ABSTRACT
Understanding the involvement of the insulin-GH-IGF-axis in the different phases of human growth, development, and metabolism is the key to understanding human pathophysiology. The normal physiological actions of the axis optimize human growth and metabolism to impact adult height by approximately one third. IGF binding proteins modulate access of circulating IGF-I to the tissues and modulate IGF-I and -II access to the type 1 IGF receptor (IGF1R) at the cellular level. Complete lack of IGF1R signaling is most likely not compatible with a viable human fetus, while allelic haploinsufficiency impairs brain development and causes severe short stature. Lack of insulin receptor signaling in Leprechaunism may result in the rare event of an alive but severely small for gestational age baby that will only survive if treated with recombinant-IGF-1 to substitute inulin receptor signaling with IGF1R signaling via their common intracellular pathways. IGF-I gene defects result in mental retardation and severe fetal and postnatal growth failure with GH hypersecretion and marked insulin resistance. Likewise, IGF2 gene defects or imprinting defects cause severe fetal growth failure but somewhat less adverse effects on postnatal growth, more variable effects on brain development, and an absence of marked metabolic effects. GH fine-tunes insulin and IGF-I signaling with no impact on IGF-II expression and has a minor impact on fetal development and growth. GH effects on lipolysis are established in the newborn and ensure gluconeogenesis and prevents hypoglycemia after birth. The complete absence of GH expression such as in GHRHR or GH1 gene defects or absence of GH signaling in GHR or STAT5B gene defect leads to an adult height of 120-130cm if untreated, and has severe metabolic consequences. Even excess of insulin, GH, IGF-I and IGF-II signaling are associated with severe metabolic disease and excess growth and/or obesity. Malnutrition or malabsorption causes decreased insulin signaling which reduces GHR expression and blocks the GH signaling pathway leading to IGF-I expression (GHR uncoupling), while GH’s metabolic actions on lipolysis and gluconeogenesis are unaffected. GH signaling attenuate insulin actions on glucose metabolism which causes insulin resistance and hyperinsulinemia or may precipitate diabetes. However, insulin signaling pathways that enhances GHR function or suppress IGFBP-1 or SHBG production are still intact and promote anabolism, optimize growth, enhance androgen actions and play a mechanistic role in premature adrenarche and PCOS. Long-term nutritional deprivation compromises growth, while from a developmental perspective, decreased insulin signaling (leading to GHR uncoupling) prolongs life (at least in some experimental animal models) which ensures that fertile age is reached, and survival of the species is ensured. For the health of the general population, the subtle changes in insulin, GH and IGF-I signaling associated with gene polymorphisms or epigenetic changes programmed during fetal and early postnatal life and affecting gene expression are important. They determine growth and pubertal development in childhood and predispose the individual for developing the metabolic diseases and malignancies in adult life, as predicted by the Barker hypothesis. As the roles of the insulin-GH-IGF-axis in growth and metabolism, often discussed separately, are intimately linked they will be described jointly here. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
EARLY WORK DEFINING THE INSULIN-GH-IGF-AXIS
Daughaday realized that the mitogenic effect of GH in the growth plate was not direct but mediated by Insulin-like Growth Factor-I (IGF-I), at that time named sulphation factor or somatomedin C (1). Another effect of IGF-I was insulin-like and not inhibited by insulin specific antibodies (2,3,4) and therefore it was named non-suppressible Insulin-like activity (NSILA). Hall and Van Wyk purified IGF-I from human muscle extracts (5,6) and realized that these biological activities originated from the same molecule. They also identified significant quantities in blood (7). The primary structure of IGF-I and Insulin-like Growth Factor-II (IGF-II) was discovered by Froesch and coworkers as a result of their persistent work to characterize the metabolic activity of NSILA (8,9). Soon after, the mitogenic activity of the sulphation factor or somatomedin C as well as somatomedin A was shown to be identical to IGF-I (10). Rechler and Nissley demonstrated that IGF-II was identical to multiplication stimulating activity, a factor known to stimulate DNA synthesis in chick embryo fibroblasts (11).
The concept that binding proteins existed for peptide hormones like the IGFs, similar to those for steroid and thyroid hormones, were suggested by studies from Zapf and Froesch (12) and by Hintz (13), demonstrating that NSILA was present in high molecular weight complexes in serum. The binding was exclusive to IGFs and did not apply to insulin or proinsulin despite their structural similarities. High molecular weight IGF-I complexes with IGFBPs were GH dependent (14) and formed a ternary complex composed of IGFBP-3 (15), the Acid Labile Subunit (ALS) (16) and IGF-I or IGF-II. Low molecular weight complexes contained IGF-I or IGF-II bound to an insulin regulated liver derived protein IGFBP-1 (17, 18), at first called the 28 kDa binding protein or PP12 (19). The existence of other IGF binding proteins, six in total, became clear when Hossenloop (20) developed Western ligand blotting as a technique to quantify these proteins. The components of the IGF-IGFBP-system are outlined in Figure 1.

Figure 1.
The primary structures of IGF-I, IGF-II and insulin are similar. IGFs are produced by many differentiated cell types, and their bioactivity in the extracellular fluids or in the circulation are coordinated by six IGF binding proteins (IGFBP-1 through -6). IGFBP-3, the major binding protein in serum is stimulated by GH and it forms a large 150 kDa ternary complex with IGF-I or -II and the GH regulated acid labile subunit (ALS). IGFBP-5, an important supporter of bone tissue formation, also forms ternary complexes with IGF-I or -II and ALS. IGFBP-1, suppressed by insulin, is one of several binding proteins in the smaller 50 kDa binary complexes with IGF-I or –II. IGFBP-2 has inverse association with insulin under many physiological conditions. In contrast, IGFBP-4, -5 and -6 do not appear to be directly regulated by GH or insulin and are important local regulators of IGF activity in bone and the CNS. The type 1 IGF receptor (IGF1R) is the mediator of the mitogenic, anti-apoptotic, differentiating and metabolic effects of both IGF-I and -II. The structural similarity of the IGF1R with the insulin receptor (IR) explains the formation of hybrid receptors in cells that expresses both receptors such as myocytes and pre-adipocytes. Cross reactivity among the ligands and the receptors have been demonstrated, although it has minor importance under physiological conditions but may cause non-islet-cell tumor hypoglycemia due to unprocessed pro-IGFs with markedly decreased binding affinity to IGFBPs. A second receptor, exclusively binding IGF-II, work as a scavenger receptor and is identical to the mannose-6-phosphate receptor, known to direct proteins for degradation in the lysosomes. A second level of control of IGF bioactivity is exerted by IGFBP proteases which release IGF-I activity after fragmentation of IGFBPs. Specific production and regulation of IGFBP proteases at the tissue level controls processes such as ovulation and atherosclerosis. Furthermore, interaction of IGFBPs and IGFBP proteases with the extracellular matrix modify the binding affinity for the IGFs and are involved in prolonging the actions of IGFs at the tissue level. Extracellular matrix also signals though integrin receptors on the cell surface and modifies IGF-1R signaling. This figure also shows the existence of IGFBP-related proteins with markedly lower affinity for the IGFs and with physiological roles not related to their IGF binding.
ANIMAL EXPERIMENTS ESSENTIAL FOR THE UNDERSTANDING OF HUMAN INSULIN-GH-IGF AXIS PHYSIOLOGY
Insulin, IGF-I, and IGF-II and Their Receptors
Efstratiadis’ series of knock-outs of the insulin-GH-IGF-axis in mice in the early 1990s clearly confirmed its importance in fetal and postnatal growth and metabolism (21). It also predicted the phenotype of experiment of nature in humans with gene defects in the axis yet to be discovered. The studies opened new insights, not least the equal importance of IGF-I and IGF-II in fetal growth, reducing birth weight by about 60% in both Igf1 knock-out (Igf1ko) and Igf2ko animals and demonstrating that the previous perceived concept that there was a fetal (IGF-II) and a postnatal (IGF-I) form of IGF was incorrect. IGF-I and -II had actions through the type 1 IGF receptor (IGF1R) demonstrated by Igf1rko animals with 45% of wild type birth weight and no further effect when crossed with Igf1ko animals. While Igf1ko animals were viable depending on genetic background and were non-fertile, the Igf1rko animals died from respiratory failure but with an absence of apparent malformations. Interestingly, crossing Igf2ko with Igf1rko resulted in further growth retardation indicating that IGF-II had actions through an additional receptor. Another new insight came from knock-out of the ‘mysterious type 2 IGF receptor’, identical to the mannose-6 phosphate receptor (M6P-R), specifically binding IGF-II and involved in internalization of proteins for lysosomal degradation. Knock-out of the Igf2r/M6pr resulted in increased serum and tissue levels of IGF-II and fetal overgrowth (140% of wild-type birth weight) (22). This receptor works to clear IGF-II and its presence in endothelial cells may, at least partly, explain the lack of endocrine actions of IGF-II due to its proteolytic lysosomal degradation (23). Thus, IGF-II effects on fetal growth are paracrine/autocrine actions mediated by the IGF1R. Knock-out of the Igf2r/M6pr gene combined with Igf2ko/Igf1rko could partly rescue growth retardation, a finding that was explained by IGF-II actions via the insulin receptor (INSR). The formation of heterodimers, more commonly named hybrid receptors, between type A or B isoforms of the insulin receptors (INSRA or INSRB) and the IGF1R of which IRA-IGF1R are highly expressed in the fetus (and in malignant cells) and activated by IGF-II, may further point to the importance of IGF-II during the fetal period. INSRB-IGF1R hybrids comprises up to 30% of INSR and IGF-I receptors in muscle due to high expression of both and this hybrid predominantly responds to IGF-I (less to insulin) and explains the important role of IGF-I in growth and metabolism in skeletal muscle.
Postnatally, the Igf1ko mice continued to grow poorly, resulting in an adult weight 30% of wild-type and with poor gonadal function and delayed bone development. Knock-out of GH or its receptor (GHR), both expressed in the mouse fetus, did not affect birth size, indicating that the Igf1 gene is not under GH control during the fetal period. The actions of GH and its receptor on growth in mice were obvious from postnatal day 15 and largely slowed growth resulting in a 50% reduction of wild-type adult weight. On the other hand, double Ghrko/Igf1ko resulted in further postnatal growth retardation relative to Igf1ko mice completely obstructing further weight gain after postnatal day 15 and supporting previous studies suggesting that progenitor cells in the growth plate require direct GH actions (24).
IGFBPs and IGFBP Proteases
Like the above attempts to pinpoint the role of important ligands and receptors in the axis, steps to assess the role of IGF binding proteins involved in modulating IGF-I and IGF-II bioactivity were taken (reviewed by Pintar (25)). In contrast to the pronounced phenotypes caused by mutations in receptors and their ligands, the growth phenotypes of the various IGFBP knock-out animals were far less pronounced as were the metabolic changes observed (26,27,28). It was argued that there is a large degree of redundancy among the functions of the IGFBPs which to some extend contradicts their specialized functions in various tissues (29). However, this idea was to some extent supported by the finding of somewhat more pronounced phenotypes in double and triple knock-out animals (30). This is largely in accordance with the absence of reports of IGFBP gene defects causing growth retardation in humans. The most affected phenotype identified was that of Igfbp4ko mice who were growth retarded at birth and displayed poor postnatal growth (30). No such mutation has been identified in humans. IGFBP-4 is specifically degraded by the metalloproteinase PAPP-A (Pregnancy Associated Plasma Protease -A) produced by the placenta as well as bone and ovary. In Pappa knock-out animals a 20-30% reduction in body weight was reported (31). Interestingly, the growth restriction phenotype of mice null for Pappa could be rescued by disruption of IGF-II imprinting during embryonic development (32).
Endocrine Versus Paracrine Autocrine IGF-I
One of the controversies of the area has been the relative contribution to linear growth of circulating endocrine IGF-I largely produced by the liver versus peripherally produced IGF-I with major paracrine/autocrine actions on local tissues. The major importance of paracrine/autocrine IGF-I was demonstrated by liver specific Igf1ko mice (Ligf1ko) with largely unaffected longitudinal growth (33). Circulating levels were 20% of wild-type with compensatory elevation of GH, insulin resistance and hyperinsulinemia. With age the animals developed type 2 diabetes, underlining the metabolic consequences of largely elevated GH combined with circulating IGF-I deficiency (34). Somewhat unexpectedly, this animal model closely resembles children and adolescents with type 1 diabetes, as further elaborated on below.
Another model to assess the relative importance of endocrine versus paracrine/autocrine IGF-I is the liver-specific Ghrko mouse. It produces a similar phenotype but with more specific hepatic consequences of absent GH signaling (35).
INSULIN-GH-IGF AXIS PHYSIOLOGY, A PEDIATRIC PERSPECTIVE
Insulin-GH-IGF-Axis and Human Fetal Growth
IGF-I controls the pace of the cell cycle from early on in human embryogenesis. INSR and IGF1R is expressed in human pre-implantation blastocysts already from the 8-cell stage, while IGF-II is expressed already in the oocyte (36). After implantation, IGF-I is produced in the human embryo (37), but until then the source of IGF-I is thought to be the female reproductive tract, and it is known that the availability of the IGF-I ligand is important for blastocyst growth in human in vitro fertilization - IVF (38). IGF-I production is controlled by nutritional factors in the early embryo and even later during human fetal development (39). Circulating endocrine IGF-I increases with gestational age (40) and is strongly correlated with fetal growth in the second part of gestation (41,42). However, little has been reported concerning the regulatory control of the IGF1 gene in the human fetus. IGFBPs can be identified in the human fetal circulation (40), and recently the role of IGFBP-5 in regulating fetal growth was suggested by fetal growth retardation in the absence of a specific IGFBP-5 protease, PPAP-A2 (43). Insulin continues to be permissive for IGF-I production even after GH is established as the major pituitary stimuli controlling endocrine as well as paracrine/autocrine IGF-I, as described below.
Fetal Growth Restriction and Programming of the Insulin-GH-IGF-Axis Setpoint
Insulin resistance has been developmentally advantageous for mankind until very recent decades of excess food and sedentary life style. It was proposed by Barker et al (44) in his ‘fetal and infant origin of adult disease’ hypothesis that intrauterine restriction of growth compensated by excessive postnatal catch-up growth results in an increased risk of developing disease entities of the metabolic syndrome later in life. In his early epidemiological studies, he demonstrated that there is a U-shaped relationship between birth weight and risk of obesity, insulin resistance, type 1 diabetes, hypertension, dyslipidemia and ischemic heart disease, with lower birth weight (within the normal range) imposing a risk. Notably, at higher birth weights this risk rises again which may represent genetic risks of obesity and type 2 diabetes. The concept was that poor fetal nutrition would lower fetal IGF-I and program the child to a low IGF-I setpoint and slower postnatal growth, an epigenetic phenomenon that could be preserved over a few generations (45). At the same time, small for gestational age (SGA) babies becomes insulin resistant (46) and this trait is enforced by a low endocrine IGF-I setpoint (47), creating the best physiological circumstances for the storage of fat during short times of food availability in a world with limited access to food. However, in a world of plenty, this advantage would turn into a disadvantage and give fast increases in body weight, hyperinsulinemia, and the development of metabolic syndrome problems early on (reviewed by Dunger et al (48). New information even suggests that the parental nutritional state can impose epigenetic metabolic changes in the fetus (49).
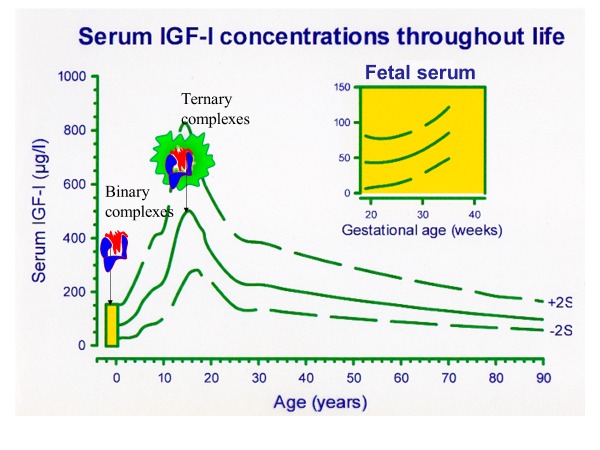
Figure 2.
In the fetus (insert) IGF-I increases with gestational age toward birth. Endocrine circulating IGF-I is strongly nutritionally dependent and correlated with birth size. Pituitary GH control of IGF-I production is not fully established during the first year of human life. The ability of serum IGF-I levels to increase during childhood is dependent on the shift from binary complexes of IGF-I with short half-life to a complete dominance of IGF-I bound in a stable ternary complex with the GH dependent proteins IGFBP-3 and ALS. Both these proteins increase, when pituitary GH control of the axis is established. During pubertal development, sex steroids change the set-point of negative IGF-I feedback and allow a peak of IGF-I in mid-puberty. Total IGF-I levels decline to low levels in senescence. Serum IGF-I reference values based on Juul (50).
Postnatal Establishment of Pituitary GH Control of IGF-I Production
In humans, full GH control of the IGFI gene, as well as the IGFBP3 and Acid Labile Subunit (ALS) genes, is developmentally regulated and established not until the first year of life. IGF-I and IGFBP-3 levels increase slowly from birth until a more rapid increase and peak during puberty, which is followed by a decline toward low levels in senescence (50, 51) (Figure 2). The late establishment of pituitary GH control of the axis is strongly supported by animal data from GHR KO mice reviewed above, and by a new model of Laron syndrome (GHR defect) in pigs (52). In accordance, newborns with mutations in the GH Releasing Hormone Receptor (GHRHR) gene resulting in an isolated GH deficiency (GHD) phenotype was associated with normal birth weight in one cohort (53) and slightly subnormal birth weight in another (54). In a subgroup of 12 children with congenital isolated GHD, birth weight (1·1 ± 0·8 SD) and length (0·5 ± 1·3 SD) was not affected (Mehta A). GH1 mutations appear to be slightly more affected with mean birth weights of −1.0 ± 0.9 (54). Studies of common polymorphisms in GH1 demonstrate dose effects of 150 and 100 grams in term newborns of normal and low birth weight, respectively (55). Somewhat contradictory to the observations in animals, Savage et al (56) reported 27 prepubertal children with severe GH insensitivity syndrome (GHIS or Severe Primary IGF Deficiency (SPIGFD)) to have a median (range) birth weight SDS of -0.72 (1.75 - (-3.29)) and birth length SDS of -1.59 (0.63 - (-3.63)). SPIGFD in these patients were defined by phenotypic and biochemical characteristics and they were treated with recombinant human (rh)IGF-1 in one of the clinical trials leading to approval of this therapy, as described later. There was no complete genetic characterization of these patients and 7 patients had a normal serum GH Binding Protein (GHBP) suggesting that the extracellular part of GHR was not affected. In a monograph, Professor Zvi Laron (57), who gave his name to this syndrome, reported that birth weight is unaffected while birth length is slightly on the shorter side. In summary, human fetal growth is only marginally affected by GH. GH is detectable and the GHR is expressed in the human fetus and the metabolic effects of GH on lipolysis are essential to maintain normal levels of glucose in the newborn.
Given this critical role of GH in adjusting metabolism to the fasting condition, it is plausible that the metabolic effects of GH are required for optimal linear growth of the human fetus and that this explains marginal effects on linear growth of the human fetus. However, strict GH control of IGF-I in the human fetus would predict severe growth retardation of the above-mentioned genetic defects comparable with the birth size observed in defects in the IGF-I gene. And that is not observed: Children with IGF1 defects suffer from far more severe fetal growth retardation with birth weight SDS around -4 and birth length SDS of -5 to -6 (reviewed in (58). In the study by Mehta et al (Mehta A), children with congenital isolated GH deficiency demonstrated growth retardation as already at 6 months of age (−2·6 ± 1·0 SD and −2·2 ± 1·3 SD in weight and length, respectively) prior to starting treatment. This suggests that the developmental GH control of IGF-I production is established early after birth. It is in accordance with data from Jensen RB and Juul A et al (Jensen RB) suggesting that low IGF-I is a marker of GH deficiency early in life. Children with combined pituitary hormone deficiencies were even more growth retarded at 6 months (Metha A).
GHR Signaling Pathway to IGF1 Gene Transcription
The important cell signaling steps associated with GH stimulated IGF1 gene activation, transcription and IGF-I production are detailed in the Endotext chapter ‘Normal Physiology of Growth Hormone in Adults´. Briefly, GH binding to preformed dimeric GHR - JAK2 complexes introduces structural changes in the receptor complex that separates JAK2 inhibitory and kinase active sites and enables trans-phosphorylation of the two JAK2 molecules (reviewed in (59). The GHR belongs to the class 1 cytokine receptors which uses STAT as one of its principal secondary messengers, and the subsequent phosphorylation of two STAT5b molecules results in a phospo-STAT5b homodimer which translocate to the cell nucleus, binds to STAT5b recognition sites on the IGF1 gene promoter, and initiates transcription (Figure 3).
Primary and Secondary IGF-I Deficiency
IGF-I deficiency may be the result of low or inadequate production of GH. This condition is known as GH deficiency (GHD) but in analogy with other pituitary deficiencies leading to peripheral hormone deficiencies the term secondary IGF-I deficiency was proposed (Rosenfeld et al). GHD is severe in GH1 or GHRH-R gene defects and in some children with congenital GHD as well as after treatment of brain tumors with radiation therapy. However, these conditions are rare and most children treated with rhGH has less severe GHD or an indication not associated with GHD. Disorders of GH in childhood is outlined in the Endotext chapter Disorders of Growth Hormone in Childhood by Murray and Clayton (132)
Primary IGF-I deficiency or GH insensitivity is severe in homozygous genetic defects in genes including the GH receptor (Laron syndrome) gene, STAT-5b gene and IGF-I gene. Less severe growth retardation is reported in children with homozygous genetic defects in the ALS gene. Treatment with rhGH do not improve growth in these cases while rhIGF-1 is efficient in SPIGFD and approved by FDA and EMA. In many patients with severe primary IGF-I deficiency (SPIGFD) defined by low IGF-I (less than – 3 SDS or the 2.5th percentile), severe short stature (Height SDS less than – 3), normal GH secretion and absence of acquired insensitivity to GH (discussed below) genetic defects may be absent. Still treatment with rhIGF-1 may be as efficient as in patients with confirmed homozygous GHR defects (131).
Insulin Enhancement of GHR Signaling to IGF1 Gene Transcription
Insulin signaling enhances the GH signaling pathway to enable IGF-I production in the fed state and promotes linear growth and other anabolic responses (60, 61, 62). Moreover, GH signaling to elicit IGF1 gene transcription is blocked in the absence of appropriate insulin signaling, a phenomenon also known as un-coupling, and resulting in growth arrest (60, 61, 63, 64) (Figure 3). This is partly a result of insulin’s effects on hepatic GHR expression, and partly a post-receptor signaling effect as unraveled by extensive animal studies (reviewed in (60). In obese individuals with hyperinsulinemia, hepatic GHR expression is enhanced as indicated by elevated GHBP levels reflecting proteolytic cleavage of highly expressed surface GHR and release of the extracellular part to the circulation (65). This allows obese individuals to maintain normal serum IGF-I levels despite markedly diminished GH secretion (66, 67). Consequently, obese individuals have attenuated GH responses to GH secretagogues (68).
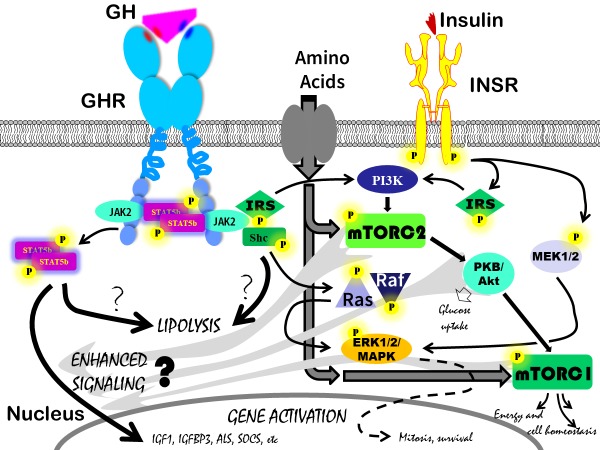
Figure 3.
Multiple, partly identical, pathways have been described to be activated by the GHR, the INSR and many other hormone kinase receptors not shown on the slide. Limiting this cartoon to the GHR and INSR, still the complexity is very high and the potential candidate hubs for crosstalk are numerous. The crosstalk that, following activation of the GHR, leads to resistance to specific signaling events from the INSR (related to glucose metabolism) is more well established and describe in detail in the Endotext chapter ‘Normal Physiology of Growth Hormone in Adults´. In the current review the focus is on the crosstalk that is executed by activation of the INSR and results in enhanced signaling from the GHR leading to gene activation of IGF1 and other GH dependent genes such as IGFBP3 and ALS. There are basically no studies addressing this crosstalk on the cellular level despite the strong evidence for INSR signaling being permissive for IGF1 transcription. Given that mTORC1 and mTORC2, downstream the INSR, are essential hubs for substrate and energy sensing and thus controlling the switch between cell anabolism and catabolism, they appear to be strong candidates to determine whether IGF1 should be on or off. A further argument for their candidate role is the so far limited evidence of mTORC1 and mTORC2 involvement in branched chain amino acid sensing directly enhancing IGF1 transcription. The unique role of the Jak2, STAT5b pathway in connecting the GHR with IGF1 gene activation has not been challenged and is thus the major candidate pathway to be affected by INSR signaling crosstalk. It is less clear which of the signaling pathways from the GHR that results in enhanced lipolysis although the STAT5b pathways has been implicated. This is particularly interesting given that GHR induced lipolysis does not require INSR signaling crosstalk. The reader is encouraged to seek specific information regarding other GHR and INSR pathways depicted in the cartoon but not further discussed in this review.
While short-term fasting decreases serum IGF-I but does not affect GHBP (69), the triad of IGF-I deficiency, poor growth and pubertal delay/arrest in long term nutritional deficiency such as in anorexia nervosa is associated with low GHBP that is partly restored with weight gain (63). Also, circulating IGF-I deficiency due to hepatic under-insulinization in type 1 diabetes is associated with low GHBP levels. Normal circulating IGF-I and GHBP are fully restored only after experimental intra-peritoneal (70) or intraportal (71) insulin delivery.
Functional/Acquired IGF-I Deficiency - Uncoupling of GHR Signaling to IGF1 Gene Transcription and Maintained GHR Metabolic Signaling Due to Insulin Deficiency
Increased GHR signaling in obese children does not generally result in elevated IGF-I, due to negative feedback inhibition of GH. In contrast, impairment of GH signaling due to insulin deficiency cannot generally be compensated by GH hypersecretion. This is true in fasting children (63) and adults (62). The exact mechanisms by which insulin and GH signaling crosstalk on the post-receptor level is not yet understood (Figure 3). More recent data suggesting that FGF21 plays a role in mediating these events needs further confirmation (72). Interestingly, GH signaling leading to activation of lipolysis in adipose tissue and increased hepatic glucose production via both glycogenolysis and gluconeogenesis in the liver, is not affected by the absence of insulin crosstalk (reviewed in (72)). This has important implications in securing substrate mobilization and gluconeogenesis during fasting and explains the cardinal hypoglycemic symptoms in GHD and GHIS newborns in the absence of intrauterine growth retardation (IUGR). GH signaling leading to lipolysis is thought to involve STAT5b. Most information comes from animal models and involves GH signaling in the liver, but in mouse adipose tissue GHR KO downregulates beta-3 adrenergic receptor expression and inhibits lipolysis (73). GH effects are lost if STAT5b signaling is blocked (74), gene transcript profiles of GHR KO and STAT5b KO animals overlap largely, and STAT5b controls key regulator enzymes involved in lipid metabolism (75). However, if STAT5b mediates both metabolic signaling and IGF-I production it still needs to be understood where the two pathways diverge, and why GH metabolic signaling is not blocked in the absence of insulin crosstalk. In humans, recent studies have identified new GH signaling responses involving GH downregulation of fat-specific protein (FSP27), a negative regulator of lipolysis. MEK/ERK activation and inhibition of peroxisome proliferator-activated receptor-γ (PPARγ) are involved, and this offers an alternative signaling pathway from the GHR (76).
Interactions Among Endocrine Axes
The activity of the insulin-GH-IGF-axis is dependent on the other endocrine axes which have permissive actions on GH stimulated IGF-I expression and affect IGFBPs and proteases (Figure 4). For example, thyroxine is needed to enhance GH effects on endocrine IGF-I expression and a normal GH-IGF-IGFBP-axis is needed for optimal thyroid hormone production (77). Sex steroids further enhance the function of the GH-IGF-axis, most likely by attenuating pituitary and hypothalamic sensitivity to IGF-I negative feedback (78). The pivotal role of sex steroids on the setpoint of the axis is reflected by the peak circulating levels of IGF-I and IGFBP-3 reached in mid-puberty (50,51). On the other hand, GH via its stimulation of local IGF-I is important for testicular production of testosterone and spermatogenesis (79), and the local IGF-IGFBP-axis is involved in selection and growth of the primary follicle in the ovary, estradiol production and ovulation (80, 81). Finally, cortisol has impact on the actions of the GH-IGF-axis on growth by blocking IGF1R signaling leading to apoptosis (82) despite normal endocrine IGF-I levels (83).

Figure 4.
Hypothalamic GH releasing hormone and somatostatin establish the pulsatile pituitary GH secretion that is established as the main regulator of endocrine and paracrine/autocrine IGF-I production during the first year of life in humans. Insulin is permissive for this regulation by modulating GHR signaling, and normal beta-cell release of insulin is required for normal liver derived endocrine IGF-I levels measured in serum (blue insert) that in most cases is a good marker of the local production and actions of IGF-I. During fasting the GH regulation of IGF-I is uncoupled, resulting in decreased IGF-I (and catabolism) and elevated GH secretion and maintained lipolytic signals securing gluconeogenesis and preventing hypoglycemia. Apart from insulin, the endocrine thyroid axis is important for normal GH induced IGF-I production and during pubertal development sex steroids from the gonads enhance the performance of the GH-IGF-axis presumable by relaxation of the negative IGF-I feedback on GH secretion allowing a higher set-point of the axis. Whether this is an estradiol effect is not fully elucidated but it is suggested by the fact that non-aromatizable androgens such as oxandrolone do not affect IGF-I levels. The actions of the adrenal axis are most likely local and involve actions on IGF1R signaling leading to apoptosis of stem cells in the growth plate and thus irreversible loss of height. Cortisol excess leaves endocrine IGF-I and GH levels largely unaffected.
Discordance Between Endocrine and Paracrine/Autocrine IGF-I
An important example of metabolic and mitogenic consequences of an unbalanced endocrine versus autocrine/paracrine insulin-GH-IGF-axis comes from observations in children and adolescents with type 1 diabetes (Figure 5). They suffer specifically from insulin deficiency in the hepatic portal circulation as a result of the subcutaneous delivery of insulin (reviewed by Dunger (64)). This attenuates their endocrine production of circulating IGF-I despite excessive GH secretion (84). Circulating IGF-I deficiency and GH hypersecretion induce insulin resistance which is further augmented by insufficient suppression of hepatic glucose output. To overcome this, higher subcutaneous insulin doses are needed to maintain glycemic control, and this results in aggravated systemic hyperinsulinemia. The importance of local tissue hyperinsulinemia and GH hypersecretion in generating high paracrine/autocrine IGF-I production and promoting mitogenic vascular events leading to diabetic long-term complications should not be underestimated. Based on this insight, a promising new drug targeting the alphaVbeta3 integrin affecting IGF-I signaling in smooth muscle cells has been found to inhibit the development of atherosclerotic lesions in diabetic pigs (85). Another consequence of a compensatory increased in local IGF-I activity is the finding of normal childhood and pubertal linear growth despite endocrine IGF-I deficiency in type 1 diabetes (86). It is interesting that the endocrine and paracrine/autocrine changes in the insulin-GH-IGF-axis observed in children with type 1 diabetes closely resembles those observed in liver IGF-I KO mice which eventually leads to diabetes in the KO mice. Given that portal delivery of insulin, which has the potential to completely restore IGF-I levels in type 1 diabetes (70, 71, 87), remains an experimental treatment, rhIGF-1 treatment to restore circulating IGF-I and suppress GH and decrease insulin needs appears to be the most feasible approach to take (88). In a 6-month clinical trial of a single daily injection of rhIGF-1 improved glycemic control in adolescents with type 1 diabetes were found (89). Long-term studies on diabetic vascular complications have yet to be performed.
If paracrine/autocrine IGF-I production is lost in addition to liver-derived IGF-I, the metabolic consequences become obvious. This situation was first reported in a boy with a deletion of exon 4 of the IGF-I gene (90) resulting in severe fetal and post-natal growth arrest, poor brain development and extreme insulin resistance with compensatory hyperinsulinemia and acanthosis nigricans. A short trial of treatment with rhIGF-1 resulted in normalization of circulating IGF-I, suppression of GH hypersecretion and a markedly decreased insulin response to a meal tolerance test (91). In this example and in type 1 diabetes, it has been discussed whether the normalization of glucose metabolism following rhIGF-I therapy is most importantly associated with insulin-like effects of IGF-I on glucose uptake in muscle or suppression of GH hypersecretion? Although most studies support the importance of GH suppression, prolonged actions of IGF-I similar to that of long-acting insulin analogs in type 1 diabetic patients are important. IGF-I is equipotent with insulin in stimulating glucose uptake in human muscle but has less effects in fat and liver (92). Reports that newborns with a complete lack of insulin effects due to inactivating defects in the INSR gene can now survive for extended time into adolescence when treated with recombinant IGF-I, that stimulate glucose uptake via the IGF1R sharing common signaling pathways with the INSR, support an important direct role of IGF-1 signaling on metabolism (93).

Figure 5.
Changes in liver derived endocrine IGF-I measured in the circulation and paracrine/autocrine IGF-I are in most cases concordant. In the absence of practical and validated methods to measure IGF-I at the tissue site of action, paracrine/autocrine IGF-I activity is assessed by determining known physiological actions of IGF-I such as growth or glucose metabolism. Type 1 diabetes is a condition with discordant changes in endocrine vs. paracrine/autocrine changes in IGF-I that in many ways resembles those reported in a mouse model of conditional knock-out of IGF-1 expression in the liver. In type 1 diabetes, insulin deficiency in the liver, caused by a systemic rather than a portal insulin replacement therapy, results in a functional GHR signaling defect to IGF-I transcription (uncoupling). Low endocrine IGF-I production decreases circulating IGF-I and results in decreased negative pituitary feedback and GH hypersecretion. The lack of direct IGF-I effects on glucose uptake in muscle and the diabetogenic effects of GH (including maintained signaling to lipolysis) decreases insulin actions on glucose metabolism (known as insulin resistance). The portal insulin deficiency also fails to suppress hepatic glucose production. In other to maintain glycemic control, the increased insulin requirement can only be met by more subcutaneous insulin leading to systemic hyperinsulinemia. There is no direct information about local paracrine/autocrine IGF-I activity, but there are several indications that tissue hyperinsulinemia and GH hypersecretion results in a compensatory increase of tissue IGF-I activity. Firstly, linear growth is not impaired in children and adolescents with Type 1 Diabetes despite of their low endocrine IGF-I (comparable to levels in short stature children), indicating a compensatory upregulation of local IGF-I activity (IGF-I being the most important stimulator of longitudinal growth). Secondly, it is plausible that increased local IGF-I activity contributes to diabetes complications known to be tightly associated with increased rather than decreased IGF-I activity. While type 1 diabetes is not generally associated with increased risk of cancer, the increase in local production of IGF-I in obesity and type 2 diabetes may contribute.
Liver Disease and Endocrine Versus Paracrine/Autocrine IGF-1 Production
In children with severe liver disease, there may be similar discrepancies between circulating endocrine levels of IGF-I and IGF-I activity in peripheral levels contributed by paracrine/autocrine secretion of IGF-I (94). However, less is known about peripheral IGF-I activity and it is possible that there are more secondary metabolic and nutritional issues that could lower local IGF-I production and impact on linear growth. Particularly in liver cirrhosis associated with thalassemia major, which concomitantly can impair pituitary GH secretion, there is no secondary upregulation of local IGF-I and linear growth failure is common (95). Recently, increased IGF-I expression was reported in obese children with non-alcoholic fatty liver disease (NAFLD) and it was combined with upregulation of IGF1R (96), not expressed in the normal liver but involved in liver repair, such as after liver resection in a mouse model (97).
GH and Cytokine Crosstalk
STAT5b phosphorylation is also mediated by activation of other members of the cytokine receptor family and has an impact on immunological function: this is evident from the finding of immune deficient symptoms in children with STAT5b genetic defects (98) but these are not found in GHD and GHIS children.
Negative Control of GHR Signaling
Control of the GH signaling cascade is also under inhibitory control, principally by two mechanisms. Firstly, tyrosine phosphatases including PTP dephosphorylate GHR associated molecules. In Noonan’s syndrome genetic defects in the PTPN11 gene may affect this pathway and has been implicated in poor growth and poor response to GH therapy, although reports are conflicting (99, 100). In addition, the SOCS gene is activated by GH signaling and works as a short intracellular negative feedback loop which rapidly down-regulate GHR activity by internalization and receptor ubiquitinoylation resulting in lysosomal and proteasomal degradation (101).
Other Nutritional Signals to the GH-IGF-Axis
Nutritional supplementation increasing dietary protein intake from cow’s milk increases endocrine IGF-I, while an equal intake of animal protein from meat does not (102). It is possible that the amino-acid composition that differs depending on the dietary source may contribute, and there are other constituents in skimmed milk not present in meat. More likely, however, it is explained by a higher carbohydrate intake in the milk group (while fat content was higher with meat supplementation) and the finding that fasting insulin levels doubled (103), in accordance with the essential regulatory role of insulin on GHR signaling discussed above. There is, however, direct evidence for a regulatory role of amino-acids on IGF-I production that is independent of insulin. Branched chained amino-acids (BCAA) are known to stimulate cell growth by the activation of mTORC2, a protein complex that controls protein synthesis in cells by sensing nutrient and energy availability and is also one of the main signaling pathways of IGF-I and insulin (Figure 3). The role of BCAA has been studied in rats given a restricted diet containing high levels of BCAA, compared to a group given low levels of BCAA (104).
The availability of nutrients in the circulation might also have a direct effect on the production of IGF-I. Human breast adipocytes cultured in high glucose levels have been found to produce more IGF-I compared to adipocytes cultured in low glucose levels (105).
IGF-II
IGF-II is a paracrine/autocrine hormone which is as essential for fetal growth as IGF-I (21). IGF-II may also contribute considerable to postnatal growth although the absence of endocrine effects makes it difficult to study in humans. Less is known about the metabolic actions of IGF-II. Growth promoting actions of IGF-II is via the IGF1R and IGF1R-INSR hybrid receptors which has preference for IGF-II actions if the A isoform of the INSR receptor – expressed in the fetus and in malignantly transformed cells - pairs with the IGF1R in the hybrid (106). The human IGF2 gene is an imprinted gene (107) exclusively expressed from the paternal allele in certain tissues (reviewed by Rossignol (108). The imprinted promoter region is found in a complex configuration with the H19 gene on chromosome 11 and shares two important imprinting regions with this gene. Methylation of the imprinting region ICR1 results in expression of the paternal IGF2 allele, while H19 gene expression is suppressed. Correct imprinting should lead to expression of the paternal allele only and sufficient expression of IGF-II for normal growth. Loss of methylation of the ICR1 on the paternal allele results in the phenotype of Silver Russell syndrome (SRS) which may also arise from other genetic aberrations that have not yet been linked to the IGF-II production or signaling cascade including maternal uniparental dyssomnia of chromosome 7.
SRS is characterized by proportional IUGR with severe SGA at birth, relative sparing of the brain with close to normal head circumference at birth, severe feeding difficulties in infancy and childhood (which in contrast to Prader-Willi syndrome does not rebound into feeding obsession later in life), postnatal growth retardation and body asymmetry. As indicated by the SRS phenotype, IGF2 gene transcription of some organs are not controlled by imprinting. The relative normal development of the brain and the relative macrocephaly of SRS is explained by the lack of imprinting control of IGF-II expression in the brain. Children with genetic mutations in the expressed paternal allele of the IGF2 gene, were reported to have an SRS phenotype. They had somewhat more pronounced psychomotor developmental problems compared with the SRS phenotype, which has increased risk of autism spectrum defects including attention deficit hyperactivity disorder. In SRS, there is a normal postnatal expression of the IGF2 gene in the liver leading to normal levels of circulating IGF-II. Interestingly, the normal endocrine levels of IGF-II do not overcome the postnatal growth restriction. A similar lack of endocrine IGF-II effects on growth and metabolism was reported in the IGF-I deficient child mentioned previously with a loss of exon 4 of the IGF1 gene. He had compensatory increased GH, IGFBP-3, ALS as a result of lack of negative IGF-I feedback and secondary to the increased IGF binding capacity, increased IGF-II (see also the chapter on IGF-binding proteins below). Another observation in favor of this view is the lack of a correlation between newborn cord levels of IGF-II and birth size (41). This contrasts with a strong positive correlation between cord blood IGF-I concentrations and birth size. The role of IGF-II should be viewed in the light of Efstradiadis series of knock-out experiments in mice (21) where the Igf2ko mice had the same degree of fetal growth retardation as the igf1ko which demonstrates that IGF-II is a paracrine/autocrine and not an endocrine hormone.
It is possible that circulating IGF-II after release from the ternary complex is cleared from the circulation by binding to the IGF2R – identical to the mannose-6-phosphate receptor – which is associated with lysosomes and results in degradation of IGF-II in endothelial cells (23). In the elegant mouse KO experiments by Efstradiadis et al (21), KO of the IGF2R resulted in fetal overgrowth. However, the largely elevated IGF-II serum levels in that model are more likely a secondary finding, while the lack of clearance of paracrine/autocrine IGF-II is the explanation for the excessive growth.
IGF Binding Proteins
Six IGFBPs bind IGF-I and IGF-II inside and outside the circulation and has impact on IGF bioactivity (reviewed by Clemmons (109). The IGFBP-related proteins share some structural similarities with the six IGFBPs but have no relevant impact on IGF bioactivity. GF-I passes the endothelium intact primarily via IGF1R mediated transcytosis and this process is essential for endocrine actions of liver derived IGF-I (110). Limited experimental evidence from animal and tissue cultures suggest that IGF-I complexed with IGFBP-1 and -2 may leave the circulation, although the extent and importance is unclear (Bar 1990). After endothelial passage IGF-I redistributes to soluble IGFBPs in the extravascular fluids or IGFBPs bound on extracellular matrix or cell surfaces (111). The concentration of unbound IGF-I in the circulation is likely to be proportional to unbound IGF-I concentrations in the tissues, but they are not equal and may have different relationship in different target tissues with differentially-expressed IGFBPs.
As pointed out earlier in this review, transgenic animal studies disrupting one or more IGFBPs have not suggested that a marked growth phenotype should be expected in children and no IGFBP mutations causing growth retardation in children had been reported to the best of my knowledge. Interestingly, as predicted by the Igfbp4-ko and the pappa-ko animal models described previously, a human PAPP-A2 gene defect with growth phenotype was recently reported as detailed below (43).
Free IGF-I Assays
Assays claiming to measure free circulating IGF-I have been developed, but it is unclear to what extent different techniques are influenced by redistribution of IGF-I among IGFBPs associated with the assay procedure (112). Anyway, the fact that IGF-I redistribute among extravascular IGFBPs after passing the endothelium is likely to affect the local tissue bioavailability even more. Moreover, the fact that most data in the literature originate from one assay technique established in one single laboratory has resulted in a lack of confirmatory reports. In a few cases, measurements with different free IGF-I assays have been reported from the same study/experiment with large differences in results (113). The bottom line is that measurements of free IGF-I have not been demonstrated to better predict different physiological or pathophysiological conditions in humans and do therefore not provide any clinically important contributions (114, 112). Techniques to assess IGF-I at the tissue site of action pose practical and methodological challenges. Attempts to establish and validate a method to determine local tissue levels by microdialysis have been reported in adolescents with type 1 diabetes, where endocrine levels are a poor marker of local IGF-I activity (115).
Ternary Complex Formation
The developmental establishment of GH control over the IGF1, IGFBP3 and IGFALS genes in early childhood initiates the dominance of the ternary complex formed by IGF-I or IGF-II and IGFBP-3 (or IGFBP-5) and ALS as the quantitatively most important circulating form of IGF-I and IGF-II (reviewed by Baxter (116). In the human fetus and newborn, serum IGFBP-3 and ALS concentrations are low and ternary complex formation is absent (117). Although IGF2 gene expression is not under GH control, the circulating levels of IGF-II are largely influenced by GH status since IGF-II (as well as IGF-I and IGFBP-3) is rapidly cleared from the circulation if not bound in the ternary complex. This can be observed in children with SPIGFD, who are deficient in IGF-I as well as IGFBP-3 and ALS, and in whom sc injected rhIGF-I displays a very fast serum clearance rate (118). As mentioned, formation of the ternary complex also governs the circulating levels of IGFBP-3 which under physiological conditions is present in a 1:1 molar relationship with IGF-I plus IGF-II. ALS is a large glucoprotein that under physiological conditions are present in a two-fold molar excess (16).
Immunometric IGFBP-3 assays have been claimed to be more predictive of GH status in very young children; however, the support for that is weak. It is rather a misconception related to problems of commercial IGF-I assays at the lower end of IGF-I detection. Moreover, IGFBP-3 has been claimed to provide information about IGF-I bioavailability from calculating the molar ratio of total IGF-I to IGFBP-3. Given that both IGF-I and IGFBP-3 are rapidly cleared from the circulation if unbound, using the IGF-I/IGFBP-3 ratio and disregarding IGF-II concentrations (that are 2-3-fold those of IGF-I on a molar basis) does not make any sense. During puberty, for example, IGF-I bioactivity is increased (114). This is dependent on the 3-4-fold increase in total IGF-I (50), which consequently results in an increase in unbound IGF-I, even if the increase is matched with the same absolute molar increase in IGFBP-3 (and complexed with ALS in a ternary complex). A common view is that increased IGF-I bioactivity depends on a higher IGF-I/IGFBP-3 molar ratio during puberty. However, the increase in molar ratio is entirely explained by the fact that IGF-I and IGFBP-3 increase with the same number of moles per liter, but with a larger relative increase in IGF-I than IGFBP-3 and with IGF-II molar concentrations being unchanged (112).
IGFBP Proteolysis and Physiological Consequences
The fact that proteolytic cleavage of IGFBP-3 is common, and may result in falsely elevated IGFBP-3 immunoactivity, is the most likely reason for observing a low IGF-I/ IGFBP-3 ratio. Under certain physiological conditions first described in pregnancy (119, 120), specific proteases cleave IGFBP-3 into several proteolytic fragments of which each may retain immunoactivity and thus give rise to signals in an immunometric assay (121). This will lead to overestimations of the IGFBP-3 immunoreactivity in pregnancy, which is already truly increased due to increased placental GH tonus. It may also lead to the erroneous conclusion that IGF-I bioactivity is decreased. On the contrary, IGF-I bioactivity is increased in the maternal circulation resulting from increased total serum IGF-I and decreased binding affinity of fragmented IGFBP-3 (122). There is strong experimental evidence that IGFBP proteolysis results in lower IGF binding affinity. The finding that partial IGFBP-3 proteolysis, such as in pregnancy, does not disrupt the ternary complex, has questioned its significance. However, evidence for increased IGF-I bioactivity in a ternary complex with fragmented IGFBP-3 exists (123). IGFBP-3 proteolysis has also been described in insulin resistant states such as fasting, obesity and type 1 and 2 diabetes (62, 124, 125). While several known proteolytic enzymes such as those involved in blood clotting (126) and cancer metastasis (127, 128) have been identified as IGFBP-3 proteases, the identity of the pregnancy protease is still not resolved.
Recently, a human gene mutation of PAPP-A2, a circulating and tissue protease with IGFBP-5 (and to some extent IGFBP-3) as its primary substrate (Gaudamauskes et al), was demonstrated to have a marked growth phenotype involving fetal and post-natal growth retardation in children in a consanguineous family (43). Largely elevated levels of circulating IGF-I but as a result of absence of proteolysis of IGFBP-5 and IGFBP-3, necessary for disruption of ternary complex formation, IGF-I bioactivity in serum is low and presumably tissue bioactivity of IGF-I (and IGF-II) is low. Functionally, this is a state of severe primary IGF-I deficiency (despite of elevated total serum IGF-I) and pharmacokinetic studies suggested that sc. Injections of rhIGF-1 resulted in a fraction of unbound IGF-I in serum despite the impaired proteolysis of IGFBP-5 and IGFBP-3 (133). Attempts to improve linear growth by rhIGF-I treatment has been reported to result in some improvements in a few affected children but not all (134, 135)
It is beyond the scope of this chapter to review the overwhelming evidence from cell biology experiments demonstrating the important role of IGFBPs in modulating IGF bioactivity and the role of IGFBP proteases and their actions at the cellular level. Furthermore, IGFBPs other than IGFBP-3 may play a role in the access of IGF-I to various tissues (129).
SUMMARY
In the present review the pivotal role of nutrition and insulin in determining the regulation and actions of the GH-IGF-axis is reviewed. For the pediatrician, caring for patients in a phase of rapid growth and development, it is important to refer to normality and understand the requirements for a normal insulin-GH-IGF-axis in order to succeed in this task. In the complex work-up, treatment and management of growth disorders a thorough understanding of the normal physiology of the axis is essential in taking the right actions (130, 131). From the normal physiology of this axis, it is possible to understand the consequences of various genetic defects and disorders that affect its regulation and function. The most severe conditions associated with defects in the axis may cause a loss of adult height of approximately 1/3 and may cause severe developmental and neurological deficits and compromise pubertal maturation and fertility. Minor changes in the setpoint of the axis caused by programming of the fetus exposed to intra-uterine growth retardation may predispose the individual for poor linear growth and later metabolic disease, insights that the pediatrician should be aware of and consider in order to improve health and prevent later disease.
REFERENCES
- 1.
- Salmon WD, Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957;49:825–826. [PubMed: 13429201]
- 2.
- Leonards JR. Insulin-like activity of blood, what is it? Federation Proc. 1959;18:272.
- 3.
- Froesch ER, Bürgi WA, Ramseier EB, Bally P, Labhart A. Antibody-suppressible and nonsuppressible insulin-like activities in human serum and their physiological significance. An insulin assay with adipose tissue of increased precision and specificity. J Clin Invest 1963;42:1816– 1834. [PMC free article: PMC289464] [PubMed: 14083170]
- 4.
- Froesch ER, Muller WA, Bürgi H, Waldvogel M, Labhart A. Non-suppressible insulin-like activity of human serum. II. Biological properties of plasma extracts with non-suppressible insulin-like activity. Biochim Biophys Acta. 1966;121:360–374. [PubMed: 5962523]
- 5.
- Van Wyk JJ, Hall K, Van den Brande JL, Weaver RP. Further purification and characterization of sulfation factor and thymidine factor from acromegalic plasma. J Clin Endocrinol Metab. 1971;32:389–403. [PubMed: 5541346]
- 6.
- Van Wyk JJ, Hall K, Weaver RP. Partial purification of sulphation factor and thymidine factor from plasma. Biochim Biophys Acta 1969;192:560–562. [PubMed: 5368262]
- 7.
- Uthne K. Human Somatomedin. Purification and some studies on their biological actions. Thesis, Stockholm, 1973. [PubMed: 4542208]
- 8.
- Rinderknecht E, Humbel RE. The amino acid sequence of human insulin-like growth factor I and its structural homology with proinsulin. J Biol Chem. 1978;253:2769–2776. [PubMed: 632300]
- 9.
- Rinderknecht E, Humbel RE. Primary structure of human insulin-like growth factor II. Febs Letters. 1978;89:283–286. [PubMed: 658418]
- 10.
- Klapper DG, Svoboda ME, Van Wyk JJ. Sequence analysis of somatomedin-C: confirmation of identity with insulin-like growth factor I. Endocrinology. 1983;112:2215–2217. [PubMed: 6343062]
- 11.
- Moses AC, Nissley SP, Short PA, Rechler MM, Podskalny JM. Purification and characterization of multiplication stimulating activity. Insulin- like growth factors purified from rat-liver- cell-conditioned medium. Eur J Biochem. 1980;103:387–400. [PubMed: 6153979]
- 12.
- Zapf J, Waldvogel M, Froesch ER. Binding of nonsuppressible insulinlike activity to human serum: evidence for a carrier protein. Arch Biochem Biophys. 1975;169:638–645. [PubMed: 237487]
- 13.
- Hintz RL, Liu F. Demonstration of specific plasma protein binding sites for somatomedin. J Clin Endocrinol Metab. 1977;45:988– 495. [PubMed: 925145]
- 14.
- Hintz RL, Liu F. Demonstration of specific plasma protein binding sites for somatomedin. J Clin Endocrinol Metab. 1977;45:988– 495. [PubMed: 925145]
- 15.
- Martin JL, Baxter RC. Insulin-like growth factor-binding protein from human plasma. Purification and characterization. J Biol Chem. 1986;261:8754–8760. [PubMed: 3722172]
- 16.
- Baxter RC. Characterization of the acid-labile subunit of the growth hormone-dependent insulin-like growth factor binding protein complex. J Clin Endocrinol Metab. 1988;67:265–272. [PubMed: 2455727]
- 17.
- Póvoa G, Enberg G, Jörnvall H, Hall K. Isolation and characterization of a somatomedin-binding protein from mid-term human amniotic fluid. Eur J Biochem. 1984;144:199-204. [PubMed: 6208022]
- 18.
- Póvoa G, Roovete A, Hall K. Cross-reaction of serum somatomedin-binding protein in a radioimmunoassay developed for somatomedin-binding protein isolated from human amniotic fluid. Acta Endocrinol (Copenh). 1984;107:563-70. [PubMed: 6083690]
- 19.
- Suikkari AM, Koivisto VA, Rutanen EM, Yki Jarvinen H, Karonen SL, Seppala M. Insulin regulates the serum levels of low molecular weight insulin-like growth factor-binding protein. J Clin Endocrinol Metab. 1988;66:266–272. [PubMed: 2448329]
- 20.
- Hossenlopp P, Seurin D, Segovia B, Quinson B, Hardouin S, Binoux M. Analysis of serum insulin-like growth factor binding proteins using Western competitive binding studies. Anal Biochem 1986;154:138–143. [PubMed: 2422981]
- 21.
- Efstratiadis A. Genetics of mouse growth. Int J Dev Biol. 1998;42:955-976. [PubMed: 9853827]
- 22.
- Ludwig T, Eggenschwiler J, Fisher P, D'Ercole AJ, Davenport ML, Efstratiadis A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev Biol. 1996;177:517-535. [PubMed: 8806828]
- 23.
- Hachiya HL, Carpentier JL, King GL. Comparative studies on insulin-like growth factor II and insulin processing by vascular endothelial cells. Diabetes. 1986;35:1065-1072. [PubMed: 2944781]
- 24.
- Lindahl A, Isgaard J, Carlsson L, Isaksson OG. Differential effects of growth hormone and insulin-like growth factor I on colony formation of epiphyseal chondrocytes in suspension culture in rats of different ages. Endocrinology. 1987;121:1061-1069. [PubMed: 3622375]
- 25.
- Pintar JE, Schuller A, Cerro JA, Czick M, Grewal A, Green B. Genetic ablation of IGFBP-2 suggests functional redundancy in the IGFBP family. Prog Growth Factor Res. 1995;6:437-45. [PubMed: 8817688]
- 26.
- Wood TL, Rogler LE, Czick ME, Schuller AG, Pintar JE. Selective alterations in organ sizes in mice with a targeted disruption of the insulin-like growth factor binding protein-2 gene. Mol Endocrinol. 2000;14:1472-82. [PubMed: 10976924]
- 27.
- Murali SG, Liu X, Nelson DW, Hull AK, Grahn M, Clayton MK, Pintar JE, Ney DM. Intestinotrophic effects of exogenous IGF-I are not diminished in IGF binding protein-5 knockout mice. Am J Physiol Regul Integr Comp Physiol. 2007;292:R2144-2150. [PubMed: 17332154]
- 28.
- Ning Y, Hoang B, Schuller AG, Cominski TP, Hsu MS, Wood TL, Pintar JE. Delayed mammary gland involution in mice with mutation of the insulin-like growth factor binding protein 5 gene. Endocrinology. 2007;148:2138-2147. [PubMed: 17255210]
- 29.
- Pintar JE, Cerro JA, Wood TL. Genetic approaches to the function of insulin-like growth factor-binding proteins during rodent development. Horm Res. 1996;45:172-177. [PubMed: 8964578]
- 30.
- Ning Y, Schuller AG, Bradshaw S, Rotwein P, Ludwig T, Frystyk J, Pintar JE. Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol Endocrinol. 2006;20:2173-2186. [PubMed: 16675541]
- 31.
- Conover CA, Bale LK, Overgaard MT, Johnstone EW, Laursen UH, Füchtbauer EM, Oxvig C, van Deursen J. Metalloproteinase pregnancy-associated plasma protein A is a critical growth regulatory factor during fetal development. Development. 2004;131:1187-1194. [PubMed: 14973274]
- 32.
- Bale LK, Conover CA. Disruption of insulin-like growth factor-II imprinting during embryonic development rescues the dwarf phenotype of mice null for pregnancy-associated plasma protein-A. J Endocrinol. 2005;186:325-331. [PubMed: 16079258]
- 33.
- Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A. 1999;96:7324-7329. [PMC free article: PMC22084] [PubMed: 10377413]
- 34.
- Yu R, Yakar S, Liu YL, Lu Y, LeRoith D, Miao D, Liu JL. Liver-specific IGF-I gene deficient mice exhibit accelerated diabetes in response to streptozotocin, associated with early onset of insulin resistance. Mol Cell Endocrinol. 2003;204:31-42. [PubMed: 12850279]
- 35.
- Fan Y, Menon RK, Cohen P, Hwang D, Clemens T, DiGirolamo DJ, Kopchick JJ, Le Roith D, Trucco M, Sperling MA. Liver-specific deletion of the growth hormone receptor reveals essential role of growth hormone signaling in hepatic lipid metabolism. J Biol Chem. 2009;284:19937-19944. [PMC free article: PMC2740419] [PubMed: 19460757]
- 36.
- Lighten AD, Hardy K, Winston RM, Moore GE. Expression of mRNA for the insulin-like growth factors and their receptors in human preimplantation embryos. Mol Reprod Dev. 1997;47:134-9. [PubMed: 9136113]
- 37.
- Han VK, D'Ercole AJ, Lund PK. Cellular localization of somatomedin (insulin-like growth factor) messenger RNA in the human fetus. Science. 1987;236:193-197. [PubMed: 3563497]
- 38.
- Yu Y, Yan J, Li M, Yan L, Zhao Y, Lian Y, Li R, Liu P, Qiao J. Effects of combined epidermal growth factor, brain-derived neurotrophic factor and insulin-like growth factor-1 on human oocyte maturation and early fertilized and cloned embryo development. Hum Reprod. 2012;27:2146-59. [PubMed: 22532606]
- 39.
- Rifas-Shiman SL, Fleisch A, Hivert MF, Mantzoros C, Gillman MW, Oken E, Lighten AD. First and second trimester gestational weight gains are most strongly associated with cord blood levels of hormones at delivery important for glycemic control and somatic growth. Metabolism. 2017;69:112-119. [PMC free article: PMC5354298] [PubMed: 28285640]
- 40.
- Bang P, Westgren M, Schwander J, Blum WF, Rosenfeld RG, Stangenberg M. Ontogeny of insulin-like growth factor-binding protein-1, -2, and -3: quantitative measurements by radioimmunoassay in human fetal serum. Pediatr Res. 1994;36:528-536. [PubMed: 7529395]
- 41.
- Lassarre C, Hardouin S, Daffos F, Forestier F, Frankenne F, Binoux M. Serum insulin-like growth factors and insulin-like growth factor binding proteins in the human fetus. Relationships with growth in normal subjects and in subjects with intrauterine growth retardation. Pediatr Res. 1991;29:219-225. [PubMed: 1709729]
- 42.
- Östlund E, Bang P, Hagenäs L, Fried G. Insulin-like growth factor I in fetal serum obtained by cordocentesis is correlated with intrauterine growth retardation. Hum Reprod. 1997;12:840-844. [PubMed: 9159453]
- 43.
- Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, Desikan V, Pozo J, Muzumdar R, Martos-Moreno GÁ, Hawkins F, Jasper HG, Conover CA, Frystyk J, Yakar S, Hwa V, Chowen JA, Oxvig C, Rosenfeld RG, Pérez-Jurado LA, Argente J. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016;8:363-374. [PMC free article: PMC4818753] [PubMed: 26902202]
- 44.
- Barker DJ, Osmond C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet. 1986;1:1077-1081. [PubMed: 2871345]
- 45.
- Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, Slieker RC, Stok AP, Thijssen PE, Müller F, van Zwet EW, Bock C, Meissner A, Lumey LH, Eline Slagboom P, Heijmans BT. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592. [PMC free article: PMC4246417] [PubMed: 25424739]
- 46.
- Veening MA, Van Weissenbruch MM, Delemarre-Van De Waal HA. Glucose tolerance, insulin sensitivity, and insulin secretion in children born small for gestational age. J Clin Endocrinol Metab. 2002;87:4657-4661. [PubMed: 12364453]
- 47.
- Sandhu MS, Heald AH, Gibson JM, Cruickshank JK, Dunger DB, Wareham NJ. Circulating concentrations of insulin-like growth factor-I and development of glucose intolerance: a prospective observational study. Lancet. 2002;359:1740-1745. [PubMed: 12049864]
- 48.
- Dunger D, Yuen K, Ong K. Insulin-like growth factor I and impaired glucose tolerance. Horm Res. 2004;62 Suppl 1:101-107. [PubMed: 15761241]
- 49.
- Öst A, Lempradl A, Casas E, Weigert M, Tiko T, Deniz M, Pantano L, Boenisch U, Itskov PM, Stoeckius M, Ruf M, Rajewsky N, Reuter G, Iovino N, Ribeiro C, Alenius M, Heyne S, Vavouri T, Pospisilik JA. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell. 2014;159:1352-1364. [PubMed: 25480298]
- 50.
- Juul A, Bang P, Hertel NT, Main K, Dalgaard P, Jørgensen K, Müller J, Hall K, Skakkebaek NE. Serum insulin-like growth factor-I in 1030 healthy children, adolescents, and adults: relation to age, sex, stage of puberty, testicular size, and body mass index. J Clin Endocrinol Metab. 1994;78:744-752. [PubMed: 8126152]
- 51.
- Juul A, Dalgaard P, Blum WF, Bang P, Hall K, Michaelsen KF, Müller J, Skakkebaek NE. Serum levels of insulin-like growth factor (IGF)-binding protein-3 (IGFBP-3) in healthy infants, children, and adolescents: the relation to IGF-I, IGF-II, IGFBP-1, IGFBP-2, age, sex, body mass index, and pubertal maturation. J Clin Endocrinol Metab. 1995;80:2534-42. [PubMed: 7543116]
- 52.
- Hinrichs A, Kessler B, Kurome M, Blutke A, Kemter E, Bernau M, Scholz AM, Rathkolb B, Renner S, Bultmann S, Leonhardt H, de Angelis MH, Nagashima H, Hoeflich A, Blum WF, Bidlingmaier M, Wanke R, Dahlhoff M, Wolf E. Growth hormone receptor-deficient pigs resemble the pathophysiology of human Laron syndrome and reveal altered activation of signaling cascades in the liver. Mol Metab. 2018;11:113-128 [PMC free article: PMC6001387] [PubMed: 29678421]
- 53.
- Alba M, Salvatori R. Familial Growth Hormone Deficiency and Mutations in the GHRH Receptor Gene. Vitam Horm. 2004;69:209-220. [PubMed: 15196883]
- 54.
- Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, Aylwin S, Crowne EC, Christesen HT, Hertel NT, Trainer PJ, Savage MO, Raza J, Banerjee K, Sinha SK, Ten S, Mushtaq T, Brauner R, Cheetham TD, Hindmarsh PC, Mullis PE, Dattani MT. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009;94:3191-3199. [PubMed: 19567534]
- 55.
- The role of growth hormone in determining birth size and early postnatal growth, using congenital growth hormone deficiency (GHD) as a model. Mehta A, Hindmarsh PC, Stanhope RG, Turton JP, Cole TJ, Preece MA, Dattani MT.Clin Endocrinol (Oxf). 2005 Aug;63(2):223-31. doi: .10.1111/j.1365-2265.2005.02330.x [PubMed: 16060918] [CrossRef]
- 56.
- Savage MO, Blum WF, Ranke MB, Postel-Vinay MC, Cotterill AM, Hall K, Chatelain PG, Preece MA, Rosenfeld RG. Clinical features and endocrine status in patients with growth hormone insensitivity (Laron syndrome). J Clin Endocrinol Metab. 1993;77:1465-1471. [PubMed: 7505286]
- 57.
- Laron Z. Lessons from 50 years of study of Laron Syndrome. Endocr Pract. 2015;21:1395-1402. Jensen RB, Jeppesen KA, Vielwerth S, Michaelsen KM, Main KM, Skakkebaek NE, Juul A. Insulin-like growth factor I (IGF-I) and IGF-binding protein 3 as diagnostic markers of growth hormone deficiency in infancy Horm Res 2005;63(1):15-21. doi: . Epub 2004 Nov 30.10.1159/000082456 [PubMed: 15583470] [CrossRef]
- 58.
- Waters MJ, Hoang HN, Fairlie DP, Pelekanos RA, Brown RJ. New insights into growth hormone action. J Mol Endocrinol. 2006;36:1-7. [PubMed: 16461922]
- 59.
- Maes M, Maiter D, Thissen JP, Underwood LE, Ketelslegers JM. Contributions of growth hormone receptor and postreceptor defects to growth hormone resistance in malnutrition. Trends Endocrinol Metab. 1991;2:92-97. [PubMed: 18411172]
- 60.
- Argente J, Caballo N, Barrios V, Pozo J, Muñoz MT, Chowen JA, Hernández M. Multiple endocrine abnormalities of the growth hormone and insulin-like growth factor axis in prepubertal children with exogenous obesity: effect of short- and long-term weight reduction. J Clin Endocrinol Metab. 1997;82:2076-2083. [PubMed: 9215275]
- 61.
- Bang P, Brismar K, Rosenfeld RG, Hall K. Fasting affects serum insulin-like growth factors (IGFs) and IGF-binding proteins differently in patients with noninsulin-dependent diabetes mellitus versus healthy nonobese and obese subjects. J Clin Endocrinol Metab. 1994;78:960-967. [PubMed: 7512573]
- 62.
- Argente J, Caballo N, Barrios V, Muñoz MT, Pozo J, Chowen JA, Morandé G, Hernández M. Multiple endocrine abnormalities of the growth hormone and insulin-like growth factor axis in patients with anorexia nervosa: effect of short- and long-term weight recuperation. J Clin Endocrinol Metab. 1997;82:2084-2092. [PubMed: 9215276]
- 63.
- Dunger DB, Acerini CL. IGF-I and diabetes in adolescence. Diabetes Metab. 1998;24:101-107. [PubMed: 9592633]
- 64.
- Frystyk J, Skjaerbaek C, Vestbo E, Fisker S, Orskov H. Circulating levels of free insulin-like growth factors in obese subjects: the impact of type 2 diabetes. Diabetes Metab Res Rev. 1999;15:314-322. [PubMed: 10585616]
- 65.
- Van Dam EW, Roelfsema F, Helmerhorst FH, Frölich M, Meinders AE, Veldhuis JD, Pijl H. Low amplitude and disorderly spontaneous growth hormone release in obese women with or without polycystic ovary syndrome. J Clin Endocrinol Metab. 2002;87:4225-4230. [PubMed: 12213875]
- 66.
- Roemmich JN, Clark PA, Weltman A, Veldhuis JD, Rogol AD. Pubertal alterations in growth and body composition: IX. Altered spontaneous secretion and metabolic clearance of growth hormone in overweight youth. Metabolism. 2005;54:1374-1383. [PubMed: 16154439]
- 67.
- Stanley TL, Levitsky LL, Grinspoon SK, Misra M. Effect of body mass index on peak growth hormone response to provocative testing in children with short stature. J Clin Endocrinol Metab. 2009;94:4875-4881. [PMC free article: PMC2795667] [PubMed: 19890023]
- 68.
- Salgin B, Marcovecchio ML, Hill N, Dunger DB, Frystyk J. The effect of prolonged fasting on levels of growth hormone-binding protein and free growth hormone. Growth Horm IGF Res. 2012;22:76-81. [PubMed: 22386777]
- 69.
- Hanaire-Broutin H, Sallerin-Caute B, Poncet MF, Tauber M, Bastide R, Chalé JJ, Rosenfeld R, Tauber JP. Effect of intraperitoneal insulin delivery on growth hormone binding protein, insulin-like growth factor (IGF)-I, and IGF-binding protein-3 in IDDM. Diabetologia. 1996;39:1498-1504. [PubMed: 8960832]
- 70.
- Shishko PI, Dreval AV, Abugova IA, Zajarny IU, Goncharov VC. Insulin-like growth factors and binding proteins in patients with recent-onset type 1 (insulin-dependent) diabetes mellitus: influence of diabetes control and intraportal insulin infusion. Diabetes Res Clin Pract. 1994;25:1-12. [PubMed: 7530621]
- 71.
- Chia DJ. Mechanisms of growth hormone-mediated gene regulation. Mol Endocrinol. 2014;28:1012-1025. [PMC free article: PMC4075164] [PubMed: 24825400]
- 72.
- Heffernan M1, Summers RJ, Thorburn A, Ogru E, Gianello R, Jiang WJ, Ng FM. The effects of human GH and its lipolytic fragment (AOD9604) on lipid metabolism following chronic treatment in obese mice and beta(3)-AR knock-out mice. Endocrinology. 2001;142:5182-5189. [PubMed: 11713213]
- 73.
- Fain JN, Ihle JH, Bahouth SW. Stimulation of lipolysis but not of leptin release by GH is abolished in adipose tissue from STAT5a and b knockout mice. Biochem Biophys Res Commun. 1999;263:201–205. [PubMed: 10486277]
- 74.
- Clodfelter KH, Holloway MG, Hodor P, Park SH, Ray WJ, Waxman DJ. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol Endocrinol. 2006;20:1333-1351. [PubMed: 16469768]
- 75.
- Sharma VM, Vestergaard ET, Jessen N, Kolind-Thomsen P, Nellemann B, Nielsen TS, Vendelbo MH, Møller N, Sharma R, Lee KY, Kopchick JJ, Jørgensen JOL, Puri V. Growth hormone acts along the PPARγ-FSP27 axis to stimulate lipolysis in human adipocytes. Am J Physiol Endocrinol Metab. 2019;316:E34-E42. [PMC free article: PMC6417689] [PubMed: 30325658]
- 76.
- Näntö-Salonen K, Muller HL, Hoffman AR, Vu TH, Rosenfeld RG. Mechanisms of thyroid hormone action on the insulin-like growth factor system: all thyroid hormone effects are not growth hormone mediated. Endocrinology. 1993;132:781-788. [PubMed: 7678799]
- 77.
- Veldhuis JD, Keenan DM, Bailey JN, Adeniji A, Miles JM, Paulo R, Cosma M, Soares-Welch C. Testosterone supplementation in older men restrains insulin-like growth factor's dose-dependent feedback inhibition of pulsatile growth hormone secretion. J Clin Endocrinol Metab. 2009;94:246-254. [PMC free article: PMC2630862] [PubMed: 18984660]
- 78.
- Colón E, Svechnikov KV, Carlsson-Skwirut C, Bang P, Soder O. Stimulation of steroidogenesis in immature rat Leydig cells evoked by interleukin-1alpha is potentiated by growth hormone and insulin-like growth factors. Endocrinology. 2005;146:221-30. [PubMed: 15486223]
- 79.
- Giudice LC. Insulin-like growth factor family in Graafian follicle development and function. J Soc Gynecol Investig. 2001;8(1 Suppl Proceedings):S26-9. [PubMed: 11223367]
- 80.
- Conover CA, Faessen GF, Ilg KE, Chandrasekher YA, Christiansen M, Overgaard MT, Oxvig C, Giudice LC. Pregnancy-associated plasma protein-a is the insulin-like growth factor binding protein-4 protease secreted by human ovarian granulosa cells and is a marker of dominant follicle selection and the corpus luteum. Endocrinology. 2001;142:2155-2161. [PubMed: 11316785]
- 81.
- Chrysis D, Zaman F, Chagin AS, Takigawa M, Sävendahl L. Dexamethasone induces apoptosis in proliferative chondrocytes through activation of caspases and suppression of the Akt-phosphatidylinositol 3'-kinase signaling pathway. Endocrinology. 2005;146:1391-1397. [PubMed: 15576458]
- 82.
- Bang P, Degerblad M, Thorén M, Schwander J, Blum W, Hall K. Insulin-like growth factor (IGF) I and II and IGF binding protein (IGFBP) 1, 2 and 3 in serum from patients with Cushing's syndrome. Acta Endocrinol (Copenh). 1993;128:397-404. [PubMed: 7686320]
- 83.
- Öberg D, Salemyr J, Örtqvist E, Juul A, Bang P. A longitudinal study of serum insulin-like growth factor-I levels over 6 years in a large cohort of children and adolescents with type 1 diabetes mellitus: A marker reflecting diabetic retinopathy. Pediatr Diabetes. 2018;19:972-978. [PubMed: 29663652]
- 84.
- Maile LA, Busby WH, Nichols TC, Bellinger DA, Merricks EP, Rowland M, Veluvolu U, Clemmons DR. A monoclonal antibody against alphaVbeta3 integrin inhibits development of atherosclerotic lesions in diabetic pigs. Sci Transl Med. 2010;2:18ra11. [PMC free article: PMC3938114] [PubMed: 20371482]
- 85.
- Vella A, Bouatia-Naji N, Heude B, Cooper JD, Lowe CE, Petry C, Ring SM, Dunger DB, Todd JA, Ong KK. Association analysis of the IGF1 gene with childhood growth, IGF-1 concentrations and type 1 diabetes. Diabetologia. 2008;51:811-815. [PMC free article: PMC2292425] [PubMed: 18317720]
- 86.
- Hedman CA, Frystyk J, Lindström T, Oskarsson P, Arnqvist HJ. Intraperitoneal insulin delivery to patients with type 1 diabetes results in higher serum IGF-I bioactivity than continuous subcutaneous insulin infusion. Clin Endocrinol (Oxf). 2014;81:58-62. [PubMed: 23865977]
- 87.
- Saukkonen T, Shojaee-Moradie F, Williams RM, Amin R, Yuen KC, Watts A, Acerini CL, Umpleby AM, Dunger DB. Effects of recombinant human IGF-I/IGF-binding protein-3 complex on glucose and glycerol metabolism in type 1 diabetes. Diabetes. 2006;55:2365-2370. [PubMed: 16873702]
- 88.
- Acerini CL, Patton CM, Savage MO, Kernell A, Westphal O, Dunger DB. Randomised placebo-controlled trial of human recombinant insulin-like growth factor I plus intensive insulin therapy in adolescents with insulin-dependent diabetes mellitus. Lancet. 1997;350:1199-1204. [PubMed: 9652560]
- 89.
- Woods KA, Camacho-Hübner C, Savage MO, Clark AJ. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N Engl J Med. 1996;335:1363-1367. [PubMed: 8857020]
- 90.
- Woods KA, Camacho-Hübner C, Bergman RN, Barter D, Clark AJ, Savage MO. Effects of insulin-like growth factor I (IGF-I) therapy on body composition and insulin resistance in IGF-I gene deletion. J Clin Endocrinol Metab. 2000;85:1407-1411. [PubMed: 10770174]
- 91.
- Dohm GL, Elton CW, Raju MS, Mooney ND, DiMarchi R, Pories WJ, Flickinger EG, Atkinson SM Jr, Caro JF. IGF-I--stimulated glucose transport in human skeletal muscle and IGF-I resistance in obesity and NIDDM. Diabetes. 1990;39:1028-1032. [PubMed: 2166697]
- 92.
- Nakae J, Kato M, Murashita M, Shinohara N, Tajima T, Fujieda K. Long-term effect of recombinant human insulin-like growth factor I on metabolic and growth control in a patient with leprechaunism. J Clin Endocrinol Metab. 1998;83:542-549. [PubMed: 9467572]
- 93.
- Dehghani SM, Karamifar H, Hamzavi SS, Haghighat M, Malek-Hosseini SA. Serum insulinlike growth factor-1 and its binding protein-3 levels in children with cirrhosis waiting for a liver transplant. Exp Clin Transplant. 2012;10:252-257. [PubMed: 22631062]
- 94.
- Moayeri H, Oloomi Z. Prevalence of growth and puberty failure with respect to growth hormone and gonadotropins secretion in beta-thalassemia major. Arch Iran Med. 2006;9:329-334. [PubMed: 17061604]
- 95.
- Alisi A, Pampanini V, De Stefanis C, Panera N, Deodati A, Nobili V, Cianfarani S. Expression of insulin-like growth factor I and its receptor in the liver of children with biopsy-proven NAFLD. PLoS One. 2018;13:e0201566. [PMC free article: PMC6067746] [PubMed: 30063751]
- 96.
- E CY, Cai Y, Sun BZ, Guan LY, Jiang T. Hepatic insulin-like growth factor receptor is upregulated by activation of the GSK3B-FOXO3 pathway after partial hepatectomy. J Biol Regul Homeost Agents. 2017;31:549-555. [PubMed: 28952285]
- 97.
- Kofoed EM, Hwa V, Little B, Woods KA, Buckway CK, Tsubaki J, Pratt KL, Bezrodnik L, Jasper H, Tepper A, Heinrich JJ, Rosenfeld RG. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349:1139-1147. [PubMed: 13679528]
- 98.
- Binder G, Neuer K, Ranke MB, Wittekindt NE. PTPN11 mutations are associated with mild growth hormone resistance in individuals with Noonan syndrome. J Clin Endocrinol Metab. 2005;90:5377-81. [PubMed: 15985475]
- 99.
- De Rocca Serra-Nédélec A, Edouard T, Tréguer K, Tajan M, Araki T, Dance M, Mus M, Montagner A, Tauber M, Salles JP, Valet P, Neel BG, Raynal P, Yart A. Noonan syndrome-causing SHP2 mutants inhibit insulin-like growth factor 1 release via growth hormone-induced ERK hyperactivation, which contributes to short stature. Proc Natl Acad Sci U S A. 2012;109:4257-4262. [PMC free article: PMC3306697] [PubMed: 22371576]
- 100.
- Vesterlund M, Zadjali F, Persson T, Nielsen ML, Kessler BM, Norstedt G, Flores-Morales A. The SOCS2 ubiquitin ligase complex regulates growth hormone receptor levels. PLoS One. 2011;6:e25358. [PMC free article: PMC3183054] [PubMed: 21980433]
- 101.
- Hoppe C, Mølgaard C, Juul A, Michaelsen KF. High intakes of skimmed milk, but not meat, increase serum IGF-I and IGFBP-3 in eight-year-old boys. Eur J Clin Nutr. 2004;58):1211-1216. [PubMed: 15054433]
- 102.
- Hoppe C, Mølgaard C, Vaag A, Barkholt V, Michaelsen KF. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur J Clin Nutr. 2005;59:393-398. [PubMed: 15578035]
- 103.
- Mogami H, Yura S, Itoh H, Kawamura M, Fujii T, Suzuki A, Aoe S, Ogawa Y, Sagawa N, Konishi I, Fujii S. Isocaloric high-protein diet as well as branched-chain amino acids supplemented diet partially alleviates adverse consequences of maternal undernutrition on fetal growth. Growth Horm IGF Res. 2009;19:478-485. [PubMed: 19395294]
- 104.
- D'Esposito V, Passaretti F, Hammarstedt A, Liguoro D, Terracciano D, Molea G, Canta L, Miele C, Smith U, Beguinot F, Formisano P. Adipocyte-released insulin-like growth factor-1 is regulated by glucose and fatty acids and controls breast cancer cell growth in vitro. Diabetologia. 2012;55:2811-2822. [PMC free article: PMC3433668] [PubMed: 22798065]
- 105.
- Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586-623. [PubMed: 19752219]
- 106.
- DeChiara TM, Robertson EJ, Efstratiadis A. Parental imprinting of the mouse insulin-like growth factor II gene. Cell. 1991;64:849-859. [PubMed: 1997210]
- 107.
- Rossignol S, Netchine I, Le Bouc Y, Gicquel C. Epigenetics in Silver-Russell syndrome. Best Pract Res Clin Endocrinol Metab. 2008;22:403-414. [PubMed: 18538282]
- 108.
- Clemmons DR. Role of IGF-binding proteins in regulating IGF responses to changes in metabolism. J Mol Endocrinol. 2018;61:T139-T169. [PubMed: 29563157]
- 109.
- Banskota NK, Carpentier JL, King GL. Processing and release of insulin and insulin-like growth factor I by macro- and microvascular endothelial cells. Endocrinology. 1986;119:1904-1913. [PubMed: 3533517]
- 110.
- Maile LA, Badley-Clarke J, Clemmons DR. The association between integrin-associated protein and SHPS-1 regulates insulin-like growth factor-I receptor signaling in vascular smooth muscle cells. Mol Biol Cell. 2003;14:3519-3528. [PMC free article: PMC196546] [PubMed: 12972543]
- 111.
- Bang P, Ahlsén M, Berg U, Carlsson-Skwirut C. Free insulin-like growth factor I: are we hunting a ghost? Horm Res. 2001;55 Suppl 2:84-93. [PubMed: 11684884]
- 112.
- Bang P, Thorell A, Carlsson-Skwirut C, Ljungqvist O, Brismar K, Nygren J. Free dissociable IGF-I: Association with changes in IGFBP-3 proteolysis and insulin sensitivity after surgery. Clin Nutr. 2016;35:408-413. [PubMed: 25817945]
- 113.
- Juul A, Holm K, Kastrup KW, Pedersen SA, Michaelsen KF, Scheike T, Rasmussen S, Müller J, Skakkebaek NE. Free insulin-like growth factor I serum levels in 1430 healthy children and adults, and its diagnostic value in patients suspected of growth hormone deficiency. J Clin Endocrinol Metab. 1997;82:2497-2502. [PubMed: 9253324]
- 114.
- Ekström K, Pulkkinen MA, Carlsson-Skwirut C, Brorsson AL, Ma Z, Frystyk J, Bang P. Tissue IGF-I Measured by Microdialysis Reflects Body Glucose Utilization After rhIGF-I Injection in Type 1 Diabetes.J Clin Endocrinol Metab. 2015;100:4299-4306. [PubMed: 26331550]
- 115.
- Baxter RC. Insulin-like growth factor binding proteins in the human circulation: a review. Horm Res. 1994;42:140-144. [PubMed: 7532612]
- 116.
- Bang P, Stangenberg M, Westgren M, Rosenfeld RG. Decreased ternary complex formation and predominance of a 29 kDa IGFBP-3 fragment in human fetal serum. Growth Regul. 1994;4:68-76. [PubMed: 7524886]
- 117.
- Ekström K, Carlsson-Skwirut C, Ritzén EM, Bang P. Insulin-like growth factor-I and insulin-like growth factor binding protein-3 cotreatment versus insulin-like growth factor-I alone in two brothers with growth hormone insensitivity syndrome: effects on insulin sensitivity, body composition and linear growth. Horm Res Paediatr. 2011;76:355-366. [PubMed: 21968387]
- 118.
- Giudice LC, Farrell EM, Pham H, Lamson G, Rosenfeld RG. Insulin-like growth factor binding proteins in maternal serum throughout gestation and in the puerperium: effects of a pregnancy-associated serum protease activity. J Clin Endocrinol Metab. 1990;71:806-816. [PubMed: 1698200]
- 119.
- Hossenlopp P, Segovia B, Lassarre C, Roghani M, Bredon M, Binoux M. Evidence of enzymatic degradation of insulin-like growth factor-binding proteins in the 150K complex during pregnancy. J Clin Endocrinol Metab. 1990;71:797-805. [PubMed: 1698199]
- 120.
- Diamandi A, Mistry J, Krishna RG, Khosravi J. Immunoassay of insulin-like growth factor-binding protein-3 (IGFBP-3): new means to quantifying IGFBP-3 proteolysis. J Clin Endocrinol Metab. 2000;85:2327-2333. [PubMed: 10852472]
- 121.
- Ahlsén M, Carlsson-Skwirut C, Jonsson AP, Cederlund E, Bergman T, Bang P. A 30-kDa fragment of insulin-like growth factor (IGF) binding protein-3 in human pregnancy serum with strongly reduced IGF-I binding. Cell Mol Life Sci. 2007;64:1870-1880. [PMC free article: PMC11136294] [PubMed: 17593323]
- 122.
- Yan X, Payet LD, Baxter RC, Firth SM. Activity of human pregnancy insulin-like growth factor binding protein-3: determination by reconstituting recombinant complexes. Endocrinology. 2009;150:4968-4976. [PubMed: 19734278]
- 123.
- Zachrisson I, Brismar K, Carlsson-Skwirut C, Dahlquist G, Wallensteen M, Bang P. Increased 24 h mean insulin-like growth factor binding protein-3 proteolytic activity in pubertal type 1 diabetic boys. Growth Horm IGF Res. 2000;10:324-331. [PubMed: 11161963]
- 124.
- Bang P, Brismar K, Rosenfeld RG. Increased proteolysis of insulin-like growth factor-binding protein-3 (IGFBP-3) in noninsulin-dependent diabetes mellitus serum, with elevation of a 29-kilodalton (kDa) glycosylated IGFBP-3 fragment contained in the approximately 130- to 150-kDa ternary complex. J Clin Endocrinol Metab. 1994;78:1119-1127. [PubMed: 7513716]
- 125.
- Bang P, Fielder PJ. Human pregnancy serum contains at least two distinct proteolytic activities with the ability to degrade insulin-like growth factor binding protein-3. Endocrinology. 1997;138:3912-3917. [PubMed: 9275081]
- 126.
- Rajah R, Katz L, Nunn S, Solberg P, Beers T, Cohen P. Insulin-like growth factor binding protein (IGFBP) proteases: functional regulators of cell growth. Prog Growth Factor Res. 1995;6:273-84. [PubMed: 8817670]
- 127.
- Martin DC, Fowlkes JL, Babic B, Khokha R. Insulin-like growth factor II signaling in neoplastic proliferation is blocked by transgenic expression of the metalloproteinase inhibitor TIMP-1. J Cell Biol. 1999;146:881-892. [PMC free article: PMC2156132] [PubMed: 10459021]
- 128.
- Gaidamauskas E, Gyrup C, Boldt HB, Schack VR, Overgaard MT, Laursen LS, Oxvig C.IGF dependent modulation of IGF binding protein (IGFBP) proteolysis by pregnancy-associated plasma protein-A (PAPP-A): multiple PAPP-A-IGFBP interaction sites. Biochim Biophys Acta. 2013 Mar;1830(3):2701-9. doi: .10.1016/j.bbagen.2012.11.002 [PubMed: 23671931] [CrossRef]
- 129.
- Bang P, Ahmed SF, Argente J, Backeljauw P, Bettendorf M, Bona G, Coutant R, Rosenfeld RG, Walenkamp MJ, Savage MO. Identification and management of poor response to growth-promoting therapy in children with short stature. Clin Endocrinol (Oxf). 2012;77:169-181. [PubMed: 22540980]
- 130.
- Bang P, Polak M, Woelfle J, Houchard A; EU IGFD Registry Study Group. Effectiveness and Safety of rhIGF-1 Therapy in Children: The European Increlex® Growth Forum Database Experience. Horm Res Paediatr. 2015;83:345-357. [PubMed: 25824333]
- 131.
- Murray and Clayton. Disorders of growth hormone in childhood. MDText.com 2022
- 132.
- Cabrera-Salcedo C, Mizuno T, Tyzinski L, Andrew M, Vinks AA, Frystyk J, Wasserman H, Gordon CM, Hwa V, Backeljauw P, Dauber A. Pharmacokinetics of IGF-1 in PAPP-A2-Deficient Patients, Growth Response, and Effects on Glucose and Bone Density. J Clin Endocrinol Metab. 2017 Dec 1;102(12):4568-4577. doi: .10.1210/jc.2017-01411 [PMC free article: PMC5718699] [PubMed: 29029190] [CrossRef]
- 133.
- Muthuvel G, Dauber A, Alexandrou E, Tyzinski L, Andrew M, Hwa V, Backeljauw P. Five-Year Therapy with Recombinant Human Insulin-Like Growth Factor-1 in a Patient with PAPP-A2 Deficiency. Horm Res Paediatr. 2023;96(5):449-457. doi: . Epub 2023 Jan 16.10.1159/000529071 [PubMed: 36646053] [CrossRef]
- 134.
- Muñoz-Calvo MT, Barrios V, Pozo J, Chowen JA, Martos-Moreno GÁ, Hawkins F, Dauber A, Domené HM, Yakar S, Rosenfeld RG, Pérez-Jurado LA, Oxvig C, Frystyk J, Argente J. Treatment With Recombinant Human Insulin-Like Growth Factor-1 Improves Growth in Patients With PAPP-A2 Deficiency. J Clin Endocrinol Metab. 2016 Nov;101(11):3879-3883. doi: . Epub 2016 Sep 20.10.1210/jc.2016-2751 [PMC free article: PMC5393598] [PubMed: 27648969] [CrossRef]
Publication Details
Author Information and Affiliations
Publication History
Last Update: April 4, 2024.
Copyright
This electronic version has been made freely available under a Creative Commons (CC-BY-NC-ND) license. A copy of the license can be viewed at http://creativecommons.org/licenses/by-nc-nd/2.0/.
Publisher
MDText.com, Inc., South Dartmouth (MA)
NLM Citation
Bang P. Pediatric Implications of Normal Insulin-GH-IGF Axis Physiology. [Updated 2024 Apr 4]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.