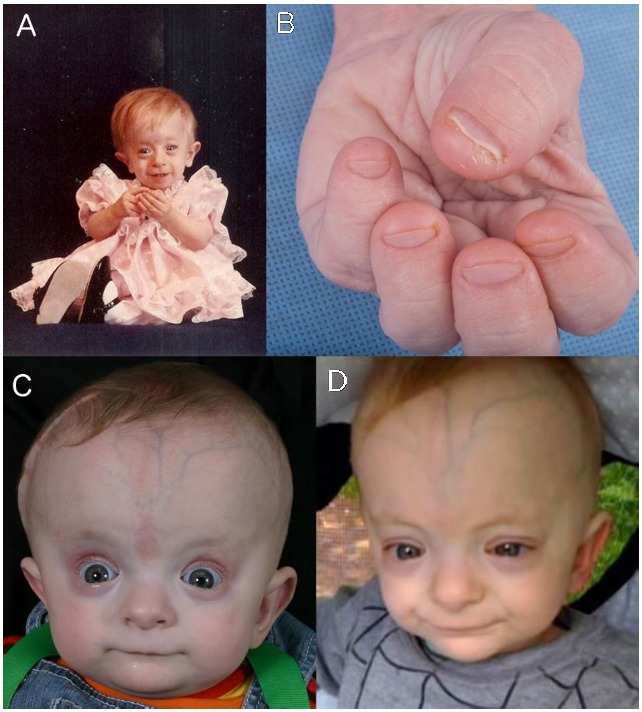
Clinical Description
Saul-Wilson syndrome is a skeletal dysplasia characterized by profound short stature, distinctive craniofacial features, short distal phalanges of fingers and toes, and often clubfoot. Early development (primarily speech) is delayed; cognition is normal. Other findings can include hearing loss (conductive, sensorineural, and mixed), lamellar cataracts, and/or rod-cone retinal dystrophy.
A total of 16 individuals with Saul-Wilson syndrome have been reported to date. Saul-Wilson syndrome was first described in a small-for-gestational-age infant with bulging fontanelles, clubfoot, blue sclerae, and blunted fingertips; over time, growth was delayed and the child developed bilateral cataracts, and hearing loss as the result of frequent otitis media [Saul 1982]. Three additional individuals with similar features were reported [Saul & Wilson 1990, Hersh et al 1994]. Subsequently the diagnosis of Saul-Wilson syndrome was entertained in a child without typical facial features [Chinen et al 2015], but the diagnosis could not be confirmed by molecular genetic testing. Fourteen individuals were described in 2018, including two originally reported in the 1990s [Ferreira et al 2018]. Since that publication, a few additional individuals with Saul-Wilson syndrome worldwide have been diagnosed [Author, personal observation]. The clinical findings discussed in this section are based on these reports.
Growth
Individuals with Saul-Wilson syndrome show impaired postnatal growth, and several also had intrauterine growth restriction (IUGR).
Mean length, weight, and head circumference at birth:
Length. 44.1 cm (range: 38.0-49.0)
Weight. 2.09 kg (range: 1.45-2.80)
Head circumference. 31.7 cm (range: 29.0-34.0)
Z scores at birth:
Length. -2.3 (1.5 SD; range: -0.4 to -5.1)
Weight. -2.4 (0.7 SD; range: -1.2 to -3.8)
Head circumference: -2.0 (0.9 SD; range: -0.8 to -3.9)
Z scores decline sharply over the first few months of life. At last examination:
Stature. -6.3 (1.8 SD; range: -3.5 to -9.8)
Weight. -4.0 (1.2 SD; range: -1.1 to -5.8)
Head circumference. -1.7 (1.7 SD; range: 0.8 to -5.0)
Based on data available from three adults
Mean height, weight, and head circumference at skeletal maturity:
Z scores at skeletal maturity:
Height. -8.9 (0.8 SD; range: -8.3 to -9.8)
Weight. -4.3 (0.6 SD; range: -3.6 to -4.8)
Head circumference. -3.9 (1.6 SD; range: -2.7 to -5.0)
Growth charts for clinical use are currently under development.
Despite absolute microcephaly, head circumferences exceed the height by more than 2 SD, with consequent relative macrocephaly.
Development
Speech delay (8/11) and motor delay (12/14) are common, probably related to the presence of hearing loss and skeletal deformities, respectively; cognitive development does not appear to be affected.
Ophthalmologic Features
The majority of affected individuals develop lamellar cataracts during the first few years of life (10/13), and several developed retinal involvement (6/9). Retinal pigmentary changes can be seen in the periphery as early as the toddler years. During adolescence and early adulthood, a rod-cone dystrophy (5/9) becomes evident with constricted visual fields and night blindness.
Macular cystic changes were also described (2/4). One individual had myelinated retinal nerve fibers [Ferreira et al 2020].
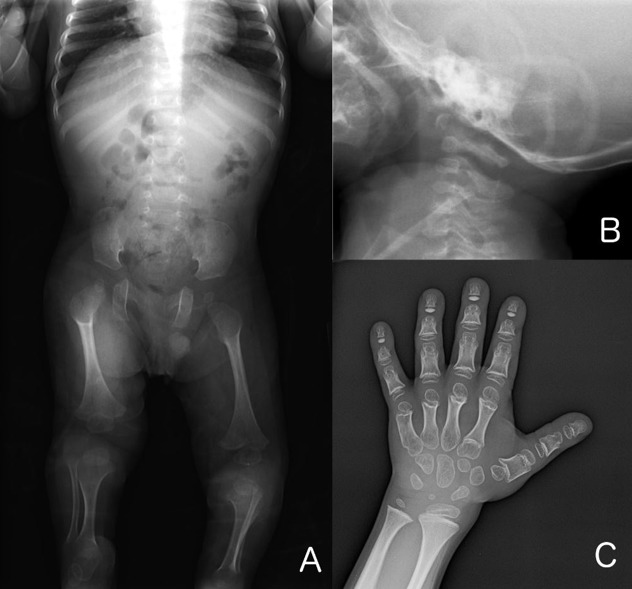
Skeletal Features
Shortening of the distal phalanges of fingers and toes is appreciated on physical examination (12/14). This finding, apparent at birth, did not progress over time. The majority of individuals (10/14) had clubfoot, and in some cases residual deformity even after multiple attempts at surgical repair [Ferreira et al 2020]. Pectus deformity (5/14) and cervical spinal cord compression (3/7) have also been reported.
Bone fragility has been suggested, as several individuals (4/14) developed fractures with minimal or no known trauma [Ferreira et al 2020]. One of these individuals had poor fracture healing, which was also described in the original patient [Saul & Wilson 1990]. Nonunion with pseudoarthrosis has been seen in two individuals [Saul & Wilson 1990, Ferreira et al 2020]. Although DXA scans for two individuals reported bone mineral density (BMD) <2 SD below the mean, the height-adjusted BMD [Zemel et al 2010, Zemel et al 2011] was normal in both [Author, personal observation].
Osteoarticular pain was reported by all three adults. Two had confirmed degenerative joint disease, leading to joint replacement surgeries in one individual in her 20s [Ferreira et al 2020].
The combination of megaepiphyses with coxa valga, leading to acetabulum-femoral epiphyseal incongruence, may contribute to premature osteoarthropathy of the hip.
Other
Hearing loss, seen in the majority of affected individuals over time, can be conductive, sensorineural, or mixed. In one child hearing impairment associated with inner-ear malformations was detected on newborn hearing screen.
MRI findings. Ventriculomegaly was seen in 5/9 and spinal cord syrinx in 1/4 individuals. Spinal cord compression was observed in 3/7: in one child with soft-tissue pannus surrounding the odontoid process, it was seen as early as age four years, and in another child with cervical spine instability, as late as age 14 years.
Intermittent neutropenia, though not appreciated in the first two months of life, was seen in all 12 individuals subsequently evaluated for this finding [Ferreira et al 2020]. The earliest known age of onset is three months, and it still occurred in adults. While intermittent neutropenia could be one possible explanation for the frequent (although rarely life-threatening) respiratory infections experienced in the first years of life, the neutropenia persisted into adulthood whereas the number of respiratory infections decreased over time.
Asymptomatic elevation of liver transaminases. Elevated aspartate aminotransferase was observed in 6/8 individuals and elevated alanine aminotransferase in 3/8, without abnormalities in other liver function tests, such as serum albumin, coagulation parameters, alkaline phosphatase, and bilirubin [Ferreira et al 2020].