

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Dahabreh IJ, Wieland LS, Adam GP, et al. Core Needle and Open Surgical Biopsy for Diagnosis of Breast Lesions: An Update to the 2009 Report [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2014 Sep. (Comparative Effectiveness Reviews, No. 139.)

Core Needle and Open Surgical Biopsy for Diagnosis of Breast Lesions: An Update to the 2009 Report [Internet].
Show detailsThis report updates a previously completed Comparative Effectiveness Review on core needle and open surgical biopsy methods for the diagnosis of breast cancer. To update the report we performed a systematic review of the published scientific literature using established methodologies as outlined in the Agency for Healthcare Research and Quality’s (AHRQ) “Methods Guide for Comparative Effectiveness Reviews,” which is available at: http://effectivehealthcare.ahrq.gov. 7 The main sections in this chapter reflect the elements of the protocol that guided this review. We have followed the reporting requirements of the “Preferred Reporting Items for Systematic Reviews and Meta-analyses” (PRISMA) checklist.8 All key methodological decisions were made a priori. The protocol was developed with input from external clinical and methodological experts, in consultation with the AHRQ task order officer (TOO), and was posted online to solicit additional public comments. Its PROSPERO registration number is CRD42013005690.
AHRQ Task Order Officer
The AHRQ Task Order Officer (TOO) was responsible for overseeing all aspects of this project. The TOO facilitated a common understanding among all parties involved in the project, resolved ambiguities, and fielded all Evidence-based Practice Center (EPC) queries regarding the scope and processes of the project. The TOO and other staff at AHRQ helped to establish the Key Questions and protocol and reviewed the report for consistency, clarity, and to ensure that it conforms to AHRQ standards.
External Stakeholder Input
A new panel of experts was convened to form the Technical Expert Panel (TEP). The TEP included representatives of professional societies, experts in the diagnosis and treatment of breast cancer (including radiologists and surgeons), and a patient representative. The TEP provided input to help further refine the Key Questions and protocol, identify important issues, and define the parameters for the review of evidence. Discussions among the EPC, TOO, and the TEP occurred during a series of teleconferences and via email.
Key Questions
The final Key Questions are listed at the end of the Background section. The refinement of the Key Questions took into account the patient populations, interventions, comparators, outcomes, and study designs that are clinically relevant for core needle biopsies.
Analytic Framework
We used an analytic framework (Figure 1) that maps the Key Questions within the context of populations, interventions, comparators, and outcomes of interest. The framework was adapted from that used in the original 2009 CER. It depicts the chain of logic that links the test performance of core needle biopsy for the diagnosis of breast abnormalities (Key Question 1) with patient-relevant outcomes (Key Question 3) and adverse events of testing (Key Question 2).
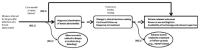
Figure 1
Analytic framework. KQ = Key Question.
Scope of the Review
Populations and Conditions of Interest
The population of interest for all Key Questions was women who have been referred for biopsy for the diagnosis of primary breast cancer (including multifocal and bilateral disease) following self-examination, physical examination, or screening mammography. Studies carried out in women who had been previously diagnosed with breast cancer and were being examined for recurrence or to assess the extent of disease (staging) were excluded. The original report excluded studies carried out in women at high risk of breast cancer; however, MRI-guided biopsy is used mainly in this subset of patients. For this reason, following extensive discussions with the TEP, we decided to broaden the scope of the review to include studies carried out in women at high baseline risk of breast cancer (e.g., on the basis of BRCA genetic testing or family history of breast cancer). Of note, studies often do not provide information on the risk of cancer among included patients. Thus we grouped studies into two categories: (1) studies that explicitly reported that more than 15 percent of included patients were at high risk of cancer; (2) studies that either enrolled less than 15 percent of patients at high risk of cancer or did not provide information on baseline risk. Throughout this report, we refer to the latter group as “studies of women at average risk of cancer”; however, we acknowledge that it may include studies enrolling patients at higher-than-average cancer risk but failing to report the relevant information.
Interventions
For all Key Questions, the interventions of interest were core needle and open biopsy done to evaluate whether a breast lesion is malignant. Other uses of biopsy techniques (e.g., use of biopsy to examine the sentinel lymph nodes in women with an established diagnosis of breast cancer) were not considered. Studies were required to have used biopsy instrumentation that is currently commercially available, as studies of discontinued devices are not applicable to current practice.
Comparators (Index and Reference Standard Tests)
For test performance outcomes (Key Question 1) the reference standard was either open surgical biopsy, or followup by clinical examination and/or mammography for at least six months. The diagnostic performance of each core biopsy technique (each index test) was quantified versus the reference standard. Most assessments of diagnostic performance quantify the sensitivity and the specificity of each index test – here each needle core biopsy technique. Sensitivity and specificity are probabilities conditional on true disease status, and are noncomparative in nature. The reference standard is used in their definition, and is not a “comparator test”. The comparative diagnostic performance of alternative needle core biopsy techniques was also evaluated. For adverse events and patient-relevant outcomes (outcomes other than diagnostic performance; Key Questions 2 and 3) the comparators of interest were: open surgical biopsy, followup by clinical examination and/or mammography for at least six months, or alternative core needle biopsy methods (e.g., stereotactic mammography versus ultrasound to locate the breast lesion; use versus non-use of vacuum-assistance to extract tissue samples).
Outcomes
For Key Question 1, the outcome of interest was test performance, as assessed by sensitivity (proportion of cancers detected by the reference standard that are also detected by core needle biopsy); specificity (proportion of negative findings according to core needle biopsy that were classified as negative by the reference standard; equal to one minus the false positive rate); underestimation rate for high risk lesions (most often atypical ductal hyperplasia, ADH), defined as the proportion of core needle biopsy findings of high risk lesions that are found to be malignant according to the reference standard); and underestimation rate for ductal carcinoma in situ (DCIS), defined as the proportion of core needle biopsy findings of DCIS that are found to be invasive according to the reference standard.
For Key Question 2 we looked for the following outcomes: rate of inconclusive biopsy findings (e.g. inadequate sampling of lesion); comparisons of repeat biopsy rates between core needle and open surgical biopsy; subsequent false positive and false negative rates on mammography (impact of breast biopsy on future mammographic examinations); dissemination or displacement of cancerous cells along the needle track; and patient-centered outcomes (including bruising, bleeding or hematomas, pain, use of pain medication, infections, fainting or near fainting, time to recover). Because adverse events were not consistently defined across studies, we accepted the definitions used in the individual studies (when available).
For Key Question 3, we considered patient-relevant outcomes (patient preferences for specific procedures, cosmetic results, quality of life, anxiety and other psychological outcomes, time to complete tumor removal [for women with cancer], recurrence rate [for women with cancer, including local, regional, and distant recurrence], cancer-free survival and overall survival); resource use and logistics (costs, resource utilization other than cost [number of additional surgical procedures, procedural time], subsequent surgical procedures, wait time for test results); and availability of technology and relevant expertise (physician experience, availability of equipment, availability of [qualified] pathologists to evaluate biopsy samples).
Timing
We required that the duration of clinical and/or mammography followup was at least six months in studies where open surgical biopsy was not performed.
Setting
Studies in all geographic locations and care settings were evaluated, including general hospitals, academic medical centers, and ambulatory surgical centers, among others.
Study Design and Additional Criteria
We required that studies had been published in peer-reviewed journals as full articles. For all Key Questions, studies were required to have been published in English. Restricting included studies to those published in English, which was also an inclusion criterion in the original review, was deemed unlikely to bias the results of the review and avoids the resource-intensive translation of research articles published in languages other than English.
For Key Question 1 eligible studies were prospective or retrospective cohort studies or randomized controlled trials. Retrospective case studies (“case series”9) and other studies sampling patients on the basis of outcomes (e.g. diagnostic case-control studies, or studies selecting cases on the basis of specific histological findings) were excluded. Empirical evidence from meta-epidemiological studies suggests that diagnostic case-control studies may overestimate test performance. Studies were required to report information on the sensitivity, specificity, positive or negative predictive value of tests, or to include data that allow the calculation of one or more of these outcomes. Specifically, studies needed to provide adequate information to reconstruct 2×2 tables of test performance of the index against the reference standard. Table 1 illustrates how index and reference standard results were used to construct such 2×2 tables.
Table 1
Definitions of diagnostic groups based on index and reference standard test results.
Two issues related to the definition of diagnostic test categories merit additional description. First, occasionally core needle biopsy removes the entire target lesion that is being biopsied, rendering subsequent surgical biopsies unable to confirm the findings of the index test procedure. In such cases of core needle diagnoses of malignancy, we considered the core needle results to be true positive. This operational definition was adopted by several of the primary studies we reviewed and the original ECRI report. Second, in our primary analysis (and consistent with the 2009 ECRI report) core needle biopsy identified high risk lesions that on subsequent surgery (or followup) are not found to be associated with malignant disease were considered false positive. To assess the impact of this operational definition on our findings we performed a sensitivity analysis where high risk lesions on index core needle biopsy found to be non-malignant (high risk or benign) on subsequent open biopsy or surgery were excluded from the analyses.
Noncomparative studies of test performance (i.e. studies of a single index test) were required to have enrolled at least 10 participants per arm or per comparison group. This inclusion criterion was intended to reduce the risk of bias from non-representative participants in small studies. Further, smaller studies do not produce precise estimates of test performance and as such are unlikely to substantially affect results. Studies were also required to have followed at least fifty percent of participants to completion. This criterion was intended to reduce the risk of bias from high rates of attrition.
Key Question 2 was addressed by extracting harm-related information for core needle biopsy and open surgical biopsy from studies meeting the criteria for Key Question 1. In addition, we included studies that met all other selection criteria for Key Question 1 except for the use of a reference standard and the reporting of information on test performance outcomes. This allowed us to consider additional sources of evidence that assess adverse events. Finally, for this Key Question, we also reviewed primary research articles, regardless of design (i.e., case reports and case series, case-control studies, cohort studies, randomized trials), that address the dissemination or displacement of cancer cells by the biopsy procedure, a relatively rare harm that is specific to core biopsy.
The original report did not use formal criteria for study selection for Key Question 3. Based on the findings of the original report, we used the same PICOTS criteria described above and considered the following study designs:
- Randomized controlled trials, cohort studies, and cross-sectional studies on patient preferences, cosmetic results of biopsy procedures, physician experience (including studies of the “learning curve” for different biopsy methods and tools).
- Cost studies, including cost-minimization and cost-consequence analyses, were used to obtain information on resource utilization and unit costs. Given the large variability of cost information among different jurisdictions, we only considered studies conducted in the U.S. setting and published after 2004.10
- Cost-effectiveness/cost-utility analyses based on primary trials of breast biopsy interventions were used to obtain information on unit costs and resource utilization.11 Specifically, we considered the components of cost and resource use but did not use cost-effectiveness ratios or other summary measures of cost-effectiveness/utility. As for cost studies, we only considered primary cost-effectiveness/-utility studies conducted in the US setting and published after 2004.10 We did not use model-based cost-effectiveness results.
- Studies of pathologist qualifications for interpreting core needle biopsy results; including interlaboratory initiatives to standardize diagnostic criteria (e.g., proficiency testing) or minimal competency requirements.
- Surveys of the availability of equipment for obtaining core needle biopsies and of qualified pathologists to examine biopsy samples.
Literature Search and Abstract Screening
We searched MEDLINE®, Embase®, the Cochrane Central Register of Controlled Trials (CENTRAL), the Cochrane Database of Systematic Reviews, the Database of Abstracts of Reviews of Effects (DARE), the Health Technology Assessment Database (HTA), the U.K. National Health Service Economic Evaluation Database (NHS EED), the U.S. National Guideline Clearinghouse (NGC), and the Cumulative Index to Nursing and Allied Health Literature (CINAHL®); last search on December 16, 2013. Appendix A describes the search strategy we employed which is a revision and expansion of the search strategy used in the original report. Of note, the original report used a search filter for studies of diagnostic tests to increase search specificity; this is a reasonable approach given the large volume of literature on studies on diagnostic biopsy methods for breast cancer. Because this update covered a short time period (from 2009 to 2013) we opted to not use this filter, in order to increase search sensitivity.12, 13 Our searches covered the time period from six months before the most recent search date in the original report, to ensure adequate overlap.
To identify studies excluded from the original report because they enrolled women at high risk for cancer, the set of abstracts screened for the original report was obtained and rescreened for potentially eligible studies of high risk women. In addition, the list of studies excluded from the original report following full text review was checked to identify studies excluded because they included women at high risk for cancer. We also performed a search for systematic reviews on the topic and used their reference lists of included studies to validate our search strategy and to make sure we identified all relevant studies.
All reviewers screened a common set of 200 abstracts (in 2 pilot rounds, each with 100 abstracts), and discussed discrepancies, in order to standardize screening practices and ensure understanding of screening criteria. The remaining citations were split into nonoverlapping sets, each screened by two reviewers independently. Discrepancies were resolved by consensus involving a third investigator.
We asked the TEP to provide citations of potentially relevant articles. Additional studies were identified through the perusal of reference lists of eligible studies, published clinical practice guidelines, relevant narrative and systematic reviews, Scientific Information Packages from manufacturers, and a search of U.S. Food and Drug Administration databases. All articles identified through these sources were screened for eligibility against the same criteria as for articles identified through literature searches. We sent the final list of included studies to the TEP to ensure that no key publications had been missed.
Study Selection and Eligibility Criteria
Potentially eligible citations were obtained in full text and reviewed for eligibility on the basis of the predefined inclusion criteria. A single reviewer screened each potentially eligible article in full-text to determine eligibility; reviewers were instructed to be inclusive. A second reviewer verified all relevant articles. Disagreements regarding article eligibility were resolved by consensus involving a third reviewer. Appendix B lists all the studies excluded after full-text screening and the reason for exclusion.
Data Abstraction and Management
Data was extracted using electronic forms and entered into the Systematic Review Data Repository (SRDR; http://srdr.ahrq.gov/). The basic elements and design of these forms is similar to those we have used for other reviews of diagnostic tests and includes elements that address population characteristics, sample size, study design, descriptions of the index and reference standard tests of interest, analytic details, and outcome data. Prior to data extraction, forms were customized to capture all elements relevant to the Key Questions. We used separate sections in the extraction forms for Key Questions related to short-term outcomes, including classification of breast abnormalities, intermediate outcomes (such as clear surgical margins), patient-relevant outcomes (such as quality of life), and factors affecting (modifying) test performance. We pilot-tested the forms on several studies extracted by multiple team members to ensure consistency in operational definitions.
A single reviewer extracted data from each eligible study. At least one other team member reviewed and confirmed all data (data verification). Disagreements were resolved by consensus including a third reviewer. We contacted authors (1) to clarify information reported in the papers that is hard to interpret (e.g., inconsistencies between tables and text); and (2) to verify suspected overlap between study populations in publications from the same group of investigators.
Assessment of the Risk of Bias of Individual Studies
We assessed the risk of bias for each individual study using the assessment methods detailed in the AHRQ Methods Guide for Effectiveness and Comparative Effectiveness Review hereafter referred to as the Methods Guide. We used elements from the Quality Assessment for Diagnostic Accuracy Studies instrument (QUADAS version 2), to assess the risk of bias (methodological quality or internal validity) of the diagnostic test studies included in the review (these studies comprise the majority of the available studies).14–17 The tool assesses four domains of risk of bias related to patient selection, index test, reference standard test, and patient flow and timing. For studies of other designs we used appropriate sets of items to assess risk of bias or methodological “quality”: for nonrandomized cohort studies we used items from the Newcastle-Ottawa scale,18 for randomized controlled trials we used items from the Cochrane Risk of Bias tool,19 and for studies of resource utilization and costs we used items from the checklist proposed by Drummond et al.20
We assessed and reported methodological quality items (as “Yes”, “No”, or “Unclear/Not Reported”) for each eligible study. We then rated each study as being of low, intermediate, or high risk of bias on the basis of adherence to accepted methodological principles. Generally, studies with low risk of bias have the following features: lowest likelihood of confounding due to comparison to a randomized controlled group; a clear description of the population, setting, interventions, and comparison groups; appropriate measurement of outcomes; appropriate statistical and analytic methods and reporting; no reporting inconsistencies; clear reporting of dropouts and a low dropout rate; and no other apparent sources of bias. Studies with moderate risk of bias are susceptible to some bias but not sufficiently to invalidate results. They do not meet all the criteria for low risk of bias owing to some deficiencies, but none are likely to introduce major bias. Studies with moderate risk of bias may not be randomized or may be missing information, making it difficult to assess limitations and potential problems. Studies with high risk of bias are those with indications of bias that may invalidate the reported findings (e.g., observational studies not adjusting for any confounders, studies using historical controls, or studies with very high dropout rates). These studies have serious errors in design, analysis, or reporting and contain discrepancies in reporting or have large amounts of missing information. We discuss the handling of high risk of bias studies in evidence synthesis in the following sections. Studies of different designs were graded within the context of their study design.
Data Synthesis
We summarized included studies qualitatively and presented important features of the study populations, designs, tests used, outcomes, and results in summary tables. Population characteristics of interest included age, race/ethnicity, and palpability of lesion. Design characteristics included methods of population selection and sampling, and followup duration. Test characteristics included imaging-guided versus not imaging-guided, and vacuum-assisted versus not vacuum-assisted methods. We looked for information on test performance, adverse events, patient preferences, and resource utilization including costs.
Statistical analyses were conducted using methods currently recommend for use in Comparative Effectiveness Reviews of diagnostic tests.21, 22 For all outcomes we assessed heterogeneity graphically (e.g. by inspecting a scatterplot of studies in the receiver operating characteristic, ROC, space) and by examining the posterior distribution of between-study variance parameters.
For Key Question 1 we performed meta-analysis on studies that were deemed sufficiently similar. Based on the technical characteristics of the different tests, and the findings of the original Evidence Report, we developed a mixed effects binomial-bivariate normal regression model that accounted for different imaging methods (e.g. US, stereotactic mammography, MRI), the use of vacuum (yes vs. not), the baseline of risk of cancer of included patients (high versus average risk), and residual (unexplained) heterogeneity.23–25 This model allowed us to estimate the test performance of alternative diagnostic tests, and perform indirect comparisons among them.23 Furthermore, it allowed us to model the correlation between sensitivity and specificity and to derive meta-analytic ROC curves.24, 25 A univariate mixed effects logistic regression (binomial-normal) model was used for the meta-analysis of DCIS and high risk lesion underestimation rates.26
We performed meta-regression analyses (e.g. to evaluate the impact of study risk of bias items, or the effect of other study-level characteristics) by extending the model to include additional appropriately coded terms in the regression equations.27, 28 Such analyses were planned for patient and breast lesion factors (e.g., age, density of breast tissue, microcalcifications, and palpability of the lesions), biopsy procedure factors (e.g., needle size, imaging guidance, vacuum extraction, and number of samples), clinician and facility-related factors (e.g., training of the operator, country were the study was conducted), and risk of bias items. We performed additional sensitivity analyses (e.g., leave-one-out meta-analysis and comparisons of studies added in the update versus studies included in the original report).29
For Key Question 2, we found that adverse events were inconsistently reported (across studies) and that the methods for ascertaining their occurrence were often not presented in adequate detail. For this reason we refrained from performing meta-analyses for these outcomes. Instead, we calculated descriptive statistics (medians, 25th and 75th percentiles, minimum and maximum values) across all studies and for specific test types. For Key Question 3, because of the heterogeneity of research designs and outcomes assessed, for all outcomes except the number of surgical procedures, we did not perform meta-analysis but instead chose to summarize the data qualitatively. We performed a meta-analysis comparing core needle and open surgical biopsies with respect to the number of patients who required one versus more than one surgical procedures for treatment, after the establishment of breast cancer diagnosis. This analysis used a standard univariate normal random effects model with a binomial distribution for the within-study likelihood of each biopsy group (core needle vs. open).
All statistical analyses were performed using Bayesian methods; models were fit using Markov Chain Monte Carlo methods and noninformative prior distributions. Theory and empirical comparisons suggest that, when the number of studies is large, this approach produces results similar to those of maximum likelihood methods (which do not require the specification of priors).30 Results were summarized as medians of posterior distributions with associated 95 percent central credible intervals (CrIs). A CrI denotes a range of values within which the parameter value is expected to fall with 95% probability.
Grading the Strength of Evidence
We followed the Methods Guide7 to evaluate the strength of the body of evidence for each Key Question with respect to the following domains: risk of bias, consistency, directness, precision, and reporting bias.7, 31 Generally, strength of evidence was downgraded when risk of bias was not low, in the presence of inconsistency, when evidence was indirect or imprecise, or when we suspected that results were affected by selective analysis or reporting.
We determined risk of bias (low, medium, or high) on the basis of the study design and the methodological quality. We assessed consistency on the basis of the direction and magnitude of results across studies. We considered the evidence to be indirect when we had to rely on comparisons of biopsy methods across different studies (i.e., indirect comparisons). We considered studies to be precise if the CrI was narrow enough for a clinically useful conclusion, and imprecise if the CrI was wide enough to include clinically distinct conclusions. The potential for reporting bias (“suspected” vs. “not suspected”) was evaluated with respect to publication, selective outcome reporting, and selective analysis reporting. We made qualitative dispositions rather than perform formal statistical tests to evaluate differences in the effect sizes between more precise (larger) and less precise (smaller) studies because such tests cannot distinguish between “true” heterogeneity between smaller and larger studies, other biases, and chance.32, 33 Therefore, instead of relying on statistical tests, we evaluated the reported results across studies qualitatively, on the basis of completeness of reporting, number of enrolled patients, and numbers of observed events. Judgment on the potential for selective outcome reporting bias was based on reporting patterns for each outcome of interest across studies. We acknowledge that both types of reporting bias are difficult to reliably detect on the basis of data available in published research studies. We believe that our searches (across multiple databases), combined with our plan for contacting test manufacturers (for additional data) and the authors of published studies (for data clarification) limited the impact of reporting and publication bias on our results, to the extent possible.
Finally, we rated the body of evidence using four strength of evidence levels: high, moderate, low, and insufficient.7 These describe our level of confidence that the evidence reflects the true effect for the major comparisons of interest.
Assessing Applicability
We followed the Methods Guide7 in evaluating the applicability of included studies to patient populations of interest. Applicability to the population of interest was also judged separately on the basis of patient characteristics (e.g., age may affect test performance because the consistency of the breast tissue changes over time), method by which suspicion is established (e.g., mammography vs. other methods may affect test performance through spectrum effects), baseline risk of cancer (“average risk” vs. “high risk” women may affect estimated test performance because of differences in diagnostic algorithms), outcomes (e.g., prevalence of breast cancers diagnosed upon biopsy may also be a marker of spectrum effects), and setting of care (because differences in patient populations, diagnostic algorithms, and available technologies may affect test results).
Peer Review
The initial draft report was pre-reviewed by the TOO and an AHRQ Associate Editor (a senior member of another EPC). Following revisions, the draft report was sent to invited peer reviewers and was simultaneously uploaded to the AHRQ Web site where it was available for public comment for 30 days. All reviewer comments (both invited and from the public) were collated and individually addressed. The revised report and the EPC’s responses to invited and public reviewers’ comments were again reviewed by the TOO and Associate Editor prior to completion of the report. The authors of the report had final discretion as to how the report was revised based on the reviewer comments, with oversight by the TOO and Associate Editor.
- AHRQ Task Order Officer
- External Stakeholder Input
- Key Questions
- Analytic Framework
- Scope of the Review
- Literature Search and Abstract Screening
- Study Selection and Eligibility Criteria
- Data Abstraction and Management
- Assessment of the Risk of Bias of Individual Studies
- Data Synthesis
- Grading the Strength of Evidence
- Assessing Applicability
- Peer Review
- Methods - Core Needle and Open Surgical Biopsy for Diagnosis of Breast LesionsMethods - Core Needle and Open Surgical Biopsy for Diagnosis of Breast Lesions
Your browsing activity is empty.
Activity recording is turned off.
See more...