Summary
Clinical characteristics.
Maternal 15q duplication syndrome (maternal dup15q) is characterized by hypotonia and motor delays, intellectual disability, autism spectrum disorder (ASD), and epilepsy including infantile spasms. Rarely, maternal dup15q may also be associated with psychosis or sudden unexplained death. Those with a maternal isodicentric 15q11.2-q13.1 supernumerary chromosome are typically more severely affected than those with an interstitial duplication.
Diagnosis/testing.
The diagnosis of maternal dup15q is established by detection of at least one extra maternally derived copy of the Prader-Willi/Angelman critical region, a region approximately 5 Mb long within chromosome region 15q11.2-q13.1. The extra copy or copies most commonly arise by one of two mechanisms:
- A maternal isodicentric 15q11.2-q13.1 supernumerary chromosome – idic(15) – typically comprising two extra copies of 15q11.2-q13.1 and resulting in tetrasomy for 15q11.2-q13.1 (~60-80%);
- A maternal interstitial 15q11.2-q13.1 duplication that typically includes one extra copy of 15q11.2-q13.1 within chromosome 15, resulting in trisomy for 15q11.2-q13.1 (~20-40%).
Management.
Treatment of manifestations: Multidisciplinary team evaluation of motor and speech development and to assist in referrals for appropriate educational programs. Supportive care may include: feeding therapy, occupational and physical therapy, alternative and augmentative communication, behavioral therapy (e.g., applied behavioral analysis therapy), psychotropic medications for behavioral manifestations, and standard management for seizures.
Surveillance: Growth and nutritional assessment at each visit. Periodic: neurodevelopmental and/or developmental/behavioral assessments, and monitoring for evidence of seizures and/or change in seizure type.
Agents/circumstances to avoid: Seizure triggers (e.g., sleep deprivation, stress).
Evaluation of relatives at risk: Consider genetic testing of sibs of a proband (known to be at increased risk for an inherited maternal interstitial 15q11.2-q13.1 duplication) in order to refer those with the interstitial duplication promptly for multidisciplinary team evaluation and developmental support.
Genetic counseling.
Maternal dup15q caused by:
- Maternal idic(15). De novo in all affected individuals reported to date; thus, risk to sibs is low, but presumed to be marginally greater than in the general population because of the possibility of maternal germline mosaicism;
- Maternal interstitial 15q11.2-q13.1 duplication. De novo in approximately 85% of probands and inherited from the mother in approximately 15%. If the mother has the 15q interstitial duplication, the risk to each child of inheriting the duplication is 50%.
Prenatal testing or preimplantation genetic testing using chromosomal microarray (CMA) will detect the 15q interstitial duplication; however, prenatal test results cannot reliably predict the severity of the phenotype even in a pregnancy known to be at increased risk for maternal dup15q.
GeneReview Scope
Table
Maternal isodicentric 15q11.2-q13.1 supernumerary chromosome [idic(15)] resulting in tetrasomy or hexasomy for 15q11.2-q13.1 Maternal interstitial 15q11.2-q13.1 duplication or triplication
Diagnosis
Suggestive Findings
Maternal 15q duplication syndrome (maternal dup15q) should be suspected in individuals with the following clinical features:
- Moderate-to-severe hypotonia in infancy and early delays in reaching motor milestones
- Developmental delay that includes not only motor but also language
- Intellectual disability (ID)
- Autism spectrum disorder (ASD)
- Seizures, particularly infantile spasms
- Dysmorphic features including upturned nose, epicanthal folds, and downslanting palpebral fissures [Urraca et al 2013]
- Behavioral difficulties including hyperactivity, anxiety, or emotional lability [Al Ageeli et al 2014]
- A characteristic EEG biomarker involving excessive beta oscillations (12–30 Hz) [Frohlich et al 2016, Saravanapandian et al 2020]
Establishing the Diagnosis
The diagnosis of maternal dup15q is established in a proband by detection of at least one extra maternally derived copy of the Prader-Willi/Angelman critical region (PWACR), a region approximately 5 Mb long within chromosome 15q11.2-q13.1 (see Table 1).
The proximal 15q region includes five regions of segmental duplications or low copy repeats (designated by breakpoints [BPs]), which result in increased susceptibility to genomic rearrangements [Hogart et al 2010]. These five regions are termed BP1 through BP5. The PWACR lies between BP2 and BP3 (see Figure 1) and is always included in the interstitial duplications or the idic(15) that causes maternal dup15q. The PWACR is imprinted: maternally derived increases in copy number cause maternal dup15q (the topic of this GeneReview) while paternally derived increases are typically associated with more variable and sometimes different neurodevelopmental phenotypes (see Genetically Related Disorders) [Cook et al 1997, Urraca et al 2013].

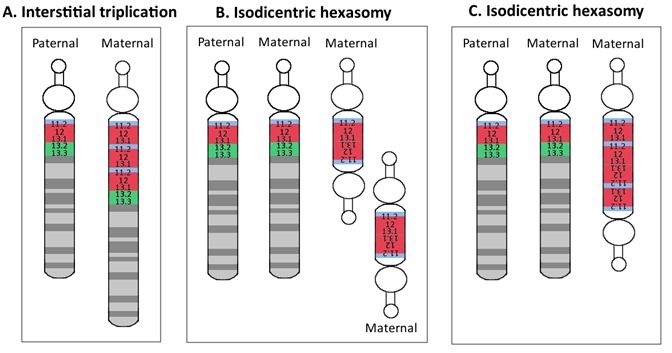
Figure 1
A. Schematic of the normal paternal and maternal chromosome 15 B. & C. The most common causes of maternal dup15q:
The extra copy or copies of the PWACR most commonly arise by one of two mechanisms (Figure 1):
- A maternal isodicentric 15q11.2-q13.1 supernumerary chromosome – idic(15) – that typically comprises two extra copies of 15q11.2-q13.1, resulting in tetrasomy for 15q11.2-q13.1 (~60-80%)OR
- A maternal interstitial 15q11.2-q13.1 duplication that typically includes one extra copy of 15q11.2-q13.1 within chromosome 15, resulting in trisomy for 15q11.2-q13.1 (~20-40%)
For this GeneReview, maternal dup15q is defined as the presence of one or more extra copies of 15q11.2-q13.1 that include the PWACR at the approximate position of chr15:22782170-28134728 in the reference genome (NCBI Build GRCh38/hg38, seen here). Duplications may vary in size and have been seen up to 12 Mb long (as seen here) but must contain the PWACR to be causative of dup15q.
Although several genes of interest (e.g., ATP10A, CYFIP1, MAGEL2, NECDIN, SNRPN, UBE3A, snoRNAs, and a cluster of genes encoding GABAA receptor subunits) are within the 4.5- to 12-Mb recurrent duplication, no single gene that – when duplicated – causes maternal dup15q has been identified. (See Molecular Genetics for genes of interest in the duplicated region.)
Genomic testing methods that determine the copy number of sequences can include chromosomal microarray analysis (CMA) or targeted duplication analysis. Note: (1) Interstitial 15q11.2-q13.1 duplications cannot typically be identified by routine analysis of G-banded chromosomes or other conventional cytogenetic banding techniques; however, idic(15) and large interstitial duplications (>5 Mb) that extend beyond the PWACR can be identified through cytogenetic analysis. (2) Mosaicism has been reported for idic(15), suggesting some degree of mitotic instability [Wang et al 2008], which may affect the phenotype and the sensitivity of genomic testing strategies used for diagnosis.
CMA using oligonucleotide or SNP arrays can detect increases in copy number of the 15q11.2-q13.1 region in a proband. The ability to size the duplication depends on the type of microarray used and the density of probes in the 15q11.2-q13.1 region. CMA cannot reliably differentiate between idic(15) and interstitial triplication of 15q11.2-q13.1.
Note: (1) Most individuals with maternal dup15q are identified by CMA performed in the context of evaluation for developmental delay, intellectual disability, or autism spectrum disorder. (2) FISH or a cytogenetic study is required to determine whether the duplication is supernumerary or interstitial and to determine whether there is evidence for mosaicism.
Targeted duplication analysis. FISH analysis, quantitative PCR (qPCR), multiplex ligation-dependent probe amplification (MLPA), or other targeted quantitative methods may be used to test relatives of a proband who is known to have the 15q11.2-q13.1 recurrent duplication.
Note: (1) Targeted duplication testing is not appropriate for an individual in whom the 15q11.2-q13.1 recurrent duplication was not detected by CMA designed to target this region. (2) It is not possible to size the duplication routinely by use of targeted methods.
Maternal origin of the 15q11.2-q13.1 duplication is identified by either of the following:
- Genotyping or methylation analysis, including PCR-based methylation analysis [Zielinski et al 1988, Urraca et al 2010]
- Identification of a 15q11.2-q13.1 interstitial duplication in a maternal sample
Table 1.
Genomic Testing used in Maternal 15q Duplication Syndrome
Clinical Characteristics
Clinical Description
Maternal 15q duplication syndrome (maternal dup15q) is characterized by hypotonia and motor delays, intellectual disability, autism spectrum disorder (ASD), and epilepsy including infantile spasms. These clinical findings differ significantly between people with a maternal interstitial duplication and those with a maternal isodicentric supernumerary chromosome, or idic(15) (Table 2). Those with a maternal idic(15) are typically more severely affected than those with an interstitial duplication. However, severity varies even among individuals who have increased dosage by the same genetic mechanism. Some phenotypic features, such as ASD, are more consistently observed in individuals with a maternal idic(15) or large (>5-Mb) interstitial duplications that extend beyond the PWACR [Hogart et al 2010].
Table 2.
Maternal 15q Interstitial Duplication and Idic(15): Comparison of Clinical Features
Hypotonia and motor skills. Hypotonia in newborns and infants with maternal dup15q is associated with feeding difficulties and gross motor delays [Depienne et al 2009, Hogart et al 2010, Urraca et al 2013]. Hypotonia also contributes to gastrointestinal issues in maternal dup15q, such as constipation.
Although childhood hypotonia impairs motor development, most children achieve independent walking after age two to three years (younger in children with an interstitial duplication) [Hogart et al 2010, Piard et al 2010, Al Ageeli et al 2014].
A wide-based or ataxic gait is common [Bundey et al 1994]. Delays and persistent impairment in both fine and gross motor skills affect adaptive living skills and distinguish children with maternal dup15q from children with nonsyndromic autism spectrum disorder [DiStefano et al 2016].
Developmental delay and intellectual disability. Developmental delay in early childhood is nearly universal. This can be more specifically diagnosed as intellectual disability after age five years.
In addition to motor delays, speech and language development is particularly affected, with universal delays ranging from moderate to severe [Hogart et al 2010]. Some individuals exhibit echolalia, pronoun reversal, and stereotyped utterances, while others may lack functional speech [Battaglia et al 1997, Battaglia 2008].
Most children and adults with maternal dup15q function in the moderate-to-severe range of intellectual disability; however, there is some variability, with a higher range of cognitive abilities seen in those with an interstitial duplication [DiStefano et al 2020].
Individuals with maternal dup15q who have a diagnosis of epilepsy have lower verbal, daily living, socialization, fine motor, and gross motor skills compared to individuals with maternal dup15q who do not have epilepsy [DiStefano et al 2016, DiStefano et al 2020].
Autism spectrum disorder (ASD). Most children and adults with maternal dup15q meet criteria for ASD. In one study, 25/27 individuals with a maternal idic(15) met criteria for autism on the ADOS, while the remaining 2/27 met criteria for ASD [DiStefano et al 2020]. Among individuals with a maternal interstitial dup15q duplication, 10/12 met criteria for autism, one individual met criteria for ASD, and one individual did not meet criteria for either. Compared to other CNVs known to cause ASD, maternal dup15q confers the greatest risk (odds ratio ≥2.6) [Malhotra & Sebat 2012, Moreno-De-Luca et al 2013]. Manifestations of ASD, particularly difficulties with social interaction, may increase from early to late childhood [Simon et al 2010].
Compared to children with nonsyndromic ASD, children with maternal dup15q-ASD demonstrate a distinctive behavioral profile, including preserved responsive social smile and directed facial expressions towards others – features that may inform behavioral interventions [DiStefano et al 2016].
Epilepsy. More than half of individuals with maternal dup15q have epilepsy, usually involving multiple seizure types including infantile spasms and myoclonic, tonic-clonic, absence, and/or focal seizures [Conant et al 2014]. Seizures most often begin between ages six months and nine years [Battaglia 2008]. Incidence of epilepsy is higher in individuals with maternal dup15q resulting from an idic(15) compared to those with maternal dup15q resulting from interstitial duplication (57% and 6% respectively) [DiStefano et al 2020].
Maternal dup15q is one of the most common known causes of infantile spasms [Conant et al 2014]. Infantile spasms in individuals with maternal dup15q often progress to Lennox Gastaut syndrome and other complex seizure patterns that may be difficult to control. As many as 40% of individuals with seizures present initially with infantile spasms; of this group, approximately 90% subsequently develop other seizure types.
Intractable epilepsy in individuals with maternal dup15q may result in disabling secondary effects, including falls or developmental regression. This occurs in more than half of individuals with frequent, uncontrolled seizures or nonconvulsive status epilepticus [Battaglia et al 1997].
Children with epilepsy have been found to have lower cognitive and adaptive function than those without epilepsy [DiStefano et al 2020].
Dysmorphic features. Minor dysmorphic features often reported in individuals with maternal dup15q include flat occiput, downslanting palpebral fissures, depressed nasal bridge, short nose with an upturned nasal tip, low-set ears, long philtrum, high-arched palate, thick vermilion of the upper and lower lips, and micrognathia [Battaglia et al 1997, Borgatti et al 2001, Hogart et al 2010, Urraca et al 2013]. These features are typically subtle and may be missed in infancy.
Psychosis. Although maternal idic(15) has been reported in schizophrenia cohorts [Rees et al 2014], psychosis is not a commonly ascertained comorbidity in maternal dup15q – a finding that may reflect the difficulty of recognizing and diagnosing psychosis in individuals with low cognitive functioning and limited verbal skills. For instance, psychosis is a common comorbidity in Prader-Willi syndrome caused by uniparental disomy, which similarly involves a duplication of the maternally contributed 15q11.2-13.1 [Bassett 2011]. These individuals tend to have higher cognitive and verbal abilities than individuals with maternal dup15q. Conversely, with a high rate of ASD in individuals with maternal dup15q, psychosis related to mood disorder may be misdiagnosed as schizophrenia.
Sudden unexpected death in epilepsy (SUDEP) occurs in a small but significant minority of individuals with maternal dup15q [Friedman et al 2016, Devinsky 2011, Wegiel et al 2012]. These deaths almost always occur during sleep and most (though not all) have occurred in teenagers and young adults with epilepsy. Nonambulatory status and poor seizure control appear to be risk factors for SUDEP in individuals with maternal dup15q [Friedman et al 2016].
The mechanism underlying SUDEP is not well understood; however, available evidence suggests that in most instances a tonic-clonic seizure is followed by a shutdown of brain function and cardio-respiratory arrest. SUDEP occurs in 9% of individuals with epilepsy; the incidence of SUDEP in individuals with maternal dup15q is unknown.
Penetrance
In maternal idic(15) penetrance is 100%.
In maternal interstitial 15q11.2-q13.1 duplication penetrance appears to be complete, although some individuals may have mild features and appear unaffected.
Penetrance is the same for males and females.
Nomenclature
Terms used to refer to maternal 15q duplication syndrome and related disorders:
- 15q11.2-q13.1 duplication syndrome
- Dup15q syndrome
- Inverted duplication 15 (inv dup15)
- Partial trisomy 15
- Isodicentric chromosome 15 syndrome [Idic(15)]
- Supernumerary marker chromosome 15 (SMC15)
- Partial tetrasomy 15q
Prevalence
Maternal dup15q is one of the most common cytogenetic anomalies in persons with ASD, occurring in approximately 1:522 persons with ASD [Depienne et al 2009].
Genetically Related Disorders
Table 3.
Genetically Related Disorders
Differential Diagnosis
Phenotypic features associated with maternal 15q duplication syndrome (maternal dup15q) are not sufficient for diagnosis. All chromosome anomalies and genes known to be associated with intellectual disability (ID) should be included in the differential diagnosis of maternal dup15q. See OMIM Autosomal Dominant, Autosomal Recessive, Nonsyndromic X-Linked, and Syndromic X-Linked Intellectual Developmental Disorder Phenotypic Series.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with maternal 15q duplication syndrome (maternal dup15q), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 4
Recommended Evaluations Following Initial Diagnosis in Individuals with Maternal 15q Duplication Syndrome
Treatment of Manifestations
A multidisciplinary team evaluation is recommended beginning in early infancy to evaluate motor and speech development and later to assist in referrals for appropriate educational programs.
Table 5.
Treatment of Manifestations in Individuals with Maternal 15q Duplication Syndrome
Seizure management is important in preventing secondary complications, including (in the most severe cases) brain damage, developmental regression, and sudden unexpected death in epilepsy (SUDEP) [Devinsky 2011].
Approximately half of seizure-related deaths are not due to SUDEP, but to other causes including status epilepticus, drowning, falls, and accidents. Many of these are preventable. For example, status epilepticus may be prevented with the use of rescue medications such as rectal diazepam or nasal midazolam. Some evidence suggests that prompt identification of a seizure and basic care (e.g., repositioning a person on the side instead of face down) after a seizure may help prevent SUDEP [Ryvlin et al 2013]. However, the only known preventive therapy is the best possible seizure control [Ryvlin et al 2011]. Although a variety of monitors can help detect SUDEP (e.g., wrist and mattress accelerometers), none can prevent it [Devinsky 2011].
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary by country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction. Physical therapy is recommended to maximize mobility.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech & language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder (ADHD), when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Table 6.
Recommended Surveillance for Individuals with Maternal 15q Duplication Syndrome
Agents/Circumstances to Avoid
Seizure triggers (e.g., sleep deprivation, stress, and failure to follow medication regimen) should be avoided.
Evaluation of Relatives at Risk
Consider genetic testing of sibs of a proband who is known to have an inherited maternal interstitial 15q11.2-q13.1 duplication in order to refer sibs with an interstitial duplication promptly for developmental evaluation and early intervention services.
Note: Recurrence is extremely rare in the sibs of probands with a maternal isodicentric 15q11.2-q13.1 supernumerary chromosome – idic(15).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Maternal 15q duplication syndrome (maternal dup15q) is an autosomal dominant disorder typically caused by a de novo genetic alteration.
Risk to Family Members
Maternal Isodicentric 15q11.2-q13.1 Supernumerary Chromosome – Idic(15)
Parents of a proband
- All probands reported to date with maternal dup15q caused by idic(15) whose parents have undergone genomic testing have had the disorder as the result of a de novo genetic alteration.
- Evaluation of the mother by genomic testing to determine if she has the genetic alteration present in the proband is recommended to confirm her genetic status and to allow reliable recurrence risk counseling. Note: Maternal transmission of supernumerary partial trisomy of the region has been reported in one instance [Michelson et al 2011].
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's mother:
- The risk to sibs is presumed to be low as the idic(15) has occurred as a de novo alteration in all affected individuals reported to date.
- If the idic(15) identified in the proband is not identified in a parent, the risk to sibs is presumed to be marginally greater than in the general population because of the theoretic possibility of maternal germline mosaicism for the idic(15).
Offspring of a proband. Individuals with idic(15) are not known to reproduce.
Other family members. Given that all probands with maternal dup15q caused by idic(15) reported to date have the disorder as a result of a de novo genetic alteration, the risk to other family members is presumed to be low.
Maternal 15q Interstitial Duplication
Parents of a proband
- Approximately 85% of individuals with maternal dup15q caused by a maternal interstitial 15q11.2-q13.1 duplication have a de novo genetic alteration.
- Approximately 15% of individuals with maternal dup15q caused by a maternal interstitial 15q11.2-q13.1 duplication inherited the genetic alteration from the mother.If the mother of a proband inherited a 15q interstitial duplication from her father (i.e., the maternal grandfather of the proband), the mother will not have maternal dup15q syndrome. Instead, she may appear to be unaffected or have features associated with paternal duplications, which – although distinct from those of the proband – may share some similarities (see Genetically Related Disorders).
- Evaluation of the mother by genomic testing to determine if she has the genetic alteration present in the proband is recommended to confirm her genetic status and to allow reliable recurrence risk counseling
- If the maternal 15q interstitial duplication found in the proband cannot be detected in maternal leukocyte DNA, the most likely explanation is a de novo 15q interstitial duplication in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's mother:
- If the mother of the proband has the 15q interstitial duplication, the risk to each sib of inheriting the duplication is 50%.
- It is not possible to reliably predict the severity of the phenotype in sibs who inherit a maternal interstitial 15q11.2-q13.1 duplication (see Penetrance).
- If the maternal 15q interstitial duplication identified in the proband cannot be detected in maternal leukocyte DNA, the risk to sibs is presumed to be marginally greater than in the general population because of the theoretic possibility of maternal germline mosaicism for the duplication.
Offspring of a proband. Each child of an individual with a 15q interstitial duplication has a 50% chance of inheriting the duplication.
- If the proband is female, offspring who inherit a 15q interstitial duplication are at risk for maternal dup15q.
- If the proband is male, offspring who inherit a 15q interstitial duplication are at risk for features associated with paternal interstitial duplications of 15q11.2-q13.1 (see Genetically Related Disorders).
Other family members. The risk to other family members depends on the genetic status of the proband's mother: if the mother of the proband has the 15q interstitial duplication, her family members may also have the duplication.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy. Similarly, decisions about testing to determine the genetic status of at-risk family members are best made before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are at risk of having a child with maternal dup15q.
Prenatal Testing and Preimplantation Genetic Testing
Maternal idic(15). Risk to future pregnancies is presumed to be low, as to date all reported instances of idic(15) have been de novo. However, couples may wish to consider prenatal testing or preimplantation genetic testing, as risk may be slightly greater than in the general population due to the possibility of maternal germline mosaicism.
Maternal 15q interstitial duplication. Once a maternal 15q interstitial duplication has been identified in the proband, prenatal testing and preimplantation genetic testing using CMA that will detect the 15q interstitial duplication are possible.
Pregnancies not known to be at increased risk for idic(15) or a 15q interstitial duplication. CMA performed in a pregnancy not known to be at increased risk for maternal dup15q may detect increased copy numbers of 15q11.2-q13.1 due to an interstitial duplication or idic(15).
Note: Prenatal test results cannot reliably predict the severity of the phenotype (see Clinical Description) regardless of whether the pregnancy is known or not known to be at increased risk for maternal dup15q.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Dup15q AlliancePO Box 674Fayetteville NY 13066Phone: 855-DUP-15QAEmail: info@dup15q.org
- Unique: Understanding Rare Chromosome and Gene DisordersUnited KingdomPhone: +44 (0) 1883 723356Email: info@rarechromo.org
- Linking Angelman and Dup15q Data for Expanded Research (LADDER) RegistryEmail: contact@laddertotreatment.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Maternal 15q Duplication Syndrome: Genes and Databases
Table B.
OMIM Entries for Maternal 15q Duplication Syndrome (View All in OMIM)
Molecular Pathogenesis
The extra copy or copies of the PWACR most commonly arise by one of two mechanisms (Figure 1):
- A maternal isodicentric 15q11.2-q13.1 supernumerary chromosome – idic(15) – that typically comprises two extra copies of 15q11.2-q13.1, resulting in tetrasomy for 15q11.2-q13.1 (~60%-80%) (Dup15q Alliance International Registry, 3-8-21 and 3-14-14)OR
- A maternal interstitial 15q11.2-q13.1 duplication that typically includes one extra copy of 15q11.2-q13.1 within chromosome 15, resulting in trisomy for 15q11.2-q13.1 (~20%-40%) (Dup15q Alliance International Registry, 3-8-21 and 3-14-14)
The proximal 15q region includes five regions of segmental duplications or low copy repeats (designated by breakpoints [BPs]), which result in increased susceptibility to genomic rearrangements [Christian et al 1999]. These five regions are termed BP1 through BP5. The Prader-Willi/Angelman critical region (PWACR) lies between BP2 and BP3 (see Figure 1) and is always included in the interstitial duplications or the idic(15) that cause maternal dup15q. Duplications that extend from BP1 to BP3 are referred to as Class I duplications; those that span BP2 and BP3 only are Class II duplications. The PWACR is imprinted: maternally derived increases in copy number cause maternal dup15q while paternally derived increases are typically associated with more variable and sometimes different neurodevelopmental phenotypes [Cook et al 1997, Urraca et al 2013].
The maternal isodicentric 15q11.2-q13.1 – or idic(15) – is typically a bisatellited chromosome thought to arise from U-type exchange during meiosis. Idic(15), which typically includes two mirrored copies of 15pter-q13.1 (p arm of chromosome 15, centromere, and 15q11.2-q13.1) [Roberts et al 2003], is sometimes referred to as inv dup(15). The distal breakpoint is typically BP3 (approximate position [hg38] chr15:28300000) on both sides of truly isodicentric chromosomes and BP4 and BP5 (approximate positions [hg19] chr15:30600000 and [hg38] chr15:32200000) for asymmetric supernumerary chromosomes (see Figure 2) [Hogart et al 2010]. Idic(15) usually results in tetrasomy for 15q11.2-q13.1.

Figure 2.
Asymmetry in dup15q, as seen in: A. Interstitial triplication of 15q11.2-13.1; and
Interstitial duplications leading to maternal dup15q arise by nonallelic homologous recombination (NAHR) between two different breakpoint regions (e.g., BP1 and BP3). The distal breakpoint for maternal interstitial duplications is typically BP3 (approximate position [hg38] chr15:28300000), and the proximal breakpoint is typically either BP1 or BP2 (approximate positions [hg38] chr15:22300000 or [hg38] chr15:23300000, respectively). Interstitial duplications usually result in trisomy for 15q11.2-q13.1.
Variations of these primary mechanisms include the following:
- Interstitial 15q11.2-q13.1 triplication, which results in tetrasomy of 15q11.2-q13.1. This phenotype tends to be more severe than that of maternal interstitial 15q11.2-q13.1 duplication and more like that of maternal isodicentric 15q11.2-q13.1 supernumerary chromosome [Ungaro et al 2001, Hogart et al 2010] (see Figure 3).
- Isodicentric 15q hexasomy. The phenotype is severe, including profound intellectual disability, intractable epilepsy, and more prominent dysmorphic features (myopathic facies and low-set ears) [Mann et al 2004] (Figure 3).
- Asymmetric isodicentric or interstitial maternal triplications, which typically result in tetrasomy for 15q11.2-q13.2 and trisomy for 15q13.2-13.3. These asymmetric copy number variations are observed in approximately 10%-15% of isodicentric and interstitial chromosomes (Dup15q Alliance International Registry, 3-14-14) (see Figure 2).
- Ring chromosome 15. Rarely, supernumerary ring chromosomes that include the PWACR have been seen. These are typically mosaic, indicating the unstable nature of ring chromosomes [Wang et al 2008].

Figure 3.
Uncommon variations in copy number seen in dup15q A. Interstitial triplication of 15q11.2-13.1
Genes of interest in this region
- UBE3A, the gene implicated in Angelman syndrome, is also thought to be the primary gene responsible for the intellectual impairment, anxiety, and reduced seizure threshold of maternal dup15q [Copping et al 2017], as well as contributing to the autism features seen in the disorder [Glessner et al 2009, Greer et al 2010, LaSalle et al 2015]. Transgenic mice with overexpression of UBE3A exhibit learning deficits, anxiety-like behaviors, and reduced seizure thresholds, with normal social interactions and no increased motor stereotypies [Copping et al 2017]. UBE3A is imprinted with maternal-specific expression in postnatal neurons, and thus expressed at a higher dosage in brain from individuals with a maternally derived duplication [Hogart et al 2010, Urraca et al 2013].
- GABRB3, GABRA5, and GABRG3, genes that encode GABAA receptor subunits, are implicated in the seizures observed in maternal dup15q [Menold et al 2001, Samaco et al 2005, Hogart et al 2007, Frohlich et al 2019]. Knockout mouse models of these genes develop neurologic problems including seizures [DeLorey et al 1998, DeLorey et al 2008]. In addition, individuals with maternal dup15q have a characteristic EEG pattern that is similar to the EEG pattern seen in typically developing children after being administered midazolam, a GABAA modulator [Frohlich et al 2019].GABRB3 may also contribute to the autism phenotype in maternal dup15q [Conant et al 2014], as single-nucleotide polymorphisms in this gene are associated with autism [Menold et al 2001] and expression is reduced in brain tissue samples of individuals with ASD [Samaco et al 2005, Hogart et al 2007]. GABRB3 has been implicated in ASD by genome-wide de novo single-nucleotide variant studies [Sanders et al 2015].
- HERC2 is an E3 ubiquitin ligase. Individuals with biallelic HERC2 pathogenic variants can have intellectual disability [Puffenberger et al 2012] or a severe Angelman syndrome-like neurodevelopmental disorder [Harlalka et al 2013]. This gene has increased expression in the neurons of individuals with maternal dup15q [Urraca et al 2018].
Chapter Notes
Acknowledgments
The authors are indebted to the Dup15q Alliance for its efforts to advance research into maternal dup15q. Many thanks go to Christa L Martin, PhD, Geisinger Health System, for technical assistance in the preparation of this review.
Author History
Dimitrios Arkilo, MD; Minnesota Epilepsy Group (2016-2021)
Guy Calvert, DPhil; Dup15q Alliance (2016-2021)
Stormy Chamberlain, PhD; University of Connecticut Health Center (2016-2021)
Edwin H Cook, MD; University of Illinois at Chicago (2016-2021)
Orrin Devinsky, MD; New York University Langone Medical Center (2016-2021)
Scott Dindot, PhD; Texas A&M University (2016-2021)
Charlotte DiStefano, PhD (2021-present)
Brenda M Finucane, MS, LGC; Geisinger Health System (2016-2021)
Shafali Jeste, MD (2016-present)
Janine M LaSalle, PhD; University of California Davis School of Medicine (2016-2021)
Kadi Luchsinger, BS, PT; Dup15q Alliance (2016-2021)
Laina Lusk, MMSc, CGC (2016-present)
Lawrence T Reiter, PhD; University of Tennessee Health Science Center (2016-2021)
N Carolyn Schanen, MD, PhD; Dup15q Alliance (2016-2021)
Sarah J Spence, MD, PhD; Children's Hospital Boston (2016-2021)
Ronald L Thibert, DO, MSPH; Massachusetts General Hospital (2016-2021)
Vanessa Vogel-Farley, BA (2021-present)
Revision History
- 15 July 2021 (sw) Comprehensive update posted live
- 16 June 2016 (bp) Review posted live
- 23 September 2015 (ll) Original submission
References
Literature Cited
- Al Ageeli E, Drunat S, Delanoë C, Perrin L, Baumann C, Capri Y, Fabre-Teste J, Aboura A, Dupont C, Auvin S, El Khattabi L, Chantereau D, Moncla A, Tabet AC, Verloes A. Duplication of the 15q11-q13 region: clinical and genetic study of 30 new cases. Eur J Med Genet. 2014;57:5-14. [PubMed: 24239951]
- Bassett AS. Parental origin, DNA structure, and the schizophrenia spectrum. Am J Psychiatry. 2011;168:350-3. [PMC free article: PMC3276592] [PubMed: 21474594]
- Battaglia A, Gurrieri F, Bertini E, Bellacosa A, Pomponi MG, Paravatou-Petsotas M, Mazza S, Neri G. The inv dup(15) syndrome: a clinically recognizable syndrome with altered behavior, mental retardation and epilepsy. Neurology. 1997;48:1081-6. [PubMed: 9109904]
- Battaglia A. The inv dup (15) or idic(15) syndrome (tetrasomy 15q). Orphanet J Rare Dis. 2008;3:30. [PMC free article: PMC2613132] [PubMed: 19019226]
- Battaglia A, Parrini B, Tancredi R. The behavioural phenotype of idic(15) syndrome. Am J Med Genet Part C Semin Med Genet. 2010;154C:448-55. [PubMed: 20981774]
- Borgatti R, Piccinelli P, Passoni D, Dalprà L, Miozzo M, Micheli R, Gagliardi C, Balottin U. Relationship between clinical and genetic features in "inverted duplicated chromosome 15" patients. Pediatr Neurol. 2001;24:111-6. [PubMed: 11275459]
- Bundey S, Hardy C, Vickers S, Kilpatrick MW, Corbett JA. Duplication of the 15q11-13 region in a patient with autism, epilepsy and ataxia. Dev Med Child Neurol. 1994;36:736-42. [PubMed: 8050626]
- Burnside RD, Pasion R, Mikhail FM, Carroll AJ, Robin NH, Youngs EL, Gadi IK, Keitges E, Jaswaney VL, Papenhausen PR, Potluri VR, Risheg H, Rush B, Smith JL, Schwartz S, Tepperberg JH, Butler MG. Microdeletion/microduplication of proximal 15q11.2 between BP1 and BP2: a susceptibility region for neurological dysfunction including developmental and language delay. Hum Genet. 2011;130:517-28. [PMC free article: PMC6814187] [PubMed: 21359847]
- Chaste P, Sanders SJ, Mohan KN, Klei L, Song Y, Murtha MT, Hus V, Lowe JK, Willsey AJ, Moreno-De-Luca D, Yu TW, Fombonne E, Geschwind D, Grice DE, Ledbetter DH, Lord C, Mane SM, Martin DM, Morrow EM, Walsh CA, Sutcliffe JS, State MW, Martin CL, Devlin B, Beaudet AL, Cook EH Jr, Kim SJ. Modest impact on risk for autism spectrum disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res. 2014;7:355-62. [PMC free article: PMC6003409] [PubMed: 24821083]
- Christian SL, Fantes JA, Mewborn SK, Huang B, Ledbetter DH. Large genomic duplicons map to sites of instability in the Prader-Willi/Angelman syndrome chromosome region (15q11-q13). Hum Mol Genet. 1999;8:1025-37. [PubMed: 10332034]
- Conant KD, Finucane B, Cleary N, Martin A, Muss C, Delany M, Murphy EK, Rabe O, Luchsinger K, Spence SJ, Schanen C, Devinsky O, Cook EH, LaSalle J, Reiter LT, Thibert RL. A survey of seizures and current treatments in 15q duplication syndrome. Epilepsia. 2014;55:396-402. [PubMed: 24502430]
- Cook EH Jr, Lindgren V, Leventhal BL, Courchesne R, Lincoln A, Shulman C, Lord C, Courchesne E. Autism or atypical autism in maternally but not paternally derived proximal 15q duplication. Am J Hum Genet. 1997;60:928-34. [PMC free article: PMC1712464] [PubMed: 9106540]
- Copping NA, Christian SGB, Ritter DJ, Islam MS, Buscher N, Zolkowska D, Pride MC, Berg EL, LaSalle JM, Ellegood J, Lerch JP, Reiter LT, Silverman JL, Dindot SV. Neuronal overexpression of Ube3a isoform 2 causes behavioral impairments and neuroanatomical pathology relevant to 15q11.2-q13.3 duplication syndrome. Hum Mol Genet. 2017;26:3995-4010. [PMC free article: PMC5886211] [PubMed: 29016856]
- Cox DM, Butler MG. The 15q11.2 BP1-BP2 microdeletion syndrome: a review. Int J Mol Sci. 2015;16:4068-82. [PMC free article: PMC4346944] [PubMed: 25689425]
- DeLorey TM, Handforth A, Anagnostaras SG, Homanics GE, Minassian BA, Asatourian A, Fanselow MS, Delgado-Escueta A, Ellison GD, Olsen RW. Mice lacking the beta3 subunit of the GABAA receptor have the epilepsy phenotype and many of the behavioral characteristics of Angelman syndrome. J Neurosci 1998;18:8505-14. [PMC free article: PMC6792844] [PubMed: 9763493]
- DeLorey TM, Sahbaie P, Hashemi E, Homanics GE, Clark JD. Gabrb3 gene deficient mice exhibit impaired social and exploratory behaviors, deficits in non-selective attention and hypoplasia of cerebellar vermal lobules: a potential model of autism spectrum disorder. Behav Brain Res 2008;187:207-20. [PMC free article: PMC2684890] [PubMed: 17983671]
- Depienne C, Moreno-De-Luca D, Heron D, Bouteiller D, Gennetier A, Delorme R, Chaste P, Siffroi JP, Chantot-Bastaraud S, Benyahia B, Trouillard O, Nygren G, Kopp S, Johansson M, Rastam M, Burglen L, Leguern E, Verloes A, Leboyer M, Brice A, Gillberg C, Betancur C. Screening for genomic rearrangements and methylation abnormalities of the 15q11-q13 region in autism spectrum disorders. Biol Psychiatry. 2009;66:349-59. [PubMed: 19278672]
- Devinsky O. Sudden, unexpected death in epilepsy. N Engl J Med. 2011;365:1801-11. [PubMed: 22070477]
- DiStefano C, Gulsrud A, Huberty S, Kasari C, Cook E, Reiter L, Thibert R, Jeste SS. Identification of a distinct developmental and behavioral profile in children with dup15q syndrome. J Neurodev Disord. 2016;8:19. [PMC free article: PMC4858912] [PubMed: 27158270]
- DiStefano C, Wilson RB, Hyde C, et al. Behavioral characterization of dup15q syndrome: Toward meaningful endpoints for clinical trials. Am J Med Genet A. 2020;182:71-84. [PMC free article: PMC7334030] [PubMed: 31654560]
- Friedman D, Thaler A, Thaler J, Rai S, Cook E, Schanen C, Devinsky O. Mortality in isodicentric chromosome 15 syndrome: The role of SUDEP. Epilepsy Behav. 2016;61:1-5. [PubMed: 27218684]
- Frohlich J, Reiter LT, Saravanapandian V, DiStefano C, Huberty S, Hyde C, Chamberlain S, Bearden CE, Golshani P, Irimia A, Olsen RW, Hipp JF, Jeste SS. Mechanisms underlying the EEG biomarker in Dup15q syndrome. Mol Autism. 2019;10:29. [PMC free article: PMC6609401] [PubMed: 31312421]
- Frohlich J, Senturk D, Saravanapandian V, Golshani P, Reiter LT, Sankar R, Thibert RL, DiStefano C, Huberty S, Cook EH, Jeste SS. A quantitative electrophysiological biomarker of duplication 15q11.2-q13.1 syndrome. PLoS One. 2016;11:e0167179. [PMC free article: PMC5157977] [PubMed: 27977700]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, Zhang H, Estes A, Brune CW, Bradfield JP, Imielinski M, Frackelton EC, Reichert J, Crawford EL, Munson J, Sleiman PM, Chiavacci R, Annaiah K, Thomas K, Hou C, Glaberson W, Flory J, Otieno F, Garris M, Soorya L, Klei L, Piven J, Meyer KJ, Anagnostou E, Sakurai T, Game RM, Rudd DS, Zurawiecki D, McDougle CJ, Davis LK, Miller J, Posey DJ, Michaels S, Kolevzon A, Silverman JM, Bernier R, Levy SE, Schultz RT, Dawson G, Owley T, McMahon WM, Wassink TH, Sweeney JA, Nurnberger JI, Coon H, Sutcliffe JS, Minshew NJ, Grant SF, Bucan M, Cook EH, Buxbaum JD, Devlin B, Schellenberg GD, Hakonarson H. Autism genome-wide copy number variation reveal ubiquitin and neuronal genes. Nature. 2009;459:569-73. [PMC free article: PMC2925224] [PubMed: 19404257]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, Ploegh HL, Chowdhury S, Worley PF, Steen J, Greenberg ME. The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell. 2010;140:704-16. [PMC free article: PMC2843143] [PubMed: 20211139]
- Harlalka GV, Baple EL, Cross H, Kühnle S, Cubillos-Rojas M, Matentzoglu K, Patton MA, Wagner K, Coblentz R, Ford DL, Mackay DJ, Chioza BA, Scheffner M, Rosa JL, Crosby AH. Mutation of HERC2 causes developmental delay with Angelman-like features. J Med Genet. 2013;50:65-73. [PubMed: 23243086]
- Hogart A, Nagarajan RP, Patzel KA, Yasui DH, Lasalle JM. 15q11-13 GABAA receptor genes are normally biallelically expressed in brain yet are subject to epigenetic dysregulation in autism-spectrum disorders. Hum Mol Genet. 2007;16:691-703. [PMC free article: PMC1934608] [PubMed: 17339270]
- Hogart A, Wu D, LaSalle JM, Schanen NC. The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis. 2010;38:181-91. [PMC free article: PMC2884398] [PubMed: 18840528]
- Kaminsky EB, Kaul V, Paschall J, Church DM, Bunke B, Kunig D, Moreno-De-Luca D, Moreno-De-Luca A, Mulle JG, Warren ST, Richard G, Compton JG, Fuller AE, Gliem TJ, Huang S, Collinson MN, Beal SJ, Ackley T, Pickering DL, Golden DM, Aston E, Whitby H, Shetty S, Rossi MR, Rudd MK, South ST, Brothman AR, Sanger WG, Iyer RK, Crolla JA, Thorland EC, Aradhya S, Ledbetter DH, Martin CL. An evidence-based approach to establish the functional and clinical significance of copy number variants in intellectual and developmental disabilities. Genet Med. 2011;13:777-84. [PMC free article: PMC3661946] [PubMed: 21844811]
- LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics. 2015;7:1213-28. [PMC free article: PMC4709177] [PubMed: 26585570]
- Malhotra D, Sebat J. CNVs: Harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223-41. [PMC free article: PMC3351385] [PubMed: 22424231]
- Mann SM, Wang NJ, Liu DH, Wang L, Schultz RA, Dorrani N, Sigman M, Schanen NC. Supernumerary tricentric derivative chromosome 15 in two boys with intractable epilepsy: another mechanism for partial hexasomy. Hum Genet. 2004;115:104-11. [PubMed: 15141347]
- Menold MM, Shao Y, Wolpert CM, Donnelly SL, Raiford KL, Martin ER, Ravan SA, Abramson RK, Wright HH, Delong GR, Cuccaro ML, Pericak-Vance MA, Gilbert JR. Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J Neurogenet. 2001;15:245-59. [PubMed: 12092907]
- Michelson M, Eden A, Vinkler C, Leshinsky-Silver E, Kremer U, Lerman-Sagie T, Lev D. Familial partial trisomy 15q11-13 presenting as intractable epilepsy in the child and schizophrenia in the mother. Eur J Paediatr Neurol. 2011;15:230-3. [PubMed: 21145272]
- Miller DT, Shen Y, Weiss LA, Korn J, Anselm I, Bridgemohan C, Cox GF, Dickinson H, Gentile J, Harris DJ, Hegde V, Hundley R, Khwaja O, Kothare S, Luedke C, Nasir R, Poduri A, Prasad K, Raffalli P, Reinhard A, Smith SE, Sobeih MM, Soul JS, Stoler J, Takeoka M, Tan WH, Thakuria J, Wolff R, Yusupov R, Gusella JF, Daly MJ, Wu BL. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242-8. [PMC free article: PMC4090085] [PubMed: 18805830]
- Moreno-De-Luca D, Sanders SJ, Willsey AJ, Mulle JG, Lowe JK, Geschwind DH, State MW, Martin CL, Ledbetter DH. Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry. 2013;18:1090-5. [PMC free article: PMC3720840] [PubMed: 23044707]
- Piard J, Philippe C, Marvier M, Beneteau C, Roth V, Valduga M, Béri M, Bonnet C, Grégoire MJ, Jonveaux P, Leheup B. Clinical and molecular characterization of a large family with an interstitial 15q11q13 duplication. Am J Med Genet A. 2010;152A:1933-41. [PubMed: 20635369]
- Puffenberger EG, Jinks RN, Wang H, Xin B, Fiorentini C, Sherman EA, Degrazio D, Shaw C, Sougnez C, Cibulskis K, Gabriel S, Kelley RI, Morton DH, Strauss KA. A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum Mutat. 2012;33:1639-46. [PubMed: 23065719]
- Rees E, Walters JTR, Georgieva L, Isles AR, Chambert KD, Richards AL, Mahoney-Davies G, Legge SE, Moran JL, McCarroll SA, O'Donovan MC, Owen MJ, Kirov G. Analysis of copy number variations at 15 schizophrenia-associated loci. Br J Psychiatry. 2014;204:108-14. [PMC free article: PMC3909838] [PubMed: 24311552]
- Roberts SE, Maggouta F, Thomas NS, Jacobs PA, Crolla JA. Molecular and fluorescence in situ hybridization characterization of the breakpoints in 46 large supernumerary marker 15 chromosomes reveals an unexpected level of complexity. Am J Hum Genet. 2003;73:1061-72. [PMC free article: PMC1180486] [PubMed: 14560400]
- Ryvlin P, Cucherat M, Rheims S. Risk of sudden unexpected death in epilepsy in patients given adjunctive antiepileptic treatment for refractory seizures: a meta-analysis of placebo-controlled randomised trials. Lancet Neurol. 2011;10:961-8. [PubMed: 21937278]
- Ryvlin P, Nashef L, Tomson T. Prevention of sudden unexpected death in epilepsy: a realistic goal? Epilepsia. 2013;54:23-8. [PubMed: 23646967]
- Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483-92. [PMC free article: PMC1224722] [PubMed: 15615769]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, Goldberg AP, Jinlu C, Keaney JF III, Klei L, Mandell JD, Moreno-De-Luca D, Poultney CS, Robinson EB, Smith L, Solli-Nowlan T, Su MY, Teran NA, Walker MF, Werling DM, Beaudet AL, Cantor RM, Fombonne E, Geschwind DH, Grice DE, Lord C, Lowe JK, Mane SM, Martin DM, Morrow EM, Talkowski ME, Sutcliffe JS, Walsh CA, Yu TW, Ledbetter DH, Martin CL, Cook EH, Buxbaum JD, Daly MJ, Devlin B, Roeder K, State MW, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215-33. [PMC free article: PMC4624267] [PubMed: 26402605]
- Saravanapandian V, Frohlich J, Hipp JF, Hyde C, Scheffler AW, Golshani P, Cook EH, Reiter LT, Senturk D, Jeste SS. Properties of beta oscillations in Dup15q syndrome. J Neurodev Disord. 2020;12:22. [PMC free article: PMC7425173] [PubMed: 32791992]
- Simon EW, Haas-Givler B, Finucane B. A longitudinal follow-up study of autistic symptoms in children and adults with duplications of 15q11-13. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:463-7. [PubMed: 19548260]
- Ungaro P, Christian SL, Fantes JA, Mutirangura A, Black S, Reynolds J, Malcolm S, Dobyns WB, Ledbetter DH. Molecular characterisation of four cases of intrachromosomal triplication of chromosome 15q11-q14. J Med Genet. 2001;38:26-34. [PMC free article: PMC1734721] [PubMed: 11134237]
- Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, Schanen NC, Esmer C, Lamport D, Reiter LT. The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res. 2013;6:268-79. [PMC free article: PMC3884762] [PubMed: 23495136]
- Urraca N, Davis L, Cook EH, Schanen NC, Reiter LT. A single-tube quantitative high-resolution melting curve method for parent-of-origin determination of 15q duplications. Genet Test Mol Biomarkers. 2010;14:571-6. [PMC free article: PMC3064527] [PubMed: 20642357]
- Urraca N, Hope K, Victor AK, Belgard TG, Memon R, Goorha S, Valdez C, Tran QT, Sanchez S, Ramirez J, Donaldson M, Bridges D, Reiter LT. Significant transcriptional changes in 15q duplication but not Angelman syndrome deletion stem cell-derived neurons. Mol Autism. 2018;9:6. [PMC free article: PMC5787244] [PubMed: 29423132]
- van Bon BW, Mefford HC, Menten B, Koolen DA, Sharp AJ, Nillesen WM, Innis JW, de Ravel TJ, Mercer CL, Fichera M, Stewart H, Connell LE, Ounap K, Lachlan K, Castle B, Van der Aa N, van Ravenswaaij C, Nobrega MA, Serra-Juhé C, Simonic I, de Leeuw N, Pfundt R, Bongers EM, Baker C, Finnemore P, Huang S, Maloney VK, Crolla JA, van Kalmthout M, Elia M, Vandeweyer G, Fryns JP, Janssens S, Foulds N, Reitano S, Smith K, Parkel S, Loeys B, Woods CG, Oostra A, Speleman F, Pereira AC, Kurg A, Willatt L, Knight SJ, Vermeesch JR, Romano C, Barber JC, Mortier G, Pérez-Jurado LA, Kooy F, Brunner HG, Eichler EE, Kleefstra T, de Vries BB. Further delineation of the 15q13 microdeletion and duplication syndromes: a clinical spectrum varying from non-pathogenic to a severe outcome. J Med Genet. 2009;46:511-23. [PMC free article: PMC3395372] [PubMed: 19372089]
- Vanlerberghe C, Petit F, Malan V, Vincent-Delorme C, Bouquillon S, Boute O, Holder-Espinasse M, Delobel B, Duban B, Vallee L, Cuisset JM, Lemaitre MP, Vantyghem MC, Pigeyre M, Lanco-Dosen S, Plessis G, Gerard M, Decamp M, Mathieu M, Morin G, Jedraszak G, Bilan F, Gilbert-Dussardier B, Fauvert D, Roume J, Cormier-Daire V, Caumes R, Puechberty J, Genevieve D, Sarda P, Pinson L, Blanchet P, Lemeur N, Sheth F, Manouvrier-Hanu S, Andrieux J. 15q11.2 microdeletion (BP1-BP2) and developmental delay, behaviour issues, epilepsy and congenital heart disease: a series of 52 patients. Eur J Med Genet. 2015;58:140-7. [PubMed: 25596525]
- Wang NJ, Parokonny AS, Thatcher KN, Driscoll J, Malone BM, Dorrani N, Sigman M, LaSalle JM, Schanen NC. Multiple forms of atypical rearrangements generating supernumerary derivative chromosome 15. BMC Genet. 2008;9:2. [PMC free article: PMC2249594] [PubMed: 18177502]
- Wegiel J, Schanen NC, Cook EH, Sigman M, Brown WT, Kuchna I, Nowicki K, Wegiel J, Imaki H, Yong Ma S, Marchi E, Wierzba-Bobrowski T, Chauhan A, Chauhan V, Cohen IL, London E, Flory M, Lach B, Wisnewski T. Differences between the pattern of developmental abnormalities in autism associated with duplications 15q11.2-q13 and idiopathic autism. J Neuropathol Exp Neurol. 2012;71:382-97. [PMC free article: PMC3612833] [PubMed: 22487857]
- Zielinski C, Müller C, Smolen J. Use of plasmapheresis in therapy of systemic lupus erythematosus: a controlled study. Acta Med Austriaca. 1988;15:155-8. [PubMed: 3064527]
Publication Details
Author Information and Affiliations
Philadelphia, Pennsylvania
Highland Park, Illinois
Los Angeles, California
Publication History
Initial Posting: June 16, 2016; Last Update: July 15, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Lusk L, Vogel-Farley V, DiStefano C, et al. Maternal 15q Duplication Syndrome. 2016 Jun 16 [Updated 2021 Jul 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.