Feingold Syndrome 1
Synonyms: Oculodigitoesophagoduodenal Syndrome, ODED Syndrome
Carlo LM Marcelis, MD
Department of Human Genetics
Radboud University Nijmegen Medical Center
Nijmegen, The Netherlands
Arjan PM de Brouwer, PhD
Assistant Professor, Department of Human Genetics
Radboud University Nijmegen Medical Center
Nijmegen, The Netherlands
Initial Posting: June 30, 2009; Last Update: April 4, 2019.
Estimated reading time: 17 minutes
Summary
Clinical characteristics.
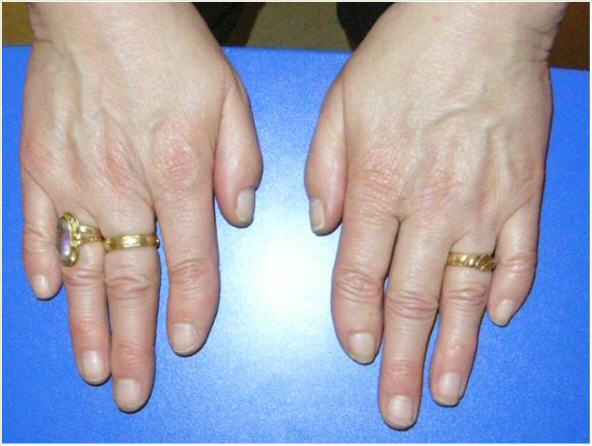
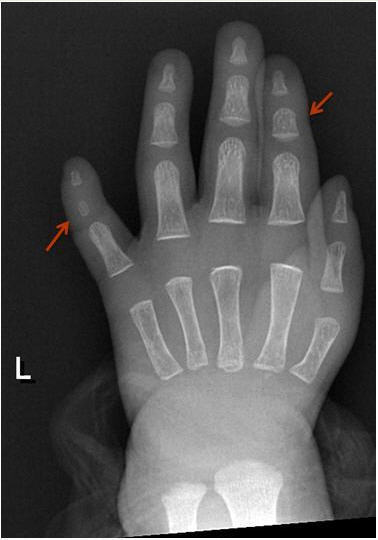
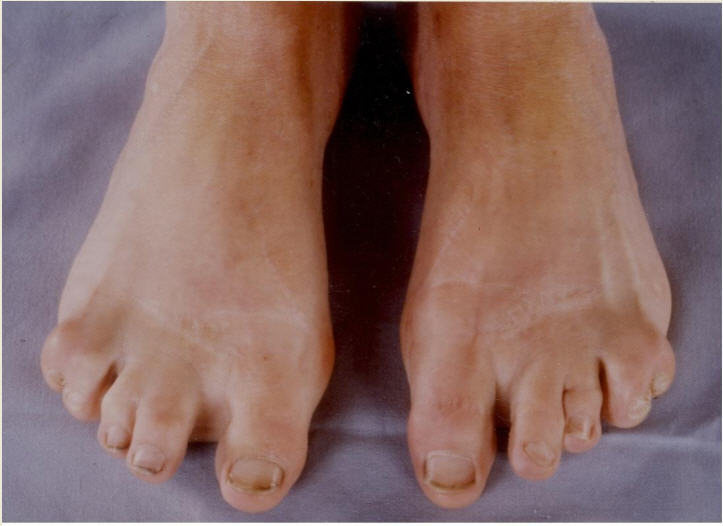
Feingold syndrome 1 (referred to as FS1 in this GeneReview) is characterized by digital anomalies (shortening of the 2nd and 5th middle phalanx of the hand, clinodactyly of the 5th finger, syndactyly of toes 2-3 and/or 4-5, thumb hypoplasia), microcephaly, facial dysmorphism (short palpebral fissures and micrognathia), gastrointestinal atresias (primarily esophageal and/or duodenal), and mild-to-moderate learning disability.
Diagnosis/testing.
The diagnosis of FS1 is established in a proband with suggestive clinical findings and a heterozygous pathogenic variant in MYCN identified by molecular genetic testing.
Management.
Treatment of manifestations: Gastrointestinal atresia is treated surgically. Mild-to-moderate learning disabilities are treated in the usual manner.
Genetic counseling.
FS1 is inherited in an autosomal dominant manner. Approximately 60% of individuals with Feingold syndrome 1 have an affected parent; the proportion of FS1 caused by a de novo
MYCN pathogenic variant is unknown. Each child of an individual with FS1 has a 50% chance of inheriting the MYCN pathogenic variant. When the MYCN pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
Feingold syndrome 1 (FS1) should be suspected in probands with the following clinical findings [Marcelis et al 2008].
Digital anomalies (brachymesophalangy, thumb hypoplasia, toe syndactyly)
Microcephaly (occipito-frontal circumference <10th centile)
Short palpebral fissures
Gastrointestinal atresias, especially esophageal and duodenal, diagnosed pre- or postnatally by imaging studies (usually ultrasound examination, possibly MRI)
Establishing the Diagnosis
The diagnosis of FS1 is established in a proband with suggestive clinical findings and a heterozygous pathogenic (or likely pathogenic) variant in MYCN identified by molecular genetic testing (see Table 1). Note: (1) Large contiguous-gene deletions encompassing MYCN and other genes have been reported in individuals with features of FS1 but more complex phenotypes (see Genetically Related Disorders). Of note, most individuals with these large deletions are identified by chromosomal microarray analysis (CMA) performed in the context of evaluation for multiple congenital anomalies. (2) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (3) Identification of a heterozygous MYCN variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (chromosomal microarray analysis, exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with atypical findings are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic findings suggest the diagnosis of FS1, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
Single-gene testing. Sequence analysis of MYCN detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
Note: Gene-targeted methods will detect single-exon up to whole-gene deletions; however, breakpoints of large deletions and/or deletion of adjacent genes may not be determined.
An intellectual disability
multigene panel that includes
MYCN and other genes of interest (see
Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this
GeneReview. Of note, given the rarity of FS1, some panels for intellectual disability may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see
Table 1).
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Option 2
When the diagnosis of FS1 is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is an option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible. If exome sequencing is not diagnostic – and particularly when evidence supports autosomal dominant inheritance – exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Feingold Syndrome 1 (FS1)
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
MYCN
| Sequence analysis 3 | 59% 4 |
| Gene-targeted deletion/duplication analysis 5 | 9% 6 |
| Chromosomal microarray (CMA) 7 | See footnote 8. |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications. Gene-targeted deletion/duplication testing will detect deletions ranging from a single exon to the whole gene; however, breakpoints of large deletions and/or deletion of adjacent genes (e.g., those described by van Bokhoven et al [2005], Blaumeiser et al [2008], Marcelis et al [2008], and Cognet et al [2011]) may not be detected by these methods.
- 6.
- 7.
Chromosomal microarray analysis (CMA) uses oligonucleotide or SNP arrays to detect genome-wide large deletions/duplications (including MYCN) that cannot be detected by sequence analysis. The ability to determine the size of the deletion/duplication depends on the type of microarray used and the density of probes in the 2p24.3 region. CMA designs in current clinical use target the 2p24.3 region. Deletions reported in van Bokhoven et al [2005], Blaumeiser et al [2008], Marcelis et al [2008], and Cognet et al [2011] would be detected by CMA.
- 8.
Larger deletions of MYCN and adjacent genes detected by CMA associated with features of FS1 with additional clinical findings are not included in this table (see Genetically Related Disorders).
Clinical Characteristics
Clinical Description
Feingold syndrome 1 (FS1) as described by Feingold [1975] and Brunner & Winter [1991] is characterized by digital anomalies, microcephaly, facial dysmorphism, gastrointestinal atresias, and learning disability. To date, 69 families with 116 affected individuals having three or more of the core features of FS1 (brachymesophalangy, toe syndactyly, microcephaly, short palpebral fissures, and intestinal atresia) have been reported [Blaumeiser et al 2008, Marcelis et al 2008, Cognet et al 2011].
Features are summarized in Table 2.
Table 2.
Features in Feingold Syndrome 1 (FS1)
View in own window
| Feature | % of Persons w/Feature |
|---|
|
Digital anomalies
| Brachymesophalangy | 100% |
| Toe syndactyly | 93% |
| Thumb hypoplasia | 17% |
|
Microcephaly
| 86% |
|
Facial dysmorphism
| Short palpebral fissures | 73% |
| Micrognathia | 30% |
|
Atresia
| Esophageal | 35% |
| Duodenal | 27% |
| Jejunal | 3% |
| Anal | 2% |
| Multiple | 12% |
|
Mild learning deficit
| 56% |
|
Stature <10th centile
| 60% |
|
Other
| Renal abnormalities | 19% |
| Cardiac abnormalities | 15% |
| Hearing loss | 10% |
Digital anomalies include brachymesophalangy (shortening of the 2nd and 5th middle phalanx of the hand with clinodactyly of the 5th finger) and thumb hypoplasia ( and ). Toe syndactyly refers to syndactyly of toes 2-3 and/or 4-5 ().
Gastrointestinal atresia (esophageal and/or duodenal) is a cause of major medical concern in FS1 and requires immediate surgical intervention (see Management).
Mild learning deficit is frequent in FS1; most affected individuals are able to live an independent life. Clear intellectual disability is rare, but intelligence is below average when compared to the general population and healthy, unaffected family members.
Some reports show that growth is impaired in FS1 [Shaw-Smith et al 2005]. Short stature (height <3rd centile) is uncommon but average height is below that in the general population.
Associated features that occur in fewer than 50% of affected individuals include the following:
Renal abnormalities. Horseshoe kidneys, dysplastic kidneys, hydronephrosis and pelvic dilatation, chronic nephritis, and vesicourethral reflux leading to renal dysplasia and renal failure
Cardiac abnormalities. Patent ductus arteriosus, multiple ventricular septal defects (VSD), tricuspid valve stenosis and VSD plus tricuspid atresia, and interrupted aortic arch
Hearing loss. Variable and can include conductive and sensorineural hearing loss; the latter is more commonly reported
Genotype-Phenotype Correlations
No significant differences are observed among individuals with deletions or missense, nonsense, or frameshift variants.
Penetrance
The penetrance for major features of FS1, especially digital abnormalities, appears to be 100% but clinical expression can vary considerably.
Nomenclature
Terms used in the past for Feingold syndrome 1:
Microcephaly-oculo-digito-esophageal-duodenal syndrome
Microcephaly mesobrachyphalangy tracheoesophageal fistula syndrome
Microcephaly-digital anomalies-normal intelligence syndrome
Differential Diagnosis
Table 3.
Other Genes of Interest in the Differential Diagnosis of Feingold Syndrome 1 (FS1)
View in own window
| Gene(s) | Disorder | MOI | Clinical Features of Differential Diagnosis Disorder |
|---|
| Overlapping w/FS1 | Distinguishing from FS1 |
|---|
|
CHD7
|
CHARGE syndrome
| AD | Esophageal atresia Heart defects Renal abnormalities
| Coloboma Genital abnormalities Ear anomalies
|
| MIR17HG 1 | Feingold syndrome 2
(OMIM 614326) | AD |
| Absence of gastrointestinal abnormalities 2 |
| 21 genes 3 |
Fanconi anemia
| AR
AD
XL | Thumb hypoplasia Microcephaly Growth restriction Intestinal/anal atresia Renal abnormalities
|
|
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; XL = X-linked
- 1.
Feingold syndrome 2 is caused by hemizygous deletions of chromosome 13q31.3 including MIR17HG [Tassano et al 2013].
- 2.
- 3.
BRCA2, BRIP1, ERCC4, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, MAD2L2, PALB2, RAD51, RAD51C, RFWD3, SLX4, UBE2T, XRCC2
VACTERL association (vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula with esophageal atresia, renal and limb abnormalities) shows considerable overlap with FS1, but the two should be distinguishable by the presence of microcephaly, brachymesophalangy, and toe syndactyly in FS1. The molecular basis of VACTERL association is unknown.
The brachymesophalangy observed in FS1 is very similar to brachydactyly type A4 (BDA4). The molecular basis of BDA4 is known. Although no pathogenic variants in MYCN have been identified in BDA4, molecular genetic testing of MYCN could be considered in families with BDA4.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Feingold syndrome 1 (FS1), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Feingold Syndrome 1 (FS1)
View in own window
| System/Concern | Evaluation | Comment |
|---|
|
Hands/Feet
| Assess for digital anomalies. |
|
|
Gastrointestinal tract
| Assess for gastrointestinal atresia (incl esophageal, duodenal, jejunal, anal). | Gastroenterologist / GI surgeon |
|
Development
| Developmental assessment | Incl eval of motor, speech-language, general cognitive, & vocational skills |
|
Renal abnormalities
| Renal ultrasound eval | Assess for renal anomalies. |
|
Cardiac abnormalities
| Assess for congenital heart defects. | Pediatric cardiologist |
|
Hearing loss
| Audiologic eval 1 | Conductive & sensorineural |
|
Miscellaneous
| Consultation w/clinical geneticist &/or genetic counselor | |
Treatment of Manifestations
Appropriate treatment includes a multidisciplinary approach to address the following possible concerns:
Surgical treatment of gastrointestinal atresia
Occupational therapy / surgical intervention for finger/toe anomalies
Treatment of cardiac and/or renal anomalies as per standard practice
Developmental or educational intervention for children with learning difficulties
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States (US); standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states and provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center-based; however, for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life. Some issues to consider:
IEP services are for those who require specially designed instruction / related services.
IEP services will be reviewed annually to determine if any changes are needed.
As required by special education law, children should be in the least restrictive environment at school and included in general education as much as possible and when appropriate.
Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
As a child enters teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
In the US:
Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Surveillance
Table 5.
Recommended Surveillance for Individuals with Feingold Syndrome 1
View in own window
| System/Concern | Evaluation | Frequency |
|---|
|
Hands/Feet
| Hand function / need for OT | Per OT |
|
GI tract atresia
| As specified by GI consultants | Per GI consultants |
Development/
Education
| Monitor developmental progress & educational needs. | Routinely, per developmental pediatrician &/or school |
|
Renal
| As specified by renal consultants | Per renal consultants |
|
Cardiac
| As specified by cardiac consultants | Per cardiologist |
|
Hearing
| Audiologic reexamination to determine type & extent of hearing loss & success w/hearing habilitation | Per treating audiologist |
GI = gastrointestinal; OT = occupational therapist/therapy
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Feingold syndrome 1 (FS1) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Many individuals (~60%) diagnosed with FS1 have an affected parent.
A proband with FS1 may have the disorder as the result of a
de novo
MYCN pathogenic variant.
De novo pathogenic variants were observed in approximately 40% of individuals with FS1 [
Marcelis et al 2008; Author, personal communication].
Molecular genetic testing is recommended for the parents of a proband with an apparent de novo pathogenic variant.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or germline mosaicism in a parent. Though theoretically possible, no instances of germline mosaicism have been reported.
The family history of some individuals diagnosed with FS1 may appear to be negative because of failure to recognize the disorder in a family member with a milder phenotypic presentation. Therefore, an apparently negative family history cannot be confirmed until appropriate evaluations and/or molecular genetic testing have been performed.
Note: If the parent is the family member in whom the pathogenic variant first occurred, the parent may have somatic mosaicism for the pathogenic variant and may be mildly/minimally affected.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents.
If a parent of the proband is affected and/or known to have the MYCN pathogenic variant identified in the proband, the risk to the sibs is 50%.
If the proband has a known pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents have not been tested for the MYCN pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for FS1 because of the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with FS1 has a 50% chance of inheriting the MYCN pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the MYCN pathogenic variant, the parent's family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the MYCN pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Because of the risk for major congenital abnormalities of the gastrointestinal tract, heart, and kidney, high-resolution ultrasound investigations (including fetal echocardiogram) are advised in any pregnancy in which the fetus is known to have an MYCN pathogenic variant or in which the genetic status of a fetus at 50% risk is unknown.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Children's Craniofacial Association
Phone: 800-535-3643
Email: contactCCA@ccakids.com
Face Equality International
United Kingdom
Learning Disabilities Association of America
4156 Library Road
Pittsburgh PA 15234-1349
Phone: 412-341-1515
Fax: 412-344-0224
Medline Plus
REACH
Helping children with upper limb differences live life without limits.
United Kingdom
Phone: 0845 1306 225; 020 3478 0100
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Feingold Syndrome 1: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
The Myc family of proteins is well known for its role in oncogenic processes. Members of this family include c-Myc, N-Myc, and L-Myc. The gene N-myc was identified by homology with c-Myc in amplified sequences of neuroblastomas.
c-Myc and N-Myc also play a crucial role in development. Mice deficient for either of these Myc genes die before embryonic day 11.5. Inactivation of murine N-myc demonstrates its developmental importance [Charron et al 2002, Knoepfler et al 2002, Ota et al 2007]. N-myc plays a role in branching morphogenesis in the lung and development of the mesonephric tubules, neuroepithelium, sensory ganglia, gut, heart, and limb, many of which are also affected in FS1.
Mechanism of disease causation. Feingold syndrome 1 is caused by a loss-of-function variant resulting in haploinsufficiency of MYCN.
MYCN-specific laboratory considerations.
MYCN encodes three exons. Although exon 1 contains a potential translation start codon, initiation of MYCN protein synthesis commences at the first ATG codon in exon 2 because of an internal ribosome entry site in the 5'-untranslated region [Jopling & Willis 2001]. A missense nucleotide change in exon 1, which introduced a termination codon, was determined to be benign [Marcelis et al 2008]. The effect of this variant on alternative transcripts and their role in pathogenesis of FS1 is unknown.
Table 6.
View in own window
| Reference Sequences | DNA Nucleotide Change | Predicted Protein Change | Comment |
|---|
NM_001293231.1
NP_001280160.1
| c.64C>T | p.Gln22Ter | Benign variant 1 |
NM_005378.4
NP_005369.2
| c.964C>T | p.Arg322Ter | Recurrent pathogenic variants 1 |
| c.1105_1106dupAG | p.Ser369ArgfsTer3 |
| c.1117C>T | p.Arg373Ter |
| c.1178G>A | p.Arg393His |
| c.1181G>A | p.Arg394His |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Cancer and Benign Tumors
Somatic amplification of MYCN is found in up to 25% of a diverse range of cancers, such as neuroblastoma and skin, lung, and prostate cancer. This invariably leads to overexpression of MYCN resulting in a poor prognosis [Ruiz-Pérez et al 2017]. This is in sharp contrast to the variants causing FS1, which are germline variants that without exception result in a loss of function.
Chapter Notes
Revision History
4 April 2019 (bp) Comprehensive update posted live
6 September 2012 (me) Comprehensive update posted live
30 June 2009 (me) Review posted live
23 April 2009 (cm) Original submission