

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Walker HK, Hall WD, Hurst JW, editors. Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition. Boston: Butterworths; 1990.

Clinical Methods: The History, Physical, and Laboratory Examinations. 3rd edition.
Show detailsDefinition
Uric acid is the ultimate catabolite of purine metabolism in humans and higher primates. It is a weak organic acid that under physiologic conditions exists mainly as a monosodium salt. At a pH less than 5.75, as may occur in the urine, the predominant form is nonionized uric acid. The solubility of monosodium urate is about 18 times greater than uric acid in aqueous solutions. This solubility differential provides the therapeutic rationale for alkalinization of the urine pH to greater than 6.0 in patients forming uric acid stones.
The upper limit of plasma uric acid may be defined by a statistical range in a normal population. Epidemiologic studies in the United States have generally accepted 7.0 mg/dl as the upper limit in adult men and 6.0 mg/dl in women.
The physiochemical definition of hyperuricemia may be considered 7.0 mg/dl measured by the specific uricase method. This represents the solubility limit of urate in plasma at 37°C. Levels beyond 7.0 result in supersaturated solutions that are prone to crystal formation.
Uric acid levels are influenced by age and sex. Prior to puberty, the average serum uric acid is 3.6 mg/dl for males and females. Following puberty, values rise to adult levels with women typically 1 mg/dl less than men. This lower level in women apparently reflects estrogen-related enhancement of renal urate clearance and disappears at the menopause. Many additional factors, including exercise, diet, drugs, and state of hydration, may result in transient fluctuations of uric acid levels.
Technique
Currently, two methods are widely utilized to quantify uric acid. A colorimetric method depends on the reduction of a chromogen such as sodium tungstate by uric acid to produce a measurable color change. This technique has been commonly employed in automated hospital screening (SMA systems). The method measures materials other than urate, such as ascorbic acid. Colorimetric determinations are generally considered an overestimation of true uric acid levels, and the normal range is usually 1 mg/dl higher than the more specific enzymatic techniques.
Enzymatic determination of uric acid results from the specific oxidation of uric acid by uricase, which converts its substrate to allantoin. The differential absorbance of these substances at 293 nm allows quantification.
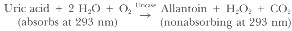
Although traditionally a more expensive technique, uricase methods are currently available for SMA systems at comparable costs and are gradually replacing the less specific colorimetric method.
Basic Science
Serum uric acid reflects the interactions of four major processes: dietary purine intake, endogenous purine metabolism, urinary urate excretion, and intestinal uricolysis.
The typical American diet may provide a significant purine load. Humans do not depend on exogenous purines to serve as precursors of tissue nucleic acids, and nearly all of this dietary component is directly converted into uric acid. Xanthine oxidase, the enzyme responsible for conversion of oxypurines to uric acid, is found in abundance in the liver and the mucosa of the small intestine. Most dietary nucleic acids are ingested in the form of nucleoproteins and can be metabolized to uric acid at the level of the gut mucosa.
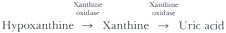
Dietary manipulation was once the mainstay of antihyperuricemic therapy. Nevertheless, even rigid purine-free diets result in only modest reductions of serum uric acid, generally in the range of 1 mg/dl. The advent of potent antihyperuricemic drugs has resulted in diminished emphasis on the therapeutic role of diet. Nonetheless, certain foods, such as organ meats, are rich in purines and can cause a significant flux in serum uric acid if ingested in sufficient quantities.
Endogenous purines are derived from de novo biosynthesis and breakdown of tissue nucleic acids. Measurement of the true rate of endogenous purine turnover requires isotopic dilution techniques carried out on patients severely restricted in dietary purines. A more practical approach in patients with normal renal function who are not taking uricosuric agents involves the measurement of a 24-hour urine collection for uric acid. This provides a crude estimate of the rate of purine production. On purine-restricted diets, a commonly accepted upper limit of uric acid excretion is 600 mg/24 hr. On an unrestricted diet, the limit is raised to 800 mg/24 hr.
A minority of patients with primary hyperuricemia excrete excessive amounts of uric acid. In only a small fraction of these patients has a specific regulatory enzyme defect been identified. Hyperuricemia secondary to hematologic diseases characterized by increased cellular turnover, such as hemolytic and myeloproliferative disorders, are associated with increased urinary excretion of uric acid. The accelerated turnover of nucleic acids in these disorders results in a compensatory increase in the rate of purine biosynthesis. Finally, certain exogenous substances, such as methylene blue and fructose, may stimulate purine biosynthesis.
The kidney is the major site for removal of uric acid and accounts for two-thirds to three-fourths of the daily losses. Urate excretion is believed to depend on a system that includes four components: glomerular filtration, proximal tubular reabsorption, secretion, and postsecretory reabsorption. In primary hyperuricemia and gout, most patients demonstrate a defect in the renal handling of uric acid. In theory, failure of any of the model's components could be involved in the development of hyperuricemia in these patients. At present, the exact site of the defect remains unresolved.
Diminished glomerular filtration rate and renal failure can lead to secondary hyperuricemia. Diuretics increase serum uric acid by multiple mechanisms including an increase in tubular reabsorption. The secretory mechanism for uric acid may be inhibited by a number of organic acids including lactate, betahydroxybutyrate, and acetoacetate. This accounts, in part, for the hyperuricemia seen in diabetic ketoacidosis and lactic acidosis. Salicylates in low dose (less than 2.5 gm/day) impair the secretory mechanism and raise serum uric acid levels. Higher doses inhibit urate absorption, producing a urate diuresis and reduction in serum uric acid.
About one-quarter to one-third of uric acid is normally disposed of by intestinal uricolysis carried out by enzymes of the gut bacterial flora. Uric acid reaches the intestines through alimentary secretions including saliva, bile, gastric, pancreatic, and intestinal juices. Hyperuricemia secondary to failure of intestinal uricolysis has not been recognized. The role of intestinal degradation expands in patients with renal insufficiency and may account for as much as 80% of urate elimination.
Clinical Significance
Hyperuricemia
Hyperuricemia may be conveniently divided into two major categories. Symptomatic hyperuricemia is manifested by gout, nephrolithiasis, and uric acid nephropathy. A larger group of patients have asymptomatic hyperuricemia. Some of these patients will eventually become symptomatic.
The risk of acute gouty arthritis increases with the level of serum uric acid and the duration of hyperuricemia. Acute fluctuations in serum uric acid may be associated with the precipitation of acute gouty arthritis. Sudden reductions in serum uric acid may accompany the introduction of antihyperuricemic therapy; hence, these patients often simultaneously begin prophylactic doses of colchicine.
Nephrolithiasis may accompany gouty arthropathy or occur as an independent problem. Uric acid forms radiolucent stones or may contribute to the formation of calcium stones. In patients with gout, the risk of stone formation rises with the level of serum uric acid. A better correlation exists between stone formation and urinary uric acid excretion. One study found the prevalence of stones to be 50% in gouty patients excreting greater than 1100 mg of uric acid per 24 hours.
Acute uric acid nephropathy results from the precipitation of uric acid crystals within the collecting tubules and ureters. It is a severe form of acute renal failure and is classically associated with the chemotherapy of leukemias and lymphomas. It may also occur following strenuous exercise and epileptic seizures. Hyperuricosuria, aciduria, and urine concentration seem to act in concert to produce this syndrome. The diagnosis can be made by the demonstration of a uric acid to creatinine ratio greater than 1 in the setting of acute renal failure.
Routine screening of hospitalized patients will identify a substantial number with elevated serum uric acid and no related symptoms. Most of these patients will remain asymptomatic throughout their lives. A complete discussion of the management of asymptomatic hyperuricemia is beyond the scope of this chapter. Suffice it to say that the weight of current evidence speaks against the normalization of uric acid in asymptomatic patients. Regardless of the level of uric acid, little seems to be lost by awaiting the onset of the first bout of arthritis or kidney stone.
Hypouricemia
Hypouricemia is commonly defined as a serum urate concentration of 2 mg/dl or less. A low serum urate concentration may result from decreased production or increased excretion. Quantification of urinary uric acid can facilitate a distinction between these two mechanisms. Patients with hypouricemia secondary to impaired production will have little or no urinary uric acid.
Hypouricemia can be found in about 1% of hospitalized patients. In most cases the cause is related to drugs, including salicylates, allopurinol, x-ray contrast agents, and glyceryl guaiacholate. Forced diuresis, used mainly in the treatment of suicide-attempt patients and renal colic, may result in hypouricemia. Total parenteral nutrition can cause profound hypouricemia in some patients.
Several malignant diseases have been associated with hypouricemia, including Hodgkin's disease, sarcoma, glioblastoma, and a variety of carcinomas. In multiple myeloma, light chains most likely cause tubular epithelial damage and Fanconi syndrome. Other malignancies have also been associated with tubular dysfunction and increased renal clearance of urate. The inappropriate secretion of antidiuretic hormone that accompanies some malignancies may lower serum uric acid. Decreased xanthine oxidase activity, acquired after the development of a malignancy, appears to be an uncommon mechanism.
Hypouricemia produces no symptoms or known morbidity. Its fortuitous discovery on automated chemistry screening requires no therapy but should alert the physician to search for an underlying cause.
References
- Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the normative aging study. Am J Med. 1987;82:421–26. [PubMed: 3826098]
- Duffy WB, Senekjian HO, Knight TF, Weinman EF. Management of asymptomatic hyperuricemia. JAMA. 1981;246:2215–16. [PubMed: 7289015]
- Dwosh JL, Roncari DAK, Marliss E. et al. Hypouricemia in disease: study of different mechanisms. J Lab Clin Med. 1977;90:153–61. [PubMed: 874366]
- Lansford HG, Blaufox MD, Borhani NO. et al. Is thiazide-produced uric acid elevation harmful? Analysis of data from the Hypertension Detection and Follow-up Program. Arch Intern Med. 1987;147:645–49. [PubMed: 3827451]
- Liagn MH, Fries JF. Asymptomatic hyperuricemia: the case for conservative management. Ann Intern Med. 1978;88:666–70. [PubMed: 646260]
- Palella TD, Kelley WN. Purine and deoxypurine metabolism. In: Kelley WN, Harris ED Jr, Ruddy S, Sledge C, eds. Textbook of rheumatology. 2d ed. Philadelphia: W.B. Saunders, 1985;337–52.
- Peretz A, Decaux G, Famaly JP. Hypouricemia and intravenous infusions. J Rheum. 1983;10:66–70. [PubMed: 6842488]
- Ramsdell MC, Kelley WN. The clinical significance of hypouricemia. Ann Intern Med. 1973;78:239–42. [PubMed: 4683752]
- Wyngaarden JB, Kelley WN. Gout. In: Stanbury JB, Wyngaarden JB, Frederickson DS, Goldstein JL, Brown MS, eds. The metabolic basis of inherited disease. 5th ed. New York: McGraw-Hill, 1983;1043–1105.
- Yu TF, Gutman AB. Uric acid nephrolithiasis in gout: predisposing factors. Ann Intern Med. 1967;67:1133–48. [PubMed: 6061930]
- PubMedLinks to PubMed
- Solubility of uric acid and supersaturation of monosodium urate: why is uric acid so highly soluble in urine?[J Urol. 1989]Solubility of uric acid and supersaturation of monosodium urate: why is uric acid so highly soluble in urine?Iwata H, Nishio S, Yokoyama M, Matsumoto A, Takeuchi M. J Urol. 1989 Oct; 142(4):1095-8.
- Review Is it time to revise the normal range of serum uric acid levels?[Eur Rev Med Pharmacol Sci. 2014]Review Is it time to revise the normal range of serum uric acid levels?Desideri G, Castaldo G, Lombardi A, Mussap M, Testa A, Pontremoli R, Punzi L, Borghi C. Eur Rev Med Pharmacol Sci. 2014; 18(9):1295-306.
- Studies on the impaired metabolism of uric acid in obese subjects: marked reduction of renal urate excretion and its improvement by a low-calorie diet.[Int J Obes. 1986]Studies on the impaired metabolism of uric acid in obese subjects: marked reduction of renal urate excretion and its improvement by a low-calorie diet.Yamashita S, Matsuzawa Y, Tokunaga K, Fujioka S, Tarui S. Int J Obes. 1986; 10(4):255-64.
- Uricosuric therapy and urate solubility in blood and urine.[Postgrad Med J. 1979]Uricosuric therapy and urate solubility in blood and urine.Gröbner W, Zöllner N. Postgrad Med J. 1979; 55 Suppl 3:26-31.
- Review [Physiology and dynamics of uric acid in hyperuricemia].[Nihon Rinsho. 2008]Review [Physiology and dynamics of uric acid in hyperuricemia].Takagi K, Nakamura T, Ueda T. Nihon Rinsho. 2008 Apr; 66(4):669-74.
- Uric Acid - Clinical MethodsUric Acid - Clinical Methods
Your browsing activity is empty.
Activity recording is turned off.
See more...