Clinical Description
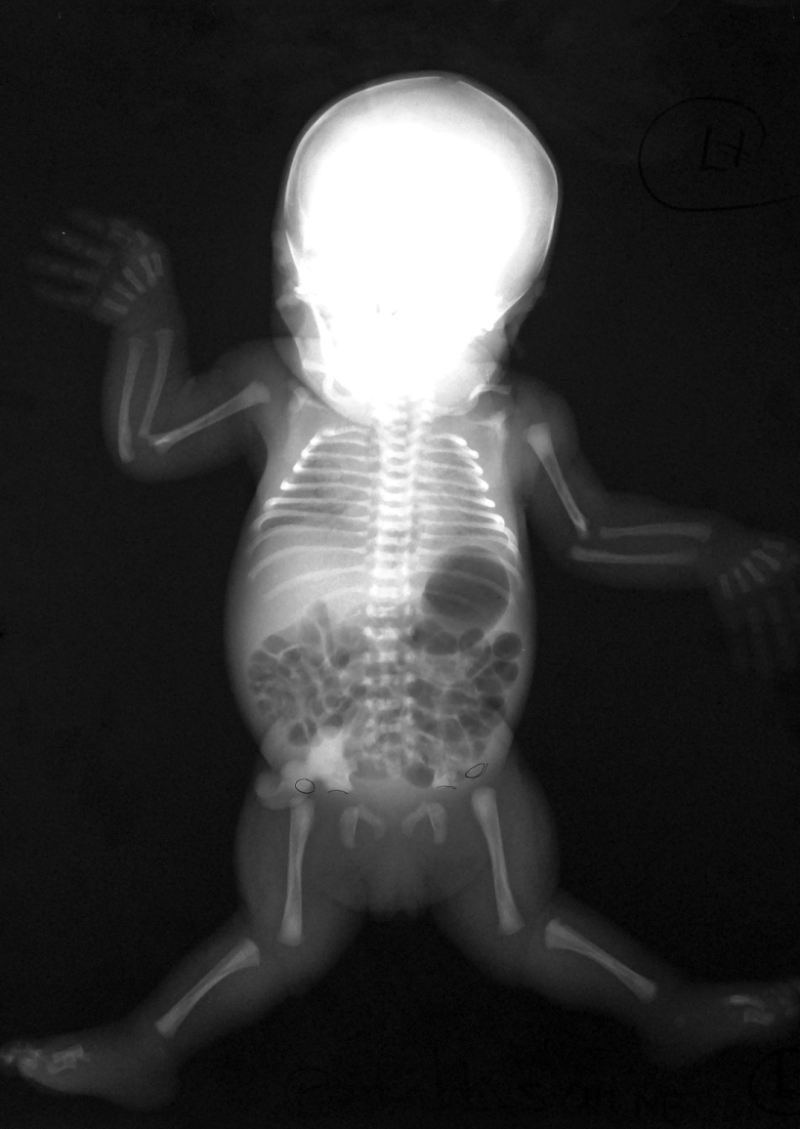
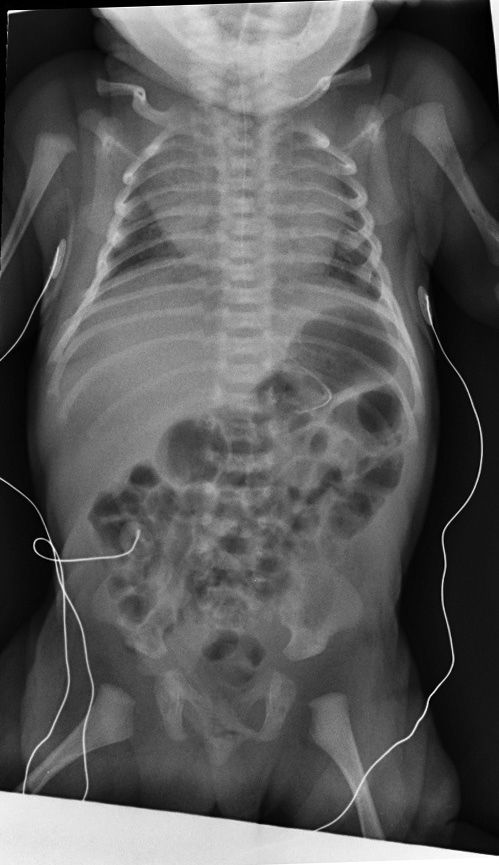
Most children with chondrodysplasia with congenital joint dislocations, CHST3-related (CDCJD-CHST3) are identified at birth as having a generalized skeletal disorder. The features of this disorder are generally limited to the skeleton and joints and are progressive in nature.
Occasionally, short stature and knee dislocations are seen on prenatal ultrasound examination [Unger et al 2010]. The prenatal presentation may be that of arthrogryposis [Muys et al 2017].
At birth, affected infants are noted to have short stature (birth length: 39-44 cm) and joint dislocations; the large majority have bilateral knee luxation or subluxation. The radial heads and hips are the next most commonly affected joints. Clubfeet are also frequently seen. Despite the congenital joint dislocations, the overall phenotype is one of restricted movement, and many children undergo multiple corrective procedures with only limited success [Rajab et al 2004, Unger et al 2010, Searle et al 2014].
In a large family reported from Oman, the adult heights ranged from 110 cm to 130 cm [Rajab et al 2004], while in a large Pakistani family the mean adult height was 84 cm [Waryah et al 2016]. A review article included information on three adults with heights of 117 cm, 121 cm, and 134.5 cm [Unger et al 2010]. Adult height appears to be severely affected in all individuals with CDCJD-CHST3, but with some intrafamilial variability and large interfamilial variability.
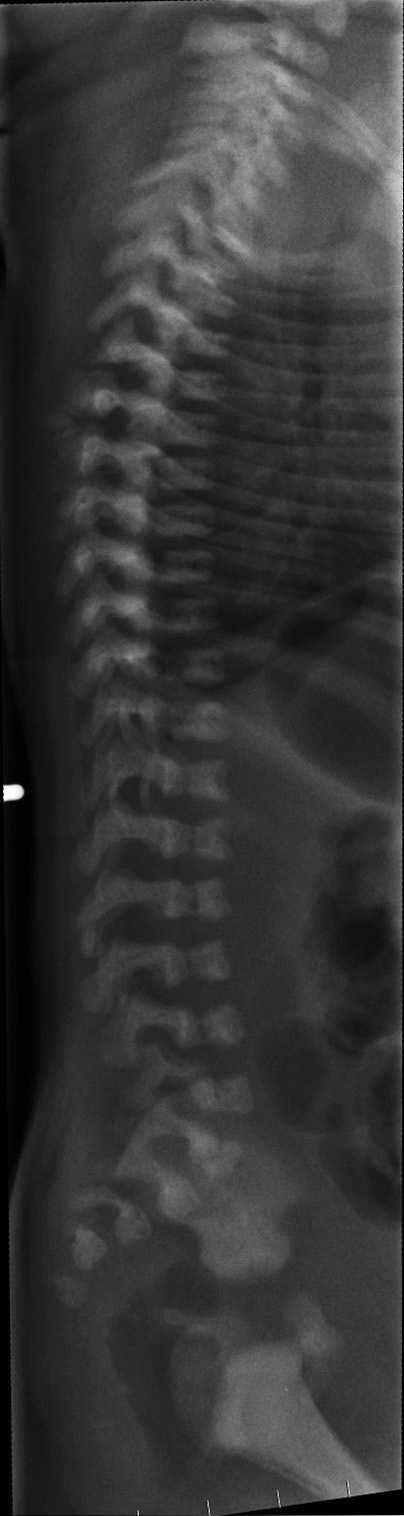
Many adults develop arthritic-type changes. They also develop spinal kyphosis, frequently in the cervical spine, and (rarely) scoliosis.
Cardiac manifestations. Minor heart valve dysplasia with valvular insufficiency has been described in multiple reports, including one in the kindred from Oman [Hall 1997, Rajab et al 2004, Tuysuz et al 2009, Otaify et al 2023, Singh et al 2024]. There are occasional reports of structural cardiac malformations such as atrial septal defect, but it is not yet clear if CHST3 pathogenic variants are causally linked [Otaify et al 2023].
Tooth anomalies (microdontia, delayed eruption) have been reported in the large Omani kindred as well as several individuals from Egypt [Rajab et al 2004, Otaify et al 2023].
Intellect, vision, and hearing is usually normal [Unger et al 2010, Singh et al 2024]. However, in a large Pakistani kindred, at least six affected individuals also had mixed hearing loss; thus, this may be an associated feature [Waryah et al 2016]. One additional individual of Pakistani origin was reported to have mild bilateral hearing loss on audiometry but no clinical signs of hearing impairment [Kausar et al 2022].
Other. Sagittal craniosynostosis has been reported in a single individual [Searle et al 2014], while "sclerosis of sutures" was reported in three unrelated individuals [Srivastava et al 2017].
Nomenclature
In 1950, Dr LJ Larsen described autosomal dominant Larsen syndrome, now known to be caused by pathogenic variants in FLNB. Larsen syndrome is characterized by multiple joint dislocations, dysmorphic facial features, spatulate thumbs, and accelerated carpal ossification (see FLNB-Related Disorders). Following the delineation of autosomal dominant Larsen syndrome, several reports of "autosomal recessive Larsen syndrome" and other similar disorders were published. By careful reevaluation of affected individuals and through recruitment of individuals with autosomal recessive Larsen syndrome, several investigators showed that, in fact, all the various reports of autosomal recessive Larsen syndrome, humerospinal dysostosis, and spondyloepiphyseal dysplasia (SED), Omani type could be attributed to CHST3 deficiency and that the different names had arisen from the part of the phenotype the various authors had emphasized [Hermanns et al 2008, Unger et al 2010]; that is, they were describing the same condition from different viewpoints.
Humerospinal dysostosis was described by
Kozlowski et al [1974] in two brothers with joint dislocations and radiographic abnormalities (bifid humeri and coronal clefts). Because the brothers were reported to be half-sibs, autosomal dominant inheritance was suspected and no link was made to autosomal recessive Larsen syndrome.
Mégarbané and Ghanem [2004] described "a newly recognized chondrodysplasia with joint dislocations." Although they made the link to humerospinal dysostosis, they rejected that diagnosis because the evidence strongly suggested autosomal recessive inheritance.
Rajab et al [2004] described a large family originating from Oman with what they termed "a new recessive type of SED with progressive spinal involvement." The same group went on to demonstrate that the disorder was caused by CHST3 deficiency and renamed the disorder SED, Omani type [
Thiele et al 2004].
The name "chondrodysplasia with congenital joint dislocations, CHST3-related (CDCJD-CHST3)" has been proposed as an unbiased and inclusive designation for this disorder and is the current designation used in the Nosology of Genetic Skeletal Disorders [Unger et al 2023]. However, an argument could also be made for retaining the name "autosomal recessive Larsen syndrome," as the joint dislocations are the presenting feature and "Larsen syndrome" is usually the first diagnosis considered; thus, the continued use of this designation is open to debate. The term "recessive Larsen syndrome" is more appropriate for CDCJD-CHST3 than for the B4GALT7-associated linkeropathy, as bilateral knee dislocation at birth is much more common in CHST3 deficiency than in B4GALT7 deficiency.