

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Pediatric primary or monogenic dyslipidemias are a heterogeneous group of disorders, characterized by severe elevation of cholesterol, triglycerides, or rarely a combination of the two. Monogenic hypercholesterolemias have elevated low-density lipoprotein-cholesterol (LDL-C) levels and very high risk of premature atherosclerotic disease. They are caused by mutations in genes involved in the receptor-mediated uptake of LDL by the LDL receptor (LDLR) in hepatocytes. Autosomal dominant familial hypercholesterolemia results from mutations in LDLR, apolipoprotein B-100 (APOB), or proprotein convertase subtilisin-like kexin type 9 (PCSK9). Autosomal recessive hypercholesterolemia is caused by mutations in the LDLR adaptor protein 1 (LDLRAP1) gene. Type 1 hyperlipoproteinemia (Familial Chylomicronemia Syndrome) have severe fasting hypertriglyceridemia secondary to accumulation of triglyceride (TG)-rich lipoproteins, especially chylomicrons. It results from mutations in one or more genes that compromise chylomicron lipolysis and clearance. It has autosomal recessive inheritance caused by mutations in lipoprotein lipase (LPL), Apolipoprotein C-II(APOCII), Lipase maturation factor 1(LMF-1), Apolipoprotein A-V(APOAV), Glycosylphosphatidylinositolanchored high-density lipoprotein-binding protein 1(GPIHBP1). Familial combined hypercholesterolemia is a complex genetic disease and primarily a disorder of adults. There is strong evidence demonstrating a log-linear relationship between total cholesterol levels and coronary heart disease risk. Severe hypertriglyceridemia has an increased risk of acute pancreatitis. Universal lipid screening with measurement of non-fasting non-HDL cholesterol should be performed in all children ages 9 –11 years and 17–21 years. Advanced genetic testing and counseling play very important role in patients with genetic dyslipidemia. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Dyslipidemias are heterogeneous group of disorders characterized by abnormal levels of circulating lipids and lipoproteins. These abnormalities include elevations in cholesterol (hypercholesterolemia, Fredrickson Class IIa), triglycerides (hypertriglyceridemia, Frederickson Classes I, IV and V), or a combination of the two (Fredrickson Classes III or IIb). Genetic disorders of high-density lipoprotein or hypocholesterolemias are extremely rare and discussed in other Endotext chapters.
The etiology of genetic disorders are very complex, and can encompass from rare monogenic disorders due to single gene defects to complex polygenic basis (1). Meta-analysis of genome-wide association study identified 95 loci associated with abnormal total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG) (2). Recent studies have shown that most patients with HTG have a complex genetic etiology consisting of multiple genetic variants ranging in both frequency and effect. Patients with TG concentration of 200-1000 mgl/dL typically have polygenic or multigenic HTG. The genome-wide association (GWA) studies re-discovered associations known from prior genetic studies: that of HDL-C with CETP, and of LDL-C with APOE, and eventually identified more than 30 chromosomal loci with common variants associated with lipid levels. Thus polygenic TG results from complex interplay of rare heterozygous variants with relatively large effects in APOA5, GCKR, LPL, APOB, APOE, CREBH, GPIHBP1 and rare variants in more than 30 genes together with secondary factors (3). Polygenic risk scores use weighted summations of single nucleotide variants and are proposed as tools to improve the prediction of cardiovascular disease events independent of LDL-C, and their usefulness in clinical applications requires further studies (4).
Secondary dyslipidemias are multifactorial – combining underlying genetic predispositions with disease states such as diabetes, thyroid disease, or drug-related changes in lipid metabolism. Only monogenic disorders are discussed in this chapter.
MONOGENIC HYPERCHOLESTEROLEMIA
Monogenic hypercholesterolemias are a group of single gene defects with Mendelian transmission characterized by elevated low-density lipoprotein-cholesterol (LDL-C) levels and very high risk of premature atherosclerotic disease (5) (Table 1).
Table 1.
Monogenic Causes of Hypercholesterolemia (5)
| Inheritance | Disease | Gene | Prevalence | Mechanism |
|---|---|---|---|---|
| Autosomal Dominant | ||||
| Familial Hypercholesterolemia (FH) | LDLR (6,7) | 1 in 270 (8) (heterozygous) 1 in 1.6 to 3 X 105 (9-12) (homozygous) | ↓LDL Clearance | |
| Familial defective apo B-100 | APOB (13) | 1:1000 (10) (heterozygous) 1 in 4 X 106 (homozygous) | ↓LDL Clearance | |
| FH3 | PCSK9(14) | <1 in 10,000 | ↑Degradation of LDLR | |
| Autosomal Recessive | ||||
| Autosomal recessive hypercholesterolemia | LDLRAP1 (15) | <1 in 1 X 106 (16) | ↓LDL Clearance | |
| Sitosterolemia | ABCG5/ABCG8 (17) | < 1 in 5x 106 | ↓cholesterol excretion ↓LDL Clearance | |
| Cerebrotendinous xanthomatosis | CYP27A1 | 3-5 in 1X105 | ↓ conversion of cholesterol to chenodeoxycholic acid (CDCA) and cholic acid | |
| Lysosomal Acid Lipase Deficiency | LIPA ( 18 ) | 1 in 4 to 30 X 104 | ↓ hydrolysis of cholesterol esters and triglycerides |
Autosomal Dominant Hypercholesterolemia
Autosomal dominant hypercholesterolemia (ADH) is characterized by severe life-long elevations in low-density lipoprotein-cholesterol (LDL-C) with a concomitant 10-20 fold-increased risk of premature coronary heart disease (CHD) compared with the general population (11). Autosomal dominant hypercholesterolemia is primarily caused by mutations in genes involved in the receptor-mediated uptake of LDL by the LDL receptor (LDLR) in hepatocytes (Figure 2).
Thus far, three genes have been found to cause the disorder: LDLR (Online Mendelian Inheritance in Man [OMIM] # 143890, referred to as having familial hypercholesterolemia [FH]), apolipoprotein B-100 (APOB, OMIM # 107730, referred to as familial defective APOB), and proprotein convertase subtilisin-like kexin type 9 (PCSK9, OMIM # 603776, referred to as FH3) (5). In ADH cohorts, mutation detection rates vary - as high as 90% in ethnically homogenous populations (19-23) and as low as 40% in a multiethnic US cohort (24).
FAMILIAL HYPERCHOLESTEROLEMIA
Brown and Goldstein (6) first demonstrated that autosomal dominant hypercholesterolemia is due to dysfunctional LDLR. Pathogenic changes in LDLR result in impaired uptake and processing of LDL particles, which leads to decreased LDL clearance and elevated serum cholesterol levels. Over 1700 mutations in LDLR have been described thus far, and roughly about 1000 are likely to be pathogenic (7,25-28). Mutations can be predicted to be pathogenic using scoring tools such as Sorting Intolerant from Tolerant (SIFT) (29), Polymorphism Phenotyping v2 (PolyPhen-2) (30), or Combined Annotation Dependent Depletion (CADD) (31). Guo et al (32) recently developed a prediction model using structural modeling and bioinformatics algorithm called “Structure-based Functional Impact Prediction for Mutation Identification” (SFIP-MutID) for FH with LDLR single missense mutations. Among autosomal dominant hypercholesterolemia patients with detectable mutations, LDLR mutations represent ~90% of cases, and recent large-scale exome sequencing studies have identified LDLR mutations as the most common genetic defect among all individuals with premature CHD (33).
FH can occur as either homozygous (or compound heterozygous) or heterozygous, with a gene dosage effect. Homozygous FH is rare with a frequency of 1 in 1,000,000, whereas heterozygous FH affects 1 in 250-500. Higher frequencies have been reported in homogenous ethnicities such as the Danish, French Canadians, South African Afrikaners, and Christian Lebanese (34,35). As expected, homozygotes are more severely affected than heterozygotes, with LDL-C that are typically > 500 mg/dL (36) (Figure 1). Heterozygotes have LDL-C between 190 and 500 mg/dL. Recent literature has suggested that FH is more common and complex than previously thought and many patients have polygenic susceptibility rather than a monogenic cause (1).
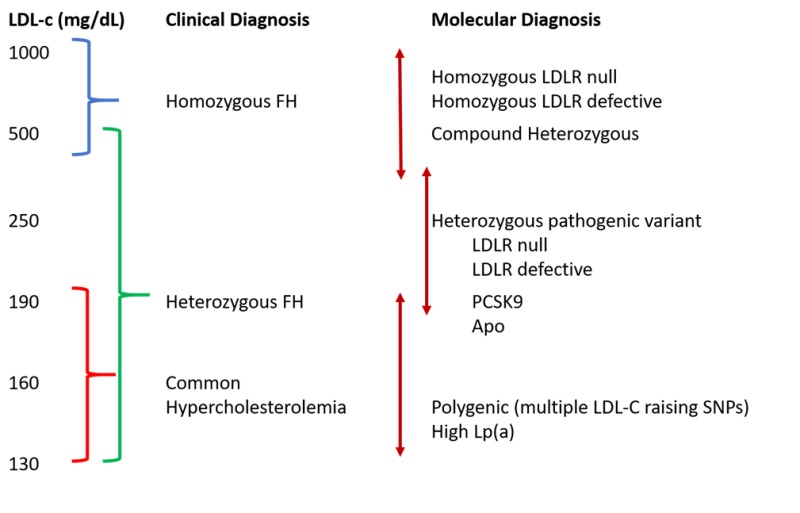
Figure 1.
Phenotypic Spectrum of Familial Hypercholesterolemia (FH). Clinical diagnosis of FH can be variable due to different underlying molecular mutations and additional genetic characteristics. LDL, low-density lipoprotein; APO, apolipoprotein B; PCSK9, pro-protein convertase subtilisin/kexin type 9; Lp(a), lipoprotein a; SNP = single nucleotide polymorphism. (Adapted from Strum, A.C., et al., Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018; 72(6):662-680 (9)).
FAMILIAL DEFECTIVE APO B-100 (FDB)
APOB-100 is the major apolipoprotein on LDL particles and helps the LDL-receptor bind LDL. FDB was first described phenotypically by Innerarity et al. in 1987 (37) after investigation by Vega and Grundy suggested that reduced binding of LDL to LDLR played a causative role in hypercholesterolemia. Mutations can occur in the ApoB domain involved in the binding of APOB to the LDLR, reducing clearance of LDL from plasma and causing hypercholesterolemia (13). Mutations in ApoB account for approximately 5% of the FH cases (27). Approximately 0.1% of the Northern Europeans and US Caucasians are known to carry p.Arg3500Gln variant in ApoB, whereas p.Arg3500Trp variant in ApoB is seen among East Asians (38-40). The p.Arg3500Gln variant raises plasma LDL-C by approximately 60 to 70 mg/dL and thus have a milder effect on plasma LDL-c than mutations in LDLR or PCSK9, but has been associated with increased coronary artery calcification, and earlier coronary artery disease, likely due to increase in small dense LDL particles (41).
PRO-PROTEIN CONVERTASE SUBTILISIN/KEXIN TYPE 9 (PCSK9)
PCSK9 was discovered in 2003 as a serine protease that degrades hepatic LDLRs in the endosomes thereby reducing receptor availability. PCSK9 gain-of-function (GOF) mutations cause increased LDRr degradation and reduced recycling to the cell surface, causing reduced LDL uptake and an increase in LDL-C concentration (42). Interestingly, functional studies show that different variants have different mechanisms to achieve the enhanced degradation of LDLr (43-46). Mutations upregulating activation of the PCSK9 gene were discovered in three French families with autosomal dominant hypercholesterolemia but no mutations in LDLR or ApoB (47). PCSK9 GOF mutations represent less than 1% of cases, with approximately 30 variants described to date (48). Currently there are two FDA approved human monoclonal antibodies to PCSK9: alirocumab and evolocumab. They were approved in 2015 and work by neutralizing PCSK9, inhibiting the interaction between PCSK9 and the LDLR, leading to an increase in the number of LDL receptors and, finally, enhancing uptake of LDL particles.
Autosomal Recessive Hypercholesterolemia (ARH)
ARH is caused by bi-allelic mutations in the LDLR adaptor protein 1 (LDLRAP1) gene. LDLR adaptor protein (LDLRAP1 or ARH) promotes the clustering of LDLRs into the clathrin-coated pits on the basolateral surface of hepatocytes by coupling the cytoplasmic tail of LDLR to structural components of the clathrin-coated pit and thus is essential for LDLR-mediated endocytosis. Inactivating mutations in LDLRAP1 lead to retention of LDLRs on the apical surface, thus severely reducing LDL uptake (15).
Sitosterolemia, Lysosomal Acid Lipase Deficiency, and Cerebrotendinous Xanthomatosis are discussed in other Endotext chapters.
Clinical Features
FH should be suspected in any child with elevated LDL-C along with family history of elevated LDL-C, tendon xanthomas, premature CHD, or sudden premature cardiac death. Cholesterol esters deposit in peripheral tissues like Achilles and extensor tendons giving rise to tendon xanthomas and their accumulation in arterial walls lead to development of plaques and atherosclerosis. Xanthomas are rarely seen in children and adolescents. However atherosclerosis is present from early childhood, and children with FH have endothelial dysfunction and increased carotid intima-media thickness (49).
There are three diagnostic tools available for FH (Figure 2-4):
- 1.
The US MedPed Program diagnostic criteria (50): It utilizes total cholesterol levels specific to an individual’s age and family history. The levels were derived from mathematical modeling using published cholesterol levels for FH individuals in the United States and Japan (Figure 2).
- 2.
The Simon Broome Register Group criteria (51): It utilizes cholesterol levels, clinical characteristics, molecular diagnosis, and family history (Figure 3).
- 3.
The Dutch Lipid Clinic Network criteria (52): It utilizes family history of hyperlipidemia or heart disease, clinical characteristics such as tendinous xanthomata, elevated LDL cholesterol, and/or an identified mutation (Figure 4).

Figure 2.
US MedPed Program Diagnostic Criteria.
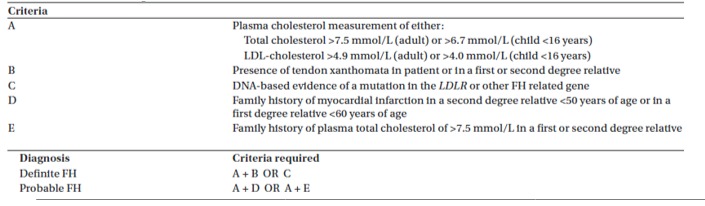
Figure 3.
The Simon Broome Register Criteria.

Figure 4.
The Dutch Lipid Clinic Network Criteria.
LIPOPROTEIN(a)
Lipoprotein (a) [Lp(a)] consists of an LDL particle and apolipoprotein(a) [apo(a)] and has been shown to be associated with increased risk of atherosclerotic cardiovascular disease including CHD, myocardial infarction and ischemic strokes. An Lp(a) level >100 nmol/L) in Caucasians and >150 nmol/L in African American is considered a risk enhancing factor. National Lipid Association recommends measurement of Lp(a) in youth (< 20 years) with FH; family history of first-degree relatives with premature ASCVD; unknown cause of ischemic stroke; or a parent or sibling with elevated Lp(a) (53). Lp(a) is discussed in another Endotext chapter.
FAMILIAL CHYLOMICRONEMIA SYNDROME (FCS) (TYPE 1 HYPERLIPOPROTEINEMIA)
Type 1 hyperlipoproteinemia (T1HLP, OMIM# 238600) or familial chylomicronemia syndrome is characterized by severe fasting hypertriglyceridemia secondary to accumulation of triglyceride (TG)-rich lipoproteins, especially chylomicrons. It results from mutations in one or more genes that compromise chylomicron lipolysis and clearance; mostly due to biallelic loss of function mutations in lipoprotein lipase (LPL) gene (3,54-56), or rarely due to mutations in apolipoprotein CII (APOC2), lipase maturation factor 1 (LMF1), glycosyl-phosphatidylinositol anchored high-density lipoprotein-binding protein 1 (GPIHBP1), and apolipoprotein AV (APOA5) (57,58). These disorders typically show autosomal recessive inheritance with published estimates of prevalence of ~1:1,000,000. A recent study estimates that population prevalence could be as high as 1 in 300,000 (59).
Genetics
Table 2.
Genetic Basis of Familial Chylomicronemia Syndrome
| Gene | Homozygote prevalence | Gene product function | Age of onset |
|---|---|---|---|
| LPL | 1 in 1 million (95% cases) | Hydrolysis of TG, peripheral uptake of FFA | Infancy or childhood |
| APOC2 | 20 families | Required cofactor of LPL | Childhood or adolescence |
| LMF1 | 2 families | Chaperone molecule required for proper LPL folding and/or expression | Late adulthood |
| APOA5 | 5 families | Enhancer of LPL activity | Late adulthood |
| GPIHBP1 | 15 families | Anchors LPL on capillary endothelium. Stabilizes binding of chylomicrons near LPL, supports lipolysis | Infancy or childhood |
Lipoprotein Lipase (LPL) Deficiency
FCS most commonly results from lipolytic defects due to deficiency of LPL. LPL is produced primarily by adipocytes and myocytes and binds to heparan sulfate, located at the heparin-binding site on the surface of capillary endothelial cells, allowing LPL to extend into the plasma and participate in the hydrolysis of TG carried in chylomicrons and very-low-density lipoproteins. Bi-allelic LPL mutations account for about 95% cases of FCS. More than 114 mutations in LPL have been described, and almost all of these have been shown to reduce or eliminate LPL activity in the homozygous state, preventing hydrolysis, and resulting in accumulation of triglyceride-rich lipoproteins, primarily chylomicrons (3,60).
Apolipoprotein C-II (APOC2) Mutations
APOC2 encodes for apolipoprotein (apo) C-II which is found on high-density lipoproteins (HDL), chylomicrons, and very-low-density lipoproteins, and acts as a key cofactor and an activator for LPL (61,62). Twenty families with disease causing mutations in ApoC2 have been reported in the literature.
Lipase Maturation Factor 1 (LMF1) Mutations
LMF1 serves as a chaperone in the endoplasmic reticulum and is required for the posttranslational activation of LPL, thus playing a regulatory role in lipase activation and lipid metabolism (63). Two families with disease causing mutations in LMF1 have been reported in literature.
Apolipoprotein A-V (APOAV) Mutation
Apo A-V is believed to stabilize the lipoprotein–enzyme complex and to enhance lipolysis; thus, when Apo A‑V is defective or absent, the efficiency of LPL-mediated lipolysis is decreased (64,65). Five patients with disease causing mutations in APOAV have been reported in literature.
Glycosylphosphatidylinositol-Anchored High-Density Lipoprotein-Binding Protein 1 (GPIHBP1) Mutation
GPIHBP1 is a glycosylphosphatidylinositol-anchored protein on capillary endothelial cells, which transports LPL into capillaries (66). GPIHBP1 directs the transendothelial transport of LPL, helps anchor chylomicrons to the endothelial surface, and enhances lipolysis (67). Mutations in mutations in GPIHBP1 have been reported in 15 families.
Clinical Features
FCS usually presents by adolescence although cases are often unrecognized until adulthood (60). Often, patients don’t get diagnosed until after developing pancreatitis (60,68), at which time triglycerides are noted to be severely elevated (at least > 1000 mg/dL). Other clinical features include eruptive or tuberous xanthomas, recurrent pancreatitis, lipemia retinalis, and hepatosplenomegaly. Some rare cases may present with failure to thrive, intestinal bleeding, anemia, or encephalopathy (69-71). Unique clinical features like neonatal transient obstructive jaundice due to xanthomas in pancreatic head region and asymptomatic renal xanthomas have been recently described (72,73).
Several physical exam findings characterize FCS. On fundoscopic exam, a pale pink appearance of vessels can be noted, referred to as lipemia retinalis. Lipemia retinalis occurs due to light scattering of large chylomicron particles. Eruptive xanthomas - crops of discrete yellow papules on an erythematous base – can manifest on the back, buttocks, and extensor aspects of elbows and knees. The eruptive xanthomas clear as triglycerides decrease. Hepatosplenomegaly occurs due to triglyceride accumulation in the liver and spleen.
Severe hypertriglyceridemia is an increased risk of acute pancreatitis, a serious condition often complicated by the systemic inflammatory response syndrome, multiorgan failure, pancreatic necrosis, and mortality rates as high as 20%. Even when not having pancreatitis episodes, some FCS patients suffer from bouts of abdominal pain.
Diagnostic Approach
FCS should be suspected in patients with severe hypertriglyceridemia (> 1000 mg/dL) without any secondary cause (e.g., uncontrolled diabetes, alcohol use, etc.). Gene sequencing to look for homozygous or compound heterozygous mutations in known genes such as LPL, APOC2, APOA5, LMF1 and GPIHBP1 may be performed. Although not always clinically available, several research labs can do sequencing or these genes can be included as part of targeted next-generation sequencing diagnostic panel for monogenic dyslipidemias. A molecular diagnosis aids in the early identification of at-risk family members. It might also help to establish candidacy for emerging therapies that target primary LPL deficiency, especially for patients who present at a young age. Treatment of these patients poses a significant challenge, as the current medications for hypertriglyceridemia such as fibrates, niacin, and omega-3 fatty acids are ineffective (55,74). The only effective therapy is extremely low-fat diet (55,75). Recent clinical trial of the gastric and pancreatic lipase inhibitor, orlistat, reduced serum triglycerides by greater than 50% in two patients with FCS due to GPIHBP1 mutations and was shown to be safe and highly efficacious in lowering serum triglycerides in children with FCS (76). Alipogene tiparvovec (Glybera®; AMT-011, AAV1-LPL(S447X)) is an adeno-associated virus serotype 1-based gene therapy, which was approved in Europe for adult patients with familial LPL deficiency in 2012 but has been subsequently withdrawn from the market in April 2017 (77). Volanesorsen, an antisense oligonucleotide against APOC3 mRNA, is approved to treat individuals with familial chylomicronemia syndrome in Europe but not the US. In a pooled analysis of four studies comparing 139 patients treated with volanesorsen a significant reduction in triglycerides was observed compared to placebo [TG level (MD: -73.9%; 95%CI: -93.5%, -54.2; p < .001) (77A).
FAMILIAL COMBINED HYPERLIPIDEMIA (FCHL)
FCHL is the most common inherited form on dyslipidemia. Its prevalence is estimated to be about 1 in 100 and thus is of importance for cardiovascular metabolic health of the population (78). A nomogram was created in 2004 to calculate probability of being affected by FCHL using three variables: age and gender adjusted triglyceride, total cholesterol, and absolute apoB levels. Points are calculated on point scale, translated into probabilities. The individual is considered as affected by FCHL if probability is at least 60%, in the setting of one other family member with FCH phenotype, and at least one individual in the family with premature cardiovascular disease (CVD) (79) . No single gene has yet been identified as a causative factor. It is a complex genetic disease and the features are determined by interaction of multiple FCHL susceptibility genes with environmental factors. The genes most frequently reported to be associated with FCHL are functionally related to plasma lipid metabolism and clearance, such as USF1, HL, PPARG, TNFRSF1B, LPL, LIPC, APOA1/CIII/AIV/AV and APOE (80). Overproduction of VLDL particles and hepatic fat accumulation are both central aspects of FCHL. Increased free fatty acid flux (from dysfunctional adipose tissue) towards the liver, increased hepatic de novo lipogenesis, and impaired β oxidation results in hepatic fat accumulation (80). FCHL is typically a diagnosis of adults. Its diagnosis is very complex in children due to lack of long-term data linking lipid values measured in children to the expression of the disease in the adult state or in older people. Hyperapo B in children may be a precursor of other lipid abnormalities, and thus it is suggested as a good marker of early diagnosis of FCH (81).
FAMILIAL HYPERTRIGLYCERIDEMIA (FHTG)
Similar to FCHL, FHTG is a complex genetic disease and the features are determined by the interaction of multiple susceptibility genes that increase triglyceride levels with environmental factors. Triglyceride levels are between 250-1000 mg/dL and LDL-c and apoB levels are not elevated. It is often accompanied by obesity and insulin resistance.
FAMILIAL DYSBETALIPOPROTEINEMIA
Dysbetalipoproteinemia is characterized by accumulation of remnant particles due to homozygous apoE2 genotype. The estimated prevalence is from 0.12% to 0.40% (82). A secondary insult such as insulin resistance, obesity, diabetes, hypothyroidism, or estrogen use decreases remnant clearance, increasing VLDL production. Patients have elevated total cholesterol (250-500 mg/dL) and triglyceride levels (250- 600 mg/dL), often with decreased HDL-C and LDL-C. This disorder is suspected when TG/apoB ratio is <10.0 and the diagnosis can be confirmed by VLDL-C/ plasma TG >0.69 plus an apoE2/E2 genotype (83).
LIPODYSTROPHY
Generalized and partial lipodystrophy syndromes are frequently associated with hypertriglyceridemia from late childhood and are discussed in details in another Endotext chapter (84,85).
SCREENING
There is strong evidence demonstrating a log-linear relationship between total cholesterol levels and coronary heart disease (CHD) risk. Thus the National Heart, Lung, and Blood Institute (NHLBI) along with the American Academy, issued integrated recommendations for cardiovascular (CV) risk reduction, including guidelines for management of hypertension, obesity, and hyperlipidemia (86). Universal lipid screening should be performed with measurement of non-fasting non-HDL cholesterol in all children ages 9 –11 years and 17–21 years. Those with abnormal levels should have two additional fasting lipid profiles measured 2 weeks to 3 months apart and averaged. Abnormal levels are then stratified by LDL cholesterol, TG levels, and risk factors. One of the important goals of the universal screening is identifying patients with FH. FH affects 1 in 250 population, and patients develop severe coronary artery disease and other vascular complications at a young age if not recognized and treated. Current evidence suggests that early detection of FH and cascade screening are required. Among heterozygous patients the long latent period before the expected onset of coronary artery disease provides an opportunity for initiating effective drug and lifestyle changes improving the prognosis of the disease (87,88). Universal screening in youth can also provide means of identifying affected family members through reverse cascade screening (89).
With decreasing cost and increasing accessibility, incidentally identified variants are becoming common and the ACMG (American College of Medical Genetics and Genomics) recently published guidance on clinically actionable genes. LDLRR, APOB and PCSK9 are amongst these genes. The Centers for Disease Control and Prevention has devised a 3-tier system for actionable genomic applications; with tier 1 genes backed by strong evidence that supports that identification should alter management to prevent the disease. Currently, the hyperlipidemia–associated genes represent the Centers for Disease Control and Prevention tier 1 list (90,91).
Cost-Effectiveness
Multiple studies have reported cost-effectiveness of screening. Goldman et al (92) showed the use of low-to-moderate doses of hydroxymethylglutaryl coenzyme A (HMG CoA) reductase inhibitor for primary prevention in patients with heterozygous FH was cost effective. Statins are now very inexpensive and generic. A detailed study from the United Kingdom compared the identification and treatment of FH patients by universal screening, opportunistic screening in primary care, screening of premature myocardial infarction admissions, and tracing family members of affected patients. They concluded that screening family members of people with familial hypercholesterolemia is the most cost effective option for detecting cases across the whole population (93). Another study showed that the cost-effectiveness of a family based screening program for FH in the Netherlands is between 25·5- and 32-thousand Euros per year of life gained (94). A recent study showed cost effectiveness if searching primary care databases for high-risk population of FH followed by cascade testing as only half of the carriers are identified by cascade screening at this time (95).
GENETIC COUNSELING
FH has an autosomal dominant inheritance with a gene dosage effect and the impact of diagnosis is likely to extend beyond the affected patient to multiple relatives across multiple generations. Identifying at-risk individuals is very important to prevent morbidity and mortality due to premature CVD. Given the complicated nature of genetic testing, there is significant role of genetic counseling for professionals treating hypercholesterolemic patients. Genetic counseling should begin when the proband is suspected to have diagnosis of FH. The discussion should include an explanation of inheritance patterns, information about genetic testing, including potential benefits, risks, and potential for incidental or uncertain findings. Once results are obtained, genetic counseling helps the patient in their interpretation. Genetic counselors should discuss the genetic tests results and interpretations and need to test family members in families with positive results. They also need to discuss that about 20–40% of FH patients do not have any unidentifiable mutations in Sanger sequencing (first line testing), and might benefit from new testing modalities like whole exome sequencing. FCS has autosomal recessive inheritance and genetic testing of the families help identify at risk individuals. Early identification of subjects at risk for developing HTG could prompt early lifestyle modification or evidence- based pharmacological intervention to reduce risk of clinical end points. Individuals that are heterozygous for LPL defects are at increased risk of developing hypertriglyceridemia, particularly in response to environmental insults such as obesity, diabetes, ETOH, etc. FCHL on the other hand is a complex disorder that both genetics and environment can play a role in its pathogenesis which can be explained to the families.
REFERENCES
- 1.
- Berberich AJ, Hegele RA. The complex molecular genetics of familial hypercholesterolaemia. Nat Rev Cardiol. 2019;16:9–20. [PubMed: 29973710]
- 2.
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y, Lee JY, Park T, Kim K, Sim X, Twee-Hee Ong R, Croteau-Chonka DC, Lange LA, Smith JD, Song K, Hua Zhao J, Yuan X, Luan J, Lamina C, Ziegler A, Zhang W, Zee RY, Wright AF, Witteman JC, Wilson JF, Willemsen G, Wichmann HE, Whitfield JB, Waterworth DM, Wareham NJ, Waeber G, Vollenweider P, Voight BF, Vitart V, Uitterlinden AG, Uda M, Tuomilehto J, Thompson JR, Tanaka T, Surakka I, Stringham HM, Spector TD, Soranzo N, Smit JH, Sinisalo J, Silander K, Sijbrands EJ, Scuteri A, Scott J, Schlessinger D, Sanna S, Salomaa V, Saharinen J, Sabatti C, Ruokonen A, Rudan I, Rose LM, Roberts R, Rieder M, Psaty BM, Pramstaller PP, Pichler I, Perola M, Penninx BW, Pedersen NL, Pattaro C, Parker AN, Pare G, Oostra BA, O'Donnell CJ, Nieminen MS, Nickerson DA, Montgomery GW, Meitinger T, McPherson R, McCarthy MI, McArdle W, Masson D, Martin NG, Marroni F, Mangino M, Magnusson PK, Lucas G, Luben R, Loos RJ, Lokki ML, Lettre G, Langenberg C, Launer LJ, Lakatta EG, Laaksonen R, Kyvik KO, Kronenberg F, Konig IR, Khaw KT, Kaprio J, Kaplan LM, Johansson A, Jarvelin MR, Janssens AC, Ingelsson E, Igl W, Kees Hovingh G, Hottenga JJ, Hofman A, Hicks AA, Hengstenberg C, Heid IM, Hayward C, Havulinna AS, Hastie ND, Harris TB, Haritunians T, Hall AS, Gyllensten U, Guiducci C, Groop LC, Gonzalez E, Gieger C, Freimer NB, Ferrucci L, Erdmann J, Elliott P, Ejebe KG, Doring A, Dominiczak AF, Demissie S, Deloukas P, de Geus EJ, de Faire U, Crawford G, Collins FS, Chen YD, Caulfield MJ, Campbell H, Burtt NP, Bonnycastle LL, Boomsma DI, Boekholdt SM, Bergman RN, Barroso I, Bandinelli S, Ballantyne CM, Assimes TL, Quertermous T, Altshuler D, Seielstad M, Wong TY, Tai ES, Feranil AB, Kuzawa CW, Adair LS, Taylor HA Jr, Borecki IB, Gabriel SB, Wilson JG, Holm H, Thorsteinsdottir U, Gudnason V, Krauss RM, Mohlke KL, Ordovas JM, Munroe PB, Kooner JS, Tall AR, Hegele RA, Kastelein JJ, Schadt EE, Rotter JI, Boerwinkle E, Strachan DP, Mooser V, Stefansson K, Reilly MP, Samani NJ, Schunkert H, Cupples LA, Sandhu MS, Ridker PM, Rader DJ, van Duijn CM, Peltonen L, Abecasis GR, Boehnke M, Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. [PMC free article: PMC3039276] [PubMed: 20686565]
- 3.
- Johansen CT, Hegele RA. Genetic bases of hypertriglyceridemic phenotypes. Curr Opin Lipidol. 2011;22:247–253. [PubMed: 21519249]
- 4.
- O'Sullivan JW, Raghavan S, Marquez-Luna C, Luzum JA, Damrauer SM, Ashley EA, O'Donnell CJ, Willer CJ, Natarajan P. American Heart Association Council on G, Precision M, Council on Clinical C, Council on Arteriosclerosis T, Vascular B, Council on Cardiovascular R, Intervention, Council on L, Cardiometabolic H, Council on Peripheral Vascular D. Polygenic Risk Scores for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation. 2022;146:e93–e118. [PMC free article: PMC9847481] [PubMed: 35862132]
- 5.
- Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J Clin Invest. 2003;111:1795–1803. [PMC free article: PMC161432] [PubMed: 12813012]
- 6.
- Goldstein JL, Brown MS. The LDL receptor locus and the genetics of familial hypercholesterolemia. Annu Rev Genet. 1979;13:259–289. [PubMed: 231932]
- 7.
- Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Ann Hum Genet. 2008;72:485–498. [PubMed: 18325082]
- 8.
- Wald DS, Bestwick JP, Morris JK, Whyte K, Jenkins L, Wald NJ. Child-Parent Familial Hypercholesterolemia Screening in Primary Care. N Engl J Med. 2016;375:1628–1637. [PubMed: 27783906]
- 9.
- Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM, Baum SJ, Bourbon M, Carrie A, Cuchel M, de Ferranti SD, Defesche JC, Freiberger T, Hershberger RE, Hovingh GK, Karayan L, Kastelein JJP, Kindt I, Lane SR, Leigh SE, Linton MF, Mata P, Neal WA, Nordestgaard BG, Santos RD, Harada-Shiba M, Sijbrands EJ, Stitziel NO, Yamashita S, Wilemon KA, Ledbetter DH, Rader DJ. Convened by the Familial Hypercholesterolemia F. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018;72:662–680. [PubMed: 30071997]
- 10.
- Sjouke B, Hovingh GK, Kastelein JJ, Stefanutti C. Homozygous autosomal dominant hypercholesterolaemia: prevalence, diagnosis, and current and future treatment perspectives. Curr Opin Lipidol. 2015;26:200–209. [PubMed: 25950706]
- 11.
- Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Boren J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg-Hansen A. European Atherosclerosis Society Consensus P. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a. [PMC free article: PMC3844152] [PubMed: 23956253]
- 12.
- Garg A, Garg V, Hegele RA, Lewis GF. Practical definitions of severe versus familial hypercholesterolaemia and hypertriglyceridaemia for adult clinical practice. Lancet Diabetes Endocrinol. 2019;7:880–886. [PubMed: 31445954]
- 13.
- Soria LF, Ludwig EH, Clarke HR, Vega GL, Grundy SM, McCarthy BJ. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci U S A. 1989;86:587–591. [PMC free article: PMC286517] [PubMed: 2563166]
- 14.
- Maxwell KN, Fisher EA, Breslow JL. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc Natl Acad Sci U S A. 2005;102:2069–2074. [PMC free article: PMC546019] [PubMed: 15677715]
- 15.
- Pisciotta L, Priore Oliva C, Pes GM, Di Scala L, Bellocchio A, Fresa R, Cantafora A, Arca M, Calandra S, Bertolini S. Autosomal recessive hypercholesterolemia (ARH) and homozygous familial hypercholesterolemia (FH): a phenotypic comparison. Atherosclerosis. 2006;188:398–405. [PubMed: 16343504]
- 16.
- D'Erasmo L, Minicocci I, Nicolucci A, Pintus P, Roeters Van Lennep JE, Masana L, Mata P, Sanchez-Hernandez RM, Prieto-Matos P, Real JT, Ascaso JF, Lafuente EE, Pocovi M, Fuentes FJ, Muntoni S, Bertolini S, Sirtori C, Calabresi L, Pavanello C, Averna M, Cefalu AB, Noto D, Pacifico AA, Pes GM, Harada-Shiba M, Manzato E, Zambon S, Zambon A, Vogt A, Scardapane M, Sjouke B, Fellin R, Arca M. Autosomal Recessive Hypercholesterolemia: Long-Term Cardiovascular Outcomes. J Am Coll Cardiol. 2018;71:279–288. [PubMed: 29348020]
- 17.
- Lu K, Lee MH, Hazard S, Brooks-Wilson A, Hidaka H, Kojima H, Ose L, Stalenhoef AF, Mietinnen T, Bjorkhem I, Bruckert E, Pandya A, Brewer HB Jr, Salen G, Dean M, Srivastava A, Patel SB. Two genes that map to the STSL locus cause sitosterolemia: genomic structure and spectrum of mutations involving sterolin-1 and sterolin-2, encoded by ABCG5 and ABCG8, respectively. Am J Hum Genet. 2001;69:278–290. [PMC free article: PMC1201544] [PubMed: 11452359]
- 18.
- Anderson RA, Rao N, Byrum RS, Rothschild CB, Bowden DW, Hayworth R, Pettenati M. In situ localization of the genetic locus encoding the lysosomal acid lipase/cholesteryl esterase (LIPA) deficient in Wolman disease to chromosome 10q23.2-q23.3. Genomics. 1993;15:245–247. [PubMed: 8432549]
- 19.
- Khachadurian AK, Uthman SM. Experiences with the homozygous cases of familial hypercholesterolemia. A report of 52 patients. Nutrition and metabolism. 1973;15:132–140. [PubMed: 4351242]
- 20.
- Jenkins T, Nicholls E, Gordon E, Mendelsohn D, Seftel HC, Andrew MJ. Familial hypercholesterolaemia--a common genetic disorder in the Afrikaans population. S Afr Med J. 1980;57:943–947. [PubMed: 7404060]
- 21.
- van der Graaf A, Avis HJ, Kusters DM, Vissers MN, Hutten BA, Defesche JC, Huijgen R, Fouchier SW, Wijburg FA, Kastelein JJ, Wiegman A. Molecular basis of autosomal dominant hypercholesterolemia: assessment in a large cohort of hypercholesterolemic children. Circulation. 2011;123:1167–1173. [PubMed: 21382890]
- 22.
- Moorjani S, Roy M, Gagne C, Davignon J, Brun D, Toussaint M, Lambert M, Campeau L, Blaichman S, Lupien P. Homozygous familial hypercholesterolemia among French Canadians in Quebec Province. Arteriosclerosis. 1989;9:211–216. [PubMed: 2923577]
- 23.
- Seftel HC, Baker SG, Jenkins T, Mendelsohn D. Prevalence of familial hypercholesterolemia in Johannesburg Jews. Am J Med Genet. 1989;34:545–547. [PubMed: 2624266]
- 24.
- Ahmad Z, Adams-Huet B, Chen C, Garg A. Low Prevalence of Mutations in Known Loci for Autosomal Dominant Hypercholesterolemia in a Multi-Ethnic Patient Cohort. Circ Cardiovasc Genet. 2012;5:666–675. [PMC free article: PMC3774009] [PubMed: 23064986]
- 25.
- 26.
- Bruikman CS, Hovingh GK, Kastelein JJ. Molecular basis of familial hypercholesterolemia. Curr Opin Cardiol. 2017
- 27.
- Henderson R, O'Kane M, McGilligan V, Watterson S. The genetics and screening of familial hypercholesterolaemia. J Biomed Sci. 2016;23:39. [PMC free article: PMC4833930] [PubMed: 27084339]
- 28.
- Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, Gidding SS, de Ferranti SD, Ito MK, McGowan MP, Moriarty PM, Cromwell WC, Ross JL, Ziajka PE. National Lipid Association Expert Panel on Familial H. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. Journal of clinical lipidology. 2011;5:S1–8. [PubMed: 21600525]
- 29.
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. [PMC free article: PMC168916] [PubMed: 12824425]
- 30.
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet 2013; Chapter 7:Unit7 20.
- 31.
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–315. [PMC free article: PMC3992975] [PubMed: 24487276]
- 32.
- Guo J, Gao Y, Li X, He Y, Zheng X, Bi J, Hou L, Sa Y, Zhang M, Yin H, Jiang L. Systematic prediction of familial hypercholesterolemia caused by low-density lipoprotein receptor missense mutations. Atherosclerosis. 2019;281:1–8. [PubMed: 30583242]
- 33.
- Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, Guella I, Asselta R, Lange LA, Peloso GM, Auer PL, Project NES, Girelli D, Martinelli N, Farlow DN, DePristo MA, Roberts R, Stewart AF, Saleheen D, Danesh J, Epstein SE, Sivapalaratnam S, Kees Hovingh G, Kastelein JJ, Samani NJ, Schunkert H, Erdmann J, Shah SH, Kraus WE, Davies R, Nikpay M, Johansen CT, Wang J, Hegele RA, Hechter E, Marz W, Kleber ME, Huang J, Johnson AD, Li M, Burke GL, Gross M, Liu Y, Assimes TL, Heiss G, Lange EM, Folsom AR, Taylor HA, Olivieri O, Hamsten A, Clarke R, Reilly DF, Yin W, Rivas MA, Donnelly P, Rossouw JE, Psaty BM, Herrington DM, Wilson JG, Rich SS, Bamshad MJ, Tracy RP, Adrienne Cupples L, Rader DJ, Reilly MP, Spertus JA, Cresci S, Hartiala J, Wilson Tang WH, Hazen SL, Allayee H, Reiner AP, Carlson CS, Kooperberg C, Jackson RD, Boerwinkle E, Lander ES, Schwartz SM, Siscovick DS, McPherson R, Tybjaerg-Hansen A, Abecasis GR, Watkins H, Nickerson DA, Ardissino D, Sunyaev SR, O'Donnell CJ, Altshuler D, Gabriel S, Kathiresan S. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2014
- 34.
- Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160:407–420. [PubMed: 15321837]
- 35.
- Goldstein JL HH, Brown MS. . Familial hypercholesterolemia.: New York, NY: McGraw-Hill Companies, Inc,.
- 36.
- Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ. European Atherosclerosis Society Consensus Panel on Familial H. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–2157. [PMC free article: PMC4139706] [PubMed: 25053660]
- 37.
- Innerarity TL, Weisgraber KH, Arnold KS, Mahley RW, Krauss RM, Vega GL, Grundy SM. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc Natl Acad Sci U S A. 1987;84:6919–6923. [PMC free article: PMC299196] [PubMed: 3477815]
- 38.
- Andersen LH, Miserez AR, Ahmad Z, Andersen RL. Familial defective apolipoprotein B-100: A review. Journal of clinical lipidology. 2016;10:1297–1302. [PubMed: 27919345]
- 39.
- Peloso GM, Auer PL, Bis JC, Voorman A, Morrison AC, Stitziel NO, Brody JA, Khetarpal SA, Crosby JR, Fornage M, Isaacs A, Jakobsdottir J, Feitosa MF, Davies G, Huffman JE, Manichaikul A, Davis B, Lohman K, Joon AY, Smith AV, Grove ML, Zanoni P, Redon V, Demissie S, Lawson K, Peters U, Carlson C, Jackson RD, Ryckman KK, Mackey RH, Robinson JG, Siscovick DS, Schreiner PJ, Mychaleckyj JC, Pankow JS, Hofman A, Uitterlinden AG, Harris TB, Taylor KD, Stafford JM, Reynolds LM, Marioni RE, Dehghan A, Franco OH, Patel AP, Lu Y, Hindy G, Gottesman O, Bottinger EP, Melander O, Orho-Melander M, Loos RJ, Duga S, Merlini PA, Farrall M, Goel A, Asselta R, Girelli D, Martinelli N, Shah SH, Kraus WE, Li M, Rader DJ, Reilly MP, McPherson R, Watkins H, Ardissino D, Project NGES, Zhang Q, Wang J, Tsai MY, Taylor HA, Correa A, Griswold ME, Lange LA, Starr JM, Rudan I, Eiriksdottir G, Launer LJ, Ordovas JM, Levy D, Chen YD, Reiner AP, Hayward C, Polasek O, Deary IJ, Borecki IB, Liu Y, Gudnason V, Wilson JG, van Duijn CM, Kooperberg C, Rich SS, Psaty BM, Rotter JI, O'Donnell CJ, Rice K, Boerwinkle E, Kathiresan S, Cupples LA. Association of low-frequency and rare coding-sequence variants with blood lipids and coronary heart disease in 56,000 whites and blacks. Am J Hum Genet. 2014;94:223–232. [PMC free article: PMC3928662] [PubMed: 24507774]
- 40.
- Chiou KR, Charng MJ. Genetic diagnosis of familial hypercholesterolemia in Han Chinese. Journal of clinical lipidology. 2016;10:490–496. [PubMed: 27206935]
- 41.
- Hoogeveen RC, Gaubatz JW, Sun W, Dodge RC, Crosby JR, Jiang J, Couper D, Virani SS, Kathiresan S, Boerwinkle E, Ballantyne CM. Small dense low-density lipoprotein-cholesterol concentrations predict risk for coronary heart disease: the Atherosclerosis Risk In Communities (ARIC) study. Arterioscler Thromb Vasc Biol. 2014;34:1069–1077. [PMC free article: PMC3999643] [PubMed: 24558110]
- 42.
- Zhang DW, Lagace TA, Garuti R, Zhao Z, McDonald M, Horton JD, Cohen JC, Hobbs HH. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J Biol Chem. 2007;282:18602–18612. [PubMed: 17452316]
- 43.
- Cameron J, Holla OL, Ranheim T, Kulseth MA, Berge KE, Leren TP. Effect of mutations in the PCSK9 gene on the cell surface LDL receptors. Hum Mol Genet. 2006;15:1551–1558. [PubMed: 16571601]
- 44.
- Lipari MT, Li W, Moran P, Kong-Beltran M, Sai T, Lai J, Lin SJ, Kolumam G, Zavala-Solorio J, Izrael-Tomasevic A, Arnott D, Wang J, Peterson AS, Kirchhofer D. Furin-cleaved proprotein convertase subtilisin/kexin type 9 (PCSK9) is active and modulates low density lipoprotein receptor and serum cholesterol levels. J Biol Chem. 2012;287:43482–43491. [PMC free article: PMC3527935] [PubMed: 23135270]
- 45.
- Bottomley MJ, Cirillo A, Orsatti L, Ruggeri L, Fisher TS, Santoro JC, Cummings RT, Cubbon RM, Lo Surdo P, Calzetta A, Noto A, Baysarowich J, Mattu M, Talamo F, De Francesco R, Sparrow CP, Sitlani A, Carfi A. Structural and biochemical characterization of the wild type PCSK9-EGF(AB) complex and natural familial hypercholesterolemia mutants. J Biol Chem. 2009;284:1313–1323. [PubMed: 19001363]
- 46.
- Sanchez-Hernandez RM, Di Taranto MD, Benito-Vicente A, Uribe KB, Lamiquiz-Moneo I, Larrea-Sebal A, Jebari S, Galicia-Garcia U, Novoa FJ, Boronat M, Wagner AM, Civeira F, Martin C, Fortunato G. The Arg499His gain-of-function mutation in the C-terminal domain of PCSK9. Atherosclerosis. 2019;289:162–172. [PubMed: 31518966]
- 47.
- Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, Derre A, Villeger L, Farnier M, Beucler I, Bruckert E, Chambaz J, Chanu B, Lecerf JM, Luc G, Moulin P, Weissenbach J, Prat A, Krempf M, Junien C, Seidah NG, Boileau C. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. [PubMed: 12730697]
- 48.
- Dron JS, Hegele RA. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr Opin Lipidol. 2017;28:161–169. [PubMed: 28157721]
- 49.
- Wiegman A, de Groot E, Hutten BA, Rodenburg J, Gort J, Bakker HD, Sijbrands EJ, Kastelein JJ. Arterial intima-media thickness in children heterozygous for familial hypercholesterolaemia. Lancet. 2004;363:369–370. [PubMed: 15070569]
- 50.
- Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–176. [PubMed: 8328379]
- 51.
- Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ. 1991;303:893–896. [PMC free article: PMC1671226] [PubMed: 1933004]
- 52.
- World Health Organization. Familial hypercholesterolemia—report of a second WHO Consultation. Geneva SWHO, 1999. (WHO publication no. WHO/HGN/FH/CONS/99.2).
- 53.
- Wilson DP, Jacobson TA, Jones PH, Koschinsky ML, McNeal CJ, Nordestgaard BG, Orringer CE. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. Journal of clinical lipidology. 2019;13:374–392. [PubMed: 31147269]
- 54.
- Brunzell JD, Deeb SS. Familial Lipoprotein Lipase Deficiency, Apo C-II Deficiency, and Hepatic Lipase Deficiency. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, eds. Online Metabolic and Molecular Bases of Inherited Disease: McGraw-Hill; 2006:1-60.
- 55.
- Brunzell JD. Familial lipoprotein lipase deficiency. Gene reviews [Internet] 2014.;
- 56.
- Brahm AJ, Hegele RA. Chylomicronaemia-current diagnosis and future therapies. Nat Rev Endocrinol. 2015;11:352–362. [PubMed: 25732519]
- 57.
- Peterfy M, Ben-Zeev O, Mao HZ, Weissglas-Volkov D, Aouizerat BE, Pullinger CR, Frost PH, Kane JP, Malloy MJ, Reue K, Pajukanta P, Doolittle MH. Mutations in LMF1 cause combined lipase deficiency and severe hypertriglyceridemia. Nat Genet. 2007;39:1483–1487. [PubMed: 17994020]
- 58.
- Beigneux AP, Franssen R, Bensadoun A, Gin P, Melford K, Peter J, Walzem RL, Weinstein MM, Davies BS, Kuivenhoven JA, Kastelein JJ, Fong LG, Dallinga-Thie GM, Young SG. Chylomicronemia With a Mutant GPIHBP1 (Q115P) That Cannot Bind Lipoprotein Lipase. Arterioscler Thromb Vasc Biol. 2009
- 59.
- Patni N, Li X, Adams-Huet B, Garg A. The prevalence and etiology of extreme hypertriglyceridemia in children: Data from a tertiary children's hospital. Journal of clinical lipidology. 2018;12:305–310. [PubMed: 29454679]
- 60.
- Chokshi N, Blumenschein SD, Ahmad Z, Garg A. Genotype-phenotype relationships in patients with type I hyperlipoproteinemia. Journal of clinical lipidology. 2014;8:287–295. [PubMed: 24793350]
- 61.
- Fojo SS, Brewer HB. Hypertriglyceridaemia due to genetic defects in lipoprotein lipase and apolipoprotein C-II. J Intern Med. 1992;231:669–677. [PubMed: 1619390]
- 62.
- Johansen CT, Hegele RA. The complex genetic basis of plasma triglycerides. Curr Atheroscler Rep. 2012;14:227–234. [PubMed: 22528520]
- 63.
- Peterfy M. Lipase maturation factor 1: a lipase chaperone involved in lipid metabolism. Biochim Biophys Acta. 2012;1821:790–794. [PMC free article: PMC3288453] [PubMed: 22063272]
- 64.
- Nilsson SK, Heeren J, Olivecrona G, Merkel M. Apolipoprotein A-V; a potent triglyceride reducer. Atherosclerosis. 2011;219:15–21. [PubMed: 21831376]
- 65.
- Calandra S, Priore Oliva C, Tarugi P, Bertolini S. APOA5 and triglyceride metabolism, lesson from human APOA5 deficiency. Curr Opin Lipidol. 2006;17:122–127. [PubMed: 16531747]
- 66.
- Davies BS, Beigneux AP, Barnes RH 2nd, Tu Y, Gin P, Weinstein MM, Nobumori C, Nyren R, Goldberg I, Olivecrona G, Bensadoun A, Young SG, Fong LG. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010;12:42–52. [PMC free article: PMC2913606] [PubMed: 20620994]
- 67.
- Beigneux AP, Davies BS, Gin P, Weinstein MM, Farber E, Qiao X, Peale F, Bunting S, Walzem RL, Wong JS, Blaner WS, Ding ZM, Melford K, Wongsiriroj N, Shu X, de Sauvage F, Ryan RO, Fong LG, Bensadoun A, Young SG. Glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab. 2007;5:279–291. [PMC free article: PMC1913910] [PubMed: 17403372]
- 68.
- Ahmad Z, Wilson DP. Familial chylomicronemia syndrome and response to medium-chain triglyceride therapy in an infant with novel mutations in GPIHBP1. Journal of clinical lipidology. 2014;8:635–639. [PubMed: 25499947]
- 69.
- Feoli-Fonseca JC, Levy E, Godard M, Lambert M. Familial lipoprotein lipase deficiency in infancy: clinical, biochemical, and molecular study. J Pediatr. 1998;133:417–423. [PubMed: 9738727]
- 70.
- Wilson CJ, Priore Oliva C, Maggi F, Catapano AL, Calandra S. Apolipoprotein C-II deficiency presenting as a lipid encephalopathy in infancy. Ann Neurol. 2003;53:807–810. [PubMed: 12783430]
- 71.
- Rahalkar AR, Hegele RA. Monogenic pediatric dyslipidemias: classification, genetics and clinical spectrum. Mol Genet Metab. 2008;93:282–294. [PubMed: 18023224]
- 72.
- Servaes S, Bellah R, Verma R, Pawel B. Lipoprotein lipase deficiency with visceral xanthomas. Pediatr Radiol. 2010;40:1440–1442. [PubMed: 20143060]
- 73.
- Patni N, Brothers J, Xing C, Garg A. Type 1 hyperlipoproteinemia in a child with large homozygous deletion encompassing GPIHBP1. Journal of clinical lipidology. 2016
- 74.
- Dyslipidaemia Durrington P. Lancet. 2003;362:717–731. [PubMed: 12957096]
- 75.
- Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128 Suppl 5:S213–256. [PMC free article: PMC4536582] [PubMed: 22084329]
- 76.
- Patni N, Quittner C, Garg A. Orlistat Therapy for Children With Type 1 Hyperlipoproteinemia: A Randomized Clinical Trial. J Clin Endocrinol Metab. 2018;103:2403–2407. [PMC free article: PMC6456945] [PubMed: 29659879]
- 77.
- Senior M. After Glybera's withdrawal, what's next for gene therapy? Nat Biotechnol. 2017;35:491–492. [PubMed: 28591128]
- 78.
- Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. II. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J Clin Invest. 1973;52:1544–1568. [PMC free article: PMC302426] [PubMed: 4718953]
- 79.
- Veerkamp MJ, de Graaf J, Hendriks JC, Demacker PN, Stalenhoef AF. Nomogram to diagnose familial combined hyperlipidemia on the basis of results of a 5-year follow-up study. Circulation. 2004;109:2980–2985. [PubMed: 15184285]
- 80.
- Brouwers MC, van Greevenbroek MM, Stehouwer CD, de Graaf J, Stalenhoef AF. The genetics of familial combined hyperlipidaemia. Nat Rev Endocrinol. 2012;8:352–362. [PubMed: 22330738]
- 81.
- Gaddi A, Cicero AF, Odoo FO, Poli AA, Paoletti R. Atherosclerosis, Metabolic Diseases Study G. Practical guidelines for familial combined hyperlipidemia diagnosis: an up-date. Vasc Health Risk Manag. 2007;3:877–886. [PMC free article: PMC2350131] [PubMed: 18200807]
- 82.
- Koopal C, Marais AD, Visseren FL. Familial dysbetalipoproteinemia: an underdiagnosed lipid disorder. Curr Opin Endocrinol Diabetes Obes. 2017;24:133–139. [PubMed: 28098593]
- 83.
- Sniderman A, Tremblay A, Bergeron J, Gagne C, Couture P. Diagnosis of type III hyperlipoproteinemia from plasma total cholesterol, triglyceride, and apolipoprotein B. Journal of clinical lipidology. 2007;1:256–263. [PubMed: 21291689]
- 84.
- Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11:522–534. [PMC free article: PMC7605893] [PubMed: 26239609]
- 85.
- Patni N, Li X, Adams-Huet B, Vasandani C, Gomez-Diaz RA, Garg A. Regional Body Fat Changes and Metabolic Complications in Children With Dunnigan Lipodystrophy-Causing LMNA Variants. J Clin Endocrinol Metab. 2019;104:1099–1108. [PMC free article: PMC6382455] [PubMed: 30418556]
- 86.
- Expert Panel on Integrated Guidelines for Cardiovascular H. Risk Reduction in C, Adolescents, National Heart L, Blood I. Expert panel on integrated guidelines for cardiovascular health and risk reduction in children and adolescents: summary report. Pediatrics. 2011;128 Suppl 5:S213–256. [PMC free article: PMC4536582] [PubMed: 22084329]
- 87.
- Mortality in treated heterozygous familial hypercholesterolaemia: implications for clinical management. Scientific Steering Committee on behalf of the Simon Broome Register Group. Atherosclerosis. 1999;142:105–112. [PubMed: 9920511]
- 88.
- Brett T, Qureshi N, Gidding S, Watts GF. Screening for familial hypercholesterolaemia in primary care: Time for general practice to play its part. Atherosclerosis. 2018;277:399–406. [PubMed: 30270077]
- 89.
- Vinson A, Guerra L, Hamilton L, Wilson DP. Fnla. Reverse Cascade Screening for Familial Hypercholesterolemia. J Pediatr Nurs. 2019;44:50–55. [PubMed: 30683281]
- 90.
- Landstrom AP, Chahal AA, Ackerman MJ, Cresci S, Milewicz DM, Morris AA, Sarquella-Brugada G, Semsarian C, Shah SH, Sturm AC. American Heart Association Data S, Precision Medicine Committee of the Council on G, Precision M, Council on Clinical C, Council on C, Stroke N, Council on H, Council on Lifelong Congenital Heart D, Heart Health in the Y, Council on Peripheral Vascular D, Stroke C. Interpreting Incidentally Identified Variants in Genes Associated With Heritable Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circ Genom Precis Med. 2023;16:e000092. [PubMed: 36970980]
- 91.
- Miller DT, Lee K, Abul-Husn NS, Amendola LM, Brothers K, Chung WK, Gollob MH, Gordon AS, Harrison SM, Hershberger RE, Klein TE, Richards CS, Stewart DR, Martin CL. documents@acmg.net ASFWGEa. ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2022;24:1407–1414. [PubMed: 35802134]
- 92.
- Goldman L, Goldman PA, Williams LW, Weinstein MC. Cost-effectiveness considerations in the treatment of heterozygous familial hypercholesterolemia with medications. Am J Cardiol. 1993;72:75D–79D.
- 93.
- Marks D, Wonderling D, Thorogood M, Lambert H, Humphries SE, Neil HA. Cost effectiveness analysis of different approaches of screening for familial hypercholesterolaemia. BMJ. 2002;324:1303. [PMC free article: PMC113765] [PubMed: 12039822]
- 94.
- Marang-van de Mheen PJ, ten Asbroek AH, Bonneux L, Bonsel GJ, Klazinga NS. Cost-effectiveness of a family and DNA based screening programme on familial hypercholesterolaemia in The Netherlands. Eur Heart J. 2002;23:1922–1930. [PubMed: 12473254]
- 95.
- Crosland P, Maconachie R, Buckner S, McGuire H, Humphries SE, Qureshi N. Cost-utility analysis of searching electronic health records and cascade testing to identify and diagnose familial hypercholesterolaemia in England and Wales. Atherosclerosis. 2018;275:80–87. [PMC free article: PMC6116897] [PubMed: 29879685]
- Review Update on the molecular biology of dyslipidemias.[Clin Chim Acta. 2016]Review Update on the molecular biology of dyslipidemias.Ramasamy I. Clin Chim Acta. 2016 Feb 15; 454:143-85. Epub 2015 Nov 4.
- Review Familial Hypercholesterolemia: Genes and Beyond.[Endotext. 2000]Review Familial Hypercholesterolemia: Genes and Beyond.Warden BA, Fazio S, Shapiro MD. Endotext. 2000
- Review Homozygous familial hypercholesterolaemia: update on management.[Paediatr Int Child Health. 2016]Review Homozygous familial hypercholesterolaemia: update on management.France M. Paediatr Int Child Health. 2016 Nov; 36(4):243-247.
- Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms.[Circulation. 2014]Proprotein convertase subtilisin kexin type 9 promotes intestinal overproduction of triglyceride-rich apolipoprotein B lipoproteins through both low-density lipoprotein receptor-dependent and -independent mechanisms.Rashid S, Tavori H, Brown PE, Linton MF, He J, Giunzioni I, Fazio S. Circulation. 2014 Jul 29; 130(5):431-41. Epub 2014 May 23.
- Review [Congenital disorders of lipoprotein metabolism].[Herz. 2017]Review [Congenital disorders of lipoprotein metabolism].März W, Grammer TB, Delgado G, Kleber ME. Herz. 2017 Aug; 42(5):449-458.
- Genetics and Dyslipidemia - EndotextGenetics and Dyslipidemia - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...