

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010-.

Probe Development Efforts to Identify Novel Inhibitors of Protein Phosphatase Methylesterase-1 (PME-1)
DA Bachovchin, AE Speers, SJ Brown, TP Spicer, Fernandez-Vega, V, J Ferguson, JT Mohr, J Murphy, GC Fu, BF Cravatt, PS Hodder, and H Rosen.
Author Information and Affiliations
Received: August 31, 2010; Last Update: December 12, 2011.
Reversible protein phosphorylation networks play essential roles in most cellular processes. While over 500 kinases catalyze protein phosphorylation, only two enzymes, PP1 and PP2A, are responsible for more than 90% of all serine/threonine phosphatase activity. Phosphatases, unlike kinases, achieve substrate specificity through complex subunit assembly and post-translational modifications rather than number. Mutations in several of the PP2A subunits have been identified in human cancers, suggesting that PP2A may act as a tumor suppressor. Adding further complexity, several residues of the catalytic subunit of PP2A can be reversibly phosphorylated, and the C-terminal leucine residue can be reversibly methylated. Protein phosphatase methylesterase-1 (PME-1) is specifically responsible for demethylation of the carboxyl terminus. Methylesterification is thought to control the binding of different subunits to PP2A, but little is known about physiological significance of this post-translational modification in vivo. Recently, PME-1 has been identified as a protector of sustained ERK pathway activity in malignant gliomas. PME-1 knockout mice generated by targeted gene disruption result in perinatal lethality, underscoring the importance of PME-1 but hindering biological studies. The Scripps Research Institute Molecular Screening Center (SRIMSC), part of the Molecular Libraries Probe Production Centers Network (MLPCN), identified a potent and selective PME-1 inhibitor probe, ML174, by high-throughput screening using fluorescence polarization-activity-based protein profiling (FluoPol-ABPP). ML174, with an IC50 of 10 nM, is based on the aza-beta-lactam scaffold and is selective for PME-1 among serine hydrolases in human cell line proteomes as assessed by gel-based competitive-activity-based protein profiling. Among more than 30 serine hydrolase anti-targets, ML174 is selective at 1 μM. Additionally, ML174 was shown in situ to be highly active against PME-1 and to result in 85% reduction of demethylated PP2A. We previously reported a modestly potent 500 nM inhibitor that was selective for PME-1, the first reported selective PME-1 inhibitor. ML174 is 50 times more potent and from an entirely different structural and mechanistic class of inhibitors. Due to its much higher potency, ML174 has greater potential for use in long time-course in situ studies, and is a much better candidate for in vivo applications.
Assigned Assay Grant #: 1 R01 CA132630-01
Screening Center Name & PI: The Scripps Research Institute Molecular Screening Center (SRIMSC), H Rosen
Chemistry Center Name & PI: SRIMSC, H Rosen
Assay Submitter & Institution: BF Cravatt, TSRI, La Jolla
PubChem Summary Bioassay Identifier (AID): 2143
Probe Structure & Characteristics

| CID/ML# | Target Name | IC50 (nM) [SID, AID] | Anti-target Name(s) | IC50*(μM) [SID, AID] | Fold Selective† | Secondary Assay(s) Name: IC50 (nM) [SID, AID] |
|---|---|---|---|---|---|---|
| CID 24856225/ML174 | PME-1 | 10 [SID 99206500, AID 463124] | > 30 serine hydrolases, including APEH, PREP, CES, FAS, ABHD10 | > 1 [SID 99206500, AID 463149] | >100 | LC-MS/MS assay: [SID 99206500, AID 463090] Cytox assay: [SID 99206500, AID 463091] Gel-based ABPP IC50: 10 nM [SID 99206500, AID 463124] Cell-based assay: [SID 99206500, AID 463131] Endogenous enzyme assay: [SID 99206500, AID 463149; SID 92709579, AID 463146] Demethylation assay: [SID 99206500, AID 463132] |
- *
IC50 of the anti-target is defined as greater than the test compound concentration at which less than or equal to 50% inhibition of the anti-target is observed, which is reported in AID 463149. For SID 99206500, no anti-targets were observed at 1 μM in AID 463149, so the IC50 is reported as > 1 μM.
- †
Fold-selectivity was calculated as: > IC50 for anti-target/IC50 for target
Recommendations for scientific use of the probe
Protein phosphatase methylesterase-1 (PME-1)-mediated methylesterification is thought to control the binding of different subunits to protein phosphatase 2A (PP2A), which, along with protein phosphatase 1 (PP1), is responsible for >90% of all serine/threonine phosphatase activity. However, little is known about the physiological significance of this post-translational modification in living systems [1]. Recently, PME-1 has been identified as a protector of sustained ERK pathway activity in malignant gliomas [1]. Because PME-1 deletion results in perinatal lethality in genetically disrupted (knockout) mice [2], temporal control over PME-1 activity with a selective inhibitor would be of significant utility for biochemical and cell biological studies. Previously, we reported a modestly potent 500 nM inhibitor that was selective for PME-1 [3]. The probe in this report is 50 times more potent and from an entirely different structural and mechanistic class of inhibitors. Due to its much higher potency, the probe described in this report has greater potential for use in long time-course in situ studies, and is a much better candidate for in vivo applications. This inhibitor is intended for inhibition of PME-1 for primary research studies and, if research merits, for analysis of this enzyme as a potential prototype therapeutic agent.
1. Introduction
Reversible protein phosphorylation networks play essential roles in most cellular processes. While over 500 kinases catalyze protein phosphorylation, only two enzymes, PP1 and PP2A, are responsible for more than 90% of all serine/threonine phosphatase activity [4]. Phosphatases, unlike kinases, achieve substrate specificity through complex subunit assembly and post-translational modifications rather than number. PP2A, for example, typically exists as heterotrimer with diverse subunits that may combinatorially make as many as 70 different holoenzyme assemblies [5]. Mutations in several of these PP2A subunits have been identified in human cancers, suggesting that PP2A may act as a tumor suppressor [6]. Adding further complexity, several residues of the catalytic subunit of PP2A can be reversibly phosphorylated, and the C-terminal leucine residue can be reversibly methylated [7–8]. PME-1 is specifically responsible for demethylation of the carboxyl terminus [9].
Methylesterification is thought to control the binding of different subunits to PP2A, but little is known about physiological significance of this post-translational modification in vivo [10]. Recently, PME-1 has been identified as a protector of sustained ERK pathway activity in malignant gliomas [1]. In order to further elucidate the role of PP2A methylation in vivo, our lab has generated mice that lack PME-1 (PME-1 knockout mice) by targeted gene disruption [2]. Unfortunately, PME-1 deletion resulted in perinatal lethality, underscoring the importance of PME-1 but hindering biological studies. Biochemical elucidation of PME-1 would thus greatly benefit from the development of potent and selective chemical inhibitors.
As a serine hydrolase, catalytically active PME-1 is readily labeled by fluorescent activity-based protein profiling (ABPP) probes bearing a fluorophosphonate (FP) reactive group [11]. This reactivity can be exploited for inhibitor discovery using a competitive-ABPP platform, whereby small molecule enzyme inhibition is assessed by the ability to out-compete ABPP probe labeling [12]. Competitive ABPP has also been configured to operate in a high-throughput manner via fluorescence polarization readout, FluoPol-ABPP [13]. In conjunction with the Southern Research Institute Molecule Screening Center (SRIMSC), we previously applied FluoPol-ABPP to PME-1 inhibitor discovery, and reported a PME-1 inhibitor based on a sulfonyl acrylonitrile scaffold (SID 87457340) [14]. While selective, the compound exhibited an IC50 of only 0.5 μM, which made the compound suitable for some basic in situ and in vitro studies, but not a good candidate for more elaborate studies with longer time courses and eventual in vivo studies. While the potency of SID 87457340 was a significant (>20-fold) improvement over the initial lead structure found by uHTS, extensive SAR analysis suggested that we were unlikely to obtain a more potent derivative based on the sulfonyl acrylonitrile scaffold. As such, we endeavored to derive a second inhibitor based on an entirely different scaffold from the uHTS screening results, and can now report a potent and selective probe compound (SID 99206500) based on the aza-beta-lactam scaffold with an IC50 of 10 nM. The sulfonyl acrylonitrile probe was the first reported selective PME-1 inhibitor, and no other compounds have been reported to date.
2. Materials and Methods
Solubility, stability, and reactivity with glutathione analyses were conducted in accordance with NIH guidelines. All reagents for chemical synthesis were obtained from ThermoFisher or SigmaAldrich. All other protocols are reported in the AIDs summarized below.
2.1. Assays
Primary uHTS assay to identify PME-1 inhibitors (AID 2130)
Assay Overview: The purpose of this assay was to identify compounds that act as PME-1 inhibitors. This competitive activity-based protein profiling (ABPP) assay uses fluorescence polarization to investigate enzyme-substrate functional interactions based on active site-directed molecular probes [11–12]. A fluorophosphonate-rhodamine (FP-Rh) probe, which broadly targets enzymes from the serine hydrolase family [15] was used to label PME-1 in the presence of test compounds. The reaction was excited with linear polarized light and the intensity of the emitted light was measured as the polarization value (mP). As designed, test compounds that act as PME-1 inhibitors will prevent PME-1-probe interactions, thereby increasing the proportion of free (unbound) fluorescent probe in the well, leading to low fluorescence polarization. Omission of enzyme (which gives the same result as use of a catalytically-dead enzyme) serves as a positive control. Compounds were tested at a nominal concentration of 5.9 μM.
Protocol Summary: Prior to the start of the assay, Assay Buffer (4.0 μL; 0.01% Pluronic acid, 50 μM Tris HCl pH 8.0, 150 μM NaCl, 1 μM DTT) containing PME-1 protein (1.25 μM) was dispensed into 1536-well microtiter plates. Next, test compound (30 nL in DMSO) or DMSO alone (0.59% final concentration) was added to the appropriate wells and incubated for 30 minutes at 25 degrees Celsius. The assay was started by dispensing FP-Rh probe (1.0 μL of 375 nM in Assay Buffer) to all wells. Plates were centrifuged and, after 45 minutes of incubation at 25 degrees Celsius, fluorescence polarization was read on a Viewlux microplate reader (PerkinElmer, Turku, Finland) using a BODIPY TMR FP filter set and a BODIPY dichroic mirror (excitation = 525 nm, emission = 598 nm). Fluorescence polarization was read for 15 seconds for each polarization plane (parallel and perpendicular). The well fluorescence polarization value (mP) was obtained via the PerkinElmer Viewlux software. Assay Cutoff: compounds that inhibited PME-1 greater than 26.13% were considered active.
Confirmation uHTS assay to identify PME-1 inhibitors (AID 2171)
Assay Overview: The purpose of this assay was to confirm activity of compounds identified as active in the primary uHTS screen (AID 2130). In this assay, the FP-Rh probe was used to label PME-1 in the presence of test compounds and analyzed as described above (AID 2130). Compounds were tested in triplicate at a nominal concentration of 5.9 μM.
Protocol Summary: The assay was performed as described above (AID 2130), except that compounds were tested in triplicate. Assay Cutoff: compounds that inhibited PME-1 greater than 26.13% were considered active.
Inhibition of endogenous PME-1 by aza-beta lactams from DPI (AID 463146)
Assay Overview: The purpose of this gel-based competitive-ABPP assay was to determine whether or not test compounds can inhibit PME-1 in a complex proteomic lysate. Complex proteomes were incubated with test compound followed by reaction with a rhodamine-conjugated fluorophosphonate (FP-Rh) activity-based probe. The reaction products were separated by SDS-PAGE and visualized in-gel using a flatbed fluorescence scanner. The percentage activity remaining was determined by measuring the integrated optical density (IOD) of the bands. As designed, test compounds that act as PME-1 inhibitors will prevent PME-1-probe interactions, thereby decreasing the proportion of bound fluorescent probe, giving lower fluorescence intensity in the band in the gel. Percent inhibition was calculated relative to a DMSO (no compound) control.
Protocol Summary: The following proteomic sources and test compound concentrations were used: 1) HeLa soluble proteome (1 mg/mL in DPBS) with 0.1, 1, 10, 100, 1000, and 10000 nM test compound; 2) MDA-MB-231 soluble proteome (1 mg/mL in DPBS) with 0.1, 1, 10, 100, 1000, and 10000 nM test compound. All test compounds were incubated for 30 min at 25 degrees Celsius (50 μL reaction volume). FP-Rh (1 μL of 50x stock in DMSO) was added to a final concentration of 1 μM. The reactions were incubated for 20 min at 25 degrees Celsius, quenched with 2x SDS-PAGE loading buffer, separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the integrated optical density of the PME-1 band relative to a DMSO-only (no compound) control. Assay Cutoff: compounds that inhibited PME-1 at least 90% at 1 μM were considered active.
Inhibition of endogenous PME-1: compound potency and selectivity analysis (AID 463149)
Assay Overview: The purpose of this gel-based competitive-ABPP assay was to determine whether or not powder samples of test compounds can inhibit PME-1 activity in a complex proteomic lysate and to estimate compound selectivity. A complex proteome was incubated with test compound followed by reaction with a rhodamine-conjugated fluorophosphonate (FP-Rh) activity-based probe. The reaction products were analyzed by gel as described above (AID 463146).
Protocol Summary: HeLa soluble proteome (1 mg/mL in DPBS) was treated with 0.1 μM, 1 μM, or 20 μM test compound (1 μL of a 50x stock in DMSO). Test compounds were incubated for 30 min at 25 degrees Celsius (50 μL reaction volume). FP-Rh (1 μL of 50x stock in DMSO) was added to a final concentration of 1 μM. The reaction was incubated for 20 min at 25 degrees Celsius, quenched with 2x SDS-PAGE loading buffer, separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the integrated optical density of the PME-1 band relative to a DMSO-only (no compound) control. Anti-targets with greater than 50% inhibition were scored as hits. Observed anti-targets were carboxylesterase (CES), fatty acid synthase (FAS), N-acylaminoacyl-peptide hydrolase (APEH), prolyl endopeptidase (PREP), and ABHD10. Assay Cutoff: compounds that inhibited PME-1 at least 50% at 0.1 μM with no anti-targets at 1 μM were considered active.
Inhibition of endogenous PME-1 in situ (AID 463131)
Assay Overview: The purpose of this gel-based competitive-ABPP assay was to determine whether or not powder samples of test compounds can inhibit PME-1 activity in situ. Cultured HeLa cells were incubated with test compound. Cells were harvested and the soluble fraction isolated and reacted with a FP-Rh activity-based probe. The reaction products were analyzed by gel as described above (AID 463146).
Protocol Summary: HeLa cells in media (5 mL total volume; supplemented with FCS) were treated with 500 nM test compound (5 μL of a 1000x stock in DMSO) for 1 hour at 37 degrees Celsius. Cells were harvested, washed 4 times with 10 mL DPBS, and homogenized by sonication in DPBS. The soluble fraction was isolated by centrifugation (100K × g, 45 min) and the protein concentration was adjusted to 1 mg/mL with DPBS. FP-Rh (1 μL of 50x stock in DMSO) was added to a final concentration of 1 μM in 50 μL total reaction volume. The reaction was incubated for 20 min at 25 degrees Celsius, quenched with 2x SDS-PAGE loading buffer, separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the integrated optical density of the PME-1 band relative to a DMSO-only (no compound) control. Assay Cutoff: compounds that inhibited PME-1 at least 90% were considered active.
Determination of IC50 values (AIDs 463130 and 463124)
Assay Overview: The purpose of this assay was to determine the IC50 values of powder samples of test compounds for PME-1 inhibition in a complex proteome. An FP-Rh activity-based probe was used to label PME-1 in the presence of test compounds, and the reaction products were analyzed by gel as described above (AID 463146).
Protocol Summary: MDA-MB-231 soluble proteome (1 mg/mL in DPBS) was incubated with DMSO or compound for 30 min at 37 degrees Celsius before the addition of FP-Rh at a final concentration of 1 μM in 25 μL total reaction volume. The reaction was incubated for 20 min at 25 degrees Celsius, quenched with 2x SDS-PAGE loading buffer, separated by SDS-PAGE and visualized by in-gel fluorescent scanning. The percentage activity remaining was determined by measuring the integrated optical density of the bands. IC50 values for inhibition of PME-1 was determined from dose-response curves from three trials at each inhibitor concentration in an 8-point dilution series (from 100 μM to 1.3 nM for AID 463130 or 1 μM to 0.06 nM for AID 463124). For each test compound, percent inhibition was plotted against the log of the compound concentration. A three parameter equation describing a sigmoidal dose-response curve was then fitted using GraphPad Prism (GraphPad Software Inc). The software-generated IC50 values were reported. Assay Cutoff: compounds with an IC50 <1 μM were considered active.
Inhibition of PME-1-mediated PP2A demethylation (AID 463132)
Assay Overview: The purpose of this assay was to determine the effect of powder samples of test compounds on the demethylation of PP2A by endogenous PME-1 in cultured HeLa cells. HeLa cells were incubated with test compounds, homogenized, and proteins separated by SDS-PAGE. Demethylated PP2A was detected using an antibody linked to horseradish peroxidase. As designed, test compounds that act as PME-1 inhibitors will inhibit demethylation of PP2A, resulting in a decrease in the demethylated PP2A signal.
Protocol Summary: HeLa cells in culture (5 mL media supplemented with FCS) were treated with 500 nM inhibitor (5 μL of a 1000x stock in DMSO) for 1 hour at 37 degrees Celsius. Cells were washed 4 times in DPBS, harvested in DPBS and homogenized by sonication. The protein concentration was adjusted to 1 mg/mL with DPBS and proteins separated by SDS-PAGE. PP2A demethylation was visualized by chemi-luminescent dectection using HRP-antibodies specific to C-terminal demethylated PP2A. As designed, test compounds that act as PME-1 inhibitors will inhibit demethylation of PP2A, resulting in a decrease in the demethylated PP2A signal. Activity Cutoff: compounds that inhibit PP2A demethylation greater than 50% were considered active in situ PME-1 inhibitors.
Analysis of Cytotoxicity (AID 463091)
Assay Overview: The purpose of this assay was to determine cytotoxicity of powder samples of inhibitor compounds belonging to aza-beta lactam scaffold. HeLa cells in either serum-free media (Assay 1) or media containing FCS (Assay 2) were incubated with test compounds, followed by determination of cell viability. The assay utilizes the CellTiter-Glo luminescent reagent to measure intracellular ATP in viable cells. Luciferase present in the reagent catalyzes the oxidation of beetle luciferin to oxyluciferin and light in the presence of cellular ATP. Well luminescence is directly proportional to ATP levels and cell viability. As designed, compounds that reduce cell viability will reduce ATP levels, luciferin oxidation and light production, resulting in decreased well luminescence. Compounds were tested in quadruplicate in a 7-point 1:5 dilution series starting at a nominal test concentration of 50 μM.
Protocol Summary: This assay was started by dispensing HeLa cells in RPMI media (15 μL, 5 × 103 cells/well) into a 384-well plate. Both serum-free media (Assay 1) and media supplemented with fetal calf serum (Assay 2) were tested. Compound (5 μL of 0–200 μM in media containing 5% DMSO) was added to each well, giving final compound concentrations of 0–50 μM. Cells were incubated for 48 hours at 37 degrees Celsius in a humidified incubator and cell viability was determined by the CellTitre-Glo assay (Promega) according to manufacturer instructions. Assay Cutoff: compounds with a CC50 value of less than 10 μM were considered active (cytotoxic).
LC-MS/MS Analysis of Inhibitor Binding Mode (AID 463090)
Assay Overview: The purpose of this assay was to assess the covalent nature PME-1 inhibition by the aza-beta-lactam probe compound 1 (SID 99206500) and determine whether or not the probe labels the active site serine of PME-1. Purified enzyme was reacted with inhibitor compound, digested with trypsin, and the resulting peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The resulting data were analyzed to identify sites of covalent labeling.
Protocol Summary: Two aliquots (25 μL) of 50 μM PME-1 in DPBS were prepared. To one aliquot was added compound 1 (0.5 μL of 10 μM in DMSO), giving a final concentration of 200 μM. To the second (control) aliquot was added DMSO (0.5 μL). Reactions were gently vortexed and incubated at room temperature for 30 minutes. To each reaction was added solid urea (50 mg), followed by freshly prepared aqueous ammonium bicarbonate (75 μL of 25 μM). The reactions were vortexed until the urea was dissolved. Final urea concentration was approximately 8 M. To each reaction was added freshly prepared TCEP (5 μL of 100 μM in water), and the reactions were incubated at 30 degrees Celsius for 30 minutes. To each reaction was then added freshly prepared IAA (10 μL of 100 μM in water), and the reactions were incubated for 30 minutes at room temperature in the dark. Aqueous ammonium bicarbonate (375 μL of 25 μM) was added to reduce the urea concentration to 2 M. To each reaction was added sequencing grade modified trypsin (1 μg), and reactions were incubated at 37 degrees Celsius for 12 hours. The solvent was removed under reduced pressure in a SpeedVac, and peptides resuspended in ammonium bicarbonate containing 0.1% TFA.
An Agilent 1200 series quaternary HPLC pump and Thermo Scientific LTQ-Orbitrap mass spectrometer were used for sample analysis. A fraction (10 μL) of the protein digest for each sample was pressure-loaded onto a 100 micron fused-silica column (with a 5 micron in-house pulled tip) packed with 10 cm of Aqua C18 reversed-phase packing material. Chromatography was carried out using an increasing gradient of aqueous acetonitrile containing 0.1% formic acid over 125 minutes. Mass spectra were acquired in a data-dependent mode with dynamic exclusion enabled.
The MS/MS spectra generated for each run were searched against a human protein database concatenated to a reversed decoy database using Sequest. A static modification of +57.021 was specified cysteine, and a variable modification of +332.137 was specified for serine to account for possible probe labeling by SID 99206500. The resulting peptide identifications were assembled into protein identifications using DTA Select, and filters were adjusted to maintain a false discovery rate (as determined by number of hits against the reversed database) of less than 1%. Any modified peptides also identified in the DMSO-treated sample were discarded as spurious hits. Assay Cutoff: compounds observed to covalently modify the PME-1 active site serine were considered active.
2.2. Probe Chemical Characterization
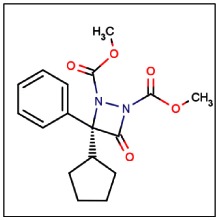
CID 24856225
SID 99206500
ML174
The probe structure was verified by NMR and high resolution MS (calc for Na+: 355.1264, found 355.1258). Purity was assessed to be greater than 99% by NMR, with an enantiomeric excess (ee) of 99.9% by chiral HPLC (Figure 1). Solubility in PBS (137 μM NaCl, 2.7 μM KCl, 10 μM sodium phosphate dibasic, 2 μM potassium phosphate monobasic, pH 7.4) at room temperature was determined to be 165 μM, and the probe has a half-life of 22 hours in PBS at room temperature (tested at 10 μM concentration, see Figure 2).

Figure 1
UV/Vis chiral LC trace of probe (SID 99206500) at 5 different wavelengths. Minor enantiomer (S) elutes at 26.5 min, major enantiomer (R) elutes at 35 min.

Figure 2
Stability of Probe in PBS.
Table 1Compounds Submitted to the SMR Collection
| Designation | Compound Number | Compound Lab Name | CID | SID | SRID | MLS |
|---|---|---|---|---|---|---|
| Probe | 1 | 127A | CID 24856225 | SID 99206500 | SR-01000786812-5 | MLS003126535 |
| Analog 1 | 13 | 227A | CID 24856236 | SID 99206510 | SR-01000786824-3 | MLS003126536 |
| Analog 2 | 15 | 105A | CID 24856234 | SID 99206511 | SR-01000786825-3 | MLS003126537 |
| Analog 3 | 17 | 107B | CID 24856222 | SID 99206506 | SR-01000786819-3 | MLS003126538 |
| Analog 4 | 20 | 111A | CID 24856321 | SID 99206516 | SR-01000808498-3 | MLS003126539 |
| Analog 5 | 21 | 113C | CID 24856226 | SID 99206513 | SR-01000808495-4 | MLS003126540 |
2.3. Probe Preparation
Synthesis of probe compound, as it appears below, has been reported in the literature [15]. Phenyl cyclopentyl ketene [16], catalyst (−)-C [17], and dimethyl azodicarboxylate [18] were prepared according to the reported procedure. CH2Cl2 was purified by passage through activated alumina. Other chemicals were purchased from commercial suppliers and used as received. All reactions were carried out in oven-dried glassware with magnetic stirring.

(R)-Dimethyl 3-cyclopentyl-4-oxo-3-phenyl-1,2-diazetidine-1,2-dicarboxylate (1). In a glove box, a solution of phenyl cyclopentyl ketene A (127mg, 0.68 mmol) and dimethyl azodicarboxylate B (100 mg, 0.68 mmol) in CH2Cl2 (49 mL) was prepared, as well as a solution of catalyst (−)-C (13 mg, 0.035 mmol) in CH2Cl2 (0.8 mL). Solutions were removed from the glove box and placed in a −20 °C bath. After 10 minutes, the solution of the catalyst (−)-C was added (via syringe) to the solution of A and B. The reaction was stirred at −20 °C (2 hours), and the solvent removed by rotary evaporation under reduced pressure. The resulting residue was purified by column chromatography (20% EtOAc/hexanes), which furnished 1 as a viscous, colorless oil (188 mg, 83%). The ee was improved from ~86% to 99.9% through chiral HPLC purification (Chiralcel AD, 1 hour gradient).
1H NMR (500 MHz, CDCl3) δ 7.55 (2H, d, J = 7.5 Hz), 7.38–7.33 (3H, m), 3.89 (3H, s), 3.77 (3H, s), 2.91–2.89 (1H, m), 1.77 (1H, br s), 1.66 (4H, br s), 1.56 (1H, br s), 1.43 (2H, br s)
3. Results
Compound 1 (see Table 2) satisfies the goals for the probe characteristics identified at the project’s outset: 1) probes should be selective for PME-1 among serine hydrolases in human cell line proteomes as assessed by gel-based competitive-activity-based protein profiling (ABPP), and 2) probes should exhibit an IC50 of <10 μM and preferably <1 μM. An IC50 value of 10 nM was derived from gel-based competitive ABPP data (see Section 3.2). A search of more than 30 serine hydrolases yielded data for target/anti-target selectivity. Among this group of anti-targets, which includes carboxylesterase (CES), fatty acid synthase (FAS), N-acylaminoacyl-peptide hydrolase (APEH), prolyl endopeptidase (PREP), and ABHD10, compound 1 is selective at 1 μM. Among observed anti-targets carboxylesterase (CES), fatty acid synthase (FAS), N-acylaminoacyl-peptide hydrolase (APEH), prolyl endopeptidase (PREP), and ABHD10, compound 1 is selective at 0.1 μM. Additionally, compound 1 was shown to be highly active in situ against PME-1 in HeLa cells and to result in 85% reduction of demethylated PP2A (see Section 3.5).
3.1. Summary of Screening Results
In the primary FluoPol uHTS assay (AID 2130), ~302K compounds were screened by FluoPol-ABPP with the FP-Rh probe. A total of 1683 compounds (0.6%) were active, passing the set threshold of 26.13% PME-1 inhibition. For the confirmation uHTS screen (AID 2171), 1514 active compounds were retested in triplicate, and 1068 compounds (70.5%) were confirmed as active (Figure 3).
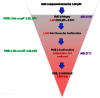
Figure 3
Flow chart describing uHTS screening results.
One of the most interesting chemotypes to emerge from the FluoPol uHTS campaign was the aza-beta-lactam. There were 26 aza-beta lactams in the screening library, and only 2 of them inhibited more than 50% of PME-1 activity. These two compounds, 1 (SID 92709579) and 3 (SID 92709580), were obtained from DPI as liquids for secondary screening. Initially, these compounds were screened against endogenous PME-1 in soluble proteomes derived from two different human cancer cell lines (Hela and MDA-MB-231) by gel-based competitive-ABPP with the FP-Rh activity-based probe (AID 463146). As shown in Figure 4, both compounds were highly active against PME-1, inhibiting 100% of PME-1 activity at 100 nM. In the HeLa soluble proteome both compounds inhibited PME-1 at 50% at 10 μM concentration; in the MDA-MB-231 soluble proteome Compound 3 was somewhat less effective, inhibiting only 25% of PME-1 activity. Neither compound showed evidence of serine hydrolase anti-targets except at the highest (10 μM) dose as compared to the DMSO control (Figure 4).

We then obtained powder samples of 24 of the aza-beta-lactams from Dr. Greg Fu (MIT), the chemist who initially deposited the compounds into the MLSMR Library. These compounds were screened by gel-based competitive-ABPP against the HeLa soluble proteome at 0.1, 1, and 20 μM concentrations to simultaneously assess potency against PME-1 and serine hydrolase anti-target inhibition (see Figure 5 and AID 463149). Anti-targets were scored as hits and included in Table 2 if they were inhibited by at least 50% for a given test compound concentration. With a threshold of ≥50% inhibition of PME-1, of the 24 synthetic compounds tested, 22 compounds were active at 20 μM, 15 compounds were active at 1 μM, and 4 compounds were active at 0.1 μM. In terms of selectivity, 17 compounds had one or more anti-targets at 20 μM, 9 compounds had anti-targets at 1 μM, and no compounds showed evidence of anti-target effects at 0.1 μM. There were 9 compounds that exhibited both potency (≥50% inhibition PME-1) and selectivity (<50% inhibition anti-targets) at 1 μM, and 3 compounds that were both potent and selective at 0.1 μM (the AID 463149 assay cutoff): Compounds 1 (SID 99206500), 2 (SID 99206508), and 3 (SID 99206503)

3.2. Dose Response Curves for Probe
IC50 values were obtained from gel-based competitive-ABPP data (Figure 6).
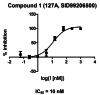
Figure 6
IC50 Curve for Probe Compound as determined by gel-based competitive-ABPP with FP-Rh (AID 463124).
3.3. Scaffold/Moiety Chemical Liabilities
The probe compound was determined to covalently modify the catalytic serine (Ser156) of PME-1 (AID 463090). The observed mass shift of the active site peptide corresponds to the adduct depicted in Figure 7 below, formed by serine nucleophilic attack at the carbonyl to open the lactam ring.

Figure 7
Covalent modification of PME-1 by Probe Compound 1 (SID 99206500).
The probe compound showed no reactivity with glutathione (100 μM), indicating that it is not generally cysteine reactive, but rather has a tempered electrophilicity and specific structural elements that direct reactivity towards PME-1.
An irreversible probe has some distinct advantages over reversible analogs. Targets can be readily characterized by methods such as mass spectrometry and click chemistry-ABPP, required dosing is often lower, irreversible compounds are not as sensitive to pharmacokinetic parameters, and administration can induce long-lasting inhibition [19]. In the case of the EGFR inhibitor PD 0169414, its irreversibility and high selectivity were credited with producing prolonged inhibition of the target, alleviating concerns over short plasma half-lives and reducing the need for high peak plasma levels, thus minimizing potential nonspecific toxic effects [20].
Indeed, over a third of enzymatic drug targets are irreversibly inhibited by currently marketed drugs [21]. Examples of covalent enzyme-inhibitor pairs include serine type D-Ala-D-Ala carboxypeptidase, which is covalently modified by all B-lactam antibiotics, acetylcholinesterase, whose active site serine undergoes covalent modification by pyridostigmine, prostaglandin-endoperoxide synthase, which is the target of the ubiquitously prescribed aspirin, aromatase, which is irreversibly modified by exemestane, monoamine oxidase, which is covalently modified by L-deprenyl, thymidylate synthase, which is covalently modified by floxuridine, H+/K+ ATPase, which undergoes covalent modification by omeprazole, esomeprazole, and lansoprazole, and triacylglycerol lipase, whose serine nucleophile is targeted by orlistat [21].
3.4. SAR Tables
Structures for the 24 aza-beta-lactam structures tested appear in the SAR Table (Table 2).
Table 2SAR Analysis for Target†
 | Potency and Selectivity Analysis‡ | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| In Vitro % INH PME-1 (AID 463146 and 463149) | IC50 (nM) (AID 463124 and 463130) | CC50 (μM) (AID 463091) | Anti-Target(s) Inhibited>50% (AID 463091)¶ | Target to anti-target fold selectivity (AID 463149)** | |||||||||||||||
| Entry | lab name | CID | SID | SRID | § | % ee | * | R1 | R2 | R3 | 0.1 μM | 1 μM | 20 μM | 0.1 μM | 1 μM | 20 μM | |||
| 1 | 127 A | CID 24856225 | SID 92709579 | SR-01000786812-2 | P | 97 | R | H | cyclopentyl | Me | 100 | 100 | NT | NT | NT | NT | NT | NT | NT |
| SID 99206500 | SR-01000786812-5 | Sy | 99.9 | 100 | 100 | 100 | 10 | >50 | 0 | 0 | FAS, APEH | >100 | |||||||
| 2 | 127 B | CID 24856313 | SID 99206508 | SR-01000786822-3 | Sy | 97 | S | H | cyclopentyl | Me | 50 | 100 | 100 | NT | NT | 0 | 0 | 0 | >200 |
| 3 | 103 A | CID 24856231 | SID 92709580 | SR-01000786815-2 | P | 97 | R | H | cyclohexyl | Me | 100 | 100 | NT | NT | NT | NT | NT | NT | NT |
| SID 99206503 | SR-01000786815-4 | Sy | 97 | 90 | 100 | 100 | NT | >10 | 0 | 0 | FAS | >10 | |||||||
| 4 | 103 B | CID 24856324 | SID 99206499 | SR-01000786811-4 | Sy | 97 | S | H | cyclohexyl | Me | 0 | 100 | 100 | NT | NT | 0 | 0 | 0 | >20 |
| 5 | 91A | CID 24856227 | SID 99206507 | SR-01000786820-4 | Sy | 98 | S | H | Et | Me | 0 | 75 | 100 | NT | NT | 0 | CES | CES | <10 |
| 6 | 91C | CID 24856229 | SID 99206515 | SR-01000808497-3 | Sy | 83 | R | H | Et | Me | 0 | 0 | 100 | NT | NT | 0 | 0 | CES, PREP, ABHD10 | NT |
| 7 | 117 A | CID 24856326 | SID 99206498 | SR-01000786810-4 | Sy | 87 | R | m-Me | Et | Me | 0 | 0 | 100 | NT | NT | 0 | CES | CES, FAS, PREP, ABHD10 | NT |
| 8 | 117 B | CID 24856232 | SID 99206514 | SR-01000808496-3 | Sy | 83 | S | m-Me | Et | Me | 0 | 50 | 100 | NT | NT | 0 | CES | CES, FAS | <10 |
| 9 | 123 A | CID 24856235 | SID 99206519 | SR-01000808501-4 | Sy | 69 | R | o-Me | Et | Me | 0 | 50 | 100 | NT | NT | 0 | CES | CES | <10 |
| 10 | 123 B | CID 46829344 | SID 99206521 | SR-02000000386-2 | Sy | 66 | S | o-Me | Et | Me | 0 | 0 | 100 | NT | >50 | 0 | 0 | CES | NT |
| 11 | 143 A | CID 24856325 | SID 99206509 | SR-01000786823-3 | Sy | 80 | R | H | Et | Et | 0 | 0 | 0 | NT | NT | 0 | 0 | CES, PREP, ABHD10 | NT |
| 12 | 143 B | CID 24856314 | SID 99206504 | SR-01000786817-4 | Sy | 84 | S | H | Et | Et | 0 | 0 | 100 | NT | NT | 0 | CES | CES, FAS | NT |
| 13 | 227 A | CID 24856236 | SID 99206510 | SR-01000786824-3 | Sy | 96 | R | H | iPr | Me | 0 | 100 | 100 | 195 | NT | 0 | 0 | 0 | >20 |
| 14 | 227 B | CID 24856238 | SID 99206505 | SR-01000786818-4 | Sy | 97 | S | H | iPr | Me | 0 | 0 | 100 | NT | NT | 0 | 0 | CES | NT |
| 15 | 105 A | CID 24856234 | SID 99206511 | SR-01000786825-3 | Sy | 98 | R | p-OMe | iPr | Me | 0 | 100 | 100 | 69 | NT | 0 | 0 | FAS | >10 |
| 16 | 105 B | CID 24856237 | SID 99206501 | SR-01000786813-4 | Sy | 98 | S | p-OMe | iPr | Me | 0 | 0 | 50 | NT | NT | 0 | 0 | CES | NT |
| 17 | 107 B | CID 24856222 | SID 99206506 | SR-01000786819-3 | Sy | 92 | R | p-Cl | iPr | Me | 0 | 100 | 100 | 315 | NT | 0 | 0 | 0 | >20 |
| 18 | 145 A | CID 24856323 | SID 99206512 | SR-01000808494-3 | Sy | 87 | R | p-Cl | iPr | Et | 0 | 50 | 100 | NT | NT | 0 | 0 | 0 | >20 |
| 19 | 145 B | CID 24856233 | SID 99206502 | SR-01000786814-4 | Sy | 86 | S | p-Cl | iPr | Et | 0 | 0 | 0 | NT | >10 | 0 | 0 | 0 | NT |
| 20 | 111 A | CID 24856321 | SID 99206516 | SR-01000808498-3 | Sy | 96 | R | 3-thiophene†† | iPr | Me | 0 | 50 | 100 | 10745 | NT | 0 | 0 | 0 | >20 |
| 21 | 113 C | CID 24856226 | SID 99206513 | SR-01000808495-4 | Sy | 82 | R | H | iBu | Me | 0 | 100 | 100 | 227 | NT | 0 | CES | CES, FAS, APEH | <10 |
| 22 | 113 B | CID 24856224 | SID 99206520 | SR-01000808502-3 | Sy | 82 | S | H | iBu | Me | 100 | 100 | 100 | NT | NT | 0 | CES, FAS | CES, FAS, APEH | >1 |
| 23 | 115 A | CID 24856228 | SID 99206518 | SR-01000808500-3 | Sy | 85 | R | H | Bz | Me | 0 | 0 | 50 | NT | NT | 0 | CES | CES, ABHD10 | NT |
| 24 | 115 B | CID 24856230 | SID 99206517 | SR-01000808499-4 | Sy | 84 | S | H | Bz | Me | 0 | 75 | 100 | NT | NT | 0 | CES | CES, FAS, APEH | <10 |
- †
Color scheme: enantiomers are grouped by pairs (alternating light and dark blue)
- ‡
Color scheme: green = active (for CC50 data, green = non cytotoxic), red = inactive, grey = not tested (NT), orange = anti-target. Anti-targets: carboxylesterase (CES), fatty acid synthase (FAS), N-acylaminoacyl-peptide hydrolase (APEH), prolyl endopeptidase (PREP), ABHD10
- §
P=obtained from DPI as a liquid; Sy=synthesized
- *
Chiral center: R=phenyl down, R2 is up; S=phenyl is up, R2 is down, the most active enantiomer is highlighted in yellow
- ¶
Anti-targets are reported as hits if ≥50% inhibition is observed at the given test compound concentration (0.1, 1 or 20 μM) as reported in AID 463149.
- **
Fold-selectivity is reported as > Conc_<50%_INH_Anti-target/Conc_≥50%_INH_PME-1 (AID 463149) where
- Conc_<50%_INH_Anti-target is the test compound concentration at which less than 50% inhibition of the anti-target is observed (AID 463149).
- Conc_≥50%_INH_PME-1 is the test compound concentration at which greater than or equal to 50% inhibition of PME-1 is observed (AID 463149).
If Conc_<50%_INH_Anti-target is 0.1 μM and Conc_≥50%_INH_PME-1 is 1 μM, fold selectivity is reported as <10 (AID 463149), except in cases where the target IC50 has been determined, in which case fold-selectivity is reported as: > Conc_<50%_INH_Anti-target/IC50_target, where
- Conc_<50%_INH_Anti-target is the test compound concentration at which less than 50% inhibition of the anti-target is observed (AID 463149).
- IC50_target is the IC50 against PME-1 (AID 463130, AID 463124).
- ††
3-thiophene replaces phenyl
SAR of Chiral Center: With the exception of compounds 17 and 20, all compounds in the SAR Table (Table 2) are also represented by their enantiomeric counterpart (grouped by color – light blue or dark blue). In all cases, one enantiomer (highlighted in yellow) is significantly more active than the other (compare in vitro % inhibition at 0.1, 1, or 20 μM). The gel-based competitive-ABPP comparison for two enantiomeric pairs is shown in Figure 8 (AID 463146). At 10 nM concentration, both R enantiomers (1 and 3) inhibit PME-1 by 50%, whereas the S enantiomers (2 and 4) show no inhibition. Indeed, some inhibition of PME-1 by 2 and 4 may be due to contamination by the active enantiomer (97% ee). As evident from Figure 8 and the number of anti-target hits in the SAR Table (Table 2), the selectivity of certain enantiomeric pairs also differs dramatically (e.g., 1 vs. 2 and 3 vs. 4 at 10000 nM concentration, Figure 8).

Figure 8
Comparative profiling of enantiomeric pairs: Compounds 1 vs. 2 and 3 vs. 4. Fluorescent image shown in grey scale; DMSO (no compound) control in lane 1 of both gels; bands corresponding to PME-1 indicated with arrows.
SAR of R2: In cases where R2 is di-substituted at the alpha lactam carbon (cyclopenyl, cyclohexyl, or isopropyl) the most active enantiomer is the R configuration (designated phenyl down, R2 up). However, in cases where the alpha lactam carbon is a methylene (ethyl, isobutyl and benzyl), the more potent enantiomer is S (designated phenyl up, R2 down). The only exception to this rule is compound 9, which has an ethyl substituent and R as the more potent enantiomer. However, it is the only analog with an ortho substituent at R1, which could impose some sort of structural differentiation (note: it is also the least enantiomerically pure (<70%) of all of the compounds, which could complicate comparative analysis).
The R2 position also serves as a differentiating factor in terms of selectivity (as judged by the number of anti-target hits in SAR Table [Table 2]), with compounds with di-substituted alpha lactam carbons being more selective than compounds where the carbon alpha to the lactam is a methylene; indeed, most of the alpha-C methylene compounds potently inhibit the anti-target carboxylesterase (CES), a common, anti-target for serine hydrolase inhibitors. In contrast to R vs. S potency, compound 9 groups with its methylene class in terms of selectivity.
Compared to selectivity, slightly different groupings distinguish the compounds based on potency. Compounds with comparatively large alkyl substituents at R2 (cyclopentyl for 1, cyclohexyl for 3, isobutyl for 22) are the most potent analogs (vs. ethyl for 5, 6 and isopropyl for 13, 14). However, placing a larger, aromatic group at R2 (benzyl for 23, 24) is also deleterious for potency.
SAR of R1: Based on the R2 analysis above, since both compound 20 (with 3-thiophene instead of phenyl) and 17 (para-chloro at R1) are R enantiomers with di-substituted carbons alpha to the lactam, they should be the more potent of their respective R/S pairs (this assumption, however, would require validation by testing the S configuration compounds, which weren’t available for analysis). If this is indeed the case, then the thiophene (20) in place of phenyl (13) is detrimental to potency, and a para-chloro subsitition at R1 (17) instead of hydrogen (13) has little effect on potency. Similarly, para-methoxy substitution (15) also does not seem to affect potency (vs. hydrogen, 13). The only other substituents tested at R1 were meta-and ortho-substituted methyl groups. Both had similar potency to each other, however, as mentioned above, presence of an ortho substituent changes the most active enantiomer from the expected S to R. Unfortunately, there is no suitable comparison with an R1=H analog, so it is unclear what type of effect an ortho- or meta- methyl substitution would have on the probe compound 1. This subject would be a good focus of future SAR exploration.
SAR of R3: Most derivatives, including the probe compound 1, have methyl at R3, and replacing methyl with ethyl reduces potency (compound 5 vs. 12 and 17 vs. 18). Due to synthetic constraints, no other substitutions have been explored at this position; however, exploration of R3 will likely be the subject of future SAR efforts.
Summary: R enantiomers of compounds with small cyclic (and hence di-substituted at the alpha lactam carbon) structures at R2, methyl at R3, and hydrogen (or possibly para-chloro, or para-methoxy) at R1 – compounds 1 and 3 – are the most potent and selective.
3.5. Cellular Activity
Probe compound 1 and compound 3 are highly active against PME-1 in situ (AID 463131), completely inhibiting enzymatic activity at 500 nM compound concentration after 1 hour as assayed by gel-based competitive ABPP (Figure 9A). This inhibition of activity was confirmed by monitoring levels of PP2A demethylation (AID 463132). As shown in Figure 9B, administration of 500nM compound 1 or 3 to Hela cells in culture (1 hour) also results in a dramatic (85%) reduction of demethylated PP2A as compared to DMSO (no compound) control. Taken together, these results indicate that both the probe compound 1 and compound 3 are free to cross cell membranes and inhibit their target in the cytoplasm of cells.
The probe compound 1 and three representative analogs (compounds 3, 10, and 19) were evaluated for cell toxicity (AID 463091) using both serum-free and serum-supplemented media. As shown in Figure 10 and as reported in the Table 2, compounds 3 and 19 have CC50s >10 μM and compounds 1 and 10 have CC50s >50 μM, which is, respectively, 20-fold and 100-fold above the concentration (500 nM) necessary for complete inhibition of PME-1 in situ. Given these results, compound 1 was selected as the probe over compound 3.

Figure 10
Cytotoxicity of probe and probe analogs in serum-free (A) and serum-supplemented (B) media.
3.6. Profiling Assays
To date, the probe compound 1 (CID 24856225) has been tested in 149 other bioassays deposited in PubChem, and has shown activity in only 2 of those assays, giving a hit rate of 1.3%, indicating that this compound is not generally active across a broad range of cell-based and non-cell based assays.
4. Discussion
Compound 1 (SID 99206500) was identified as a highly potent and selective covalent inhibitor of the target enzyme PME-1. The probe has an IC50 of 10 nM, and greater than 100-fold selectivity against potential serine hydrolase anti-targets as assessed by competitive ABPP profiling. This compound was chosen over the other two leading probe candidates (2 and 3) because it was more potent than 2 (Table 2 and Figure 8) and had a 5-fold higher CC50 than 3 (50 μM vs. 10 μM, Figure 10). The probe has been shown to inhibit both PME-1 activity and PME-1-mediated PP2A demethylation in situ. Overall, this molecule has favorable stability for in vitro and in situ assays (half-life of 22 hours in PBS), solubility (165 μM in PBS), and general reactivity (not reactive with glutathione, low [1.3%] activity in other PubChem bioassays) properties. Taken together, these findings suggest that it is very possible to develop potent and selective probes based on tempered electrophilic scaffolds, and that compound 1 will be a highly successful probe for biochemical investigation of PME-1.
We have recently shown that treatment of mice with compound 1 for two hours results in complete inactivation of PME-1 with no substantial reductions in any of approximately 40 other brain serine hydrolases [22]. An approximate 35% reduction in the amount of demethylated PP2A in brain tissue from these mice was also observed [22], confirming that compound 1 can selectively inhibit PME-1 in mice and that this inhibition alters the methylation state of brain PP2A.
The structure of compound 1 is based on per-carbamolylated aza-beta-lactam, a chemically reactive moiety. However, we observed greater than 100-fold selectivity against potential anti-targets. The somewhat limited half-life of 22 hours in PBS raises concern about whether or not this probe can provide sustained inhibition of PME-1 as required for extended studies in living systems. In an extended probe development phase, more extensive SAR studies will be pursued to optimize probe half-life, and anti-target characterization will be expanded.
4.1. Comparison to existing art and how the new probe is an improvement
In conjunction with the SRIMSC, we previously reported a PME-1 inhibitor based on a sulfonyl acrylonitrile scaffold (SID 87457340) [14]. While selective, the compound exhibited an IC50 of only 0.5 μM, which made the compound suitable for some basic in situ and in vitro studies, but not a good candidate for more elaborate studies with longer time courses and eventual in vivo application. While the potency of SID 87457340 was a significant (>20-fold) improvement over the initial lead structure found by uHTS, extensive SAR analysis suggested that we were unlikely to obtain a more potent derivative based on the sulfonyl acrylonitrile scaffold. The probe compound 1 (SID 99206500) detailed in this report is based on an entirely different scaffold, the aza-beta-lactam, and is 50-fold more potent (IC50 10 nM) than the previously reported probe.
4.2. Mechanism of Action Studies
As determined from LC-MS/MS analysis (AID 463090), the probe is an activity-based inhibitor that covalently labels the active site serine nucleophile, Ser156, of PME-1. The observed mass shift of the active site peptide suggests that reaction occurs via serine nucleophilic attack on the carbonyl to open the lactam ring (Figure 7). Because of the secondary screening methodology employed – competitive-ABPP profiling with active-site directed activity-based probes – and the direct assessment on the methylation state of PP2A, a substrate of PME-1, we can directly link inhibition of PME-1 by the probe compound 1 to a corresponding loss of enzymatic activity.
4.3. Planned Future Studies
We plan to use probe compound 1 (SID 99206500) to investigate the link between PME-1, PP2A methylation, and cancer progression by 1) globally profiling phosphorylation events differentially regulated in cancer cells with hypermethylated PP2A, and 2) evaluating the role of PME-1 in cancer pathogenesis using assays that measure cancer cell proliferation, migration, invasion, and tumor growth in mouse xenograft models. Additionally, as the chemistry for synthesis of aza-beta-lactams advances, we will continue to investigate the SAR, especially at positions R1 and R3, to optimize probe half-life for sustained PME-1 inhibition and extend anti-target characterization.
5. References
- 1.
- Puustinen P, et al. PME-1 protects extracellular signal-regulated kinase pathway activity from protein phosphatase 2A-mediated inactivation in human malignant glioma. Cancer Res. 2009;69(7):2870–2877. [PMC free article: PMC2810347] [PubMed: 19293187]
- 2.
- Ortega-Gutierrez S, et al. Targeted disruption of the PME-1 gene causes loss of demethylated PP2A and perinatal lethality in mice. PLoS One. 2008;3(7):e2486. [PMC free article: PMC2438471] [PubMed: 18596935]
- 3.
- Liu Y, Patricelli MP, Cravatt BF. Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A. 1999;96(26):14694–14699. [PMC free article: PMC24710] [PubMed: 10611275]
- 4.
- Oliver CJ, Shenolikar S. Physiologic importance of protein phosphatase inhibitors. Front. Biosci. 1998;3:D961–972. [PubMed: 9727084]
- 5.
- Janssens Va, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem. J. 2001;353(Pt 3):417–439. [PMC free article: PMC1221586] [PubMed: 11171037]
- 6.
- Janssens V, Goris J, Van Hoof C. PP2A: the expected tumor suppressor. Curr. Opin. Genet. Dev. 2005;15(1):34–41. [PubMed: 15661531]
- 7.
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257(5074):1261–1264. [PubMed: 1325671]
- 8.
- Favre B, et al. The catalytic subunit of protein phosphatase 2A is carboxyl-methylated in vivo. J. Biol. Chem. 1994;269(23):16311–16317. [PubMed: 8206937]
- 9.
- Lee J, et al. A specific protein carboxyl methylesterase that demethylates phosphoprotein phosphatase 2A in bovine brain. Proc. Natl. Acad. Sci. U.S.A. 1996;93(12):6043–6047. [PMC free article: PMC39185] [PubMed: 8650216]
- 10.
- Wu J, et al. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. EMBO J. 2000;19(21):5672–5681. [PMC free article: PMC305778] [PubMed: 11060018]
- 11.
- Jessani N, et al. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc Natl Acad Sci U S A. 2002;99(16):10335–10340. [PMC free article: PMC124915] [PubMed: 12149457]
- 12.
- Leung D, et al. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21(6):687–691. [PubMed: 12740587]
- 13.
- Bachovchin DA, et al. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 2009;27(4):387–394. [PMC free article: PMC2709489] [PubMed: 19329999]
- 14.
- Bachovchin DA. MLPCN probe report. 2010. Probe Report for PME-1 Inhibitors.
- 15.
- Berlin JM, Fu GC. Enantioselective nucleophilic catalysis: the synthesis of aza-beta-lactams through [2+2] cycloadditions of ketenes with azo compounds. Angew. Chem. Int. Ed. Engl. 2008;47(37):7048–7050. [PMC free article: PMC2790040] [PubMed: 18668500]
- 16.
- Allen AD, et al. Cyclopropylketenes: preparation and nucleophilic additions. Can. J. Chem. 1991;69(1):138–145.
- 17.
- Wurz RP, et al. Synthesis and resolution of planar-chiral derivatives of 4-(Dimethylamino)pyridine. Adv. Syn. Catal. 2007;349(14–15):2345–2352.
- 18.
- Mackay D, McIntyre DD, Wigle ID. Anomalous shielding and hidden partner chemical exchanges in the 1H NMR spectra of the bisurethane diazetidines, the 1,2-diaryl-3,5-dialkyl-6,7-dialkoxycarbonyl-4-oxo-6,7-diazabicyclo[3.2.0]hept-2-enes. J. Chem. Soc.-Perkin Trans. 1989;2(12):1999–2010.
- 19.
- Johnson DS, Weerapana E, Cravatt BF. Strategies for discovering and derisking covalent, irreversible enzyme inhibitors. Future Med Chem. 2010;2(6):949–964. [PMC free article: PMC2904065] [PubMed: 20640225]
- 20.
- Vincent PW, et al. Anticancer efficacy of the irreversible EGFr tyrosine kinase inhibitor PD 0169414 against human tumor xenografts. Cancer Chemother Pharmacol. 2000;45(3):231–238. [PubMed: 10663641]
- 21.
- Robertson JG. Mechanistic basis of enzyme-targeted drugs. Biochemistry. 2005;44(15):5561–5571. [PubMed: 15823014]
- 22.
- Bachovchin DA, et al. Academic cross-fertilization by public screening yields a remarkable class of protein phosphatase methylesterase-1 inhibitors. Proc Natl Acad Sci U S A. 2011;108(17):6811–6816. [PMC free article: PMC3084096] [PubMed: 21398589]
- PMCPubMed Central citations
- PubChem BioAssay for Chemical ProbePubChem BioAssay records reporting screening data for the development of the chemical probe(s) described in this book chapter
- PubChem SubstanceRelated PubChem Substances
- PubMedLinks to PubMed
- Review Probe Development Efforts to Identify Novel Inhibitors of ABHD10.[Probe Reports from the NIH Mol...]Review Probe Development Efforts to Identify Novel Inhibitors of ABHD10.Zuhl AM, Mohr JT, Speers AE, Bachovchin DA, Berlin JM, Spicer T, Fernandez-Vega V, Brown SJ, Ferguson J, Fu GC, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Targeted disruption of the PME-1 gene causes loss of demethylated PP2A and perinatal lethality in mice.[PLoS One. 2008]Targeted disruption of the PME-1 gene causes loss of demethylated PP2A and perinatal lethality in mice.Ortega-Gutiérrez S, Leung D, Ficarro S, Peters EC, Cravatt BF. PLoS One. 2008 Jul 2; 3(7):e2486. Epub 2008 Jul 2.
- A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A.[J Biol Chem. 1999]A protein phosphatase methylesterase (PME-1) is one of several novel proteins stably associating with two inactive mutants of protein phosphatase 2A.Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. J Biol Chem. 1999 May 14; 274(20):14382-91.
- Discovery and optimization of sulfonyl acrylonitriles as selective, covalent inhibitors of protein phosphatase methylesterase-1.[J Med Chem. 2011]Discovery and optimization of sulfonyl acrylonitriles as selective, covalent inhibitors of protein phosphatase methylesterase-1.Bachovchin DA, Zuhl AM, Speers AE, Wolfe MR, Weerapana E, Brown SJ, Rosen H, Cravatt BF. J Med Chem. 2011 Jul 28; 54(14):5229-36. Epub 2011 Jun 30.
- Review Probe Report for PME-1 Inhibitors.[Probe Reports from the NIH Mol...]Review Probe Report for PME-1 Inhibitors.Bachovchin DA, Speers AE, Zuhl AM, Brown SJ, Cravatt BF, Fernandez-Vega V, Spicer T, Mercer BA, Ferguson J, Hodder P, et al. Probe Reports from the NIH Molecular Libraries Program. 2010
- Probe Development Efforts to Identify Novel Inhibitors of Protein Phosphatase Me...Probe Development Efforts to Identify Novel Inhibitors of Protein Phosphatase Methylesterase-1 (PME-1) - Probe Reports from the NIH Molecular Libraries Program
Your browsing activity is empty.
Activity recording is turned off.
See more...