense (121). Some variants, such as p.Q170X, p.E198D, c.1398+2T>A, p. F233fsX*295, p.L462R, p.Q337X and p.Q272W, were identified in patients without an ASD classification subtype (121). A majority of mutations led to complete loss of enzyme activity, while in some mutations, such as V386A and R181W, double homozygosity was required for clinical phenotype (112,113,121).
Some patients with ASD1 (CMO I deficiency) have a homozygous 5 nucleotide deletion in exon 1 which leads to a frameshift and premature stop codon, resulting in the complete lack of enzyme production (109,110). A male Caucasian patient with ASD1 (CMO I deficiency) had a homozygous point mutation causing a R384P substitution, resulting in complete loss of 11 β- and 18-hydroxylase activity (111) (Figure 5). This suggests that the arginine-384 in aldosterone synthase is highly conserved and apparently quite important for enzyme activity.
A male infant of Turkish parents who presented with ASD1 had a homozygous missense mutation (L451F) in exon 8 of CYP11B2 gene. The L451F mutant protein in vitro showed complete aldosterone deficiency with 11-deoxycirticosterone or corticosterone as substrates. The L451F mutation located immediately adjacent to the highly conserved heme-binding C450 of the cytochrome P450 (117). Computer modeling of the molecule suggested that this substitute my lead a steric effect resulting in preventing the activity of CYP11B2 (117).
Three siblings of Pakistan origin who presented with ASD1 had a homozygous mutation (S308P) in exon 5 of CYP11B2 gene. The S308P mutant protein in vitro showed complete loss of enzyme activity. This mutated residue is likely to locate within the a-helix I, close to the heme-binding, active site of the enzyme. This structural change may be the cause of this disorder in this family (118).
A large number of kindreds with ASD2 (CMO II deficiency) have been identified among Jews originally from Isfahan, Iran. Such patients are all homozygous for two mutations, R181W in exon 3 and V386A in exon 7 (109,112,113) (Figure 5). These mutations together reduce aldosterone synthase activity to 0.2% of normal without affecting 11 β-hydroxylase activity (112,113). However, one non-Iranian patient with ASD2 (CMO II deficiency) carries mutations in the paternal allele, including V386A and T318A mutations, and maternal allele, including R181W and a deletion/frameshift mutation, resulting in complete loss of enzyme activity (113). This suggests that the high levels of 18-hydroxycorticosterone seen in ASD2 (CMO II deficiency) can be synthesized by CYP11B1, which has some 18-hydroxylase activity, and not by CYP11B2. A patient with apparent ASD 1 was homozygous for the mutations E198A and V386A, yet when assayed in vitro the double mutant enzyme behaved similarly to the mutant enzyme found in the Iranian Jewish ASD 2 patients (127). Thus, a difference in expression of CYP11B1 rather than allelic variation of CYP11B2 may be involved in the mechanism underlying the different levels of 18-hydroxycorticosterone between ASD1 and 2 (CMO I and CMO II deficiency). The distinction between ASD 1 and ASD 2 is not precise, and these disorders should be regarded as different degrees of severity on a continuous clinical spectrum.
A male Japanese patient with ASD1 (CMO I) was a compound heterozygous for W56X in exon 1 and R384W in exon 7. W56X was inherited from his mother and R384X was from his father. Since both alleles contain nonsense mutations, a lack of CYP11B2 activity was speculated to cause his condition (125).
Two male Japanese patients with ASD2 (CMO II) had homozygous missense mutation (G435S) in the exon 8 of CYP11B2 gene. The expression studies indicated that the steroid 18-hydroxylase/oxidase activities of mutant enzyme were substantially reduced.
A female infant of Albanian origin with ASD2 (CMO II) revealed homozygosity for a pathogenic T185I mutation in Exon 3 of the CYP11B2 gene and two other homozygous polymorphisms F168F and K1738 in Exon3 (128). Both healthy parents revealed heterozygous for all three substitutions.
Another female Italian Caucasian patient was diagnosed with a compound heterozygous mutation located in exon 4 causing a premature stop codon (E255X) and a further mutation in exon 5, also causing a premature stop codon (Q272X). The patient’s CYP11B2 encoded two truncated forms of aldosterone synthase predicted to be inactive because they lack critical active site residues as well as the hormone-binding site. However, this case displays biochemical features intermediate between those of ASD1 and 2 (CMO I and II).
Some cases of ASD without causative mutations in CYP11B2 have also been reported (116,119).
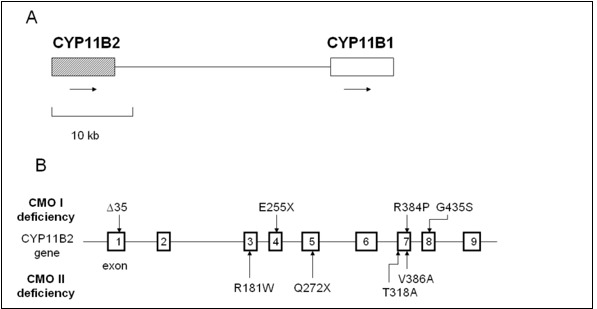
Figure 5.
Relative positions of CYP11B1 and CYP11B2 on chromosome 8 and mutations of CYP11B2. A, The relative positions of CYP11B1 and CYP11B2 on chromosome 8q22. Arrows indicate direction of transcription. B, Mutations of CYP11B2 in reported patients with CYP11B2 deficiency are summarized in the figure (109,121,126,128).
ACQUIRED FORMS OF PRIMARY HYPOALDOSTERONISM
Several conditions may be associated with aldosterone biosynthetic defects. The administration of heparin causes natriuresis and hyperkalemia (129). Heparin preparations suppress aldosterone synthesis, leading to a compensatory rise in plasma renin activity. However, it has been demonstrated that this suppression of enzyme activity is attributable to chlorbutol (1,1,1-trichloro-2-methyl-2-propanol), the preservative used in commercial heparin, rather than to pure heparin (130).
Persistently hypotensive, critically ill patients with sepsis, pneumonia, peritonitis, cholangitis and liver failure, also have inappropriately low plasma aldosterone concentrations in relation to elevated plasma renin activity (131). The defect is at the level of the adrenal but has not been associated with any particular disease or therapy. Plasma cortisol levels are high, reflecting the stressed state. The response to angiotensin infusion is impaired, and the ratio of plasma 18-hydroxycorticosterone to aldosterone is increased, suggesting selective insufficiency of CMO II. It is possible that the hypoxia causes a relative zona glomerulosa insufficiency (132).
ALDOSTERONE RESISTANCE
Pseudohypoaldosteronism (PHA) Type 1
Mineralocorticoid resistance (pseudohypoaldosteronism type 1, PHA1) results from inability of aldosterone to exert its effect on its target tissues and was first reported by Cheek and Perry as a sporadic occurrence in 1958 (133). This disease, usually presents in infancy with severe salt-wasting and failure to thrive, accompanied by profound urinary sodium loss, severe hyponatremia, hyperkalemia, acidosis, hyperreninemia, and paradoxically markedly elevated plasma and urinary aldosterone concentrations. Usually, renal and adrenal functions are normal. This disease has been reported in over 70 patients (134). The prevalence, as estimated from recruitment through a genetic laboratory at the Hôpital Européen Georges Pompidou in France, which is a national reference center for a rare disease, is ~1 per 80,000 newborns (135)(136). Approximately one fifth of these cases are familial, and both an autosomal dominant and a recessive form of genetic transmission have been observed. A previous study found that all patients had renal tubular unresponsiveness to aldosterone, while some had involvement of other mineralocorticoid target-tissues, including the sweat and salivary glands, and the colonic epithelium, as well. Autosomal recessive PHA1 presents in the neonatal period with hyponatremia caused by multi-organ salt loss, including kidney, colon, and sweat and salivary glands. Autosomal recessive PHA1 persists into adulthood and shows no improvement over time. However, literature regarding follow-up of these patients after diagnosis is insufficient. In contrast, autosomal dominant PHA1 is characterized by an isolated renal resistance to aldosterone, leading to renal salt loss. Particularly autosomal dominant form of PHA1 typically shows a gradual clinical improvement during childhood, allowing the cessation of sodium supplementation.
PATHOPHYSIOLOGY
The mechanism(s) by which aldosterone controls sodium transport in its target tissues involves the mineralocorticoid receptor (MR) and proteins that are associated with the amiloride-sensitive sodium channel (ENaC). The latter proteins are expressed in the apical membrane of epithelial cells of the distal convoluted tubule and in the membranes of cells of other tissues involved in the conservation of salt, such as colon, sweat gland, lung and tongue. Thus, the MR and the ENaC were considered as potential candidate molecules for the pathogenesis of PHA1. In fact, mutations of α- and β-subunits of the ENaC were reported in PHA patients from autosomal recessive kindreds (61,137). Mutations of the MR were also reported in the patients with autosomal dominant PHA1 (138,139). However, no molecular defects were found in either MR or ENaC in some patients with PHA1, especially in those with the sporadic form PHA1, which suggests molecular heterogeneity in PHA1 (79,140–144).
DIAGNOSIS
Electrolyte profiles suggest mineralocorticoid deficiency or end-organ resistance, along with hyperkalemia, hyponatremia and metabolic acidosis associated with profound urinary salt loss. Renal and adrenal function is normal. The diagnosis is confirmed by the markedly elevated plasma aldosterone concentrations and plasma renin activity.
The differential diagnosis of PHA1 includes salt-wasting states due to hypoaldosteronism, including several forms of congenital adrenal hyperplasia, isolated hypoaldosteronism due to corticosterone methyloxidase (CMO) I and II deficiencies and congenital adrenal hypoplasia. Normal cortisol and excessive aldosterone responses to adrenocorticotropin (ACTH) are expected in patients with congenital PHA.
THERAPY
The standard treatment of PHA has been replacement with high doses of salt, with a variable response among patients (134). Recently, carbenoxolone, an 11β-hydroxysteroid dehydrogenase inhibitor, was employed as therapy in PHA1 and an ameliorating effect was observed which was attributed to mediation by the MR (140). We studied a 17-yr-old male patient with congenital multifocal target-organ resistance to aldosterone. We examined his clinical response to carbenoxolone, expected to increase the intracellular level of cortisol in the kidney by preventing local conversion of cortisol to cortisone, and to high doses of fludrocortisone, a synthetic mineralocorticoid. Subsequently, and for a brief period of time, we administered dexamethasone, which has no intrinsic salt-retaining activity, in addition to carbenoxolone, to suppress endogenous cortisol, along with its intrinsic mineralocorticoid activity.
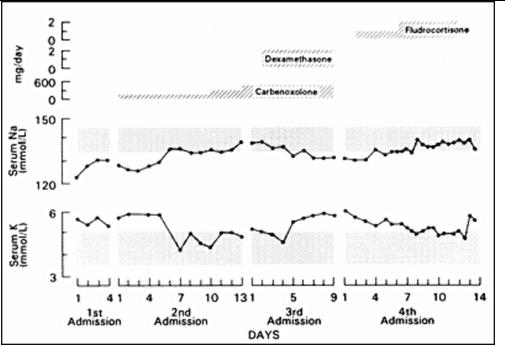
Figure 6.
Effect of carbenoxolone, carbenoxolone plus dexamethasone, and fludrocortisone (top panel) on the serum sodium (middle panel) and potassium (bottom panel) concentrations of a patient with PHA. Carbenoxolone normalized plasma electrolytes, addition of dexamethasone reversed this effect, while fludrocortisone at high doses also normalized plasma electrolytes (140).
Carbenoxolone normalized the patient's serum electrolyte concentrations and decreased his urinary excretion of sodium within a week (Figure 6). Subsequent long-term therapy of this patient with carbenoxolone (450 mg/day p.o.) maintained his electrolyte concentrations within the normal range. His urinary 24 h free cortisol was increased during carbenoxolone therapy. Addition of dexamethasone suppressed his urinary free cortisol excretion and reversed the beneficial effect of carbenoxolone on serum and urinary electrolytes (Figure 6). These data suggest that an increase in urinary free cortisol observed during carbenoxolone therapy was due to a localized effect of this drug on the kidney rather than on tissues involved in the negative feedback effect of glucocorticoids. The effect of carbenoxolone does not seem to be mediated by GR but seems to be exerted purely via the MR (Figure 7). There were no adverse effects of long-term carbenoxolone therapy in this patient. He also reported increased stamina, a better ability to concentrate and less anxiety. On treatment, the patient grew 6 cm/y and progressed from -4SD to -3SD scores for mean height for age. He also progressed in his pubertal development from Tanner stage III to IV for pubic hair, while his bone age advanced from 12 to 14 y.
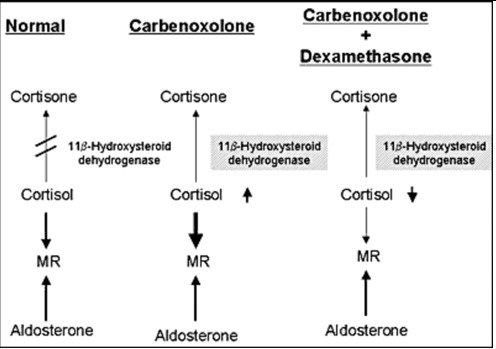
Figure 7.
Mechanism of the effect of carbenoxolone. Carbenololone inhibits of conversion of cortisol to cortisone in the kidney, resulting in the enhancement of the effect of cortisol as a ligand for MR. Dexamethasone suppressed cortisol production and reversing the beneficial effect of carbenoxolone in our patient with PHA1.
Both carbenoxolone and fludrocortisone normalized the serum electrolytes of our patient, suggesting the presence of a functional, albeit possibly defective, renal MR. Interestingly, the same patient was unresponsive to intravenous infusion of aldosterone and fludrocortisone (up to 3 mg/day) when studied in infancy (145), suggesting that the clinical improvement that has been noted in the majority of PHA patients with age may be related to changes in their responsiveness to mineralocorticoid.
On the other hand, another study reported that carbenoxolone did not show any significant salt-retaining effect in two patients with multiple PHA, while carbenoxolone significantly suppressed the renin-aldosterone system in a patient with renal-form PHA (146). This difference of responsiveness to carbenoxolone may be due to an age-dependent change on mineralocorticoid responsiveness. Additionally, the different mineralocorticoid responsiveness of renal and multisystem PHA patients indicates a difference in their MR function. The partial response to carbenoxolone in renal PHA suggests that there is at least a partly functional MR. This is also supported by the observation that spironolactone, a mineralocorticoid antagonist, aggravated sodium loss in several patients with renal PHA (147).
MOLECULAR MECHANISM(S) OF PSEUDOHYPOALDOSTERONISM TYPE 1
In 1996, a study reported homozygous mutations introducing a stop codon or frame shift in the αENaC gene of affected members of families with autosomal recessive PHA (61). To date, worldwide more than 40 different mutations have been described in the coding region of ENaC subunit genes (148–150). The majority of mutations appear in the αENaC gene SCNN1A, most frequently in exon 8 (61,150–152). Mutations are nonsense, single base deletions or insertions, or splice-site mutations, leading to abnormal length of mRNA and protein. Few missense mutations in αENaC gene have also been reported (149,153). Only a few cases of mutations in β and gamma ENaC genes have been reported (149,154,155). Phenotype and genotype correlations have been noted with more severe phenotype in nonsense, frameshift, and abnormal splicing mutations than patients with missense mutations (148,154,155).
A Swedish study regarding families with autosomal recessive PHA, homozygous or compound heterozygous mutations showed that a stop codon or a frame shift in the αENaC gene was associated with pulmonary disease as well (150). The truncation caused by these mutations influenced the PY motif at the N-terminal region of the molecule. This motif is responsible for the binding of the channel subunits with Nedd4, a carrier protein facilitating clearance of the channel (60). Moreover, a point mutation of the αENaC gene, located close to the N-terminal of the protein, was reported to cause a decrease of the probability of an open sodium channel, resulting in defective reabsorption (61,153). In the other four families with autosomal recessive PHA, insertion of a T in exon 8 and nonsense mutation (R508X) in exon 11 of the αENaC gene, resulting in a truncated αENaC subunit, was found (156). A splice site mutation in intron 12 of the βENaC gene, which preventing correct splicing of the mRNA was found in a Scottish patient (156). Also, other autosomal recessive families with PHA had a homozygous splice-site mutation in the γENaC, while a Japanese sporadic patient with the systemic form of PHA was a compound heterozygote for mutations in the αENaC, which resulted in the generation of a truncated channel subunit (137,157) . Compound heterozygous mutations (Q217X in exon 4 and Y306X in exon 6) of βENaC have been reported in the patient with multi-organ PHA1 of Ashkenazi family in Israel (154). These mutations produce shortened βENaC subunits with 253 and 317 residues respectively instead of the 640 residues present in βENaC subunit. Expression of cRNA carrying these mutations in Xenopas oocytes showed that the either mutation drastically reduced to only 3% of normal ENaC activity (154). An African American female with PHA, who had persistent and symptom hyperkalemia, had compound heterozygous mutation in the βENaC gene: c.1288delC in exon 9, a one-base deletion that generated a frameshift mutation, and c.1466+1 G>A, an intronic base substitution in intron11 that leaded to a splice site mutation (158).
To date more than 50 different mutations in the human MR gene (NR3C2) causing autosomal dominant PHA1 have been described. NR3C2 mutations were found in 62% of patients with renal PHA1 referred to a genetics laboratory at the Hôpital Européen Georges Prompidou in France (135). Nonsense mutations, frameshift mutations, splice site mutations, and deletions of whole or part of the gene lead to gross change of the MR protein. Nonsense mutations are found in all exons and lead to truncated MR protein. A past study. reported families with autosomal dominant PHA, who had molecular defects of the MR resulting in non-expression of one of the 2 alleles (138) (Figure 8). In addition, another study reported a sporadic patient with PHA who had a heterozygous mutation in exon 9 of the MR that introduced a premature stop codon (144) (Figure 8). These results, may suggest that expression of only one allele of the MR is insufficient to prevent salt loss. Another case study did not identify any abnormalities of the MR in PHA patients from two families with the autosomal dominant form of the disease (144), while other authors reported a heterozygous missense mutation in exon 8 of the MR gene identified in PHA patients from a Japanese autosomal dominant family (139) (Figure 8). A heterozygous nonsense mutation in exon 2 (S163X, C436X) and in exon 9 (R947X) of the MR, leading to a premature stop codon of the MR gene were found in other patients with autosomal dominant PHA (159–161). It was previously reported a heterozygous splice acceptor site mutation, which results in exon 7 skipping and subsequently in premature termination in exon 8 of MR with Japanese female patients with PHA1 (162). This study showed that RT-PCR products of mRNA with that patient showed both wiled-type and mutated mRNA, suggesting that haploinsufficiency due to nonsense mediated mRNA decay with premature termination is not sufficient to give rise to the PHA phenotype (162). It was also reported that Q776R mutation in exon 5 or L979P mutation in exon 9, which is located in the ligand-binding domain of the MR, presented reduced or absent aldosterone binding, respectively (163). Three-dimensional structure of MR suggests that the residue Q776 is located in helix 3 and is locking aldosterone in the ligand-binding pocket (163). A study examined patients with PHA1 presenting isolated renal salt loss from six families in Italy and Germany and found one nonsense mutation (E378X), one frameshift mutation (A958R) and two missense mutations (S818L and E972G) (164). S818L does not bind aldosterone or activate transcription or translocate into the nucleus. Three-dimensional molecular structure showed that S818 was located in helix H5 and S818 was speculated to be necessary to stabilize helix H5 and the β-sheet 1 via hydrogen bond to Y828. E972G mutation showed a significantly lower ligand-binding affinity and only 9% of wild-type transcriptional activity. Three-dimensional molecular structure showed that E972 is involved in a hydrogen-bond network with R947 anchoring helix H12 to H10. Thus, substitute of E972G suggested to be open up the hydrophobic core and displace helix H10, causing the decreased ligand-binding ability (164).
A Japanese study reported four sporadic patients and two siblings with a renal form of PHA (165). Two siblings and one sporadic patient had R651X of NR3C2 (MR) gene. One sporadic patient had R947X, another two patients had 603A deletion and 304-305CG deletion, respectively, both resulting in frameshift mutations (165).
Another study reported two female Japanese infants with the renal form of PHA1 and identified two heterozygous mutations. One had a c.4932_493insTT in Exon 2, resulting in a premature stop codon (p.Met166 LeufsX8) and another had a nonsense mutation of R861X in exon 7 (166). These mutations resulted in haploinsufficiency of the MR and were the cause of aldosterone resistance in the kidney.
From the study of the genetics laboratory at the Hôpital Européen Georges Pompidou in France, 20 mutations were found in exon 2; all of them led to truncated receptors, Of the 22 mutations identified in exon 3 and 4, coding for the MR DBD, 11 were nonsense or frameshift mutations, the reminder missense mutations. Thirty variants were located in exon 5-9 and affected LBD; the majority were missense mutations. Nine were splice variants in different introns, 19 were large deletions encompassing single or multiple exons and the flanking intronic regions of the NR3C2 gene (135) (figure 8).
These studies suggest major molecular heterogeneity in PHA.
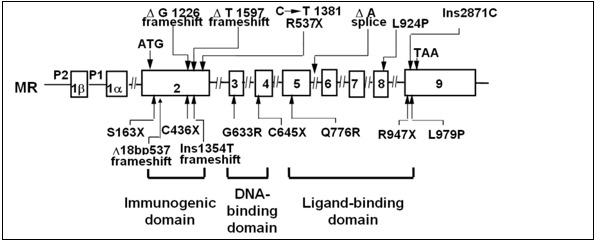
Figure 8.
Another study investigated 5 unrelated cases of sporadic PHA (79,140,143). The researchers found a nonconservative homozygous mutation (A241V) in the MR of 4 of the patients and a conservative heterozygous mutation (I180V) in one of these patients and his asymptomatic father, while no abnormalities were found in the DNA- or ligand-binding domains of the MR. The Val241 and Val180 substitutions were found also in the norm 6al population. The heterozygosity and homozygosity frequencies of the Val241 and Val180 mutations were 48%, 38%, 22% and 1.5%, respectively. Another finding was a nonconservative amino acid substitution (T663A) in the αENaC, which was located close to the C-terminal (79). Of the 5 patients, 2 were homozygous and 3 heterozygous for this variation, respectively. This amino acid substitution was also present at high frequency in apparently normal controls. The homozygosity and heterozygosity frequencies of the αENaC Ala663 were 31% and 64%, respectively. Three of the 4 (75%) patients with multiple tissue resistance to aldosterone had both αENaC (heterozygous or homozygous) and MR (homozygous) mutations as described above, while only 7% of our controls with apparently normal salt conservation had the same concurrent abnormalities (Table 2, p < 0.025).
Table 2.
MR and aENaC Polymorphisms in PHA and Normal Subjects
| MR | αENaC | Target organ | |||||
|---|---|---|---|---|---|---|---|
| I180V | A241V | T663A | |||||
| Homo | hetero | homo | Hetero | homo | Hetero | ||
| Pt.1 | + | + | + | Multiple | |||
| Pt.2 | + | Multiple | |||||
| Pt.3 | + | + | Multiple | ||||
| Pt.4 | + | Multiple | |||||
| Pt.5 | + | + | Isolated | ||||
| controls | 1.5% | 22% | 38% | 48% | 31% | 64% | |
| controls | + | + | + | ||||
| controls | + | + | |||||
| controls | + | + | |||||
(79) with permission
The researchers identified, in a Japanese patient with sporadic PHA, three homozygous substitutions in the MR gene: G215C, I180V or A241V, which had previously reported to occur in healthy populations. Luciferase activities induced by MR with either G215C, I180V or A241V substitution were significantly lower than those for wild-type MR with aldosterone at concentrations ranging from 10-11 to 10-9 M, 10-8M, or 10-11 to 10-6M, respectively. A homozygous A to G substitution of the donor splice site of αENaC intron 4 was found in the patient. These results suggest that each of three MR polymorphisms identified in our patient is functionally and structurally heterogeneous (167).
The authors suggested that the above polymorphisms may confer vulnerability in salt conservation, which might be expressed fully only when concurrently present with other genetic defects of the MR or other proteins that participate in sodium homeostasis, such as Nedd4 (168). This hypothesis, if true, would be compatible with a sporadic presentation or a digenic or multigenic expression and heredity as previously described in retinitis pigmentosa (169). In this case, hereditary transmission might be complex and appear either as a dominant and/or recessive trait with variable penetrance.
Secondary Pseudohypoaldosteronism (PHA)
Secondary PHA is a form of renal resistance to aldosterone. The cause of secondary PHA is either renal disease or medication. The clinical and laboratory findings resemble those of a transient PHA. Since Rodriguez-Soriano et al. reported the first case in 1983 (169), more than 68 cases have been reported. Secondary PHA may occur mainly in neonates and young infants with urinary tract infections, such as pyelonephritis, and/or malformation of urinary system causing obstructive uropathy, tubulointerstitial nephritis, sickle cell nephropathy, and systemic lupus erythematosus(170). Secondary PHA has been also related to drugs like non-steroidal anti-inflammatory agents and potassium-sparing diuretics (170–172). This state occurs in male infants more frequently than female infants because of the higher incidence of urinary tract infections and obstructive uropathy in male infants rather than in female infants(169). Patients present poor feeding, poor weight gain or failure to thrive, vomiting, diarrhea, polyuria, and dehydration. Acute worsening of their general condition may occur, with severe weight loss, peripheral circulatory failure, rise in serum urea and creatinine levels, and occasional life-threatening hyperkalemia (169). The laboratory features are hyponatremia, hyperkalemia, metabolic acidosis, elevation of plasma aldosterone concentrations and plasma renin activity, and inappropriately increased sodium and decreased potassium excretion in urine (173). The aldosterone resistance of secondary PHA is transient and usually reverts with the resolution of the infection.
PATHOPHYSIOLOGY
The very high ratio of plasma aldosterone to potassium, together with diminished urinary K/Na values, strongly suggests that hyponatremia and hyperkalemia result from a lack of response of the renal tubule to endogenous mineralocorticoids (174). The intrarenal expression of several cytokines, such as tumor necrosis factor alpha, interleukin (IL) 1, IL-6, transforming growth factor beta-1, angiotensin II, endothelin, thromboxane A2, and prostaglandins, are increased in cases of urinary tract infections. These changes result in inhibition of aldosterone action through reduction of its expression and/or impairment of its receptor, vasoconstriction and reduction of glomerular filtration rate, increased natriuresis and/or decreased Na+-K+-ATPase activity(173) . A past study found that the number of mineralocorticoid receptors in obstructive uropathy were low in the acute phase but returned to normal after successful surgical correction of the obstruction (175). This suggests that a reduced aldosterone effect can also reflect down-regulation of the receptor sites, due to highly elevated aldosterone levels (175).
THERAPY
The clinical and laboratory findings improve within one or two days and disappear after the completion of medical treatment of urinary tract infection and/or surgical correction of obstructive uropathy, usually within a few days to one week after beginning of treatment (173). However, in some patients, sodium bicarbonate and/or sodium chloride supplementation may be necessary for a week or month (173)
REFERENCES
- 1.
- Miller WL. Molecular biology of steroid hormone synthesis. Endocrine Reviews. 1988;9(3):295–318. [PubMed: 3061784]
- 2.
- Chung BC, Matteson KJ, Voutilainen R, Mohandas TK, Miller WL. Human cholesterol side-chain cleavage enzyme, P450scc: cDNA cloning, assignment of the gene to chromosome 15, and expression in the placenta. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(23):8962–8966. [PMC free article: PMC387054] [PubMed: 3024157]
- 3.
- White PC, Chaplin DD, Weis JH, Dupont B, New MI, Seidman JG. Two steroid 21-hydroxylase genes are located in the murine S region. Nature. 1984;312(5993):465–467. [PubMed: 6095106]
- 4.
- White PC, New MI, Dupont B. HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proceedings of the National Academy of Sciences of the United States of America 1984;81(23 I):7505–7509. [PMC free article: PMC392175] [PubMed: 6334310]
- 5.
- Curnow KM, Tusie-Lunaf MT, Pascoe L, Natarajan R, Gu JL, Nadler JL, Whitef PC. The product of the CYP11B2 gene is required for aldosterone biosynthesis in the human adrenal cortex. Molecular Endocrinology. 1991;5(10):1513–1522. [PubMed: 1775135]
- 6.
- Kawamoto T, Mitsuuchi Y, Toda K, Yokoyama Y, Miyahara K, Miura S, Ohnishi T, Ichikawa Y, Nakao K, Imura H, Ulick S, Shizuta Y. Role of steroid 11β-hydroxylase and steroid 18-hydroxylase in the biosynthesis of glucocorticoids and mineralocorticoids in humans. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(4):1458–1462. [PMC free article: PMC48470] [PubMed: 1741400]
- 7.
- Chua SC, Szabo P, Vitek A, Grzeschik KH, John M, White PC. Cloning of cDNA encoding steroid 11 beta-hydroxylase (P450c11). Proceedings of the National Academy of Sciences of the United States of America. 1987;84(20):7193–7197. [PMC free article: PMC299256] [PubMed: 3499608]
- 8.
- Rainey WE. Adrenal zonation: Clues from 11β-hydroxylase and aldosterone synthase. Molecular and Cellular Endocrinology. 1999;151(1–2):151–160. [PubMed: 10411330]
- 9.
- Demura M, Bulun SE. CpG dinucleotide methylation of the CYP19 I.3/II promoter modulates cAMP-stimulated aromatase activity. Molecular and Cellular Endocrinology. 2008;283(1–2):127–132. [PubMed: 18201819]
- 10.
- Takeda Y, Demura M, Wang F, Karashima S, Yoneda T, Kometani M, Hashimoto A, Aono D, Horike SI, Meguro-Horike M, Yamagishi M, Takeda Y. Epigenetic regulation of aldosterone synthase gene by sodium and angiotensin II. Journal of the American Heart Association. 2018;7(10) [PMC free article: PMC6015301] [PubMed: 29739797] [CrossRef]
- 11.
- Gibbons GH, Dzau VJ, Farhi ER, Barger AC. Interaction of Signals Influencing Renin Release. Annual Review of Physiology. 1984;46(1):291–308. [PubMed: 6370111]
- 12.
- Quinn SJ, Williams GH. Regulation of aldosterone secretion. Annual Review of Physiology. 1988;50:409–426. [PubMed: 3288099]
- 13.
- Kramer E, Gallant S, Brownie AC. Actions of Angiotensin 11 on Aldosterone Adrenal Cortex Biosynthesis in the Rat AND LATE PATHWAY* Animals and Tissue Preparation-Female Sprague-Dawley rats.
- 14.
- Bassett MH, White PC, Rainey WE. The regulation of aldosterone synthase expression. In: Molecular and Cellular Endocrinology.Vol 217. Mol Cell Endocrinol; 2004:67–74. [PubMed: 15134803]
- 15.
- Kojima I, Kojima K, Rasmussen H. Role of calcium and cAMP in the action of adrenocorticotropin on aldosterone secretion. Journal of Biological Chemistry. 1985;260(7):4248–4256. [PubMed: 2579947]
- 16.
- Woodcock EA, McLeod JK, Johnston CI. Vasopressin stimulates phosphatidylinositol turnover and aldosterone synthesis in rat adrenal glomerulosa cells: Comparison with angiotensin ii. Endocrinology. 1986;118(6):2432–2436. [PubMed: 2938936]
- 17.
- Hollenberg NK, Chenitz WR, Adams DF, Williams GH. Reciprocal influence of salt intake on adrenal glomerulosa and renal vascular responses to angiotensin II in normal man. Journal of Clinical Investigation. 1974;54(1):34–42. [PMC free article: PMC301522] [PubMed: 4365595]
- 18.
- Carey RM. Acute Dopaminergic Inhibition of Aldosterone Secretion Is Independent of Angiotensin II and Adrenocorticotropin. Journal of Clinical Endocrinology and Metabolism. 1982;54(2):463–469. [PubMed: 6274907]
- 19.
- Missale C, Liberini P, Memo M, Carruba MO, Spano P. Characterization of dopamine receptors associated with aldosterone secretion in rat adrenal glomerulosa. Endocrinology. 1986;119(5):2227–2232. [PubMed: 3533523]
- 20.
- Chartier L, Schiffrin EL. Role of calcium in effects of atrial natriuretic peptide on aldosterone production in adrenal glomerulosa cells. American Journal of Physiology - Endocrinology and Metabolism. 1987;252(4 (15/4)) [PubMed: 2436485] [CrossRef]
- 21.
- Jorgensen PL. Structure, function and regulation of Na,K-ATPase in the kidney. Kidney International. 1986;29(1):10–20. [PubMed: 2421041]
- 22.
- Oguchi A, Ikeda U, Kanbe T, Tsuruya Y, Yamamoto K, Kawakami K, Medford RM, Shimada K. Regulation of Na-K-ATPase gene expression by aldosterone in vascular smooth muscle cells. American Journal of Physiology - Heart and Circulatory Physiology 1993;265(4 34-4). doi: 10.1152/ajpheart.1993.265.4.h1167. [PubMed: 8238401] [CrossRef]
- 23.
- Pearce D. SGK1 regulation of epithelial sodium transport. Cellular Physiology and Biochemistry. 2003;13(1):13–20. [PubMed: 12649598]
- 24.
- Bhalla V, Daidié D, Li H, Pao AC, LaGrange LP, Wang J, Vandewalle A, Stockand JD, Staub O, Pearce D. Serum- and glucocorticoid-regulated kinase 1 regulates ubiquitin ligase neural precursor cell-expressed, developmentally down-regulated protein 4-2 by inducing interaction with 14-3-3. Molecular Endocrinology. 2005;19(12):3073–3084. [PubMed: 16099816]
- 25.
- Kornel L, Smoszna-Konaszewska B. Aldosterone (ALDO) increases transmembrane influx of Na+ in vascular smooth muscle (VSM) cells through increased synthesis of Na+ channels. Steroids. 1995;60(1):114–119. [PubMed: 7792795]
- 26.
- Mick VE, Itani OA, Loftus RW, Husted RF, Schmidt TJ, Thomas CP. The α-Subunit of the Epithelial Sodium Channel Is an Aldosterone-Induced Transcript in Mammalian Collecting Ducts, and This Transcriptional Response Is Mediated via Distinct cis -Elements in the 5′-Flanking Region of the Gene . Molecular Endocrinology. 2001;15(4):575–588. [PubMed: 11266509]
- 27.
- Yoo D, Kim BY, Campo C, Nance L, King A, Maouyo D, Welling PA. Cell surface expression of the ROMK (Kir 1.1) channel is regulated by the aldosterone-induced kinase, SGK-1, and protein kinase A. Journal of Biological Chemistry. 2003;278(25):23066–23075. [PubMed: 12684516]
- 28.
- Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD. GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension. 2011;57(3):442–451. [PMC free article: PMC6626623] [PubMed: 21242460]
- 29.
- Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. American Journal of Physiology - Cell Physiology. 2013;304(6) [PubMed: 23283935] [CrossRef]
- 30.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: The second decade. Cell. 1995;83(6):835–839. [PMC free article: PMC6159888] [PubMed: 8521507]
- 31.
- Hellal-Levy C, Fagart J, Souque A, Rafestin-Oblin ME. Mechanistic aspects of mineralocorticoid receptor activation. In: Kidney International.Vol 57. Blackwell Publishing Inc.; 2000:1250–1255. [PubMed: 10760050]
- 32.
- Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: Structural and functional kinship with the glucocorticoid receptor. Science. 1987;237(4812):268–275. [PubMed: 3037703]
- 33.
- Fan YS, Eddy RL, Byers MG, Haley LL, Henry WM, Novvak NJ, Shows TB. The human mineralocorticoid receptor gene (MLR) is located on chromosome 4 at q31.2. Cytogenetic and Genome Research. 1989;52(1–2):83–84. [PubMed: 2558856]
- 34.
- Morrison N, Harrap SB, Arriza JL, Boyd E, Connor JM. Regional chromosomal assignment of the human mineralocorticoid receptor gene to 4q31.1. Human Genetics. 1990;85(1):130–132. [PubMed: 2162806]
- 35.
- Zennaro MC, Keightley MC, Kotelevtsev Y, Conway GS, Soubrier F, Fuller PJ. Human mineralocorticoid receptor genomic structure and identification of expressed isoforms. Journal of Biological Chemistry. 1995;270(36):21016–21020. [PubMed: 7673127]
- 36.
- Zennaro MC, Farman N, Bonvalet JP, Lombès M. Tissue-specific expression of α and β messenger ribonucleic acid isoforms of the human mineralocorticoid receptor in normal and pathological states. Journal of Clinical Endocrinology and Metabolism. 1997;82(5):1345–1352. [PubMed: 9141514]
- 37.
- Arai K, Zachman K, Shibasaki T, Chrousos GP. Polymorphisms of Amiloride-Sensitive Sodium Channel Subunits in Five Sporadic Cases of Pseudohypoaldosteronism: Do They Have Pathologic Potential? 1 . The Journal of Clinical Endocrinology & Metabolism. 1999;84(7):2434–2437. [PubMed: 10404817]
- 38.
- Beato M, Sánchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocrine Reviews. 1996;17(6):587–609. [PubMed: 8969970]
- 39.
- Bamberger CM, Bamberger AM, Wald M, Chrousos GP, Schulte HM. Inhibition of mineralocorticoid activity by the β isoform of the human glucocorticoid receptor. Journal of Steroid Biochemistry and Molecular Biology. 1997;60(1–2):43–50. [PubMed: 9182857]
- 40.
- Arai K, Chrousos GP. Aldosterone Deficiency and Resistance. MDText.com, Inc.; 2000. Available at: http://www
.ncbi.nlm.nih .gov/pubmed/25905305. Accessed August 18, 2020. [PubMed: 25905305] - 41.
- Funder JW, Pearce PT, Myles K, Roy LP. Apparent mineralocorticoid excess, pseudohypoaldosteronism, and urinary electrolyte excretion: toward a redefinition of mineralocorticoid action. The FASEB Journal. 1990;4(14):3234–3238. [PubMed: 2172062]
- 42.
- Albiston AL, Obeyesekere VR, Smith RE, Krozowski ZS. Cloning and tissue distribution of the human 1 lβ-hydroxysteroid dehydrogenase type 2 enzyme. Molecular and Cellular Endocrinology. 1994;105(2) [PubMed: 7859916] [CrossRef]
- 43.
- Obeyesekere VR, Li KXZ, Ferrari P, Krozowski Z. Truncation of the N- and C-terminal regions of the human 11β-hydroxysteroid dehydrogenase type 2 enzyme and effects on solubility and bidirectional enzyme activity. Molecular and Cellular Endocrinology. 1997;131(2):173–182. [PubMed: 9296376]
- 44.
- Bujalska I, Shimojo M, Howie A, Stewart PM. Human 11β-hydroxysteroid dehydrogenase: Studies on the stably transfected isoforms and localization of the type 2 isozyme within renal tissue. In: Steroids.Vol 62. Elsevier Inc.; 1997:77–82. [PubMed: 9029719]
- 45.
- Murphy BEP. Specificity of human 11β-hydroxysteroid dehydrogenase. Journal of Steroid Biochemistry. 1981;14(8):807–809. [PubMed: 6946265]
- 46.
- Frey FJ. Kinetics and dynamics of prednisolone. Endocrine Reviews. 1987;8(4):453–473. [PubMed: 3319533]
- 47.
- Ferrari P, Smith RE, Funder JW, Krozowski ZS. Substrate and inhibitor specificity of the cloned human 11β-hydroxysteroid dehydrogenase type 2 isoform. American Journal of Physiology - Endocrinology and Metabolism. 1996;270(5) [PubMed: 8967481] [CrossRef]
- 48.
- Souness GW, Morris DJ. The antinatriuretic and kaliuretic effects of the glucocorticoids corticosterone and cortisol following pretreatment with carbenoxolone sodium (a liquorice derivative) in the adrenalectomized rat. Endocrinology. 1989;124(3):1588–1590. [PubMed: 2917528]
- 49.
- Canessa CM, Horisberger JD, Rossier BC. Epithelial sodium channel related to proteins involved in neurodegeneration. Nature. 1993;361(6411):467–470. [PubMed: 8381523]
- 50.
- Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367(6462):463–467. [PubMed: 8107805]
- 51.
- Voilley N, Lingueglia E, Champigny G, Mattéi MG, Waldmann R, Lazdunski M, Barbry P. The lung amiloride-sensitive Na+ channel: Biophysical properties, pharmacology, ontogenesis, and molecular cloning. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(1):247–251. [PMC free article: PMC42924] [PubMed: 8278374]
- 52.
- McDonald FJ, Price MP, Snyder PM, Welsh MJ. Cloning and expression of the β- and γ-subunits of the human epithelial sodium channel. American Journal of Physiology - Cell Physiology 1995;268(5 37-5). doi: 10.1152/ajpcell.1995.268.5.c1157. [PubMed: 7762608] [CrossRef]
- 53.
- Snyder PM. Minireview: Regulation of epithelial Na+ channel trafficking. Endocrinology. 2005;146(12):5079–5085. [PubMed: 16150899]
- 54.
- Masilamani S, Kim GH, Mitchell C, Wade JB, Knepper MA. Aldosterone,mediated regulation of ENaC α, β, and γ subunit proteins in rat kidney. Journal of Clinical Investigation. 1999;104(7) [PMC free article: PMC408561] [PubMed: 10510339] [CrossRef]
- 55.
- Snyder PM, Steines JC, Olson DR. Relative Contribution of Nedd4 and Nedd4-2 to ENaC Regulation in Epithelia Determined by RNA Interference. Journal of Biological Chemistry. 2004;279(6):5042–5046. [PubMed: 14645220]
- 56.
- Rotin D, Bar-Sagi D, O’Brodovich H, Merilainen J, Lehto VP, Canessa CM, Rossier BC, Downey GP. An SH3 binding region in the epithelial Na+ channel (alpha rENaC) mediates its localization at the apical membrane. The EMBO journal. 1994;13(19):4440–50. [PMC free article: PMC395375] [PubMed: 7925286]
- 57.
- Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Ulick S. Milora R v., Findling JW, Canessa CM, Rossier BC, Lifton RP. Liddle’s syndrome: Heritable human hypertension caused by mutations in the β subunit of the epithelial sodium channel. Cell. 1994;79(3):407–414. [PubMed: 7954808]
- 58.
- Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel γ subunit: Genetic heterogeneity of Liddle syndrome. Nature Genetics. 1995;11(1):76–82. [PubMed: 7550319]
- 59.
- Hansson JH, Schild L, Lu Y, Wilson TA, Gautschi I, Shimkets R, Nelson-Williams C, Rossier BC, Lifton RP. A de novo missense mutation of the β subunit of the epithelial sodium channel causes hypertension and Liddle syndrome, identifying a proline-rich segment critical for regulation of channel activity. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(25):11495–11499. [PMC free article: PMC40428] [PubMed: 8524790]
- 60.
- Tamura H, Schild L, Enomoto N, Matsui N, Marumo F, Rossier BC, Sasaki S. Liddle disease caused by a missense mutation of β subunit of the epithelial sodium channel gene. Journal of Clinical Investigation. 1996;97(7):1780–1784. [PMC free article: PMC507244] [PubMed: 8601645]
- 61.
- Schild L, Lu Y, Gautschi I, Schneeberger E, Lifton RP, Rossier BC. Identification of a PY motif in the epithelial Na channel subunits as a target sequence for mutations causing channel activation found in Liddle syndrome. EMBO Journal. 1996;15(10):2381–2387. [PMC free article: PMC450168] [PubMed: 8665845]
- 62.
- Chang SS, Grunder S, Hanukoglu A, Rösler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nature Genetics. 1996;12(3):248–253. [PubMed: 8589714]
- 63.
- McCormick JA, Bhalla V, Pao AC, Pearce D. SGK1: A rapid aldosterone-induced regulator of renal sodium reabsorption. Physiology. 2005;20(2):134–139. [PubMed: 15772302]
- 64.
- Muller OG, Parnova RG, Centeno G, Rossier BC, Firsov D, Horisberger JD. Mineralocorticoid effects in the kidney: Correlation between αENaC, GILZ, and Sgk-1 mRNA expression and urinary excretion of Na+ and K+. Journal of the American Society of Nephrology. 2003;14(5):1107–1115. [PubMed: 12707381]
- 65.
- Ziera T, Irlbacher H, Fromm A, Latouche C, Krug SM, Fromm M, Jaisser F, Borden SA. Cnksr3 is a direct mineralocorticoid receptor target gene and plays a key role in the regulation of the epithelial sodium channel. The FASEB Journal. 2009;23(11):3936–3946. [PubMed: 19567370]
- 66.
- Booth RE, Stockand JD. Targeted degradation of ENaC in response to PKC activation of the ERK1/2 cascade. American Journal of Physiology - Renal Physiology 2003;284(5 53-5). doi: 10.1152/ajprenal.00373.2002. [PubMed: 12540365] [CrossRef]
- 67.
- Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K + homeostasis. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(10):4025–4029. [PMC free article: PMC1803763] [PubMed: 17360471]
- 68.
- Valinsky WC, Touyz RM, Shrier A. Aldosterone, SGK1, and ion channels in the kidney. Clinical Science. 2018;132(2):173–183. [PMC free article: PMC5817097] [PubMed: 29352074]
- 69.
- Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Münster C, Chraïbi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na+ channel cell surface expression. EMBO Journal. 2001;20(24):7052–7059. [PMC free article: PMC125341] [PubMed: 11742982]
- 70.
- Arteaga MF, Wang L, Ravid T, Hochstrasser M, Canessa CM. An amphipathic helix targets serum and glucocorticoid-induced kinase 1 to the endoplasmic reticulum-associated ubiquitin-conjugation machinery. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(30):11178–11183. [PMC free article: PMC1544061] [PubMed: 16847254]
- 71.
- Robert-Nicoud M, Flahaut M, Elalouf JM, Nicod M, Salinas M, Bens M, Doucet A, Wincker P, Artiguenave F, Horisberger JD, Vandewalle A, Rossier BC, Firsov D. Transcriptome of a mouse kidney cortical collecting duct cell line: Effects of aldosterone and vasopressin. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2712–2716. [PMC free article: PMC30204] [PubMed: 11226305]
- 72.
- Soundararajan R, Melters D, Shih IC, Wang J, Pearce D. Epithelial sodium channel regulated by differential composition of a signaling complex. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):7804–7809. [PMC free article: PMC2683131] [PubMed: 19380724]
- 73.
- Soundararajan R, Wang J, Melters D, Pearce D. Glucocorticoid-induced leucine zipper 1 stimulates the epithelial sodium channel by regulating serum- and glucocorticoid-induced kinase 1 stability and subcellular localization. Journal of Biological Chemistry. 2010;285(51):39905–39913. [PMC free article: PMC3000972] [PubMed: 20947508]
- 74.
- Loffing J, Zecevic M, Féraille E, Kaissling B, Asher C, Rossier BC, Firestone GL, Pearce D, Verrey F. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: Possible role of SGK. American Journal of Physiology - Renal Physiology 2001;280(4 49-4). doi: 10.1152/ajprenal.2001.280.4.f675. [PubMed: 11249859] [CrossRef]
- 75.
- Soundararajan R, Pearce D, Ziera T. The role of the ENaC-regulatory complex in aldosterone-mediated sodium transport. Molecular and Cellular Endocrinology. 2012;350(2):242–247. [PMC free article: PMC3270213] [PubMed: 22101317]
- 76.
- Zhang W, Xia X, Jalal DI, Kuncewicz T, Xu W, Lesage GD, Kone BC. Aldosterone-sensitive repression of ENaCα transcription by a histone H3 lysine-79 methyltransferase. American Journal of Physiology - Cell Physiology. 2006;290(3) [PMC free article: PMC3009459] [PubMed: 16236820] [CrossRef]
- 77.
- Zhang W, Xia X, Reisenauer MR, Hemenway CS, Kone BC. Dot1a-AF9 complex mediates histone H3 Lys-79 hypermethylation and repression of ENaCα in an aldosterone-sensitive manner. Journal of Biological Chemistry. 2006;281(26):18059–18068. [PMC free article: PMC3015183] [PubMed: 16636056]
- 78.
- Reisenauer MR, Anderson M, Huang L, Zhang Z, Zhou Q, Kone BC, Morris AP, LeSage GD, Dryer SE, Zhang W. AF17 competes with AF9 for binding to Dot1a to up-regulate transcription of epithelial Na+ channel α. Journal of Biological Chemistry. 2009;284(51):35659–35669. [PMC free article: PMC2790997] [PubMed: 19864429]
- 79.
- Zhang W, Xia X, Reisenauer MR, Rieg T, Lang F, Kuhl D, Vallon V, Kone BC. Aldosterone-induced Sgk1 relieves Dot1a-Af9-mediated transcriptional repression of epithelial Na+ channel α. Journal of Clinical Investigation. 2007;117(3):773–783. [PMC free article: PMC1804379] [PubMed: 17332896]
- 80.
- Arai K, Zachman K, Shibasaki T, Chrousos GP. Polymorphisms of Amiloride-Sensitive Sodium Channel Subunits in Five Sporadic Cases of Pseudohypoaldosteronism: Do They Have Pathologic Potential? 1 . The Journal of Clinical Endocrinology & Metabolism. 1999;84(7):2434–2437. [PubMed: 10404817]
- 81.
- Kowarski A, Katz H, Mlgeon CJ. Plasma aldosterone concentration in normal subjects from infancy to adulthood. Journal of Clinical Endocrinology and Metabolism. 1974;38(3):489–491. [PubMed: 4815176]
- 82.
- van Acker KJ, Scharpe SL, Deprettere AJR, Neels HM. Renin-angiotensin-aldosterone system in the healthy infant and child. Kidney International. 1979;16(2):196–203. [PubMed: 513506]
- 83.
- Laetitia M, Eric P, Laurence FLH, Francois P, Claudine C, Pascal B, Lombès M. Physiological partial aldosterone resistance in human newborns. Pediatric Research. 2009;66(3):323–328. [PMC free article: PMC2919537] [PubMed: 19542911]
- 84.
- Bizzarri C, Pedicelli S, Cappa M, Cianfarani S. Water balance and “salt wasting” in the first year of life: The role of aldosterone-signaling defects. Hormone Research in Paediatrics. 2016;86(3):143–153. [PubMed: 27598420]
- 85.
- Coulter CL, Jaffe RB. Functional maturation of the primate fetal adrenal in vivo: 3. Specific zonal localization and developmental regulation of CYP21A2 (P450c21) and CYP11B1/CYP11B2 (P450c11/aldosterone synthase) lead to integrated concept of zonal and temporal steroid biosynthesis. Endocrinology. 1998;139(12):5144–5150. [PubMed: 9832454]
- 86.
- Martinerie L, Viengchareun S, Delezoide AL, Jaubert F, Sinico M, Prevot S, Boileau P, Meduri G, Lombes M. Low renal mineralocorticoid receptor expression at birth contributes to partial aldosterone resistance in neonates. Endocrinology. 2009;150(9):4414–4424. [PMC free article: PMC3201843] [PubMed: 19477942]
- 87.
- LANDAU RL. LUGIBIHL K. Inhibition of the sodium-retaining influence of aldosterone by progesterone. The Journal of clinical endocrinology and metabolism. 1958;18(11):1237–1245. [PubMed: 13587641]
- 88.
- HUDSON JB. CHOBANIAN A v., RELMAN AS. Hypoaldosteronism; a clinical study of a patient with an isolated adrenal mineralocorticoid deficiency, resulting in hyperkalemia and Stokes-Adams attacks. The New England journal of medicine. 1957;257(12):529–536. [PubMed: 13464977]
- 89.
- Schambelan M, Stockigt JR, Biglieri EG. Isolated Hypoaldosteronism in Adults: A Renin-Deficiency Syndrome. New England Journal of Medicine. 1972;287(12):573–578. [PubMed: 4341275]
- 90.
- Perez G, Siegel L, Schreiner GE. Selective hypoaldosteronism with hyperkalemia. Annals of internal medicine. 1972;76(5):757–763. [PubMed: 4337136]
- 91.
- Sebastian A, Schambelan M, Lindenfeild S, Morris RC. Amelioration of Metabolic Acidosis with Fludrocortisone Therapy in Hyporeninemic Hypoaldosteronism. New England Journal of Medicine. 1977;297(11):576–583. [PubMed: 18672]
- 92.
- Perez GO, Lespier L, Jacobi J, Oster JR, Katz FH, Vaamonde CA, Fishman LM. Hyporeninemia and Hypoaldosteronism in Diabetes Mellitus. Archives of Internal Medicine. 1977;137(7):852–855. [PubMed: 879919]
- 93.
- Kalin MF, Poretsky L, Seres DS, Zumoff B. Hyporeninemic hypoaldosteronism associated with acquired immune deficiency syndrome. The American Journal of Medicine. 1987;82(5):1035–1038. [PubMed: 3555065]
- 94.
- Onozaki A, Katoh T, Watanabe T. Hyporeninemic hypoaldosteronism associated with Sjogren’s syndrome. American Journal of Medicine. 2002;112(3):245–246. [4] [PubMed: 11893359]
- 95.
- DeFronzo RA. Hyperkalemia and hyporeninemic hypoaldosteronism. Kidney International. 1980;17(1):118–134. [PubMed: 6990088]
- 96.
- Nadler JL, Lee FO, Hsueh W, Horton R. Evidence of Prostacyclin Deficiency in the Syndrome of Hyporeninemic Hypoaldosteronism. New England Journal of Medicine. 1986;314(16):1015–1020. [PubMed: 3515183]
- 97.
- Tuck ML, Sambhi MP, Levin L. Hyporeninemic hypoaldosteronism in diabetes mellitus. Studies of the autonomic nervous system’s control of renin release. Diabetes. 1979;28(3):237–241. [PubMed: 446908]
- 98.
- Perez GO, Lespier LE, Oster JR, Vaamonde CA. Effect of alterations of sodium intake in patients with hyporeninemic hypoaldosteronism. Nephron. 1977;18(5):259–265. [PubMed: 865654]
- 99.
- Escarce JJ. Hyporeninemic Hypoaldosteronism in a Patient With Cirrhosis and Ascites. Archives of Internal Medicine. 1986;146(12):2407–2408. [PubMed: 3535721]
- 100.
- Nakamoto Y, Imai H, Hamanaka S, Yoshida K, Akihama T, Miura AB. IgM monoclonal gammopathy accompanied by nodular glomerulosclerosis, urine-concentrating defect, and hyporeninemic hypoaldosteronism. American Journal of Nephrology. 1985;5(1):53–58. [PubMed: 3918448]
- 101.
- Yoshino M, Amerian R, Brautbar N. Hyporeninemic hypoaldosteronism in sickle cell disease. Nephron. 1982;31(3):242–244. [PubMed: 6750423]
- 102.
- Kiley J, Zager P. Hyporeninemic Hypoaldosteronism in Two Patients With Systemic Lupus Erythematosus. American Journal of Kidney Diseases. 1984;4(1):39–43. [PubMed: 6377883]
- 103.
- Masud T, Winocour P, Clarke F. Reversible hyporeninaemic hypoaldosteronism and life-threatening cardiac dysrhythmias: The interaction of non-steroidal anti-inflammatory drugs and autonomic dysfunction. Postgraduate Medical Journal. 1993;69(813):593–594. [PMC free article: PMC2399870] [PubMed: 8415355]
- 104.
- Motoo Y, Sawabu N, Takemori Y, Ohta H, Okai T, Ikeda K, Yokoyama H. Long-Term Follow-Up of Mitomycin C Nephropathy. Internal Medicine. 1994;33(3):180–184. [PubMed: 8061399]
- 105.
- Sunderlin FF, Anderson GH, Streeten DHP, Blumenthal SA. The renin-angiotensin-aldosterone system in diabetic patients with hyperkalemia. Diabetes. 1981;30(4):335–340. [PubMed: 7009278]
- 106.
- Deleiva A, Christlieb AR, Melby JC, Graham CA, Day RP, Luetscher JA, Zager PG. Big Renin and Biosynthetic Defect of Aldosterone in Diabetes Mellitus. New England Journal of Medicine. 1976;295(12):639–643. [PubMed: 184384]
- 107.
- FitzGerald GA, Hossmann V, Hummerich W, Konrads A. The renin - kallikrein - prostaglandin system: Plasma active and inactive renin and urinary kallikrein during prostacyclin infusion in man. Prostaglandines and Medicine. 1980;5(6):445–456. [PubMed: 7008062]
- 108.
- Ulick S, Wang JZ, Morton DH. The biochemical phenotypes of two inborn errors in the biosynthesis of aldosterone. Journal of Clinical Endocrinology and Metabolism. 1992;74(6):1415–1420. [PubMed: 1592889]
- 109.
- Ulick S. Correction of the nomenclature and mechanism of the aldosterone biosynthetic defects. The Journal of Clinical Endocrinology & Metabolism. 1996;81(3):1299–1300. [PubMed: 8772616]
- 110.
- Mitsuuchi Y, Kawamoto T, Miyahara K, Ulick S, Morton DH, Naiki Y, Kuribayashi I, Toda K, Hara T, Orii T, Yasuda K, Miura K, Yamamoto Y, Imura H, Shizuta Y. Congenitally defective aldosterone biosynthesis in humans: Inactivation of the P450C18 gene (CYP11B2) due to nucleotide deletion in CMO I deficient patients. Biochemical and Biophysical Research Communications. 1993;190(3):864–869. [PubMed: 8439335]
- 111.
- Shizuta Y, Kawamoto T, Mitsuuchi Y, Miyahara K, Rösler A, Ulick S, Imura H. Inborn errors of aldosterone biosynthesis in humans. Steroids. 1995;60(1):15–21. [PubMed: 7792802]
- 112.
- Geley S, Jöhrer K, Peter M, Denner K, Bernhardt R, Sippell WG, Kofler R. Amino acid substitution R384P in aldosterone synthase causes corticosterone methyloxidase type I deficiency. Journal of Clinical Endocrinology and Metabolism. 1995;80(2):424–429. [PubMed: 7852500]
- 113.
- Pascoe L, Curnow KM, Slutsker L, Rosler A, White PC. Mutations in the human CYP11B2 (aldosterone synthase) gene causing corticosterone methyloxidase II deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(11):4996–5000. [PMC free article: PMC49215] [PubMed: 1594605]
- 114.
- Zhang G, Rodriguez H, Fardella CE, Harris DA, Miller WL. Mutation T318M in the CYP11B2 gene encoding P450c11AS (aldosterone synthase) causes corticosterone methyl oxidase II deficiency. American Journal of Human Genetics. 1995;57(5):1037–1043. [PMC free article: PMC1801390] [PubMed: 7485152]
- 115.
- Kuribayashi I, Kuge H, Santa RJ, Mutlaq AZ, Yamasaki N, Furuno T, Takahashi A, Chida S, Nakamura T, Endo F, Doi Y, Onishi S, Shizuta Y. A missense mutation (GGC[435Gly]→AGC[ser]) in exon 8 of the CYP11B2 gene inherited in Japanese patients with congenital hypoaldosteronism. Hormone Research. 2003;60(5):255–260. [PubMed: 14614232]
- 116.
- Williams TA, Mulatero P, Bosio M, Lewicka S, Palermo M, Veglio F, Armanini D. A particular phenotype in a girl with aldosterone synthase deficiency. In: Journal of Clinical Endocrinology and Metabolism.Vol 89. J Clin Endocrinol Metab; 2004:3168–3172. [PubMed: 15240589]
- 117.
- Leshinsky-Silver E, Landau Z, Unlubay S, Bistrizer T, Zung A, Tenenbaum-Rakover Y, DeVries L, Lev D, Hanukoglu A. Congenital hyperreninemic hypoaldosteronism in Israel: Sequence analysis of CYP11B2 gene. Hormone Research. 2006;66(2):73–78. [PubMed: 16733366]
- 118.
- Nguyen HH, Hannemann F, Hartmann MF, Wudy SA, Bernhardt R. Aldosterone synthase deficiency caused by a homozygous L451F mutation in the CYP11B2 gene. Molecular Genetics and Metabolism. 2008;93(4):458–467. [PubMed: 18178501]
- 119.
- Løvås K, McFarlane I, Nguyen HH, Curran S, Schwabe J, Halsall D, Bernhardts R, Wallace AM, Chatterjee VKK. A novel CYP11b2 gene mutation in an asian family with aldosterone synthase deficiency. Journal of Clinical Endocrinology and Metabolism. 2009;94(3):914–919. [PubMed: 19116236]
- 120.
- White PC. Aldosterone synthase deficiency and related disorders. In: Molecular and Cellular Endocrinology.Vol 217. Mol Cell Endocrinol; 2004:81–87. [PubMed: 15134805]
- 121.
- Rösler A. The natural history of salt-wasting disorders of adrenal and renal origin. Journal of Clinical Endocrinology and Metabolism. 1984;59(4):689–700. [PubMed: 6384251]
- 122.
- Miao H, Yu Z, Lu L, Zhu H, Auchus RJ, Liu J, Jiang J, Pan H, Gong F, Chen S, Lu Z. Analysis of novel heterozygous mutations in the CYP11B2 gene causing congenital aldosterone synthase deficiency and literature review. Steroids. 2019;150 [PubMed: 31302112] [CrossRef]
- 123.
- Peter M, Partsch CJ, Sippell WG. Multisteroid analysis in children with terminal aldosterone biosynthesis defects. Journal of Clinical Endocrinology and Metabolism. 1995;80(5):1622–1627. [PubMed: 7745009]
- 124.
- Ulick S. Cortisol as mineralocorticoid. The Journal of Clinical Endocrinology & Metabolism. 1996;81(4):1307–1308. [PubMed: 8636321]
- 125.
- Klomchan T, Supornsilchai V, Wacharasindhu S, Shotelersuk V, Sahakitrungruang T. Novel CYP11B2 mutation causing aldosterone synthase (P450c11AS) deficiency. European Journal of Pediatrics. 2012;171(10):1559–1562. [PubMed: 22801770]
- 126.
- Kondo E, Nakamura A, Homma K, Hasegawa T, Yamaguchi T, Narugami M, Hattori T, Aoyagi H, Ishizu K, Tajima T. Two novel mutations of the CYP11B2 gene in a Japanese patient with aldosterone deficiency type 1. Endocrine Journal. 2013;60(1):51–55. [PubMed: 23018980]
- 127.
- Hui E, Yeung MCW, Cheung PT, Kwan E, Low L, Tan KCB, Lam KSL, Chan AOK. The clinical significance of aldosterone synthase deficiency: Report of a novel mutation in the CYP11B2 gene. BMC Endocrine Disorders. 2014;14 [PMC free article: PMC3976226] [PubMed: 24694176] [CrossRef]
- 128.
- Portrat-Doyen S, Tourniaire J, Richard O, Mulatero P, Aupetit-Faisant B, Curnow KM, Pascoe L, Morel Y. Isolated Aldosterone Synthase Deficiency Caused by Simultaneous E198D and V386A Mutations in the CYP11B2 Gene 1 . The Journal of Clinical Endocrinology & Metabolism. 1998;83(11):4156–4161. [PubMed: 9814506]
- 129.
- Jessen CL, Christensen JH, Birkebæk NH, Rittig S. Homozygosity for a mutation in the CYP11B2 gene in an infant with congenital corticosterone methyl oxidase deficiency type II. Acta Paediatrica. International Journal of Paediatrics. 2012;101(11) [PubMed: 22931312] [CrossRef]
- 130.
- O’Kelly R, Magee F, McKenna J. Routine heparin therapy inhibits adrenal aldosterone production. Journal of Clinical Endocrinology and Metabolism. 1983;56(1):173–176. [PubMed: 6336617]
- 131.
- Sequeira SJ, McKenna TJ. Chlorbutol, a new inhibitor of aldosterone biosynthesis identified during examination of heparin effect on aldosterone production. Journal of Clinical Endocrinology and Metabolism. 1986;63(3):780–784. [PubMed: 3734043]
- 132.
- Zipser RD, Davenport MW, Martin KL, Tuck ML, Warner NE, Swinney RR, Davis CL, Horton R. Hyperreninemic hypoaldosteronism in the critically 111: A new entity. Journal of Clinical Endocrinology and Metabolism. 1981;53(4):867–873. [PubMed: 6270176]
- 133.
- Davenport MW, Zipser RD. Association of hypotension with hyperreninemic hypoaldosteronism in the critically ill patient. Archives of internal medicine. 1983;143(4):735–7. [PubMed: 6301395]
- 134.
- Cheek DB, Perry JW. A salt wasting syndrome in infancy. Archives of Disease in Childhood. 1958;33(169):252–256. [PMC free article: PMC2012226] [PubMed: 13545877]
- 135.
- Speiser PW, Stoner E, New MI. Pseudohypoaldosteronism: a review and report of two new cases. Advances in experimental medicine and biology. 1986;196:173–195. [PubMed: 3012977]
- 136.
- Zennaro MC, Fernandes-Rosa F. Mineralocorticoid receptor mutations. Journal of Endocrinology. 2017;234(1):T93–T106. [PubMed: 28348114]
- 137.
- Zennaro MC, Lombès M. Mineralocorticoid resistance. Trends in Endocrinology and Metabolism. 2004;15(6):264–270. [PubMed: 15358279]
- 138.
- Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the γ subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nature Genetics. 1996;13(2):248–250. [PubMed: 8640238]
- 139.
- Geller DS, Rodriguez-Soriano J, Vallo Boado A, Schifter S, Bayer M, Chang SS, Lifton RP. Mutations in the mineralocorticoid receptor gene cause autosomal dominant pseudohypoaldosteronism type I. Nature Genetics. 1998;19(3):279–281. [PubMed: 9662404]
- 140.
- Tajima T, Kitagawa H, Yokoya S, Tachibana K, Adachi M, Nakae J, Suwa S, Katoh S, Fujieda K. A novel missense mutation of mineralocorticoid receptor gene in one Japanese family with a renal form of pseudohypoaldosteronism type 1. Journal of Clinical Endocrinology and Metabolism. 2000;85(12):4690–4694. [PubMed: 11134129]
- 141.
- Arai K, Tsigos C, Suzuki Y, Irony I, Karl M, Listwak S, Chrousos GP. Physiological and molecular aspects of mineralocorticoid receptor action in pseudohypoaldosteronism: a responsiveness test and therapy. The Journal of Clinical Endocrinology & Metabolism. 1994;79(4):1019–1023. [PubMed: 7962269]
- 142.
- Zennaro MC, Borensztein P, Jeunemaitre X, Armanini D, Soubrier F. No alteration in the primary structure of the mineralocorticoid receptor in a family with pseudohypoaldosteronism. The Journal of Clinical Endocrinology & Metabolism. 1994;79(1):32–38. [PubMed: 8027248]
- 143.
- Komesaroff PA, Verity K, Fuller PJ. Pseudohypoaldosteronism: molecular characterization of the mineralocorticoid receptor. The Journal of Clinical Endocrinology & Metabolism. 1994;79(1):27–31. [PubMed: 8027241]
- 144.
- Arai K, Tsigos C, Suzuki Y, Listwak S, Zachman K, Zangeneh F, Rapaport R, Chanoine JP, Chrousos GP. No apparent mineralocorticoid receptor defect in a series of sporadic cases of pseudohypoaldosteronism. Journal of Clinical Endocrinology and Metabolism. 1995;80(3):814–817. [PubMed: 7883835]
- 145.
- Viemann M, Peter M, López-Siguero JP, Simic-Schleicher G, Sippell WG. Evidence for genetic heterogeneity of pseudohypoaldosteronism type 1: Identification of a novel mutation in the human mineralocorticoid receptor in one sporadic case and no mutations in two autosomal dominant kindreds. Journal of Clinical Endocrinology and Metabolism. 2001;86(5):2056–2059. [PubMed: 11344206]
- 146.
- Oberfield SE, Levine LS, Carey RM, Bejar R, New MI. Pseudohypoaldosteronism: multiple target organ unresponsiveness to mineralocorticoid hormones. The Journal of clinical endocrinology and metabolism. 1979;48(2):228–34. [PubMed: 218983]
- 147.
- Hanukoglu A, Omana J, Steinitz M, Rosler A, Hanukoglu I. Pseudohypoaldosteronism due to renal and multisystem resistance to mineralocorticoids respond differently to carbenoxolone. Journal of Steroid Biochemistry and Molecular Biology. 1997;60(1–2):105–112. [PubMed: 9182864]
- 148.
- Postel-Vinay MC, Alberti GM, Ricour C, Limal JM, Rappaport R, Royer P. Pseudohypoaldosteronism: Persistence of hyperaldosteronism and evidence for renal tubular and intestinal responsiveness to endogenous aldosterone. Journal of Clinical Endocrinology and Metabolism. 1974;39(6):1038–1044. [PubMed: 4372243]
- 149.
- Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: Phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579(2):95–132. [PMC free article: PMC4756657] [PubMed: 26772908]
- 150.
- Dirlewanger M, Huser D, Zennaro MC, Girardin E, Schild L, Schwitzgebel VM. A homozygous missense mutation in SCNN1A is responsible for a transient neonatal form of pseudohypoaldosteronism type 1. American Journal of Physiology - Endocrinology and Metabolism. 2011;301(3) [PubMed: 21653223] [CrossRef]
- 151.
- Schaedel C, Marthinsen L, Kristoffersson AC, Kornfält R, Nilsson KO, Orlenius B, Holmberg L. Lung symptoms in pseudohypoaldosteronism type 1 are associated with deficiency of the α-subunit of the epithelial sodium channel. Journal of Pediatrics. 1999;135(6):739–745. [PubMed: 10586178]
- 152.
- Wang J, Yu T, Yin L, Li J, Yu L, Shen Y, Yu Y, Shen Y, Fu Q. Novel Mutations in the SCNN1A Gene Causing Pseudohypoaldosteronism Type 1. PLoS ONE. 2013;8(6) [PMC free article: PMC3675083] [PubMed: 23762408] [CrossRef]
- 153.
- Edelheit O, Hanukoglu I, Gizewska M, Kandemir N, Tenenbaum-Rakover Y, Yurdakök M, Zajaczek S, Hanukoglu A. Novel mutations in epithelial sodium channel (ENaC) subunit genes and phenotypic expression of multisystem pseudohypoaldosteronism. Clinical Endocrinology. 2005;62(5):547–553. [PubMed: 15853823]
- 154.
- Gründer S, Firsov D, Chang SS, Jaeger NF, Gautschi I, Schild L, Lifton RP, Rossier BC. A mutation causing pseudohypoaldosteronism type 1 identifies a conserved glycine that is involved in the gating of the epithelial sodium channel. EMBO Journal. 1997;16(5):899–907. [PMC free article: PMC1169690] [PubMed: 9118951]
- 155.
- Edelheit O, Hanukoglu I, Shriki Y, Tfilin M, Dascal N, Gillis D, Hanukoglu A. Truncated beta epithelial sodium channel (ENaC) subunits responsible for multi-system pseudohypoaldosteronism support partial activity of ENaC. Journal of Steroid Biochemistry and Molecular Biology. 2010;119(1–2):84–88. [PubMed: 20064610]
- 156.
- Hanukoglu A, Edelheit O, Shriki Y, Gizewska M, Dascal N, Hanukoglu I. Renin-aldosterone response, urinary Na/K ratio and growth in pseudohypoaldosteronism patients with mutations in epithelial sodium channel (ENaC) subunit genes. Journal of Steroid Biochemistry and Molecular Biology. 2008;111(3–5):268–274. [PubMed: 18634878]
- 157.
- Saxena A, Hanukoglu I, Saxena D, Thompson RJ, Mark Gardiner R, Hanukoglu A. Novel mutations responsible for autosomal recessive multisystem pseudohypoaldosteronism and sequence variants in epithelial sodium channel α-, β-, and γ-subunit genes. Journal of Clinical Endocrinology and Metabolism. 2002;87(7):3344–3350. [PubMed: 12107247]
- 158.
- Adachi M, Tachibana K, Asakura Y, Abe S, Nakae J, Tajima T, Fujieda K. Compound Heterozygous Mutations in the γ Subunit Gene of ENaC (1627delG and 1570-1G→A) in One Sporadic Japanese Patient with a Systemic Form of Pseudohypoaldosteronism Type 1. The Journal of Clinical Endocrinology & Metabolism. 2001;86(1):9–12. [PubMed: 11231969]
- 159.
- Nobel YR, Lodish MB, Raygada M, del Rivero J, Faucz FR, Abraham SB, Lyssikatos C, Belyavskaya E, Stratakis CA, Zilbermint M. Pseudohypoaldosteronism type 1 due to novel variants of SCNN1B gene. Endocrinology, Diabetes and Metabolism Case Reports. 2016;2016 [PMC free article: PMC4722246] [PubMed: 26807262] [CrossRef]
- 160.
- Riepe FG, Krone N, Morlot M, Ludwig M, Sippell WG, Partsch CJ. Identification of a novel mutation in the human mineralocorticoid receptor gene in a german family with autosomal-dominant pseudohypoaldosteronism type 1: Further evidence for marked interindividual clinical heterogeneity. Journal of Clinical Endocrinology and Metabolism. 2003;88(4):1683–1686. [PubMed: 12679457]
- 161.
- Riepe FG, Krone N, Morlot M, Peter M, Sippell WG, Partsch CJ. Autosomal-Dominant Pseudohypoaldosteronism Type 1 in a Turkish Family Is Associated with a Novel Nonsense Mutation in the Human Mineralocorticoid Receptor Gene. Journal of Clinical Endocrinology and Metabolism. 2004;89(5):2150–2152. [PubMed: 15126534]
- 162.
- Nyström AM, Bondeson ML, Skanke N, Mårtensson J, Strömberg B, Gustafsson J, Annerén G. A Novel Nonsense Mutation of the Mineralocorticoid Receptor Gene in a Swedish Family with Pseudohypoaldosteronism Type I (PHA1). Journal of Clinical Endocrinology and Metabolism. 2004;89(1):227–231. [PubMed: 14715854]
- 163.
- Kanda K, Nozu K, Yokoyama N, Morioka I, Miwa A, Hashimura Y, Kaito H, Iijima K, Matsuo M. Autosomal dominant pseudohypoaldosteronism type 1 with a novel splice site mutation in MR gene. BMC Nephrology. 2009;10(1) [PMC free article: PMC2779785] [PubMed: 19912655] [CrossRef]
- 164.
- Sartorato P, Lapeyraque AL, Armanini D, Kuhnle U, Khaldi Y, Salomon R, Abadie V, di Battista E, Naselli A, Racine A, Bosio M, Caprio M, Poulet-Young V, Chabrolle JP, Niaudet P, de Gennes C, Lecornec MH, Poisson E, Fusco AM, Loli P, Lombès M, Zennaro MC. Different inactivating mutations of the mineralocorticoid receptor in fourteen families affected by type I pseudohypoaldosteronism. Journal of Clinical Endocrinology and Metabolism. 2003;88(6):2508–2517. [PubMed: 12788847]
- 165.
- Riepe FG, Finkeldei J, de Sanctis L, Einaudi S, Testa A, Karges B, Peter M, Viemann M, Grötzinger J, Sippell WG, Fejes-Toth G, Krone N. Elucidating the underlying molecular pathogenesis of NR3C2 mutants causing autosomal dominant pseudohypoaldosteronism type 1. Journal of Clinical Endocrinology and Metabolism. 2006;91(11):4552–4561. [PubMed: 16954160]
- 166.
- Hatta Y, Nakamura A, Hara S, Kamijo T, Iwata J, Hamajima T, Abe M, Okada M, Ushio M, Tsuyuki K, Tajima T. Clinical and molecular analysis of six Japanese patients with a renal form of pseudohypoaldosteronism type 1. Endocrine Journal. 2013;60(3):299–304. [PubMed: 23197115]
- 167.
- Morikawa S, Komatsu N, Sakata S, Nakamura-Utsunomiya A, Okada S, Tajima T. Two Japanese patients with the renal form of pseudohypoaldosteronism type 1 caused by mutations of NR3C2. Clinical Pediatric Endocrinology. 2015;24(3):135–138. [PMC free article: PMC4639533] [PubMed: 26594094]
- 168.
- Arai K, Nakagomi Y, Iketani M, Shimura Y, Amemiya S, Ohyama K, Shibasaki T. Functional polymorphisms in the mineralocorticoid receptor and amirolide-sensitive sodium channel genes in a patient with sporadic pseudohypoaldosteronism. Human Genetics. 2003;112(1):91–97. [PubMed: 12483305]
- 169.
- Warnock DG. Accessory factors and the regulation of epithelial sodium channel activity. Journal of Clinical Investigation. 1999;103(5):593. [PMC free article: PMC408133] [PubMed: 10074474]
- 170.
- Rodríguez-Soriano J, Vallo A, Oliveros R, Castillo G. Transient pseudohypoaldosteronism secondary to obstructive uropathy in infancy. The Journal of Pediatrics. 1983;103(3):375–380. [PubMed: 6350553]
- 171.
- Bizzarri C, Olivini N, Pedicelli S, Marini R, Giannone G, Cambiaso P, Cappa M. Congenital primary adrenal insufficiency and selective aldosterone defects presenting as salt-wasting in infancy: A single center 10-year experience. Italian Journal of Pediatrics. 2016;42(1) [PMC free article: PMC4971679] [PubMed: 27485500] [CrossRef]
- 172.
- Geller DS. Mineralocorticoid resistance. Clinical Endocrinology. 2005;62(5):513–520. [PubMed: 15853818]
- 173.
- Riepe FG. Clinical and Molecular Features of Type 1 Pseudohypoaldosteronism. Hormone Research. 2009;72(1):1–9. [PubMed: 19571553]
- 174.
- Bogdanović R, Stajić N, Putnik J, Paripović A. Transient type 1 pseudo-hypoaldosteronism: Report on an eight-patient series and literature review. Pediatric Nephrology. 2009;24(11):2167–2175. [PubMed: 19714368]
- 175.
- Adam WR. Hypothesis: A simple algorithm to distinguish between hypoaldosteronism and renal aldosterone resistance in patients with persistent hyperkalemia. Nephrology. 2008;13(6):459–464. [PubMed: 18518934]
- 176.
- Kuhnle U, Guariso G, Sonega M, Hinkel GK, Hubl W, Armanini D. Transient pseudohypoaldosteronism in obstructive renal disease with transient reduction of lymphocytic aldosterone receptors. Hormone Research in Paediatrics. 1993;39(3–4):152–155. [PubMed: 8262477]