

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Pregnancy and lactation require women to provide calcium to the fetus and neonate in amounts that may exceed their normal daily intake. Specific adaptations are invoked within each time period to meet the fetal, neonatal, and maternal calcium requirements. During pregnancy, intestinal calcium and phosphate absorption more than double, and this appears to be the main adaptation to meet the fetal demand for mineral. During lactation, intestinal calcium absorption is normal. Instead, the maternal skeleton is resorbed through the processes of osteoclast-mediated bone resorption and osteocytic osteolysis, in order to provide most of the calcium content of breast milk. In women this lactational loss of bone mass and strength is not suppressed by higher dietary intakes of calcium. After weaning, the skeleton appears to be restored to its prior bone density and strength, together with concomitant increases in bone volumes and cross-sectional diameters that may offset any effect of failure to completely restore the trabecular microarchitecture. These maternal adaptations during pregnancy and lactation also influence the presentation, diagnosis, and management of disorders of calcium, phosphorus, and bone metabolism such as primary hyperparathyroidism, hypoparathyroidism, vitamin D deficiency, and phosphate disorders. Pregnancy and lactation can also cause pseudohyperparathyroidism, a form of hypercalcemia that is mediated by parathyroid hormone-related protein, produced in the breasts or placenta during pregnancy, and by the breasts alone during lactation. Although rarely women may experience fragility fractures during pregnancy or lactation, for most women parity and lactation do not affect the long-term risks of low bone density, osteoporosis, or fracture. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
By the end of full-term gestation, the average fetus accretes about 30 g of calcium, 20 g of phosphorus, and 0.8 g of magnesium to mineralize its skeleton and maintain normal physiological processes. The suckling neonate obtains more than this amount of calcium in breast milk during six months of exclusive lactation. The adaptations through which women meet these calcium demands differ between pregnancy and lactation (Figure 1). Although providing this extra calcium to the offspring could conceivably jeopardize the ability of the mother to maintain her own calcium homeostasis and skeletal mineralization, as this review will make clear, pregnancy and lactation normally do not cause any adverse long-term consequences to the maternal skeleton. The reader is referred to several comprehensive reviews for more details and extensive reference lists for the material covered in this chapter (1-7).

Figure 1.
Schematic illustration contrasting calcium homeostasis in human pregnancy and lactation, as compared to normal. The thickness of arrows indicates a relative increase or decrease with respect to the normal and non-pregnant state. Although not illustrated, the serum (total) calcium is decreased during pregnancy, while the ionized calcium remains normal during both pregnancy and lactation. Adapted from ref. (8), © 1997, The Endocrine Society.
MINERAL PHYSIOLOGY DURING PREGNANCY
Calcium provided from the maternal decidua aids in fertilization of the egg and implantation of the blastocyst; from that point onward the rate of transfer from mother to offspring increases substantially. About 80% of the calcium and phosphate present in the fetal skeleton at the end of gestation crossed the placenta during the third trimester and is mostly derived from the maternal diet during pregnancy. Intestinal calcium and phosphate absorption doubles during pregnancy, driven by 1,25-dihydroxyvitamin D (calcitriol) and other factors, and this appears to be the main adaptation through which women meet the mineral demands of pregnancy.
Mineral Ions
There are several characteristic changes in maternal serum chemistries and calciotropic hormones during pregnancy (Figure 2), which can easily be mistaken as indicating the presence of a disorder of calcium and bone metabolism, especially since it is not common for clinicians to measure calcium, phosphate, and calciotropic hormones during pregnancy (1). The serum albumin and hemoglobin fall during pregnancy due to hemodilution; the albumin remains low until parturition. In turn that fall in albumin causes the total serum calcium to decline to values that can be well below the normal range. The total calcium includes albumin-bound, bicarbonate-and-citrate-complexed, and ionized or free fractions of calcium. The ionized calcium, the physiologically important fraction, remains constant during pregnancy, which confirms that the fall in total calcium is but an artifact that can usually be ignored. However, that artifactual decline in total calcium means that the serum calcium cannot be relied upon to detect hypercalcemia or hypocalcemia. The ionized calcium should be measured or the albumin-corrected total calcium should be calculated to resolve any uncertainty about what the true serum calcium level is in a pregnant woman. Serum phosphate and magnesium concentrations remain normal during pregnancy.
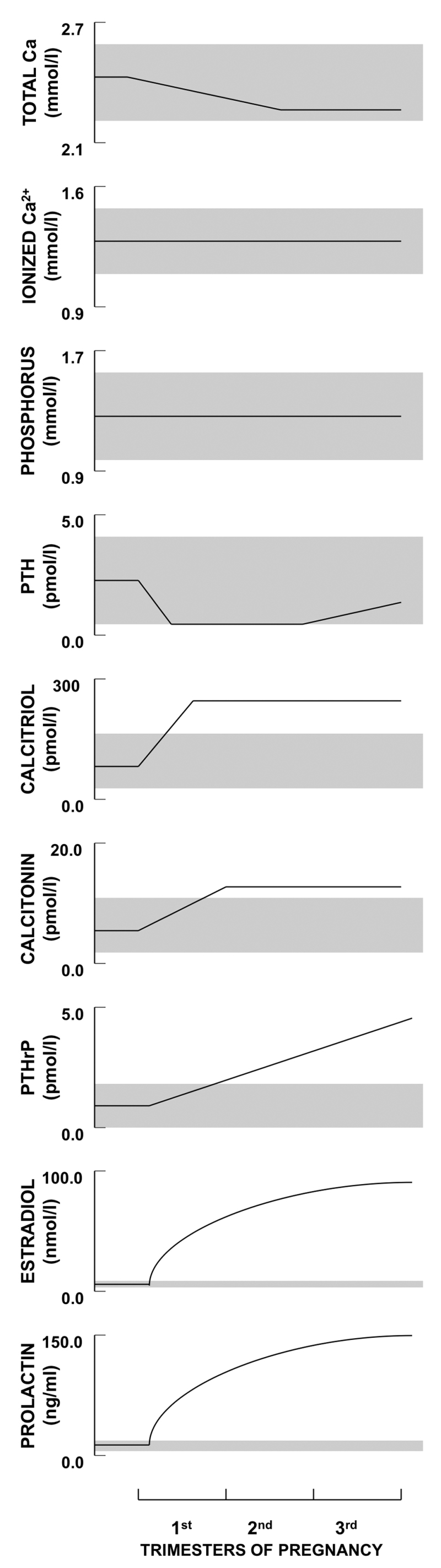
Figure 2.
Schematic depiction of longitudinal changes in calcium, phosphorus, and calciotropic hormone levels during human pregnancy. Shaded regions depict the approximate normal ranges. PTH does not decline in women with low calcium or high phytate intakes and may even rise above normal. Calcidiol (25OHD) values are not depicted; most longitudinal studies indicate that the levels are unchanged by pregnancy but may vary due to seasonal variation in sunlight exposure and changes in vitamin D intake. FGF23 values cannot be plotted due to paucity of data. Reproduced with permission from (1).
Parathyroid Hormone
Parathyroid hormone (PTH) was first measured with assays that reported high circulating levels during pregnancy. The finding of a low total serum calcium and an apparently elevated PTH led to the concept of “physiological secondary hyperparathyroidism in pregnancy.” This erroneous concept persists in some textbooks even today. Those early-generation PTH assays measured many biologically inactive fragments of PTH. When measured with 2-site “intact” assays or the more recent “bio-intact” PTH assays, PTH falls during pregnancy to the low-normal range (i.e. 0-30% of the mean non-pregnant value) during the first trimester and may increase back to the mid-normal range by term. Most of these recent studies of PTH during pregnancy have examined women from North America and Europe who also consumed calcium-replete diets. In contrast, in women from Asia and Gambia who have very low dietary calcium intakes (and often high intakes of phytate that blocks dietary calcium absorption), the PTH level does not suppress during pregnancy and in some cases it has been found to increase above normal (1).
Vitamin D Metabolites
25-hydroxyvitamin D or calcifediol (25OHD) readily crosses the rodent hemochorial placenta (9) and appears to cross hemochorial human placentas just as easily because cord blood 25OHD levels generally range from 75% to near 100% of the maternal value (1,5). A common concern is that the placenta and fetus might deplete maternal 25OHD stores, but this does not appear to be the case. Even in severely vitamin D deficient women there was no significant change in maternal 25OHD levels during pregnancy (1,4,10,11).
Total calcitriol levels increase two to five-fold early in pregnancy and stay elevated until parturition, whereas measured free calcitriol levels were reported to be increased only in the third trimester (12). However, when the 20-40% increase in vitamin D binding protein and the decline in serum albumin during pregnancy are considered, the calculated free calcitriol should be increased in all three trimesters (11,13-16).There are several unusual aspects about this situation. PTH is normally the main stimulator of the renal 1α-hydroxylase (CYP27B1); consequently, elevated calcitriol values are usually driven by high PTH concentrations. An exception to this is the ectopic expression of an autonomously functioning 1α-hydroxylase by such conditions as sarcoidosis and other granulomatous diseases. Another exception is pregnancy because the rise in calcitriol occurs when PTH levels are typically falling or quite low. Moreover, this increase in calcitriol occurs despite the ability of high levels of fibroblast growth factor-23 (FGF23) to suppress the synthesis and increase the catabolism of calcitriol, as shown in animal models of X-linked hypophosphatemic rickets (17-19). Evidence from additional animal models suggest that it is not PTH but other factors, such as PTH-related protein (PTHrP), estradiol, prolactin, and placental lactogen, which drive the 1α-hydroxylase to synthesize calcitriol (1).
The placenta expresses 1α-hydroxylase and it is often assumed that autonomous placental production of calcitriol explains why the maternal calcitriol level doubles; other sources such as maternal decidua and the fetus itself could conceivably contribute to the maternal value. However, it appears that any contributions of placenta and other extra-renal sources to the maternal calcitriol level are trivial. Animal studies indicate that the maternal renal 1α-hydroxylase is markedly upregulated during pregnancy (20,21) and that placental expression of 1α-hydroxylase is many-fold less than in the maternal kidneys (17). Clinical studies have revealed that anephric women on dialysis have very low circulating calcitriol levels before and during pregnancy (1,22), confirming that maternal kidneys must be the main source of the normal 2 to 5-fold increase in calcitriol during normal pregnancy. Rodent studies, including pregnancies in mice that lack the 1α-hydroxylase, have confirmed that there is a small contribution of fetal or placental calcitriol to the maternal circulation (1,23,24). However, it is not enough to account for the marked increase in maternal calcitriol that normally occurs during pregnancy.
Calcitonin
Serum calcitonin levels are increased during pregnancy and may derive from maternal thyroid, breast, decidua, and placenta. The importance of these extrathyroidal sites of calcitonin synthesis has been shown by serum calcitonin levels rising from undetectable to normal values in totally thyroidectomized women who become pregnant (25). Whether calcitonin plays an important role in the physiological responses to the calcium demands of pregnancy is unknown. It has been proposed to protect the maternal skeleton against excessive resorption during times of increased calcium demand; however, there are no clinical studies that have addressed this question. Study of pregnant women who lack the gene for calcitonin or the calcitonin receptor would be informative, but no such women have been identified. On the other hand, mice that lack the gene for calcitonin have normal calcium and bone metabolism during pregnancy (26,27).
Parathyroid Hormone-related Protein
PTHrP concentrations steadily increase in the maternal circulation, reaching the highest levels in the third trimester (1,11). The assays most commonly used in these studies detected PTHrP peptides encompassing amino acids 1-86, but PTHrP is a prohormone. It is cleaved into multiple N-terminal, mid-molecule, and C-terminal peptides, which differ in their biological activities and specificities. None of these peptides have been systematically measured during pregnancy. The commonly available PTHrP1-86 assays do not measure PTHrP1-34, which is likely the most abundant of the active, PTH-like, N-terminal forms of this protein. Moreover, in many clinical studies and case reports it is evident that inappropriate blood samples were used for assaying PTHrP. Special collection and handling are required because PTHrP is rapidly cleaved and degraded in serum. Blood samples should be collected in tubes containing EDTA and aprotinin (a protease inhibitor), kept chilled, and then centrifuged, separated, and frozen within 15 minutes of sample collection. Even with these rigorous standards, PTHrP has been found to begin degrading by 15 minutes after sample collection (28). Many studies did not use this method of sample collection and preparation but instead used sera that had been allowed to clot at room temperature for up to 60 minutes. This likely explains why such studies found undetectable serum concentrations of PTHrP, as compared to those that studied the plasma concentration of PTHrP during pregnancy. Individual case reports are also fraught with this problem, since standard blood collection protocols for hospital laboratories do not use the special handling described above.
PTHrP is produced by many tissues in the fetus and mother; consequently, it is uncertain which source(s) account for the rise in PTHrP in the maternal circulation. However, the placenta and breasts are likely the major sources of PTHrP. Whether circulating PTHrP has a role in maternal physiology during pregnancy is unclear, but its rise may stimulate the renal 1α-hydroxylase and contribute to the increase in calcitriol and, indirectly, the suppression of PTH. However, PTHrP appears less potent than PTH in stimulating the 1α-hydroxylase (29,30), which is why its contribution to the rise in calcitriol during pregnancy is uncertain. On the other hand, several case reports have clearly implicated breast- and placental-derived PTHrP as a cause of maternal hypercalcemia with elevated PTHrP and undetectable PTH, a condition called pseudohyperparathyroidism of pregnancy (see below). Since breasts and placenta were sources of excess PTHrP in these cases, those two tissues seem likely to be dominant sources of PTHrP during normal pregnancy. Moreover, since excess PTHrP impacted maternal calcium homeostasis to cause hypercalcemia in these cases, it is possible that the more modest elevations in circulating PTHrP seen during normal pregnancy also affect maternal calcium homeostasis.
A carboxyl-terminal form of PTHrP (so-called “osteostatin”) has been shown to inhibit osteoclastic bone resorption in vitro, and thus the notion arises that PTHrP may play a role in protecting the maternal skeleton from excessive resorption during pregnancy (31). Animal studies have shown that PTHrP has other roles during gestation such as regulating placental calcium transport in the fetus (1,32). Maternally produced PTHrP is not likely to regulate placental calcium transport since the protein should not be able to cross the placenta (1,5); instead, it is PTHrP produced within the fetus and placenta that is responsible for regulating placental calcium transport.
Fibroblast Growth Factor-23 (FGF23)
Intact FGF23 doubles its concentration in the mother’s circulation during rodent pregnancies (17-19), but whether it increases during human pregnancy is not clear. A small longitudinal study of 12 women found that intact FGF23 doubled in the third trimester over values in the first and second trimesters (33), whereas a larger longitudinal study of 81 women found no difference in intact FGF23 values at 36 weeks of gestation and 3-6 months post-weaning (34). Within 24 hours after delivery in another study, mean intact FGF23 did not differ between postpartum women and non-pregnant women (35).
Other Hormones
This section has focused on changes in static concentrations of minerals and the known calciotropic hormones; there are no studies testing hormonal reserves or response to challenges such as hypocalcemia or hypophosphatemia. Pregnancy also induces significant changes in other hormones known to affect calcium and bone metabolism, including sex steroids, prolactin, placental lactogen, oxytocin, leptin, and IGF-1. Each of these – and possibly other hormones not normally associated with mineral and bone metabolism – may have direct or indirect effects on mineral homeostasis during pregnancy. However, this aspect of the physiology of pregnancy has been largely unexplored to date.
Estradiol increases to about 100-fold the levels obtained during normal menstrual cycles and may be influencing bone metabolism. As noted earlier, estradiol has been postulated to be one of the stimulators of the 1α-hydroxylase, which synthesizes high levels of calcitriol during normal pregnancy, and even in the absence of PTH (1).
Prolactin and placental lactogen both increase during pregnancy and activate prolactin receptors. Osteoblasts express prolactin receptors, and prolactin receptor deficient mice show decreased bone formation (36). Suppressing the prolactin level with bromocriptine blunted a pregnancy-related gain in bone mineral content in rats (37). These data are consistent with the notion that prolactin or placental lactogen regulate skeletal metabolism during pregnancy. Furthermore, prolactin can indirectly affect skeletal metabolism by stimulating PTHrP synthesis and release from the breasts (38-40).
Circulating oxytocin levels also rise during pregnancy (41), and the oxytocin receptor is expressed by osteoclasts and osteoblasts (42). Male and female mice lacking oxytocin or its receptor have an osteoporotic phenotype with low bone formation (43). Oxytocin has been shown to stimulate osteoblast differentiation and function, stimulate osteoclast formation, but inhibit osteoclast function and skeletal resorption (43,44). Taken together, these data predict that oxytocin may regulate bone metabolism during pregnancy, but this has not been directly studied in vivo.
Intestinal Calcium and Phosphate Absorption
Intestinal absorption of calcium doubles as early as 12 weeks of human pregnancy, as shown by clinical studies that used stable isotopes of calcium, and by other calcium balance studies (1). This increase in calcium absorption appears to be the major maternal adaptation to meet the fetal need for calcium. It has been generally believed that the doubling or tripling of calcitriol levels explains the increased intestinal calcium absorption and concurrent increases in the intestinal expression of calbindin9k-D (S100G), TRPV6, Ca2+-ATPase (PMCA1), and other genes and proteins involved in calcium transport. However, intestinal calcium absorption doubles in the first trimester, well before the rise in free calcitriol levels during the third trimester. Animal studies have indicated that placental lactogen, prolactin, and other factors may stimulate intestinal calcium absorption (1) and that calcitriol or the vitamin D receptor are not required for intestinal calcium absorption to increase during pregnancy (1,23,45-48).
The peak fetal demand for calcium does not occur until the third trimester, and so it is unclear why intestinal calcium absorption should be upregulated in the first trimester. It may allow the maternal skeleton to store calcium in advance of the peak demands for calcium that occur later in pregnancy and lactation; some studies in rodents have shown this to be the case with the bone mineral content rising significantly before term (17,26,47). Women have also been found to be in a positive calcium balance by mid-pregnancy (49), likely due to the effect of increased intestinal calcium absorption on skeletal mineralization.
Intestinal phosphate absorption also undergoes a doubling during rodent and other mammalian pregnancies (1), and presumably human pregnancy as well. However, no clinical studies have studied this.
Renal Handling of Calcium
The doubling of intestinal calcium absorption in the first trimester means that the extra calcium must be passed to the fetus, deposited in the maternal skeleton, or excreted in the urine. Renal calcium excretion is increased as early as the 12th week of gestation, and 24-hour urine values (corrected for creatinine excretion) often exceed the normal range. Conversely, fasting urine calcium values are normal or low, confirming that this hypercalciuria is a consequence of the enhanced intestinal calcium absorption (1). This is absorptive hypercalciuria and will not be reliably detected by spot or fasting urine samples that have been corrected for creatinine concentration. Absorptive hypercalciuria contributes to the increased risk of kidney stones during pregnancy. That women commonly develop hypercalciuria in pregnancy is an indication that they normally absorb more calcium than needed by the fetus, provided that their calcium intake is not low, or that the gastrointestinal absorption of calcium is not impaired by high phytate consumption or malabsorptive disorders.
This absorptive hypercalciuria also renders nomograms of fractional calcium excretion invalid for the diagnosis of familial hypocalciuric hypercalcemia during pregnancy (50,51).
Pharmacological doses of calcitonin promote renal calcium excretion, but whether the physiologically elevated levels of calcitonin during pregnancy promote renal calcium excretion is unknown.
Hypocalciuria during pregnancy has been associated with pre-eclampsia, pregnancy-induced hypertension, and low (equal to non-pregnant values) serum calcitriol (52-55). These changes appear largely secondary to disturbed renal function and reduced creatinine clearance, rather than being causes of the hypertension. However, calcium supplementation reduces the risk of pre-eclampsia in women within the lowest quintile of calcium intake (see section J. Low and High Calcium Intake, below), and so there is a pathophysiological link between calcium metabolism and pregnancy-induced hypertension (1).
Skeletal Calcium Metabolism and Bone Density/Bone Marker Changes
As mentioned earlier, some studies in rodents indicate that bone mineral content increases during pregnancy, and other studies have shown that histomorphometric parameters of bone turnover are increased at this time. Systematic studies of bone histomorphometry from pregnant women have not been done. However, one study of 15 women who electively terminated a pregnancy at 8-10 weeks found bone biopsy evidence of increased bone resorption, including increased resorption surface and increased numbers of resorption cavities (56). These findings were not present in biopsies obtained from 13 women at term, or in the non-pregnant controls. This study bears repeating but it does suggest that early pregnancy induces skeletal resorption.
Bone turnover markers – by-products of bone formation and resorption that can be measured in the serum or urine – have been systematically studied during pregnancy in multiple studies (1). In the non-pregnant adult with osteoporosis these bone markers are fraught with significant intra- and inter-individual variability which limit their utility on an individual basis. There are additional problems with the use of bone markers during pregnancy, including lack of pre-pregnancy baseline values; hemodilution; increased GFR; altered creatinine excretion; placental, uterine and fetal contributions; degradation and clearance by the placenta; and lack of diurnally timed or fasted specimens. Bone resorption has been assessed using urinary (deoxypyridinoline, pyridinoline, and hydroxyproline) and serum (C-telopeptide) markers, and the consistent finding is that bone resorption appears increased from early or mid-pregnancy (1). Conversely, bone formation has been assessed by serum markers (osteocalcin, procollagen I N-terminal propeptide, and bone specific alkaline phosphatase) that were generally not corrected for hemodilution or increased GFR. These bone formation markers are decreased in early or mid-pregnancy from pre-pregnancy or non-pregnant values and rise to normal or above before term (1). The lack of correction for hemodilution and increased GFR means that the apparent decline in bone formation markers may not indicate a true decline in bone formation; it could mask no change or even an increase in bone formation. It should be noted that total alkaline phosphatase rises early in pregnancy due to the placental fraction and is not a useful marker of bone formation during pregnancy.
Overall, the scant bone biopsy data and the results of bone turnover markers suggest that bone resorption is increased from as early as the 10th week of pregnancy, whereas bone formation may be suppressed (if the bone formation marker results are correct) or normal (if the bone formation markers are artifactually suppressed due to the aforementioned confounding factors) (1). Notably there is little maternal-fetal calcium transfer occurring in the first trimester, nor is there a marked increase in turnover markers during the third trimester when maternal-fetal calcium transfer is at a peak. These findings may simply underscore that resorption of the maternal skeleton is a minor contributor to calcium homeostasis during pregnancy, whereas the upregulation of intestinal calcium absorption is the main mechanism through which the fetal demand for calcium is met.
Another way of assessing whether the maternal skeleton contributes to calcium regulation during pregnancy is to measure bone mineral content or density. A few sequential areal bone density (aBMD) studies have been done using older techniques (single and/or dual-photon absorptiometry, i.e., SPA and DPA), and none with newer techniques (DXA or qCT) due to concerns about fetal radiation exposure. Studies of aBMD are known to be confounded by changes in body composition, weight and skeletal volumes, and all three of these factors change during normal pregnancy. The longitudinal studies used SPA or DPA and found no significant change in cortical or trabecular aBMD during pregnancy (1). Most recent studies examined 16 or fewer subjects with DXA prior to planned pregnancy (range 1-18 months prior, but not always stated) and after delivery (range 1-6 weeks postpartum) [studies reviewed in detail in (57)]. One study found no change in lumbar spine aBMD measurements obtained pre-conception and within 1-2 weeks post-delivery, whereas the other studies reported 4-5% decreases in lumbar aBMD with the postpartum measurement taken between 1-6 weeks post-delivery. A larger study from Denmark obtained DXA measurements of hip, spine, and radius at baseline (up to 8 months before pregnancy) and again within 15 days of delivery in 73 women (58). DXA of the radius was also obtained once each trimester. aBMD decreased between pre-pregnancy and post-pregnancy by 1.8% at the lumbar spine, 3.2% at the total hip, 2.4% at the whole body, 4% at the ultradistal forearm, and 1% at the total forearm, whereas it increased by 0.5% at the proximal 1/3 forearm (58). All women went on to breastfeed, which means that the final aBMD values were confounded by lactation-induced bone loss (see lactation section). These changes in aBMD were statistically significant when compared to 57 non-pregnant controls who also had serial measurements done, but the magnitudes of change were small, and would not be considered statistically significant for an individual woman.
Ultrasound measurements of the os calcis and fingers have been examined in other longitudinal studies, which reported a progressive decrease in indices that correlate with volumetric BMD (1,57). Whether observed changes in the os calcis accurately indicate a true or clinically meaningful decrease in volumetric BMD or imply that losses of BMD are occurring in the spine or hip during pregnancy, is not known. The reliability or relevance of data obtained from ultrasound is questionable since this technique failed to detect any change in volumetric BMD at the os calcis during lactation (59), even though substantial bone loss occurs at the spine and hip during lactation (see lactation section).
Overall, the existing studies have insufficient power to allow a firm conclusion as to the extent of bone loss that might occur during pregnancy, but it seems likely (especially when data from the Danish study are considered) that modest bone loss occurs, which would be difficult to discern on an individual basis. In the long term, pregnancy does not impair skeletal strength or lead to reduced bone density. Several dozen epidemiological studies of osteoporotic and osteopenic women have failed to find a significant association of parity with bone density or fracture risk (1,60), and many have shown a protective effect of parity (61-78).
DISORDERS OF CALCIUM AND BONE METABOLISM DURING PREGNANCY
Osteoporosis in Pregnancy (and Especially Lactation)
In much of the following, the discussion encompasses osteoporosis that may present in pregnancy, the puerperium, or during lactation, so called pregnancy and lactation-associated osteoporosis (PLO). There can be a continuum of changes in bone metabolism from pre-pregnancy, during pregnancy, in the puerperium, and into the breastfeeding and post-weaning intervals. Most (80-90%) of fractures occur in women during lactation, which indicates that the changes during lactation can be more critical than those that happen during pregnancy. For simplicity, most of the discussion occurs in this section, with a shorter discussion in the lactation section of this chapter. This is also necessary because much of the literature does not distinguish between osteoporosis of pregnancy versus lactation, and because osteoporosis presenting in lactation may have been caused in part by bone loss that occurred during pregnancy.
Rarely woman will present with a fragility fracture (most commonly a vertebral fracture, but appendicular fractures also occur) during the third trimester or puerperium, and especially during lactation. Low bone mineral density is usually then confirmed on a subsequent DXA (79). In most cases an aBMD value prior to pregnancy is not available because it was never indicated to be done in women who were until then thought to be healthy. Therefore, the extent of bone loss that occurred during pregnancy or lactation is unknown in most cases, and it is not possible to exclude that low bone density or skeletal fragility preceded pregnancy. In favor of a genetic predisposition is the report that among 35 women who presented with pregnancy associated osteoporosis, there was a higher than expected prevalence of fragility fractures in their mothers (80). A positive family history of osteoporosis has been found in about one-third of patients presenting with vertebral fractures in association with pregnancy or lactation (81-83). Whole genome screening has been done in other case series with pathogenic mutations found in 25-30% of women with PLO and involving such genes as COL1A1, LRP5, and WNT (79,84). Women with genetic mutations tended to have a more severe PLO, as indicated by lower aBMD or a higher number of fractures (82).
It is conceivable that pregnancy may induce significant skeletal losses in some women and, thereby, predispose to fracture. The normal pregnancy-induced changes in mineral metabolism may cause excessive resorption of the skeleton in selected cases, and other factors such as low dietary calcium intake and vitamin D insufficiency may contribute to skeletal losses (79). If calcium intake is very low or a malabsorptive disorder is present, skeletal resorption must occur to maintain the calcium supply to the fetus and placenta. A high rate of bone turnover is an independent risk factor for fragility fractures outside of pregnancy, and so the apparently increased bone resorption observed during pregnancy may increase fracture risk. In favor of pregnancy inducing fragility through excess skeletal losses is an observational study of 13 women with pregnancy-associated osteoporosis who were followed for up to eight years. Since the bone mineral density at the spine and hip increased significantly during follow-up in these women, the investigators concluded that significant bone loss must have occurred during the pregnancy (85). Other case series have documented spontaneous 10-20% increases in bone density in women after they fractured during pregnancy or the puerperium (1,86). Taken together, fragility fractures in pregnancy or the puerperium may result from the combination of abnormal skeletal microarchitecture or fragility preceding pregnancy, and increased bone resorption that occurred during pregnancy. In other words, the woman may have entered pregnancy with a normal skeleton that then experienced excessive resorption. Alternatively, she may have had lower bone density and bone strength prior to pregnancy, and her skeleton could not tolerate the increased weight bearing, lumbar lordosis, and physiological changes in bone metabolism that occur during pregnancy.
Osteoporosis in association with pregnancy or lactation is likely under-recognized and under-reported. Approximately 75% of vertebral compression fractures in older women are clinically silent and discovered only through radiological surveys; the same is likely true for reproductive age women. Back pain may signal a vertebral fracture, but it may be readily dismissed as a common symptom of normal pregnancy. Consistent with this, an online survey found that women who suffered compression fractures during pregnancy or lactation experienced a mean delay of 3 months before a diagnostic radiograph was done (87). The medical literature is biased toward reports of women who suffered a frightening cascade of multiple compression fractures in association with pregnancy (79,83), whereas how commonly a single vertebral compression fracture might occur and be detected during pregnancy is unknown. Although the literature has focused on vertebral compression fractures occurring with pregnancy, one report suggested that ankle and other lower limb fractures are more common (88).
Osteoporosis usually presents in association with a first pregnancy (especially during lactation) with many but not all reports suggesting that there is a low risk of recurrence in subsequent pregnancies, and no risk conferred by higher parity (79,80,85,89-92). This may indicate that reversible factors such as nutrition were corrected after the first pregnancy, or that all structurally compromised vertebrae collapsed under the load of the first pregnancy. About 60% of patients present with lower thoracic or lumbar pain that may be quite debilitating due to vertebral collapse (85,91,92). Most cases show normal serum chemistries and calciotropic hormone levels, but in a few, secondary causes of bone loss may be identified, including low calcium intake, anorexia nervosa, celiac disease, hyperparathyroidism, osteogenesis imperfecta, inactivating mutations in LRP5, premature ovarian failure, and corticosteroid or heparin therapy (79,80,86,91-95). For example, a woman’s habitual calcium intake of only 229 mg daily was not enough to meet maternal and fetal demands for calcium, and likely contributed to a cascade of vertebral compression fractures occurring late in pregnancy and the puerperium (79). Bone biopsies have only occasionally been done in women having fractures associated with pregnancy or lactation. Most have confirmed osteoporosis and the absence of osteomalacia, while DXA as shown aBMD Z-scores are often in the low bone mass or osteoporotic ranges (85,91,92).
Pain from vertebral compression fractures resolves spontaneously over several weeks in most cases while the bone density has been reported to substantially improve in most women following pregnancy, including those who fractured. Fractures tend not to recur in subsequent pregnancies. Thus, although myriad medical treatments (bisphosphonates, estrogen, testosterone, calcitonin, teriparatide, denosumab) and surgical interventions (kyphoplasty, vertebroplasty, spinal fusion) have been used in individual cases of pregnancy-associated osteoporosis (79), the tendency for this condition to spontaneously improve may make pharmacological treatment unjustified except for the severest cases. At the least, it may be prudent to wait 12-18 months to determine the extent to which the aBMD recovers on its own after a pregnancy-associated vertebral fracture (79,86).
A recent study examined women with self-reported PLO that resulted in fractures occurring in pregnancy or, more commonly, during lactation (96). They were compared cross-sectionally to two groups of historical controls, healthy premenopausal women and women with known idiopathic osteoporosis (IOP) (96). Women with PLO had lower aBMD and reduced HR-pQCT parameters of cortical and trabecular bone than in healthy women and women with IOP (96). However, women with PLO who were assessed >12 months postpartum (“distant”) had higher aBMD than women who were assessed <12 months postpartum (“early”), which implies that recovery had occurred in the “distant” group (96). Moreover, the aBMD of women in the “distant” group was no different than that of women with IOP (96), which suggests that (at least in this cohort) many women with PLO have low bone mass and strength that precedes pregnancy.
A distinct condition is focal, transient osteoporosis of the hip (79). This is rare, self-limited, and probably not a manifestation of altered calciotropic hormone levels or mineral balance during pregnancy. Instead, it may be a consequence of local factors. A variety of theories have been offered to explain this condition, including femoral venous stasis due to pressure from the pregnant uterus, Sudeck’s atrophy or reflex sympathetic dystrophy (causalgia), ischemia, trauma, viral infections, marrow hypertrophy, immobilization, and fetal pressure on the obturator nerve. These patients present with unilateral or bilateral hip pain, limp and/or hip fracture in the third trimester or puerperium (79,97-99). Radiographs and DXA indicate radiolucency and reduced bone density of the symptomatic femoral head and neck, while MRI demonstrates increased water content of the femoral head and the marrow cavity; a joint effusion may also be present. The differential diagnosis of this condition includes inflammatory joint disorders, avascular necrosis of the hip, bone marrow edema, and reflex sympathetic dystrophy. It is a self-limiting condition with both symptoms and radiological appearance resolving within two to six months post-partum; conservative measures including bed rest are usually all that is required during the symptomatic phase (79). Of course, fractures of an involved femur require urgent arthroplasty or hip replacement. The condition recurs in about 40% of cases (not necessarily during pregnancy), unlike osteoporosis involving the spine, and this has prompted prophylactic hip arthroplasty to be done in a few cases where the opposite hip appears to be affected.
Vertebral compression fractures and transient osteoporosis of the hip are not always distinct entities; both have occurred in a few women in association with pregnancy (100-103).
TREATMENT CONSIDERATIONS
For fragility fractures occurring in association with pregnancy, treatment should include optimization of calcium and vitamin D intake, encouraging judicious weight-bearing physical activity, correction of nutritional deficiencies, and treatment of any reversible causes of bone loss or fragility. A supportive corset may provide short-term pain relief. Breastfeeding is not contraindicated but its relative safety should be discussed, since it will lead to progressive loss in aBMD and a transient further increase in fracture risk (see lactation section, below). The potential to rush in with pharmacotherapy should be tempered by the realization that aBMD normally increases 20-70% during the subsequent six to twelve months in women who fractured but received no interventions (1,79,80,85,86,104-112). Therefore, it seems prudent to delay any use of pharmacotherapy for 12-18 months until the extent of spontaneous recovery has been assessed. The extent of spontaneous recovery of lumbar spine aBMD at 12–18 months should be assessed by DXA. HR-pQCT will underestimate the extent of recovery unless the parameters are adjusted to detect and to capture the newly formed bone (osteoid and under-mineralized bone) (86).
Documented pharmacotherapies for PLO have included calcitonin, bisphosphonates, denosumab, strontium ranelate, and teriparatide, using the same regimens as for post-menopausal osteoporosis but with treatment durations from 6 months to as much as 10 years (1,79,83,93,113-117). These reports are observational and lacked controls to determine whether any improvements in aBMD exceeded what would have been observed with spontaneous recovery (i.e., use of calcium and vitamin D replacement only). A recent systematic review found that teriparatide increased aBMD by 8-37% and bisphosphonates by 3 to 43% (118). Notably, these ranges overlap with the bone density increases of 20-70% achieved by women in other reports who did not receive pharmacotherapy. In the few case series where pharmacologically treated women were compared to women who received calcium and vitamin D supplementation only, the final aBMD did not differ between groups (83,109,119,120). Of greater concern is that in the largest of these studies (107 women), recurrent fractures occurred twice as often in women who received teriparatide or alendronate (or both), as compared to those who received only calcium and vitamin D supplementation (109). That finding suggests that pharmacotherapy might be harmful, but better controlled studies are needed to be certain of the benefits vs. risks of pharmacotherapy in this setting.
Vertebroplasty and kyphoplasty have also been used to treat painful vertebral fractures post-partum, but their overall efficacy is uncertain, given that blinded randomized trials have found no superiority over sham surgery or medical approaches in older subjects (121).
For transient osteoporosis of the hip that has not yet resulted in a fracture, the main consideration is whether to prophylactically rod the affected femur(s), or to observe the patient with the expectation that full spontaneous recovery will occur.
Primary Hyperparathyroidism
This is an uncommon condition but there are no firm data available on its prevalence. Hypercalcemia has been found in 0.03% of routinely screened reproductive age women, while two case series indicated that 1% of all parathyroidectomies were done during pregnancy (122,123). There are at least several hundred cases in the medical literature. The diagnosis will be obscured by the normal pregnancy-induced changes that lower the total serum calcium and suppress PTH; however, finding the ionized or albumin-corrected calcium to be increased, and PTH to be detectable, should indicate primary hyperparathyroidism in most cases (note the exception of FHH in the next section).
Physiological changes of pregnancy described earlier increase intestinal calcium absorption and bone resorption, and cause hypercalciuria. In turn these developments can worsen primary hyperparathyroidism and may lead to more severe hypercalcemia, pancreatitis, and kidney stones. The potential for worsening of hypercalcemia is also offset in part by active transfer of calcium across the placenta into the developing fetus.
Primary hyperparathyroidism during pregnancy has been reported to cause a variety of symptoms that are not specific to hypercalcemia and cannot be distinguished from those occurring in normal pregnancy (nausea, vomiting, renal colic, malaise, muscle aches and pains, etc.). Conversely the literature has associated primary hyperparathyroidism with an alarming rate of adverse outcomes in the fetus and neonate, including a 10-30% rate for each of spontaneous abortion, stillbirth, and perinatal death, and 30-50% incidence of neonatal tetany (123-127). These high rates were reported in older literature; more recent case series suggest that the rates of stillbirth and neonatal death are each about 2%, while neonatal tetany occurred in 15% (124). The adverse postnatal outcomes are thought to result from suppression of the fetal and neonatal parathyroid glands; this suppression may be prolonged after birth for 3-5 months (124) and in some cases it has been permanent (124,126,128).
To prevent these adverse outcomes, surgical correction of primary hyperparathyroidism during the second trimester has been almost universally recommended. Several case series have found elective surgery to be well tolerated, and to dramatically reduce the rate of adverse events when compared to the earlier cases reported in the literature. In a series of 109 mothers with hyperparathyroidism during pregnancy who were treated medically (N=70) or surgically (N=39), there was a 53% incidence of neonatal complications and 16% incidence of neonatal deaths among medically treated mothers, as opposed to a 12.5% neonatal complications and 2.5% neonatal deaths in mothers who underwent parathyroidectomy (123). A systematic review of 382 cases found that neonatal deaths and infant morbidity were lower in surgically treated vs. medically treated mothers (9.1 vs. 38.9%) (129). Furthermore, among surgically treated mothers, neonatal death and infant morbidity significant reduced with surgery done in the second versus third trimesters (4.5 vs. 21.1%) (129). Choosing the second trimester allows organogenesis to be complete in the fetus and to avoid the poorer surgical outcomes and risk of preterm birth associated with surgery during the third trimester (124,127,130,131).
Many women in the earliest published cases had a more severe form of primary hyperparathyroidism that is not often seen today (symptomatic, with nephrocalcinosis and renal insufficiency). While mild, asymptomatic primary hyperparathyroidism during pregnancy has been followed conservatively with successful outcomes, complications continue to occur, so that, in the absence of definitive data, surgery during the second trimester remains the most common recommendation (132). An analysis of 1,057 reproductive-aged women with primary hyperparathyroidism found that the rate of C-sections was doubled but there was no difference in the incidence of spontaneous abortions; no data were available on other pregnancy outcomes or neonatal complications (133). In another study of 134 pregnancies in women with primary hyperparathyroidism compared to 431 pregnancies in normocalcemic women, there were no differences in pregnancy-related complications or spontaneous abortions, but neonatal complications were not reported (134). Other recent cases are consistent with lower rates of still birth, neonatal death, and neonatal tetany as compared to the older literature. Therefore, it is reasonable that milder cases diagnosed during the third trimester may be observed until delivery; however, rapid and severe postpartum worsening of the hypercalcemia can occur (131,135-138). This postpartum “parathyroid crisis” occurs because the placental calcium outflow has been lost, while surging PTHrP production in the breasts means an additional factor stimulating bone resorption.
TREATMENT CONSIDERATIONS
The main consideration is whether to operate electively in the second trimester or observe the patient in the hope that surgical intervention can be delayed until after delivery.
Of the five international consensus conferences on the management of primary hyperparathyroidism, only the most recent one commented on pregnancy (139). There are no definitive medical management guidelines for hyperparathyroidism during pregnancy apart from ensuring adequate hydration and correction of electrolyte abnormalities (132). There is some consensus that surgery is indicated for a persistent serum calcium above 2.80 mmol/L (11.1 mg/dL), or an ionized calcium above 1.4 mmol/L (5.6 mg/dL) (140,141). However, another review suggested a higher level 3.00 mmol/L (12.0 mg/dL) (142). If surgery is undertaken, a bilateral approach is often warranted because of the lack of preoperative imaging to localize the adenoma.
Pharmacologic agents to treat hypercalcemia have not been adequately studied in pregnancy, and follow-up on the babies has been brief (if at all). Calcitonin does not cross the placenta and has been used safely (132). Oral phosphate has also been used but is limited by diarrhea, hypokalemia, and risk of soft tissue calcifications. Bisphosphonates are relatively contraindicated because of their potential adverse effects on fetal endochondral bone development, although a review of 78 cases of bisphosphonate use in pregnancy found no obvious problems in most cases (112). Denosumab crosses the placenta and has been shown to cause an osteopetrotic-like phenotype in fetal cynomolgus monkeys and rats (143,144), and so it should be avoided in human pregnancy. High-dose magnesium has been proposed as a therapeutic alternative which should decreases serum PTH and calcium levels by activating the calcium sensing-receptor, but it has not been adequately studied for this purpose (145,146). The calcium receptor agonist cinacalcet, which is used to suppress PTH and calcium in nonpregnant subjects with primary or secondary hyperparathyroidism and parathyroid carcinoma, has also been tried in pregnancy (147-150). However, since the calcium receptor is expressed in the placenta and regulates fetal-placental calcium transfer (151), the possibility of adverse effects of cinacalcet on the fetus and neonate remain a concern. In 6 case reports, use of calcimimetics resulted in neonatal hypocalcemia in half of them (142). Heparin-free hemodialysis can lower the serum calcium before surgery (152). The recent consensus conference on management of primary hyperparathyroidism advised against the use of bisphosphonates and denosumab and cautioned that data on use of cinacalcet are very limited (139). Nevertheless, these agents have been used when there is a hypercalcemic crisis and surgery isn’t possible; the author is aware of cases treated pharmacologically that have not been reported in the literature.
In any case that was followed medically, parathyroidectomy is recommended to be done postpartum, with monitoring in place to detect a postpartum hypercalcemic crisis. Since these women are presenting young with primary hyperparathyroidism, genetic testing may be indicated to rule out inherited causes.
Familial Hypocalciuric Hypercalcemia (FHH)
Inactivating mutations in the calcium-sensing receptor cause this autosomal dominant condition which presents with hypercalcemia and hypocalciuria (153). During pregnancy there will be persistent hypercalcemia with non-suppressed PTH, and the serum calcium may progressively rise across the trimesters. As noted above, fractional excretion of calcium is not reduced during pregnancy in this condition, because it is overridden by the physiological increase in intestinal calcium absorption that in turn causes hypercalciuria (50,51,154). Consequently, FHH presenting during pregnancy can be easily mistaken for primary hyperparathyroidism. Unfortunately, at least one pregnant woman with FHH was mistaken to have primary hyperparathyroidism because of worsening hypercalcemia and hypercalciuria and underwent a three-and-a-half gland parathyroidectomy during the second trimester. FHH was only recognized when her hypercalcemia persisted and her neonate was found to be hypercalcemic too (50).
Pregnancy in women with familial hypocalciuric hypocalcemia should be uneventful for the mother, but the maternal hypercalcemia has caused fetal and neonatal parathyroid suppression with subsequent tetany in both normal and hemizygous children (5,155,156). A hemizygous neonate will later develop benign hypercalcemia, but if the baby has two inactivating mutations of the calcium receptor (most commonly from both parents being hemizygous for FHH), then the neonate may suffer a life-threatening hypercalcemic crisis (5).
TREATMENT CONSIDERATIONS
Hypercalcemia is a normal state of affairs for women with FHH and it should not be treated or mistaken for primary hyperparathyroidism. Instead, the newborn should be watched for postnatal hypocalcemia and for the later development of hypercalcemia as a sign that it inherited the mutation.
Hypoparathyroidism
Hypoparathyroidism during pregnancy usually presents as a pre-existing condition that the clinician is challenged to manage. The natural history of hypoparathyroidism during pregnancy is confusing due to seemingly conflicting case reports in the literature [reviewed in (1,3,157,158)]. Early in pregnancy, some hypoparathyroid women have fewer hypocalcemic symptoms and require less supplemental calcium. This is consistent with a limited role for PTH in the pregnant woman and suggests that an increase in calcitriol and/or increased intestinal calcium absorption occurs in the absence of PTH. However, other case reports clearly indicate that some pregnant hypoparathyroid women required increased calcitriol replacement in order to avoid worsening hypocalcemia. Adding to the confusion is that in some case reports, it appears that the normal, artifactual decrease in total serum calcium during pregnancy was the parameter that led to treatment with increased calcium and calcitriol supplementation; fewer cases reported that dose increments in calcitriol and calcium were made because of maternal symptoms of hypocalcemia or tetany, or objective evidence of true hypocalcemia (low ionized or albumin-corrected calcium).
In a well-documented case, calcitriol was stopped and the woman required only supplemental calcium during the third trimester; she developed hypocalcemia within 48 hours of delivery, which implicates loss of placental PTHrP as contributing to her normalization during pregnancy (159). In a series of ten cases of hypoparathyroidism, the ionized calcium remained normal during pregnancy with no need for calcitriol (160).
Among these and other recent cases, it is clear that hypoparathyroidism may improve, stay the same, or even worsen during pregnancy (159,161-163). It is not possible to know in advance who will improve and who will worsen during pregnancy; the task is to maintain the albumin-corrected serum calcium or ionized calcium in the normal range for the duration of pregnancy. Maternal hypocalcemia due to hypoparathyroidism must be avoided because it has been associated with intrauterine fetal hyperparathyroidism and fetal death. Conversely, overtreatment must be avoided because maternal hypercalcemia is associated with the fetal and neonatal complications described above under Primary Hyperparathyroidism. Calcitriol and 1α-calcidiol are recommended due to their shorter half-lives, lower risk of toxicity, and the clinical experience with these agents.
Late in pregnancy, hypercalcemia may occur in hypoparathyroid women unless the calcitriol dosage and supplemental calcium are substantially reduced or discontinued. This effect appears to be mediated by the increasing levels of PTHrP in the maternal circulation in late pregnancy. Conversely, one case report of hypoparathyroidism in pregnancy found that there was a transient interval of increased requirement for calcitriol immediately after delivery and before lactation was fully underway (159). This may be the result of loss of placental sources of PTHrP followed by a surge in production of PTHrP by the lactating breast (see lactation section, below).
TREATMENT CONSIDERATIONS
The albumin-corrected serum calcium should be maintained in the mid-normal range in order to insure adequate delivery of calcium to the fetus. This short-term recommendation differs from the common recommendation for non-pregnant adults of maintaining the albumin-corrected serum calcium near or just below the lower end of normal, which reduces the renal filtered load and may slow the progression of nephrocalcinosis over the long term. As discussed above, management during pregnancy may not require any change in pre-existing doses of calcium and calcitriol or 1α-calcidiol, or it may require increases or decreases in both the calcium and the active vitamin D analog.
Pseudohypoparathyroidism
Pseudohypoparathyroidism is a genetic disorder causing resistance to PTH and manifest by hypocalcemia, hypophosphatemia, and high PTH levels. The two main subtypes include type I, which has blunted PTH-induced phosphaturia and renal production of cyclic AMP, while type II has blunting of PTH-induced phosphaturia only. They are managed similarly to hypoparathyroidism.
The published experience with pseudohypoparathyroidism during pregnancy is similar to that of hypoparathyroidism, with a mix of cases that improved, worsened, or had no change. Type I pseudohypoparathyroidism improved during four pregnancies as shown by fewer hypocalcemic symptoms, achievement of normocalcemia, lowering of PTH to near-normal, calcitriol increasing several-fold, urinary calcium excretion normalizing, and supplemental vitamin D, calcitriol, or analogs no longer required (164). These findings are consistent with PTH-independent increases in intestinal calcium absorption and calcitriol synthesis occurring during pregnancy that in turn improve calcium homeostasis; endogenous serum calcitriol did double mid-pregnancy in two women in whom supplemental calcitriol had been discontinued. However, in seven other pregnancies in women with types I and II pseudohypoparathyroidism there was subjective worsening of hypocalcemia-like symptoms, or the apparent need to increase the doses of calcium, calcitriol, or 1α-calcidiol (1,165-168). Another case reported that no change in calcium or calcitriol dosages were required during pregnancy (169). Lastly, a more recent series of 5 patients reported variability of improved, worsened, or no change in the condition during pregnancy (170).
If maternal hypocalcemia persists during pregnancy, pseudohypoparathyroidism can lead to the same adverse fetal outcomes that have been associated with maternal hypoparathyroidism, including parathyroid hyperplasia, skeletal demineralization, and fractures (171,172). The maternal calcium concentration must be maintained in the normal range to avoid these fetal outcomes.
TREATMENT CONSIDERATIONS
Maintain the albumin-corrected serum calcium in the mid-normal range. As with hypoparathyroidism, this may not require any change in pre-existing doses of calcium and calcitriol or 1α-calcidiol, or it may require increases or decreases in both the calcium and active vitamin D analog.
Pseudohyperparathyroidism
As mentioned above, pseudohyperparathyroidism is hypercalcemia that is caused by physiological release of PTHrP driving increased skeletal resorption, akin to how PTHrP also causes hypercalcemia of malignancy. In several cases the breasts were the confirmed source of PTHrP because the hypercalcemia and elevated PTHrP did not abate until a bilateral reduction mammoplasty was carried out (173-175). The condition has occurred in women who simply have large breasts (175-177). In another case the hypercalcemia, elevated PTHrP, and suppressed PTH reversed within a few hours of an urgent C-section, thereby confirming the placenta as the source (178). In all cases of pseudohyperparathyroidism, it should be anticipated that the cord blood calcium will also be increased, and that the baby is at risk for fetal and neonatal hypoparathyroidism with hypocalcemic tetany.
TREATMENT CONSIDERATIONS
The diagnosis may not be clear until after delivery, when the serum calcium rapidly normalizes (indicating placental PTHrP was the cause) or stays elevated (indicating production of PTHrP by the breasts is the cause). Prior to delivery, medical management is similar to that for primary hyperparathyroidism.
Vitamin D Deficiency and Insufficiency
There are no comprehensive studies of the effects of vitamin D deficiency or insufficiency on human pregnancy, but the available data from small clinical trials of vitamin D supplementation, observational studies, and case reports suggest that, consistent with animal studies, vitamin D insufficiency and deficiency is not associated with any worsening of maternal calcium homeostasis (this topic is reviewed in detail in (1,4,7). Maternal hypocalcemia is milder with vitamin D deficiency due to the effects of secondary hyperparathyroidism to increase skeletal resorption and renal calcium reabsorption. Consequently, hypocalcemia due to vitamin D deficiency has not been clearly associated with the same adverse fetal outcomes that maternal hypoparathyroidism causes (reviewed in detail in (5,179)). The fetal effects of vitamin D deficiency, inability to form calcitriol, and absence of the vitamin D receptor have been examined across several animal species and all have indicated that the fetus will have a normal serum calcium and fully mineralized skeleton at term (reviewed in detail in (5,179)). Neonatal hypocalcemia and rickets can occur in infants born of mothers with severe vitamin D deficiency, but it is usually in the weeks to months after birth that this presents, after intestinal calcium absorption becomes dependent on calcitriol.
There has been much interest in studies that have inconsistently associated third-trimester measurements of 25OHD, or estimated vitamin D intakes during pregnancy or the first year after birth, with possible extraskeletal benefits in the mother (reduced bacterial vaginosis, pre-eclampsia, pre-term delivery) or in the offspring (lower incidence of type 1 diabetes, greater skeletal mineralization, etc.). These associational studies won’t be discussed in detail (some are cited in: (1,5,180)) because they are confounded by factors which contribute to lower 25OHD levels (maternal overweight/obesity, lower socioeconomic status, poor nutrition, lack of exercise, etc.). It is necessary to test these associations in randomized clinical trials that compare higher versus lower intakes of vitamin D during pregnancy. At present the results of the associational studies are insufficient to warrant prescribing higher intakes of vitamin D during pregnancy to prevent these postulated outcomes.
Among many clinical trials of vitamin D supplementation that have been carried out (1), only a few have included over a 100 study participants who were vitamin D deficient at entry, while other recent studies that gained press attention did not include many vitamin D deficient subjects at all.
Among the trials with over 100 participants (14,181-189), the two largest were from Bangladesh and UK with over 1,000 participants (187,188). Baseline maternal 25OHD levels were lowest (20-29 nmol/L) in trials from Bangladesh, UK, Iran, and UAE, and in the 40-60 nmol/L range in the remainder. Interventions consisted of placebo/no treatment versus low dose (400 IU/day) or high dose (1,000-5,000 IU/day) vitamin D supplementation, initiated before mid-pregnancy, and maintained until delivery. For most trials the primary outcomes were simply maternal and neonatal-cord blood 25OHD and calcium. The most recent and largest study was from Bangladesh, and the primary outcome was pre-specified as infant length-for-age z-scores at 1 year of age (188). Offspring anthropometric parameters and/or bone mineral content were pre-specified only in a few of the remaining studies (184,186,187).
In all studies vitamin D supplementation increased maternal serum and cord blood 25OHD, but there was no overall effect on maternal or cord blood calcium, except one trial that showed a small but significant difference in maternal calcium at delivery (181). The largest achieved difference in maternal 25OHD was over 160 nmol/L (60 ng/mL) in a single study: 16 nmol/L (6.4 ng/mL) in untreated and 168 nmol/L (67 ng/mL) in vitamin D-supplemented mothers at term; however, there was no obstetrical or fetal benefit (181). In that study, the incidence of neonatal hypocalcemia was reduced in offspring of vitamin D treated mothers, reflecting the role of calcitriol to stimulate postnatal intestinal calcium absorption (181). Three trials (mean baseline 25OHD of 20-56 nmol/L) showed a significantly higher maternal PTH level in the low dose vitamin D/placebo arms compared to the high dose arms (182-184), while another trial (mean baseline 25OHD 58-60 nmol/L) did not show any effect of vitamin D supplementation on maternal PTH (14). Vitamin D supplementation also had no effect on maternal aBMD parameters in the post-partum period (14,186,190). Four trials assessed obstetrical outcomes (gestational hypertension, pre-eclampsia, gestational diabetes, infection, post-partum hemorrhage, preterm labor and others), and found no effect of vitamin D supplementation (183,187-189). However, one trial from Iran that showed a 54% risk reduction in gestational diabetes when vitamin D was taken at a dose of 50,000 IU every 2 weeks and compared to 400 IU per day (189).
In the large Bangladesh study, there were no significant differences in infant anthropometrics or any other fetal, neonatal or maternal outcomes (188). In one US-based study there was no benefit on mode of delivery, gestational age at delivery, and preterm birth (14), while in another there was no benefit on mode of delivery, C-section rates, adverse events, hypertension, infection, gestational diabetes, still birth, gestational age at delivery, or combinations of these outcomes (183). The UK MAVIDOS trial reported no obstetrical benefit, and no benefit to any of the primary (neonatal bone area, BMC, and aBMD within the first 10-14 days after birth) or secondary outcomes (anthropometric and body composition parameters within 48 hours of birth). However, it received much publicity for a demonstrated increase in BMC and aBMD in winter-born neonates of vitamin D-supplemented vs. placebo-treated mothers (187). Because the neonatal skeleton accretes 100 mg/day of mineral content after birth, this result may reflect improved intestinal mineral delivery over 14 days after birth, rather than a prenatal effect on skeletal mineralization (1,191,192). Curiously, autumn-born neonates of vitamin D-supplemented vs. placebo-treated mothers showed an adverse trend of similar magnitude on BMC and aBMD, which suggests possible harm from vitamin D supplementation, or chance findings due to small numbers within the sub-groups (192). These sub-group analyses of treatment by season interaction were not specified outcomes in the trial registries (ISRCTN 82927713 and EUDRACT 2007-001716-23). In the UK study that achieved the greatest difference in 25OHD levels between untreated and vitamin D-treated mothers and babies, there was a trend for more small for gestational age babies born to mothers who did not receive antenatal vitamin D supplementation (28% vs. 15%, p<0.1), but the study was not powered for this outcome (181). In studies from the UAE, and Iran there was also no benefit on obstetrical outcomes (variably, mode of delivery, C-section rates, adverse events, stillbirths, gestational age at delivery) or neonatal anthropometric measurements and bone mass measurements (182,184,186).
The lack of any beneficial effect on maternal, immediate fetal/neonatal and neonatal outcomes (anthropometrics and cord blood calcium), even in studies that included mothers with some of the lowest 25OHD levels (181,184,186,188), suggests that vitamin D supplementation during pregnancy confers no benefit to the neonate. The most recent study was well-powered to demonstrate a beneficial effect on infant length and other fetal/neonatal outcomes, but did not yield any significant results, despite low vitamin D levels in the mothers at study entry (188).
Multiple systematic reviews have assessed the effect of vitamin D supplementation during pregnancy on maternal outcomes (193-198). The reviews differed significantly in their methodology, eligibility criteria, intervention (vitamin D alone or combined with calcium), and inclusion of trials. The Cochrane systematic review showed no significant effect of vitamin D supplementation alone on obstetrical outcomes, but there was a significant reduction in preterm birth and low birth weight (194). Conversely, the combination of vitamin D with calcium was found to reduce the risk of pre-eclampsia, but at the cost of a potential increase in preterm births (194). The other reviews yield variable and inconsistent results among them (193,195-198).
An update to the Cochrane systematic review compared vitamin D regimens during pregnancy (≤600 IU versus >600 IU per day; <4000 IU versus ≥ 4000 IU per day) (199). There was no significant effect on pre-eclampsia, preterm birth and low birth weight (low/very low quality of the evidence) (199). The risk of gestational diabetes was reduced with vitamin D supplementation in only one comparison (moderate quality evidence), favoring a dose >600 IU/d (RR 0.54 (0.34 to 0.86). The reviewers noted a concern that safety data were often not reported in the trials that included high doses of vitamin D (199).
In summary, the few large RCTs reported to date do not provide evidence for a beneficial effect of high dose vitamin D supplementation (1,000-5,000 IU/day), on maternal and neonatal outcomes. A recent large trial makes it unlikely that vitamin D supplementation in deficient women would yield any beneficial effect on infant length (188). Other studies had low power, baseline maternal serum 25OHD levels that were often not low, and lack of pre-specification of obstetrical and neonatal outcomes. A potential protective effect of vitamin D on neonatal BMC observed in the MAVIDOS trial (187) makes physiological sense because the intestines become the route of calcium delivery after birth, and the finding is consistent with the benefit observed in some RCTs of prenatal vitamin D supplementation reducing the incidence of neonatal hypocalcemia.
Overall, available data are insufficient from the individual clinical trials or these systematic reviews to conclude that vitamin D supplementation during pregnancy confers any proven obstetrical benefits, especially with respect to calcium and bone metabolism. However, much interest remains in determining whether vitamin D prevents adverse non-skeletal events in mother and baby. In the meantime, one should always ensure that any pregnant woman is vitamin D sufficient prior to pregnancy, or soon after her pregnancy is confirmed. This ensures that the baby is born with adequate 25OHD so that calcitriol can be upregulated postnatally to stimulate calcium and phosphate absorption in the neonatal intestines.
TREATMENT CONSIDERATIONS
Vitamin D supplementation is not harmful to the nonpregnant adult unless excessive doses are administered that cause hypervitaminosis D (typically in excess of 10,000 IU daily); however, the maximal level of maternal intake that is safe for the developing fetus has not been established. Clinical trials in pregnant women have safely administered doses ranging from 400 to 5,000 IU of vitamin D daily without obvious adverse effects to mother or offspring. All pregnant women should have their vitamin D intake optimized. This should prevent any non-skeletal outcomes that may be caused by vitamin D deficiency and will also ensure that the newborn has sufficient vitamin D stores to be able to normalize mineral homeostasis in the hours to days after birth, when it switches from being dependent upon the placenta for mineral delivery to relying on its maturing intestines to absorb that mineral from milk.
Genetic Vitamin D Resistance Syndromes
Case reports and series have provided insight into the effect of pregnancy on genetic disorders of vitamin physiology. Pregnancies have generally been unremarkable in women with vitamin D-dependent rickets type 1 (VDDR-I) which is due to absence of Cyp27b1, and in women with VDDR-II that is due to absence of functional VDRs (200-202). In one such uneventful VDR-II pregnancy, the pre-pregnancy intake of supplemental calcium (800 mg) and high-dose calcitriol were maintained until her clinicians increased the dose of calcitriol later in pregnancy “because of the knowledge that the circulating 1,25-(OH)2D concentration normally rises during pregnancy,” and not because of any change in albumin-adjusted serum calcium (201). Consequently, it’s unclear whether any change was needed. However, it is reasonable to increase the dose of calcitriol to mirror the increase that happens during normal pregnancy. In women with VDDR-I, the dose of calcitriol was unchanged in one-third of pregnancies but increased 1.5 to 2-fold in others (200).
TREATMENT CONSIDERATIONS
Maintain a normal albumin-corrected serum calcium with adjustments to oral calcium and calcitriol dosing as needed based on serial monitoring of blood chemistries.
24-Hydroxylase Deficiency
Genetic loss of the catabolic effects of 24-hydroxylase (CYP24A1) causes high calcitriol, mild hypercalcemia, and nephrolithiasis in non-pregnant adults, which may be asymptomatic (203). But during pregnancy in affected individuals, the physiological 2 to 5-fold increase in calcitriol is unopposed by catabolism, which causes an exaggerated increase in calcitriol, followed by severe and potentially life-threatening hypercalcemia. This has been confirmed by study of a mouse model of Cyp24a1 ablation that mimics the human condition (204). In affected women, hypercalcemia can be quite marked, with suppressed or undetectable PTH, and calcitriol concentrations that exceed what is expected for pregnancy (205-207). Pregnant patients may also present with nephrolithiasis or acute pancreatitis (207,208).
TREATMENT CONSIDERATIONS
Treatment of hypercalcemia is difficult because the agents that could be used are not approved for pregnancy. Increased intestinal calcium absorption is the direct cause, and so use of increased hydration and a modestly restricted calcium diet, combined with phosphate supplementation to bind dietary calcium, are relatively safe management approaches. If PTH increases above normal, then dietary calcium restriction should be lessened to prevent maternal bone resorption and fetal secondary hyperparathyroidism. Other pharmacologic therapy should be reserved for the most severe cases and used with caution. This includes oral glucocorticoids to suppress intestinal calcium absorption, loop diuretics, calcitonin, and bisphosphonates; denosumab should not be used because of teratogenic effects observed in cynomolgus monkeys and mice (143,144). Cinacalcet will not be useful because PTH will already be suppressed due to the combined effects of pregnancy and hypercalcemia.
More targeted treatments include ketoconazole or other azoles to inhibit calcitriol synthesis (209), or rifampin to stimulate catabolism of calcitriol via the 23-hydroxylase pathway (209,210). These drugs have been used in pregnancy to treat other conditions, and so there are data to support their relative safety. However, no case reports have yet involved use of these drugs to treat 24-hydroxylase deficiency in pregnancy.
Low or High Calcium Intake
Through the doubling of intestinal calcium absorption during pregnancy, women have the ability to adapt to wide ranges of calcium intakes and still meet the fetal demand for calcium. It is conceivable that extremely low maternal calcium intakes could impair maternal calcium homeostasis and fetal mineral accretion, but there are scant clinical data examining this possibility (211). One prospective study found that a dietary calcium intake of less than 800 mg daily during the third trimester was associated with significantly lower aBMD at 5 years post-pregnancy (212). Among women with low dietary calcium intake, there are differing results as to whether or not calcium supplementation during pregnancy improved maternal or neonatal bone density (213). There is short term evidence that bone turnover markers were reduced when 1.2 gm of supplemental calcium was given for 20 days to 31 Mexican woman at 25-30 weeks of gestation; their mean dietary calcium intake was 1 gm (214). In a double-blind study conducted in 256 pregnant women, 2 gm of calcium supplementation improved bone mineral content only in the infants of supplemented mothers who were in the lowest quintile of calcium intake (215). Among cases of fragility fractures presenting during pregnancy, some women had very low calcium intakes (<300 mg per day), and in such cases substantial maternal skeletal resorption must be invoked in order to meet the fetal calcium requirement and maintain the maternal serum calcium concentration (79).
Overall the physiological changes in calcium and bone metabolism that usually occur during pregnancy and lactation are likely to be sufficient for fetal bone growth and breast-milk production in women with reasonably sufficient calcium intake (216). However, the use of calcium supplementation for pregnant women with low calcium intake can be defended by the links between low calcium intake and both preeclampsia and hypertension in the offspring (211). Clinical trials and meta-analyses have also demonstrated the supplemental calcium will reduce the risk of preeclampsia in women with low dietary calcium intakes, but not in those with adequate intake (217-220).
High calcium intake, similar to primary hyperparathyroidism, can cause increased intestinal calcium absorption, maternal hypercalcemia, increased transplacental flow of calcium, and suppression of the fetal parathyroids. Cases of neonatal hypoparathyroidism have been reported wherein women consumed 3 to 6 grams of elemental calcium daily as antacids or antinauseants (1).
TREATMENT CONSIDERATIONS
Very low calcium intake must be avoided because it increases the risk of pre-eclampsia, maternal skeletal resorption, and inadequate mineralization of the fetal skeleton. Conversely, high calcium intake must be avoided because it increases the risk of maternal hypercalcemia and suppression of the fetal parathyroids. The Institute of Medicine advises that pregnant women require the same calcium intake as non-pregnant women, a value that ranges from 1,000 to 1,200 mg daily, depending on age (221).
Hypercalcemia of Malignancy
Hypercalcemia of malignancy is usually a terminal condition. When it has been diagnosed during pregnancy, in some cases the baby has been spared from chemotherapy, whereas in other cases the pregnancy was terminated (or ignored) so that chemotherapy could be administered in an attempt to prolong the woman’s life. Half of published case reports haven’t even mentioned the baby’s outcome. A baby born of a mother with humoral hypercalcemia of malignancy may have a high concentration of calcium in cord blood and is at high risk for fetal and neonatal hypoparathyroidism with hypocalcemic tetany.
FGF-23 Disorders
X-linked hypophosphatemic rickets (XLH) is caused by inactivating mutations in the PHEX gene, which lead to high circulating levels of FGF23. In turn this causes hypophosphatemia with rickets or osteomalacia. Pregnancies were normal in a mouse model of XLH. In particular, despite very high circulating levels of FGF23, which normally downregulate calcitriol synthesis and increase its catabolism, maternal serum calcitriol increased to the high levels normally seen during pregnancy (19,222). This rise in calcitriol should contribute to increased intestinal calcium and phosphate absorption. Several case reports documented persistent hypophosphatemia during pregnancy in women with XLH, but no adverse outcomes (223,224). Nevertheless, it is generally recommended to supplement with calcitriol and phosphate to keep the serum phosphate near normal during pregnancy.
Hyperphosphatemic disorders due to loss of FGF23 action have not been studied during human pregnancy, and animal data are also lacking because these conditions are lethal before sexual maturity. Renal insufficiency or failure causes hyperphosphatemia, and both animal and human data indicate that such renal disorders increase the risks of gestational hypertension, pre-eclampsia, eclampsia, and maternal mortality. However, the extent to which hyperphosphatemia contributes to these risks is unknown.
TREATMENT CONSIDERATIONS
For XLH and other FGF-23 mediated disorders that lead to hypophosphatemia, the serum phosphate should be kept near normal with the use of phosphate supplements and calcitriol if needed. Burosumab is a new anti-FGF23 antibody that corrects hypophosphatemia in XLH and tumor-induced hypophosphatemia (oncogenic osteomalacia). It should not be used in pregnancy because preclinical studies have shown that it crosses the placenta and causes toxicity in cynomolgus monkeys (placental mineralization, late fetal loss, shortened gestation, and preterm births) (225).
For hyperphosphatemic disorders due to inadequate FGF23 action, there are no data to guide potential treatment guidelines. However, judicious use of phosphate binders may be of value, with avoidance of any that may be harmful to the fetus.
MINERAL PHYSIOLOGY DURING LACTATION AND POST-WEANING
As lactation begins the mother is faced with another demand for calcium in order to make milk. The average daily loss of calcium into breast milk is 210 mg, although daily losses as great as 1000 mg calcium have been reported is some women nursing twins (1). Although women meet the calcium demands of pregnancy by upregulating intestinal calcium absorption and serum concentrations of calcitriol, a different adaptation occurs during lactation. A temporary resorption and demineralization of the maternal skeleton appears to be the main mechanism by which breastfeeding women meet these calcium requirements. This adaptation does not appear to require PTH or calcitriol but is regulated by the combined effects of increased circulating concentrations of PTHrP and low estradiol levels. Characteristic changes in serum minerals are calciotropic hormones are depicted in Figure 3.

Figure 3.
Schematic depiction of longitudinal changes in calcium, phosphorus, and calciotropic hormone levels during lactation and post-weaning skeletal recovery in women. Normal adult values are indicated by the shaded areas. PTH does not decline in women with low calcium or high phytate intakes and may even rise above normal. Calcidiol (25OHD) values are not depicted; most longitudinal studies indicate that the levels are unchanged by lactation but may vary due to seasonal variation in sunlight exposure and changes in vitamin D intake. PTHrP and prolactin surge with each suckling episode, and this is represented by upward spikes. FGF23 values cannot be plotted due to lack of data. Very limited data suggest that calcitriol and PTH may increase during post-weaning, and the lines are dashed to reflect the uncertainty. Reproduced with permission from (1).
Mineral Ions
The albumin-corrected serum calcium and ionized calcium are both normal during lactation, but longitudinal studies have shown that both are increased slightly over the non-pregnant values. Serum phosphate levels are also higher and may exceed the normal range. Since reabsorption of phosphate by the kidneys appears to be increased, the increased serum phosphate levels may, therefore, reflect the combined effects of increased flux of phosphate into the blood from diet and from skeletal resorption, in the setting of decreased renal phosphate excretion.
Parathyroid Hormone
PTH, as measured by 2-site “intact” or newer “bio-intact” assays, may be undetectable or in the lower quarter of the normal range during the first several months of lactation in women from North America and Europe who consume adequate calcium. PTH rises to normal by the time of weaning, and in two case series was found to rise above normal post-weaning. In contrast, and similar to findings during pregnancy, PTH did not suppress in several studies of women from Asia and Gambia who consumed diets that were low in calcium or high in phytate. The low PTH concentrations are an indication that PTH isn’t required for mineral homeostasis during lactation, and this is confirmed by hypoparathyroid and aparthyroid women in whom mineral and skeletal homeostasis normalize while they continue to breastfeed (see Hypoparathyroidism, below). The same is true of mice that lack the gene for parathyroid hormone. They are hypocalcemic and hyperphosphatemic when non-pregnant but maintain normal serum calcium and phosphate concentrations while lactating and for a time during post-weaning (17).
Vitamin D Metabolites
A common concern has been that the suckling neonate will deplete maternal 25OHD stores, but this is not the case. 25OHD should not decline because it does not enter breast milk; conversely, although vitamin D can enter milk, it is present at very low concentrations because appreciable amounts exist in the maternal circulation for only a short postprandial interval. In observational studies and in the placebo arms of several clinical trials, there was either no change or at most a nonsignificant decline in maternal 25OHD levels during lactation, even in severely vitamin D deficient women (4). Calcitriol levels were twice normal during pregnancy but both free and bound calcitriol levels fall to normal within days of parturition and remain there in breastfeeding women (a single study found that women breastfeeding twins had higher calcitriol concentrations than women nursing singletons) (226). Animal studies show that severely vitamin D deficient rodents and mice lacking the vitamin D receptor are able to lactate and provide normal milk (4,47), thereby indicating that vitamin D and calcitriol are not required for lactation to proceed normally (at least in rodents). However, a more recent study found that mice lacking calcitriol produced milk with a lower calcium content (23).
Calcitonin
Calcitonin levels fall to normal during the first six weeks postpartum in women. Mice lacking the gene that encodes calcitonin lose twice the normal amount of bone mineral content during lactation, which indicates that physiological levels of calcitonin may protect the maternal skeleton from excessive resorption during this time period (26). Whether calcitonin plays a similar role in human physiology is unknown. Totally thyroidectomized women are not calcitonin deficient during lactation due to substantial production of calcitonin by the breasts, which in turn leads to systemic calcitonin concentrations that are the same as in women with intact thyroids (25). Consequently, study of totally thyroidectomized women is not the equivalent of studying a calcitonin-null state when they are breastfeeding.
PTHrP
Plasma PTHrP concentrations are significantly higher in lactating women than in non-pregnant controls. The source of PTHrP appears to be the breast, which secretes PTHrP into breast milk at concentrations that are 1,000 to 10,000 times the level found in the blood of patients with hypercalcemia of malignancy or in normal human controls. The circulating PTHrP concentration also increases after suckling (227,228). Additional evidence that the breasts are the source of PTHrP include that ablation of the PTHrP gene selectively from mammary tissue resulted in reduced circulating levels of PTHrP in lactating mice (229). PTHrP also has an intimate association with breast tissue: in animals it has been shown to regulate mammary development and blood flow, and the calcium and water content of milk in rodents, whereas in humans it is commonly expressed by breast cancers.
Furthermore, as described in more detail below, during lactation PTHrP reaches the maternal circulation from the lactating breast to cause resorption of calcium from the maternal skeleton, renal tubular reabsorption of calcium, and (indirectly) suppression of PTH. In support of this hypothesis, deletion of the PTHrP gene from mammary tissue at the onset of lactation resulted in more modest losses of bone mineral content during lactation in mice (229). In humans, PTHrP correlates with the amount of bone mineral density lost, negatively with serum PTH, and positively with the ionized calcium of lactating women (227,230,231). Lastly, clinical observations in hypoparathyroid and aparathyroid women demonstrate the physiological importance of PTHrP to regulate calcium and skeletal homeostasis during lactation (see Hypoparathyroidism, below).
Prolactin
Prolactin is persistently elevated during early lactation and spikes further upward with suckling. Later during lactation basal prolactin levels are normal but continue to spike with suckling. Prolactin is important for initiating and maintaining milk production (232), but it also alters bone metabolism by stimulating PTHrP production in lactating mammary tissue, inhibiting GnRH and ovarian function, and possibly (as noted earlier) through direct actions in osteoblasts that express the prolactin receptor.
Oxytocin
Oxytocin induces milk ejection by contracting myoepithelial cells within mammary tissue. If milk is not ejected, the pressure of milk stasis causes apoptosis of mammary cells, and lactation ceases. Oxytocin spikes in the maternal circulation within 10 minutes after the start of suckling (233). As noted earlier, the oxytocin receptor is expressed in osteoblasts and osteoclasts. But whether oxytocin plays a role in bone metabolism during lactation has proven difficult to determine because oxytocin null mice cannot lactate due to the lack of milk ejection (234).
Estradiol
In lactating women, estradiol levels fall to menopausal levels or below. This stimulates RANKL and inhibits osteoprotegerin production by osteoblasts, thereby stimulating osteoclast proliferation, function, and bone resorption. Studies in mice have shown that increasing the serum estradiol concentration to 7 times the virgin level blunts the magnitude of bone loss during lactation (235), which confirms that estradiol deficiency plays a role in the skeletal resorption that occurs during lactation.
FGF23
A single longitudinal study found that intact FGF23 approximately doubled between late pregnancy and weeks 14 and 26 of lactation (34). An increase in FGF23 may occur in response to the increased bone resorption during lactation, which leads to a higher serum phosphorus (1).
Other Hormones
Serotonin appears to be involved in regulating PTHrP and its effect to resorb the maternal skeleton (236,237). Lactation induces changes in myriad other hormones, such as luteinizing and follicle stimulating hormone, progesterone, testosterone, inhibins, and activins. Whether these play roles in regulating skeletal metabolism during lactation has not been investigated.
Intestinal Absorption of Calcium and Phosphate
Although intestinal calcium absorption was upregulated during pregnancy, it quickly decreases post-partum to the non-pregnant rate. This also corresponds to the fall in calcitriol levels to normal. This differs from rodents which maintain increased intestinal calcium absorption during lactation; their large litters sizes mandate the need to provide some of the calcium for milk production through this route.
Intestinal phosphate absorption has not been measured during human lactation, whereas in rodents it remains increased (1).
Renal Handling of Calcium and Phosphate
Renal excretion of calcium is typically reduced to about 50 mg per 24 hours or lower, and the glomerular filtration rate is also decreased. These findings suggest that the tubular reabsorption of calcium must be increased to conserve calcium, perhaps through the actions of PTHrP.
Renal tubular phosphate reabsorption is increased during lactation. Despite this, urine phosphate excretion may be increased, likely due to the large efflux of phosphate from resorbed bone, which exceeds what is needed for milk production.
Skeletal Calcium Metabolism and Bone Density/Bone Marker Changes
Histomorphometric data from lactating animals have consistently shown increased bone turnover, and losses of 35% or more of bone mineral are achieved during 2-3 weeks of normal lactation in rodents [reviewed in (1)]. There are no histomorphometric data from lactating women; instead, biochemical markers of bone formation and resorption have been assessed in numerous cross-sectional and prospective studies. Confounding factors discussed earlier for pregnancy need to be considered when assessing bone turnover markers in lactating women; in particular, opposing changes from pregnancy include that the glomerular filtration rate is reduced and the intravascular volume is now contracted. Serum and urinary (24-hr collection) markers of bone resorption are elevated 2-3-fold during lactation and are higher than the levels attained in the third trimester. Serum markers of bone formation (not adjusted for hemoconcentration or reduced GFR) are generally high during lactation and increased over the levels attained during the third trimester. The most marked increase is in the bone resorption markers, suggesting that bone turnover becomes negatively uncoupled, with bone resorption markedly exceeding bone formation, and thereby causing net bone loss. Total alkaline phosphatase falls immediately postpartum due to loss of the placental fraction but may still remain above normal due to elevation of the bone-specific fraction. Overall, these bone marker results are compatible with a significantly increased bone resorption occurring during lactation.
Serial measurements of aBMD during lactation (by SPA, DPA or DXA) have shown that bone mineral content falls 3 to 10.0% in women after two to six months of lactation at trabecular sites (lumbar spine, hip, femur and distal radius), with smaller losses at cortical sites and whole body (1,60). These aBMD changes are in accord with studies in rats, mice, and primates in which the skeletal resorption has been shown to occur largely at trabecular surfaces and to a lesser degree in cortical bone, and as much as 25-30% of bone mass or aBMD is lost during three weeks of lactation in normal rodents. The loss in women occurs at a peak rate of 1-3% per month, far exceeding the 1-3% per year that can occur in postmenopausal women who are considered to be losing bone rapidly. This bone resorption is an obligate consequence of lactation and cannot be prevented by increasing the calcium intake in women. Several randomized trials and other studies have shown that calcium supplementation does not significantly reduce the amount of bone lost during lactation (238-241). Not surprisingly, the lactational decrease in bone mineral density correlates with the amount of calcium lost in the breast milk (242).
The skeletal losses are due in part to the low estradiol levels during lactation which stimulate osteoclast number and activity. However, low estradiol is not the sole cause of the accelerated bone resorption or other changes in calcium homeostasis that occur during lactation. It is worth noting what happens to reproductive-age women who have marked estrogen deficiency induced by GnRH agonist therapy in order to treat endometriosis, fibroids, or severe acne. Six months of GnRH-induced estrogen deficiency caused 1-4% losses in trabecular (but not cortical) aBMD, increased urinary calcium excretion, and suppression of calcitriol and PTH (Figure 4) [reviewed in (1,8)]. In contrast, during lactation women are not as estrogen deficient but lose more aBMD (at both trabecular and cortical sites), have normal (as opposed to low) calcitriol levels, and have reduced (as opposed to increased) urinary calcium excretion (Figure 4). The difference between isolated GnRH-induced estrogen deficiency and lactation appears to be explained by PTHrP. It stimulates osteoclast-mediated bone resorption and stimulates renal calcium reabsorption; by so doing, it complements the effects of low estradiol during lactation. Stimulated in part by suckling and high prolactin levels, PTHrP and estrogen deficiency combine to cause marked skeletal resorption during lactation (Figure 5).

Figure 4.
Comparison of the effects of acute estrogen deficiency vs. lactation on calcium and bone metabolism. Acute estrogen deficiency (e.g. GnRH analog therapy) increases skeletal resorption and raises the blood calcium; in turn, PTH is suppressed and renal calcium losses are increased. During lactation, the combined effects of PTHrP (secreted by the breast) and estrogen deficiency increase skeletal resorption, reduce renal calcium losses, and raise the blood calcium, but calcium is directed into breast milk. Reprinted from ref. (8), © 1997, The Endocrine Society.

Figure 5.
Brain-Breast-Bone Circuit. The breast is a central regulator of skeletal demineralization during lactation. Suckling and prolactin both inhibit the hypothalamic gonadotropin-releasing hormone (GnRH) pulse center, which in turn suppresses the gonadotropins (luteinizing hormone [LH] and follicle-stimulating hormone [FSH]), leading to low levels of the ovarian sex steroids (estradiol and progesterone). PTHrP production and release from the breast is controlled by several factors, including suckling, prolactin, and the calcium receptor. PTHrP enters the bloodstream and combines with systemically low estradiol levels to markedly upregulate bone resorption. Increased bone resorption releases calcium and phosphate into the blood stream, which then reaches the breast ducts and is actively pumped into the breast milk. PTHrP also passes into milk at high concentrations, but whether swallowed PTHrP plays a role in regulating calcium physiology of the neonate is unknown. Calcitonin (CT) may inhibit skeletal responsiveness to PTHrP and low estradiol. Not depicted are that direct effects of oxytocin and prolactin on bone cells are also possible. Adapted from ref. (26) © 2006, The Endocrine Society.
The mechanism through which the skeleton is resorbed has been shown in rodents to involve two processes, both osteoclast-mediated bone resorption (1) and osteocytic osteolysis, in which osteocytes function like osteoclasts to resorb the bone matrix that surrounds them (243). Both of these processes are dependent upon PTHrP. Conditional deletion of the PTHrP gene from mammary tissue reduced the amount of bone resorbed during lactation, whereas conditional deletion of the PTH/PTHrP receptor from osteocytes appeared to eliminate osteocytic osteolysis (244). Moreover, osteocyte-specific deletion of the PTH/PTHrP receptor resulted in a 50% blunting of the amount of aBMD lost during lactation (244), which may indicate that osteocytic osteolysis and osteoclast-mediated bone resorption each contribute about half of the net bone loss achieved during lactation. To date no studies have examined whether osteocytic osteolysis occurs in lactating women.
The lactational bone density losses in women are substantially and completely reversed during six to twelve months following weaning (1,60,239). This corresponds to a gain in bone density of 0.5 to 2% per month in a woman who has weaned her infant. The mechanism for this restoration of bone density is unknown, but studies in mice have shown that it is not dependent upon calcitriol, calcitonin, PTH, or PTHrP (17,23,26,47,245,246); nor is it fully explained by restoration of estradiol levels to normal (1). The remarkable ability of the skeleton to recover is exemplified by mice lacking the gene that encodes calcitonin. They lose up to 55% of trabecular mineral content from the spine during lactation but completely restore it within 18 days after weaning (26).
Although aBMD appears to be completely restored after weaning in women and all animals that have been studied, more detailed examination of microarchitecture by µCT has shown variable completeness of recovery of microarchitecture by skeletal site. In rodents, the vertebrae recover completely while persistent loss of trabeculae is evident in the long bones (247). Studies in women have similarly shown that the trabecular content of the long bones also appears to be incompletely restored (1,60,239,248,249). However, in both women (77,249,250) and rodents (26,251,252) the cross-sectional diameters and volumes of the long bones may be significantly increased after post-weaning. Such structural changes potentially compensate for any reduction in strength that loss of trabecular microarchitecture might induce, because an increased cross-sectional diameter increases the ability of a hollow shaft to resist bending (cross-sectional moment of inertia) and torsional stress (polar moment of inertia). This is supported by the finding that the breaking strength of rodent bones returns to pre-pregnant values after weaning (1,245), and limited clinical studies that correlated the increased bone volumes achieved after reproductive cycles with increased bone strength (77,250). In women, the vast majority of several dozen epidemiologic studies of pre- and postmenopausal women have found no adverse effect of a history of lactation on peak bone mass, bone density, or hip fracture risk (1,7,57,60). In fact, multiple studies have suggested a protective effect of lactation on the future risk of low aBMD or fragility fractures. Consequently, although lactational bone loss can transiently increase risk of fracture (see next section), it is likely unimportant in the long run for most women, in whom the skeleton is restored to its prior mineral content and strength.
DISORDERS OF CALCIUM AND BONE METABOLISM DURING LACTATION
Osteoporosis of Lactation
On occasion a woman will suffer one or more fragility fractures during lactation, and osteoporotic bone density will be found by DXA (79). As with osteoporosis presenting during pregnancy, this may represent a coincidental, unrelated disease; the woman may have had low bone density and abnormal skeletal microarchitecture prior to pregnancy, such that the normal bone loss incurred by lactation could not be tolerated. Alternatively, it is likely that some cases represent an exacerbation of the normal degree of skeletal demineralization that occurs during lactation, and a continuum from the changes in bone density and bone turnover that occurred during pregnancy. For example, the skeleton may have been normal pre-pregnancy, lost mineral content during pregnancy due to low calcium intake, and experienced the expected further loss during lactation (79). It may be somewhat artificial, therefore, to separate “osteoporosis of lactation” from “osteoporosis of pregnancy.” But since lactation normally causes a significant net loss of bone whereas pregnancy does not, it seems more likely for lactation to cause a subset of women to develop low-trauma fractures. For example, excessive PTHrP release from the lactating breast into the maternal circulation could conceivably cause excessive bone resorption, osteoporosis, and fractures. PTHrP levels were high in one case of lactational osteoporosis, and remained elevated for months after weaning (253).
The literature can be confusing because “pregnancy-associated osteoporosis” is the term often used for bone loss and fractures that present during or after pregnancy, despite such fractures being more likely to have resulted from the bone loss during lactation. In fact, multiple case series have demonstrated that about 80-90% of the fragility fractures associated with reproductive cycles occur during lactation, with the remaining 10-20% occurring either during pregnancy or in the puerperium for women who do not breastfeed. Therefore, the term “pregnancy and lactation-associated osteoporosis” (PLO) is a more suitable one to use.
The earlier, longer discussion about osteoporosis of pregnancy should be reviewed for more details since everything in that section applies to osteoporosis presenting during lactation. The skeleton may be normal or abnormal prior to pregnancy, bone loss may have occurred during pregnancy, and bone loss will certainly occur during lactation. The magnitude of bone loss during lactation correlates with the volume of breast milk produced, which in turn correlates with the duration of near-exclusive or exclusive breast feeding (i.e., that most or all of the baby’s nutrition comes from breast milk).
TREATMENT CONSIDERATIONS
The diagnostic and treatment considerations described earlier for osteoporosis of pregnancy also apply to women who are lactating (79). Case series have revealed that a spontaneous 20-70% increase in bone density occurs in women who fractured while breastfeeding (1,79,80,85,86,104-112). Therefore, pharmacological therapy may be best avoided for 12 to 18 months to determine the extent of spontaneous recovery, and then decide if additional treatment is necessary (79,86). The extent of spontaneous recovery of lumbar spine aBMD at 12–18 months should be assessed by DXA. As noted in the pregnancy section, HR-pQCT will underestimate the extent of recovery at this early stage unless the parameters are adjusted to detect under-mineralized bone and osteoid.
The mechanism through which post-weaning recovery occurs is not established, but a theoretical concern is that anti-remodeling agents such as bisphosphonates or denosumab might blunt spontaneous recovery since they suppress bone formation. Furthermore, none of the available pharmacotherapies are indicated for use in premenopausal women, and especially not in women who continue to breastfeed. As noted in the earlier section on osteoporosis associated with pregnancy, individual case reports and series have described marked increases in bone mass in association with pharmacotherapy use after lactation, but in each of these cases the magnitude of increases were in keeping with that achieved through spontaneous recovery (86). A few reports compared to women treated with vitamin D and calcium alone, and there was no difference in the final aBMD achieved with pharmacotherapy (usually teriparatide or bisphosphonates) vs. spontaneous recovery (83,109,119,120). One large case series of 107 women found that subsequent fractures were twice as likely to occur in women who had received pharmacotherapy, which suggests that pharmacotherapy may lead to weaker bone (109). Consequently, it is unclear whether early use of pharmacotherapy after lactation-induced bone loss achieved any added benefit.
Primary Hyperparathyroidism
When surgical correction of primary hyperparathyroidism is not possible or advisable during pregnancy, it is normally carried out in the postpartum interval. A hypercalcemic crisis is possible soon after delivery due in part to loss of the placental calcium infusion, which represented a drain on the serum calcium. If a woman with untreated primary hyperparathyroidism chooses to breastfeed, the serum calcium should be monitored closely for significant worsening due to the effects of secretion of PTHrP from the breasts being added to the high concentrations of PTH already in the circulation. The potential impact of this is even more evident in women with hypoparathyroidism, as discussed below.
TREATMENT CONSIDERATIONS
The albumin-corrected serum calcium may subside after pregnancy due to intestinal calcium absorption returning to normal after the pregnancy-induced increase. Consequently, there may be no urgency for parathyroidectomy to be done unless a parathyroid crisis occurs. If future pregnancies are planned, then it will be prudent for a neck exploration to be done to correct the condition in advance of another pregnancy. Breastfeeding may require that surgery and required localization procedures be delayed.
Familial Hypocalciuric Hypercalcemia
The calcium-sensing receptor is expressed in mammary epithelial ducts, and it modulates the production of PTHrP and calcium transport into milk during lactation in mice (254,255). Inactivating calcium-sensing receptor mutations increased mammary tissue production of PTHrP but decreased the calcium content of milk (255). These opposing changes meant that there was a further increase in bone resorption during lactation as compared to normal mice, and the serum calcium also became higher because of reduced output of calcium into milk. Conversely, a calcimimetic drug (similar to cinacalcet) caused increased milk calcium content (255). These data predict that women with FHH will have more marked skeletal resorption during lactation, lower milk calcium content, higher serum calcium, and a greater loss of aBMD during lactation as compared to normal women. However, the effect of breastfeeding on mineral and skeletal homeostasis in women with FHH has not yet been described.
TREATMENT CONSIDERATIONS
Women with FHH can be expected to breastfeed normally and do not require any treatment. It would be of interest for observational studies to be done to clarify if they experience excess bone loss and produce milk with lower calcium content, as compared to women without FHH.
Hypoparathyroidism
As noted earlier, in the first day or two after parturition the requirement for supplemental calcium and calcitriol may transiently increase in hypoparathyroid women before secretion of PTHrP surges in the breast tissue (159). The onset of lactation induces an important change in skeletal metabolism because the breasts produce PTHrP at high levels, some of which escapes into the maternal circulation to stimulate bone resorption and raise the serum calcium level. In women who lack parathyroid glands, the release of PTHrP into the circulation during lactation can temporarily restore calcium and bone homeostasis to normal. Levels of calcitriol and calcium supplementation required for treatment of hypoparathyroid women fall early and markedly after the onset of lactation, and hypercalcemia can occur if the calcitriol dosage and calcium intake are not substantially reduced (256-259). This decreased need for calcium and calcitriol occurs at a time when circulating PTHrP levels are high in the maternal circulation (256,259,260). As illustrated in one case, this is consistent with PTHrP reaching the maternal circulation in amounts sufficient to allow stimulation of calcitriol synthesis, and maintenance of normal (or slightly increased) maternal serum calcium (260).
TREATMENT CONSIDERATIONS
Management of hypoparathyroidism during lactation requires monitoring the albumin-corrected calcium or ionized calcium, reducing or stopping the calcitriol and calcium as indicated, and planning to reinstitute both supplements in escalating doses as lactation wanes. However, production of PTHrP doesn’t necessarily promptly cease around the time of weaning. The author is aware of a woman with hypoparathyroidism who required no supplemental calcium or calcitriol at all for about a year after her baby had been weaned. She thought that her hypoparathyroidism had been permanently cured by breastfeeding, until the abrupt recurrence of symptomatic hypocalcemia, and the need for pre-pregnancy doses of calcium and calcitriol, signaled the end of PTHrP production by her breasts. In another woman, lactation appeared to permanently cure her hypoparathyroidism (261), likely because of persistent production of PTHrP by her breasts.
Pseudohypoparathyroidism
The management of pseudohypoparathyroidism during lactation has been less well documented. Since these patients are likely resistant to the renal actions of PTHrP, and the placental sources of calcitriol are lost at parturition, the calcitriol requirements might well increase and may require further adjustments during lactation. Conversely, these patients do not have skeletal resistance to PTH, and so it is possible that calcium and calcitriol requirements may decrease secondary to enhanced skeletal resorption caused by the combined effects of high PTH levels, PTHrP release from the breast, and lactation-induced estrogen deficiency. Thus, women with pseudohypoparathyroidism might lose more bone density than normal during lactation, but this has not been studied.
TREATMENT CONSIDERATIONS
In the absence of data, it would be best to monitor the albumin-corrected serum calcium to determine if any adjustments are needed in the doses of oral calcium and calcitriol.
Pseudohyperparathyroidism
Severe, PTHrP-mediated hypercalcemia during lactation was first noted to occur in women with large breasts, but it has also developed in women with average-sized breasts in whom milk let-down took place but the baby’s illness prevented breastfeeding (176). This represents an exaggeration of normal lactational physiology, which benefits hypoparathyroid women, but in some normal women can overwhelm the normal regulatory pathways and cause potentially severe hypercalcemia.
TREATMENT CONSIDERATIONS
Cessation of lactation should reverse the condition, aided by use of breast-binders, and bromocriptine or cabergoline to suppress prolactin. However, a reduction mammoplasty or mastectomy has proved necessary for recalcitrant hypercalcemia in some cases.
Vitamin D Deficiency and Insufficiency, and Genetic Vitamin D Disorders
The mother andata from small clinical trials, observational studies and case reports indicate that lactation proceeds normally regardless of vitamin D status, and breast milk calcium content is unaffected by vitamin D deficiency or supplementation in doses as high as 6,400 IU per day given to the mother, which achieved maternal 25OHD blood levels of 168 nmol/L (topic reviewed in detail in (1,4,5,7,179)). This is likely because maternal calcium homeostasis is dominated by skeletal resorption induced by estrogen deficiency and PTHrP, with vitamin D/calcitriol playing no substantial role in lactational mineral homeostasis. It is the neonate who will suffer the consequences of being born of a vitamin D deficient mother. This is especially true if the infant is exclusively breast fed, since both vitamin D and 25-hyroxyvitamin D are normally present at very low concentrations in breast milk.
The high-dose (6,400 IU) vitamin D supplementation strategy raises the maternal vitamin D concentration substantially for hours and, in turn, this increases the penetration of vitamin D into milk. Consequently, breastfed babies whose mothers consumed 6,400 IU per day achieved the same 25OHD level as babies who received a 300 IU dose of vitamin D directly (262). The potential advantage of this approach is that all of the neonate’s nutrition can then come from breast milk, rather than requiring that breastfed babies receive a vitamin D supplement. Further study is needed regarding the safety of this approach for the mothers and their babies. A Cochrane review also concluded there is no established benefit from high-dose supplementation of breastfeeding women, as compared to the normal route of giving a lower dose of vitamin D directly to a breastfed baby (263).
A misconception about vitamin D and milk often arises because marketed forms of cow’s and goat’s milk contain approximately 100 IU of vitamin D per standard serving, but that is a synthetic vitamin D supplement which is added to the milk after the pasteurization stage. It is not put there by the cow or goat.
Given that vitamin D deficiency does not affect breast milk content in humans, it is likely that genetic absence of VDR or calcitriol also does not affect milk calcium, but this has not been studied.
Whether vitamin D deficiency impairs the ability of the maternal skeleton to recover post-weaning has not been examined in any clinical study. However, studies in mice lacking the vitamin D receptor or Cyp27b1 to synthesize calcitriol, indicate that these mice are able to fully remineralize their skeletons after lactation (23,47).
TREATMENT CONSIDERATIONS
Breastfeeding women have the same vitamin D intake requirements as non-pregnant and pregnant women (221). Therefore, no change in dose of any oral supplements is needed. Penetrance of vitamin D into breast milk is poor, and so breastfed babies need oral vitamin D supplementation to prevent vitamin D deficiency, until such time as vitamin D-supplemented infant nutrition is taken.
24-Hydroxylase Deficiency
Hereditary absence of Cyp24a1 reduces calcitriol catabolism, which can lead to very high calcitriol concentrations and marked maternal hypercalcemia during pregnancy. But calcitriol production falls to non-pregnant levels during normal lactation, and the same should be true in women with 24-hydroxylase deficiency. Consistent with this, in one affected woman who breastfed, calcitriol was normal and hypercalcemia was milder compared to pregnancy (205).
TREATMENT CONSIDERATIONS
Breastfeeding appears unlikely to require any intervention in women with 24-hydroxylase deficiency.
Low and High Calcium Intakes
The calcium content of milk appears to be largely derived from skeletal resorption during lactation, a process that cannot be suppressed in women by consuming greater amounts of calcium (however, it can be suppressed in rodents by high calcium intakes). It shouldn’t be surprising, therefore, that low calcium intake does not impair breast milk quality, nor does it accentuate maternal bone loss (216). Even in women with very low calcium intakes, the same amount of mineral was lost during lactation from the skeleton as compared to women who had supplemented calcium intakes, and the breast milk calcium content was unaffected by calcium intake or vitamin D status (264-266). Conversely, since randomized trials and cohort studies have shown that high calcium intakes do not affect the degree of skeletal demineralization that occurs during lactation (238-241), it is unlikely that increasing calcium supplementation well above normal would affect skeletal demineralization either.
There is a lingering concern that adolescent mothers with low calcium intakes may not achieve normal peak bone mass as a consequence of lactation-induced bone loss. In fact the adolescent skeleton appears to recover fully from lactation (267), and adolescent women who breastfed have higher aBMD than those who did not breastfeed or had not been pregnant as adolescents (268). However, it remains reasonable to give a calcium supplement to adolescents who lactate in order to ensure that the needs of adolescent growth are met and that peak bone mass is achieved (216,267).
TREATMENT CONSIDERATIONS
A normal calcium intake of 1,000-1,200 mg daily, as suggested by the Institute of Medicine (221), is recommended for breastfeeding women. Low and high calcium intake should be avoided. It is well established that breastfeeding women do not require increased calcium intake.
FGF23 Disorders
There is very limited information available about the effect of FGF23-related disorders on mineral homeostasis during lactation, and milk production. Lactation normally causes the serum phosphorus to rise due to the increased release of phosphate from the resorbing skeleton. Indeed in one case report, serum phosphorus increased into the normal range in a breastfeeding woman with XLH (223). Curiously, the phosphate content of expressed milk was reduced to 50% of normal in two cases of XLH (223,224), which differs from findings in animal models of XLH in which milk phosphate content was normal (269). Use of oral phosphate supplementation normalized the milk phosphate content in one case where this intervention was studied (223).
Milk phosphate content has not been studied in disorders in which FGF23 activity is reduced; however, given the findings with XLH, it is conceivable that milk phosphate content will be increased.
TREATMENT CONSIDERATIONS
The mineral content of milk is not normally analyzed. Given the limited findings from two cases of XLH, it may be prudent to recommend that oral phosphate supplementation be maintained while breastfeeding, even if the serum phosphorus has spontaneously normalized. If the baby develops hypophosphatemia, this may indicate inadequate milk phosphate content or that the baby inherited XLH. Use of burosumab has not been described in breastfeeding women, but this large protein should have low penetrance into milk and is likely to be destroyed in the infant’s gastrointestinal tract (270).
IMPLICATIONS
During pregnancy and lactation, novel regulatory systems specific to these settings complement the usual regulators of mineral homeostasis. Intestinal calcium absorption more than doubles from early in pregnancy in order to meet the fetal demand for calcium. In comparison, skeletal calcium resorption is a dominant mechanism by which calcium is supplied to the breast milk, while renal calcium conservation is also apparent. Calcium supplementation during pregnancy will result in a woman absorbing more calcium, but it is clear from clinical trials and observational studies that calcium supplements have little or no impact on the amount of bone lost during lactation.
The skeleton appears to recover promptly from lactation to achieve the pre-pregnancy bone mass through mechanisms that remain unclear. The transient loss of bone mass during lactation can at least temporarily compromise skeletal strength and rarely lead to fragility fractures. Furthermore, full recovery of mineral content and bone strength may not always be achieved after weaning. But the majority of women can be assured that the changes in calcium and bone metabolism during pregnancy and lactation are normal, healthy, temporary, and without adverse consequences in the long-term.
REFERENCES
- 1.
- Kovacs CS. Maternal Mineral and Bone Metabolism During Pregnancy, Lactation, and Post-Weaning Recovery. Physiol Rev 2016; 96:449-547 [PubMed: 26887676]
- 2.
- Wysolmerski JJ. Conversations between breast and bone: physiological bone loss during lactation as evolutionary template for osteolysis in breast cancer and pathological bone loss after menopause. BoneKEy 2007; 4:209-225
- 3.
- Kovacs CS. Calcium and bone metabolism disorders during pregnancy and lactation. Endocrinol Metab Clin North Am 2011; 40:795-826 [PubMed: 22108281]
- 4.
- Kovacs CS. The role of vitamin D in pregnancy and lactation: insights from animal models and clinical studies. Annu Rev Nutr 2012; 32:97-123 [PubMed: 22483092]
- 5.
- Kovacs CS. Bone development and mineral homeostasis in the fetus and neonate: roles of the calciotropic and phosphotropic hormones. Physiol Rev 2014; 94:1143-1218 [PubMed: 25287862]
- 6.
- Kovacs CS. Physiology of Calcium, Phosphorus, and Bone Metabolism During Pregnancy, Lactation, and Postweaning. In: Kovacs CS, Deal CL, eds. Maternal-Fetal and Neonatal Endocrinology. San Diego: Academic Press; 2020:61-73.
- 7.
- Kovacs CS, Chakhtoura M, El-Hajj Fuleihan G. Disorders of Mineral and Bone Metabolism During Pregnancy and Lactation. In: Kovacs CS, Deal CL, eds. Maternal-Fetal and Neonatal Endocrinology. San Diego: Academic Press; 2020:329-370.
- 8.
- Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev 1997; 18:832-872 [PubMed: 9408745]
- 9.
- Haddad JG, Jr., Boisseau V, Avioli LV. Placental transfer of vitamin D3 and 25-hydroxycholecalciferol in the rat. J Lab Clin Med 1971; 77:908-915 [PubMed: 4327020]
- 10.
- Hillman LS, Slatopolsky E, Haddad JG. Perinatal vitamin D metabolism. IV. Maternal and cord serum 24,25-dihydroxyvitamin D concentrations. J Clin Endocrinol Metab 1978; 47:1073-1077 [PubMed: 318093]
- 11.
- Ardawi MS, Nasrat HA, HS BAA. Calcium-regulating hormones and parathyroid hormone-related peptide in normal human pregnancy and postpartum: a longitudinal study. Eur J Endocrinol 1997; 137:402-409 [PubMed: 9368509]
- 12.
- Bikle DD, Gee E, Halloran B, Haddad JG. Free 1,25-dihydroxyvitamin D levels in serum from normal subjects, pregnant subjects, and subjects with liver disease. J Clin Invest 1984; 74:1966-1971 [PMC free article: PMC425383] [PubMed: 6549014]
- 13.
- Zhang JY, Lucey AJ, Horgan R, Kenny LC, Kiely M. Impact of pregnancy on vitamin D status: a longitudinal study. Br J Nutr 2014; 112:1081-1087 [PubMed: 25159824]
- 14.
- Hollis BW, Johnson D, Hulsey TC, Ebeling M, Wagner CL. Vitamin D supplementation during pregnancy: double-blind, randomized clinical trial of safety and effectiveness. J Bone Miner Res 2011; 26:2341-2357 [PMC free article: PMC3183324] [PubMed: 21706518]
- 15.
- Markestad T, Ulstein M, Aksnes L, Aarskog D. Serum concentrations of vitamin D metabolites in vitamin D supplemented pregnant women. A longitudinal study. Acta Obstet Gynecol Scand 1986; 65:63-67 [PubMed: 3487197]
- 16.
- Ritchie LD, Fung EB, Halloran BP, Turnlund JR, Van Loan MD, Cann CE, King JC. A longitudinal study of calcium homeostasis during human pregnancy and lactation and after resumption of menses. Am J Clin Nutr 1998; 67:693-701 [PubMed: 9537616]
- 17.
- Kirby BJ, Ma Y, Martin HM, Buckle Favaro KL, Karaplis AC, Kovacs CS. Upregulation of calcitriol during pregnancy and skeletal recovery after lactation do not require parathyroid hormone. J Bone Miner Res 2013; 28:1987-2000 [PubMed: 23505097]
- 18.
- Cooke-Hubley S, Kirby BJ, Suppiah S, Simmonds CS, Karaplis AC, Kovacs CS. Circulating FGF23 is regulated by PTH and calcitriol during fetal development but low FGF23 does not significantly alter fetal phosphorus metabolism. IBMS BoneKEy 2013; 10:S95
- 19.
- Ma Y, Samaraweera M, Cooke-Hubley S, Kirby BJ, Karaplis AC, Lanske B, Kovacs CS. Neither absence nor excess of FGF23 disturbs murine fetal-placental phosphorus homeostasis or prenatal skeletal development and mineralization. Endocrinology 2014; 155:1596-1605 [PMC free article: PMC3990847] [PubMed: 24601885]
- 20.
- Fenton E, Britton HG. 25-hydroxycholecalciferol 1 alpha-hydroxylase activity in the kidney of the fetal, neonatal and adult guinea pig. Biol Neonate 1980; 37:254-256 [PubMed: 7388079]
- 21.
- Kubota M, Ohno J, Shiina Y, Suda T. Vitamin D metabolism in pregnant rabbits: differences between the maternal and fetal response to administration of large amounts of vitamin D3. Endocrinology 1982; 110:1950-1956 [PubMed: 6280980]
- 22.
- Turner M, Barre PE, Benjamin A, Goltzman D, Gascon-Barre M. Does the maternal kidney contribute to the increased circulating 1,25-dihydroxyvitamin D concentrations during pregnancy? Miner Electrolyte Metab 1988; 14:246-252 [PubMed: 3211093]
- 23.
- Gillies BR, Ryan BA, Tonkin BA, Poulton IJ, Ma Y, Kirby BJ, St-Arnaud R, Sims NA, Kovacs CS. Absence of Calcitriol Causes Increased Lactational Bone Loss and Lower Milk Calcium but Does Not Impair Post-lactation Bone Recovery in Cyp27b1 Null Mice. J Bone Miner Res 2018; 33:16-26 [PubMed: 28686309]
- 24.
- Ryan BA, Alhani K, Sellars KB, Kirby BJ, St-Arnaud R, Kaufmann M, Jones G, Kovacs CS. Mineral Homeostasis in Murine Fetuses Is Sensitive to Maternal Calcitriol but Not to Absence of Fetal Calcitriol. J Bone Miner Res 2019; 34:669-680 [PubMed: 30508318]
- 25.
- Bucht E, Telenius-Berg M, Lundell G, Sjoberg HE. Immunoextracted calcitonin in milk and plasma from totally thyroidectomized women. Evidence of monomeric calcitonin in plasma during pregnancy and lactation. Acta Endocrinol (Copenh) 1986; 113:529-535 [PubMed: 3788423]
- 26.
- Woodrow JP, Sharpe CJ, Fudge NJ, Hoff AO, Gagel RF, Kovacs CS. Calcitonin plays a critical role in regulating skeletal mineral metabolism during lactation. Endocrinology 2006; 147:4010-4021 [PubMed: 16675524]
- 27.
- Woodrow JP. Calcitonin modulates skeletal mineral loss during lactation through interactions in mammary tissue and directly though osteoclasts in bone [PhD thesis] [PhD]. St. John's, Newfoundland: Biomedical Sciences, Faculty of Medicine, Memorial University of Newfoundland; 2009.
- 28.
- Hutchesson AC, Hughes SV, Bowden SJ, Ratcliffe WA. In vitro stability of endogenous parathyroid hormone-related protein in blood and plasma. Ann Clin Biochem 1994; 31 (Pt 1):35-39 [PubMed: 7512317]
- 29.
- Horwitz MJ, Tedesco MB, Sereika SM, Hollis BW, Garcia-Ocana A, Stewart AF. Direct comparison of sustained infusion of human parathyroid hormone-related protein-(1-36). J Clin Endocrinol Metab 2003; 88:1603-1609 [PubMed: 12679445]
- 30.
- Horwitz MJ, Tedesco MB, Sereika SM, Syed MA, Garcia-Ocana A, Bisello A, Hollis BW, Rosen CJ, Wysolmerski JJ, Dann P, Gundberg C, Stewart AF. Continuous PTH and PTHrP infusion causes suppression of bone formation and discordant effects on 1,25(OH)2 vitamin D. J Bone Miner Res 2005; 20:1792-1803 [PubMed: 16160737]
- 31.
- Cornish J, Callon KE, Nicholson GC, Reid IR. Parathyroid hormone-related protein-(107-139) inhibits bone resorption in vivo. Endocrinology 1997; 138:1299-1304 [PubMed: 9048639]
- 32.
- Kovacs CS, Lanske B, Hunzelman JL, Guo J, Karaplis AC, Kronenberg HM. Parathyroid hormone-related peptide (PTHrP) regulates fetal-placental calcium transport through a receptor distinct from the PTH/PTHrP receptor. Proc Natl Acad Sci U S A 1996; 93:15233-15238 [PMC free article: PMC26386] [PubMed: 8986793]
- 33.
- Zhao S, Zhou J, Chen R, Zhou W, Geng H, Huang Y, Shi S, Yuan L, Wang Z, Wang D. Decreased FGF23 inhibits placental angiogenesis via the ERK1/2-EGR-1 signaling pathway in preeclampsia. Cytokine 2024; 176:156508 [PubMed: 38266461]
- 34.
- Nabwire F, Hamill MM, Fowler MG, Byamugisha J, Kekitiinwa A, Prentice A. Biochemical Markers of Calcium and Bone Metabolism during and after Lactation in Ugandan Women with HIV on Universal Maternal Antiretroviral Therapy. J Bone Miner Res 2023; 38:1296-1311 [PMC free article: PMC10947145] [PubMed: 37306529]
- 35.
- Ohata Y, Arahori H, Namba N, Kitaoka T, Hirai H, Wada K, Nakayama M, Michigami T, Imura A, Nabeshima Y, Yamazaki Y, Ozono K. Circulating levels of soluble alpha-Klotho are markedly elevated in human umbilical cord blood. J Clin Endocrinol Metab 2011; 96:E943-947 [PubMed: 21411554]
- 36.
- Clement-Lacroix P, Ormandy C, Lepescheux L, Ammann P, Damotte D, Goffin V, Bouchard B, Amling M, Gaillard-Kelly M, Binart N, Baron R, Kelly PA. Osteoblasts are a new target for prolactin: analysis of bone formation in prolactin receptor knockout mice. Endocrinology 1999; 140:96-105 [PubMed: 9886812]
- 37.
- Suntornsaratoon P, Wongdee K, Goswami S, Krishnamra N, Charoenphandhu N. Bone modeling in bromocriptine-treated pregnant and lactating rats: possible osteoregulatory role of prolactin in lactation. Am J Physiol Endocrinol Metab 2010; 299:E426-436 [PubMed: 20551289]
- 38.
- Thiede MA. The mRNA encoding a parathyroid hormone-like peptide is produced in mammary tissue in response to elevations in serum prolactin. Mol Endocrinol 1989; 3:1443-1447 [PubMed: 2608067]
- 39.
- Ratcliffe WA, Thompson GE, Care AD, Peaker M. Production of parathyroid hormone-related protein by the mammary gland of the goat. J Endocrinol 1992; 133:87-93 [PubMed: 1517711]
- 40.
- Stiegler C, Leb G, Kleinert R, Warnkross H, Ramschak-Schwarzer S, Lipp R, Clarici G, Krejs GJ, Dobnig H. Plasma levels of parathyroid hormone-related peptide are elevated in hyperprolactinemia and correlated to bone density status. J Bone Miner Res 1995; 10:751-759 [PubMed: 7639111]
- 41.
- Dawood MY, Ylikorkala O, Trivedi D, Fuchs F. Oxytocin in maternal circulation and amniotic fluid during pregnancy. J Clin Endocrinol Metab 1979; 49:429-434 [PubMed: 468976]
- 42.
- Colucci S, Colaianni G, Mori G, Grano M, Zallone A. Human osteoclasts express oxytocin receptor. Biochem Biophys Res Commun 2002; 297:442-445 [PubMed: 12270111]
- 43.
- Tamma R, Colaianni G, Zhu LL, DiBenedetto A, Greco G, Montemurro G, Patano N, Strippoli M, Vergari R, Mancini L, Colucci S, Grano M, Faccio R, Liu X, Li J, Usmani S, Bachar M, Bab I, Nishimori K, Young LJ, Buettner C, Iqbal J, Sun L, Zaidi M, Zallone A. Oxytocin is an anabolic bone hormone. Proc Natl Acad Sci U S A 2009; 106:7149-7154 [PMC free article: PMC2678458] [PubMed: 19369205]
- 44.
- Liu X, Shimono K, Zhu LL, Li J, Peng Y, Imam A, Iqbal J, Moonga S, Colaianni G, Su C, Lu Z, Iwamoto M, Pacifici M, Zallone A, Sun L, Zaidi M. Oxytocin deficiency impairs maternal skeletal remodeling. Biochem Biophys Res Commun 2009; 388:161-166 [PubMed: 19653998]
- 45.
- Halloran BP, DeLuca HF. Calcium transport in small intestine during pregnancy and lactation. Am J Physiol 1980; 239:E64-68 [PubMed: 6249126]
- 46.
- Brommage R, Baxter DC, Gierke LW. Vitamin D-independent intestinal calcium and phosphorus absorption during reproduction. Am J Physiol 1990; 259:G631-638 [PubMed: 2221074]
- 47.
- Fudge NJ, Kovacs CS. Pregnancy up-regulates intestinal calcium absorption and skeletal mineralization independently of the vitamin D receptor. Endocrinology 2010; 151:886-895 [PubMed: 20051486]
- 48.
- Ryan BA, McGregor NE, Kirby BJ, Al-Tilissi A, Poulton IJ, Sims NA, Kovacs CS. Calcitriol-Dependent and -Independent Regulation of Intestinal Calcium Absorption, Osteoblast Function, and Skeletal Mineralization during Lactation and Recovery in Mice. J Bone Miner Res 2022; 37:2483-2497 [PubMed: 36128890]
- 49.
- Heaney RP, Skillman TG. Calcium metabolism in normal human pregnancy. J Clin Endocrinol Metab 1971; 33:661-670 [PubMed: 5093770]
- 50.
- Walker A, Fraile JJ, Hubbard JG. "Parathyroidectomy in pregnancy"-a single centre experience with review of evidence and proposal for treatment algorithim. Gland Surg 2014; 3:158-164 [PMC free article: PMC4139131] [PubMed: 25207208]
- 51.
- Morton A. Altered calcium homeostasis during pregnancy may affect biochemical differentiation of hypercalcaemia. Intern Med J 2004; 34:655-656; author reply 656-657 [PubMed: 15546467]
- 52.
- Pedersen EB, Johannesen P, Kristensen S, Rasmussen AB, Emmertsen K, Moller J, Lauritsen JG, Wohlert M. Calcium, parathyroid hormone and calcitonin in normal pregnancy and preeclampsia. Gynecol Obstet Invest 1984; 18:156-164 [PubMed: 6489849]
- 53.
- Frenkel Y, Barkai G, Mashiach S, Dolev E, Zimlichman R, Weiss M. Hypocalciuria of preeclampsia is independent of parathyroid hormone level. Obstet Gynecol 1991; 77:689-691 [PubMed: 1849625]
- 54.
- Seely EW, Wood RJ, Brown EM, Graves SW. Lower serum ionized calcium and abnormal calciotropic hormone levels in preeclampsia. J Clin Endocrinol Metab 1992; 74:1436-1440 [PubMed: 1592891]
- 55.
- Lalau JD, Jans I, el Esper N, Bouillon R, Fournier A. Calcium metabolism, plasma parathyroid hormone, and calcitriol in transient hypertension of pregnancy. Am J Hypertens 1993; 6:522-527 [PubMed: 8343236]
- 56.
- Purdie DW, Aaron JE, Selby PL. Bone histology and mineral homeostasis in human pregnancy. Br J Obstet Gynaecol 1988; 95:849-854 [PubMed: 3191057]
- 57.
- Kovacs CS, El-Hajj Fuleihan G. Calcium and bone disorders during pregnancy and lactation. Endocrinol Metab Clin North Am 2006; 35:21-51, v [PubMed: 16310641]
- 58.
- Moller UK, Vieth Streym S, Mosekilde L, Rejnmark L. Changes in bone mineral density and body composition during pregnancy and postpartum. A controlled cohort study. Osteoporos Int 2012; 23:1213-1223 [PubMed: 21607805]
- 59.
- To WW, Wong MW. Changes in bone mineral density of the os calcis as measured by quantitative ultrasound during pregnancy and 24 months after delivery. Aust N Z J Obstet Gynaecol 2011; 51:166-171 [PubMed: 21466520]
- 60.
- Sowers M. Pregnancy and lactation as risk factors for subsequent bone loss and osteoporosis. J Bone Miner Res 1996; 11:1052-1060 [PubMed: 8854240]
- 61.
- Kreiger N, Kelsey JL, Holford TR, O'Connor T. An epidemiologic study of hip fracture in postmenopausal women. Am J Epidemiol 1982; 116:141-148 [PubMed: 7102649]
- 62.
- Aloia JF, Vaswani AN, Yeh JK, Ross P, Ellis K, Cohn SH. Determinants of bone mass in postmenopausal women. Arch Intern Med 1983; 143:1700-1704 [PubMed: 6615091]
- 63.
- Aloia JF, Cohn SH, Vaswani A, Yeh JK, Yuen K, Ellis K. Risk factors for postmenopausal osteoporosis. Am J Med 1985; 78:95-100
- 64.
- Hreshchyshyn MM, Hopkins A, Zylstra S, Anbar M. Associations of parity, breast-feeding, and birth control pills with lumbar spine and femoral neck bone densities. Am J Obstet Gynecol 1988; 159:318-322 [PubMed: 3407686]
- 65.
- Dequeker J, Tobing L, Rutten V, Geusens P. Relative risk factors for osteoporotic fracture: a pilot study of the MEDOS questionnaire. Clin Rheumatol 1991; 10:49-53 [PubMed: 2065507]
- 66.
- Feldblum PJ, Zhang J, Rich LE, Fortney JA, Talmage RV. Lactation history and bone mineral density among perimenopausal women. Epidemiology 1992; 3:527-531 [PubMed: 1420520]
- 67.
- Cumming RG, Klineberg RJ. Breastfeeding and other reproductive factors and the risk of hip fractures in elderly women. Int J Epidemiol 1993; 22:684-691 [PubMed: 8225744]
- 68.
- Berning B, van Kuijk C, Schutte HE, Kuiper JW, Drogendijk AC, Fauser BC. Determinants of lumbar bone mineral density in normal weight, non-smoking women soon after menopause. A study using clinical data and quantitative computed tomography. Bone Miner 1993; 21:129-139 [PubMed: 8358250]
- 69.
- Cure CC, Ramirez PC, Lopez-Jaramillo P. Osteoporosis, pregnancy, and lactation. Lancet 1998; 352:1227-1228
- 70.
- Cure-Cure C, Cure-Ramirez P, Teran E, Lopez-Jaramillo P. Bone-mass peak in multiparity and reduced risk of bone-fractures in menopause. Int J Gynaecol Obstet 2002; 76:285-291 [PubMed: 11880132]
- 71.
- Bjornerem A, Ahmed LA, Jorgensen L, Stormer J, Joakimsen RM. Breastfeeding protects against hip fracture in postmenopausal women: the Tromso study. J Bone Miner Res 2011; 26:2843-2850 [PubMed: 21898594]
- 72.
- Chapman DJ. Longer cumulative breastfeeding duration associated with improved bone strength. Journal of human lactation : official journal of International Lactation Consultant Association 2012; 28:18-19
- 73.
- Canal-Macias ML, Roncero-Martin R, Moran JM, Lavado-Garcia JM, Costa-Fernandez Mdel C, Pedrera-Zamorano JD. Increased bone mineral density is associated with breastfeeding history in premenopausal Spanish women. Archives of medical science : AMS 2013; 9:703-708 [PMC free article: PMC3776181] [PubMed: 24049532]
- 74.
- Heidari B, Heidari P, Nourooddini HG, Hajian-Tilaki KO. Relationship between parity and bone mass in postmenopausal women according to number of parities and age. J Reprod Med 2013; 58:389-394 [PubMed: 24050027]
- 75.
- Schnatz PF, Marakovits KA, O'Sullivan DM. Assessment of postmenopausal women and significant risk factors for osteoporosis. Obstet Gynecol Surv 2010; 65:591-596 [PubMed: 21144090]
- 76.
- Schnatz PF, Barker KG, Marakovits KA, O'Sullivan DM. Effects of age at first pregnancy and breast-feeding on the development of postmenopausal osteoporosis. Menopause (New York, NY) 2010; 17:1161-1166
- 77.
- Specker B, Binkley T. High parity is associated with increased bone size and strength. Osteoporos Int 2005; 16:1969-1974 [PubMed: 16091837]
- 78.
- Crandall CJ, Liu J, Cauley J, Newcomb PA, Manson JE, Vitolins MZ, Jacobson LT, Rykman KK, Stefanick ML. Associations of Parity, Breastfeeding, and Fractures in the Women's Health Observational Study. Obstet Gynecol 2017; 130:171-180 [PMC free article: PMC5484587] [PubMed: 28594759]
- 79.
- Kovacs CS, Ralston SH. Presentation and management of osteoporosis presenting in association with pregnancy or lactation. Osteoporos Int 2015; 26:2223-2241 [PubMed: 25939309]
- 80.
- Dunne F, Walters B, Marshall T, Heath DA. Pregnancy associated osteoporosis. Clin Endocrinol (Oxf) 1993; 39:487-490 [PubMed: 8287577]
- 81.
- Gehlen M, Lazarescu AD, Hinz C, Schwarz-Eywill M, Pfeifer M, Balasingam S, Maier A. Long-term outcome of patients with pregnancy and lactation-associated osteoporosis (PLO) with a particular focus on quality of life. Clin Rheumatol 2019; 38:3575-3583 [PubMed: 31468224]
- 82.
- Butscheidt S, Tsourdi E, Rolvien T, Delsmann A, Sturznickel J, Barvencik F, Jakob F, Hofbauer LC, Mundlos S, Kornak U, Seefried L, Oheim R. Relevant genetic variants are common in women with pregnancy and lactation-associated osteoporosis (PLO) and predispose to more severe clinical manifestations. Bone 2021; 147:115911 [PubMed: 33716164]
- 83.
- Laroche M, Talibart M, Cormier C, Roux C, Guggenbuhl P, Degboe Y. Pregnancy-related fractures: a retrospective study of a French cohort of 52 patients and review of the literature. Osteoporos Int 2017; 28:3135-3142 [PubMed: 28879474]
- 84.
- Butscheidt S, Delsmann A, Rolvien T, Barvencik F, Al-Bughaili M, Mundlos S, Schinke T, Amling M, Kornak U, Oheim R. Mutational analysis uncovers monogenic bone disorders in women with pregnancy-associated osteoporosis: three novel mutations in LRP5, COL1A1, and COL1A2. Osteoporos Int 2018; 29:1643-1651 [PubMed: 29594386]
- 85.
- Phillips AJ, Ostlere SJ, Smith R. Pregnancy-associated osteoporosis: does the skeleton recover? Osteoporos Int 2000; 11:449-454 [PubMed: 10912848]
- 86.
- Kovacs CS. Complex clinical encounter series: osteoporosis presenting during pregnancy and lactation: wait and reassess. J Bone Miner Res 2024; 39:197-201 [PMC free article: PMC11207916] [PubMed: 38477812]
- 87.
- Kondapalli AV, Kamanda-Kosseh M, Williams JM, Shiau S, Bucovsky M, Colon I, Shane E, Cohen A. Clinical characteristics of pregnancy and lactation associated osteoporosis: An online survey study. Osteoporos Int 2023; 34:1477-1489 [PubMed: 37204454]
- 88.
- Herath M, Wong P, Trinh A, Allan CA, Wallace EM, Ebeling PR, Fuller PJ, Milat F. Minimal-trauma ankle fractures predominate during pregnancy: a 17-year retrospective study. Archives of osteoporosis 2017; 12:86 [PubMed: 28965301]
- 89.
- Dent CE, Friedman M. Pregnancy and idiopathic osteoporosis. Q J Med 1965; 34:341-357 [PubMed: 5838264]
- 90.
- Nordin BE, Roper A. Post-pregnancy osteoporosis; a syndrome? Lancet 1955; 268:431-434 [PubMed: 13234409]
- 91.
- Khovidhunkit W, Epstein S. Osteoporosis in pregnancy. Osteoporos Int 1996; 6:345-354 [PubMed: 8931028]
- 92.
- Smith R, Athanasou NA, Ostlere SJ, Vipond SE. Pregnancy-associated osteoporosis. Quarterly Journal of Medicine 1995; 88:865-878
- 93.
- Campos-Obando N, Oei L, Hoefsloot LH, Kiewiet RM, Klaver CC, Simon ME, Zillikens MC. Osteoporotic vertebral fractures during pregnancy: be aware of a potential underlying genetic cause. J Clin Endocrinol Metab 2014; 99:1107-1111 [PubMed: 24423337]
- 94.
- Cook FJ, Mumm S, Whyte MP, Wenkert D. Pregnancy-associated osteoporosis with a heterozygous deactivating LDL receptor-related protein 5 (LRP5) mutation and a homozygous methylenetetrahydrofolate reductase (MTHFR) polymorphism. J Bone Miner Res 2014; 29:922-928 [PubMed: 24014470]
- 95.
- Khastgir G, Studd JW, King H, Abdalla H, Jones J, Carter G, Alaghband-Zadeh J. Changes in bone density and biochemical markers of bone turnover in pregnancy-associated osteoporosis. Br J Obstet Gynaecol 1996; 103:716-718 [PubMed: 8688403]
- 96.
- Agarwal S, El-Najjar D, Kondapalli AV, Kil N, Kamanda-Kosseh M, Bucovsky M, Colon I, Lappe JM, Stubby J, Recker RR, Guo XE, Shane E, Cohen A. HR-pQCT Reveals Marked Trabecular and Cortical Structural Deficits in Women with Pregnancy and Lactation associated Osteoporosis (PLO). J Bone Miner Res 2024; 39:zjae167
- 97.
- Lakhanpal S, Ginsburg WW, Luthra HS, Hunder GG. Transient regional osteoporosis. A study of 56 cases and review of the literature. Ann Intern Med 1987; 106:444-450 [PubMed: 3813239]
- 98.
- Arayssi TK, Tawbi HA, Usta IM, Hourani MH. Calcitonin in the treatment of transient osteoporosis of the hip. Semin Arthritis Rheum 2003; 32:388-397 [PubMed: 12833247]
- 99.
- Curtiss PH, Jr., Kincaid WE. Transitory demineralization of the hip in pregnancy. A report of three cases. The Journal of bone and joint surgery American volume 1959; 41-A:1327-1333 [PubMed: 13849487]
- 100.
- Baki ME, Uygun H, Ari B, Aydin H. Bilateral femoral neck insufficiency fractures in pregnancy. Eklem hastaliklari ve cerrahisi = Joint diseases & related surgery 2014; 25:60-62 [PubMed: 24650388]
- 101.
- Aynaci O, Kerimoglu S, Ozturk C, Saracoglu M. Bilateral non-traumatic acetabular and femoral neck fractures due to pregnancy-associated osteoporosis. Arch Orthop Trauma Surg 2008; 128:313-316 [PubMed: 17828410]
- 102.
- Bonacker J, Janousek M, Krober M. Pregnancy-associated osteoporosis with eight fractures in the vertebral column treated with kyphoplasty and bracing: a case report. Arch Orthop Trauma Surg 2014; 134:173-179 [PubMed: 24357025]
- 103.
- Pallavi P, Padma S, Vanitha Anna Selvi D. Transient osteoporosis of hip and lumbar spine in pregnancy. Journal of obstetrics and gynaecology of India 2012; 62:8-9 [PMC free article: PMC3632689] [PubMed: 24293857]
- 104.
- Iwamoto J, Sato Y, Uzawa M, Matsumoto H. Five-year follow-up of a woman with pregnancy and lactation-associated osteoporosis and vertebral fractures. Therapeutics and clinical risk management 2012; 8:195-199 [PMC free article: PMC3333459] [PubMed: 22547939]
- 105.
- Anai T, Tomiyasu T, Arima K, Miyakawa I. Pregnancy-associated osteoporosis with elevated levels of circulating parathyroid hormone-related protein: a report of two cases. The journal of obstetrics and gynaecology research 1999; 25:63-67 [PubMed: 10067016]
- 106.
- Tran HA, Petrovsky N. Pregnancy-associated osteoporosis with hypercalcaemia. Intern Med J 2002; 32:481-485 [PubMed: 12380703]
- 107.
- Segal E, Hochberg I, Weisman Y, Ish-Shalom S. Severe postpartum osteoporosis with increased PTHrP during lactation in a patient after total thyroidectomy and parathyroidectomy. Osteoporos Int 2011; 22:2907-2911 [PubMed: 21243337]
- 108.
- Liel Y, Atar D, Ohana N. Pregnancy-associated osteoporosis: preliminary densitometric evidence of extremely rapid recovery of bone mineral density. South Med J 1998; 91:33-35 [PubMed: 9438399]
- 109.
- Kyvernitakis I, Reuter TC, Hellmeyer L, Hars O, Hadji P. Subsequent fracture risk of women with pregnancy and lactation-associated osteoporosis after a median of 6 years of follow-up. Osteoporos Int 2018; 29:135-142 [PubMed: 28965212]
- 110.
- Kyvernitakis I, Reuter TC, Hellmeyer L, Hars O, Hadji P. Correction to: Subsequent fracture risk of women with pregnancy and lactation-associated osteoporosis after a median of 6 years of follow-up. Osteoporos Int 2023; 34:2143-2144 [PubMed: 37855888]
- 111.
- Ferrari S, Bianchi ML, Eisman JA, Foldes AJ, Adami S, Wahl DA, Stepan JJ, de Vernejoul MC, Kaufman JM. Osteoporosis in young adults: pathophysiology, diagnosis, and management. Osteoporos Int 2012; 23:2735-2748 [PubMed: 22684497]
- 112.
- Stathopoulos IP, Liakou CG, Katsalira A, Trovas G, Lyritis GG, Papaioannou NA, Tournis S. The use of bisphosphonates in women prior to or during pregnancy and lactation. Hormones (Athens, Greece) 2011; 10:280-291 [PubMed: 22281884]
- 113.
- Ijuin A, Yoshikata H, Asano R, Tsuburai T, Kikuchi R, Sakakibara H. Teriparatide and denosumab treatment for pregnancy and lactation-associated osteoporosis with multiple vertebral fractures: A case study. Taiwanese journal of obstetrics & gynecology 2017; 56:863-866 [PubMed: 29241936]
- 114.
- Hong N, Kim JE, Lee SJ, Kim SH, Rhee Y. Changes in bone mineral density and bone turnover markers during treatment with teriparatide in pregnancy- and lactation-associated osteoporosis. Clin Endocrinol (Oxf) 2018; 88:652-658 [PubMed: 29389010]
- 115.
- Grizzo FM, da Silva Martins J, Pinheiro MM, Jorgetti V, Carvalho MD, Pelloso SM. Pregnancy and Lactation-Associated Osteoporosis: Bone Histomorphometric Analysis and Response to Treatment with Zoledronic Acid. Calcif Tissue Int 2015; 97:421-425 [PubMed: 26108650]
- 116.
- Chan B, Zacharin M. Maternal and infant outcome after pamidronate treatment of polyostotic fibrous dysplasia and osteogenesis imperfecta before conception: a report of four cases. J Clin Endocrinol Metab 2006; 91:2017-2020 [PubMed: 16551739]
- 117.
- Tanriover MD, Oz SG, Sozen T. Ten-year follow-up in pregnancy and lactation-associated osteoporosis: sequential therapy with strontium ranelate and ibandronate. Spine J 2015; 15:1164-1165 [PubMed: 25925624]
- 118.
- Anagnostis P, Lampropoulou-Adamidou K, Bosdou JK, Trovas G, Galanis P, Chronopoulos E, Goulis DG, Tournis S. Comparative Effectiveness of Therapeutic Interventions in Pregnancy and Lactation-Associated Osteoporosis: A Systematic Review and Meta-analysis. J Clin Endocrinol Metab 2024; 109:879-901 [PMC free article: PMC10876413] [PubMed: 37708365]
- 119.
- Orhadje E, Berg K, Hauser B, Ralston SH. Clinical Features, Incidence and Treatment Outcome in Pregnancy-Associated Osteoporosis: A Single-Centre Experience over Two Decades. Calcif Tissue Int 2023; 113:591-596 [PMC free article: PMC10673946] [PubMed: 37819437]
- 120.
- Lampropoulou-Adamidou K, Trovas G, Triantafyllopoulos IK, Yavropoulou MP, Anastasilakis AD, Anagnostis P, Toulis KA, Makris K, Gazi S, Balanika A, Tournis S. Teriparatide Treatment in Patients with Pregnancy- and Lactation-Associated Osteoporosis. Calcif Tissue Int 2021; 109:554-562 [PubMed: 34132853]
- 121.
- Rodriguez AJ, Fink HA, Mirigian L, Guanabens N, Eastell R, Akesson K, Bauer DC, Ebeling PR. Pain, Quality of Life, and Safety Outcomes of Kyphoplasty for Vertebral Compression Fractures: Report of a Task Force of the American Society for Bone and Mineral Research. J Bone Miner Res 2017; 32:1935-1944 [PubMed: 28513888]
- 122.
- Kort KC, Schiller HJ, Numann PJ. Hyperparathyroidism and pregnancy. Am J Surg 1999; 177:66-68 [PubMed: 10037311]
- 123.
- Kelly TR. Primary hyperparathyroidism during pregnancy. Surgery 1991; 110:1028-1033; discussion 1033-1034 [PubMed: 1745971]
- 124.
- Shangold MM, Dor N, Welt SI, Fleischman AR, Crenshaw MC, Jr. Hyperparathyroidism and pregnancy: a review. Obstet Gynecol Surv 1982; 37:217-228 [PubMed: 7041021]
- 125.
- Wagner G, Transhol L, Melchior JC. Hyperparathyroidism and pregnancy. Acta Endocrinol (Copenh) 1964; 47:549-564 [PubMed: 14250391]
- 126.
- Ludwig GD. Hyperparathyroidism in relation to pregnancy. N Engl J Med 1962; 267:637-642 [PubMed: 14467191]
- 127.
- Delmonico FL, Neer RM, Cosimi AB, Barnes AB, Russell PS. Hyperparathyroidism during pregnancy. Am J Surg 1976; 131:328-337 [PubMed: 769583]
- 128.
- Bruce J, Strong JA. Maternal hyperparathyroidism and parathyroid deficiency in the child, with account of effect of parathyroidectomy on renal function, and of attempt to transplant part of tumor. Q J Med 1955; 24:307-319 [PubMed: 13290019]
- 129.
- Sandler ML, Ho R, Xing MH, Gidumal S, Spitzer H, Levy JC, Chai RL. Primary Hyperparathyroidism During Pregnancy Treated With Parathyroidectomy: A Systematic Review. The Laryngoscope 2021; 131:1915-1921 [PubMed: 33751589]
- 130.
- Kristoffersson A, Dahlgren S, Lithner F, Jarhult J. Primary hyperparathyroidism in pregnancy. Surgery 1985; 97:326-330 [PubMed: 3975852]
- 131.
- Carella MJ, Gossain VV. Hyperparathyroidism and pregnancy: case report and review. J Gen Intern Med 1992; 7:448-453 [PubMed: 1506954]
- 132.
- Schnatz PF, Curry SL. Primary hyperparathyroidism in pregnancy: evidence-based management. Obstet Gynecol Surv 2002; 57:365-376 [PubMed: 12140371]
- 133.
- Abood A, Vestergaard P. Pregnancy outcomes in women with primary hyperparathyroidism. Eur J Endocrinol 2014; 171:69-76 [PubMed: 24743398]
- 134.
- Hirsch D, Kopel V, Nadler V, Levy S, Toledano Y, Tsvetov G. Pregnancy outcomes in women with primary hyperparathyroidism. J Clin Endocrinol Metab 2015; 100:2115-2122 [PubMed: 25751112]
- 135.
- Matthias GS, Helliwell TR, Williams A. Postpartum hyperparathyroid crisis. Case report. Br J Obstet Gynaecol 1987; 94:807-810 [PubMed: 3663537]
- 136.
- Clark D, Seeds JW, Cefalo RC. Hyperparathyroid crisis and pregnancy. Am J Obstet Gynecol 1981; 140:840-842 [PubMed: 7258268]
- 137.
- Salem R, Taylor S. Hyperparathyroidism in pregnancy. Br J Surg 1979; 66:648-650 [PubMed: 497655]
- 138.
- Nilsson IL, Adner N, Reihner E, Palme-Kilander C, Edstrom G, Degerblad M. Primary hyperparathyroidism in pregnancy: a diagnostic and therapeutic challenge. J Womens Health (Larchmt) 2010; 19:1117-1121 [PubMed: 20469964]
- 139.
- Bilezikian JP, Khan AA, Silverberg SJ, Fuleihan GE, Marcocci C, Minisola S, Perrier N, Sitges-Serra A, Thakker RV, Guyatt G, Mannstadt M, Potts JT, Clarke BL, Brandi ML. Evaluation and Management of Primary Hyperparathyroidism: Summary Statement and Guidelines from the Fifth International Workshop. J Bone Miner Res 2022; 37:2293-2314 [PubMed: 36245251]
- 140.
- Han ES, Fritton K, Bacon P, Slodzinski MK, Argani C. Preterm Parturient with Polyhydramnios and Pancreatitis: Primary Presentation of Hyperparathyroidism. Case reports in obstetrics and gynecology 2018; 2018:2091082 [PMC free article: PMC5828412] [PubMed: 29607233]
- 141.
- Herrera-Martinez AD, Bahamondes-Opazo R, Palomares-Ortega R, Munoz-Jimenez C, Galvez-Moreno MA, Quesada Gomez JM. Primary hyperparathyroidism in pregnancy: a two-case report and literature review. Case reports in obstetrics and gynecology 2015; 2015:171828 [PMC free article: PMC4393898] [PubMed: 25893124]
- 142.
- Ali DS, Dandurand K, Khan AA. Primary Hyperparathyroidism in Pregnancy: Literature Review of the Diagnosis and Management. Journal of clinical medicine 2021; 10 [PMC free article: PMC8745487] [PubMed: 35011750]
- 143.
- Boyce RW, Varela A, Chouinard L, Bussiere JL, Chellman GJ, Ominsky MS, Pyrah IT. Infant cynomolgus monkeys exposed to denosumab in utero exhibit an osteoclast-poor osteopetrotic-like skeletal phenotype at birth and in the early postnatal period. Bone 2014; 64:314-325 [PubMed: 24727159]
- 144.
- Okamatsu N, Sakai N, Karakawa A, Kouyama N, Sato Y, Inagaki K, Kiuchi Y, Oguchi K, Negishi-Koga T, Takami M. Biological effects of anti-RANKL antibody administration in pregnant mice and their newborns. Biochem Biophys Res Commun 2017; 491:614-621 [PubMed: 28760341]
- 145.
- Rajala B, Abbasi RA, Hutchinson HT, Taylor T. Acute pancreatitis and primary hyperparathyroidism in pregnancy: treatment of hypercalcemia with magnesium sulfate. Obstet Gynecol 1987; 70:460-462 [PubMed: 3627603]
- 146.
- Dahan M, Chang RJ. Pancreatitis secondary to hyperparathyroidism during pregnancy. Obstet Gynecol 2001; 98:923-925 [PubMed: 11704205]
- 147.
- Horjus C, Groot I, Telting D, van Setten P, van Sorge A, Kovacs CS, Hermus A, de Boer H. Cinacalcet for hyperparathyroidism in pregnancy and puerperium. J Pediatr Endocrinol Metab 2009; 22:741-749 [PubMed: 19845125]
- 148.
- Gonzalo Garcia I, Robles Fradejas M, Martin Macias MLA, Biain Ciganda A, Bustinza Beaskoetxea Z, Ruiz Perez E, Fernandez Matia G, Martinez Guisasola J. Primary hyperparathyroidism in pregnancy treated with cinacalcet: a case report. J Obstet Gynaecol 2018; 38:132-134 [PubMed: 28760052]
- 149.
- Vera L, Oddo S, Di Iorgi N, Bentivoglio G, Giusti M. Primary hyperparathyroidism in pregnancy treated with cinacalcet: a case report and review of the literature. Journal of medical case reports 2016; 10:361 [PMC free article: PMC5175373] [PubMed: 27998296]
- 150.
- Nadarasa K, Bailey M, Chahal H, Raja O, Bhat R, Gayle C, Grossman AB, Druce MR. The use of cinacalcet in pregnancy to treat a complex case of parathyroid carcinoma. Endocrinol Diabetes Metab Case Rep 2014; 2014:140056 [PMC free article: PMC4174590] [PubMed: 25298882]
- 151.
- Kovacs CS, Ho-Pao CL, Hunzelman JL, Lanske B, Fox J, Seidman JG, Seidman CE, Kronenberg HM. Regulation of murine fetal-placental calcium metabolism by the calcium-sensing receptor. J Clin Invest 1998; 101:2812-2820 [PMC free article: PMC508872] [PubMed: 9637715]
- 152.
- Zeng H, Li Z, Zhang X, Wang N, Tian Y, Wang J. Anesthetic management of primary hyperparathyroidism during pregnancy: A case report. Medicine (Baltimore) 2017; 96:e9390 [PMC free article: PMC5758246] [PubMed: 29390544]
- 153.
- Brown EM, Pollak M, Seidman CE, Seidman JG, Chou YH, Riccardi D, Hebert SC. Calcium-ion-sensing cell-surface receptors. N Engl J Med 1995; 333:234-240 [PubMed: 7791841]
- 154.
- Jones AR, Hare MJ, Brown J, Yang J, Meyer C, Milat F, Allan CA. Familial Hypocalciuric Hypercalcemia in Pregnancy: Diagnostic Pitfalls. JBMR Plus 2020; 4:e10362 [PMC free article: PMC7285754] [PubMed: 32537548]
- 155.
- Powell BR, Buist NR. Late presenting, prolonged hypocalcemia in an infant of a woman with hypocalciuric hypercalcemia. Clin Pediatr (Phila) 1990; 29:241-243 [PubMed: 2331838]
- 156.
- Thomas BR, Bennett JD. Symptomatic hypocalcemia and hypoparathyroidism in two infants of mothers with hyperparathyroidism and familial benign hypercalcemia. J Perinatol 1995; 15:23-26 [PubMed: 7650548]
- 157.
- Kovacs CS. Hypoparathyroidism during pregnancy, lactation, and fetal/neonatal development. In: Brandi ML, Brown EM, eds. Hypoparathyroidism. Milan: Springer; 2015:249-269.
- 158.
- Khan AA, Clarke B, Rejnmark L, Brandi ML. MANAGEMENT OF ENDOCRINE DISEASE: Hypoparathyroidism in pregnancy: review and evidence-based recommendations for management. Eur J Endocrinol 2019; 180:R37-R44 [PubMed: 30444723]
- 159.
- Sweeney LL, Malabanan AO, Rosen H. Decreased calcitriol requirement during pregnancy and lactation with a window of increased requirement immediately post partum. Endocr Pract 2010; 16:459-462 [PubMed: 20061285]
- 160.
- Graham WP, III., Gordon CS, Loken HF, Blum A, Halden A. Effect of pregnancy and of the menstrual cycle on hypoparathyroidism. J Clin Endocrinol Metab 1964; 24:512-516 [PubMed: 14187290]
- 161.
- Hatswell BL, Allan CA, Teng J, Wong P, Ebeling PR, Wallace EM, Fuller PJ, Milat F. Management of hypoparathyroidism in pregnancy and lactation - A report of 10 cases. Bone Rep 2015; 3:15-19 [PMC free article: PMC5365205] [PubMed: 28377963]
- 162.
- Hartogsohn EAR, Khan AA, Kjaersulf LU, Sikjaer T, Hussain S, Rejnmark L. Changes in treatment needs of hypoparathyroidism during pregnancy and lactation: A case series. Clin Endocrinol (Oxf) 2020; 93:261-268 [PubMed: 32350890]
- 163.
- Wang JJ, Wang O, Wang YB, Yang J, Song A, Jiang Y, Li M, Xia WB, Xing XP. Changes in serum calcium and treatment of hypoparathyroidism during pregnancy and lactation: A single-center case series. J Clin Endocrinol Metab 2021;
- 164.
- Breslau NA, Zerwekh JE. Relationship of estrogen and pregnancy to calcium homeostasis in pseudohypoparathyroidism. J Clin Endocrinol Metab 1986; 62:45-51 [PubMed: 3753557]
- 165.
- O'Donnell D, Costa J, Meyers AM. Management of pseudohypoparathyroidism in pregnancy. Case report. Br J Obstet Gynaecol 1985; 92:639-641 [PubMed: 4005206]
- 166.
- Saito H, Saito M, Saito K, Terauchi A, Kobayashi T, Tominaga T, Hosoi E, Senoo M, Saito T. Subclinical pseudohypoparathyroidism type II: evidence for failure of physiologic adjustment in calcium metabolism during pregnancy. Am J Med Sci 1989; 297:247-250 [PubMed: 2539718]
- 167.
- Seki K, Osada H, Yasuda T, Sekiya S. Pseudohypoparathyroidism type 1b in pregnancy. Gynecol Obstet Invest 1999; 47:278-280 [PubMed: 10352393]
- 168.
- Ochiai D, Uchino H, Ikeda T, Yakubo K, Fukuiya T. Pseudohypoparathyroidism type 1a in pregnancy. J Obstet Gynaecol 2013; 33:900 [PubMed: 24219739]
- 169.
- Guedes Ramallo P, Boix Carreno E. Management of pseudohypoparathyroidism in pregnancy: a case report. J Obstet Gynaecol 2017; 37:519-520 [PubMed: 27981863]
- 170.
- Wang JJ, Yang Y, Wang YB, Song A, Jiang Y, Li M, Xia WB, Liu YP, Wang O, Xing XP. Pseudohypoparathyroidism during pregnancy and the postpartum period: A case series of five patients. Frontiers in endocrinology 2022; 13:1050305 [PMC free article: PMC9709395] [PubMed: 36465610]
- 171.
- Glass EJ, Barr DG. Transient neonatal hyperparathyroidism secondary to maternal pseudohypoparathyroidism. Arch Dis Child 1981; 56:565-568 [PMC free article: PMC1627360] [PubMed: 7271292]
- 172.
- Vidailhet M, Monin P, Andre M, Suty Y, Marchal C, Vert P. [Neonatal hyperparathyroidism secondary to maternal hypoparathyroidism]. Arch Fr Pediatr 1980; 37:305-312 [PubMed: 7469705]
- 173.
- Khosla S, van Heerden JA, Gharib H, Jackson IT, Danks J, Hayman JA, Martin TJ. Parathyroid hormone-related protein and hypercalcemia secondary to massive mammary hyperplasia. N Engl J Med 1990; 322:1157
- 174.
- Jackson IT, Saleh J, van Heerden JA. Gigantic mammary hyperplasia in pregnancy associated with pseudohyperparathyroidism. Plastic and reconstructive surgery 1989; 84:806-810 [PubMed: 2813591]
- 175.
- Jodeh W, Sparks PJ, Higgins JM, Tom A, Anilovich N, Moit H, Korff L, Hadad I, Wang X, Imel EA, Donegan DM. Parathyroid hormone-related peptide induced hypercalcemia of pregnancy due to mammary hyperplasia. JBMR Plus 2024; 8:ziae083 [PMC free article: PMC11260271] [PubMed: 39035786]
- 176.
- Sato K. Hypercalcemia during pregnancy, puerperium, and lactation: review and a case report of hypercalcemic crisis after delivery due to excessive production of PTH-related protein (PTHrP) without malignancy (humoral hypercalcemia of pregnancy). Endocr J 2008; 55:959-966 [PubMed: 18614854]
- 177.
- Marx SJ, Zusman RM, Umiker WO. Benign breast dysplasia causing hypercalcemia. J Clin Endocrinol Metab 1977; 45:1049-1052 [PubMed: 925130]
- 178.
- Eller-Vainicher C, Ossola MW, Beck-Peccoz P, Chiodini I. PTHrP-associated hypercalcemia of pregnancy resolved after delivery: a case report. Eur J Endocrinol 2012; 166:753-756 [PubMed: 22247017]
- 179.
- Kovacs CS, Ward LM. Disorders of Calcium, Phosphorus, and Bone Metabolism During Fetal and Neonatal Development. In: Kovacs CS, Deal CL, eds. Maternal-Fetal and Neonatal Endocrinology. San Diego: Academic Press; 2020:755-782.
- 180.
- Rosen CJ, Adams JS, Bikle DD, Black DM, Demay MB, Manson JE, Murad MH, Kovacs CS. The nonskeletal effects of vitamin D: an Endocrine Society scientific statement. Endocr Rev 2012; 33:456-492 [PMC free article: PMC3365859] [PubMed: 22596255]
- 181.
- Brooke OG, Brown IR, Bone CD, Carter ND, Cleeve HJ, Maxwell JD, Robinson VP, Winder SM. Vitamin D supplements in pregnant Asian women: effects on calcium status and fetal growth. Br Med J 1980; 280:751-754 [PMC free article: PMC1600591] [PubMed: 6989438]
- 182.
- Roth DE, Al Mahmud A, Raqib R, Akhtar E, Perumal N, Pezzack B, Baqui AH. Randomized placebo-controlled trial of high-dose prenatal third-trimester vitamin D3 supplementation in Bangladesh: the AViDD trial. Nutrition journal 2013; 12:47 [PMC free article: PMC3641012] [PubMed: 23587190]
- 183.
- Wagner CL, McNeil R, Hamilton SA, Winkler J, Rodriguez Cook C, Warner G, Bivens B, Davis DJ, Smith PG, Murphy M, Shary JR, Hollis BW. A randomized trial of vitamin D supplementation in 2 community health center networks in South Carolina. Am J Obstet Gynecol 2013; 208:137 e131-113
- 184.
- Dawodu A, Saadi HF, Bekdache G, Javed Y, Altaye M, Hollis BW. Randomized controlled trial (RCT) of vitamin D supplementation in pregnancy in a population with endemic vitamin D deficiency. J Clin Endocrinol Metab 2013; 98:2337-2346 [PubMed: 23559082]
- 185.
- Grant CC, Stewart AW, Scragg R, Milne T, Rowden J, Ekeroma A, Wall C, Mitchell EA, Crengle S, Trenholme A, Crane J, Camargo CA, Jr. Vitamin D during pregnancy and infancy and infant serum 25-hydroxyvitamin D concentration. Pediatrics 2014; 133:e143-153 [PubMed: 24344104]
- 186.
- Vaziri F, Dabbaghmanesh MH, Samsami A, Nasiri S, Shirazi PT. Vitamin D supplementation during pregnancy on infant anthropometric measurements and bone mass of mother-infant pairs: A randomized placebo clinical trial. Early Hum Dev 2016; 103:61-68 [PubMed: 27513714]
- 187.
- Cooper C, Harvey NC, Bishop NJ, Kennedy S, Papageorghiou AT, Schoenmakers I, Fraser R, Gandhi SV, Carr A, D'Angelo S, Crozier SR, Moon RJ, Arden NK, Dennison EM, Godfrey KM, Inskip HM, Prentice A, Mughal MZ, Eastell R, Reid DM, Javaid MK, Group MS. Maternal gestational vitamin D supplementation and offspring bone health (MAVIDOS): a multicentre, double-blind, randomised placebo-controlled trial. The Lancet Diabetes & Endocrinology 2016; 4:393-402 [PMC free article: PMC4843969] [PubMed: 26944421]
- 188.
- Roth DE, Morris SK, Zlotkin S, Gernand AD, Ahmed T, Shanta SS, Papp E, Korsiak J, Shi J, Islam MM, Jahan I, Keya FK, Willan AR, Weksberg R, Mohsin M, Rahman QS, Shah PS, Murphy KE, Stimec J, Pell LG, Qamar H, Al Mahmud A. Vitamin D Supplementation in Pregnancy and Lactation and Infant Growth. N Engl J Med 2018; 379:535-546 [PMC free article: PMC6004541] [PubMed: 30089075]
- 189.
- Mojibian M, Soheilykhah S, Fallah Zadeh MA, Jannati Moghadam M. The effects of vitamin D supplementation on maternal and neonatal outcome: A randomized clinical trial. Iranian journal of reproductive medicine 2015; 13:687-696 [PMC free article: PMC4695683] [PubMed: 26730243]
- 190.
- Wei W, Shary JR, Garrett-Mayer E, Anderson B, Forestieri NE, Hollis BW, Wagner CL. Bone mineral density during pregnancy in women participating in a randomized controlled trial of vitamin D supplementation. Am J Clin Nutr 2017; 106:1422-1430 [PMC free article: PMC5698834] [PubMed: 29046301]
- 191.
- Kovacs CS, Ward LM. Physiology of Calcium, Phosphorus, and Bone Metabolism During Fetal and Neonatal Development. In: Kovacs CS, Deal CL, eds. Maternal-Fetal and Neonatal Endocrinology. San Diego: Academic Press; 2020:573-586.
- 192.
- Kovacs CS. Analysis of the MAVIDOS trial. The Lancet Diabetes & Endocrinology 2017; 5:328
- 193.
- Khaing W, Vallibhakara SA, Tantrakul V, Vallibhakara O, Rattanasiri S, McEvoy M, Attia J, Thakkinstian A. Calcium and Vitamin D Supplementation for Prevention of Preeclampsia: A Systematic Review and Network Meta-Analysis. Nutrients 2017; 9
- 194.
- De-Regil LM, Palacios C, Lombardo LK, Pena-Rosas JP. Vitamin D supplementation for women during pregnancy. Cochrane Database Syst Rev 2016; 1:CD008873
- 195.
- Hypponen E, Cavadino A, Williams D, Fraser A, Vereczkey A, Fraser WD, Banhidy F, Lawlor D, Czeizel AE. Vitamin D and pre-eclampsia: original data, systematic review and meta-analysis. Annals of nutrition & metabolism 2013; 63:331-340 [PubMed: 24603503]
- 196.
- Thorne-Lyman A, Fawzi WW. Vitamin D during pregnancy and maternal, neonatal and infant health outcomes: a systematic review and meta-analysis. Paediatric and perinatal epidemiology 2012; 26 Suppl 1:75-90 [PMC free article: PMC3843348] [PubMed: 22742603]
- 197.
- Roth DE, Leung M, Mesfin E, Qamar H, Watterworth J, Papp E. Vitamin D supplementation during pregnancy: state of the evidence from a systematic review of randomised trials. BMJ 2017; 359:j5237 [PMC free article: PMC5706533] [PubMed: 29187358]
- 198.
- Harvey NC, Holroyd C, Ntani G, Javaid K, Cooper P, Moon R, Cole Z, Tinati T, Godfrey K, Dennison E, Bishop NJ, Baird J, Cooper C. Vitamin D supplementation in pregnancy: a systematic review. Health Technol Assess 2014; 18:1-190
- 199.
- Palacios C, Kostiuk LK, Pena-Rosas JP. Vitamin D supplementation for women during pregnancy. Cochrane Database Syst Rev 2019; 7:Cd008873 [PMC free article: PMC6659840] [PubMed: 31348529]
- 200.
- Edouard T, Alos N, Chabot G, Roughley P, Glorieux FH, Rauch F. Short- and long-term outcome of patients with pseudo-vitamin D deficiency rickets treated with calcitriol. J Clin Endocrinol Metab 2011; 96:82-89 [PubMed: 20926527]
- 201.
- Marx SJ, Swart EG, Jr., Hamstra AJ, DeLuca HF. Normal intrauterine development of the fetus of a woman receiving extraordinarily high doses of 1,25-dihydroxyvitamin D3. J Clin Endocrinol Metab 1980; 51:1138-1142 [PubMed: 6893458]
- 202.
- Malloy PJ, Tiosano D, Feldman D. Hereditary 1,25-Dihydroxyvitamin-D-Resistant Rickets. In: Feldman D, Pike JW, Adams JS, eds. Vitamin D, Third Edition. San Diego, CA: Academic Press; 2011:1197-1232.
- 203.
- Carpenter TO. CYP24A1 loss of function: Clinical phenotype of monoallelic and biallelic mutations. J Steroid Biochem Mol Biol 2017; 173:337-340 [PubMed: 28093352]
- 204.
- Maekawa AS, Walker EC, Bennin D, Hartery SA, Kirby BJ, St-Arnaud R, Sims NA, Kovacs CS. Maternal loss of 24-hydroxylase causes increased intestinal calcium absorption and hypercalcemia during pregnancy but reduced skeletal resorption during lactation in mice. J Bone Miner Res 2024; 39:zjae166
- 205.
- Shah AD, Hsiao EC, O'Donnell B, Salmeen K, Nussbaum R, Krebs M, Baumgartner-Parzer S, Kaufmann M, Jones G, Bikle DD, Wang Y, Mathew AS, Shoback D, Block-Kurbisch I. Maternal Hypercalcemia Due to Failure of 1,25-Dihydroxyvitamin-D3 Catabolism in a Patient With CYP24A1 Mutations. J Clin Endocrinol Metab 2015; 100:2832-2836 [PMC free article: PMC4524985] [PubMed: 26097993]
- 206.
- Dinour D, Davidovits M, Aviner S, Ganon L, Michael L, Modan-Moses D, Vered I, Bibi H, Frishberg Y, Holtzman EJ. Maternal and infantile hypercalcemia caused by vitamin-D-hydroxylase mutations and vitamin D intake. Pediatr Nephrol 2015; 30:145-152 [PubMed: 25194629]
- 207.
- Woods GN, Saitman A, Gao H, Clarke NJ, Fitzgerald RL, Chi NW. A Young Woman With Recurrent Gestational Hypercalcemia and Acute Pancreatitis Caused by CYP24A1 Deficiency. J Bone Miner Res 2016; 31:1841-1844 [PMC free article: PMC5071127] [PubMed: 27105398]
- 208.
- Kwong WT, Fehmi SM. Hypercalcemic Pancreatitis Triggered by Pregnancy With a CYP24A1 Mutation. Pancreas 2016; 45:e31-32 [PubMed: 27295538]
- 209.
- Brancatella A, Cappellani D, Kaufmann M, Semeraro A, Borsari S, Sardella C, Baldinotti F, Caligo MA, Jones G, Marcocci C, Cetani F. Long-term Efficacy and Safety of Rifampin in the Treatment of a Patient Carrying a CYP24A1 Loss-of-Function Variant. J Clin Endocrinol Metab 2022; 107:e3159-e3166 [PubMed: 35569070]
- 210.
- Hawkes CP, Li D, Hakonarson H, Meyers KE, Thummel KE, Levine MA. CYP3A4 Induction by Rifampin: An Alternative Pathway for Vitamin D Inactivation in Patients With CYP24A1 Mutations. J Clin Endocrinol Metab 2017; 102:1440-1446 [PMC free article: PMC5443336] [PubMed: 28324001]
- 211.
- Prentice A. Micronutrients and the bone mineral content of the mother, fetus and newborn. J Nutr 2003; 133:1693S-1699S [PubMed: 12730486]
- 212.
- O'Brien EC, Geraghty AA, Kilbane MT, McKenna MJ, McAuliffe FM. Bone resorption and dietary calcium in pregnancy-a window to future maternal bone health. Osteoporos Int 2021; 32:1803-1814 [PubMed: 33659997]
- 213.
- Prentice A. Pregnancy and lactation. In: Glorieux FH, Petifor JM, Jüppner H, eds. Pediatric bone: biology & diseases. New York: Academic Press; 2003:249-269.
- 214.
- Janakiraman V, Ettinger A, Mercado-Garcia A, Hu H, Hernandez-Avila M. Calcium supplements and bone resorption in pregnancy: a randomized crossover trial. Am J Prev Med 2003; 24:260-264 [PubMed: 12657345]
- 215.
- Koo WW, Walters JC, Esterlitz J, Levine RJ, Bush AJ, Sibai B. Maternal calcium supplementation and fetal bone mineralization. Obstet Gynecol 1999; 94:577-582 [PubMed: 10511362]
- 216.
- Prentice A. Calcium in pregnancy and lactation. Annu Rev Nutr 2000; 20:249-272 [PubMed: 10940334]
- 217.
- Hofmeyr GJ, Atallah AN, Duley L. Calcium supplementation during pregnancy for preventing hypertensive disorders and related problems. Cochrane Database Syst Rev 2006; 3:CD001059
- 218.
- Kumar A, Devi SG, Batra S, Singh C, Shukla DK. Calcium supplementation for the prevention of pre-eclampsia. Int J Gynaecol Obstet 2009; 104:32-36 [PubMed: 18851852]
- 219.
- Hiller JE, Crowther CA, Moore VA, Willson K, Robinson JS. Calcium supplementation in pregnancy and its impact on blood pressure in children and women: follow up of a randomised controlled trial. Aust N Z J Obstet Gynaecol 2007; 47:115-121 [PubMed: 17355300]
- 220.
- Villar J, Abdel-Aleem H, Merialdi M, Mathai M, Ali MM, Zavaleta N, Purwar M, Hofmeyr J, Nguyen TN, Campodonico L, Landoulsi S, Carroli G, Lindheimer M. World Health Organization randomized trial of calcium supplementation among low calcium intake pregnant women. Am J Obstet Gynecol 2006; 194:639-649 [PubMed: 16522392]
- 221.
- Ross AC, Abrams SA, Aloia JF, Brannon PM, Clinton SK, Durazo-Arvizu RA, Gallagher JC, Gallo RL, Jones G, Kovacs CS, Manson JE, Mayne ST, Rosen CJ, Shapses SA. Dietary Reference Intakes for Calcium and Vitamin D. Washington, DC: Institute of Medicine, 2011.
- 222.
- Ohata Y, Yamazaki M, Kawai M, Tsugawa N, Tachikawa K, Koinuma T, Miyagawa K, Kimoto A, Nakayama M, Namba N, Yamamoto H, Okano T, Ozono K, Michigami T. Elevated fibroblast growth factor 23 exerts its effects on placenta and regulates vitamin D metabolism in pregnancy of Hyp mice. J Bone Miner Res 2014; 29:1627-1638 [PubMed: 24470103]
- 223.
- Jonas AJ, Dominguez B. Low breast milk phosphorus concentration in familial hypophosphatemia. J Pediatr Gastroenterol Nutr 1989; 8:541-543 [PubMed: 2723946]
- 224.
- Reade TM, Scriver CR. Hypophosphatemic rickets and breast milk. N Engl J Med 1979; 300:1397
- 225.
- European Medicines Agency. Crysvita: Annex I: Summary of Product Characteristics. 2022; https://www
.ema.europa .eu/en/documents/product-information /crysvita-epar-product-information_en.pdf. Accessed 22 October 2023. - 226.
- Greer FR, Lane J, Ho M. Elevated serum parathyroid hormone, calcitonin, and 1,25-dihydroxyvitamin D in lactating women nursing twins. Am J Clin Nutr 1984; 40:562-568 [PubMed: 6548076]
- 227.
- Dobnig H, Kainer F, Stepan V, Winter R, Lipp R, Schaffer M, Kahr A, Nocnik S, Patterer G, Leb G. Elevated parathyroid hormone-related peptide levels after human gestation: relationship to changes in bone and mineral metabolism. J Clin Endocrinol Metab 1995; 80:3699-3707 [PubMed: 8530622]
- 228.
- Yamamoto M, Duong LT, Fisher JE, Thiede MA, Caulfield MP, Rosenblatt M. Suckling-mediated increases in urinary phosphate and 3',5'-cyclic adenosine monophosphate excretion in lactating rats: possible systemic effects of parathyroid hormone-related protein. Endocrinology 1991; 129:2614-2622 [PubMed: 1657580]
- 229.
- VanHouten JN, Dann P, Stewart AF, Watson CJ, Pollak M, Karaplis AC, Wysolmerski JJ. Mammary-specific deletion of parathyroid hormone-related protein preserves bone mass during lactation. J Clin Invest 2003; 112:1429-1436 [PMC free article: PMC228471] [PubMed: 14597768]
- 230.
- Kovacs CS, Chik CL. Hyperprolactinemia caused by lactation and pituitary adenomas is associated with altered serum calcium, phosphate, parathyroid hormone (PTH), and PTH-related peptide levels. J Clin Endocrinol Metab 1995; 80:3036-3042 [PubMed: 7559893]
- 231.
- Sowers MF, Hollis BW, Shapiro B, Randolph J, Janney CA, Zhang D, Schork A, Crutchfield M, Stanczyk F, Russell-Aulet M. Elevated parathyroid hormone-related peptide associated with lactation and bone density loss. JAMA 1996; 276:549-554 [PubMed: 8709404]
- 232.
- Kelly PA, Bachelot A, Kedzia C, Hennighausen L, Ormandy CJ, Kopchick JJ, Binart N. The role of prolactin and growth hormone in mammary gland development. Mol Cell Endocrinol 2002; 197:127-131 [PubMed: 12431805]
- 233.
- Dawood MY, Khan-Dawood FS, Wahi RS, Fuchs F. Oxytocin release and plasma anterior pituitary and gonadal hormones in women during lactation. J Clin Endocrinol Metab 1981; 52:678-683 [PubMed: 6782115]
- 234.
- Nishimori K, Young LJ, Guo Q, Wang Z, Insel TR, Matzuk MM. Oxytocin is required for nursing but is not essential for parturition or reproductive behavior. Proc Natl Acad Sci U S A 1996; 93:11699-11704 [PMC free article: PMC38121] [PubMed: 8876199]
- 235.
- VanHouten JN, Wysolmerski JJ. Low estrogen and high parathyroid hormone-related peptide levels contribute to accelerated bone resorption and bone loss in lactating mice. Endocrinology 2003; 144:5521-5529 [PubMed: 14500568]
- 236.
- Hernandez LL, Gregerson KA, Horseman ND. Mammary gland serotonin regulates parathyroid hormone-related protein and other bone-related signals. Am J Physiol Endocrinol Metab 2012; 302:E1009-1015 [PMC free article: PMC3774078] [PubMed: 22318950]
- 237.
- Horseman ND, Hernandez LL. New concepts of breast cell communication to bone. Trends Endocrinol Metab 2014; 25:34-41 [PubMed: 24055165]
- 238.
- Kolthoff N, Eiken P, Kristensen B, Nielsen SP. Bone mineral changes during pregnancy and lactation: a longitudinal cohort study. Clin Sci (Lond) 1998; 94:405-412 [PubMed: 9640346]
- 239.
- Polatti F, Capuzzo E, Viazzo F, Colleoni R, Klersy C. Bone mineral changes during and after lactation. Obstet Gynecol 1999; 94:52-56 [PubMed: 10389717]
- 240.
- Kalkwarf HJ, Specker BL, Bianchi DC, Ranz J, Ho M. The effect of calcium supplementation on bone density during lactation and after weaning. N Engl J Med 1997; 337:523-528 [PubMed: 9262495]
- 241.
- Cross NA, Hillman LS, Allen SH, Krause GF. Changes in bone mineral density and markers of bone remodeling during lactation and postweaning in women consuming high amounts of calcium. J Bone Miner Res 1995; 10:1312-1320 [PubMed: 7502702]
- 242.
- Laskey MA, Prentice A, Hanratty LA, Jarjou LM, Dibba B, Beavan SR, Cole TJ. Bone changes after 3 mo of lactation: influence of calcium intake, breast-milk output, and vitamin D-receptor genotype. Am J Clin Nutr 1998; 67:685-692 [PubMed: 9537615]
- 243.
- Teti A, Zallone A. Do osteocytes contribute to bone mineral homeostasis? Osteocytic osteolysis revisited. Bone 2009; 44:11-16 [PubMed: 18977320]
- 244.
- Qing H, Ardeshirpour L, Pajevic PD, Dusevich V, Jahn K, Kato S, Wysolmerski J, Bonewald LF. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J Bone Miner Res 2012; 27:1018-1029 [PMC free article: PMC3770147] [PubMed: 22308018]
- 245.
- Kirby BJ, Ardeshirpour L, Woodrow JP, Wysolmerski JJ, Sims NA, Karaplis AC, Kovacs CS. Skeletal recovery after weaning does not require PTHrP. J Bone Miner Res 2011; 26:1242-1251 [PMC free article: PMC3179289] [PubMed: 21308774]
- 246.
- Collins JN, Kirby BJ, Woodrow JP, Gagel RF, Rosen CJ, Sims NA, Kovacs CS. Lactating Ctcgrp nulls lose twice the normal bone mineral content due to fewer osteoblasts and more osteoclasts, whereas bone mass is fully restored after weaning in association with up-regulation of Wnt signaling and other novel genes. Endocrinology 2013; 154:1400-1413 [PMC free article: PMC3678150] [PubMed: 23462960]
- 247.
- Liu XS, Ardeshirpour L, VanHouten JN, Shane E, Wysolmerski JJ. Site-specific changes in bone microarchitecture, mineralization, and stiffness during lactation and after weaning in mice. J Bone Miner Res 2012; 27:865-875 [PubMed: 22189918]
- 248.
- Bjornerem A, Ghasem-Zadeh A, Wang X, Bui M, Walker SP, Zebaze R, Seeman E. Irreversible Deterioration of Cortical and Trabecular Microstructure Associated With Breastfeeding. J Bone Miner Res 2017; 32:681-687 [PubMed: 27736021]
- 249.
- Brembeck P, Lorentzon M, Ohlsson C, Winkvist A, Augustin H. Changes in cortical volumetric bone mineral density and thickness, and trabecular thickness in lactating women postpartum. J Clin Endocrinol Metab 2015; 100:535-543 [PubMed: 25387262]
- 250.
- Wiklund PK, Xu L, Wang Q, Mikkola T, Lyytikainen A, Volgyi E, Munukka E, Cheng SM, Alen M, Keinanen-Kiukaanniemi S, Cheng S. Lactation is associated with greater maternal bone size and bone strength later in life. Osteoporos Int 2012; 23:1939-1945 [PubMed: 21927916]
- 251.
- Bowman BM, Miller SC. Skeletal adaptations during mammalian reproduction. J Musculoskelet Neuronal Interact 2001; 1:347-355 [PubMed: 15758485]
- 252.
- Vajda EG, Bowman BM, Miller SC. Cancellous and cortical bone mechanical properties and tissue dynamics during pregnancy, lactation, and postlactation in the rat. Biol Reprod 2001; 65:689-695 [PubMed: 11514329]
- 253.
- Reid IR, Wattie DJ, Evans MC, Budayr AA. Post-pregnancy osteoporosis associated with hypercalcaemia. Clin Endocrinol (Oxf) 1992; 37:298-303 [PubMed: 1424213]
- 254.
- VanHouten J, Dann P, McGeoch G, Brown EM, Krapcho K, Neville M, Wysolmerski JJ. The calcium-sensing receptor regulates mammary gland parathyroid hormone-related protein production and calcium transport. J Clin Invest 2004; 113:598-608 [PMC free article: PMC338258] [PubMed: 14966569]
- 255.
- Ardeshirpour L, Dann P, Pollak M, Wysolmerski J, VanHouten J. The calcium-sensing receptor regulates PTHrP production and calcium transport in the lactating mammary gland. Bone 2006; 38:787-793 [PubMed: 16377269]
- 256.
- Caplan RH, Beguin EA. Hypercalcemia in a calcitriol-treated hypoparathyroid woman during lactation. Obstet Gynecol 1990; 76:485-489 [PubMed: 2381632]
- 257.
- Salle BL, Berthezene F, Glorieux FH, Delvin EE, Berland M, David L, Varenne JP, Putet G. Hypoparathyroidism during pregnancy: treatment with calcitriol. J Clin Endocrinol Metab 1981; 52:810-813 [PubMed: 6894151]
- 258.
- Sadeghi-Nejad A, Wolfsdorf JI, Senior B. Hypoparathyroidism and pregnancy. Treatment with calcitriol. JAMA 1980; 243:254-255 [PubMed: 7350374]
- 259.
- Shomali ME, Ross DS. Hypercalcemia in a woman with hypoparathyroidism associated with increased parathyroid hormone-related protein during lactation. Endocr Pract 1999; 5:198-200 [PubMed: 15251676]
- 260.
- Mather KJ, Chik CL, Corenblum B. Maintenance of serum calcium by parathyroid hormone-related peptide during lactation in a hypoparathyroid patient. J Clin Endocrinol Metab 1999; 84:424-427 [PubMed: 10022395]
- 261.
- Rideout KL. When endocrine disorders disrupt pregnancy: perspectives of affected mothers. In: Kovacs CS, Deal CL, eds. Maternal-Fetal and Neonatal Endocrinology: Physiology, Pathophysiology, and Clinical Management. San Diego: Academic Press; 2019:xxxvii-xli.
- 262.
- Wagner CL, Hulsey TC, Fanning D, Ebeling M, Hollis BW. High-dose vitamin D3 supplementation in a cohort of breastfeeding mothers and their infants: a 6-month follow-up pilot study. Breastfeed Med 2006; 1:59-70 [PubMed: 17661565]
- 263.
- Tan ML, Abrams SA, Osborn DA. Vitamin D supplementation for term breastfed infants to prevent vitamin D deficiency and improve bone health. Cochrane Database Syst Rev 2020; 12:Cd013046 [PMC free article: PMC8812278] [PubMed: 33305822]
- 264.
- Prentice A, Jarjou LM, Cole TJ, Stirling DM, Dibba B, Fairweather-Tait S. Calcium requirements of lactating Gambian mothers: effects of a calcium supplement on breast-milk calcium concentration, maternal bone mineral content, and urinary calcium excretion. Am J Clin Nutr 1995; 62:58-67 [PubMed: 7598067]
- 265.
- Prentice A, Jarjou LM, Stirling DM, Buffenstein R, Fairweather-Tait S. Biochemical markers of calcium and bone metabolism during 18 months of lactation in Gambian women accustomed to a low calcium intake and in those consuming a calcium supplement. J Clin Endocrinol Metab 1998; 83:1059-1066 [PubMed: 9543117]
- 266.
- Prentice A, Yan L, Jarjou LM, Dibba B, Laskey MA, Stirling DM, Fairweather-Tait S. Vitamin D status does not influence the breast-milk calcium concentration of lactating mothers accustomed to a low calcium intake. Acta Paediatr 1997; 86:1006-1008 [PubMed: 9343285]
- 267.
- Bezerra FF, Mendonca LM, Lobato EC, O'Brien KO, Donangelo CM. Bone mass is recovered from lactation to postweaning in adolescent mothers with low calcium intakes. Am J Clin Nutr 2004; 80:1322-1326 [PubMed: 15531682]
- 268.
- Chantry CJ, Auinger P, Byrd RS. Lactation among adolescent mothers and subsequent bone mineral density. Arch Pediatr Adolesc Med 2004; 158:650-656 [PubMed: 15237064]
- 269.
- Delzer PR, Meyer RA, Jr. Normal milk composition in lactating X-linked hypophosphatemic mice despite continued hypophosphatemia. Calcif Tissue Int 1983; 35:750-754 [PubMed: 6689137]
- 270.
- Burosumab. Drugs and Lactation Database (LactMed®). Bethesda (MD): National Institute of Child Health and Human Development; 2006.
- Calcium and bone metabolism disorders during pregnancy and lactation.[Endocrinol Metab Clin North Am...]Calcium and bone metabolism disorders during pregnancy and lactation.Kovacs CS. Endocrinol Metab Clin North Am. 2011 Dec; 40(4):795-826.
- Review Maternal Mineral and Bone Metabolism During Pregnancy, Lactation, and Post-Weaning Recovery.[Physiol Rev. 2016]Review Maternal Mineral and Bone Metabolism During Pregnancy, Lactation, and Post-Weaning Recovery.Kovacs CS. Physiol Rev. 2016 Apr; 96(2):449-547.
- Review Presentation and management of osteoporosis presenting in association with pregnancy or lactation.[Osteoporos Int. 2015]Review Presentation and management of osteoporosis presenting in association with pregnancy or lactation.Kovacs CS, Ralston SH. Osteoporos Int. 2015 Sep; 26(9):2223-41. Epub 2015 May 5.
- Review The Skeleton Is a Storehouse of Mineral That Is Plundered During Lactation and (Fully?) Replenished Afterwards.[J Bone Miner Res. 2017]Review The Skeleton Is a Storehouse of Mineral That Is Plundered During Lactation and (Fully?) Replenished Afterwards.Kovacs CS. J Bone Miner Res. 2017 Apr; 32(4):676-680. Epub 2017 Feb 28.
- Review Parathyroid Disease in Pregnancy and Lactation: A Narrative Review of the Literature.[Biomedicines. 2021]Review Parathyroid Disease in Pregnancy and Lactation: A Narrative Review of the Literature.Tsourdi E, Anastasilakis AD. Biomedicines. 2021 Apr 26; 9(5). Epub 2021 Apr 26.
- Calcium and Phosphate Metabolism and Related Disorders During Pregnancy and Lact...Calcium and Phosphate Metabolism and Related Disorders During Pregnancy and Lactation - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...