

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Glucagon is a peptide hormone secreted from the alpha cells of the pancreatic islets of Langerhans. Hypoglycemia is physiologically the most potent secretory stimulus and the best known action of glucagon is to stimulate glucose production in the liver and thereby to maintain adequate plasma glucose concentrations. However, glucagon is also involved in hepatic lipid and amino acid metabolism and may increase resting energy expenditure. Based on satiety-inducing and food intake-lowering effects of exogenous glucagon, a role for glucagon in the regulation of appetite has also been proposed. This chapter provides an overview of the structure, secretion, degradation and elimination of glucagon, and reviews the actions of glucagon including its role in glucose metabolism and its effects on lipolysis, ketogenesis, energy expenditure, appetite and food intake. Finally, the role of glucagon in the pathophysiology of diabetes, obesity and hepatic steatosis is discussed and emerging glucagon-based therapies for these conditions are outlined. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Glucagon secreted from pancreatic alpha cells in the islet of Langerhans plays an important role in maintaining glucose homeostasis by stimulating hepatic glucose production (1). Thus, in contrast to the glucose-depositing nature of insulin action, glucagon acts as a glucose-mobilizing hormone. In line with these opposed actions, high plasma glucose concentrations stimulating insulin secretion from pancreatic beta cells, inhibit glucagon secretion whereas low plasma glucose concentrations represent one of the most potent glucagon secretory stimuli. Accordingly, normal plasma glucose concentrations depend largely on the balanced secretion of insulin and glucagon from the pancreatic beta cells and alpha cells, respectively. The hyperglycemic effect of glucagon was described as early as 1922 by Kimball and Murlin who discovered a hyperglycemic factor in pancreatic extracts and called this factor “the glucose agonist”, hence the name glucagon (2). In the 1950s glucagon was purified and crystallized at Eli Lilly and Co., and shortly after, the amino acid sequence of the peptide was determined (3). This led to the development of medical use of glucagon for the treatment of severe insulin-induced hypoglycemia (4,5). The development of a radioimmunoassay for the detection of glucagon in 1959 spurred further investigations of glucagon physiology and its role in health and disease (6). It was discovered that patients with diabetes exhibit increased glucagon levels which led to the “bihormonal hypothesis” stating that the combination of hypoinsulinemia and hyperglucagonemia constitutes a central pathophysiological determinant for diabetic hyperglycemia (7). Since then it has become evident that glucagon not only acts by increasing hepatic glucose production but affects overall energy homeostasis in times of limited energy supply by stimulating lipid and protein catabolism, reducing appetite and food intake and increasing energy expenditure.
STRUCTURE AND SYNTHESIS OF GLUCAGON
Glucagon is a 29-amino acid peptide hormone predominantly secreted from the alpha cells of the pancreas. It is derived from the precursor proglucagon which can be processed into a number of related peptide hormones (Fig. 1). Proglucagon is expressed in pancreatic islet alpha cells, intestinal enteroendocrine L cells, and to a minor extent in neurons in the brain stem and hypothalamus (8,9). Processing of proglucagon is undertaken by the processing enzymes prohormone convertase 1/3 (PC1/3) and prohormone convertase 2 (PC2), respectively. In the pancreas, PC2 processes proglucagon to glucagon while processing of proglucagon in the intestine and the brain is undertaken by PC1 leading to the formation of glucagon-like peptide 1 (GLP-1) and glucagon-like peptide 2 (GLP-2) (9).
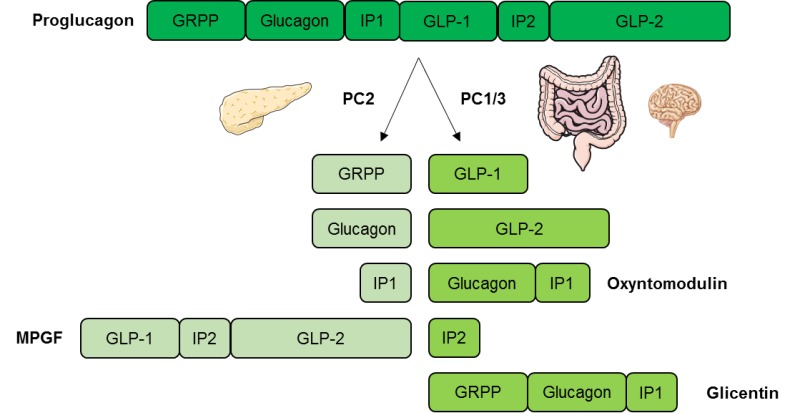
Figure 1.
Tissue specific processing of proglucagon. In the pancreas proglucagon is processed into glucagon, glicentin-related pancreatic polypeptide (GRPP), intervening peptide 1 (IP1), and major proglucagon fragment (MPGF) by the processing enzyme prohormone convertase 2 (PC2). In the intestine and in the brain proglucagon is processed by prohormone convertase 1/3 (PC1/3) into glucagon-like peptide 1 (GLP-1), glucagon-like peptide 2 (GLP-2), oxyntomodulin, intervening peptide 2 (IP2), and glicentin.
GLUCAGON SECRETION
Glucagon is secreted in response to hypoglycemia, prolonged fasting, exercise and protein-rich meals (10). Glucagon release is regulated through endocrine and paracrine pathways; by nutritional substances; and by the autonomic nervous system (11). Glucagon secretion occurs as exocytosis of stored peptide vesicles initiated by secretory stimuli of the alpha cell. Stimulatory regulators of glucagon release include hypoglycemia, amino acids and the gut hormone glucose-dependent insulinotropic peptide (GIP), whereas hyperglycemia and GLP-1 inhibit glucagon release. Additionally, glucagon release is inhibited in a paracrine fashion by factors like somatostatin, insulin, zinc and possibly amylin. Glucagon may regulate its own secretion indirectly via stimulatory effect on beta cells to secrete insulin (12,13). In contrast to glucose, non-glucose regulators of glucagon secretion seem to mediate their action through changes in cAMP levels rather than through the calcium-dependent pathway outlined below (14,15).
Regulation of Glucagon Secretion by Glucose
The most potent regulator of glucagon secretion is circulating glucose. Hypoglycemia stimulates the pancreatic alpha cell to release glucagon and hyperglycemia inhibits glucagon secretion (Fig. 2) (11). The cellular mechanism behind this glucose-dependent regulation of glucagon secretion involves uptake of glucose by the glucose transporter 1 (GLUT1) in the cell membrane of pancreatic alpha cells and subsequent glycolysis which ultimately generates adenosine triphosphate (ATP) in the mitochondria of the alpha cell. Thus, the intracellular ATP level in the alpha cell reflects plasma glucose levels. Hypoglycemia and resulting low intracellular ATP levels in the alpha cell close ATP-sensitive potassium channels (KATP-channels) whereby the efflux of potassium (K+) is reduced. This causes a depolarization of the cell membrane which, in turn, opens voltage-dependent Ca2+ channels allowing influx of Ca2+. This increases intracellular Ca2+ levels, the primary trigger for exocytosis of glucagon granules from the alpha cells (Fig. 2). Conversely, increasing circulating glucose levels increase glucose influx to the alpha cell generating an increase in intracellular ATP concentration, which opens KATP-channels. This leads to a membrane potential that closes voltage-dependent Ca2+ channels thereby preventing Ca2+ influx and glucagon secretion (12).
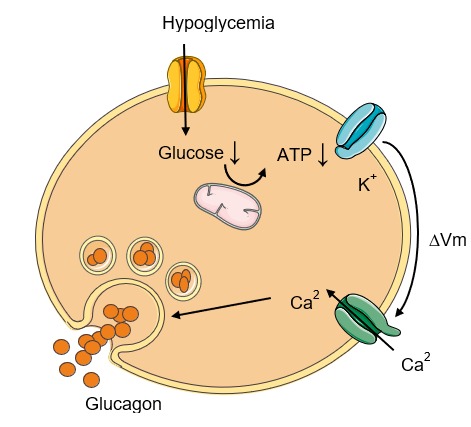
Figure 2.
Glucose-dependent glucagon secretion from the alpha cell. During hypoglycemia intracellular glucose concentration falls with a subsequent reduction in glycolysis-generated adenosine triphosphate (ATP) in the mitochondria of the cell. This closes ATP-sensitive potassium (K+) channels and the intracellular K+ concentration rises, which depolarizes the cell membrane, thus opening voltage-dependent Ca2+ channels allowing influx of Ca2+. Increase in intracellular Ca2+ concentration triggers secretion of glucagon through exocytosis. ∆Vm, change in membrane potential (i.e. depolarization of the cell membrane).
Glucagon Concentrations in The Circulation
In normal physiology, circulating glucagon concentrations are in the picomolar range. In the fasting state with plasma glucose levels around 5 mmol/l, glucagon is secreted in basal levels resulting in plasma concentrations below 20 pmol/l (16–18). Basal glucagon secretion balances the effect of basal insulin secretion resulting in a steady-state between glucose uptake and endogenous glucose production in the fasted state; i.e. stable blood glucose concentrations. During exercise or in case of hypoglycemia, circulating glucagon levels may increase dramatically to 3-4 times basal levels increasing the glucagon to insulin ratio (12,19,20) (Fig. 3).
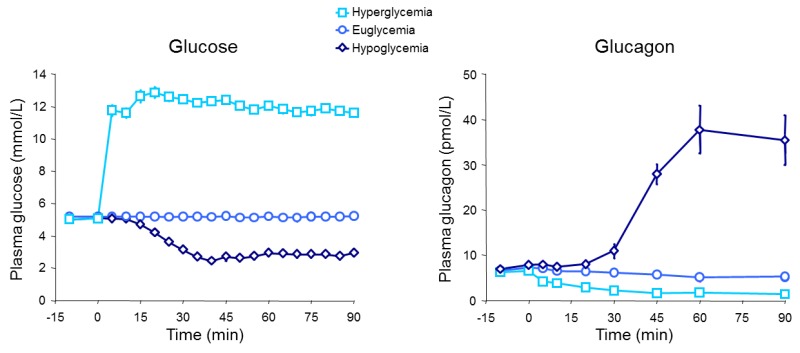
Figure 3.
Glucagon concentrations in response to hypoglycemia, euglycemia, and hyperglycemia.
GLUCAGON RECEPTOR SIGNALLING
The effects of glucagon are mediated through binding to and activation of the glucagon receptor. The glucagon receptor is a seven transmembrane G protein-coupled receptor (Fig. 4) predominantly expressed in the liver, but also found in varying amounts in the kidneys, heart (controversial), adrenal glands, adipose tissue (controversial), gastrointestinal tract, and pancreas (21). The main mode of intracellular signaling involves activation of Gs and Gq. Gs activation stimulates adenylyl cyclase which produces cyclic adenosine monophosphate (cAMP) that activates protein kinase A (PKA). The activated PKA migrates to the nucleus and activates transcription factors like cAMP response element-binding protein (CREB) through phosphorylation. This enables CREB to bind to response elements of target genes resulting in the recruitment of coactivators and ultimately promoting gene expression. Activation of Gq by glucagon leads to activation of phospholipase C (PLC) and subsequent increase in inositol 1,4,5-triphosphate (IP3), which signals to enhance release of calcium from the endoplasmic reticulum. This, in turn, activates downstream signaling cascades including CREB-regulated transcription co-activator (CRTC2) which enhance CREB-dependent gene expression. In addition to the CREB-CRTC2 pathway, glucagon may signal through various other pathways reviewed in detail elsewhere (1,12,22).
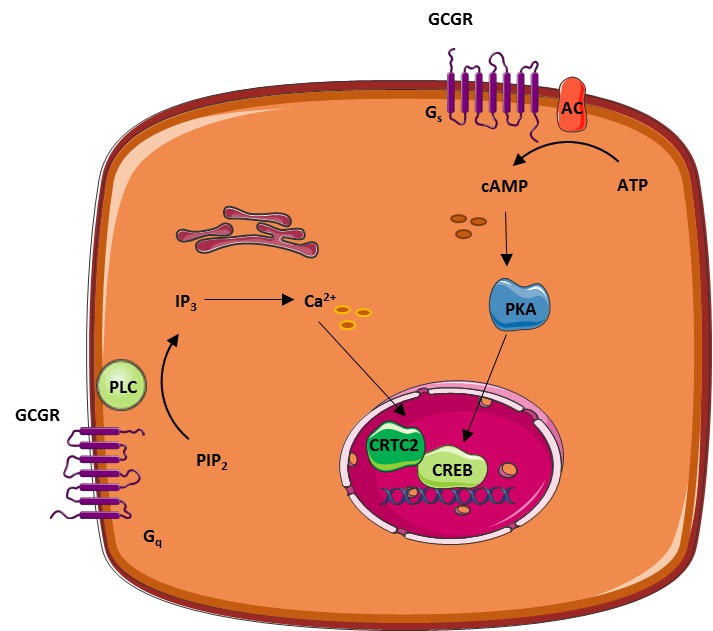
Figure 4.
Examples of the two most well-described intracellular pathways involved in glucagon-induced regulation of target gene expression: the PKA and the IP3 pathways. AC, adenylyl cyclase; CRTC2, CREB-regulated transcription co-activator; CREB, cAMP response element-binding protein; IP3, inositol 1,4,5-triphosphate; PIP2, phosphatidyl-inositol-4,5-bisphosphate; PKA, protein kinase A; PLC, phospholipase C.
DEGRADATION AND ELIMINATION OF GLUCAGON
The degradation of glucagon is mainly facilitated by receptor-mediated endocytosis and proteolysis by the ubiquitous enzyme dipeptidyl peptidase 4 (22,23). Consistent with the relative receptor expression, the liver and kidneys seem to represent the two main organs removing glucagon from the circulation. The circulating half-life of glucagon in plasma is reported to be between four to seven minutes in humans (24,25).
GLUCAGON ACTIONS
Glucagon Increases Hepatic Glucose Production
Glucagon controls plasma glucose concentrations during fasting, exercise and hypoglycemia by increasing hepatic glucose output to the circulation. Specifically, glucagon promotes hepatic conversion of glycogen to glucose (glycogenolysis), stimulates de novo glucose synthesis (gluconeogenesis), and inhibits glucose breakdown (glycolysis) and glycogen formation (glycogenesis) (Fig. 5) (26). Hepatic glucose production is rapidly enhanced in response to a physiological rise in glucagon; achieved through stimulation of glycogenolysis with minor acute changes in gluconeogenesis (27,28). This ability of glucagon is critical in the life-saving counterregulatory response to severe hypoglycemia. Additionally, it is a key factor in providing adequate circulating glucose for brain function and for working muscle during exercise (28). During prolonged fasting, glycogen stores are depleted, and gluconeogenesis takes over (29). The hyperglycemic property of glucagon is enhanced when hepatic glycogen levels are high and diminished when hepatic glycogen levels are low in conditions of fasting or liver diseases like cirrhosis (12).
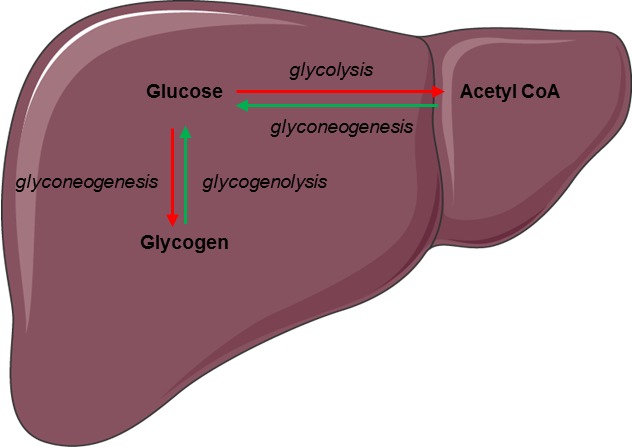
Figure 5.
Regulation of glucose metabolism by glucagon in the liver. Glucagon increases hepatic glucose production by stimulating glycogenolysis and glycogenogenesis (green arrows) while inhibiting glycolysis and glycogenesis (red arrows).
Glucagon Stimulates Break-Down of Fatty Acids and Inhibits Lipogenesis in the Liver
Glucagon promotes formation of non-carbohydrate energy sources in the form of lipids and ketone bodies. Thereby, glucagon contributes to a stable energy homeostasis during conditions where energy supply is limited (fasting) or in states of increased energy demand (e.g. exercise or cold exposure) (12). Specifically, in times of energy demand, glucagon enhances break-down of fatty acids to acetyl-coenzyme A molecules (beta-oxidation) in the liver. These intermediates are either reduced to generate ATP in the tricarboxylic acid cycle or converted to ketone bodies (ketogenesis) – a process also stimulated by glucagon. (30–34). Furthermore, glucagon signaling inhibits de novo lipogenesis by inactivating the enzyme that catalyzes the first step in fatty acid synthesis from other substrates like carbohydrates (34).
Glucagon Promotes Break-Down of Amino Acids
During prolonged fasting, glucagon stimulates formation of glucose from amino acids (via gluconeogenesis) by upregulating enzymes involved in the process. However, the rate-limiting step of the process depends on the supply of gluconeogenic amino acids from muscle or dietary intake, a process not controlled by glucagon (35). In addition to enter gluconeogenesis, amino acids are deaminated to generate ATP in the liver. Glucagon is involved in this process by promoting the conversion of ammonia – a toxic biproduct from deamination – to urea, which is excreted in the urine. Thereby glucagon reduces ammonia levels in the blood (36). Disruption of glucagon action by inhibition of the glucagon receptor (37) leads to increased plasma levels of amino acids and pancreatic alpha cell hyperplasia, which in turn, leads to glucagon hypersecretion. This suggests that glucagon and amino acids are linked in a feedback loop between the liver and the pancreatic alpha cells (35).
Glucagon Reduces Food Intake
Acute administration of glucagon has been shown to reduce food intake and diminish hunger (38,39). Conversely, preprandial inhibition of glucagon signaling increases food intake in rats (40,41) providing evidence for a role of glucagon in the regulation of appetite. It is somewhat counterintuitive that glucagon should reduce food intake given that glucagon levels are typically elevated upon fasting and decrease upon feeding. Thus, the observed effect upon glucagon administration (in supraphysiological concentrations) could partly be due to cross-reactivity with the GLP-1 receptor (which normally result in suppression of food intake) (12). The mechanism behind glucagon’s potential appetite-reducing effect is not fully understood but could arise from hepatic metabolic changes induced by glucagon or from glucagon working directly in the central nervous system (31).
Glucagon Increases Energy Expenditure
In addition to a potential effect of glucagon on food intake, evidence suggests that glucagon contributes to a negative energy balance by stimulating energy expenditure. In humans, this effect has been observed in studies in which glucagon infusion resulted in increases in resting energy expenditure (42–44). However, the effect of endogenous glucagon on resting energy expenditure remains unclear. Also, the exact mechanisms behind the increase in resting energy expenditure elicited by exogenous glucagon remain to be determined. It has been speculated that glucagon activates brown adipose tissue (12), however this was recently challenged in an in vivo study that found no direct effect of glucagon on brown adipose tissue (43). Rodent studies indicate that the actions of glucagon to increase energy expenditure might be indirectly mediated partly by fibroblast growth factor 21 (FGF21) as glucagon-induced increase in energy expenditure is abolished in animals with FGF21 receptor deletion (45).
Glucagon May Regulate Heart Rate and Contractility
Infusion of high doses of glucagon increases heart rate and cardiac contractility (46). In fact, infusion of glucagon in pharmacological doses (milligram) is often used in the treatment of acute cardiac depression caused by calcium channel antagonist or beta-blocker overdoses (47) despite limited evidence (48). In comparison, glucagon concentrations within the normal physiological range do not appear to affect heart rate or contractility (49) and any physiological role of endogenous glucagon in the regulation of pulse rate remains questionable. This is supported by studies investigating the effect of glucagon receptor antagonist for the treatment of type 2 diabetes in which no effect of pulse rate were observed (50). Nevertheless, whether increased glucagon concentrations have a sustained effect on the heart remains unknow. Of note, most studies use bolus injections of glucagon which cause only a transient increase in heart rate and contractility (potentially reflecting the rapid elimination of glucagon from circulation) (48). Taken together, it remains uncertain whether glucagon has a place in the treatment of heart failure or hold a cardioprotective effect in healthy subjects.

Figure 6.
Organ specific actions of glucagon. GIP, glucose-dependent insulinotropic polypeptide
GLUCAGON PATHOPHYSIOLOGY
Glucagon in Type 2 Diabetes
Patients with type 2 diabetes exhibit an impaired regulation of glucagon secretion which contributes importantly to diabetic hyperglycemia. Specifically, type 2 diabetes is characterized by elevated levels of glucagon during fasting while suppression of glucagon in response to oral intake of glucose is impaired or even paradoxically elevated (Fig. 7) (16,51,52). The mechanisms behind hyperglucagonemia are not fully understood but is usually explained by a diminished suppressive effect of insulin on alpha cells due to hypoinsulinemia and insulin resistance at the level of the alpha cells (53,54). Interestingly, subjects with type 2 diabetes, who exhibit a hyperglucagonemic response to oral glucose, respond with a normal suppression of glucagon after intravenous glucose administration (16). This suggests that diabetic hyperglucagonemia may not be explained by alpha cell resistance to glucose and/or insulin. Accordingly, hormones secreted from the gastrointestinal tract may play an important role (55,56). It has recently been confirmed that glucagon can be secreted from extrapancreatic tissue (demonstrated in experiments with totally pancreatectomized subjects) (17). This supports the notion that postprandial hypersecretion of glucagon in patients with type 2 diabetes might be of extrapancreatic origin.
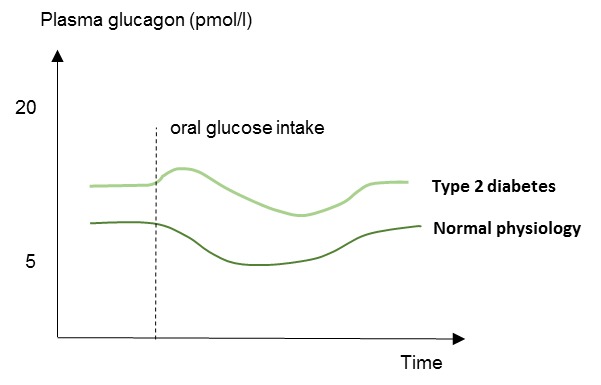
Figure 7.
Schematic illustration of plasma glucagon concentrations in patients with type 2 diabetes and in normal physiology (healthy subjects). Type 2 diabetes is characterized by elevated fasting plasma glucagon levels and impaired suppression of plasma glucagon levels in response to oral glucose.
Glucagon in Type 1 Diabetes
Traditionally type 1 diabetic hyperglycemia has been explained by selective loss of beta cell mass and resulting decrease in insulin secretion. However, emerging evidence indicate that glucagon plays a major role in type 1 diabetes pathophysiology. The glucagon dyssecretion that characterizes patients with type 1 diabetes is associated with two clinical manifestations: Postprandial hyperglucagonemia and impaired glucagon counterregulation to hypoglycemia (57). Data regarding fasting plasma glucagon concentrations in type 1 diabetes are inconsistent (57,58). Thus, the general notion that glucagon hypersecretion plays a role in type 1 diabetes hyperglycemia is mainly based on elevated postprandial glucagon concentrations (57). The explanation behind this is unclear, although a common explanation is, that in type 1 diabetes the postprandial increase in plasma glucose is not followed by an increase in insulin secretion from beta cells, which in normal physiology would inhibit glucagon secretion. The absence of that restraining signal from endogenous insulin could result in an increase in glucagon secretion from alpha cells after a meal (Fig. 8) (12,57). However, like in type 2 diabetes, subjects with type 1 diabetes preserve their ability to suppress glucagon after intravenous glucose administration. This suggest that inappropriate glucagon secretion in type 1 diabetes occurs as a consequence of the oral administration, possible via glucagonotropic signaling from the gut or due to glucagon secretion directly from the gut, rather than as a consequence of dysfunctional alpha cell sensing to hyperglycemia and/or lack of paracrine inhibition by insulin (17,55,59).
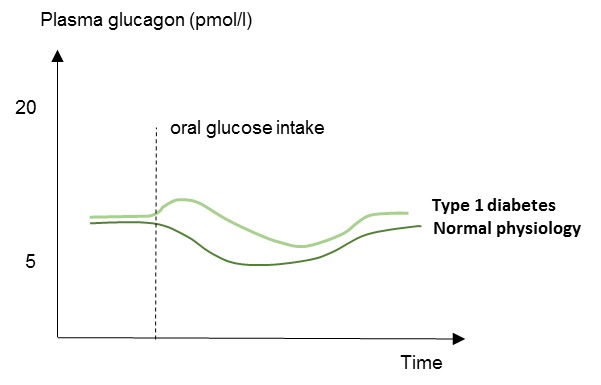
Figure 8.
Schematic illustration of plasma glucagon concentrations in patients with type 1 diabetes and in normal physiology (healthy subjects). Type 1 diabetes is characterized by elevated concentrations of glucagon in response to a meal or oral glucose intake.
Hypoglycemia is a frequent and feared side effect of insulin therapy in type 1 diabetes and it represents a common barrier in obtaining glycemic control (60). In normal physiology hypoglycemia is prevented by several mechanisms: 1) Reduced insulin secretion from beta cells diminishing glucose uptake in peripheral tissues; 2) increased glucagon secretion from alpha cells increasing hepatic glucose output; and 3) increased symphathetic neural response and adrenomedullary epinephrine secretion. The latter will stimulate hepatic glucose production and cause clinical symptoms that enables the individual to recognize hypoglycemia and ultimately ingest carbohydrates (57,61,62). In type 1 diabetes, insulin-induced hypoglycemia fails to elicit adequate glucagon responses compromising counterregulation to insulin-induced hypoglycemia; a phenomenon which seems to worsen with the duration of type 1 diabetes. This defect likely involves a combination of defective alpha cells and reduced alpha cell mass (57,63).
Glucagon in Obesity and Hepatic Steatosis
Dysregulated glucagon secretion is not only observed in patients with type 2 diabetes but also in normoglucose-tolerant individuals with obesity (64) and patients with non-alcoholic fatty liver disease (NAFLD) (65,66). This suggests that dysregulated glucagon secretion may represent hepatic steatosis rather than dysregulated glucose metabolism. Interestingly, fasting hyperglucagonemia seems to relate to circulating amino acids in addition to hepatic fat content (65). This hyperaminoacidemia suggests that impairment of amino acid turnover in the liver and ensuing elevations of circulating amino acids constitutes a feedback on the alpha cell to secrete more glucagon with increasing hepatic amino acid turnover and ureagenesis needed for clearance of toxic ammonia from the body. This feedback loop is proposed as “the liver-alpha cell axis” (35,55,65,67).
PERSPECTIVES
The implication of hyperglucagonemia in obesity and NAFLD has renewed the scientific interest in actions of glucagon and the role of glucagon in the pathophysiology of these metabolic disorders. Clearly, glucagon may represent a potential target for treatments of obesity and NAFLD. A simple way to restrain the undesirable hyperglycemic effect of glucagon while realizing its actions on lipolysis and energy expenditure could be by co-treating with a glucose-lowering drug. This may be done by mimicking the gut hormone oxyntomodulin which acts as a ligand to both the glucagon and the GLP-1 receptor. Accordingly, treatment with dual glucagon/GLP-1 receptor agonists in subjects with type 2 diabetes and obesity improves glycemic control, reduces body weight (68) and ameliorates NAFLD (69).
SUMMARY AND CONCLUSIONS
Glucagon is a glucoregulatory peptide hormone that counteracts the actions of insulin by stimulating hepatic glucose production and thereby increases blood glucose levels. Additionally, glucagon mediates several non-glucose metabolic effects of importance for maintaining whole-body energy balance in times of limited nutrient supply. These actions include mobilization of energy resources through hepatic lipolysis and ketogenesis; stimulation of hepatic amino acid turnover (and related ureagenesis). Also, glucagon has been shown to increase energy expenditure and inhibit food intake, but whether endogenous glucagon is involved in the regulation of these processes remains uncertain. Glucagon plays an important role in the pathophysiology of diabetes as elevated glucagon levels observed in these patients stimulate hepatic glucose production, thereby contributing to diabetic hyperglycemia. Investigations of glucagon’s role in non-glucose metabolism and metabolic physiology and pathophysiological processes are ongoing and may reveal new treatment targets based on glucagon actions.
ACKNOWLEDGEMENTS
Figure 1,2,4,5,6 created by adopting templates from Servier Medical Arts, Les Laboratoires Servier (https://smart.servier.com/). Used under Creative Commons License 3.0.
REFERENCES
- 1.
- Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6(12):689–697. [PMC free article: PMC3563428] [PubMed: 20957001]
- 2.
- Kimball CP, Murlin JR. Aqueous Extracts of Pancreas Iii. Some Precipitation Reactions of Insulin. J. Biol. Chem. 1923;58(1):337–346.
- 3.
- Bromer WW, Sinn LG, Staub A, Behrens OK. The amino acid sequence of glucagon. Diabetes. 1957;6(3):234–238. [PubMed: 13427628]
- 4.
- Blackman B. The use of glucagon in insulin coma therapy. Psychiatr Q. 1961;35:482–487. [PubMed: 13869689]
- 5.
- Esquibel AJ, Kurland AA, Mendelsohn D. The use of glucagon in terminating insulin coma. Dis Nerv Syst. 1958;19(11):485–486. [PubMed: 13586175]
- 6.
- Unger RH, Eisentraut AM. McCALL MS, Madison LL. Glucagon antibodies and an immunoassay for glucagon. J. Clin. Invest. 1961;40:1280–1289. [PMC free article: PMC290840] [PubMed: 13779204]
- 7.
- Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet. 1975;1(7897):14–16. [PubMed: 46337]
- 8.
- Drucker DJ, Asa S. Glucagon gene expression in vertebrate brain. J. Biol. Chem. 1988;263(27):13475–13478. [PubMed: 2901414]
- 9.
- Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J. Biol. Chem. 1986;261(25):11880–11889. [PubMed: 3528148]
- 10.
- Gerich JE, Lorenzi M, Hane S, Gustafson G, Guillemin R, Forsham PH. Evidence for a physiologic role of pancreatic glucagon in human glucose homeostasis: studies with somatostatin. Metab. Clin. Exp. 1975;24(2):175–182. [PubMed: 1113681]
- 11.
- Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr. Rev. 2007;28(1):84–116. [PubMed: 17261637]
- 12.
- Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH. The New Biology and Pharmacology of Glucagon. Physiological Reviews. 2017;97(2):721–766. [PubMed: 28275047]
- 13.
- Wewer Albrechtsen NJ, Kuhre RE, Pedersen J, Knop FK, Holst JJ. The biology of glucagon and the consequences of hyperglucagonemia. Biomarkers in Medicine. 2016;10(11):1141–1151. [PubMed: 27611762]
- 14.
- Gromada J, Chabosseau P, Rutter GA. The α-cell in diabetes mellitus. Nat Rev Endocrinol. 2018. [PubMed: 30310153] [CrossRef]
- 15.
- Hughes JW, Ustione A, Lavagnino Z, Piston DW. Regulation of islet glucagon secretion: Beyond calcium. Diabetes, Obesity and Metabolism. 20(S2):127–136. [PMC free article: PMC6148361] [PubMed: 30230183]
- 16.
- Knop FK, Vilsbøll T, Madsbad S, Holst JJ, Krarup T. Inappropriate suppression of glucagon during OGTT but not during isoglycaemic i.v. glucose infusion contributes to the reduced incretin effect in type 2 diabetes mellitus. Diabetologia. 2007;50(4):797–805. [PubMed: 17225124]
- 17.
- Lund A, Bagger JI, Albrechtsen NJW, Christensen M, Grøndahl M, Hartmann B, Mathiesen ER, Hansen CP, Storkholm JH, Hall G, van, Rehfeld JF, Hornburg D, Meissner F, Mann M, Larsen S, Holst JJ, Vilsbøll T, Knop FK. Evidence of Extrapancreatic Glucagon Secretion in Man. Diabetes. 2016;65(3):585–597. [PubMed: 26672094]
- 18.
- Miyachi A, Kobayashi M, Mieno E, Goto M, Furusawa K, Inagaki T, Kitamura T. Accurate analytical method for human plasma glucagon levels using liquid chromatography-high resolution mass spectrometry: comparison with commercially available immunoassays. Anal Bioanal Chem. 2017;409(25):5911–5918. [PubMed: 28801845]
- 19.
- Hansen JS, Pedersen BK, Xu G, Lehmann R, Weigert C, Plomgaard P. Exercise-Induced Secretion of FGF21 and Follistatin Are Blocked by Pancreatic Clamp and Impaired in Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016;101(7):2816–2825. [PubMed: 27163358]
- 20.
- Schwartz NS, Clutter WE, Shah SD, Cryer PE. Glycemic thresholds for activation of glucose counterregulatory systems are higher than the threshold for symptoms. J Clin Invest. 1987;79(3):777–781. [PMC free article: PMC424197] [PubMed: 3546378]
- 21.
- Svoboda M, Tastenoy M, Vertongen P, Robberecht P. Relative quantitative analysis of glucagon receptor mRNA in rat tissues. Mol. Cell. Endocrinol. 1994;105(2):131–137. [PubMed: 7859919]
- 22.
- Sandoval DA, D’Alessio DA. Physiology of proglucagon peptides: role of glucagon and GLP-1 in health and disease. Physiol. Rev. 2015;95(2):513–548. [PubMed: 25834231]
- 23.
- Pospisilik JA, Hinke SA, Pederson RA, Hoffmann T, Rosche F, Schlenzig D, Glund K, Heiser U, McIntosh CHS, Demuth H-U. Metabolism of glucagon by dipeptidyl peptidase IV (CD26). Regulatory Peptides. 2001;96(3):133–141. [PubMed: 11111019]
- 24.
- Pontiroli AE, Calderara A, Perfetti MG, Bareggi SR. Pharmacokinetics of intranasal, intramuscular and intravenous glucagon in healthy subjects and diabetic patients. Eur. J. Clin. Pharmacol. 1993;45(6):555–558. [PubMed: 8157042]
- 25.
- Lund A, Bagger JI, Albrechtsen NW, Christensen M, Grøndahl M, Hansen CP, Storkholm JH, Holst JJ, Vilsbøll T, Knop FK. Increased Liver Fat Content in Totally Pancreatectomized Patients. Diabetes. 2017;67 Supplement 1:2600. -pub.
- 26.
- Unger RH. Glucagon physiology and pathophysiology in the light of new advances. Diabetologia. 1985;28(8):574–578. [PubMed: 3902546]
- 27.
- Miller RA, Birnbaum MJ. Glucagon: acute actions on hepatic metabolism. Diabetologia. 2016;59(7):1376–1381. [PubMed: 27115415]
- 28.
- Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab. 2011;13 Suppl 1:118–125. [PMC free article: PMC5371022] [PubMed: 21824265]
- 29.
- Rui L. Energy Metabolism in the Liver. Compr Physiol. 2014;4(1):177–197. [PMC free article: PMC4050641] [PubMed: 24692138]
- 30.
- Geisler CE, Renquist BJ. Hepatic lipid accumulation: cause and consequence of dysregulated glucoregulatory hormones. J Endocrinol. 2017;234(1):R1–R21. [PubMed: 28428362]
- 31.
- Habegger KM, Heppner KM, Geary N, Bartness TJ, DiMarchi R, Tschöp MH. The metabolic actions of glucagon revisited. Nat Rev Endocrinol. 2010;6(12):689–697. [PMC free article: PMC3563428] [PubMed: 20957001]
- 32.
- Longuet C, Sinclair EM, Maida A, Baggio LL, Maziarz M, Charron MJ, Drucker DJ. The Glucagon Receptor Is Required for the Adaptive Metabolic Response to Fasting. Cell Metabolism. 2008;8(5):359–371. [PMC free article: PMC2593715] [PubMed: 19046568]
- 33.
- Wang H, Zhao M, Sud N, Christian P, Shen J, Song Y, Pashaj A, Zhang K, Carr T, Su Q. Glucagon regulates hepatic lipid metabolism via cAMP and Insig-2 signaling: implication for the pathogenesis of hypertriglyceridemia and hepatic steatosis. Sci Rep. 2016;6:32246. [PMC free article: PMC5007496] [PubMed: 27582413]
- 34.
- Galsgaard KD, Pedersen J, Knop FK, Holst JJ, Wewer Albrechtsen NJ. Glucagon Receptor Signaling and Lipid Metabolism. Front Physiol. 2019;10 [PMC free article: PMC6491692] [PubMed: 31068828] [CrossRef]
- 35.
- Holst JJ, Albrechtsen NJW, Pedersen J, Knop FK. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver–α-Cell Axis. Diabetes. 2017;66(2):235–240. [PubMed: 28108603]
- 36.
- Hamberg O, Vilstrup H. Regulation of urea synthesis by glucose and glucagon in normal man. Clin Nutr. 1994;13(3):183–191. [PubMed: 16843380]
- 37.
- Solloway MJ, Madjidi A, Gu C, Eastham-Anderson J, Clarke HJ, Kljavin N, Zavala-Solorio J, Kates L, Friedman B, Brauer M, Wang J, Fiehn O, Kolumam G, Stern H, Lowe JB, Peterson AS, Allan BB. Glucagon Couples Hepatic Amino Acid Catabolism to mTOR-Dependent Regulation of α-Cell Mass. Cell Rep. 2015;12(3):495–510. [PubMed: 26166562]
- 38.
- Bagger JI, Holst JJ, Hartmann B, Andersen B, Knop FK, Vilsbøll T. Effect of Oxyntomodulin, Glucagon, GLP-1, and Combined Glucagon +GLP-1 Infusion on Food Intake, Appetite, and Resting Energy Expenditure. J Clin Endocrinol Metab. 2015;100(12):4541–4552. [PubMed: 26445112]
- 39.
- Geary N, Kissileff HR, Pi-Sunyer FX, Hinton V. Individual, but not simultaneous, glucagon and cholecystokinin infusions inhibit feeding in men. Am. J. Physiol. 1992;262(6 Pt 2):R975–980. [PubMed: 1621876]
- 40.
- Langhans W, Zeiger U, Scharrer E, Geary N. Stimulation of feeding in rats by intraperitoneal injection of antibodies to glucagon. Science. 1982;218(4575):894–896. [PubMed: 7134979]
- 41.
- Le Sauter J, Noh U, Geary N. Hepatic portal infusion of glucagon antibodies increases spontaneous meal size in rats. Am. J. Physiol. 1991;261(1 Pt 2):R162–165. [PubMed: 1858943]
- 42.
- Nair KS. Hyperglucagonemia Increases Resting Metabolic Rate In Man During Insulin Deficiency. J Clin Endocrinol Metab. 1987;64(5):896–901. [PubMed: 2881943]
- 43.
- Salem V, Izzi‐Engbeaya C, Coello C, Thomas DB, Chambers ES, Comninos AN, Buckley A, Win Z, Al‐Nahhas A, Rabiner EA, Gunn RN, Budge H, Symonds ME, Bloom SR, Tan TM, Dhillo WS. Glucagon increases energy expenditure independently of brown adipose tissue activation in humans. Diabetes Obes Metab. 2016;18(1):72–81. [PMC free article: PMC4710848] [PubMed: 26434748]
- 44.
- Tan TM, Field BCT, McCullough KA, Troke RC, Chambers ES, Salem V, Gonzalez Maffe J, Baynes KCR, De Silva A, Viardot A, Alsafi A, Frost GS, Ghatei MA, Bloom SR. Coadministration of Glucagon-Like Peptide-1 During Glucagon Infusion in Humans Results in Increased Energy Expenditure and Amelioration of Hyperglycemia. Diabetes. 2013;62(4):1131–1138. [PMC free article: PMC3609580] [PubMed: 23248172]
- 45.
- Habegger KM, Stemmer K, Cheng C, Müller TD, Heppner KM, Ottaway N, Holland J, Hembree JL, Smiley D, Gelfanov V, Krishna R, Arafat AM, Konkar A, Belli S, Kapps M, Woods SC, Hofmann SM, D’Alessio D, Pfluger PT, Perez-Tilve D, Seeley RJ, Konishi M, Itoh N, Kharitonenkov A, Spranger J, DiMarchi RD, Tschöp MH. Fibroblast Growth Factor 21 Mediates Specific Glucagon Actions. Diabetes. 2013;62(5):1453–1463. [PMC free article: PMC3636653] [PubMed: 23305646]
- 46.
- Ceriello A, Genovese S, Mannucci E, Gronda E. Glucagon and heart in type 2 diabetes: new perspectives. Cardiovasc Diabetol. 2016;15(1):123. [PMC free article: PMC5002329] [PubMed: 27568179]
- 47.
- Graudins A, Lee HM, Druda D. Calcium channel antagonist and beta-blocker overdose: antidotes and adjunct therapies. Br J Clin Pharmacol. 2016;81(3):453–461. [PMC free article: PMC4767195] [PubMed: 26344579]
- 48.
- Meidahl Petersen K, Bøgevig S, Holst JJ, Knop FK, Christensen MB. Hemodynamic Effects of Glucagon - A Literature Review. J. Clin. Endocrinol. Metab. 2018. [PubMed: 29546411] [CrossRef]
- 49.
- Thuesen L, Christiansen JS, Sørensen KE, Orskov H, Henningsen P. Low-dose intravenous glucagon has no effect on myocardial contractility in normal man. An echocardiographic study. Scand. J. Clin. Lab. Invest. 1988;48(1):71–75. [PubMed: 3064277]
- 50.
- Kazda CM. Treatment with the glucagon receptor antagonist LY2409021 increases ambulatory blood pressure in patients with type 2 diabetes. DIABETES OBESITY AND METABOLISM. 2017;19(8):1071–1077. [PubMed: 28191913]
- 51.
- Lund A, Bagger JI, Christensen M, Grøndahl M, van Hall G, Holst JJ, Vilsbøll T, Knop FK. Higher Endogenous Glucose Production During OGTT vs Isoglycemic Intravenous Glucose Infusion. J Clin Endocrinol Metab. 2016;101(11):4377–4384. [PubMed: 27533305]
- 52.
- Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia throughout the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1987;64(1):106–110. [PubMed: 3536980]
- 53.
- Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr. Rev. 2007;28(3):253–283. [PubMed: 17409288]
- 54.
- Hamaguchi T, Fukushima H, Uehara M, Wada S, Shirotani T, Kishikawa H, Ichinose K, Yamaguchi K, Shichiri M. Abnormal glucagon response to arginine and its normalization in obese hyperinsulinaemic patients with glucose intolerance: importance of insulin action on pancreatic alpha cells. Diabetologia. 1991;34(11):801–806. [PubMed: 1769438]
- 55.
- Knop FK. EJE PRIZE 2018: A gut feeling about glucagon. Eur. J. Endocrinol. 2018;178(6):R267–R280. [PubMed: 29678923]
- 56.
- Lund A, Vilsbøll T, Bagger JI, Holst JJ, Knop FK. The separate and combined impact of the intestinal hormones, GIP, GLP-1, and GLP-2, on glucagon secretion in type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2011;300(6):E1038–1046. [PubMed: 21386059]
- 57.
- Cryer PE. Minireview: Glucagon in the Pathogenesis of Hypoglycemia and Hyperglycemia in Diabetes. Endocrinology. 2012;153(3):1039–1048. [PMC free article: PMC3281526] [PubMed: 22166985]
- 58.
- Li K, Song W, Wu X, Gu D, Zang P, Gu P, Lu B, Shao J. Associations of serum glucagon levels with glycemic variability in type 1 diabetes with different disease durations. Endocrine. 2018;61(3):473–481. [PubMed: 29916102]
- 59.
- Hare KJ, Vilsbøll T, Holst JJ, Knop FK. Inappropriate glucagon response after oral compared with isoglycemic intravenous glucose administration in patients with type 1 diabetes. American Journal of Physiology-Endocrinology and Metabolism. 2010;298(4):E832–E837. [PubMed: 20103744]
- 60.
- Diabetes Control and Complications Trial Research Group. Nathan DM, Genuth S, Lachin J, Cleary P, Crofford O, Davis M, Rand L, Siebert C. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993;329(14):977–986. [PubMed: 8366922]
- 61.
- Cryer PE, Gerich JE. Glucose counterregulation, hypoglycemia, and intensive insulin therapy in diabetes mellitus. N. Engl. J. Med. 1985;313(4):232–241. [PubMed: 2861565]
- 62.
- Sprague JE, Arbeláez AM. Glucose Counterregulatory Responses to Hypoglycemia. Pediatr Endocrinol Rev. 2011;9(1):463–475. [PMC free article: PMC3755377] [PubMed: 22783644]
- 63.
- Yosten GLC. Alpha cell dysfunction in type 1 diabetes. Peptides. 2018;100:54–60. [PubMed: 29412832]
- 64.
- Knop FK, Aaboe K, Vilsbøll T, Vølund A, Holst JJ, Krarup T, Madsbad S. Impaired incretin effect and fasting hyperglucagonaemia characterizing type 2 diabetic subjects are early signs of dysmetabolism in obesity. Diabetes Obes Metab. 2012;14(6):500–510. [PubMed: 22171657]
- 65.
- Albrechtsen NJW, Junker AE, Christensen M, Hædersdal S, Wibrand F, Lund AM, Galsgaard KD, Holst JJ, Knop FK, Vilsbøll T. Hyperglucagonemia correlates with plasma levels of non-branched chained amino acids in patients with liver disease independent of type 2 diabetes. American Journal of Physiology - Gastrointestinal and Liver Physiology 2017:ajpgi.00216.2017. [PubMed: 28971838]
- 66.
- Junker AE, Gluud L, Holst JJ, Knop FK, Vilsbøll T. Diabetic and nondiabetic patients with nonalcoholic fatty liver disease have an impaired incretin effect and fasting hyperglucagonaemia. J Intern Med. 2016;279(5):485–493. [PubMed: 26728692]
- 67.
- Suppli MP, Lund A, Bagger JI, Vilsbøll T, Knop FK. Involvement of steatosis-induced glucagon resistance in hyperglucagonaemia. Med. Hypotheses. 2016;86:100–103. [PubMed: 26547273]
- 68.
- Tillner J, Posch MG, Wagner F, Teichert L, Hijazi Y, Einig C, Keil S, Haack T, Wagner M, Bossart M, Larsen PJ. A novel dual glucagon-like peptide and glucagon receptor agonist SAR425899: Results of randomized, placebo-controlled first-in-human and first-in-patient trials. Diabetes Obes Metab. 2018. [PubMed: 30091218] [CrossRef]
- 69.
- Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, Tsai L-F, Robertson D, Jain M, Petrone M, Rondinone C, Hirshberg B, Jermutus L. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: a randomised, controlled, double-blind, ascending dose and phase 2a study. The Lancet. 2018;391(10140):2607–2618. [PubMed: 29945727]
- Review Inhibition of glucagon secretion.[Adv Pharmacol. 2005]Review Inhibition of glucagon secretion.Young A. Adv Pharmacol. 2005; 52:151-71.
- Glucagon Receptor Signaling and Lipid Metabolism.[Front Physiol. 2019]Glucagon Receptor Signaling and Lipid Metabolism.Galsgaard KD, Pedersen J, Knop FK, Holst JJ, Wewer Albrechtsen NJ. Front Physiol. 2019; 10:413. Epub 2019 Apr 24.
- Effects of dipeptidyl peptidase IV inhibition on glycemic, gut hormone, triglyceride, energy expenditure, and energy intake responses to fat in healthy males.[Am J Physiol Endocrinol Metab....]Effects of dipeptidyl peptidase IV inhibition on glycemic, gut hormone, triglyceride, energy expenditure, and energy intake responses to fat in healthy males.Heruc GA, Horowitz M, Deacon CF, Feinle-Bisset C, Rayner CK, Luscombe-Marsh N, Little TJ. Am J Physiol Endocrinol Metab. 2014 Nov 1; 307(9):E830-7. Epub 2014 Sep 16.
- Glucagon-like peptide-1 and islet lipolysis.[Horm Metab Res. 2004]Glucagon-like peptide-1 and islet lipolysis.Sörhede Winzell M, Ahrén B. Horm Metab Res. 2004 Nov-Dec; 36(11-12):795-803.
- Review [Glucagon and glucagon-like peptides the role in control glucose homeostasis. Part I].[Pediatr Endocrinol Diabetes Me...]Review [Glucagon and glucagon-like peptides the role in control glucose homeostasis. Part I].Otto-Buczkowska E. Pediatr Endocrinol Diabetes Metab. 2011; 17(4):215-21.
- Glucagon Physiology - EndotextGlucagon Physiology - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...