Clinical Description
Multicentric osteolysis nodulosis and arthropathy (MONA) is a skeletal dysplasia characterized by progressive osteolysis (particularly of the carpal and tarsal bones), osteoporosis, subcutaneous nodules on the palms and soles, and progressive arthropathy (contractures, pain, joint swelling/stiffness). Other manifestations include pigmented lesions on the skin, coarse facies, corneal opacities, and cardiac defects.
Most affected children are apparently normal at birth. Onset is usually between ages six months and six years [Azzollini et al 2014]; the range is from birth to 11 years [Castberg et al 2013, Bhavani et al 2016].
To date, 51 individuals have been identified with biallelic pathogenic variants in MMP2 [Martignetti et al 2001, Zankl et al 2005, Rouzier et al 2006, Phadke et al 2007, Zankl et al 2007, Tuysuz et al 2009, Gok et al 2010, Jeong et al 2010, Temtamy et al 2012, Castberg et al 2013, Azzollini et al 2014, Ekbote et al 2014, Bader-Meunier et al 2016, Bhavani et al 2016, Pichler et al 2016, Kröger et al 2019, Elsebaie et al 2021]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Multicentric Osteolysis Nodulosis and Arthropathy: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
| Joint disease | ~100% | |
| Progressive osteolysis | 100% | Onset in the first few years of life |
| Osteopenia | 100% | |
| Coarse facial features | ~86% | |
| Gingival hypertrophy | ~35% | |
| Subcutaneous nodules | ~80% | |
| Congenital heart disease | >30% | |
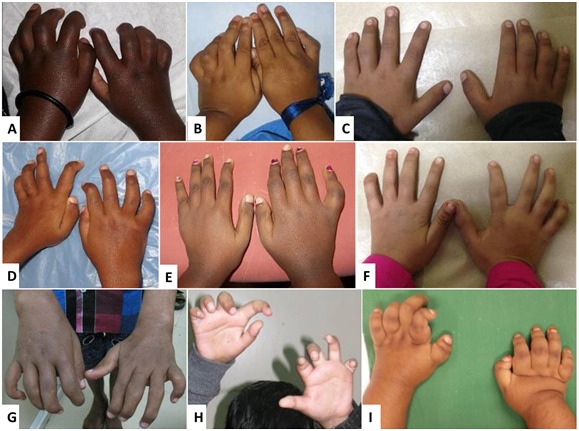
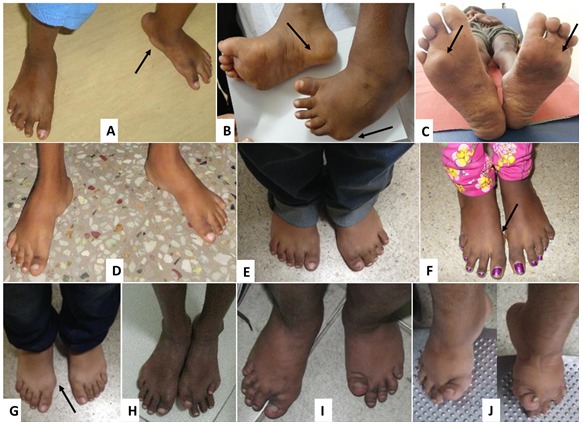
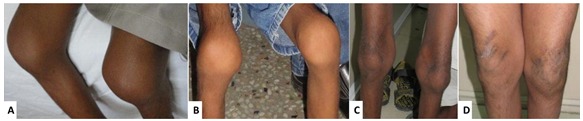
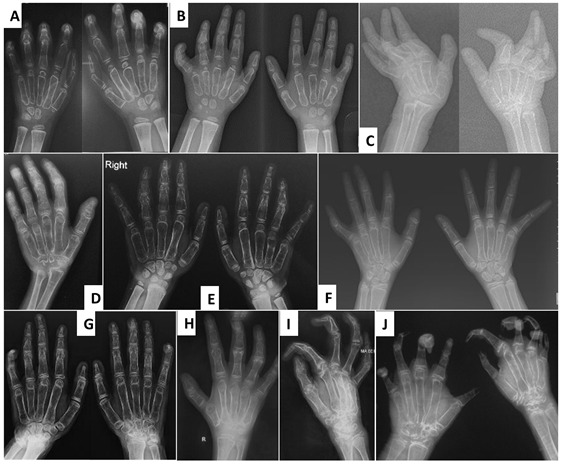
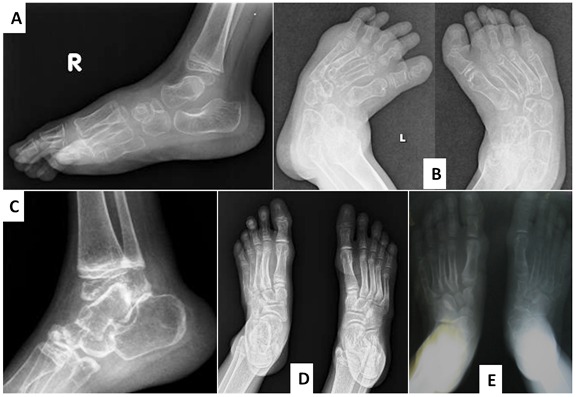
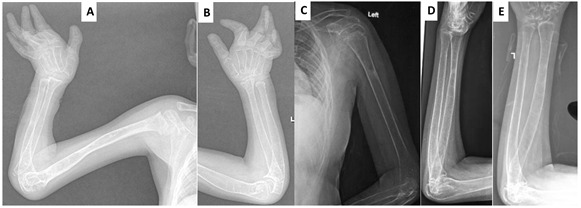
Joint manifestations. Peripheral joints are more involved than proximal joints. Small joints of the hands and feet are the most obvious sites of involvement. Progressive bone destruction leads to pain, swelling, stiffness, and later flexion contractures (, ). Foot and hand deformities and progressive shortening of digits occur with age.
Knees show swelling and contractures in the majority (); hip involvement is less severe [Bhavani et al 2016].
Wrists, ankles, and elbows are also involved.
Over time all affected individuals need assistance with mobility. Those with more severe manifestations are wheelchair bound between ages three and 12 years [Zankl et al 2005, Rouzier et al 2006, Temtamy et al 2012].
Osteopenia. Low bone density and osteoporosis may lead to an increased risk of fracture of long bones and vertebrae [Jeong et al 2010, Temtamy et al 2012].
Short stature. Most individuals have short stature [Castberg et al 2013]. Stature is normal in early childhood; progressive bone and joint deformities could be the cause of short stature.
Facial features. Coarsening of facial features, bulbous nose, proptosis, strabismus, and gingival hypertrophy have been observed in several affected individuals [Bhavani et al 2016].
Skin. Subcutaneous, firm, palpable nodules in the palms and soles are noted in the majority of (but not all) affected individuals ( and ). The presence of subcutaneous nodules may be age dependent. To date it has not been possible to determine if these nodules are painful as affected individuals have pain in their hands and feet due to osteopenia, fractures, contractures, limitation of movement, and sometimes inflammation.
Other cutaneous manifestations include hyperpigmentation and thickening. Serpiginous hyperpigmented cutaneous lesions are present in a few individuals ( and ) [Zankl et al 2005, Azzollini et al 2014, Bhavani et al 2016].
In some individuals excessive skin folds in the hands and feet have resulted from destruction of underlying bones ().
Heart. Congenital heart defects (transposition of great arteries, atrial septal defect, ventricular septal defect, bicuspid aortic valve, and mitral valve prolapse) have been observed in one third of individuals reported to date [Tuysuz et al 2009, Castberg et al 2013, Bhavani et al 2016].
Intellect is normal.
Other
Nomenclature
In addition to MONA, this phenotype has been reported in the literature as Torg syndrome, Winchester-Torg (or Torg-Winchester) syndrome, and nodulosis-arthropathy-osteolysis (NAO) syndrome. All of these conditions have been shown to be caused by biallelic pathogenic variants in MMP2 with no discernible genotype-phenotype correlation. The term MONA is therefore used throughout this GeneReview to refer to all of these conditions.
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], MONA is referred to as MMP2-related multicentric osteolysis, nodulosis, and arthropathy and is included in the osteolysis group.