Summary
Clinical characteristics.
Encephalocraniocutaneous lipomatosis (ECCL) comprises a spectrum of predominantly congenital anomalies. In its typical form, ECCL is characterized by congenital anomalies of the skin (nevus psiloliparus, patchy or streaky non-scarring alopecia, subcutaneous lipomas in the frontotemporal region, focal skin aplasia or hypoplasia on the scalp, and/or small nodular skin tags on the eyelids or between the outer canthus and tragus), eye (choristoma), and brain (in particular intracranial and spinal lipomas). To a much lesser degree, the bones and the heart can be affected. About 40% of affected individuals have bilateral abnormalities of the skin or the eyes. About one third of affected individuals have normal cognitive development, another one third have mild developmental delay (DD) or intellectual disability (ID), and the final one third have severe or unspecified DD/ID. Half of individuals have seizures. Affected individuals are at an increased (i.e., above the general population) risk of developing brain tumors, particularly low-grade gliomas such as pilocytic astrocytomas. There is evidence that oculoectodermal syndrome (OES) may constitute a clinical spectrum with ECCL, with OES on the mild end and ECCL on the more severe end of the spectrum.
Diagnosis/testing.
A clinical diagnosis of ECCL can be made in individuals with:
- Involvement of at least three systems, with major criteria in at least two of the three systems; OR
- Involvement of at least three systems, in which one major criterion is either a biopsy-proven nevus psiloliparus (NP) OR a possible NP with at least one other minor skin criterion; OR
- At least one major criterion in each of two systems, where one major criterion is either a biopsy-proven NP or a possible NP with at least one other minor skin criterion.
A molecular diagnosis can be established in a proband with suggestive findings and a mosaic activating pathogenic variant identified in either FGFR1 or KRAS. Due to the mosaic nature of the condition, molecular genetic testing on DNA derived from affected tissue has the best detection rate and molecular methods that can detect low levels of mosaicism are recommended.
Management.
Treatment of manifestations: Standard treatment for skin manifestations (when appropriate; most of the skin findings do not require active management), choristomas / eye anomalies (including community vision services, as needed), low-grade gliomas, DD/ID, and jaw/dental anomalies; standard treatment with anti-seizure medication for those with epilepsy; standard treatment of Wilms tumor per oncologist.
Surveillance: Assess for new neurologic manifestations (changes in tone, seizures), developmental progress, and educational needs at each visit; dental evaluation at least every six months; unless the individual has molecularly confirmed KRAS-related ECCL, consider performing brain MRI to screen for brain tumors annually or as clinically indicated; ophthalmologic evaluations annually in childhood and adolescence or as clinically indicated; consideration of renal ultrasound every three months until age eight years to screen for Wilms tumor in those who have a KRAS pathogenic variant that involves codon 12.
Genetic counseling.
ECCL is not known to be inherited. No confirmed vertical transmission or sib recurrence has been reported. Given the postzygotic mutational mechanism of ECCL, the risk for an affected sib would be expected to be the same as in the general population.
Diagnosis
Clinical diagnostic criteria for encephalocraniocutaneous lipomatosis (ECCL) have been published [Moog 2009] and are adapted below based on the recent literature (see Establishing the Diagnosis).
Suggestive Findings
ECCL should be suspected in individuals with at least one major criterion in each of two different systems or in individuals with a biopsy-proven or possible nevus psiloliparus (NP), and one minor criterion in a second (non-skin) system (for clinical diagnostic criteria, see Clinical Diagnosis).
Major Criteria
Skin
- Biopsy-proven nevus psiloliparus (NP) (Figure 1)
- Possible NP in addition to one or more of the other minor skin criteria listed below
- Two or more minor skin criteria listed below
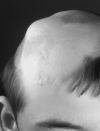
Figure 1:
Nevus psiloliparus: a smooth hairless fatty tissue nevus on the scalp Reproduced with permission from Moog [2009]
Eye. Choristoma (e.g., epibulbar dermoid) (Figure 2C)
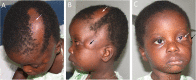
Central nervous system (CNS)
- Intracranial lipoma (Figure 3)
- Intraspinal lipoma (Figure 4)
- Low-grade glioma (if associated with other suggestive findings)
- Two minor CNS criteria listed below
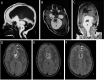
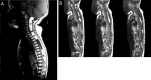
Figure 4.
Brain and spinal cord MRI in ECCL An image from one affected individual (A) shows several large lipomas ventral to the spinal cord (black asterisks) and a subcutaneous nuchal fatty mass.
Other
- Jaw tumor (osteoma, odontoma, or ossifying fibroma) (Figure 5)
- Multiple bone cysts (Figure 6)
- Aortic coarctation
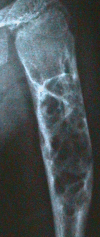
Figure 6.
Multiple lytic bone lesions in the humerus Reproduced with permission from Moog [2009]
Minor Criteria
Skin
- Possible NP
- Focal skin aplasia/hypoplasia on the scalp
- Small nodular skin tags on the eyelids or between outer canthus and tragus
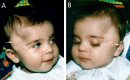
Figure 7.
An affected individual age 5 months. Note the bilateral subcutaneous fatty accumulations (white arrows). Adapted with permission from Moog et al [2007a]
Eye
- Corneal and other anterior chamber anomalies
- Ocular or eyelid coloboma
- Calcification of the globe
CNS
- Abnormal intracranial vessels (e.g., angioma, excessive vessels)
- Arachnoid cyst or other abnormality of meninges
- Complete or partial atrophy of a hemisphere
- Porencephalic cysts(s)
- Asymmetrically dilated ventricles or hydrocephalus
- Calcification not involving the basal ganglia
Establishing the Diagnosis
The clinical diagnosis of ECCL can be established in a proband based on clinical diagnostic criteria [Moog 2009], or the molecular diagnosis can be established in a proband with suggestive findings (i.e., findings consistent with ECCL as noted above but not sufficient to meet the clinical diagnostic criteria for ECCL) and a mosaic activating pathogenic variant in either FGFR1 or KRAS identified by molecular genetic testing (see Table 1).
Clinical Diagnosis
A clinical diagnosis can be made in an individual with:
- Involvement of at least three systems, with major criteria in at least two out of the three systems; OR
- Involvement of at least three systems, in which one major criterion is either a biopsy-proven NP OR a possible NP with at least one other minor skin criterion; OR
- At least one major criterion in each of two systems, where one major criterion is either a biopsy-proven NP OR a possible NP with at least one other minor skin criterion.
Molecular Diagnosis
Molecular genetic testing approaches typically include use of gene-targeted testing (single-gene testing, multigene panel), although comprehensive genomic testing (exome sequencing, genome sequencing) is available. Because all reported FGFR1 or KRAS pathogenic variants are postzygotic (and thus mosaic), more than one tissue (excluding blood, buccal swabs, and saliva) may need to be tested.
- Experience suggests that sequence analysis of DNA derived from affected tissue has the best detection rate with the highest levels detected in fibroblast culture derived from skin overlying nevus psiloliparus, non-ossifying fibromas, and dermoids. Pathogenic variants have been detected in DNA derived from skin biopsy-based fibroblast cultures; however, the impact of cell culture on pathogenic variant levels remains uncertain. DNA directly obtained from these tissues, without culture, should be prioritized for genetic testing.
- Intermediate levels of somatic pathogenic variants have been detected in lipoma, bone, and non-affected skin, but these should be used primarily when biopsies of higher-yield lesions are not available.
- The level of mosaicism for an activating variant in skin is variable, whether a lesion (aplasia, alopecia, or hyperpigmentation) is seen or not.
- Sequence analysis of DNA from blood, buccal swabs, and saliva has been uniformly normal (i.e., it did not identify a mosaic-activating pathogenic variant) ‒ with the exception of one individual in whom ultra-deep sequencing by digital droplet PCR (ddPCR) detected a pathogenic variant allele fraction (level of mosaicism) at or below 1% [Kordacka et al 2019]. Thus, these tissues should be avoided for sequencing.
Note: On analysis of DNA derived from affected tissues, the method used for testing must be sensitive enough to detect low-level mosaicism of a pathogenic variant (see Molecular Pathogenesis, Gene-specific laboratory technical considerations).
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of ECCL has not been considered may be diagnosed using genomic testing (see Option 2), if an appropriate sample is used.
Option 1
When the phenotypic findings suggest a diagnosis of ECCL, molecular genetic testing approaches can include use of a multigene panel that includes both FGFR1 and KRAS, or less often, serial single-gene testing.
- A multigene panel that includes the genes listed in Table 1 and other genes of interest (see Differential Diagnosis) on an appropriate sample (see preceding) may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- Serial single-gene testing. Because the mosaic-activating pathogenic variants in FGFR1 and KRAS are restricted to a few specific variants, targeted genetic testing for known mosaic-activating variants in FGFR1, followed by KRAS, may be considered (see Molecular Genetics). However, sequence analysis of both FGFR1 and KRAS is also available.
Notes: (1) The mosaic pathogenic variants observed in both FGFR1 and KRAS have all been associated with gain of function, so gene-targeted deletion/duplication analysis is not recommended. (2) Failure to detect an activating FGFR1 or KRAS pathogenic variant does not exclude a diagnosis of ECCL in individuals who meet the clinical diagnostic criteria, given that a low level of mosaicism is observed in many affected individuals. Indeed, far more affected individuals have a clinical diagnosis of ECCL than a diagnosis established by molecular genetic testing.
Option 2
When the diagnosis of ECCL has not been considered because an individual has atypical phenotypic features, comprehensive genomic testing on an appropriate sample (see above) may be considered.
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Encephalocraniocutaneous Lipomatosis
Clinical Characteristics
Clinical Description
To date, at least 85 individuals with a clinical and/or molecular diagnosis of encephalocraniocutaneous lipomatosis (ECCL) have been reported [Moog 2009, Bennett et al 2016, McDonell et al 2018, Valera et al 2018]. Of these, 14 individuals have been identified with a pathogenic variant in FGFR1 [Bennett et al 2016, Bavle et al 2018, Valera et al 2018, Chacon-Camacho et al 2019, Córdoba et al 2019, Kordacka et al 2019] and a few with a pathogenic variant in KRAS [Boppudi et al 2016, McDonell et al 2018, Chacon-Camacho et al 2019, Nagatsuma et al 2019]. The following description of the phenotypic features in individuals with a clinical and/or molecular diagnosis associated with ECCL is based on these reports.
Note: There is evidence that oculoectodermal syndrome (OES) may constitute a clinical spectrum with ECCL, with OES on the mild end and ECCL on the more severe end of the spectrum. Individuals who do not fulfill the clinical diagnostic criteria for ECCL but display some features have sometimes been reported as having OES (see Nomenclature).
ECCL comprises a spectrum of predominantly congenital anomalies. In its typical form, ECCL is characterized by congenital skin, eye, and brain anomalies, in particular intracranial and spinal lipomas. To a much lesser degree, the bones and the heart can be affected. About 40% of affected individuals have bilateral abnormalities of the skin or the eyes. Although variable in its extent and severity, the pattern of skin and eye findings is often consistent and recognizable.
Since 2016, ECCL has also been recognized as a tumor predisposition syndrome [Bennett et al 2016].
Table 2.
Encephalocraniocutaneous Lipomatosis: Frequency of Select Features
Skin. Non-scarring alopecia (with or without underlying fatty tissue) and subcutaneous fatty masses are the most typical skin anomalies.
- A smooth, hairless fatty tissue nevus of the scalp, the so-called nevus psiloliparus (NP) (Figure 1), is the dermatologic hallmark and found in about 75% of affected individuals.
- Rarely, subcutaneous lipomas are seen outside the craniofacial region.
- Areas of focal skin aplasia are usually not extensive (up to a few cm in size) and there have been no reports of affected individuals requiring surgical treatment, such as skin graft, for focal skin aplasia.
- Small nodular skin tags (which histologically are fibromas, lipomas, fibrolipomas, or choristomas) are found in about 70%-75% of affected individuals, most often on the eyelids or following a line from the outer canthus to the tragus.
- Some individuals have several café au lait spots. Linear hyperpigmentation following the lines of Blaschko and (rarely) asymmetric growth may be seen [Moog et al 2007b].
Eyes. Choristomas, with or without other eye anomalies, are seen in the majority of affected individuals (Figure 2C). Several affected individuals have had irregular or disrupted eyebrows.
- Choristomas are benign ocular tumors and include epibulbar or limbal dermoids (dermolipomas) derived from epidermis and dermis, and lipodermoids, which consist mainly of fat and rarely decrease vision [Hunter 2006].Dermolipomas can be associated with significant additional eye anomalies listed below.
- Most affected individuals without choristomas have one or more of the following associated eye anomalies:
- Corneal and scleral anomalies
- Other anterior chamber abnormalities
- Ocular and palpebral colobomas
- Aniridia
- Microphthalmia
- Calcification of the globe
Central nervous system (CNS) findings in 52 individuals who underwent imaging are summarized in Table 3. Note: The presence and extent of intracranial abnormalities does not correlate well with the severity of the developmental, skin, and eye findings.
- Lipomas. Intracranial lipomas (Figure 3) are the most common and specific CNS feature, found on neuroimaging in about 65% of affected individuals.
- Almost 60% of these are located in the cerebello-pontine angle.
- Spinal lipomas, which can extend over the whole length of the spinal cord (Figure 4), are also commonly seen on spinal cord imaging.Subcutaneous fatty masses may overlie the spinal lipomas or be found adjacent to the spinal cord (Figure 4).
- Vascular abnormalities. Abnormal intracranial blood vessels, such as leptomeningeal angiomatosis, thrombosed vascular malformations, and abnormal vessels, were found in nine individuals with ECCL in whom vascular imaging was performed.While this finding could represent ascertainment bias, it is suspected that these vascular abnormalities are common, as many of the other brain abnormalities observed could result from prenatal (or, less likely, postnatal) vascular perfusion defects or hemorrhage, including asymmetric brain atrophy, porencephaly, ventriculomegaly, and calcifications [Moog et al 2007a].
Table 3.
Encephalocraniocutaneous Lipomatosis: Frequency of Central Nervous System Anomalies in 52 Affected Individuals Who Underwent Imaging
Neurology. The spectrum of neurologic dysfunction is broad, ranging from normal cognitive development and no seizures to a very disabling disorder with severe intellectual disability (ID), intractable seizures, and other physical disabilities.
- Among reported individuals with ECCL, about one third have normal cognitive development, one third have mild developmental delay (DD) or ID, and the final one third have severe or unspecified DD/ID.
- About half of affected individuals had a history of typically infant- or childhood-onset epileptic seizures of different types that may be refractory or difficult to treat.
Musculoskeletal
- Jaw tumors identified as osteomas, odontomas, or ossifying fibromas have been described in affected individuals [Zielińska-Kaźmierska et al 2005, Moog 2009] (see Tumors below). These tumors are typically not malignant but can grow into the surrounding tissues, thus displacing teeth and other facial tissues.
- In severely affected individuals, possibly progressive, multiple lytic bone lesions affecting the long bones have been described (Figure 6) [Moog et al 2007b].
Tumors (cancers). Apart from lipomas and jaw tumors, ECCL is associated with increased risk for:
- Brain tumors, specifically low-grade gliomas (although high-grade gliomas have also been observed) [Phi et al 2010, Bennett et al 2016, Valera et al 2018].Pilocytic astrocytomas have been reported in six of 14 (43%) individuals with FGFR1-related ECCL; they have not been reported in those with KRAS-related ECCL (see Phenotype Correlations by Gene).
- Possibly Wilms tumor
- The first reported child with ECCL and Wilms tumor also had severe growth restriction, which is unusual in individuals with ECCL, such that Wilms tumor in this situation could represent a rare co-occurrence with ECCL [Damar et al 2017].
- Currently, insufficient evidence has accumulated to support a general recommendation for Wilms tumor screening in individuals with ECCL. Such screening may, however, be considered in individuals with KRAS-related ECCL in whom a pathogenic variant involving codon 12 is identified.
- Note: Wilms tumor and nephroblastomatosis have been reported in two individuals with postzygotic KRAS variants c.35G>A (p.Gly12Asp) and c.35G>T (p.Gly12Val), respectively, and phenotypes different from ECCL and oculoectodermal syndrome [Chang et al 2021] (see Genetically Related Disorders).
Other findings include:
- Macrocephaly (in ~20% of affected individuals);
- Congenital heart malformations, in particular aortic coarctation.
Oculoectodermal syndrome (OES; OMIM 600268) shares many features with ECCL, including focal alopecia or aplasia of the scalp, NP (possibly), periorbital skin tags, linear hyperpigmentation, ocular choristomas, macrocephaly, benign bone tumors, and coarctation of the aorta [Ardinger et al 2007]. Cranial MRI has been performed in about 60% of individuals with OES and may show arachnoid cysts, asymmetry of hemispheres, or mild dilatation of ventricles. Unlike ECCL, no lipomas or brain tumors (including pilocytic astrocytomas) have been reported in individuals with OES to date.
In at least seven individuals with a clinical diagnosis of OES, postzygotic activating variants in KRAS have been identified, many of which involve codon p.Ala146 [Boppudi et al 2016, McDonell et al 2018, Chacon-Camacho et al 2019]; pathogenic gain-of-function variants involving the p.Ala146 codon have also been identified in individuals who meet the clinical diagnostic criteria for ECCL. As noted above, recent data suggests that ECCL and OES are manifestations of a single spectrum (see Phenotype Correlations by Gene), as was first suggested in 2007 [Ardinger et al 2007, Moog et al 2007a]. Further research is needed to determine the phenotypes related to specific postzygotic activating pathogenic variants in KRAS.
Phenotype Correlations by Gene
Comparing FGFR1-related ECCL to KRAS-related ECCL, a gene-phenotype correlation emerges, especially when including individuals reported as having the OES phenotype who were found to have a KRAS pathogenic variant (see Nomenclature).
Compared to individuals with FGFR1-related ECCL, individuals with KRAS-related ECCL are:
- Less likely to have central nervous system lipomas
- Less likely to develop an astrocytoma (none reported to date)
- More likely to have:
- Body asymmetry
- Pigmentary mosaicism (hyperpigmented, hypopigmented, or both)
- Epidermal nevi
- Other malformations
Although these are preliminary considerations based on a small number of known affected individuals, the difference between FGFR1-related ECCL and KRAS-related ECCL (including those with an OES phenotype) needs to be reviewed in future, as different surveillance and management needs are likely ‒ underscoring the need for molecular confirmation of the diagnosis.
Genotype-Phenotype Correlations
To date, all individuals with molecularly confirmed ECCL have had mosaicism for one of a few known gain-of-function pathogenic variants in either FGFR1 or KRAS (see Molecular Genetics, Table 9, and Table 1).
Nomenclature
ECCL refers to the characteristic phenotypic findings involving the skin, eye, and central nervous system.
OES may be at one end of the ECCL spectrum [Ardinger et al 2007, Moog et al 2007a], and some individuals who do not fulfill the clinical diagnostic criteria for ECCL but display some features have been reported under this name. A subset of these individuals have indeed been found to have a mosaic KRAS pathogenic variant, providing further evidence that individuals with a clinical diagnosis of OES could fall within the spectrum of ECCL. However, pathogenic variants in other genes have also been found in individuals reported as having OES (see Differential Diagnosis).
Prevalence
The prevalence of ECCL is unknown. At least 85 individuals who meet the clinical diagnostic criteria or who have a molecular diagnosis of ECCL have been reported.
Genetically Related (Allelic) Disorders
Table 4 summarizes other phenotypes caused by pathogenic variants in FGFR1.
Table 4.
FGFR1 Allelic Disorders
Table 5 summarizes other phenotypes caused by pathogenic variants in KRAS.
Table 5.
KRAS Allelic Disorders
Note: Wilms tumor and nephroblastomatosis have been reported in two individuals with postzygotic KRAS variants c.35G>A (p.Gly12Asp) and c.35G>T (p.Gly12Val), respectively, and phenotypes different from encephalocraniocutaneous lipomatosis (ECCL) and oculoectodermal syndrome [Chang et al 2021].
Sporadic tumors occurring as single tumors in the absence of other findings of ECCL frequently contain a somatic variant in FGFR1 and/or KRAS that is not present in the germline. In these circumstances predisposition to these tumors is not heritable. For more information, see Cancer and Benign Tumors.
Differential Diagnosis
The differential diagnosis of encephalocraniocutaneous lipomatosis (ECCL) also includes other mosaic RASopathies (a group of syndromes caused by mosaic mutations of the Ras/MAPK signaling pathway [Hafner & Groesser 2013]):
- NRAS. A postzygotic gain-of-function variant in NRAS (c.182A>G:p.Gln61Arg) was reported in a newborn with an oculoectodermal syndrome (OES) phenotype plus a few additional features such as dark brown to violet bullae over the scalp and unilateral polymicrogyria [Richters et al 2020].
- Neurocutaneous melanosis (NCMS) (OMIM 249400) is a severe form of congenital melanocytic nevus syndrome (CMNS) (OMIM 137550) in association with abnormalities of the central nervous system possibly leading to seizures and other neurologic signs. In rare cases, NCMS may present with the skin, eye, or brain findings seen in OES or ECCL [Ahmed et al 2002, Jain et al 2014]. NCMS is caused by postzygotic activating variants in NRAS. The individual mentioned above with a postzygotic variant in NRAS and an OES phenotype showed a dark coloration of bullous scalp lesions but no further signs of CMNS [Richters et al 2020].
- Schimmelpenning-Feuerstein-Mims syndrome (SFMS, also referred to as linear sebaceous nevus syndrome and, formerly, epidermal nevus syndrome) (OMIM 163200). The dermatologic hallmark of this disorder is sebaceous nevi and other epidermal nevi following the lines of Blaschko, which are rarely seen in ECCL. SFMS is associated with congenital anomalies of the eye, brain, and skeleton, which overlap with features seen in ECCL (choristomas, coloboma, hemimegalencephaly, cortical malformations, cortical atrophy, porencephalic cysts, calcifications, lipoma [rarely], and focal bone lesions) [Wang et al 2014]. SFMS (as well as isolated sebaceous nevus) has been shown to result from postzygotic activating variants in HRAS, KRAS, or NRAS [Groesser et al 2012, Aslam et al 2014]. One individual with the postzygotic activating KRAS variant c.35G>A (p.Gly12Asp) has been reported with a phenotype that overlaps SFMS and ECCL [Nagatsuma et al 2019].
Oculocerebrocutaneous syndrome (OCCS) (OMIM 164180). Like ECCL, OCCS presents with focal skin lesions and congenital anomalies of the eyes and the brain. OCCS is characterized by hypo- or aplastic skin defects and nodular or pedunculated skin tags, cystic microphthalmia, a combination of forebrain anomalies, and a specific mid-hindbrain malformation [Moog & Dobyns 2018]. The skin anomalies primarily affect the region of the head and the neck. OCCS is hypothesized to be a mosaic disorder, but the specific cause has not yet been identified.
Management
No clinical practice guidelines for encephalocraniocutaneous lipomatosis (ECCL) have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with ECCL, the evaluations summarized in Table 6 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 6.
Recommended Evaluations Following Initial Diagnosis in Individuals with Encephalocraniocutaneous Lipomatosis
Treatment of Manifestations
Table 7.
Treatment of Manifestations in Individuals with Encephalocraniocutaneous Lipomatosis
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Surveillance
Table 8.
Recommended Surveillance for Individuals with Encephalocraniocutaneous Lipomatosis
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Encephalocraniocutaneous lipomatosis (ECCL) is not known to be inherited. No confirmed vertical transmission or sib recurrence has been reported.
Risk to Family Members
Parents of a proband. No parent of a child with ECCL has had any significant, distinctive manifestations of the disorder, nor would such a finding be expected, given the postzygotic mutational mechanism of this disorder.
Sibs of a proband. Given the postzygotic mutational mechanism of ECCL, the risk for an affected sib would be expected to be the same as in the general population.
Offspring of a proband. Reproductive outcome data on adults with ECCL are limited. There are no instances of vertical transmission of ECCL.
Other family members. The risk to other family members is presumed to be the same as in the general population.
Related Genetic Counseling Issues
Considerations in families with an apparent de novo mosaic pathogenic variant. This is a relatively new area for clinical genetics, as only a small (albeit growing) number of disorders are known to be caused by this genetic mechanism. Counseling for recurrence risks in ECCL should emphasize that, while no pregnancy is at zero risk, all evidence suggests that the risk of recurrence for this disorder is not increased compared to the general population.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). Because ECCL is a mosaic disorder, DNA derived from affected tissue should be banked in addition to DNA derived from lymphocytes. For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
As ECCL is not known to be inherited, prenatal diagnosis is usually not indicated. The authors recognize no potential role for preimplantation genetic testing in ECCL.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Born a Hero
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Encephalocraniocutaneous Lipomatosis: Genes and Databases
Table B.
OMIM Entries for Encephalocraniocutaneous Lipomatosis (View All in OMIM)
Molecular Pathogenesis
FGFR1 encodes a growth factor receptor, which, when activated, signals through the RAS/mitogen activated protein kinase (MAPK) pathway. KRAS is a component of the RAS/MAPK pathway, critical for regulating the pathway's activity level. Cell growth and cell differentiation are affected by this signaling pathway. Dysregulation of this signaling pathway during embryogenesis or later in life results in developmental malformations or malignant tumors, respectively. The pathogenic variants reported in ECCL are of postzygotic origin, but arise early during development. Dysregulation of the RAS/MAPK pathway ‒ typically, increased activation ‒ results in characteristic developmental malformations reflecting the mosaic distribution of the pathogenic variant. The spectrum of pathogenic missense variants causing developmental malformations but remaining compatible with survival is likely narrow ‒ accounting for the limited number of pathogenic variants identified in ECCL.
Mechanism of disease causation. Gain of function
Note: It is currently unknown why different gain-of-function pathogenic variants cause different, completely nonoverlapping phenotypes (e.g., FGFR1 p.Asn547Lys substitutions are associated with ECCL, but FGFR1 p.Pro252Arg substitutions cause Pfeiffer syndrome, a craniosynostosis syndrome).
Gene-specific laboratory technical considerations. In affected tissues, any sequencing method may detect ECCL-specific pathogenic variants. However, in unaffected tissues (blood, saliva, normal-appearing skin) and possibly also in affected tissues, low levels of mosaicism for the ECCL-specific pathogenic variants in FGFR1 and KRAS can only be detected using targeted approaches that yield deep sequencing data. The authors therefore recommend use of deep targeted approaches, particularly deep amplicon-based next-generation sequencing or digital droplet PCR (ddPCR) when ECCL is suspected.
Table 9.
Encephalocraniocutaneous Lipomatosis: Notable Pathogenic Variants by Gene
Cancer and Benign Tumors
FGFR1 and KRAS are well known proto-oncogenes with somatic pathogenic KRAS variants comprising the most common genetic abnormalities in cancer, particularly pancreatic, colon, and lung cancers [Jancík et al 2010, Hunter et al 2015]. However, cancer has not been reported in KRAS-related ECCL.
Somatic FGFR1 pathogenic variants have also been observed in many different cancer types, including squamous cell (lung, head and neck, esophagus) carcinoma, ovarian cancers, and osteosarcoma [Dienstmann et al 2014]. More relevant for ECCL, somatic pathogenic variants in FGFR1 are common in pilocytic astrocytomas and related midline gliomas [Jones et al 2013, Picca et al 2018], the most frequent cancer reported in individuals with ECCL.
Finally, somatic pathogenic variants in both KRAS (the same as ECCL) and FGFR1 (different from ECCL) have been described in giant cell lesions of the jaw, which can occur in individuals with ECCL [Gomes et al 2018].
Chapter Notes
Revision History
- 27 January 2022 (ma) Review posted live
- 1 March 2021 (um) Original submission
References
Literature Cited
- Ahmed I, Tope WD, Young TL, Miller DM, Bloom KE. Neurocutaneous melanosis in association with encephalocraniocutaneous lipomatosis. J Am Acad Dermatol. 2002;47:S196-200. [PubMed: 12140461]
- Ardinger HH, Horii KA, Begleiter ML. Expanding the phenotype of oculoectodermal syndrome: possible relationship to encephalocraniocutaneous lipomatosis. Am J Med Genet A. 2007;143A:2959-62. [PubMed: 17963257]
- Aslam A, Salam A, Griffiths CEM, McGrath JA. Naevus sebaceus: a mosaic RASopathy. Clin Exp Dermatol. 2014;39:1-6. [PubMed: 24341474]
- Bavle A, Shah R, Gross N, Gavula T, Ruiz-Elizalde A, Wierenga K, McNall-Knapp R. Encephalocraniocutaneous lipomatosis. J Pediatr Hematol Oncol. 2018;40:553-4. [PubMed: 29683947]
- Bennett JT, Tan TY, Alcantara D, Tétrault M, Timms AE, Jensen D, Collins S, Nowaczyk MJM, Lindhurst MJ, Christensen KM, Braddock SR, Brandling-Bennett H, Hennekam RCM, Chung B, Lehman A, Su J, Ng S, Amor DJ, Majewski J, Biesecker LG, Boycott KM, Dobyns WB, O'Driscoll M, Moog U, McDonell LM, et al. Mosaic activating mutations in FGFR1 cause encephalocraniocutaneous lipomatosis. Am J Hum Genet. 2016;98:579-87. [PMC free article: PMC4800051] [PubMed: 26942290]
- Boppudi S, Bögershausen N, Hove HB, Percin EF, Aslan D, Dvorsky R, Kayhan G, Li Y, Cursiefen C, Tantcheva-Poor I, Toft PB, Bartsch O, Lissewski C, Wieland I, Jakubiczka S, Wollnik B, Ahmadian MR, Heindl LM, Zenker M. Specific mosaic KRAS mutations affecting codon 146 cause oculoectodermal syndrome and encephalocraniocutaneous lipomatosis. Clin Genet. 2016;90:334-42. [PubMed: 26970110]
- Chacon-Camacho OF, Lopez-Moreno D, Morales-Sanchez MA, Hofmann E, Pacheco-Quito M, Wieland I, Cortes-Gonzalez V, Villanueva-Mendoza C, Zenker M, Zenteno JC. Expansion of the phenotypic spectrum and description of molecular findings in a cohort of patients with oculocutaneous mosaic RASopathies. Mol Genet Genomic Med. 2019;7:e625. [PMC free article: PMC6503218] [PubMed: 30891959]
- Chang CA, Perrier R, Kurek KC, Estrada-Veras J, Lehman A, Yip S, Hendson G, Diamond C, Pinchot JW, Tran JM, Arkin LM, Drolet BA, Napier MP, O'Neill SA, Balci TB, Keppler-Noreuil KM. Novel findings and expansion of phenotype in a mosaic RASopathy caused by somatic KRAS variants. Am J Med Genet A. 2021;185:2829-45. [PubMed: 34056834]
- Córdoba A, Graue-Hernández EO, Navas A, Chacon-Camacho OF, Zenteno JC, Ramirez-Miranda A, Bermudez-Magner JA, Ordaz-Robles T, Pérez-Solórzano S, Olivo-Payne A. Giant ocular lipodermoid cyst in encephalocraniocutaneous lipomatosis: surgical treatment and genetic analysis. Am J Case Rep. 2019;20:1566-71. [PMC free article: PMC6824416] [PubMed: 31649234]
- Damar Ç, Yaman A, Ali İkidağ M, Pekpak E, Olgaç A. Encephalocraniocutaneous lipomatosis with Wilms' tumor. Pediatr Int. 2017;59:835-6. [PubMed: 28612492]
- Dienstmann R, Rodon J, Prat A, Perez-Garcia J, Adamo B, Felip E, Cortes J, Iafrate AJ, Nuciforo P, Tabernero J. Genomic aberrations in the FGFR pathway: opportunities for targeted therapies in solid tumors. Ann Oncol. 2014;25:552-63. [PMC free article: PMC4433501] [PubMed: 24265351]
- Gomes CC, Gayden T, Bajic A, Harraz OF, Pratt J, Nikbakht H, Bareke E, Diniz MG, Castro WH, St-Onge P, Sinnett D, Han H, Rivera B, Mikael LG, De Jay N, Kleinman CL, Valera ET, Bassenden AV, Berghuis AM, Majewski J, Nelson MT, Gomez RS, Jabado N. TRPV4 and KRAS and FGFR1 gain-of-abnormal-function mutations drive giant cell lesions of the jaw. Nat Commun. 2018;9:4572. [PMC free article: PMC6212533] [PubMed: 30385747]
- Groesser L, Herschberger E, Ruetten A, Ruivenkamp C, Lopriore E, Zutt M, Langmann T, Singer S, Klingseisen L, Schneider-Brachert W, Toll A, Real FX, Landthaler M, Hafner C. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783-7. [PubMed: 22683711]
- Hafner C, Groesser L. Mosaic RASopathies. Cell Cycle. 2013;12:43-50. [PMC free article: PMC3570515] [PubMed: 23255105]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Hunter AG. Oculocerebrocutaneous and encephalocraniocutaneous lipomatosis syndromes: blind men and an elephant or separate syndromes? Am J Med Genet A. 2006;140:709-26. [PubMed: 16523517]
- Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res. 2015;13:1325-35. [PubMed: 26037647]
- Jain P, Chakrabarty B, Kumar A, Gupta N, Kabra M, Gulati S. Encephalocraniocutaneous lipomatosis with neurocutaneous melanosis. J Child Neurol. 2014;29:846-9. [PubMed: 23620525]
- Jancík S, Drábek J, Radzioch D, Hajdúch M. Clinical relevance of KRAS in human cancers. J Biomed Biotechnol. 2010;2010:150960. [PMC free article: PMC2896632] [PubMed: 20617134]
- Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, Fontebasso AM, Stütz AM, Hutter S, Zuckermann M, Sturm D, Gronych J, Lasitschka B, Schmidt S, Seker-Cin H, Witt H, Sultan M, Ralser M, Northcott PA, Hovestadt V, Bender S, Pfaff E, Stark S, Faury D, Schwartzentruber J, Majewski J, Weber UD, Zapatka M, Raeder B, Schlesner M, Worth CL, Bartholomae CC, von Kalle C, Imbusch CD, Radomski S, Lawerenz C, van Sluis P, Koster J, Volckmann R, Versteeg R, Lehrach H, Monoranu C, Winkler B, Unterberg A, Herold-Mende C, Milde T, Kulozik AE, Ebinger M, Schuhmann MU, Cho YJ, Pomeroy SL, von Deimling A, Witt O, Taylor MD, Wolf S, Karajannis MA, Eberhart CG, Scheurlen W, Hasselblatt M, Ligon KL, Kieran MW, Korbel JO, Yaspo ML, Brors B, Felsberg J, Reifenberger G, Collins VP, Jabado N, Eils R, Lichter P, Pfister SM, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927-32. [PMC free article: PMC3951336] [PubMed: 23817572]
- Kordacka J, Zakrzewski K, Gruszka R, Witusik-Perkowska M, Taha J, Sikorska B, Liberski PP, Zakrzewska M. Sensitive detection of FGFR1 N546K mosaic mutation in patient with encephalocraniocutaneous lipomatosis and pilocytic astrocytoma. Am J Med Genet A. 2019;179:1622-7. [PubMed: 31173478]
- McDonell LM, Leung GK, Daoud H, Ip J, Chim S, Luk HM, Lan L, Boycott KM, Chung BH, et al. Mosaic KRAS mutation in a patient with encephalocraniocutaneous lipomatosis and renovascular hypertension. Am J Med Genet A. 2018;176:2523-7. [PubMed: 30289595]
- Moog U. Encephalocraniocutaneous lipomatosis. J Med Genet. 2009;46:721-9. [PubMed: 19574261]
- Moog U, Dobyns WB. An update on oculocerebrocutaneous (Delleman-Oorthuys) syndrome. Am J Med Genet C Semin Med Genet. 2018;178:414-22. [PMC free article: PMC6501825] [PubMed: 30580480]
- Moog U, Jones MC, Viskochil DH, Verloes A, Van Allen MI, Dobyns WB. Brain anomalies in encephalocraniocutaneous lipomatosis. Am J Med Genet A. 2007a;143A:2963-72. [PubMed: 18000987]
- Moog U, Roelens F, Mortier GR, Sijstermans H, Kelly M, Cox GF, Robson CD, Kimonis VE. Encephalocraniocutaneous lipomatosis accompanied by the formation of bone cysts: harboring clues to pathogenesis? Am J Med Genet A. 2007b;143A:2973-80. [PubMed: 18000896]
- Nagatsuma M, Takasawa K, Yamauchi T, Nakagawa R, Mizuno T, Tanaka E, Yamamoto K, Uemura N, Kashimada K, Morio T. A postzygotic KRAS mutation in a patient with Schimmelpenning syndrome presenting with lipomatosis, renovascular hypertension, and diabetes mellitus. J Hum Genet. 2019;64:177-81. [PubMed: 30443000]
- Peacock JD, Dykema KJ, Toriello HV, Mooney MR, Scholten DJ 2nd, Winn ME, Borgman A, Duesbery NS, Hiemenga JA, Liu C, Campbell S, Nickoloff BP, Williams BO, Steensma M. Oculoectodermal syndrome is a mosaic RASopathy associated with KRAS alterations. Am J Med Genet A. 2015;167:1429-35. [PubMed: 25808193]
- Phi JH, Park SH, Chae JH, Wang KC, Cho BK, Kim SK. Papillary glioneuronal tumor present in a patient with encephalocraniocutaneous lipomatosis: case report. Neurosurgery. 2010;67:E1165-9. [PubMed: 20881536]
- Picca A, Berzero G, Bielle F, Touat M, Savatovsky J, Polivka M, Trisolini E, Meunier S, Schmitt Y, Idbaih A, Hoang-Xuan K, Delattre JY, Mokhtari K, Di Stefano AL, Sanson M. FGFR1 actionable mutations, molecular specificities, and outcome of adult midline gliomas. Neurology. 2018;90:e2086-e2094. [PubMed: 29728520]
- Richters RJH, Seyger MMB, Meeuwis KAP, Rinne T, Eijkelenboom A, Willemsen MA. Oculoectodermal syndrome - encephalocraniocutaneous lipomatosis associated with NRAS mutation. Acta Derm Venereol. 2020;100:adv00103. [PMC free article: PMC9234940] [PubMed: 31633190]
- Valera ET, McConechy MK, Gayden T, Rivera B, Jones DTW, Wittmann A, Han H, Bareke E, Nikbakht H, Mikael L, Queiroz RG, Suazo VK, Phi JH, Kim SK, Park SH, Fukaya R, Yum MS, Ko TS, de Oliveira RS, Machado HR, Brassesco MS, do Santos AC, Simão GN, Ramalho LNZ, Neder L, Scrideli CA, Tone LG, Majewski J, Jabado N. Methylome analysis and whole-exome sequencing reveal that brain tumors associated with encephalocraniocutaneous lipomatosis are midline pilocytic astrocytomas. Acta Neuropathol. 2018;136:657-60. [PMC free article: PMC6132939] [PubMed: 30143858]
- Wang SM, Hsieh YJ, Chang KM, Tsai HL, Chen CP. Schimmelpenning syndrome: a case report and literature review. Pediatr Neonatol. 2014;55:487-90. [PubMed: 23597534]
- Zielińska-Kaźmierska B, Grodecka J, Jabłońska-Polakowska L, Arkuszewski P. Mandibular osteoma in the encephalocraniocutaneous lipomatosis. J Craniomaxillofac Surg. 2005;33:286-9. [PubMed: 15975807]
Publication Details
Author Information and Affiliations
Heidelberg University
Heidelberg, Germany
Publication History
Initial Posting: January 27, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Moog U, Dobyns WB. Encephalocraniocutaneous Lipomatosis. 2022 Jan 27. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

%20and%20choristoma%20(C)%20in%20an%20affected%20individual%20age%202.&p=BOOKS&id=576966_eccl-Image002.jpg "Click on image to zoom")




