Summary
The purpose of this overview is to increase the clinician's awareness of the clinical phenotypes, genetic causes, and management of tubulinopathies, a wide, overlapping range of brain malformations caused by pathogenic variants of genes encoding different isotypes of tubulin.
The following are the goals of this overview.
Goal 2.
Review the genetic causes of tubulinopathies.
Goal 4.
Provide an evaluation strategy to identify the genetic cause of a tubulinopathy in a proband (when possible).
Goal 5.
Review general medical management of tubulinopathies.
1. Clinical Characteristics of Tubulinopathies
Tubulinopathies (or tubulin-related cortical dysgenesis) comprise a wide and overlapping range of brain malformations as well as other clinical features caused by pathogenic variants in genes encoding different isotypes of tubulin [Romero et al 2018].
Brain Malformations
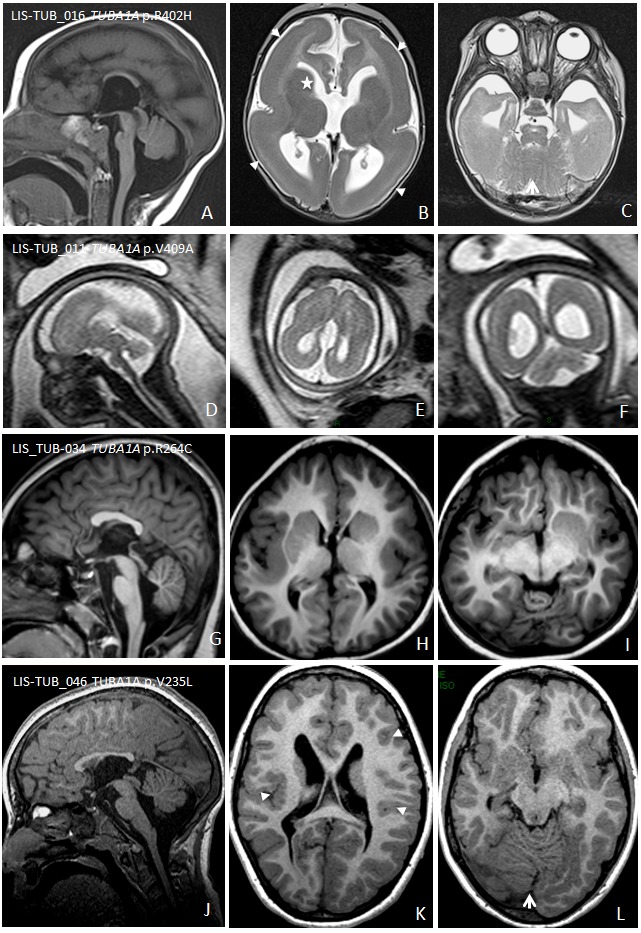
Lissencephaly ranges from a thickened cortex and complete absence of sulci (agyria) to a thickened cortex and a few shallow sulci (pachygyria) [Di Donato et al 2017].
Classic lissencephaly is characterized by marked thickening of the cortex with a posterior-to-anterior gradient of severity (i.e., more severe involvement posteriorly [parietal and occipital lobes] than anteriorly [orbitofrontal and anterior temporal regions]). Most often, cerebellar structure is normal and the basal ganglia appear normal except that the anterior limb of the internal capsule is usually not visible.
Lissencephaly with cerebellar hypoplasia. Some rare forms of lissencephaly are associated with a disproportionately small cerebellum.
Lissencephaly with agenesis of the corpus callosum. The corpus callosum in living individuals is commonly dysmorphic (missing rostrum plus flat genu and anterior body), hypoplastic, or partially absent. In contrast, all prenatally diagnosed cases show complete agenesis ( and ).
Centrally predominant pachygyria is characterized by pachygyria involving the insula and the frontal, temporal, and parietal opercula [
Bahi-Buisson et al 2008]. All have abnormalities of the basal ganglia that appear as large round structures in which the caudate, putamen, and globus pallidus are indistinguishable. Hypoplasia of the anterior limbs of the internal capsule is a major feature. Common findings are vermian hypoplasia and brain stem hypoplasia. The corpus callosum is commonly dysmorphic or hypoplastic; less frequently, it is partially or completely absent ().
Microlissencephaly refers to the most severe of the cortical dysplasias that combines extreme microcephaly and lissencephaly with hemispheres lacking primary fissures and olfactory sulci. Mainly diagnosed by prenatal MRI, findings at a median age of 25 weeks' gestation include microcephaly, absent-to-poorly operculized cortex, virtually no visible gyration, severe cerebellar hypoplasia, absent or severely hypoplastic basal ganglia, and usually complete agenesis of the corpus callosum. Associated malformations include pontocerebellar hypoplasia [Fallet-Bianco et al 2008, Lecourtois et al 2010, Fallet-Bianco et al 2014].
Dysgyria describes a cortex of normal thickness but with an abnormal gyral pattern characterized by abnormalities of sulcal depth or orientation: obliquely oriented sulci directed radially toward the center of the cerebrum and narrow gyri separated by abnormally deep or shallow sulci without imaging evidence of lissencephaly, pachygyria, cobblestone cortex, polymicrogyria, or other cortical abnormalities [Mutch et al 2016, Blumkin et al 2020]. This pattern tends to predominate in the perisylvian areas.
MRI reveals a "coarse" appearance with a thick cortex and irregular surfaces on both the pial and grey-white junction sides (, , ). Dysgyria can range in severity and distribution.
Note: Dysgyria was previously referred to as "simplified gyral pattern" or "polymicrogyria-like cortical dysplasia"; these terms are potentially confusing.
Basal ganglia, thalami, and corpus callosum. One of the key feature of tubulinopathies is dysmorphic basal ganglia. This pattern is usually asymmetric with bulbous appearance of the caudate and the thalami, with diffuse, branched or absent anterior limb of the internal capsule. The lateral ventricles have an irregular contour and abnormal rounding of the frontal horns likely related to the basal ganglia dysplasia.
The corpus callosum is variably affected, ranging from almost complete agenesis to normal.
Recognizable cerebellar and brain stem abnormalities. The most characteristic tubulinopathy-related cerebellar malformation is dysplasia of the superior cerebellum, especially the vermis (with "diagonal" folia – i.e., folia crossing the midline at an oblique angle). Less commonly, the vermis is hypoplastic with the anterior vermis more severely affected. The cerebellar hemispheres are either normal in size or mildly hypoplastic with mild asymmetry.
A large majority of affected individuals show brain stem hypoplasia that is usually asymmetric with a midline ventral indentation and asymmetric inferior and middle cerebellar peduncles [Oegema et al 2015].
Clinical Features of the Tubulinopathies
The clinical features of the tubulinopathies include motor and intellectual disabilities and epilepsy.
Motor and cognitive and impairments, present in almost all individuals with a tubulinopathy, correlate with the severity of brain malformations.
Lissencephaly, microlissencephaly, and generalized severe dysgyria: spastic tetraplegia and virtually no voluntary motor control and absent eye contact
Mild-to-moderate dysgyria: mild motor disability and intellectual disabilities
While most affected individuals have severe-to-profound intellectual disability, a minority have less extensive cortical malformations that result in only moderate intellectual disability, and a few have limited malformations that allow near-normal cognitive abilities. In the latter instance, the cortical malformation is typically less severe and less extensive on MRI.
Epilepsy varies significantly among affected individuals and is not necessarily determined by the severity of the cortical malformation, the gene involved, or the causative pathogenic variant [Romaniello et al 2019]. However:
Additional findings. Facial diplegia and strabismus suggestive of pseudobulbar palsy are often observed in central pachygyria and various forms of dysgyria [Bahi-Buisson et al 2008].
Prognosis. Individuals with the milder forms of tubulinopathies survive into adulthood, while those with the most severe forms may die at a young age as a result of complications such as seizures or pneumonia.
2. Genetic Causes of Tubulinopathies
The genetic causes of tubulinopathies and their associated complex cortical malformations are summarized in Table 1.
Table 1.
Tubulinopathies: Molecular Genetics and Complex Cortical Malformations
View in own window
AD = autosomal dominant; AR = autosomal recessive; CC = corpus callosum; CFEOM = congenital fibrosis of the extraocular muscles; CL = classic lissencephaly; MLIS = microlissencephaly; MOI = mode of inheritance; SGP = simplified gyral pattern
- 1.
Genes are in alphabetic order.
- 2.
- 3.
At the extreme severe end of the spectrum, only one fetus was reported with microlissencephaly and corpus callosum agenesis, severe brain stem and cerebellar hypoplasia, and dysmorphic basal ganglia [Poirier et al 2010] ().
3. Differential Diagnosis of Tubulinopathies
Tubulinopathies need to be distinguished clinically from other brain malformations that may resemble them (Table 2).
Table 2.
Differential Diagnosis of Tubulinopathies
View in own window
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; XL = X-linked
- 1.
Mutation of DYNC1H1 is associated with isolated polymicrogyria, nodular heterotopia, hypoplasia of the corpus callosum, abnormally shaped basal ganglia, and in some cases, evidence of peripheral neuropathy (see DYNC1H1-Related Disorders).
- 2.
As currently defined, Miller-Dieker syndrome is associated with deletions that include both PAFAH1B1 (LIS1) and YWHAE (a region of ~1.3 Mb harboring many genes) in 17p13.3 [Pilz et al 1998, Cardoso et al 2003].
- 3.
4. Evaluation Strategies to Identify the Genetic Cause of Tubulinopathy in a Proband
Establishing a specific genetic cause of a tubulinopathy:
Can aid in discussions of prognosis (which are beyond the scope of this GeneReview) and genetic counseling;
Usually involves a medical history, physical examination, family history, and molecular genetic testing.
Family history. A three-generation family history should be taken, with attention to relatives with manifestations of a tubulinopathy and documentation of relevant findings through direct examination or review of medical records, including results of molecular genetic testing.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires the clinician to hypothesize which gene(s) are likely involved, whereas genomic testing does not.
5. General Medical Management of Tubulinopathies
A pediatric neurologist with expertise in the management of children with multiple disabilities and medically refractory epilepsy is recommended for long-term management.
Supportive management, including an individualized therapy plan that includes physical therapy to manage the complications of spasticity, occupational therapy, speech therapy, and vision therapy for oculomotor deficits and/or strabismus should begin at the time of diagnosis to ensure the best possible functionality and developmental outcome. Of note, it is appropriate to institute measures early on to manage potential complications of spasticity (e.g., joint contractures or reduced range of motion), which can increase the risk for decubitus ulcers as well as affect mobility and hygiene.
Those with congenital fibrosis of the extraocular muscles may require nonsurgical and/or surgical treatment.
Nutritional needs in infants with the more severe brain malformations (e.g., lissencephaly, generalized polymicrogyria) are usually managed by nasogastric tube feedings, followed by gastrostomy tube placement as needed.
Seizures are treated with anti-seizure medications based on the specific seizure type. In general, seizures should be treated promptly by specialists, as poor seizure control frequently worsens feeding and increases both the likelihood that a gastrostomy tube will be needed and the risk for aspiration.
Education of parents regarding common seizure presentations is appropriate. For information on non-medical interventions and coping strategies for parents or caregivers of children diagnosed with epilepsy, see Epilepsy Foundation Toolbox.
For individuals with severe cortical malformations (lissencephalies, polymicrogyria-like cortical dysplasia, microlissencephaly), it is usually appropriate to discuss the level of care to be provided in the event of a severe intercurrent illness.
6. Genetic Counseling of Family Members of an Individual with a Tubulinopathy
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Tubulinopathies caused by pathogenic variants in TUBA1A, TUBB2A, TUBB2B, TUBB3, TUBB (TUBB5), or TUBG1 are inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
More than 95% of individuals diagnosed with a tubulinopathy have a de novo pathogenic variant in TUBA1A, TUBB2A, TUBB2B, TUBB3, TUBB (TUBB5), or TUBG1.
Rarely, an individual diagnosed with a tubulinopathy has an affected parent. These individuals generally have either a TUBB3 or (less frequently) TUBB2B pathogenic variant.
If the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
If the pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
The proband has a
de novo pathogenic variant. Note: A pathogenic variant is reported as "
de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed
de novo" [
Richards et al 2015].
The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Maternal germline mosaicism has been reported in two families with multiple affected offspring [
Zillhardt et al 2016]. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
* A parent with somatic and germline mosaicism for a pathogenic variant may be mildly/minimally affected. While this situation is unusual, several groups have reported somatic mosaic pathogenic variants in genes encoding tubulin [
Jamuar & Walsh 2014,
Zillhardt et al 2016].
The family history of some individuals diagnosed with a tubulinopathy may appear to be negative because of failure to recognize the disorder in family members or reduced penetrance. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
If the proband has a known tubulinopathy-related pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of the possibility of parental germline mosaicism [
Poirier et al 2013a,
Zillhardt et al 2016].
If the parents have not been tested for the pathogenic variant identified in the proband but are clinically unaffected, the risk to the sibs of the proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for a tubulinopathy because of the possibility of the possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with a tubulinopathy has a 50% chance of inheriting the pathogenic variant.
Other family members
The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members may be at risk.
The risk to other family members appears to be low given that most probands with an autosomal dominant tubulinopathy have the disorder as a result of a de novo pathogenic variant.
Prenatal Testing and Preimplantation Genetic Testing
Once the tubulinopathy-related pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
American Association on Intellectual and Developmental Disabilities (AAIDD)
Phone: 202-387-1968
American Epilepsy Society
CDC - Child Development
Phone: 800-232-4636
Epilepsy Foundation
Phone: 800-332-1000; 866-748-8008
National Institute of Neurological Disorders and Stroke (NINDS)
PO Box 5801
Bethesda MD 20824
Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
Chapter Notes
Author Notes
Nadia Bahi-Buisson is a pediatric neurologist specializing in cortical malformations and fetal neurology. Her research at Imagine Institute focuses on the genetic and pathophysiologic bases of cortical malformations. She follows more than 100 patients with diffuse cortical malformations, and has consulted on more than 500 such cases. Dr Bahi-Buisson is involved in the European consortium on cortical malformations. This work is performed in collaboration with Chérif Beldjord, MD, PhD (director of the Laboratory of Biochemical Genetics – Cochin-Port-Royal).
Acknowledgments
Catherine Fallet Bianco and Annie Laquerriere, for sharing their fetal cases and for their helpful discussion of fetal brain tubulinopathies
Chérif Beldjord, Aurelie Toussaint, and Nathalie Carion, for their help in the screening of tubulin genes for diagnosis – from Sanger sequencing to the recent development of NGS panel screening
Jamel Chelly and Karine Poirier, who allowed the first author to collaborate on the identification of tubulin genes and to define/refine the associated phenotypes; and who have contributed through constructive discussion to the understanding of the pathophysiology of tubulinopathies
Sophie Thomas and Stanislas Lyonnet (Imagine Institute), for welcoming the authors to the Laboratory of Embryology and Genetics of Congenital Malformations and supporting their continued work in the identification of genes associated with cortical malformations
Author History
Nadia Bahi-Buisson, MD, PhD (2016-present)
Mara Cavallin, MD; Paris Descartes University (2016-2021)
Camille Maillard, PhD (2021-present)
Revision History
16 September 2021 (bp) Comprehensive update posted live
24 March 2016 (bp) Review posted live
7 July 2015 (nbb) Original submission
References
Literature Cited
Amrom
D, Tanyalcin
I, Verhelst
H, Deconinck
N, Brouhard
G, Decarie
JC, Vanderhasselt
T, Das
S, Hamdan
F, Lissens
W, Michaud
J, Jansen
A. Polymicrogyria with dysmorphic basal ganglia? Think tubulin!
Clin Genet.
2014;85:178–83.
[
PubMed: 23495813]
Bahi-Buisson
N, Poirier
K, Boddaert
N, Saillour
Y, Castelnau
L, Philip
N, Buyse
G, Villard
L, Joriot
S, Marret
S, Bourgeois
M, Van Esch
H, Lagae
L, Amiel
J, Hertz-Pannier
L, Roubertie
A, Rivier
F, Pinard
JM, Beldjord
C, Chelly
J. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations.
J Med Genet.
2008;45:647–53.
[
PubMed: 18728072]
Bahi-Buisson
N, Poirier
K, Fourniol
F, Saillour
Y, Valence
S, Lebrun
N, Hully
M, Bianco
CF, Boddaert
N, Elie
C, Lascelles
K, Souville
I, et al.
The wide spectrum of tubulinopathies: what are the key features for the diagnosis?
Brain.
2014;137:1676–700.
[
PubMed: 24860126]
Blumkin
L, Leibovitz
Z, Krajden-Haratz
K, Arad
A, Yosovich
K, Gindes
L, Zerem
A, Ben-Sira
L, Lev
D, Nissenkorn
A, Kidron
D, Dobyns
WB, Malinger
G, Bahi-Buisson
N, Leventer
RJ, Lerman-Sagie
T. Autosomal dominant TUBB3-related syndrome: fetal, radiologic, clinical and morphological features.
Eur J Paediatr Neurol.
2020;26:46–60.
[
PubMed: 32169460]
Breuss
M, Heng
JI, Poirier
K, Tian
G, Jaglin
XH, Qu
Z, Braun
A, Gstrein
T, Ngo
L, Haas
M, Bahi-Buisson
N, Moutard
ML, Passemard
S, Verloes
A, Gressens
P, Xie
Y, Robson
KJ, Rani
DS, Thangaraj
K, Clausen
T, Chelly
J, Cowan
NJ, Keays
DA. Mutations in the beta-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities.
Cell Rep.
2012;2:1554–62.
[
PMC free article: PMC3595605] [
PubMed: 23246003]
Brock
S, Stouffs
K, Scalais
E, D'Hooghe
M, Keymolen
K, Guerrini
R, Dobyns
WB, Di Donato
N, Jansen
AC. Tubulinopathies continued: refining the phenotypic spectrum associated with variants in TUBG1.
Eur J Hum Genet.
2018;26:1132–42.
[
PMC free article: PMC6057922] [
PubMed: 29706637]
Cardoso
C, Leventer
RJ, Ward
HL, Toyo-Oka
K, Chung
J, Gross
A, Martin
CL, Allanson
J, Pilz
DT, Olney
AH, Mutchinick
OM, Hirotsune
S, Wynshaw-Boris
A, Dobyns
WB, Ledbetter
DH. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3.
Am J Hum Genet.
2003;72:918–30.
[
PMC free article: PMC1180354] [
PubMed: 12621583]
Cushion
TD, Dobyns
WB, Mullins
JG, Stoodley
N, Chung
SK, Fry
AE, Hehr
U, Gunny
R, Aylsworth
AS, Prabhakar
P, Uyanik
G, Rankin
J, Rees
MI, Pilz
DT. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A.
Brain.
2013;136:536–48.
[
PubMed: 23361065]
Cushion
TD, Paciorkowski
AR, Pilz
DT, Mullins
JG, Seltzer
LE, Marion
RW, Tuttle
E, Ghoneim
D, Christian
SL, Chung
SK, Rees
MI, Dobyns
WB. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy.
Am J Hum Genet.
2014;94:634–41.
[
PMC free article: PMC3980418] [
PubMed: 24702957]
Di Donato
N, Chiari
S, Mirzaa
GM, Aldinger
K, Parrini
E, Olds
C, Barkovich
AJ, Guerrini
R, Dobyns
WB. Lissencephaly: expanded imaging and clinical classification.
Am J Med Genet A.
2017;173:1473–88.
[
PMC free article: PMC5526446] [
PubMed: 28440899]
Fallet-Bianco
C, Laquerriere
A, Poirier
K, Razavi
F, Guimiot
F, Dias
P, Loeuillet
L, Lascelles
K, Beldjord
C, Carion
N, Toussaint
A, Revencu
N, Addor
MC, Lhermitte
B, Gonzales
M, Martinovich
J, Bessieres
B, Marcy-Bonniere
M, Jossic
F, Marcorelles
P, Loget
P, Chelly
J, Bahi-Buisson
N. Mutations in tubulin genes are frequent causes of various foetal malformations of cortical development including microlissencephaly.
Acta Neuropathol Commun.
2014;2:69.
[
PMC free article: PMC4222268] [
PubMed: 25059107]
Fallet-Bianco
C, Loeuillet
L, Poirier
K, Loget
P, Chapon
F, Pasquier
L, Saillour
Y, Beldjord
C, Chelly
J, Francis
F. Neuropathological phenotype of a distinct form of lissencephaly associated with mutations in TUBA1A.
Brain.
2008;131:2304–20.
[
PubMed: 18669490]
Guerrini
R, Mei
D, Cordelli
DM, Pucatti
D, Franzoni
E, Parrini
E. Symmetric polymicrogyria and pachygyria associated with TUBB2B gene mutations.
Eur J Hum Genet.
2012;20:995–8.
[
PMC free article: PMC3421113] [
PubMed: 22333901]
Hebebrand
M, Hüffmeier
U, Trollmann
R, Hehr
U, Uebe
S, Ekici
AB, Kraus
C, Krumbiegel
M, Reis
A, Thiel
CT, Popp
B. The mutational and phenotypic spectrum of TUBA1A-associated tubulinopathy.
Orphanet J Rare Dis.
2019;14:38.
[
PMC free article: PMC6371496] [
PubMed: 30744660]
Jamuar
SS, Walsh
CA. Somatic mutations in cerebral cortical malformations.
N Engl J Med.
2014;371:2038.
[
PubMed: 25409382]
Jansen
AC, Oostra
A, Desprechins
B, De Vlaeminck
Y, Verhelst
H, Regal
L, Verloo
P, Bockaert
N, Keymolen
K, Seneca
S, De Meirleir
L, Lissens
W. TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria.
Neurology.
2011;76:988–92.
[
PubMed: 21403111]
Kumar
RA, Pilz
DT, Babatz
TD, Cushion
TD, Harvey
K, Topf
M, Yates
L, Robb
S, Uyanik
G, Mancini
GM, Rees
MI, Harvey
RJ, Dobyns
WB. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins.
Hum Mol Genet.
2010;19:2817–27.
[
PMC free article: PMC2893812] [
PubMed: 20466733]
Lecourtois
M, Poirier
K, Friocourt
G, Jaglin
X, Goldenberg
A, Saugier-Veber
P, Chelly
J, Laquerriere
A. Human lissencephaly with cerebellar hypoplasia due to mutations in TUBA1A: expansion of the foetal neuropathological phenotype.
Acta Neuropathol.
2010;119:779–89.
[
PubMed: 20376468]
Oegema
R, Cushion
TD, Phelps
IG, Chung
SK, Dempsey
JC, Collins
S, Mullins
JG, Dudding
T, Gill
H, Green
AJ, Dobyns
WB, Ishak
GE, Rees
MI, Doherty
D. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes.
Hum Mol Genet.
2015;24:5313–25.
[
PMC free article: PMC4550818] [
PubMed: 26130693]
Pilz
DT, Matsumoto
N, Minnerath
S, Mills
P, Gleeson
JG, Allen
KM, Walsh
CA, Barkovich
AJ, Dobyns
WB, Ledbetter
DH, Ross
ME. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation.
Hum Mol Genet.
1998;7:2029–37.
[
PubMed: 9817918]
Poirier
K, Lebrun
N, Broix
L, Tian
G, Saillour
Y, Boscheron
C, Parrini
E, Valence
S, Pierre
BS, Oger
M, Lacombe
D, Genevieve
D, Fontana
E, Darra
F, Cances
C, Barth
M, Bonneau
D, Bernadina
BD, N'Guyen
S, Gitiaux
C, Parent
P, des Portes
V, Pedespan
JM, Legrez
V, Castelnau-Ptakine
L, Nitschke
P, Hieu
T, Masson
C, Zelenika
D, Andrieux
A, Francis
F, Guerrini
R, Cowan
NJ, Bahi-Buisson
N, Chelly
J. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly.
Nat Genet.
2013a;45:639–47.
[
PMC free article: PMC3826256] [
PubMed: 23603762]
Poirier
K, Saillour
Y, Bahi-Buisson
N, Jaglin
XH, Fallet-Bianco
C, Nabbout
R, Castelnau-Ptakhine
L, Roubertie
A, Attie-Bitach
T, Desguerre
I, Genevieve
D, Barnerias
C, Keren
B, Lebrun
N, Boddaert
N, Encha-Razavi
F, Chelly
J. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects.
Hum Mol Genet.
2010;19:4462–73.
[
PMC free article: PMC3298850] [
PubMed: 20829227]
Poirier
K, Saillour
Y, Fourniol
F, Francis
F, Souville
I, Valence
S, Desguerre
I, Marie Lepage
J, Boddaert
N, Line Jacquemont
M, Beldjord
C, Chelly
J, Bahi-Buisson
N.
Expanding the spectrum of TUBA1A-related cortical dysgenesis to Polymicrogyria.
Eur J Hum Genet.
2013b;21:381–5.
[
PMC free article: PMC3598328] [
PubMed: 22948023]
Richards
S, Aziz
N, Bale
S, Bick
D, Das
S, Gastier-Foster
J, Grody
WW, Hegde
M, Lyon
E, Spector
E, Voelkerding
K, Rehm
HL, et al.
Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med.
2015;17:405–24.
[
PMC free article: PMC4544753] [
PubMed: 25741868]
Romaniello
R, Tonelli
A, Arrigoni
F, Baschirotto
C, Triulzi
F, Bresolin
N, Bassi
MT, Borgatti
R. A novel mutation in the beta-tubulin gene TUBB2B associated with complex malformation of cortical development and deficits in axonal guidance.
Dev Med Child Neurol.
2012;54:765–9.
[
PubMed: 22591407]
Romero
DM, Bahi-Buisson
N, Francis
F. Genetics and mechanisms leading to human cortical malformations.
Semin Cell Dev Biol.
2018;76:33–75.
[
PubMed: 28951247]
Tischfield
MA, Baris
HN, Wu
C, Rudolph
G, Van Maldergem
L, He
W, Chan
WM, Andrews
C, Demer
JL, Robertson
RL, Mackey
DA, Ruddle
JB, Bird
TD, Gottlob
I, Pieh
C, Traboulsi
EI, Pomeroy
SL, Hunter
DG, Soul
JS, Newlin
A, Sabol
LJ, Doherty
EJ, de Uzcategui
CE, de Uzcategui
N, Collins
ML, Sener
EC, Wabbels
B, Hellebrand
H, Meitinger
T, de Berardinis
T, Magli
A, Schiavi
C, Pastore-Trossello
M, Koc
F, Wong
AM, Levin
AV, Geraghty
MT, Descartes
M, Flaherty
M, Jamieson
RV, Moller
HU, Meuthen
I, Callen
DF, Kerwin
J, Lindsay
S, Meindl
A, Gupta
ML
Jr, Pellman
D, Engle
EC. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance.
Cell.
2010;140:74–87.
[
PMC free article: PMC3164117] [
PubMed: 20074521]
Zillhardt
JL, Poirier
K, Broix
L, Lebrun
N, Elmorjani
A, Martinovic
J, Saillour
Y, Muraca
G, Nectoux
J, Bessieres
B, Fallet-Bianco
C, Lyonnet
S, Dulac
O, Odent
S, Rejeb
I, Jemaa
LB, Rivier
F, Pinson
L, Genevieve
D, Musizzano
Y, Bigi
N, Leboucq
N, Giuliano
F, Philip
N, Vilain
C, Van Bogaert
P, Maurey
H, Beldjord
C, Artiguenave
F, Boland
A, Olaso
R, Masson
C, Nitschke
P, Deleuze
JF, Bahi-Buisson
N, Chelly
J. Mosaic parental germline mutations causing recurrent forms of malformations of cortical development.
Eur J Hum Genet.
2016;24:611–4.
[
PMC free article: PMC4929884] [
PubMed: 26395554]