

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
The glucocorticoid receptor (GR) is an evolutionally conserved nuclear receptor superfamily protein that mediates the diverse actions of glucocorticoids as a ligand-dependent transcription factor. This receptor is a protein that shuttles from the cytoplasm to the nucleus upon binding to its ligand glucocorticoid hormone, where it modulates the transcription rates of glucocorticoid-responsive genes positively or negatively. Tremendous efforts have been made to reveal the molecular signaling actions of the GR, including intracellular shuttling, transcriptional regulation and interaction with other intracellular signaling pathways. Glucocorticoids are essential for both maintenance of the resting state and the stress response, and are pivotal in the treatment of many disorders, including autoimmune, inflammatory, allergic, and lymphoproliferative diseases. Thus, pathologic or therapeutic implications of the GR, including genetic alterations in the human GR gene, disease-associated GR regulatory molecules, and development of GR ligands with selective GR actions, are of great importance. This chapter provides an overview on such GR-related research activities. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Glucocorticoids are steroid hormones secreted by the adrenal glands. They are important for the maintenance of basal and stress-related homeostasis by acting as end products of the stress-responsive hypothalamic-pituitary-adrenal (HPA) axis (1). Glucocorticoids regulate a variety of biologic processes and exert profound influences on many physiologic functions (2,3). In pharmacologic doses, glucocorticoids are used as potent immunosuppressive agents in the therapeutic management of many inflammatory, autoimmune and lympho-proliferative diseases (4). At the cellular level, the actions of glucocorticoids are mediated by an intracellular receptor protein, the glucocorticoid receptor (GR) (its gene name is “nuclear receptor subfamily 3, group C, member 1: NR3C1”), which belongs to the steroid/sterol/thyroid/retinoid/orphan receptor superfamily of nuclear transactivating factors with over 200 members in general and over 40 in mammals currently cloned and characterized across species (5). Human GR consists of 777 amino acid residues (5). GR is ubiquitously expressed in almost all human tissues and organs including neural stem cells (6). GR functions as a hormone-dependent transcription factor that regulates the expression of glucocorticoid-responsive genes, which probably represent 3-10% of the human genome and can be influenced by the ligand-activated GR directly or indirectly (7).
EVOLUTION OF GR
Nuclear hormone receptors (NRs) form a highly conserved protein family observed even in simple metazoans. They are phylogenically differentiated into 7 subfamilies under the evolutional selection pressure, and are still active in the current human population (8). GR is a member of the steroid hormone receptor (SR) subfamily (subfamily 3) of NRs. This receptor family of vertebrates consists of six evolutionarily related SRs: two for estrogens (estrogen receptor (ER) a and ERβ) and one each for androgens (androgen receptor: AR), progestins (progesterone receptor: PR), glucocorticoids (GR), and mineralocorticoids (mineralocorticoid receptor: MR) (Figure 1). These steroid receptors are also categorized as type I receptors, based on their functional characteristics, such as cytoplasmic localization in the absence of ligand with association to the heat shock proteins, homo-dimerization and recognition of their target DNA sequence (see below), while the other NRs belong to type II to IV (5).
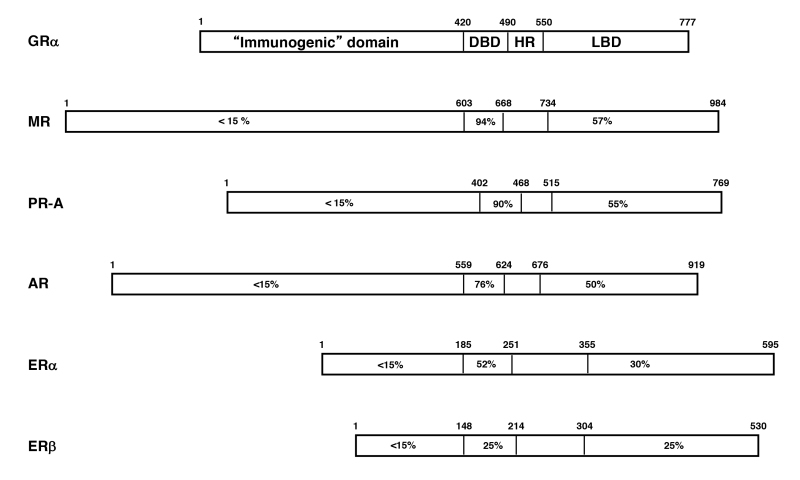
Figure 1.
Steroid hormone receptors (SRs: class I receptors) and their homologies expressed as percent identity to the protein sequence of human GR. AR; androgen receptor, ER: estrogen receptor, ER: estrogen receptor, GR; glucocorticoid receptor, MR: mineralocorticoid receptor, PR-A: progesterone receptor-A. Modified from (9).
SRs evolved in the chordate lineage after the separation of deuterostomes and protostomes, prior to or at the base of the Cambrian explosion about 540 million years ago (10,11) (Figure 2A). The receptor phylogeny suggests that two serial gene duplications of an ancestral SR gene occurred before the divergence of lamprey and jawed vertebrates (Figure 2B). The first gene duplication (duplication #1 in Figure 2B) created an estrogen receptor (ER) and a 3-ketosteroid receptor, whereas the second duplication (duplication #2 in Figure 2B) of the latter gene produced a corticoid receptor and a receptor for 3-ketogonadal steroids (androgens, progestins, or both). Therefore, the ancestral vertebrates (e.g., lamprey) had three SRs: an estrogen receptor (ER), a receptor for corticoids (corticosteroid receptor: CR) and a receptor that bound androgens, progestins or both (ancestral PR). At some later time within the gnathostome lineage, each of these three receptor genes were duplicated again (duplications #3, #4 and #5 in Figure 2B) to yield the six SRs currently found in jawed vertebrates: the ER creating ERa and ERβ, CR yielding the GR and MR, and the 3-ketogonadal steroid receptor (ancestral PR) producing the PR and AR. Therefore, the genome of ‘higher’ vertebrates is thought to be the result of three genome duplication events that occurred early in chordate evolution (10,12). Although the timing of these events is not entirely clear, it is most likely that the first 2 duplications occurred before the lamprey-gnathostome divergence and one after (10,13).
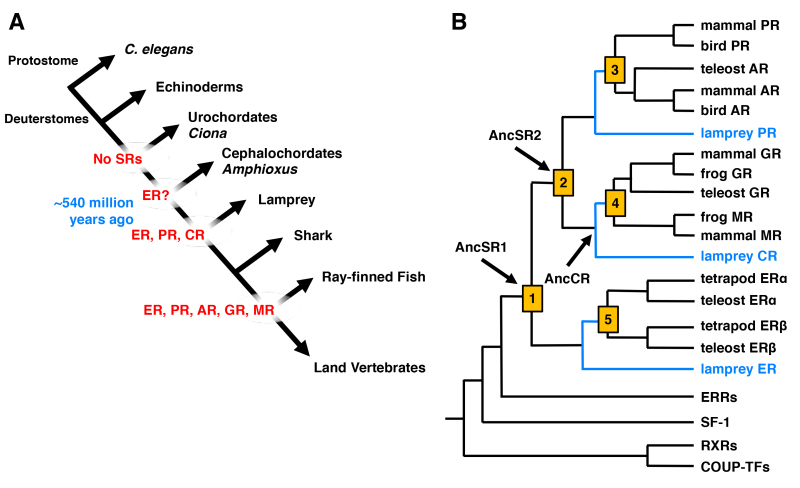
Figure 2.
Evolution of SRs including GR. A: Appearance of the SR member receptors through evolution of the chordate lineage. The first ancestral SR, which is close to the current ER, appeared ~540 million years ago. At lamprey, 3 receptors, ER, PR and CR, emerged. From the ray-finned fishes, all SR members, ER, PR, AR, GR and MR, appeared. Modified from (14). B: Phylogeny of the SR family genes. Current human SRs including GR were generated through several gene duplications (shown as orange squares). Appearance of the ancestral (Anc) SR1, SR2 and CR are shown with arrows in the phylogeny tree. Blue lines indicate the lamprey-gnathostome divergence. Modified from (10).
The GR and its closest family member MR, both descend from duplication of the ancestral CR (AncCR) gene, and emerged in the vertebrate lineage approximately 450 million years ago (12,15) (Figure 2B). The GR is activated by cortisol, while the MR is activated by aldosterone in tetrapods and by deoxycorticosterone (DOC) in teleosts. The MR is also sensitive to cortisol, though considerably less so than to aldosterone and DOC (12,15). Like the MR, the AncCR is sensitive to aldosterone, DOC and cortisol, indicating that the specificity of GR for cortisol is evolutionarily derived (12,15).
To determine how the preference of the GR for cortisol evolved, Ortlund et al. identified substitutions that occurred during the same period as the shift in GR function (16). Using maximum likelihood phylogenetics, he revealed that GR retained AncCR’s sensitivity to aldosterone, DOC and cortisol, from the common ancestor of all jawed vertebrates, but the GR from the common ancestor of bony vertebrates obtained a phenotype like that of the current GRs that respond only to cortisol. These findings indicate that the specificity of GR for cortisol evolved during the interval between these two speciation events, approximately 420 to 440 million years ago (16). Amino acid substitutions found in the modern GR compared to AncGR are not a consequence of the direct introduction of corresponding nucleotide changes, but supported by permissive mutations that enabled the intermediate receptor to tolerate insertion of the final substitutions (17).
Teleosts, one of the 3 subgroups of ray-finned fishes that covers most of the living fishes today, underwent an additional gene duplication event about 350 million years ago (18). Thus, all fishes that belong to this subclass, including carp and rainbow trout, have 2 GR genes (GR1 and 2 in rainbow trout). However, zebrafish has only one GR gene in contrast to the other teleost families, because this species lost the 2nd GR gene sometime during the last 33 million years (18).
STRUCTURE OF THE HUMAN GR GENE AND PROTEIN
All SRs including GR display a modular structure comprised of five to six regions (A-F): the amino-terminal A/B region, also called immunogenic or N-terminal domain (NTD), and the C and E regions, which correspond to the DNA- (DBD) and ligand-binding (LBD) domains, respectively (Figure 3). D region represents the hinge region (HR), while F region is located in the C-terminal end of the NRs with high variability. GR does not have a F region. The GR cDNA was isolated by expression cloning in 1985 (19). The human GR gene consists of 9 exons and is located in the long arm of the chromosome 5 (5q31.3) in an inverse orientation and spanning ~160 kbs. Alternative splicing of the human GR gene in exon 9 generates two highly homologous receptor isoforms, termed a and b. These are identical through amino acid 727, but then diverge, with human GRa having an additional 50 amino acids and human GRb having an additional, nonhomologous 15 amino acids (20). The molecular weights of these receptor isoforms are 97 and 94 kilo-Dalton, respectively. Human GRa is expressed virtually in all organs and tissues, resides primarily in the cytoplasm, and represents the classic glucocorticoid receptor that functions as a ligand-dependent transcription factor. Human GRb, also expressed ubiquitously, does not bind glucocorticoid agonists and functions as a dominant negative receptor for GRa-induced transcriptional activity (see Section 7. THE SPLICE VARIANT GR-beta ISOFORM) (21).
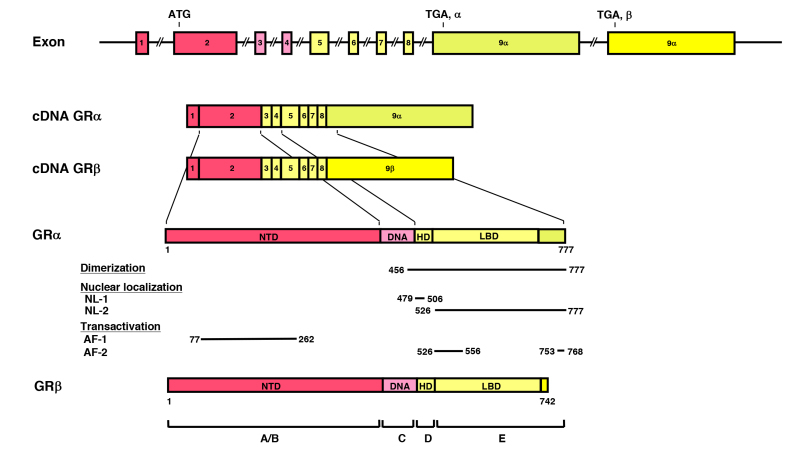
Figure 3.
Genomic and complementary DNA and protein structures of the human (h) GR with its functional distribution, and the isoforms produced through alternative splicing. The hGR (NR3C1) gene consists of 10 exons. Exon 1 is an untranslated region (UTR), exon 2 encodes for NTD (A/B), exon 3 and 4 for DBD (C), and exons 5-9 for the hinge region (D) and LBD (E). GR does not have an F region in contrast to the other steroid hormone receptors. The GR (NR3C1) gene contains two terminal exons 9 (exon 9 and 9) alternatively spliced to produce the classic GR and the nonligand-binding GR isoform. C-terminal gray-colored domains in GR and GR show their specific portions. Locations of several functional domains are also indicated. AF-1 and -2: activation function-1 and -2; DBD; DNA-binding domain; HD: hinge region; LBD: Ligand-binding domain; NTD: N-terminal domain, NL1 and 2: Nuclear translocation signal 1 and 2.
The N-terminal domain (NTD) of GRa contains a major transactivation domain, termed activation function (AF)-1, which is located between amino acids 77 and 262 of the human GRa (22,23). AF-1 belongs to a group of acidic activators, such as VP16, nuclear factor of kB (NF-kB), p65 and p53, contains four a-helices, and plays an important role in the communication between the receptor and molecules necessary for the initiation of transcription, including coactivators, chromatin modulators and basal transcription factors [RNA polymerase II, TATA-binding protein (TBP) and a host of TBP-associated proteins (TAFIIs)] (24). GRa AF-1 is relatively unfolded at the basal state, while it forms a significantly complex helical structure in response to binding to cofactors, such as TBP and p160 coactivators (25,26). TBP-induced conformational change in AF-1 facilitates association of this domain to a p160 coactivator (27).
The DNA-binding domain (DBD) of the human GRa corresponds to amino acids 420-480 and contains two C4-type zinc finger motifs through which GRa binds to specific DNA sequences, the glucocorticoid-responsive elements (GREs) (28,29). The DBD is the most highly conserved domain throughout the NR family. It has two similar zinc finger modules, each nucleated by a zinc ion coordination center held by four cysteine (C) residues and followed by a-helix (Figure 4A). The N-terminal’s first a-helix lies in the major groove of the double-stranded DNA, while the C-terminal part of the second a-helix is positioned over the minor groove (Figure 4B).
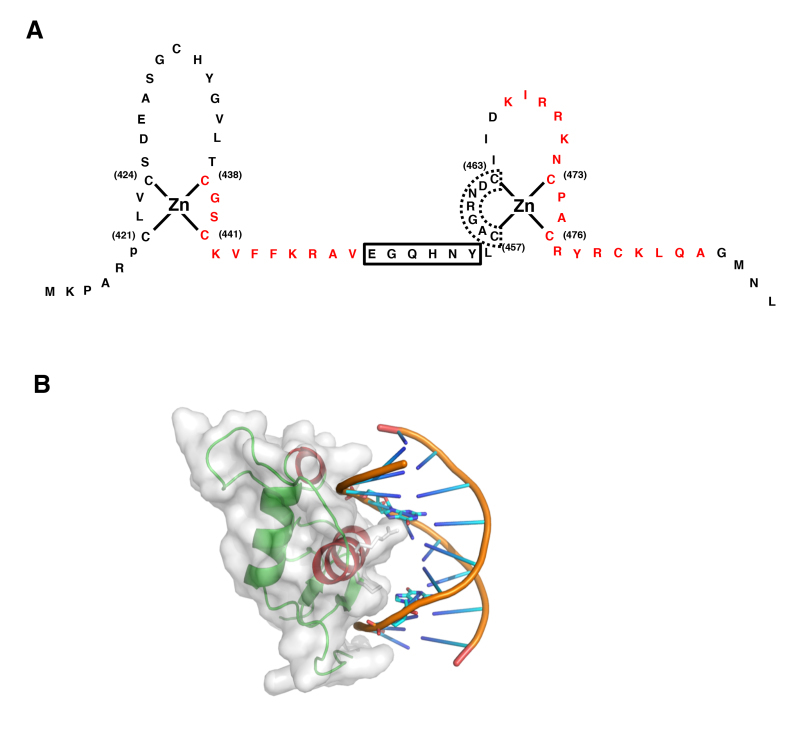
Figure 4.
Structure of GR DBD and its interaction with DNA GRE. A: Zinc finger structures in DBD of hGR. Numbered eight cysteine (C) residues chelate Zn2+ to form two separate finger structures. Red-colored amino acid residues form -helical structures. Box with bold line indicates lever arm, while that with dashed line shows D-box. Modified from (30). B: 3-Dimensional model of the physical interaction between the GR DBD and DNA GRE. The N-terminal’s first -helix of the GR DBD lies in the major groove of the double-stranded DNA, while the C-terminal part of the second -helix is positioned over the minor groove. The image was created and donated by Dr. D.E. Hurt (National Institute of Allergy and Infectious Diseases, NIH, Bethesda, MD). Box with bold line indicates lever arm, while that with dashed line shows D-box. Modified from (30).
The ligand-binding domain (LBD) of the human GRa corresponds to amino acids 481-777, binds to glucocorticoids, and plays a critical role in the ligand-induced activation of GRalpha. The crystal structure of the GRalpha LBD was successfully analyzed by using a point mutant containing a single replacement of phenylalanine at amino acid 602 by serine, and is comprised of 12 a-helices and 4 small β-sheets that fold into a three-layer helical domain (31) (Figure 5). Helices 1 and 3 form one side of a helical sandwich, while helices 7 and 10 form the other side. The middle layer of the helices (helices 4, 5, 8, and 9) is present in the top but not the bottom half of the protein. This arrangement of helices creates a cavity in the bottom half of the LBD, which is surrounded by helices 3, 4, 11 and 12, and functions as a ligand-binding pocket (31-33). Interaction of the LBD with the heat shock protein (hsp) 90 contributes to the maintenance of the protein structure that allows LBD to associate with ligand. Ligand-binding induces a conformational change in the LBD and allows GRa to communicate with several molecules, such as importin a of the nuclear import system, components of the transcription initiation complexes and other transcription factors that mediate the ligand-dependent actions of GRa. The LBD also contains one transactivation domain, termed AF-2. The activity of AF-2 is ligand-dependent.
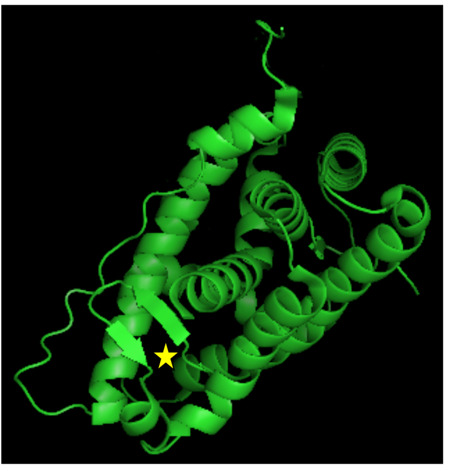
Figure 5.
Structure of the GR LBD. The GR consists of 12 -helices and 4 small β-sheets that fold into a three-layer helical domain. Helices 1 and 3 form one side of a helical sandwich, while helices 7 and 10 form the other side. The middle layer of helices 4, 5, 8, and 9 are present in the top but not in the bottom half of the protein, thus creating a ligand-binding pocket (shown as yellow star) in the bottom half of the LBD, surrounded by helices 3, 4, 11 and 12. The image was created with the MacPyMOL software using 3K22 of the RCSB Protein Data Bank (PDB) (http://www.rcsb.org/pdb/home/home.do).
TRANSCRIPTIONAL AND TRANSLATIONAL REGULATION OF GR ISOFORMS
As described above, the human GR gene expresses two mRNAs through alternative use of exon 9a and 9b, and produces two splice variants. The human GRa mRNA further expresses multiple isoforms by using at least 8 alternative translation initiation sites (34) (Figure 6). Since human GRb shares a common mRNA domain that contains the same translation initiation sites with human GRa (19), the human GRb variant mRNA seems also to be translated through the same initiation sites to a similar host of b isoforms. They are produced by ribosomal leaky scanning and/or ribosomal shunting from their alternative translation initiation sites located at amino acids 27 (GRa-B), 86 (GRa-C1), 90 (GRa-C2), 98 (GRa-C3), 316 (GRa-D1), 331 (GRa-D2) and 336 (GRa-D3), C-terminally from the classic translation start site (1: for the GRa-A) (34). Thus, they have different lengths of NTDs but the same DBDs and LBDs. Compared to GRa-A, GRa-C2 and GRa-C3 isoforms have stronger transcriptional activities on a synthetic GRE-driven promoter, while GRa-D1, GRa-D2 and GRa-D3 demonstrate weaker activities (34). GRa-B and GRa-C1, however, possess transcriptional activities similar to that of GRa-A (34). Absence of AF-1 in GRa-D isoforms may explain their reduced transcriptional activity, while ~100 amino acids (particularly 3 polar amino acids) located in the N-terminal portion of AF-1 appear to support increased transcriptional activity of GRa-C isoforms (35). All human GRa isoforms translocate into the nucleus in response to ligand, while they are differentially distributed in the cytoplasm and/or the nucleus in the absence of ligand and display distinct transactivation or transrepression patterns on global gene expression examined by cDNA microarray analyses (34). Such isoform-specific transcriptional activity is in part explained by their distinct chromatin modulatory activity, which is evident in the different potencies of the translational isoforms to induce apoptosis in T-cell Jurkat cells (36).
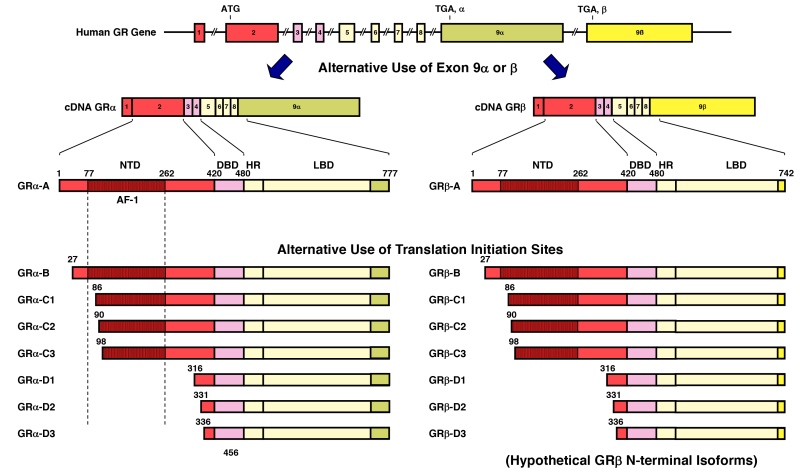
Figure 6.
GR isoforms produced through alternative splicing or use of different translational initiation sites. The human GR (NR3C1) gene contains two terminal exons 9 (9alpha and 9beta) alternatively spliced to produce the classic GR (GRalpha-A) and GRbeta-A. C-terminal dark yellow-colored domains in GRalpha-A and GRbeta-A show their specific portions. Using at least 8 different translation initiation sites located in NTD, the human GR (NR3C1) gene produces multiple GR isoforms termed A through D (A, B, C1-C3 and D1-D3) with distinct transcriptional activities on glucocorticoid-responsive genes. Since GRalpha and GRbeta share a common mRNA domain that contains the same translation initiation sites, the GRbeta variant mRNA appears to be also translated through the same initiation sites and to produce 8 isoforms with different lengths of NTD. Modified from (20,37). AF-1 and -2: activation function-1 and -2; DBD; DNA-binding domain; HD: hinge region; LBD: Ligand-binding domain
Translational Human GRa isoforms are differentially expressed in various cell lines, tissues and at different developmental stages (34). For example, GRa-D isoforms are predominant in immature bone marrow-derived dendritic cells (DCs), while GRa-A is a main isoform in mature DCs, and this characteristic expression explains maturation-specific alteration of glucocorticoid sensitivity in these cells (38). GRa-A is highly expressed in brains of toddlers to teenagers, whereas peak expression of GRa-D is observed in those of neonates (39). Thus, these N-terminal human GRa isoforms may differentially transduce glucocorticoid hormone signals to tissues, depending on their selective expression and inherent activities.
The human GR gene has eleven different promoters with their alternative first exons (1A1, 1A2, 1A3, 1B, 1C, 1D, 1E, 1F, 1H, 1I and 1J) (40,41) (Figure 7). Therefore, the human GR gene can produce eleven different transcripts from different promoters that encode the same GR proteins sharing a common exon 2 containing the translating ATG codon. 1A1, 1A2, 1A3 and 1I are located in the distal promoter region spanning ~32,000-36,000 bps upstream of the translation start site, while 1B, 1C, 1D, 1E, 1F, 1H and 1J position in the proximal promoter region located up to ~5,000 bps upstream of such a site (40). Through differential use of these promoters, expression levels of GR protein isoforms can vary considerably among tissues and disease states (40,42). DNA methylation of the human GR gene promoter area is one of the mechanisms that regulate the activity of specific GR gene promoters. Indeed, childhood trauma, which influences development of the borderline personality disorder by affecting the stress-responsive HPA axis, contributes to the alteration of DNA methylation levels of the human GR gene promoter in the brain (43). Elevated DNA methylation in the human GR gene promoter is also found in the brain hippocampus of the patients with major depression (44). Furthermore, the methylation status of the human GR gene promoter in the peripheral blood is highly altered during the perinatal period. Interestingly, preterm infants demonstrate significantly lower levels of the DNA methylation compared to full-term infants, explaining in part relative glucocorticoid insensitivity observed in preterm babies (45).
In addition to selective use/activation-inactivation of the specific GR gene promoters, alternative untranslated 1st exon transcripts differentially control stability and translational efficiency of their existing GR mRNA, and contribute to differential tissue expression of the GR proteins (46). By employing many splice/translational GR isoforms expressed from different promoters, human GR appears to form at least 256 different combinations of homo- and hetero-dimers with varying expression levels and transcriptional activities. This marked complexity in the transcription/translation of the human GR gene allows cells/tissues to respond differentially to the circulating concentrations of glucocorticoids depending on the needs of respective tissues (20).
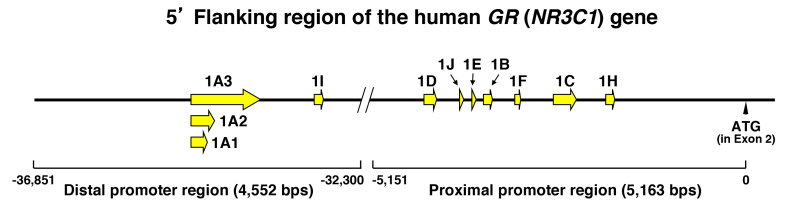
Figure 7.
The human (h) GR (NR3C1) gene has 11 different promoters with specific exon 1 sequences. The hGR (NR3C1) gene has 11 different promoters harboring specific exon 1 sequences. Alternative exon 1s are shown as yellow arrows or arrowheads. The 5’ flanking region of the hGR (NR3C1) gene has proximal and distal promoter regions, which respectively span from ~-37,000 to ~-32,000 and from ~-5,000 to ~0, upstream of the translation initiation site located in the exon 2 (shown as “ATG” and arrowhead), and contain exons 1A1, 1A2, 1A3, and 1I, and 1B, 1C, 1D, 1E, 1F and 1H, respectively. Modified from (40,41).
ACTIONS OF GR
Nucleocytoplasmic Shuttling of GRa
In the absence of ligand, GRa resides primarily in the cytoplasm of cells as part of a large multiprotein complex, which consists of the receptor polypeptide, two molecules of hsp90, and several other proteins (28,47-49) (Figure 8). Following ligand binding, the receptor dissociates from the hsps and translocates into the nucleus. The GRa contains two nuclear translocation signals (NL), NL1 and NL2 (Figure 3): NL1 contains a classic basic-type nuclear localization signal (NLS) structure that overlaps with and extends C-terminally from the DBD of GRa (50). The function of NL1 is dependent on importin a, a protein component of the nuclear translocation system, which is energy-dependent and facilitates the translocation of the activated receptor into the nucleus through the nuclear pore. NL2 spans over almost the entire LBD. In the absence of ligand, binding of hsps with the LBD of GRa masks/inactivates NL1 and NL2, thereby maintaining GRa in the cytoplasm. Inside the nucleus, GRa binds to GREs in the promoter regions of target genes. The interaction of GRa with GREs is dynamic, with the GRa binding to and dissociating from GREs in the order of seconds, while the GRE-bound receptor helps other GRas to bind DNA by increasing chromatin accessibility (the mechanism called “assisted loading”), and ultimately up-regulates their steady state association on glucocorticoid-responsive gene promoters (51,52). The above findings were obtained using the multi-copy GREs artificially inserted into the host cell chromatin, but a recent report confirmed them by examining endogenous GREs using a single molecule imaging technique (53). GRa also modulates transcriptional activity of other transcription factors by physically interacting with them. After modulating the transcription of its responsive genes, GRa dissociates from the ligand and slowly returns to the cytoplasm as a component of the heterocomplexes with hsps (54-56). The ubiquitin-proteasomal pathway degrades ligand-bound GRa in the nucleus, facilitating clearance of the receptor from GREs; thus, this system regulates the transcriptional activity of GRalpha in a negative fashion (57,58).
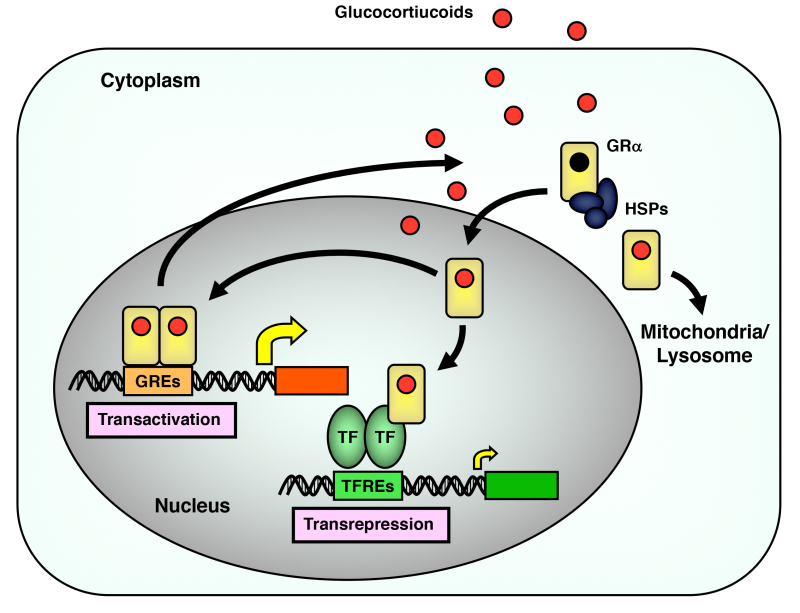
Figure 8.
Intracellular circulation of GR. Circulation of GR between the cytoplasm and the nucleus, and its transcriptional regulation on the glucocorticoid-responsive genes in the nucleus are shown in the panel. GR translocates into mitochondria or lysosomes as well. GREs: glucocorticoid responsive elements; TFREs: transcription factor responsive elements; HSPs: heat shock proteins; TF: transcription factor. From (59).
Several mechanisms have been postulated for the regulation of GRa nuclear export [27]. The Ca2+-binding protein calreticulin plays a role in the nuclear export of GRa, directly binding to the DBD of this receptor (60-62). In contrast, the CRM1/exportin and the classic nuclear export signal (NES)-mediated nuclear export machinery does not appear to be functional directly on GR (50,60,63). Rather, NES-harboring and phospho-serine/threonine-binding protein 14-3-3s can bind the human GR phosphorylated at serine 134, and segregates the nuclear GRa into the cytoplasm (64,65) (see also Section 6. FACTORS THAT MODULATE GR ACTIONS, B. Epigenetic Modulation of GRa, Phosphorylation).
In addition to translocating into the nucleus, GRa was reported to shuttle into mitochondria upon ligand activation and to stimulate mitochondrial gene expression by binding to their own DNA (66) (Figure 8). Exposure of rats to stress or corticosterone induces translocation of GRa to mitochondria and modulates mitochondrial mRNA expression (67), indicating that this activity of GRa is evident at an animal level. GRa was also shown to move into the lysosome, which leads to the negative regulation of its transcriptional activity (68).
Mechanisms of GRa-mediated Activation of Transcription
Classically, GRa exerts its transcriptional activity on glucocorticoid-responsive genes by binding to GREs located in the promoter region of these genes (69). The optimal tandem GREs is an inverted hexameric palindrome separated by 3 base pairs, PuGNACANNNTGTNCPy, on which each GRa molecule binds one of the palindromes, forming a homodimer on this binding site through multiple contacts between the 2 receptors (70,71). Recent research indicated that sequence variation of GREs, including 3 non-specific spacer nucleotides, influences the 3-dimensional structure of DBD and modulates the transcriptional activity of whole GRa molecule (72,73); Binding of GRa DBD to GRE DNA sequence induces conformational changes in the dimerization surface located in D-loop through the lever arm, which positions itself between the first a-helix and D-loop (Figure 4A). Two receptors bound on each GRE half site then communicate with each other with their GRE sequence-specific dimerization surfaces, and ultimately develop net transcriptional activity. These findings suggest that DNA GRE is a sequence-specific allosteric modulator of GRa transcriptional activity through alteration of its protein conformation, explaining in part gene-specific transcriptional effects of this receptor.
The GRE-bound GRa stimulates the transcription rates of glucocorticoid-responsive genes by facilitating formation of the transcription initiation complex on the GREs-containing promoter of these genes via its AF-1 and AF-2 transactivation domains (74) (Figure 3). Actions of AF-1 located in NTD of GRa is ligand-independent, while AF-2 is created on GRa LBD upon ligand-binding (75).
The transcription initiation complex attracted and formed on DNA-bound GRa is a mega protein structure that include over 100 proteins with different activities, such as RNA polymerase II and its ancillary factors, general transcription factors and numerous co-regulatory molecules with/without enzymatic activities (74). Research studies aimed to identify molecules interacting with GRa AF-2 have led to several proteins and protein complexes, called coactivators or cofactors, that form a bridge between DNA-bound GRa and the transcription initiation complex, and assist enzymatically with the transmission of the glucocorticoid signal to RNA synthesis promoted by the RNA polymerase II (76) (Figure 9). These include: (1) p300 and the homologous cAMP-responsive element-binding protein (CREB)-binding protein (CBP), which also serve as macromolecular docking “platforms” for transcription factors from several signal transduction cascades, including NRs, CREB, activator protein-1 (AP-1), NF-kB, p53, Ras-dependent growth factor, and signal transducers and stimulators of transcription (STATs) (77). Because of their central position in many signal transduction cascades, the p300/CBP coactivators are also called co-integrators; (2) p300/CBP-associated factor (p/CAF), originally reported as a human homologue of yeast Gcn5, which interacts with p300/CBP and is also a broad transcription coactivator (78,79); and (3) the p160 family of coactivators, which preferentially interact with SRs (80). These include the steroid receptor coactivator-1 (SRC-1), SRC-2, which consists of transcription intermediate factor-II (TIF-II) and the glucocorticoid receptor-interacting protein-1 (GRIP1), and SRC-3, which consists of the p300/CBP/co-integrator-associated protein (p/CIP), activator of thyroid receptor (ACTR) and the receptor-associated coactivator-3 (RAC3) (76,80,81). These 3 subclasses of p160 family coactivators are also called, respectively, as nuclear receptor coactivators (NCoA) 1, 2 and 3.
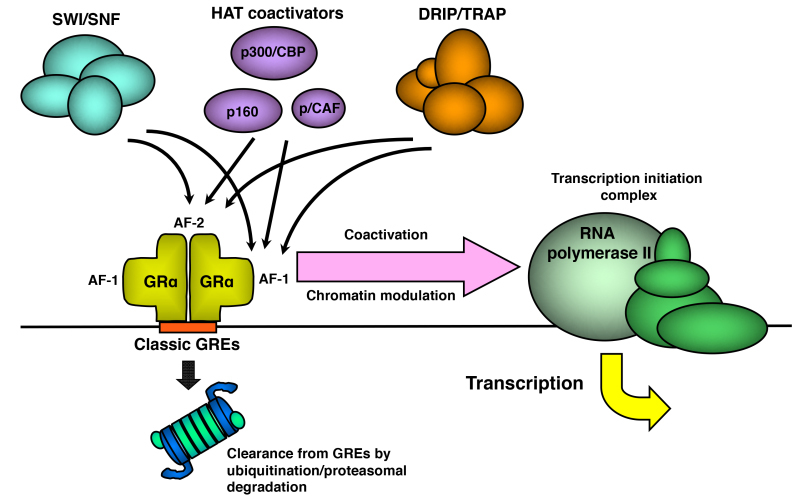
Figure 9.
Schematic model demonstrating the interaction and activity of HAT coactivators and other chromatin modulators, which are attracted by GR to the promoter region of glucocorticoid-responsive genes. Promoter-associated GR is cleared by the ubiquitin-proteasomal pathway, which regulates turnover of GR on DNA. Modified from (82). AF-1 and -2: activation function-1 and -2; CBP: cAMP-responsive element-binding protein (CREB)-binding protein; DRIP: vitamin D receptor-interacting protein; GREs: glucocorticoid response elements; p/CAF: p300/CBP-associated factor; SWI/SNF: mating-type switching/sucrose non-fermenting; TRAP: thyroid hormone receptor-associated protein.
The p160 coactivators are the first to be attracted to the DNA-bound NRs and help accumulating p300/CBP and p/CAF proteins to the promoter region, indicating that p160 proteins play a pivotal role in NR-mediated transactivation. A study using the cryoelectron microscopy demonstrated detailed attraction modes of p160 proteins and p300/CBP to DNA-bound and ligand-activated ERa (83); Each of the tandem ER response elements (EREs)-bound receptors independently attracts one p160 molecule via the transactivation surface of the receptor created by their AF-1 and AF-2. Then, the two NCoAs attracted to the receptors recruits one p300/CBP molecule to the DNA/receptors/p160s complex through multiple contacts mediated by different portions of the p160 proteins.
In addition to physical interaction and subsequent formation of the transcriptional initiating complex on the DNA-bound receptors by these coactivators (that is assembly of transcriptional initiation complex), these molecules have intrinsic histone acetyltransferase (HAT) activity through which they acetylate specific lysine residues of chromatin-bound histones, loosen the tightly assembled chromatin structure and facilitate access of other transcription factors and transcriptional complexes to the promoter region (76). These HAT coactivators also acetylate specific lysine residues of their own molecules, NRs and other transcription factors, and modulate their mutual protein-protein interaction and/or association to attracted promoters (84-86). The p160 family coactivators and p300/CBP protein contain one or more copies of the coactivator signature motif sequence LxxLL, where L is leucine and x is any amino acid (80,87). LxxLL forms a helical structure, and develops multiple hydrophobic bonds with its leucine residues to the AF-2 surface, which is created by helixes 3, 4 and 12 of the GRa LBD upon binding to ligand glucocorticoid (Figure 10A). p160-type coactivators contain 3 LxxLL motifs in its nuclear receptor-binding box (NRB) located in their central portion (76) (Figure 10B). Each of these motifs demonstrates different affinity to various NRs, indicating that specific p160 proteins participate in the transcriptional activity of particular NRs through preferential use of LxxLL motifs (88). For example, GRa preferentially interacts with GRIP1 p160 protein through C-terminally located 3rd LxxLL motif of this coactivator (89).
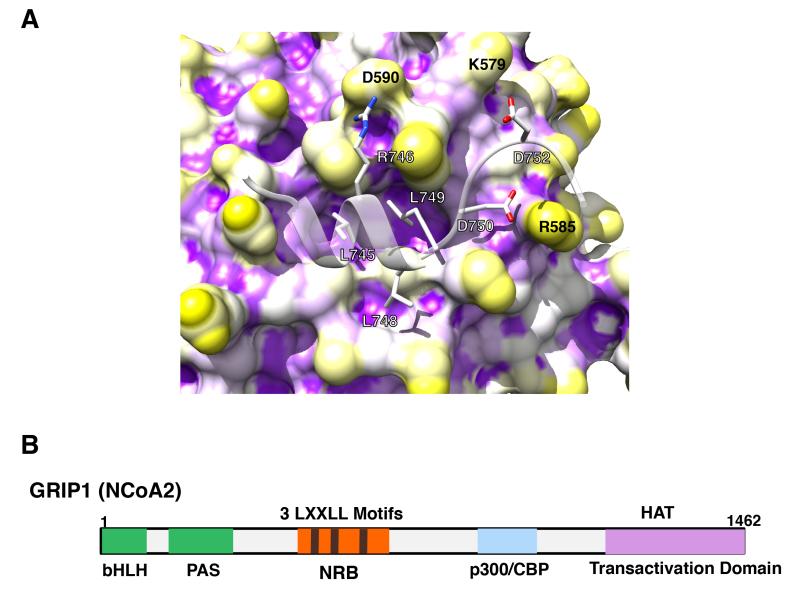
Figure 10.
p160 coactivators physically interact with its multiple LxxLL motifs to the AF-2 surface of GR. A: 3-dimensional interaction image of GR AF-2 and the LXXLL peptide. The GR AF-2 surface has three large pockets into which core leucines (L745, L748 and L749) of the LXXLL peptide deeply bury themselves. There are additional intermolecular contacts that are important for peptide binding, including the electrostatic bonds created between (i) R746 (LXXLL peptide) and D590 (receptor), (ii) D750 (LXXLL peptide) and R585 (receptor) and (iii) D752 (LXXLL peptide) and K579 (receptor). From (89). B: p160-type coactivators (NCoAs) have 3 LxxLL motifs in their NR-binding box (NRB). Linearlized GRIP1 (NCoA2) molecule with NRB located in the middle portion is shown as a representative of the p160-type coactivators (NCoAs). In addition to NRB, GRIP1 has the basic helix-loop-helix (bLHL) and the PAS domains in its N-terminal portion, and p300/CBP-binding domain and one transactivation domain containing the HAT domain in the C-terminus.
The AF-2 transactivation domain of GRa also attracts several other distinct chromatin modulators, such as the mating-type switching/sucrose non-fermenting (SWI/SNF) complex and components of the vitamin D receptor-interacting protein/thyroid hormone receptor-associated protein (DRIP/TRAP) complex (76). The SWI/SNF complex is an ATP-dependent chromatin remodeling factor with a multi-subunit structure in which the ATPase domain functions as the catalytic center (90). Depending on the energy of ATP hydrolysis, the SWI/SNF complex introduces superhelical torsion into DNA. One of its components, SNF2 binds to AF-2 of GRa directly, functioning as an interface between the GRa and the SWI/SNF complex (91). The DRIP/TRAP complex is also a multiprotein conglomerate, which consists of over 10 different proteins, including DRIP205/TRAP220/PBP and components of SMCC (76). The DRIP/TRAP complex may modulate transcription through interaction and modification of general transcription factors, such as TFIIH or the C-terminal tail of the RNA polymerase II. DRIP205/TRAP220 contains two LxxLL motifs through which it binds to the ligand-activated AF-2 directly (92). Since the DRIP/TRAP complex and p160 coactivators use the same motif for interaction with NRs, they may bind to the same site of these receptors and sequentially interact with them for full activation of transcription. Alternatively, they may interact with a particular set of NRs, or have a different effect on different tissues (76,81).
In contrast to the mechanisms of transactivation by AF-2, those of AF-1 are not as well elucidated yet. Using the yeast system, the Ada complex may act on AF-1-mediated transcriptional activation through direct interaction to this module (93). The SWI/SNF complex, TBP and the HAT coactivators, such as p160 and p300/CBP, also physically interact with AF-1 and mediate its transcriptional activity (94-97). In addition, DRIP150, a component of the DRIP/TRAP complex, and the tumor susceptibility gene 101 (TSG101) interact with the AF-1 of the GRa in a yeast two-hybrid screening (98). The RNA coactivator, steroid RNA activator (SRA), also interacts with AF-1 (99). Given that any of these molecules and complexes interact with both AF-1 and AF-2, it is likely that concurrent activation of AF-1 and AF-2 by their common and/or distinct binding partners may be necessary for optimal activation of GRa-mediated transcriptional activity (100).
Several coactivators supporting the particular actions of glucocorticoids have been identified for GRa. The PPARg coactivator-1a (PGC1a) is a ~800 amino acid single polypeptide molecule originally identified as a cofactor physically interacting with PPARg in the yeast two-hybrid screening using a brown adipocyte cDNA library (101). PGC1a has an essential role in thermogeneration and energy metabolism by controlling mitochondrial biogenesis (101). It also regulates gluconeogenesis and cholesterol metabolism, as well as blood pressure and muscle fiber determination through physical interaction with various NRs, transcriptional factors and coactivators, such as PPARa, hepatocyte nuclear factor 4, CREB, nuclear respiratory factors, and p160 and p300/CBP coactivators (101). GRa also interacts physically with PGC1a through the latter’s LxxLL motif and this interaction is important for stimulation of gluconeogenesis through transcriptional stimulation of the 2 key genes respectively encoding the glucose-6-phosphatase (G6P) and the phosphoenolpyruvate carboxykinase (PEPCK) (101). It is known that longevity-associated histone deacetylase Sirt1 regulates PGC1a activity through its deacetylase activity-dependent or -independent manner (102,103). Sirt1 is shown to interact physically with GRa as well, and PGC1a and Sirt1 cooperatively enhance GR-induced transcriptional activity of glucocorticoid-responsive genes (104).
The CREB-regulated transcription coactivator 2 (CRTC2) is a coactivator previously known to be specific to CREB, and plays a central role in the glucagon-mediated activation of gluconeogenesis in the early phase of fasting (105). This coactivator functions also as a coactivator of GRa by physically interacting with its LBD outside of AF-2, and is required for glucocorticoid-mediated early phase gluconeogenesis by supporting the transcriptional activity of GRa on the G6P and PEPCK genes, while PGC1a cooperates with GRa for maintaining a late phase of fasting-induced gluconeogenesis (106).
Presence of numerous transcriptional cofactors that interact with GRa and influence its transcriptional activity indicate that they may have functional redundancy and/or activities specific to each of them, regulating particular sets of GRa-responsive genes. A study employing knockdown of GRalpha cofactors, such as CCAR1, CCAR2, CALCOCO1 or ZNF282, has addressed this important issue: it revealed that knockdown of any of these cofactor molecules resulted in specific impact on the expression of a particular set of glucocorticoid-responsive genes (107), suggesting that each cofactor molecule has distinct transcriptional regulatory activity on GRa, thus their expression profiles in tissues/organs potentially influence the transcriptional activity of GRa in respective tissues.
Emerging Concept on GRa-mediated Transcriptional Repression
GRa has long been believed to exert its transcriptional activity by binding to the classic GREs, which consists of inverted hexameric palindrome separated by 3 base pairs. However, Surjit, et al. identified unique DNA sequences also targeted by the GRa DBD, called “negative” GREs (nGREs), which play substantial roles in gene transrepression caused by GRa (108). The consensus sequence of nGREs is an inverted quadrimeric palindrome separated by 0-2 nucleotide pairs (CTCC(N)0–2GGAGA). In the structural study employing the prototype nGREs found in the thymic stromal lymphoprotein (TSLP) promoter as a model, 2 GRa molecules bound each palindrome as a monomer with different affinity in a head-to-tail fashion, in contrast to GRa-classic GREs where 2 receptors bind DNA in a head-to-head fashion (109) (Figure 11). nGREs are ubiquitously present in the genes repressed by glucocorticoids throughout several animal species, facilitating access of the silencing mediator for retinoid and thyroid hormone receptors (SMRT)/nuclear receptor corepressor (NCoR)-repressing complexes on the agonist-associated GRa bound on these sequences. This is a new concept, indicating that direct binding of GRa through its DBD to DNA sequences distinct from those of the classic GREs mediates glucocorticoid-induced transcriptional repression. However, a genome-wide study revealed that classic GREs and the “new” nGREs both contribute to transactivation and transrepression of glucocorticoid-responsive genes, suggesting that GRa-targeting DNA sequences per se are insufficient to confer direction of transcriptional regulation, but epigenetic factors and subsequent chromatin modification may play critical roles (110).
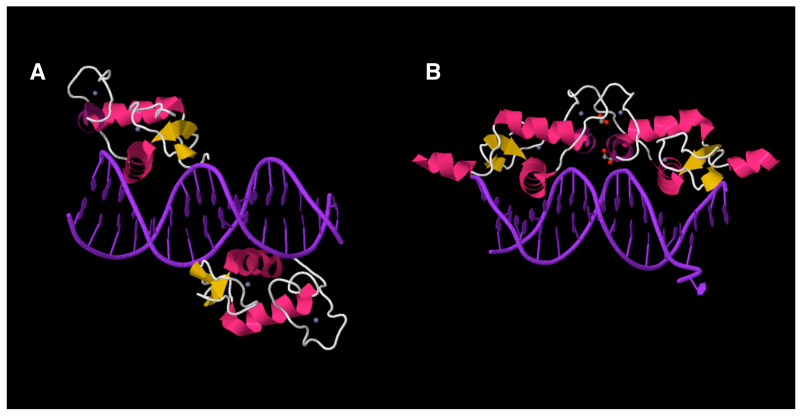
Figure 11.
GR binds nGREs as a monomer. GR binds nGREs as a monomer at each of its half site (A) in contrast to its binding as a homo-dimer to classic GREs (B). nGREs of the mouse TSLP gene is used as an example. Images are from the PDB Website (www.rcsb.org). Image data for GR interaction with nGREs and classic GREs are DOI: 10.2210/pdb4hn5/pdb and DOI: 10.2210/pdb3g9m/pdb, respectively.
Interaction of GRa with Transcription Factors
Glucocorticoids exert their diverse effects through its single receptor protein module, the GRa. In addition to direct regulation of gene expression through GRa/DNA interaction, these hormones affect other signal transduction cascades through mutual protein-protein interactions between specific transcription factors and GRa, influencing the former’s ability to stimulate or inhibit the transcription rates of the respective target genes.
The protein-protein interaction of GRa with other transcription factors may take place on the promoters that do not contain GREs (tethering mechanism), as well as on those having both GRE(s) and responsive element(s) of the transcription factors that interact with GRa (“composite promoters”) (111) (Figure 12). Repression of the transactivation activity of other transcription factors through protein-protein interaction may be particularly important in suppression of immune function and inflammation by glucocorticoids (112,113). Substantial part of the effects of glucocorticoids on the immune system may be explained by the interaction between GRa and NF-kB, AP-1 and probably STATs (114-116). It was also reported that GRa directly interacts with the transcription factors “T-box expressed in T-cells” (T-bet) and GATA-3, which play key roles respectively in the differentiation of T helper-1 and T helper-2 lymphocytes (117,118). GRa also influences indirectly the actions of the interferon regulatory factor-3 (IRF-3) through the p160 nuclear receptor GRIP1, by competing with this factor for binding to the coactivator (119). These transcription factors are important for the regulation of immune function and the above interactions may explain some GR actions on the immune system. The following subsections will discuss GRa-interacting transcription factors and their effects on GRa-induced transcriptional activity.
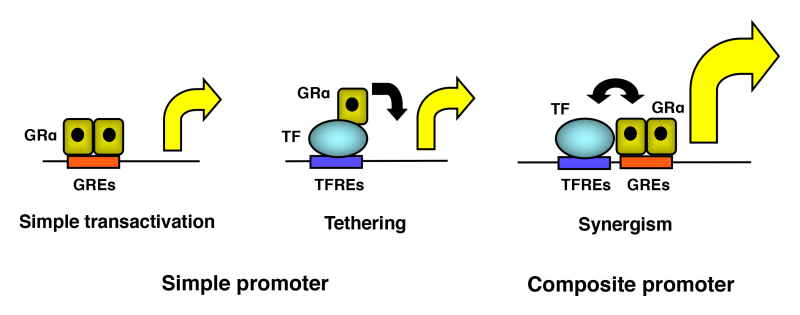
Figure 12.
Three different modes of transcriptional regulation of the glucocorticoid-responsive promoters by GR. GR may interact with other transcription factors directly or indirectly. Protein(s) or protein complex(es) may intermediate their interaction in the latter case. GREs: glucocorticoid responsive elements; TF: transcription factor; TFREs: transcription factor responsive elements
Nuclear Factor-kB (NF-kB)
NF-kB is one of the most important transcription factors that regulate inflammation and immune function. NF-kB is stimulated by many inflammatory signals and cytokines (115,120). It is a dimer of various members of the NF-kB/Rel family, including p50 (and its precursor p105), p52 (and its precursor p100), c-Rel, RelA and RelB in mammalian organisms. The heterodimer p65/p50 is a major and the most abundant form of NF-kB. In its inactive form, NF-kB creates a trimer with an additional regulatory protein, IkB in the cytoplasm. A variety of extracellular signals, such as bacterial and viral products (like lipopolysaccharide (LPS)) and several proinflammatory cytokines, induces phosphorylation of IkB by activating a cascade of kinases. The phosphorylated IkB then dissociates from NF-kB and is catabolized, while the liberated NF-kB enters into the nucleus where it binds to the kB-responsive elements in the promoter regions of its responsive genes. Ligand-activated GRa directly binds NF-kB p65 at its Rel homology domain through its DBD and suppresses the transcriptional activity of NF-kB, while NF-kB suppresses GRa-induced transactivation through GREs. Interaction with GRa inhibits binding of NF-kB to its responsive elements or neutralizes its ability to transmit an effective signal (121-124). The LBD of GRa is necessary for this suppressive action (125). GRa also suppresses NF-kB-induced transactivation by an additional mechanism, in which the GRa tethered to the kB-responsive promoters attracts histone deacetylases (HDACs) and/or modulates the phosphorylation of the RNA polymerase II C-terminal tail (126,127). In addition, ligand-activated GRa increases the synthesis of IkB by stimulating its promoter activity through classic GREs, thus segregating active NF-kB from the nucleus by forming inactive heterocomplexes with IkB in the cytoplasm (128). A study further indicated that attraction of the p160 coactivator GRIP1 together with GRa to NF-kB is required for glucocorticoid-induced repression of NF-kB-mediated cytokine gene expression in mouse primary macrophages (129).
Activator Protein-1 (AP-1)
AP-1 is a transcription factor, which regulates diverse gene expression involved in cell proliferation and differentiation (114,130,131). It acts as a dimer of the bZip protein family members, in which c-Fos and c-Jun heterodimers are most abundant. AP-1 transduces biological activities of phorbol esters, growth factors and pro-inflammatory cytokines, such as IL-1 and tumor necrosis factor (TNF) a. These compounds/cytokines stimulate different members of the mitogen-activated protein kinase (MAPK) family, e.g., p38 MAPK, extracellular signal-regulated kinase (ERK) and Jun N-terminal kinase (JNK). Once these kinases are activated, they stimulate the synthesis of specific transcription factors involved in the induction of fos and jun gene transcription, as well as enhance the transcriptional activity of both pre-existing and newly synthesized c-Fos/c-Jun proteins. AP-1 and GRa mutually interact and repress each other’s transcriptional activity on their respective responsive promoters. The LBD and DBD of GRa and the leucine zipper domain of c-Jun are necessary for this interaction (29). Inhibition of the binding of AP-1 to DNA may be one of the underlying mechanisms of GRa-induced suppression of AP-1-mediated transcriptional activity. Furthermore, GRa competes with AP-1 for the p300/CBP coactivator, which has a limited reserve, therefore, AP-1 may not have access to adequate amounts of this coactivator to exert its transcriptional activity fully (132).
cAMP Response Element-binding Protein (CREB)
CREB functions downstream of many hormones and bioactive molecules, which bind to the cell surface-located G-protein-coupled receptors that employ cAMP as their second messenger. CREB is also a member of the bZip transcription factors (133). It forms homo- and hetero-dimers with other proteins of the same family and binds to the cAMP-responsive element (CRE). Stimulation of the above receptors induces the accumulation of cAMP that leads to activation of the cAMP-dependent protein kinase A (PKA). This kinase then phosphorylates CREB at a specific serine residue and promotes recruitment of the transcriptional co-integrator CBP and its specific coactivator CRTC2 to stimulate the transcription of cAMP-responsive genes. GRa and CREB mutually repress the transcription from their simple responsive promoters (134,135). Although direct association of GRa and CREB has been reported in vitro, their physical interaction is still unclear (134,136). CRTC2 might act as a bridging factor between CREB and GRa, particularly in their synergistic activation of the composite promoters, such as that of G6P, PEPCK and the somatostatin gene, which contain both GREs and CRE sequences (106,136,137) (see also Section 5. ACTIONS OF GR, B. Mechanisms of GRalpha-mediated Activation of Transcription).
Transforming Growth Factor (TGF) b Downstream Smad6
Members of the Smad family of proteins transduce signals of transforming growth factor (TGF) b superfamily members, such as TGFb, activin and bone morphogenetic proteins (BMPs), by associating with the cytoplasmic side of the type I cell surface receptors of these hormones (138). Nine distinct vertebrate Smad family members have been identified, which are classified into three groups: receptor-regulated Smads (R-Smads), such as Smad1, 2, 3, 5 and 8, a common-partner Smad (Co-Smad), Smad4, and inhibitory Smads (I-Smads) like Smad6 and Smad7 (138).
Upon binding of TGFb, activin or BMP to their receptors, cytoplasmic R-Smads are phosphorylated by the receptor kinases, translocate into the nucleus and stimulate the transcriptional activity of TFGb-, activin- or BMP-responsive genes by binding to their response elements located in their promoter region as a hetero-trimer with Co-Smad (138). I-Smads, such as Smad6 and Smad7, act as inhibitory molecules in the TGFb family signaling, by forming stable associations with activated type I receptors, which prevent the phosphorylation of R-Smads (138). Smad6 also competes with Smad4 in the heteromeric complex formation induced by activated Smad1 (139). In addition, I-Smads directly suppress the transcriptional activity of TGFb family signaling by binding to promoter DNA and attracting HDACs and/or the C-terminal binding protein (CtBP) (140-142). Since I-Smads are produced in response to activation of the TGFb family signaling (143), they literally function in the negative feedback regulation of the Smad signaling pathways. Smad6 preferably inhibits BMP signaling, while Smad7 is a more general inhibitor, repressing TGFb and activin signaling, in addition to that of BMP (144).
We found that Smad6 physically interacts with the N-terminal domain of the GRa through its Mad-homology 2 domain and suppresses GRa-mediated transcriptional activity in vitro (145). Adenovirus-mediated Smad6 overexpression also inhibits glucocorticoid action in rat liver in vivo, preventing dexamethasone-induced elevation of blood glucose levels and hepatic mRNA expression of PEPCK, a well-known rate-limiting enzyme of hepatic gluconeogenesis (145). Smad6 suppresses GRa-induced transactivation by attracting HDAC3 to DNA-bound GRa and by antagonizing acetylation of the histones H3 and H4 induced by p160 HAT coactivators (145). Thus, Smad6 regulates glucocorticoid actions as a corepressor of GRa. It appears that the anti-glucocorticoid actions of Smad6 may contribute to the neuroprotective, anti-catabolic and pro-wound healing properties of the TGFb family of proteins through cross-talk between TGFb family members and glucocorticoids (145).
C2H2-type Zinc Finger Proteins (ZNFs)
C2H2-type ZNFs constitute the largest class of putative human transcription factors consisting of over 700 member proteins (146,147). In addition to C2H2-type zinc fingers (ZFs), these proteins harbor several structural modules, such as the Broad-Complex, Tramtrack, and Bric-a-brac (BTB)/Poxvirus and zinc finger (POZ), Krüppel-associated box (KRAB) and SCAN domains (147). These modules are usually located in the N-terminal portion, and function as platforms for protein-protein interactions, whereas ZFs are positioned in the C-terminal area and function mainly as a DNA-binding domain (147). The BTB/POZ and KRAB domains have transcriptional regulatory activity (mostly repressive), whereas the SCAN domain does not (148). Among human C2H2-type ZNFs, about 7% have a BTB/POZ domain (BTB/POZ-ZNFs), 43% harbor a KRAB domain (KRAB-ZNFs) and 7% contain a SCAN domain (SCAN-ZNFs) (146). Sixty-seven % of the human C2H2-type ZNFs have only ZFs without any of these domains (thus, they are “poly-ZNFs”) (146). Some poly-ZNFs, such as members of the specificity protein (SP)/Krüppel-like factor (KLF) family transcription factors (e.g., SP1, KLF4 and KLF11) cooperate with GRa for regulating the transcriptional activity of specific glucocorticoid-responsive genes in distinct biological pathways, such as monoamine oxidase A expression in CNS and glucocorticoid-mediated skin barrier formation in prenatal fetus (147). Furthermore, GRa stimulates the transcriptional activity of the KLF9 gene through the GREs located in the promoter region of this gene, and expressed KLF9 plays important roles in glucocorticoid-mediated survival of the newly differentiated hippocampal granule neurons (147). One poly-ZNF called CCCTC-binding factor (CTCF) is an architectural protein playing a major role in the formation of chromatin looping, which governs enhancer-gene promoter communication, and ultimately contributes to the tissue/phase-specific expression of glucocorticoid-responsive genes (149). Although direct evidence of its interaction to GRa is still missing, CTCF interacts with ERa and the thyroid hormone receptors (TRs) and regulates their transcriptional activity (150,151), thus it is highly possible that this molecule also plays roles in the regulation of GRa transcriptional activity. One KRAB-ZNF, the zinc finger protein 764 (ZNF764), which composes of a N-terminally located KRAB domain and seven C2H2-type ZF motifs in the C-terminal area, was identified as a coactivator of several SRs including GRa, possibly cooperating with other NR coactivators (152). Indeed, haploinsufficiency of the ZNF764 gene by microdeletion was associated with partial tissue insensitivity to glucocorticoids and developmental abnormalities of androgen-dependent organs in an affected boy (152). In a genome-wide binding study using ChIP-sequencing, ZNF764- and GRa-binding sites are found in close proximity, indicating that ZNF764 modulates GRa transcriptional activity by incorporated in the transcriptional complex formed on DNA-bound GR (153).
Forkhead Transcription Factors
Forkhead transcription factors are characterized by their DNA-binding domain called “Forkhead Box”, and consist of over 100 family members classified from FOXA to FOXR (154). Among them, FOXO subgroup proteins (FOXO1, 3, 4 and 6 in humans) mediate biological actions of the insulin/PI3K/Akt signaling pathway through phosphorylation of several serine/threonine residues of this subgroup proteins, acting on cell proliferation, cell cycle regulation, oxidative stress, DNA repair, energy and glucose metabolism (154). Some of forkhead transcription factors (e.g., FOXA1) can act as pioneer factors for other transcription factors including NRs, by opening DNA-binding sites of the latter molecules on the chromatin (see also Section 5. ACTIONS OF GR, E. New Findings on Genome-wide Transcriptional Regulation by GRa) (155). FOXA3 acts as a pioneer factor for GRa by facilitating the latter binding to DNA possibly through modulation of the chromatin accessibility and is required for glucocorticoid-mediated fat accumulation in adipose tissues (156).
Other Transcription Factors
Functional interaction of GRa has also been reported with other transcription factors, including the chicken ovalbumin promoter-upstream transcription factor II (COUP-TFII), HNF-6, NR4A orphan receptors (neuron-derived orphan receptor-1 (NOR-1), nuclear receptor-related 1 (NURR1) and Nur77), liver X receptors (LXRs), farnesoid X receptor (FXR), p53, T-bet, GATA-1 and -3, Oct-1 and -2, NF-1 and C/EBPb. COUP-TFII is an orphan nuclear receptor, which plays important roles in neurogenesis as well as glucose, lipid and xenobiotic metabolism. This NR physically interacts with the hinge region of GRa and suppresses GRa-induced transcriptional activity by attracting the corepressor SMRT (157). Mutual protein-protein interaction of GRa and COUP-TFII was necessary for glucocorticoid-induced enhancement of the promoter activity and the endogenous mRNA expression of the COUP-TFII-responsive PEPCK, suggesting that COUP-TFII may participate in some of the metabolic effects of glucocorticoids through direct interactions with GRa (157). The hepatocyte nuclear factor 6 (HNF6) is a transcription factor that consists of 2 different DNA binding domains (CUT and homeobox) and plays an important role in the hepatic metabolism of glucose. It represses GRa-induced transactivation by directly binding to GRa DBD (158). Interaction of another orphan nuclear receptor Nur77 and GRa is critical for the regulation of proopiomelanocortin (POMC) gene expression (159). LXRs consist of 2 isoforms LXRa and LXRb, and play a central role in the regulation of cholesterol/fatty acid metabolism by binding to their metabolites as a ligand, while FXR acts on bile acid metabolism. GRa and these NRs modulate each other’s transcriptional activity by communicating through direct protein-protein interaction (160-162). p53, a transcription factor functioning as a tumor suppressor, physically interacts with GRa in the cytoplasm along with an additional protein Hdm2. GRa and p53 mutually repress each other’s transcriptional activity by increasing their degradation rates (163,164). GRa also interacts with Oct-1 and -2 on the mouse mammary tumor virus (MMTV) promoter and the gonadotropin-releasing hormone promoter (165-169). The POU domain of Oct-1 and the DBD of GRa interact with each other in vitro. NF-1, which also stimulates the MMTV promoter, interacts with GRa and cooperatively modulates the activity of this promoter (169,170). The transcriptional activity of GATA-1, a transcription factor that plays an essential role in the erythroid differentiation is repressed by GRa at the experimental cellular levels. NTD of GRa is necessary for the interaction with GATA-1 (171). The CAAT/Enhancer-binding Protein (C/EBP) is one of the bZip family transcription factors that have diverse effects on proliferation, development and differentiation of embryonic cells/fetus, and influence functions of the liver, adipose, immune and hematopoietic tissues in adults (172). C/EBPb, also known as the nuclear factor IL-6 (NF-IL6), synergistically stimulates transcription of GRa on the composite promoter that contains both C/EBPb- and GRa-binding sites (173). GRa, on the other hand, enhances C/EBPb activity on its simple responsive promoter (173,174). Direct in vitro binding of these proteins has been reported.
GENOME-WIDE TRANSCRIPTIONAL REGULATION BY GRa
Chromatin-based Regulation of GRa Transcriptional Activity
GRa regulates expression of glucocorticoid-responsive genes by influencing their transcriptional activity through direct or indirect interaction with their enhancer/promoter regions. In eukaryotic cells, DNA is packed into chromatin by associating with numerous nuclear proteins, such as histones and chromatin-modifying factors (175,176). Double-stranded DNA wraps by 1.67 turns around a histone octamer that consists of 2 copies of each core histones H2A, H2B, H3 and H4, and forms the smallest structural unit called “nucleosome”, which is further compacted into a higher order chromatin. Nucleosome-associated histones possess a highly flexible N-terminal tail whose chemical modifications, such as acetylation and methylation at specific lysine (K) residues, modulate accessibility of GRa to its target DNA sequences residing in chromatin. Chromatin is further packed into the 3-dimensional structure called topologically associated domains (TADs) in which several protein-coding gene bodies, promoters and regulatory elements interact with each other through formation of chromatin looping, and their modes of interaction alter in different cellular circumstances. A poly-ZNF protein CTCF plays a central role in the formation of chromatin looping by cooperating with the cohesion protein complex and other accessory factors, including the transcription factor IIIC (TFIIIC), ZNF143, PR domain zinc finger protein 5 (PRDM5) and chromodomain helicase DNA-binding protein 8 (CHD8) (147,149) (Figure 13). In addition to CTCF, interaction of transcription factors, such as between GRa and NF-kB, influences formation of chromatin looping possibly through cooperation with CTCF (177). A study using a new technique called Hi-C (high throughput 3C) further revealed that even chromosomes are packed into the nucleus with some orders shared by many tissues/organs (178,179).
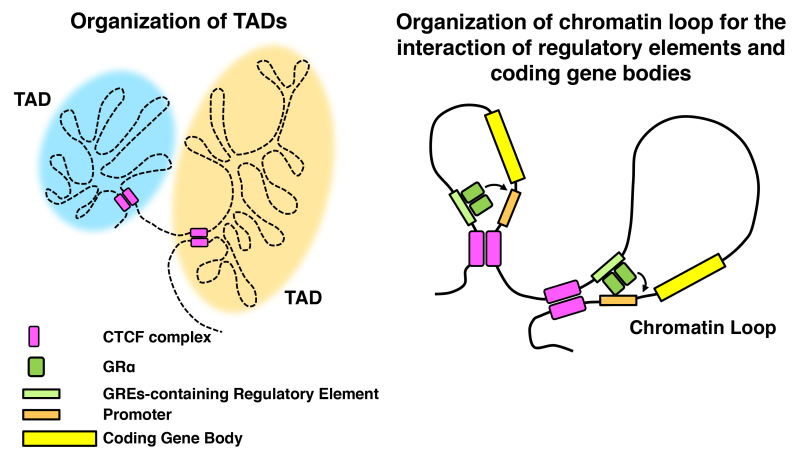
Figure 13.
Organization of the topologically associated domain (TADs) and chromatin looping promoted by CTCF for differential expression of glucocorticoid-responsive genes. CTCF organizes 3-dimensional chromatin interaction for the formation of TADs and chromatin looping, in cooperating with the cohesion protein complex and other component proteins. Chromatin loop-forming activity of CTCF is essential for differential use of enhancers/promoters by GR-responding genes, and underlies organ/tissue-specific actions of glucocorticoids. Modified from (147).
In rat liver, more than 11,000 GR-binding sites (GBSs) are identified primarily at intergenic distal and intronic regions, but only ~10% of GBSs are located in the promoter area (~2.5 kbs from the transcription start site: TSS), consistent with the fact that distantly located enhancer regions can communicate with the gene promoter through gene looping (180,181). Interestingly, ~80-90% of GRa-accessible sites exists prior to glucocorticoid addition/GRa stimulation, while their distribution is highly tissue-specific, indicating that local tissue factor(s) mainly determine(s) the sets of genes responsive to glucocorticoids by regulating chromatin accessibility (180). Indeed, some transcription factors, such as C/EBPb, AP-1 and FoxA1, have their binding sites close to GBSs (thus, composite sites) and act as tissue-specific priming factors (or pioneer factors) for the access of GRa to GBSs, respectively in murine mammary epithelial cells, rat liver and human prostate cancer cells (181-183). These pre-existing GBSs are enriched with CpG islands and are generally demethylated, further suggesting that DNA methylation also contributes negatively to the opening of GBSs (184). However, a study revealed that GRa can act as a pioneer factor for several other transcription factors previously reported to be pioneer factors for GRa (185). This report indicates that GRa can function both as a pioneer and a dependent factor based on the composition of the binding sites in the regulatory elements and/or local chromatin conditions.
Influence of Gene Variation (Single Nucleotide Polymorphisms: SNPs) to Tissue Glucocorticoid Responsiveness
Humans demonstrate variation in their responsiveness to glucocorticoids (sensitivity to glucocorticoids), which then influences the development of numerous disorders, such as hypertension, obesity, diabetes mellitus, osteoporosis and ischemic heart diseases, asthma and acute lymphoblastic leukemias. However, genetic background(s) that explain(s) such difference in glucocorticoid responsiveness among human subjects is(are) not known. To access this problem, variation of the single nucleotide polymorphisms (SNPs) in over 100 individuals was compared with glucocorticoid-induced mRNA expression profiles in subjects’ EBV-transformed lymphocytes and their secretion of some cytokines (186). The results revealed that the SNPs located close (~100 kbps) to the glucocorticoid-responsive genes were associated with variation in glucocorticoid responsiveness of their own mRNA expression, while SNPs located in the transcription factors known to regulate GRa transcriptional activity did not show statistically significant differences. These results suggest that the genetic areas close to glucocorticoid-responsive genes, possibly containing enhancer regions and/or other gene regulatory sequences, influence primarily the responsiveness of mRNA expression of their associated genes to glucocorticoids, rather than those found in the protein-coding sequence of GRa, its partner molecules or glucocorticoid-responsive genes themselves. The above results on the genetic factors determining individual glucocorticoid sensitivity are consistent with recent findings obtained in the genome-wide association studies (GWAS) in which ~70% of SNPs associated with susceptibility to common disorders and traits (thus individual variation) are found in the gene regulatory regions but not in the protein-coding sequences (187).
Tissue/Organ-specific Actions of GRa Revealed by GR Gene Knockout/Knockin Studies
Modifications of gene expression with gene knockout (deletion of existing genes) are tremendously helpful for understanding physiologic actions of endogenous GRa in glucocorticoid-target tissues. Whole body GR gene knockout revealed that GR deficient pups die just after birth due to respiratory insufficiency caused by lack of lung surfactant, indicating that GR action is essential for survival (188). By using the same mice, GR is also shown to be required for gluconeogenesis upon fasting and erythropoiesis under stress (such as erythrolysis or hypoxia) (189,190). Mice harboring forebrain-specific GR gene knockout developed a phenotype mimicking major depressive disorder in humans, including hyperactivity and impaired negative regulation of the HPA axis, indicating that alteration of GRa actions in the forebrain plays a causative role in the disease onset of major depressive disorder (191). Paraventricular nucleus (PVN) of the brain hypothalamus is the central component of the HPA axis (1), thus GR gene knockout mice in this brain region was developed and their HPA axis was evaluated. The results indicated that PVN GR is required for negative regulation of the HPA axis at a basal condition and under stress (192). GR gene knockout mice specific in the noradrenergic neurons, components of the neural circuit mediating the adaptive stress response together with the HPA axis, were also created (1,193). These mice demonstrated depressive- and anxiety-like behavior upon stress with specificity to duration and gender, indicating that GR in the noradrenergic neurons plays an important role in stress response and associated behavioral changes in addition to its actions in the HPA axis. In mice with cardiomyocyte/vascular smooth muscle cell-specific GR gene knockout, fetal heart function is impaired and causes generalized edema in embryonic day (E) 17.5. Histologically, disorganized myofibrils and cardiomyocytes are found in fetal heart, while altered expression of the genes involved in contractile function, calcium handling and energy metabolism are observed. These results suggest that GRa actions in the cardiomyocytes and vascular smooth muscle cells are important for proper functioning and maturation of the fetal heart (194). GR gene knockout specific in the vascular endothelial cells revealed that GRa in this tissue mediates a tonic effect of glucocorticoids on blood pressure, possibly by supporting autocrine or paracrine activity of this tissue for releasing vasoactive mediators in response to glucocorticoid treatment (195). GRa in this tissue is also required for the protective response against sepsis by conferring glucocorticoid-mediated suppression of cytokine and nitric oxide production (196). Challenge of vascular endothelial cell-specific GR knockout mice with LPS also revealed that GRa in this tissue is required for survival of animals against this compound by appropriately suppressing circulating levels of inflammatory cytokines (TNFa and IL-6) and release of the nitric oxide (197), indicating the important actions of the vascular endothelial cell-residing GRa for controlling otherwise overshooting inflammatory response. T-lymphocyte-specific GR gene knockout mice revealed that GRa-mediated immune suppression mainly through Th1 lymphocytes is also necessary for survival of the mice against Toxoplasma gondii infection (198). Uterine-specific GR knockout mice generated with the Cre-recombinase expressed under the PR gene promoter revealed that uterine GRa is required to establish the local cellular environment necessary for maintaining normal uterine biology and fertility (199). The GR gene knockout specific to testicular Sertoli cell identified that GRa in these cells is required to maintain normal testicular Sertoli/germ cell numbers and circulating gonadotropin levels, as well as optimal Leydig cell maturation and steroidogenesis, thus GRa in these cells is required for supporting normal male reproduction (200).
By using a knockin procedure (replacement of wild type genes with their mutants), physiologic importance of the specific GRa functions associated with introduced mutations was evaluated. For example, knockin of the mutant GRa defective in binding to classic GREs (GRdim harboring A458T replacement, which is inactive in transactivation of glucocorticoid-responsive genes harboring GREs, but active in transrepression through protein-protein interaction with other transcription factors), revealed that transactivational activity of GRa is not essential for survival (112). Indeed, mice harboring GRdim demonstrated partially active HPA axis, full activity in glucocorticoid-mediated development of adrenal medulla, and defective glucocorticoid-mediated thymocyte apoptosis. However, the GRdim mutant receptor was subsequently shown to bind GREs of the N-methyltransferase (PNMT) gene, which is a rate-limiting enzyme for the production of catecholamines in the adrenal medulla, and to activate strongly the expression of this gene (201). Thus, the GRdim mutation cannot completely abolish transactivational activity of GRa, further suggesting that this activity of GRa may be required for survival. In addition, the effect of this mutant receptor on recently identified nGREs is not known, making the original conclusion elusive.
FACTORS THAT MODULATE GR ACTIONS
New Ligands with Specific Activities
Glucocorticoids have two major activities on the transcription of glucocorticoid-responsive genes, namely transactivation and transrepression (202). The former activity is mainly mediated by binding of GRa to its DNA responsive sequences in the promoter region of glucocorticoid-responsive genes and stimulating the transcription of downstream protein-coding sequences. Mechanisms underlying the latter activity are more complex, mostly mediated by suppression of other transcription factor activities by GRa. At pharmacologic levels, the transactivation activity is well correlated with side effects of glucocorticoids, such as glucose intolerance and overt diabetes mellitus, central obesity, osteoporosis and muscle wasting (202). On the other hand, the transrepressive activity of glucocorticoids is associated mostly with their beneficial therapeutic effects, such as suppression of the inflammation and immune activity, and induction of apoptosis of several neoplastic cells/tissues. Thus, significant efforts have been put to produce dissociated glucocorticoids with transrepression but no transactivation activity (202).
RU24858, RU40066 and RU24782 were the first steroids reported to have such selectivity, having an efficient inhibitory effect on AP-1- and NF-kB-mediated gene induction with reduced transactivation activity in vitro (203). However, they did not have any therapeutic advantage when they were used in vivo. Compound Abbott-Ligand (AL)-438, a derivative of a synthetic progestin scaffold, binds GRa with similar affinity to that of prednisolone and shows the activity equivalent to prednisolone in suppressing paw-edema in a rat experimental model (204). AL-438 does not increase circulating glucose levels and bone absorption in contrast to prednisolone, indicating that this compound is a promising selective glucocorticoid. ZK216348, the (+)-enanitomer of the racemic compound ZK209614, binds GRa and demonstrates anti-inflammatory activity comparable to that of prednisolone under both systemic and topical applications with much less unwanted effects on blood glucose and skin atrophy (205). This compound, however, binds PR and AR in addition to GRa, and does not show clear selectivity between transactivation and transrepression in vitro. C108297 functions as a GRa modulator through induction of unique interaction profiles of GRa to some splice variants of the p160 coactivator SRC1. This compound potently enhances GR-mediated memory consolidation, partially suppresses hypothalamic expression of the corticotropin-releasing hormone (CRH), and antagonizes to GR-mediated inhibition of hippocampal neurogenesis (206). Cortivazol, a pyrazolosteroid, induces nuclear translocation of GRa and stimulates GRa-induced transcriptional activity (207). Another compound, AL082D06 (D06), the tri-aryl methane, specifically binds GRa with a nano-molar affinity and acts as an antagonist for GRa but not for other SRs, in contrast to RU 486 (208). CORT-108297 acts also as a competitive GRa antagonist with high affinity to GRa (Ki 0.9 nM), but almost 1000-fold lower affinity to other SRs, PR, ER, AR and MR (209,210).
Two new non-steroidal GRa ligands, GSK47867A and GSK47869A, act as potent agonists with prolonged effects (211). These compounds bind the ligand-binding pocket of GRa with high affinity and induce both transactivational and transrepressional activities at concentrations ~10-50 times less than those of dexamethasone. Interestingly, GSK47867A and GSK47869A induce very slow GRa nuclear translocation and prolonged nuclear retention that leads to delayed but prolonged activation of the receptor. In computer-based structural simulation, these compounds induce unique GRa LBD conformation at its hsp90-binding site, which may underlie their extended GRa activation by causing defective interaction to hsp90 and altered intracellular circulation of GRa. By employing high throughput screening of 3.87 million compounds with the GR fluorescence polarization binding assay, heterobiaryl sulfonamide 2 was recently identified as a potent non-steroidal GR antagonist (212). Non-steroidal compounds mapracorat (also known as BOL-303242 and ZK245186) and the plant origin ginsenide Rg1 function as selective agonists with strong anti-inflammatory effects and a better side effect profile (213,214).
Compound A (CpdA), a stable analogue of the hydroxyl phenyl aziridine precursor found in the Namibian shrub Salsola tuberculatiformis Botschantzev, exerts anti-inflammatory activity by down-regulating TNFa-induced pro-inflammatory gene expression through inhibition of the negative effects of GRa on NF-kB, but demonstrates virtually no stimulatory activity on GRa-induced transactivation (215). This compound also suppresses similarly to dexamethasone the transcriptional activity of the T-bet transcription factor, a master regulator of Th1-mediated immune response, and reduces production of the Th1 cytokine interferon g from murine primary T-cells (216). By sparing AP-1-induced transcriptional activity and subsequent activation of the JNK/MAPK signaling pathway, CpdA does not influence epithelial cell restitution, an indicator of wound healing, in contrast to regular glucocorticoids (217,218). Thus, CpdA appears to be a dissociated compound of plant origin retaining the beneficial anti-inflammatory effect of glucocorticoids, being in part devoid of some of the known side effects of these compounds. CpdA also preserves the anti-cancer effect of glucocorticoids in human T-, B- and multiple myeloma cells, and cooperates with the anti-leukemic proteasome inhibitor Brtezomib in suppressing growth and survival of these cells (219). This compound is also beneficial for the treatment of bladder cancer by suppressing cell growth by promoting transrepressive actions of GRa and partially by acting as an AR antagonist (220). CpdA does not allow GRa to bind single GRE (half-site) sites in contrast to glucocorticoids, and this activity of CpdA is beneficial for its use in the treatment of triple-negative breast cancer, as single GRE-mediated gene regulation by glucocorticoids is associated with development of chemotherapy resistance (221).
Industrial chemicals are known to influence actions of several SRs, and are major threats for the life of living organisms including humans by interfering with the physiological actions of these receptors (222,223). Recent screening of these compounds using MDA-kb2 human breast cancer cells identified bisphenol Z and its analog bis[4-(2-hydroxyethoxy(phenyl)sulfone (BHEPS) as GR agonists, binding to the ligand-binding pocket of GRa and by shifting the helix-12 to the antagonist conformation in the structural simulation (224). Phthalates, ubiquitous environmental pollutants known for their adverse effects on health, bind GRa and other ketosteroid receptors, such as AR and PR, with high binding potencies comparable to natural ligands, suggesting that they may alter transcriptional activities of these receptors (225). Although underlying mechanism(s) are still unknown, chronic low doses of ingested petroleum can alter tissue expression levels of GRa in house sparrows, and modulates the glucocorticoid-signaling system and the HPA axis (226). Tolylfluanid, a commonly detected fungicide in Europe can induce biological changes that recapitulate many features of the human metabolic syndrome in part through modulating the GRa signaling pathway in male mice (227).
In addition to the above-explained compounds with agonistic or antagonistic actions on GRa, expanding numbers of new compounds with such activities have been identified, including: 2-aryl-3-methyloctahydroohenanthrene-2,3,7-trils (228), C118335 (229), 6-(3,5-dimethylisoxazol-4-yl)-2,2,4,4-tetramethyl-2,3,4,7,8,9-hexahydro-1H-cyclopenta[h]quinolin-3-one 3d (QCA-1093) (230), several compounds containing “diazaindole” moieties (231), heterocyclic GR modulators with a 2,2-dimethyl-3-phenyl-N-(thiazol or thiadiazol-2-yl) propanamide core (232), LLY-2707 (233), trierpenes (alisol M 23-acetate and alisol A 23-acetate) (234), GSK866 analogs UAMC-1217 and UAMC-1218 (235), AZD9567 (236), 1,3-benzothiazole analogs (237), 20(R, S)-protopanaxadiol and 20(R, S)-protopanaxatriol (238) and β-Sitosterol (239).
EPIGENETIC MODULATION OF GRa
Acetylation and CLOCK-mediated Counter Regulation of Target Tissue Glucocorticoid Action against Diurnally Fluctuating Circulating Glucocorticoids
All SRs including GRa are acetylated by several acetyltransferases, such as p300, p/CAF and Tip60, and have common acetylation sites in a consensus amino acid motif, KXKK, located in their hinge region (240-242). The human GRa is acetylated at lysine 494 and 495 within an acetylation motif also located in its hinge region, and was reported to be deacetylated by the HDAC2, an effect that is required for suppression of NF-kB-induced transcriptional activity by the activated GRa (243) (Figure 14). This finding indicates that acetylation of the GRa at these lysine residues attenuates the repressive effect of GRa on this transcription factor. In agreement with these results, we recently found that the Clock transcription factor acetylates GRa at the multiple lysine cluster that includes lysines 494 and 495, and represses GRa-induced transcription of several glucocorticoid-responsive genes (244). Clock, the “circadian locomotor output cycle kaput”, and its heterodimer partner “brain-muscle-arnt-like protein 1” (Bmal1), belong to the basic helix-loop-helix (bHLH)-PER-ARNT-SIM (PAS) superfamily of transcription factors, and play an essential role in the formation of the diurnal oscillation rhythms of the circadian CLOCK system (245). The CLOCK system, located in the suprachiasmatic nucleus (SCN) of the brain hypothalamus, acts as the “master” oscillator and generator of the body’s circadian rhythm, while the peripheral CLOCK system, virtually distributed in all organs and tissues including the CNS outside the SCN, acts generally as a “slave” CLOCK under the influence of the central SCN CLOCK. The Clock transcription factor shares high amino acid and structural similarity with the activator of thyroid receptor (ACTR), a member of the p160-type nuclear receptor coactivator family with inherent histone acetyltransferase activity, and thus, has such an enzymatic function (246).
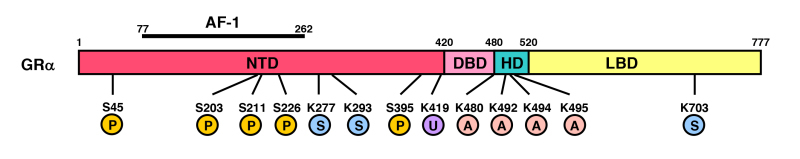
Figure 14.
Distribution of the amino acid residues of the human GR susceptible to acetylation, phosphorylation, ubiquitination or SUMOylation. Human GR has 4 acetylation sites (lysines: K at amino acid position 480, 492, 494 and 495, shown with “A”), at least 5 phosphorylation sites (serines: S at amino acid position 45, 203, 211, 226 and 395, shown with “P”), 1 ubiqitination site (Lysine: K at amino acid position 419, shown with “U”) and 3 SUMOylation sites (Lysines: K at amino acid position 277, 293 and 703, shown with “S”).
Clock physically interacts with GRa LBD through its nuclear receptor-interacting domain (NRID) in its middle portion, and acetylates human GRa at amino acids 480, 492, 494 and 495. Acetylation of GRa attenuates binding of the receptor to GREs, and hence, represses GR-induced transactivation of the GRE-driven promoters (244) (Figure 15). Since the lysine residues acetylated by Clock are located in the C-terminal extension (CTE) that follows DBD and plays a role in DNA recognition by SRs (247), it is likely that acetylation of these residues reduces binding of GRa to GREs by altering the action of CTE. The part of the hinge region acetylated by Clock also overlaps with the nuclear localization signal (NL)-1 (50,244), thus it is also possible that acetylation of GRa alters nuclear translocation of this receptor. It is well known that the central master CLOCK located in SCN creates diurnal fluctuation of circulating cortisol, therefore peripheral CLOCK-mediated repression of GRa transcriptional activity in glucocorticoid target tissues functions as a local counter regulatory mechanism for oscillating circulating cortisol (248).
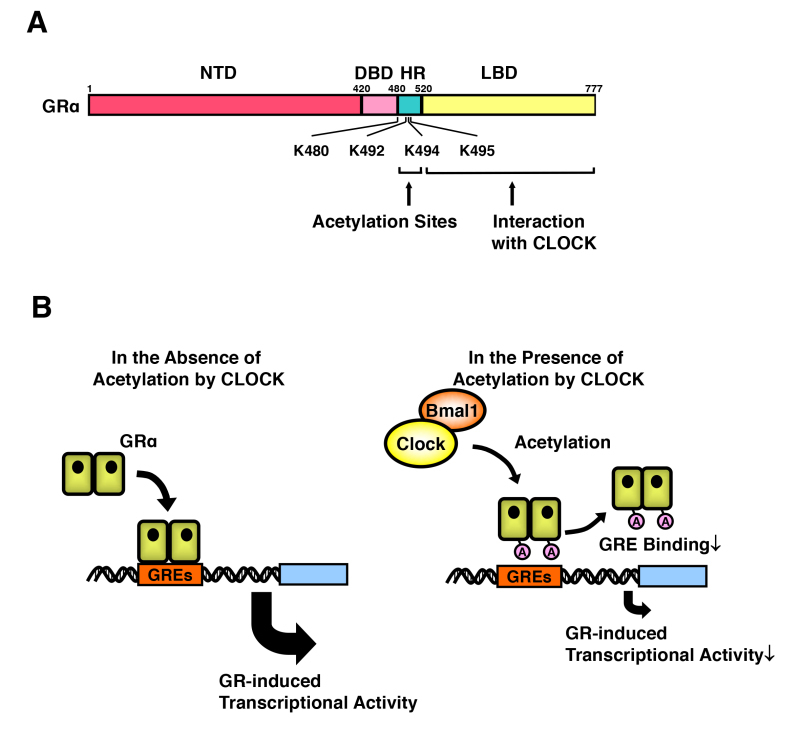
Figure 15.
Clock/Bmal1 suppresses GR-induced transcriptional activity through acetylation. Clock physically interacts with GR LBD through its nuclear receptor-interacting domain and suppresses GR-induced transcriptional activity by acetylating with its intrinsic HAT activity a lysine cluster located in the hinge region of the GR (A) through which Clock reduces affinity of GR to its cognate DNA GREs (B). A: acetylation; Bmal1: brain-muscle-arnt-like protein 1; DBD: DNA-binding domain; GREs: glucocorticoid response elements; HR: hinge region; K: lysine residue; LBD: ligand-binding domain; NTD: N-terminal domain. From (244).
In addition to the above findings obtained in in vitro cellular systems, we examined the acetylation status of human GRa and the expression of Clock-related and glucocorticoid-responsive genes in vivo and ex vivo, using peripheral blood mononuclear cells (PBMCs) from healthy adult volunteers (249). The levels of acetylated GRa were higher in the morning and lower in the evening, mirroring the fluctuations of circulating cortisol in reverse phase. All known glucocorticoid-responsive genes tested responded as expected to hydrocortisone, however, some of these genes did not show the expected diurnal mRNA fluctuations in vivo. Instead, their mRNA oscillated in a Clock- and a GRa acetylation-dependent fashion in the absence of endogenous glucocorticoid ex vivo, indicating that circulating cortisol might prevent circadian GRa acetylation-dependent effects in some glucocorticoid-responsive genes in vivo. These findings indicate that peripheral CLOCK-mediated circadian acetylation of GRa functions as a target tissue- and gene-specific counter regulatory mechanism to the actions of diurnally fluctuating cortisol, effectively decreasing tissue sensitivity to glucocorticoids in the morning and increasing it at night (36). Indeed, in another study where we measured mRNA expression of ~190 GRa action-regulating and glucocorticoid-responsive genes in subcutaneous fat biopsies from 25 obese subjects, we found that the levels of evening cortisol were much more important than those in the morning to regulate mRNA expression of glucocorticoid-responsive genes in this human tissue (250). It appears that higher sensitivity of tissues to circulating glucocorticoids in the evening due to reduced GRa acetylation by CLOCK underlies stronger impact of evening serum cortisol levels to glucocorticoid-regulated gene expression compared to morning levels.
The circadian CLOCK system and the HPA axis regulate each other’s activity through multilevel interactions in order to ultimately coordinate homeostasis against the day/night change and various unforeseen random internal and external stressors (251,252). For example, one CLOCK transcription factor Cry2 interacts with GRa and represses its transcriptional activity (253). Furthermore, GRa binds GREs located in the promoter region of the Per1, Per2 and other CLOCK components and stimulate their expression, an effect that contributes to resetting of the circadian rhythms by glucocorticoids (254,255). The peripheral CLOCK system residing in the adrenal glands contributes to the creation of circadian glucocorticoid secretion from this organ in addition to diurnally secreted ACTH from the pituitary gland (256). An important study further revealed new local factors, which also regulate circadian production of glucocorticoids in the adrenal glands: the intermediate opioid peptides secreted from the adrenal cortex influence in a paracrine fashion the amplitude of the serum corticosterone oscillations in mice through the C-X-C motif chemokine receptor 7 (CXCR7), a b-arrestin-biased G-protein-coupled receptor expressed on the adrenocortical cells (257).
Based on the above-indicated multilevel interaction between the CLOCK system and the HPA axis, uncoupling of or dysfunction in either system alters internal homeostasis and causes pathologic changes virtually in all organs and tissues, including those responsible for intermediary metabolism and immunity (248,251,252). Disrupted coupling of cortisol secretion and target tissue sensitivity to glucocorticoids may account for (1) development of central obesity and the metabolic syndrome in chronically stressed individuals, whose HPA axis circadian rhythm is characterized by blunting of the evening decreases of circulating glucocorticoids, as a result of enhanced input of higher centers upon the hypothalamic PVN’s secretion of CRH and arginine vasopressin (AVP); and (2) increased cardiometabolic risk and increased mortality of night-shift workers or subjects exposed to frequent jet-lag because of traveling across time zones (248,258). In addition, given that tissue sensitivity to glucocorticoids is increased in the evening as mentioned above (thus, evening cortisol levels have stronger impact to gene expression than those in the morning), supplemental administration of high-dose glucocorticoids at night for the treatment of adrenal insufficiency or congenital adrenal hyperplasia may increase a possibility of glucocorticoid-related side effects. Furthermore, administration of glucocorticoids at a specific period of the circadian cycle might increase their pharmacological efficacy, while at the same time reducing their unwanted side effects, because CLOCK differentially regulates transactivational and transrepressive actions of glucocorticoids, which are respectively correlated with side-effects and beneficial anti-inflammatory activities of these compounds used at pharmacological concentrations (258).
Phosphorylation
GRa has several phosphorylation sites and all of them are located in the NTD (20,259) (Figure 14). Classically, GRa is phosphorylated after binding to its ligand and this may determine target promoter specificity, cofactor interaction, strength and duration of receptor signaling and receptor stability (259,260). There are several kinases that phosphorylate GRa in vitro and in vivo (261). Yeast cyclin-dependent kinase p34CDC28 phosphorylates rat GRa at serines 224 and 232, which are orthologous to serines 203 and 211 of the human GRa, with the resultant phosphorylation enhancing rat GRa transcriptional activity in yeast (262). These residues are also phosphorylated after binding of the GRa with agonists or antagonists and the phosphorylated receptor shows reduced translocation into the nucleus and/or altered subcellular localization in mammalian cells (259,263). The p38 MAPK phosphorylates serine 211 of the human GRa, enhances its transcriptional activity and mediates GRa-dependent apoptosis (264). p38 MAPK and JNK also phosphorylate serine 226 of the human GRa and suppress its transcriptional activity by enhancing nuclear export of the receptor (63). Modulation of the molecular interactions between GRa AF-1 and cofactors through phosphorylation of these serine resides underlies in part the transcriptional regulation of this receptor by these kinases, as these serines are located within the AF-1 domain (265). Threonine 171 of the rat GRa is also phosphorylated by p38 MAPK and glycogen synthase kinase-3 (GSK3): phosphorylated rat GRa demonstrates reduced transcriptional activity in yeast and human cells, however, the human GRa does not have a threonine residue equivalent to that of the rat GRa (266,267). On the other hand, one GSK3 family protein, GSK3b, phosphorylates human GRa at serine 404 and modulates hGRa transcriptional activity including its repressive effect on NF-kB (268).
Several serine/threonine phosphatases, such as the protein phosphatase 2A (PP2A) and protein phosphatase 5, dephosphorylate human GRa at serine 203, 211 and/or 226, possibly through their association with GRa LBD (269,270). Stimulation of A549 human respiratory epithelial cells with b2 adrenergic receptor agonists increases PP2A, which in turn increases glucocorticoid sensitivity by dephosphorylating GRa at serine 226 (271). However, PP2A also regulates indirectly GRa phosphorylation by increasing dephosphorylation of JNK and subsequent activation of this kinase, as JNK directly phosphorylates GRa (272).
The cyclin-dependent kinase 5 (CDK5) physically interacts with the human GRa through its activator component p35, phosphorylates GRa at multiple serines including those at 203 and 211, and modulates GRa-induced transcriptional activity by changing accumulation of transcriptional cofactors on GRE-bound GRa (273). CDK5 and p35 are expressed mainly in neuronal cells and play important roles in embryonic brain development. Aberrant activation of CDK5 in CNS also plays a significant role in the pathogenesis of neurodegenerative disorders, such as Alzheimer’s disease and amyotrophic lateral sclerosis (274). We reported that, in addition to GRa, CDK5 phosphorylates MR and strongly suppresses its transcriptional activity (275). In brain regions, such as hippocampus and amygdala, which do not express 11b-HSD2, MR functions as a physiologic receptor for circulating glucocorticoids, and activation/suppression of MR plays an important role in glucocorticoid-related memory deficits and alterations in mood and cognition (276). Indeed, MR mediates enhancement of neuronal excitability, stabilization of synaptic transmission, and stimulation of long-term potentiation (LTP) in CA1 hippocampal cells, while MR activation is protective to hippocampal granular cell neurons. Thus, it is possible that CDK5-mediated regulation of MR might underlie development of glucocorticoid-associated pathologic conditions, such as neurodegenerative disorders and mood disorders (277,278). We examined changes of the CDK5 activity in mice under stress, and found that acute and chronic stressful stimuli differentially regulate the kinase activity together with contemporaneous alteration of the GRa phosphorylation in a brain region-specific fashion, indicating that CDK5 and its regulatory effects on GRa is an integral component of the stress response and mood disorders (279).
We also found that adenosine 5’ monophosphate-activated protein kinase (AMPK), a central regulator of energy homeostasis that plays a major role in appetite modulation and energy expenditure, indirectly phosphorylates human GRa at serine 211 through activation of p38 MAPK (280). Through phosphorylation of GRa, AMPK regulates glucocorticoid actions on carbohydrate metabolism, modifying transcription of glucocorticoid-responsive genes in a tissue- and promoter-specific fashion. Indeed, activation of AMPK in rats reverses glucocorticoid-induced hepatic steatosis and suppresses glucocorticoid-mediated stimulation of glucose metabolism. These findings indicate that the AMPK-mediated energy control system modulates glucocorticoid action at target tissues, and activation of AMPK could be a promising target for developing pharmacologic interventions in metabolic disorders in which glucocorticoids play major pathogenetic roles.
The v-akt murine thymoma viral oncogene homolog 1 (AKT1) or protein kinase B, another serine-threonine kinase known to regulate cell proliferation and survival, and aberrantly activated in various malignancies including acute leukemia, also phosphorylates human GRa at serine 134, which is located in NTD of this receptor (281). This phosphorylation of GRa retains the receptor in the cytoplasm through which activated AKT1 develops glucocorticoid resistance in acute leukemic cells, a major determinant for the prognosis of leukemic patients (281). AKT1 cooperates with phospho-serine/threonine-binding proteins 14-3-3s for regulating the transcriptional activity of GRa with 2 distinct mechanisms, one through segregation of GRa in the cytoplasm upon phosphorylation of serine 134 by AKT1 and subsequent association of 14-3-3 to GRa, and the other through direct modulation of GRa transcriptional activity in the nucleus (65). For the latter, AKT1 and 14-3-3 are attracted to DNA-bound GRa, accompanied by AKT1-dependent p300 phosphorylation, histone 3 (H3) serine (S) 10 (H3S10) phosphorylation and H3K14 acetylation at the DNA site in which 14-3-3 acts as a molecular scaffold (65). The above findings suggest that specific inhibition of the AKT1/14-3-3 activity on the cytoplasmic retention of GR but sparing the activity inside the nucleus may be a promising target for the treatment of glucocorticoid resistance observed in acute leukemia. Furthermore, they may also provide an explanation to somewhat conflicting findings previously reported for the actions of 14-3-3s on GRa (64,268,281,282).
Ubiquitination
The ubiquitin/proteasome pathway plays important roles in transcriptional regulation promoted by numerous trans-acting molecules. NRs, including GRa, ERs, PR, TRs, RARs and PPARs, as well as other transcription factors, such as p53, cJun, cMyc and E2F-1, are ubiquitinated and subsequently degraded by the proteasome (57,283). The transcriptional intermediate molecules, such as NR coactivators, chromatin remodeling factors, and some chromatin components, such as histone H1 and high mobility group (HMG) proteins, are also ubiquitinated and lysed by the proteasome (57,283,284). Moreover, the proteasome interacts with the C-terminal tail of the RNA polymerase II and is directly associated with the promoter regions of several genes, influencing their transcriptional activities (285). Thus, ubiquitination and subsequent processing of these molecules by the proteasome appear to regulate the transcriptional activity of GRa, possibly by facilitating rapid turnover of promoter-attracted and -associated GRa, ultimately down-regulating the transcriptional activity of this receptor. Indeed, mouse GRa contains a PEST motif at amino acids 407-426 (399-419 in human GRa) through which the ubiquitin-conjugating enzyme E2 and the ubiquitin-ligase enzyme E3 recognize their substrates (286). The lysine residue of the mouse GRa located at amino acid 426 (419 in human GRa) appears to be ubiquitinated, as inhibition of ubiquitination by compound MG-132 enhances the transcriptional activity of wild type GRa, while the mutant receptor with lysine to alanine replacement at amino acid 426 demonstrates elevated transcriptional activity and is insensitive to MG-132 (286) (Figure 14). Ubiquitination of GRa also influences motility of the receptor inside the nucleus, which was evaluated with the fluorescence recovery after photobleaching (FRAP) technique, possibly by changing association of the receptor to the nuclear matrix through ubiquitination (58,287,288).
SUMOylation
GRa is also SUMOylated. SUMOylation is the reaction conjugating the small ubiquitin-related modifier (SUMO) peptide (~100 amino acid peptide with molecular mass of ~11 kDa) to substrate proteins and conducted by an enzymatic cascade similar to those of ubiquitination but specific to SUMOylation (289). The human GRa has three SUMOylation sites, at lysines 277, 293 and 703 (290) (Figure 14). The first 2 sites are located in the NTD and act as major SUMOylation sites, while the last site is positioned in the LBD. SUMOylation of the former 2 sites (K277 and K293) suppresses GRa-induced transcriptional activity of a promoter containing multiple GREs, possibly by influencing the synergistic effect of multiple GRs bound on this promoter (291-293). In contrast, SUMOylation of the 3rd site (K703) enhances GRa-induced transcriptional activity, which is further enhanced by RSUME (RWD-containing SUMOylation enhancer) by changing attraction of the GRIP1 coactivator (294).
The death domain-associated protein (DAXX), a protein mediating the Fas-induced apoptosis through interacting with the death domain of Fas, was postulated to mediate SUMOylation-induced repression of GRa transcriptional activity (295). Other molecules, such as HDACs and the protein inhibitors of activated STAT (PIAS) family, which interact with SUMOylated proteins including GRa (296,297), might also participate in SUMO-mediated repression of GRa transcriptional activity, as the DAXX effect appears to be cell type- and/or cellular context-specific (298). SUMOylation of GRa is necessary for GRa-induced transrepression through the nGREs (an inverted quadrimeric palindrome separated by 0-2 nucleotide, see Section ACTIONS OF GR, Emerging Concept on GRa-mediated Transcriptional Repression) by facilitating the formation of a complex consisting of SUMOylated GRa, SMRT/NCoR1 and HDAC3 (299). It is known that phosphorylation of rat GR at amino acid position 246 (226 in the human GRa) by JNK facilitates SUMOylation of the receptor and regulates GRa-induced transcriptional activity in a target gene-specific fashion (291).
11b-Hydroxysteroid Dehydrogenases (11b-HSDs)
There are 2 types of 11b-hydroxysteroid dehydrogenases (11b-HSDs), type 1 and 2 (11b-HSD1 and 2). 11b-HSD1 catalyzes the conversion of the inactive cortisone to active cortisol, thus increases intracellular cortisol levels potentially contributing to tissue hypersensitivity to glucocorticoids. 11b-HSD1 is widely expressed, particularly in the liver, but also in the lung, adipose tissue, blood vessels, ovary and CNS (300). The transgenic mice over-expressing 11b-HSD1 in adipose tissues develop insulin-resistant diabetes mellitus, significant accumulation of visceral fat and hyperlipidemia, and increased systemic blood pressure, indicating that this enzyme may play a role in the development of visceral obesity-related metabolic syndrome by increasing availability of local cortisol in adipose tissues (301,302). 11b-HSD2, on the other hand, catalyzes the conversion of active cortisol into inactive cortisone, and is expressed in the classic mineralocorticoid-responsive tissues, such as kidney, colon and sweat glands (300). This enzyme enables these tissues to respond to the circulating mineralocorticoid aldosterone, protecting MR from binding to the excess amounts of circulating cortisol (300).
Chaperones and Co-chaperones
GRa forms a heterocomplex with several heat shock proteins (hsps), including hsp90, hsp70, hsp40 and hsp23 (69). These proteins bind many proteins and help their correct assembly and folding, therefore they are called as chaperones. In addition to hsps, there is an additional protein group called co-chaperones, such as Hop (hsp70-hsp90 organizing protein), SGTA (small glutamine-rich tetratricopeptide repeat-containing protein a), FKBP51 (FK506-binding protein 51) and FKBP52, which support folding function of hsps by forming a protein complex with the latter molecules (69). Hsps modulate the transcriptional activity of GRa by influencing maintenance, activation and intracellular circulation of this receptor (303). Specifically, hsp90, hsp70 and hsp40 organize proper folding of the GRa protein, and are required for the maintenance of its high affinity state against ligand where interaction of hsp90 to Hop as well as that between hsp70 and SGTA are required (304,305). Upon binding of the GRa to glucocorticoids, hsp90 helps the receptor to translocate close to the nuclear pore in the side of the cytoplasm by facilitating GRa’s association to microtubules through FKBP52. After GRa goes through the nuclear pore complex and enters into the nucleus, hsp90 regulates GRa-induced transactivation negatively, possibly by reducing the association of GRa to DNA GREs (306). However, there are conflicting reports indicating that hsp90 stabilizes the association of ligand-bound GRa to DNA and helps GRa stimulating the transcriptional activity of glucocorticoid-responsive genes (307). These chaperones also protect GRa from the degradation mediated by the ubiquitin-proteosomal pathway in the nucleus (308). Receptor-associating protein 46 (RAP46), another co-chaperone associated with several hsps, synergizes with hsp70 to regulate GRa transactivation negatively (309). Impact of co-chaperones on in vivo actions of GRa was evaluated in humans and mice. FKBP51 is a co-chaperon known as a negative regulator of GRa activity by reducing the latter’s affinity to glucocorticoids, and nucleotide variations in its encoding gene FKBP5 are associated with development of mood disorders and anxiety in humans possibly by skewing the GRa-signaling system (310,311). FKBP51 knockout mice demonstrate reduced basal activity of the HPA axis, a blunted response to acute stress and an enhanced recovery from this challenge (312).
Chemical Compounds
There are several chemical compounds that modulate GRa activity. 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), a wide-spread environmental contaminant that produces adverse biologic effects, such as carcinogenesis, reproductive toxicity, immune dysfunction, hepatotoxicity and teratogenesis, suppresses GRa-induced transactivation possibly by reducing the ligand-binding affinity of GRa (313-315). Geldanamycin, a benzoquinone ansamycin, which specifically binds hsp90 and disrupts its function, suppresses GRa-induced transactivation by inhibiting the translocation of GRa into the nucleus (308,316). GRa is also regulated by the cellular redox state. Thioredoxin, a compound accumulated during oxidative stress, enhances GRa transactivation, most likely due to functional replenishment of GRa (317). Ursodeoxycholic acid (UDCA), one of the hydrophilic bile acids, which acts as a bile secretagogue, cytoprotective agent and immunomodulator, and is used for the treatment of various liver diseases including primary biliary cirrhosis, induces translocation of GRa into the nucleus and causes GRa-mediated inhibition of NF-kB transactivation (318). Mizoribine (4-carbamyl-1-b-D-ribofurano-sylimidazolium-5-olate), an imidazole nucleotide with immunosuppressive activity binds to 14-3-3 and enhances 14-3-3/GRa interaction, which may further potentiate 14-3-3’s effect on GRa transactivation (319). For more details of the GRa/14-3-3 interaction, please see Section FACTORS THAT MODULATE GR ACTIONS, Epigenetic Modulation of GRa, b Phosphorylation.
NON-CODING RNAS
Human genome expresses numerous non-protein-coding RNAs in addition to protein-coding mRNAs. Indeed, over half of the genome sequence expresses RNAs in both directions either as single- or double-stranded RNAs (320,321). Classic examples of the former RNAs are ribosomal RNAs and transfer RNAs, while several distinct new members have been identified recently (322). Depending on their size, non-coding (nc) RNAs are empirically categorized as short (~200 bs) or long (>200 bs) ncRNAs. The former family includes micro (mi) RNAs, small interfering (si) RNAs, small nucleolar (sno) RNAs, piwi (pi) RNAs and transcription start site (TSS)-associated RNAs, while the latter consists of long intergenic (linc) RNAs, enhancer-associated (e) RNAs, exon-encoding long ncRNAs, circular (c) RNAs, promoter-associated RNAs and others. ncRNAs are also produced from protein-encoding mRNAs through nuclease digestion, such as 3’ UTR RNAs (323). Recently, some of these distinct classes of ncRNAs have been revealed to regulate mRNA/protein degradation and transcriptional activity of GRa and other NRs. In this subsection, new findings on miRNAs and long ncRNAs will be discussed.
Micro (mi) RNAs
miRNAs are single-stranded, ~22 b-long RNAs transcribed mainly by the RNA polymerase II either from their own genes or from the intronic sequence of the protein-coding/non-coding genes (324). Transcribed precursor miRNAs are processed by multiple reactions including digestion by the RNase III enzyme Dicer, and are liberated as mature forms into the cytoplasm. miRNAs are incorporated as binding modules for target mRNAs into the RNA-induced silencing complex (RISC), a multi-protein machinery containing Dicer, Argonaut (AGO), human immunodeficiency virus (HIV)-transactivating response RNA (TAR)-binding protein (TRBP) and the protein activator of the interferon-induced protein kinase (PACT). Binding of the RICS complex mainly to 3’UTR of the target mRNAs through the complemental 6-8 nucleotides of miRNAs leads to degradation of associated mRNAs or to inhibition of their translation to proteins. In addition to functioning inside the produced cells, miRNAs are secreted into extracellular space/circulation as components of the exosome, and act as “hormones” by influencing the functions of distant organs and tissues (325). This unique action of miRNA was confirmed in vivo using adipocyte-specific Dicer knockout mice supplemented with exosomes obtained from normal animals (326). Human genome contains over 1,000 miRNAs and some of them are known to regulate expression of the GRa protein, while glucocorticoids/GRa regulate expression of other miRNAs.
In a study exploring the miRNAs that mediate ACTH-dependent downregulation of GRa in mouse adrenal glands, 4 miRNAs, miR-96, -101a, -142-3p and -433 induced by ACTH injection, suppress GRa protein expression by ~40% (327). miR-142-3p reduces GRa expression by directly interacting with its 3’UTR region, and attenuates responsiveness to glucocorticoids in T-cell leukemia cells (328). miR-124a, -18, -18a and -124 also attenuate GRa protein expression and regulate GRa-induced transcriptional activity in various cells and tissues (329-331). miR-29a mitigates glucocorticoid-induced bone loss in part by reducing GRa expression (332). Glucocorticoids, on the other hand, modulate expression of some miRNAs, such as miR-449a, -98, and miR-155, which in turn mediate hormonal effects of these steroids (333,334). Systematic screening of glucocorticoid-responsive miRNAs in rat primary thymocytes identified over 200 miRNAs responsive to this hormone, and some validated miRNAs regulate cell death pathway (335). In myeloma cells, glucocorticoids induce miR-150-5p, which changes expression of the genes involved in cell death and cell proliferation pathways, thus this miRNA mediates in some part the therapeutic effects of glucocorticoids on multiple myeloma (336). miR-119a-5p is also glucocorticoid-responsive miRNA that mediates anti-proliferative effects of glucocorticoids on osteoblasts by affecting the WNT signaling pathway (337).
Long Non-coding (lnc) RNAs
Several lncRNAs regulate the transcriptional activity of GRa and/or other SRs. The steroid RNA coactivator (SRA) is a prototype lncRNA that regulates the transcriptional activity of several SRs (99). SRA was originally cloned in the yeast two-hybrid assay by using NTD of the PR as bait. It enhances ligand-induced transcriptional activity of AR, ER, GRa and PR. It is found in the complex containing the p160 coactivator SRC1, and regulates transcriptional activity in part by associating with the SRA stem-loop-interacting RNA binding-protein (SLIRP) and the RISC complex (99,338,339). Recently, SRA was shown to function also as a repressor of transcription, acting as a scaffold for a repressor complex (340).
The growth arrest-specific 5 (Gas5), which is a multi-exon-containing ncRNA with a poly-A tail, is accumulated in cells whose growth is arrested due to lack of nutrients or growth factors (341). Gas5 functions as a repressor of the GRa and some other SRs (342). Gas5 sensitizes cells to apoptosis by suppressing glucocorticoid-mediated induction of several responsive genes, including those encoding the cellular inhibitor of apoptosis 2 and the serum/glucocorticoid-responsive kinase. Gas5 binds GRa DBD and acts as a decoy “GRE”, thus, it competes with DNA GREs for binding to GRa (Figure 16). These findings indicate that Gas5 is a ribo-repressor of the GRa, influencing cell survival and metabolic activities during starvation by modulating the transcriptional activity of GRa. Accumulation of Gas5 upon growth arrest or starvation was previously demonstrated in a cellular context, but a study revealed that fasting of mice also accumulates Gas5 in their metabolic organs, such as liver and adipose tissues, through modulation of the mammalian target of rapamycin (mTOR) signaling pathway, but not in the brain and immune organs including thymus and spleen (343). Since basal expression levels of Gas5 in the immune organs are much higher than those of the metabolic organs, Gas5 may have a regulatory activity on GRa in the immune system independent to the nutrient/energy availability, as evidenced by the fact that Gas5 is differentially expressed in blood leukocytes of the patients with autoimmune, inflammatory or infectious diseases (343). Moreover, Gas5 has been shown to be implicated in glucocorticoid response in children with inflammatory bowel disease (344), in multiple sclerosis (345-347), in human beta cell dysfunction (348), as well as in hematologic malignancies (349,350). Similar to Gas5, PRNCR1 (also known as PCAT8) and PCGEM1 bind AR DBD and enhance the transcriptional activity of this receptor (351). These lncRNAs are highly expressed in the prostate gland, and play a role in the androgen-dependent development of prostate cancer. Their effects on GRa have not been tested as yet.
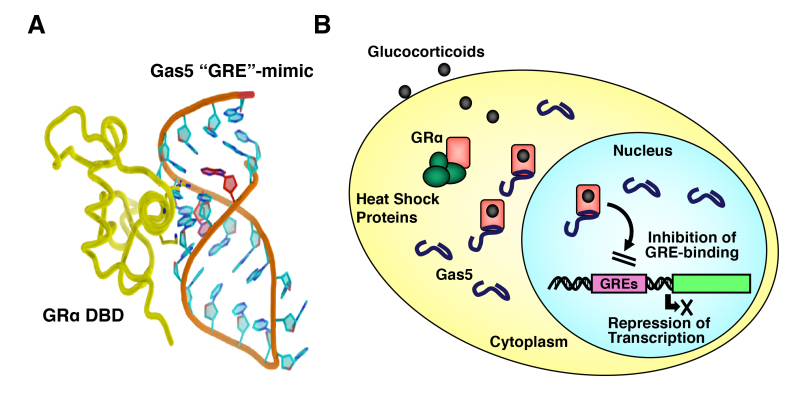
Figure 16.
Interaction model of the Gas5 RNA “GRE” to GR DBD and the molecular actions of Gas5 on GR-induced transcriptional activity. A: 3-Dimenstional structure of Gas5 “GRE”-mimic and its interaction model with GR DBD. From (342). B: Schematic model of Gas5 molecular actions on GR-induced transcriptional activity. Gas5 accumulated in response to growth arrest/starvation binds GR DBD and attenuates GR-induced transcriptional activity by competing with DNA GREs located in the promoter region of glucocorticoid-responsive genes.
THE SPLICING VARIANT GRbeta ISOFORM
The GRb isoform, which is expressed from the human GR gene through alternative use of its specific exon 9b, is known to have a dominant negative activity on classic GRa-induced transcriptional activity (21,352). This isoform was originally identified in humans, and was also reported in zebrafish, mice and rats (19,353-355). Since human (h) GRb shares the first 727 amino acids from the N-terminus with hGRa (19,356) (Figure 3), hGRb shares the same NTD and DBD with hGRa, but has a unique “LBD”. The divergence point (amino acid 727) of hGRa and hGRb is located at the C-terminal end of helix 10 in the hGRa LBD, therefore the hGRb “LBD” does not have helices 11 and 12 of the hGRa. As these helices are important for forming the ligand-binding pocket and for the creation of the AF-2 surface upon ligand binding (31), GRb cannot form an active ligand-binding pocket, does not bind glucocorticoids, and so, does not directly regulate GRE-containing, glucocorticoid-responsive gene promoters. In the absence of the hGRb “LBD”, the truncated hGR consisting of the NTD and DBD is transcriptionally active on GRE-containing promoters (357), thus the hGRb “LBD” somehow attenuates the transcriptional activity of the other subdomains of the molecule on GRE-driven promoters.
The dominant negative activity of GRb was first demonstrated in transient transfection-based reporter assays using GRE-driven reporter genes (21,358), but was subsequently confirmed on endogenous, glucocorticoid-responsive genes, such as the mitogen-activated protein kinase phosphatase-1 (MPK-1), myocilin and fibronectin (359,360). Further, GRb was shown to attenuate glucocorticoid-induced repression of the TNFa and interleukin (IL)-6 genes (359). We also confirmed this negative effect of GRb on GRa-mediated transrepression using microarray analyses (361). Several mechanisms explaining this GRb function have been reported, including (1) competition for GRE binding through their shared DBD, (2) heterodimerization with GRa and (3) coactivator squelching through the preserved AF-1 domain (21,357,358). All these different mechanisms of actions appear to be functional, depending on the promoters and the tissues affected by this GR isoform. Recently, the human GRb was shown to possess intrinsic transcriptional activity independent to its dominant negative effect on GRa-induced transcriptional activity, while the physiologic role(s) of this activity remain(s) to be examined (342,361,362) (see below). Inside the cells, hGRb can localize both in the cytoplasm and in the nucleus (363,364).
Similar to the human GR gene, the zebrafish (z) GR gene consists of 9 exons and produces zGRa and zGRb proteins, which contain 746 and 737 amino acids, respectively (353) (Figure 17). zGRa and zGRb share the N-terminal 697 amino acids, whereas they have specific C-terminal portions, which contain 47 and 40 amino acids, respectively. In contrast to hGRa and hGRb, which are produced through alternative use of specific exon 9a and 9b, zGRa and zGRb are formed as a result of intron retention (353). zGRa and zGRb use exon 1 to exon 8 for their common N-terminal 697 amino acids. zGRa uses exon 9 for its specific C-terminal portion, while zGRb continuously employs the rest of exon 8 and uses a stop codon located at the 3’ portion of this exon to express its specific C-terminal peptide (353). Protein alignment comparison of hGRb and zGRb indicated that these two molecules employ exactly the same divergence point, while their b isoform-specific C-terminal peptides show little sequence homology (353). These pieces of molecular information indicate that hGRb and zGRb evolved independently. Mouse (m), and recently, rat (r) GRb are also shown to produce in the same fashion as zGRb, indicating that intron retention may be a general mechanism for expressing this receptor isoform in organisms, while splicing-mediated expression employed by hGRb is rather unique (355,365). Nevertheless, zebrafish, mouse and rat GRb demonstrated the same functional properties as those of hGRb, namely, inability to bind glucocorticoids, a dominant negative activity on respectively zGRa-, mGRa- and rGRa-induced transactivation of GREs-drive promoters, and a strikingly similar tissue distribution as hGRb (353,365). Thus, hGRb, mGRb, rGRb and zGRb were produced through convergent evolution, most likely developed through a strong requirement of this type of GR isoform in the survival of these species. Indeed, the presence of nonligand-binding C-terminal variants is not unique to the GR. Similar to the human, mouse, rat and zebrafish GR, several other human NRs, e.g. ERb, TRa, vitamin D receptor (VDR), constitutive androstane receptor (CAR), dosage-sensitive sex reversal-1 (DAX-1), NURR-2, NOR-2, PPARα and PPARγ, all have C-terminally truncated receptor isoforms that are defective in binding to cognate ligands and have a dominant negative activity on their corresponding classic receptors (366-375). This suggests that evolution has allowed the development and retention of such alternative NRs, probably because they play important biologic roles.
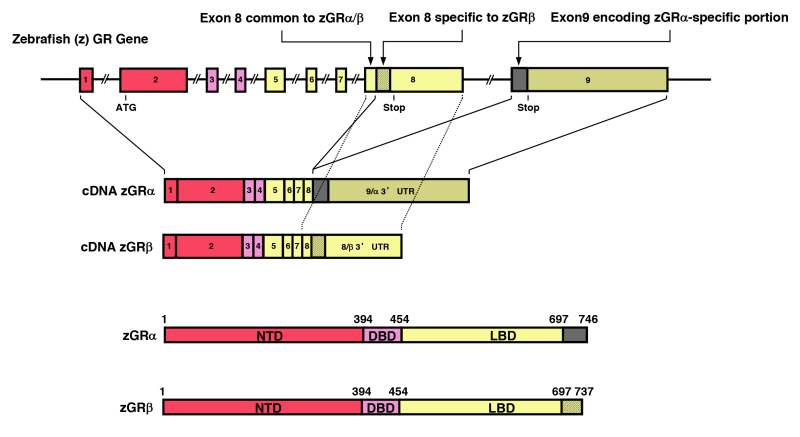
Figure 17.
Genomic and complementary DNA and protein structure of the zebrafish GR isoforms. The zebrafish (z) GR gene consists of 9 exons. The zGR gene expresses zGRa and zGRb splicing variants through intron retention (353). C-terminal gray colored and shaded domains in zGRa and zGRb show their specific portions. They are respectively encoded by exon 9 and the 3’ portion of exon 8, which are also shown in the same labeling in the genomic and complementary DNA models. Mouse and rat GR are produced with the same mechanism (355). Modified from (352). DBD: DNA-binding domain; LBD: Ligand-binding domain; NTD: N-terminal domain; UTR: untranslated region.
Biological actions of GRb and associated molecular mechanisms have been examined further during the last years. Using adeno-associated virus-based transfer of GRb to mouse liver, this isoform modulates mRNA expression of many genes in this organ including those related to endocrine system disorders, cancer, gastrointestinal diseases and immune diseases/inflammatory response both in a GRa-dependent and -independent fashions (376). Specifically, GRb attenuates GRa-dependent expression of the hepatic PEPCK gene and hepatic gluconeogenesis, while GRb stimulates expression of STAT1 through GREs located in the intergenic area close to the latter gene. The latter finding suggests that GRb can regulate gene expression by binding to classic GREs, in contrast to the previous findings obtained with GRE-driven reporter genes. In addition, GRb antagonizes to GRa-mediated suppression of bladder cancer cell migration and myogenesis of cardiomyocytes (377,378). GRb suppresses PTEN expression and enhances insulin-stimulated growth by stimulating the phosphorylation of AKT1 in a GRa-independent fashion (379). Further, GRb acts as a coactivator of T-cell factor-4 and enhances glioma cell proliferation also in a GRa-independent manner (380).
Several clinically oriented investigations suggest that GRb is responsible for the development of tissue-specific insensitivity to glucocorticoids in various disorders, most of them associated with dysregulation of immune function. They include glucocorticoid-resistant asthma, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), ankylosing spondylitis, chronic lymphocytic leukemia and nasal polyps (381-387). In these studies, various immune cells express elevated levels of GRb, which correlate with reduced sensitivity to glucocorticoids. Viral infection also stimulates GRb expression: for example, its expression in the peripheral mononuclear cells is strongly stimulated in the infants with bronchiolitis caused by the respiratory syncytial virus infection, and its expression levels are correlated with severity of the disease (388). Elevated levels of pro-inflammatory cytokines, such as IL-1, -2, -4, -7, -8 and -18, TNFa, and interferons a and g, might be responsible for increased GRb expression in cells from patients with these pathologic conditions, as these cytokines experimentally stimulate the expression of GRb in lymphocytes, neutrophils or airway smooth muscle cells (389-394). Further, presence of a single nucleotide polymorphism in the 3’ UTR of the hGRb mRNA (rs6198G allele), which increases its stability, and thus, causes elevated expression of the GRb protein, was associated with increased incidence of RA, SLE, high blood pressure, ischemic heart disease and nasal carriage of Staphylococcus aureus (382,395-397), possibly through inhibition of glucocorticoid actions by the increased concentrations of GRb. These pieces of clinical evidence further support that GRb has a dominant negative activity on GRa-induced transcription inside the human body, functioning as a negative regulator of glucocorticoid actions in local tissues.
PATHOLOGIC MODULATION OF GR ACTIVITY
Natural Pathologic GR Gene Mutations that Cause Familial/Sporadic Generalized Glucocorticoid Resistance or Chrousos Syndrome
Mutations in the human GR gene result in familial/sporadic generalized glucocorticoid resistance syndrome [see reviews (398-402)]. Since this syndrome was first reported by Chrousos et al. (403), we now call it as “Chrousos syndrome” (398,404). The condition is characterized by hypercortisolism without Cushingoid features (403,405). To overcome reduced sensitivity to glucocorticoids in tissues, affected subjects have compensatory elevations in circulating cortisol and ACTH concentrations, which maintain circadian rhythmicity and appropriate responsiveness to stressors, and resistance of the HPA axis to dexamethasone suppression, but no clinical evidence of hypercortisolism (404). Instead, the excess ACTH secretion causes increased production of adrenal steroids with mineralocorticoid activity, such as deoxycorticosterone (DOC) and corticosterone and/or androgenic activity, such as androstenedione, dehydroepiandrosterone (DHEA) and DHEA-sulfate (DHEA-S); The former accounts for symptoms and signs of mineralocorticoid excess, such as hypertension and hypokalemic alkalosis. The latter accounts for the manifestations of androgen excess, such as ambiguous genitalia and precocious puberty in children, acne, hirsutism and infertility in both sexes, male-pattern hair-loss, menstrual irregularities and oligo-anovulation in females, and adrenal rests in the testes and oligospermia in males. The clinical spectrum of the condition is broad and a large number of subjects may be asymptomatic, displaying biochemical alterations only (404).
An increasing number of kindreds and sporadic cases with abnormalities in the GR number, affinity for glucocorticoid, stability, and translocation into the nucleus have been reported (406-414). The molecular defects that have been elucidated are presented in Figure 18 and Table 1. The propositus of the original kindred was a homozygote for a single nonconservative point mutation, replacing aspartic acid with valine at amino acid 641 in the LBD of GRa; this mutation reduces the binding affinity of the affected receptor for dexamethasone by three-fold and causes loss of transactivation activity (411). The proposita of the second family had a 4-base deletion at the 3’-boundary of exon 6, removing a donor splice site. This results in complete ablation of one of the GR alleles in affected members of the family (412). Recent research employing mice with GR haploinsufficiency confirmed that ablation of one GR allele is sufficient to develop generalized glucocorticoid resistance (415). The propositus of the third kindred had a single homozygotic point mutation at amino acid 729 (valine to isoleucine: V729I) in the LBD, which reduced both the affinity and the transactivation activity of GRa (414). Several pathologic heterozygotic or homozygotic mutations of the GR gene have been recently identified in the patients as listed in Table 1 (403,411-444).
Figure 18.
Location of the known human GR mutations causing Chrousos syndrome or its mirror image, sporadic glucocorticoid hypersensitivity, in the human GR (NR3C1) gene (A) and in the linearized hGR protein molecule (B). Nucleoside numbers of the mutated sites are determined by the definition employing adenine of the translation initiation site as number 1.
Table 1.
Mutations in the NR3C1 Gene Causing Familial/Sporadic Generalized Glucocorticoid Resistance (Chrousos) or Hypersensitivity Syndromes
| Authors | cDNA* | Amino acid | Molecular Defects | Genotype | Phenotype |
|---|---|---|---|---|---|
| Chrousos et al. (403) Hurley et al. (411) | 1922A>T | Asp641Val | Transactivation: Decreased Affinity to ligand: Decreased (x3) Nuclear translocation: 22 min Abnormal interaction with GRIP1 | Homozygous | Hypertension Hypokalemic alkalosis |
| Karl et al. (412) | 4bp deletion in exon-intron 6 | GRa number: 50% reduction Inactivation of affected allele | Heterozygous | Hirsutism Male-pattern hair-loss Menstrual irregularities | |
| Malchoff et al. (414) | 2185G>A | Val729Ile | Transactivation: Decreased Affinity to ligand: Decreased (x4) Nuclear translocation: 120 min Abnormal interaction with GRIP1 | Homozygous | Precocious puberty Hyperandrogenism |
| Karl et al. (413) Kino et al. (416) | 1676T>A | Ile559Asn | Transactivation: Decreased Transdominance (+) Decrease in GR binding sites Nuclear translocation: 180< min Abnormal interaction with GRIP1 | Heterozygous | Hypertension Oligospermia Infertility |
| Ruiz et al. (418) Charmandari et al. (419) | 1430G>A | Arg477His | Transactivation: Decreased No GREs binding Decrease in GR binding sites Nuclear translocation: 20 min | Heterozygous | Hirsutism Fatigue Hypertension |
| Ruiz et al. (418) Charmandari et al. (419) | 2035G>A | Gly679Ser | Transactivation: Decreased Affinity to ligand: Decreased (x2) Nuclear translocation: 30 min Abnormal interaction with GRIP1 | Heterozygous | Hirsutism Fatigue Hypertension |
| Mendonca et al. (417) | 1712T>C | Val571Ala | Transactivation: Decreased Affinity to ligand: Decreased (x6) Nuclear translocation: 25 min Abnormal interaction with GRIP1 | Homozygous | Ambiguous genitalia Hypertension Hypokalemia Oligo-amenorrhea |
| Vottero et al. (420) | 2241T>G | Ile747Meth | Transactivation: Decreased Transdominance (+) Affinity to ligand: Decreased (x2) Nuclear translocation: Decreased Abnormal interaction with GRIP1 | Heterozygous | Cystic acne Hirsutism Oligo-amenorrhea |
| Charmandari et al. (421) | 2318T>C | Leu773Pro | Transactivation: Decreased Transdominance (+) Affinity to ligand: Decreased (x2.6) Nuclear translocation: 30 min Abnormal interaction with GRIP1 | Heterozygous | Fatigue Anxiety Acne Hirsutism Hypertension |
| Charmandari et al. (431) | 2209T>C | Phe737Leu | Transactivation: Decreased Transdominance (+) Affinity to ligand: Decreased (x1.5) Nuclear translocation: 180 min | Heterozygous | Hypertension Hypokalemia |
| Charmandari et al. (430) | 1201G>C | Asp401His | Transactivation: Increased Transdominance (-) Affinity to ligand: no change Nuclear translocation: Normal | Heterozygous | Hypertension Diabetes mellitus Accumulation of visceral fat |
| McMahon et al. (426) | 2bp (TG) deletion at 2318 and 2319 | Phe774Serfs⃰ | No transactivation activity No ligand-binding activity | Homozygous | Severe hypoglycemia developed 1 day after birth Hypertension Fatigues with feeding |
| Nader et al. (422) (443) | 2141G>A | Arg714Gln | Transactivation: Decreased Transdominance (+) Affinity to ligand: Decreased (x2.0) Nuclear translocation: 20 min Abnormal interaction with GRIP1 | Heterozygous Heterozygous | Hypoglycemia developed at age 2 years and 10 months Hypertension Accelerated bone age Mild clitoromegaly Infertility |
| Bouligand et al. (429) | 1405C>T | Arg469Ter | Transactivation: Decreased Affinity to ligand: Decreased Nuclear Translocation: No | Heterozygous | Bilateral adrenal hyperplasia Hypertension Hypokalemia |
| Zhu et al. Nicolaides et al. (423) (424) | 1667C>T | Threo556Ile | Transactivation: Decreased Transdominance: No Affinity to ligand: Decreased Nuclear translocation: 50 min | Heterozygous | Bilateral adrenal hyperplasia |
| Roberts et al. (428) | 1268T>C | Val423Ala | Transactivation: Decreased Transdominance: No Affinity to ligand: no change Nuclear translocation: 2.6-fold delay | Heterozygous | Hypertension |
| Nicolaides et al. (425) | 2177A>G | His726Arg | Transactivation: Decreased Transdominance: No Affinity to ligand: Decreased Nuclear translocation: 60 min | Heterozygous | Hirsutism Acne Alopecia Fatigue Anxiety Irregular menstrual cycle |
| Lin et al. (432) | 26C>G | Pro9Arg | Not performed | Heterozygous | Hypertension |
| Paragliola et al. (433) | 367G˃T | Glu123Ter | Not performed | Heterozygous | Chronic fatigue Anxiety Hirsutism Irregular menstrual cycles Infertility |
| Tatsi et al. (434) | 592G˃T | Glu198Ter | Not performed | Compound heterozygous | Hypertensive encephalopathy |
| Al Argan et al. (435) | 1392delC | Ile464Ilefs⃰ | Not performed | Heterozygous | Low body weight Hyperandrogenism Severe anxiety Adrenocortical hyperplasia |
| Vitellius et al. (436) | 1429C˃A | Arg477Ser | Cytoplasm to nuclear translocation: Decreased DNA binding: (-) Dominant negative effect: (-) Transactivation: (-) | Heterozygous | Obesity |
| Velayos et al. (437) | 1429C˃T | Arg477Cys | Not performed | Heterozygous | Mild hirsutism |
| Vitellius et al. (436) | 1433A˃G | Tyr478Cys | Cytoplasm to nuclear translocation: Decreased DNA binding: Weak and delayed Dominant negative effect: (-) Transactivation: Decreased | Heterozygous | Adrenal mass |
| Vitellius et al. (438) | 1471C˃T | Arg491Ter | Transactivation: (-) | Heterozygous | Bilateral adrenal hyperplasia |
| Vitellius et al. (438) | 1503G˃T | Gln501His | Transactivation: Decreased | Heterozygous | Bilateral adrenal hyperplasia |
| Ma et al. (439) | 1652C˃A | Ser551Tyr | Ligand binding: Decreased Cytoplasm to nuclear translocation: Decreased Transactivation: Decreased | Homozygous | Fatigue Hypokalemia Hypertension Polyuria |
| Velayos et al. (437) | 1762_1763insTTAC | His588Leufs⃰ | Not performed | Heterozygous | Hirsutism Chronic fatigue Anxiety |
| Cannavò et al. (440) | 1915C˃G | Leu595Val | Not performed | Not available | Hirsutism Amenorrhea Hypertension |
| Trebble et al. (441) | 1835delC | Ser612Tyrfs⃰ | Protein expression: (-) Ligand binding: (-) Cytoplasm to nuclear translocation: (-) Dominant negative effect: Yes Transactivation: (-) | Heterozygous | Fatigue |
| Vitellius et al. (442) | 1980T˃G | Tyr660Ter | Transactivation: (-) | Heterozygous | Hypertension |
| Vitellius et al. (436) | 2015T˃C | Leu672Pro | Protein expression: Decreased Ligand binding: (-) Cytoplasm to nuclear translocation: (-) DNA binding: (-) Dominant negative effect: No Transactivation: (-) | Heterozygous | Adrenal mass |
| Donner et al. (444) | 2317_2318delCT | Leu773Valfs⃰ | Protein expression: slightly reduced Transactivation: Decreased Dominant negative effect: No Ligand binding: (-) | Heterozygous | Hypertension |
We examined the impact of 10 pathologic GRa point mutations (559N, V571A, V575G, D641V, G679S, R714Q, V729I, F737L, I747M and L773P) to the molecular structure of the GRa LBD focusing on its ligand-binding pocket and AF-2 surface by using computer-based molecular simulation, and found some rules on the molecular disruption of these structural units by the mutations (89); (1) Topology of the peptide backbones is highly preserved in pathologic GRa mutant LBDs (Figure 19A). This result suggests that alteration in property and/or positioning of the side chain of replaced amino acids is rather crucial for developing molecular defects. (2) Defects in the ligand-binding pocket of the mutant receptors are driven primarily by loss/reduction (indirectly through structural changes in LBD induced by the mutations) of the electrostatic interaction formed by arginine 611 and threonine 739 of the receptor to glucocorticoid and a subsequent conformational mismatch (Figure 19B). (3) Defects of the AF-2 surface that reduce affinity to the LxxLL motif are caused mainly (also indirectly) by disruption of the electrostatic bonds to the non-core leucine residues of this peptide that determine the peptide’s specificity to GRa LBD (Figure 19C), as well as by reduced non-covalent interaction against core leucines and subsequent exposure of the AF-2 surface to solvent.
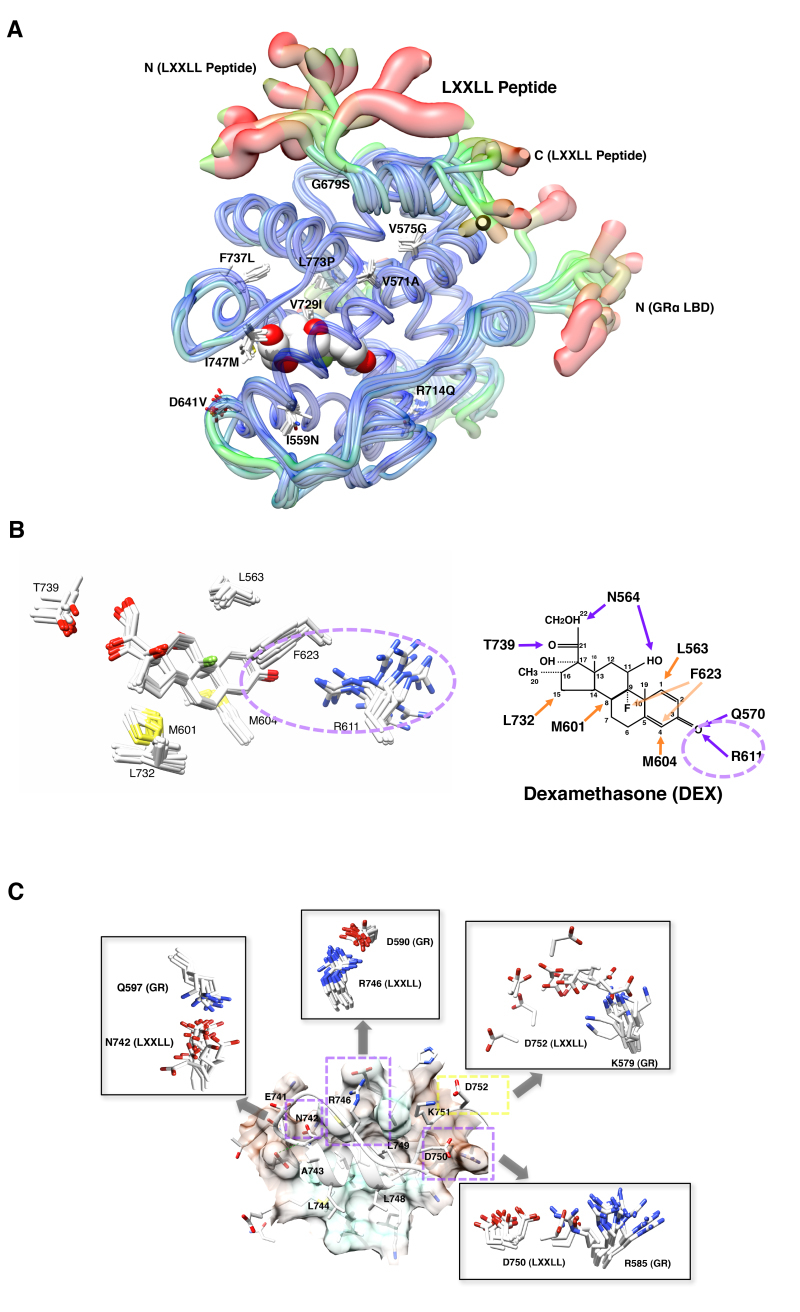
Figure 19.
Impact of pathologic GR point mutations to the molecular structure of GR LBD. A: Distribution of the pathologic GR point mutations in its LBD and their overall impact on the 3-dimensional LBD peptide backbone. Thickness and color of the overlaid C-traces of the GR mutant receptor LBDs and the wild type GR LBD indicate the areas of least (thin and blue) to most (thick and red) motion over the course of simulation. Locations and side chains of the mutated amino acids are indicated, whereas dexamethasone (shown with the white and red spheres of space-filling model) is located inside LBP. B: Alteration of the electrostatic bond formed by arginine (R) 611 and threonine (T) 739 of pathologic GR mutants to dexamethasone may largely explain the reduced affinity of many pathologic GR mutants to this steroid. The left panel demonstrates superimposed 3-dimensional interaction images of dexamethasone and the key residues of all pathologic GRa mutants. Among the key amino acids of pathologic mutants participating in interaction with dexamethasone, R611 is largely deviated in these mutant receptors, which underlies reduced/disappeared electrostatic interaction between this residue and the carbonyl oxygen at carbon-3 of dexamethasone. Q570 and N564 are omitted from these panels. Major changes observed in the electrostatic bond formed by R611 and T739 are indicated with a purple dotted circle. The right panel shows schematic molecular interaction between wild type GRa and dexamethasone. Purple and orange arrows indicate electrostatic and non-covalent bonds, respectively. DEX: dexamethasone. C: Defective non-covalent bonds formed between Q597, D590, K579 and R585 of the pathologic GRa mutants and N742, R746, D750 and D752 of the LxxLL peptide mainly explain reduced interaction of the mutant receptor AF-2s to this peptide. The panel demonstrates 3-dimensional image of the molecular interaction between the LXXLL peptide and key residues of the wild type GRa. The LxxLL peptide forms important electrostatic bonds with its non-core leucine residues (N742, R746, D750 and D752) against the receptor residues (Q597, D590, R585 and K579, respectively) as marked with purple dotted boxes. Pathologic GRa mutants demonstrate significant shift of the side chains of some of these receptor residues among which the side chain of R585 shows the most significant deviation (shown in square inserts). Modified from (89).
GR Gene Mutation-Mediated Hypersensitivity Syndrome
Only one mutation has been reported in the GRa NTD that replaces aspartic acid at amino acid 401 by histidine (D401H) (G to C replacement at nucleotide position 1201) (430). The patient harboring this heterozygous mutation presented with manifestations consistent with glucocorticoid hypersensitivity, in accordance with the in vitro results showing that the mutant receptor hGRaD401H demonstrated a 2.4-fold increase in its ability to transactivate the glucocorticoid-responsive promoters. This condition represents the mirror image of the Chrousos syndrome. Although not in humans, one porcine heterozygotic substitution that replaces alanine at amino acid 610 with valine (A610V) in LBD of the porcine GR causes a gain-of-function phenotype, shifting the titration curve of GR-transcriptional activity to leftward (this suggests increase of the receptor affinity to glucocorticoid) (445).
GR Gene Polymorphisms
Polymorphisms of the human GR gene have also been reported (446). A heterozygous polymorphism replacing aspartic acid to serine at amino acid 363 (N363S) that mildly increases transcriptional activity of the affected receptor in vitro is associated with increased sensitivity to glucocorticoids, weakly correlating with the development of central obesity, and thus, influencing the metabolic profile and the longevity of humans in a negative fashion (447-449). This polymorphism found at amino acid 363 was first described by Karl et al. (412).
The polymorphism in the human GR gene that causes arginine to lysine replacement at amino acid 23 (ER22/23EK: GAG AGG to GAA AAG) is associated with relative glucocorticoid resistance by altering the expression levels of GRa translational isoforms (450). This polymorphism increases muscle mass in males and reduces waist to hip ratio in females, and is associated with greater insulin sensitivity, and lower total and low-density lipoprotein cholesterol levels, indicating that this polymorphism causes beneficial effects on longevity by reducing glucocorticoid actions (451,452).
One recent study examined influence of N363S and ER22/23EK polymorphisms to intelligence quotient (IQ) and behavior of 344 young subjects who have been followed up from their birth (453). The study found that N363S is not associated with IQ, while ER22/23EK showed significantly higher IQ scores. Both polymorphisms did not show any effects on the behavior scores. Antenatal glucocorticoid treatment reduces IQ scores in the subjects carrying N363S or ER22/23EK polymorphism.
The BclI GR polymorphism comprises a C to G nucleotide substitution at 646 bp downstream of exon 2 in intron B of the human GR gene that creates a cutting site for the BclI restriction enzyme. G-allele of this polymorphism increases tissue sensitivity to glucocorticoids as shown by greater suppression of serum cortisol levels after dexamethasone administration (454). This polymorphism is associated with development of mood disorders, psychopathology, bronchial asthma, hypertension, hyperinsulinism and obesity (455,456). It is also associated with increased bone resorption in patients receiving glucocorticoid replacement therapy (457). Although a large study employing adolescents (15-17 years old) did not confirm the association of the BclI polymorphisms to changes in several stress-related neurological parameters (458), a study employing 460 subjects with post-traumatic stress disorder (PTSD) found that this polymorphism and another polymorphism rs258747, located in the 3’-flanking region of the human GR gene and potentially influencing stability of GR mRNA, significantly increase a risk for developing PTSD (459). In one study, the BclI GR polymorphism was associated with lower frequency of insulin resistance in the women with polycystic ovary syndrome (PCOS) in contrast to the findings obtained in normal subjects (460).
A single nucleotide polymorphism that replaces A with G at the nucleoside 3669 (A3669G) located in the 3’ end of exon 9b has been described in a European population (461). This polymorphism does not change the amino acid sequence but increases the stability of GRb mRNA and increases GRb protein expression, leading to greater inhibition of GRa-induced transcriptional activity and causing glucocorticoid resistance in tissues. The presence of the A3669G allele is associated with reduced central obesity and a more favorable lipid profile in affected subjects (461).
Viral Infection
HUMAN IMMUNODEFICIENCY VIRUS TYPE-1
Patients with the Acquired Immunodeficiency Syndrome (AIDS), which is caused by infection of the Human Immunodeficiency Virus type-1 (HIV-1), have several manifestations compatible with increased activity of GRa. They develop reduction of innate and Th1-directed cellular immunity, which is also seen in the conditions of glucocorticoid excess. Patients with AIDS often develop symptoms and signs that manifest in hypercortisolemic states, such as muscle wasting, myopathy, dyslipidemia and visceral obesity-related insulin resistance (462-466). Therefore, it is possible that some HIV-1-related factor(s) may modulate the function of GRa in patients with AIDS. Please see for more details the chapter on AIDS and the HPA Axis in the Adrenal Section of Endotext.
We have shown that one of the HIV-1 accessory proteins, Vpr, a 96-amino acid virion-associated protein with multiple functions (467,468), enhances GRa transactivation by functioning as a coactivator (469) (Figure 20). Indeed, Vpr contains a NR coactivator motif LxxLL at amino acids 64-68. This motif is used by host NR coactivators to bind NRs (80) (see Section ln ACTIONS OF GR, Mechanism of GRa-mediated Activation of Transcription). Similarly, through this motif, Vpr directly binds GRa and cooperatively enhances its activity on its responsive promoters along with host NR coactivators SRC-1 (p160-type protein, NCoA1) and p300/CBP (469). Vpr directly binds p300 at its C-terminal amino acids 2045-2191, where the p160 coactivators (NCoAs) also bind (470). Since Vpr circulates at detectable levels in HIV-1-infected individuals and is able to penetrate the cell membrane, its effects may be extended to cells not infected by HIV-1 (471,472). Indeed, extracellularly administered Vpr polypeptide regulates glucocorticoid-responsive genes, such as IL-12 p40, in the same way as the potent glucocorticoid, dexamethasone (473). In addition to regulating GRa activity, Vpr modulates the transcriptional activity of PPARb/d and PPARg, the NR family proteins important for fatty acid metabolism (474,475). Through modulating activities of GRa and the PPARs, Vpr appears to participate in the development of the characteristic AIDS-related lipodystrophy syndrome, which is quite prevalent among AIDS patients (476,477).
Figure 20.
Linearized Vpr, Tat, E1A, p300 and CtBP1 molecules and their mutual interaction domains. Vpr interacts with GR and several other NRs through its LxxLL motif located at amino acids 64 to 69. Binding sites of Vpr and p160-type HAT coactivators overlap with each other on p300. Since Vpr has a LxxLL motif similar to p160 coactivators, Vpr mimics host p160 coactivators and enhances GR transcriptional activity. Tat also binds both p300 and p160 coactivators. p300 facilitates attraction of many transcription factors, cofactors and general transcription complexes, and loosens the histone/DNA interaction through acetylation of the histone tails by its histone acetyltransferase (HAT) domain. E1A binds p300 at the latter’s C-terminal portion, while it physically interacts with the N-terminal portion of CtBP1 through its C-terminal end. The N-terminal portion of CtBP1 physically interacts with HDAC5 and Retinoblastoma protein (Rb), which have repressive activity on transcription. CtBP1 regulates its interaction to binding partners by sensing cellular NAD+ levels through its NAD+-binding domain. The HAT domain of p300 and the NAD+-binding domain of CtBP1 are indicated in grey. Modified from (478). CREB: CRE-binding protein, HAT: histone acetyltransferase, HDAC5: histone deacetylases 5, NF-B: nuclear factor-B, NAD: nicotinamide adenine dinucleotide, NR: nuclear hormone receptor, p/CAF: p300/CBP-associating factor, pTEFb: positive-acting transcription elongation factor b, Rb: retinoblastoma protein, SF-1: steroidogenic factor-1, STAT2: signal transducer and activator of transcription 2, TFIIB: transcription factor IIB.
Another HIV-1 accessory protein, Tat, which functions as a major transactivator of the HIV-1 long terminal repeat promoter (479) also potentiates GRa activity moderately by increasing accumulation of the positive transcription elongation factor b (pTEFb) (480-482) (Figure 20). Like Vpr, Tat readily penetrates the cell membranes (483) and may, therefore, modulate the transcriptional activity of GRa in the cells/tissues not infected by HIV-1.
Through Vpr and Tat, HIV-1 may facilitate the transcription of genes encoding its own proteins by directly stimulating viral proliferation. On the other hand, by enhancing transactivation of GRa and other NRs, these proteins may contribute to the viral proliferation possibly by suppressing the host immune system, while they participate in the development of several pathologic conditions associated with HIV-1 infection (480,484).
ADENOVIRUS
Adenoviruses cause illness of the respiratory system, such as common cold syndrome, pneumonia, croup and bronchitis, as well as illnesses of other organs, such as gastroenteritis, conjunctivitis and cystitis. They encode the E1A protein, which is expressed just after the infection and is necessary for the transcriptional regulation of the adenovirus-encoded genes (485). In addition to the viral genes, E1A regulates the transcriptional activity of a variety of host genes through interaction with the host transcriptional integrator p300 and its homologous molecule CBP (77,486) (Figure 20). In an in vitro system, E1A, in contrast to Vpr, blocks the actions of glucocorticoids on the transcriptional activity of genes, producing resistance to glucocorticoids (470).
E1A also interacts with the C-terminal tail-binding protein 1 (CtBP1), which functions as a transcriptional repressor for numerous transcription factors, by communicating with the class II HDACs and other inhibitory molecules like the retinoblastoma protein (Rb) (487) (Figure 20). E1A suppresses functions of p300/CBP and CtBP1 by binding to their functionally critical domains (77,487). Although there is no supportive clinical evidence, it is highly possible that adenovirus changes the peripheral action of glucocorticoids as well as of other bioactive molecules that activate NRs and directly regulates the transcriptional activity of their target genes, ultimately contributing to the pathologic states observed in adenoviral infection.
OTHER VIRUSES
We examined the impact of viral infection (murine cytomegalovirus: mCMV) on glucocorticoid-mediated modulation of gene expression in dendritic cells (488). Among 96 genes examined, the viral infection significantly enhanced dexamethasone-induced IL-10 expression. Activation of the toll-like receptors (TLRs) by the virus stimulates the extracellular signal-regulated kinase (ERK) 1/2, which in turn increases phosphorylation of the human GRa at serine 203, resulting in the enhancement of GRa transcriptional activity on the IL-10 gene promoter. Since IL-10 is a potent anti-inflammatory cytokine, it appears that the virus stimulates its own infection/propagation by enhancing GRa activity on this cytokine. Respiratory syncytial virus (RSV), which is one of the major causes of lower respiratory tract infection and hospital visits during infancy and childhood, is reported to repress the anti-inflammatory action of glucocorticoids through GRa (489-491).
ACKNOWLEDGEMENTS
This literary work was supported by the intramural fund of the Sidra Medical and Research Center to T. Kino.
REFERENCES
- 1.
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. [PubMed: 7715646]
- 2.
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44. [PubMed: 6368214]
- 3.
- Glucocorticoids, Overview. Nicolaides NC, Charmandari E, Chrousos GP. In Encyclopedia of Endocrine Diseases (2nd Edition), edited by Ilpo Huhtaniemi and Luciano Martini, New York 2018, pp. 64-71.
- 4.
- Boumpas DT, Chrousos GP, Wilder RL, Cupps TR, Balow JE. Glucocorticoid therapy for immune-mediated diseases: basic and clinical correlates. Ann Intern Med. 1993;119:1198–1208. [PubMed: 8239251]
- 5.
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–839. [PMC free article: PMC6159888] [PubMed: 8521507]
- 6.
- Androutsellis-Theotokis A, Chrousos GP, McKay RD, DeCherney AH, Kino T. Expression profiles of the nuclear receptors and their transcriptional coregulators during differentiation of neural stem cells. Horm Metab Res. 2013;45:159–168. [PMC free article: PMC3781591] [PubMed: 22990992]
- 7.
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. [PubMed: 11772937]
- 8.
- Mackeh R, Marr AK, Dargham S, Syed N, Fakhro K, Kino T. Single nucleotide variations in the human nuclear hormone receptor genes: their distribution in major domains and link to receptor gene ages and functionality. 2017 (unpublished data)
- 9.
- Kovacs WJ, Orth DN. The adrenal cortex. In: Wilson J, D., Foster DW, eds. Williams Textbook of Endocrinology. Philadelphia, PA: W.B. Saunders Company; 1998:517-750.
- 10.
- Thornton JW. Evolution of vertebrate steroid receptors from an ancestral estrogen receptor by ligand exploitation and serial genome expansions. Proc Natl Acad Sci U S A. 2001;98:5671–5676. [PMC free article: PMC33271] [PubMed: 11331759]
- 11.
- Thornton JW, Need E, Crews D. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science. 2003;301:1714–1717. [PubMed: 14500980]
- 12.
- Baker ME. Steroid receptors and vertebrate evolution. Mol Cell Endocrinol. 2019;496:110526. [PubMed: 31376417]
- 13.
- Kuraku S, Hoshiyama D, Katoh K, Suga H, Miyata T. Monophyly of lampreys and hagfishes supported by nuclear DNA-coded genes. J Mol Evol. 1999;49:729–735. [PubMed: 10594174]
- 14.
- Baker ME. Co-evolution of steroidogenic and steroid-inactivating enzymes and adrenal and sex steroid receptors. Mol Cell Endocrinol. 2004;215:55–62. [PubMed: 15026175]
- 15.
- Bridgham JT, Carroll SM, Thornton JW. Evolution of hormone-receptor complexity by molecular exploitation. Science. 2006;312:97–101. [PubMed: 16601189]
- 16.
- Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317:1544–1548. [PMC free article: PMC2519897] [PubMed: 17702911]
- 17.
- Harms MJ, Thornton JW. Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature. 2014;512:203–207. [PMC free article: PMC4447330] [PubMed: 24930765]
- 18.
- Alsop D, Vijayan M. The zebrafish stress axis: molecular fallout from the teleost-specific genome duplication event. Gen Comp Endocrinol. 2009;161:62–66. [PubMed: 18930731]
- 19.
- Hollenberg SM, Weinberger C, Ong ES, Cerelli G, Oro A, Lebo R, Thompson EB, Rosenfeld MG, Evans RM. Primary structure and expression of a functional human glucocorticoid receptor cDNA. Nature. 1985;318:635–641. [PMC free article: PMC6165583] [PubMed: 2867473]
- 20.
- Chrousos GP, Kino T. Intracellular glucocorticoid signaling: a formerly simple system turns stochastic. Sci STKE. 2005;2005:pe48. [PubMed: 16204701]
- 21.
- Bamberger CM, Bamberger AM, de Castro M, Chrousos GP. Glucocorticoid receptor b, a potential endogenous inhibitor of glucocorticoid action in humans. J Clin Invest. 1995;95:2435–2441. [PMC free article: PMC295915] [PubMed: 7769088]
- 22.
- Almlof T, Gustafsson JA, Wright AP. Role of hydrophobic amino acid clusters in the transactivation activity of the human glucocorticoid receptor. Mol Cell Biol. 1997;17:934–945. [PMC free article: PMC231819] [PubMed: 9001247]
- 23.
- Almlof T, Wallberg AE, Gustafsson JA, Wright AP. Role of important hydrophobic amino acids in the interaction between the glucocorticoid receptor tau 1-core activation domain and target factors. Biochemistry. 1998;37:9586–9594. [PubMed: 9649342]
- 24.
- Warnmark A, Gustafsson JA, Wright AP. Architectural principles for the structure and function of the glucocorticoid receptor tau 1 core activation domain. J Biol Chem. 2000;275:15014–15018. [PubMed: 10747977]
- 25.
- Kumar R, Volk DE, Li J, Lee JC, Gorenstein DG, Thompson EB. TATA box binding protein induces structure in the recombinant glucocorticoid receptor AF1 domain. Proc Natl Acad Sci U S A. 2004;101:16425–16430. [PMC free article: PMC534534] [PubMed: 15545613]
- 26.
- Khan SH, Awasthi S, Guo C, Goswami D, Ling J, Griffin PR, Simons SS Jr, Kumar R. Binding of the N-terminal region of coactivator TIF2 to the intrinsically disordered AF1 domain of the glucocorticoid receptor is accompanied by conformational reorganizations. J Biol Chem. 2012;287:44546–44560. [PMC free article: PMC3531768] [PubMed: 23132854]
- 27.
- Khan SH, Ling J, Kumar R. TBP binding-induced folding of the glucocorticoid receptor AF1 domain facilitates its interaction with steroid receptor coactivator-1. PLoS One. 2011;6:e21939. [PMC free article: PMC3131385] [PubMed: 21760925]
- 28.
- Howard KJ, Holley SJ, Yamamoto KR, Distelhorst CW. Mapping the HSP90 binding region of the glucocorticoid receptor. J Biol Chem. 1990;265:11928–11935. [PubMed: 2365707]
- 29.
- Schule R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, Yang N, Verma IM, Evans RM. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. [PubMed: 2169353]
- 30.
- Burris TP. The nuclear receptor superfamily. In: Burris TP, McCabe ERB, eds. Nuclear receptors and genetic disease. London: Academic Press; 2001:1-58.
- 31.
- Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, Consler TG, Parks DJ, Stewart EL, Willson TM, Lambert MH, Moore JT, Pearce KH, Xu HE. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. [PubMed: 12151000]
- 32.
- Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor's ligand binding domains. Proc Natl Acad Sci U S A. 1998;95:5998–6003. [PMC free article: PMC27574] [PubMed: 9600906]
- 33.
- Williams SP, Sigler PB. Atomic structure of progesterone complexed with its receptor. Nature. 1998;393:392–396. [PubMed: 9620806]
- 34.
- Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell. 2005;18:331–342. [PubMed: 15866175]
- 35.
- Bender IK, Cao Y, Lu NZ. Determinants of the heightened activity of glucocorticoid receptor translational isoforms. Mol Endocrinol. 2013;27:1577–1587. [PMC free article: PMC3753425] [PubMed: 23820903]
- 36.
- Wu I, Shin SC, Cao Y, Bender IK, Jafari N, Feng G, Lin S, Cidlowski JA, Schleimer RP, Lu NZ. Selective glucocorticoid receptor translational isoforms reveal glucocorticoid-induced apoptotic transcriptomes. Cell Death Dis. 2013;4:e453. [PMC free article: PMC3563981] [PubMed: 23303127]
- 37.
- Kino T, Chrousos GP. Tissue-specific glucocorticoid resistance-hypersensitivity syndromes: multifactorial states of clinical importance. J Allergy Clin Immunol. 2002;109:609–613. [PubMed: 11941307]
- 38.
- Cao Y, Bender IK, Konstantinidis AK, Shin SC, Jewell CM, Cidlowski JA, Schleimer RP, Lu NZ. Glucocorticoid receptor translational isoforms underlie maturational stage-specific glucocorticoid sensitivities of dendritic cells in mice and humans. Blood. 2013;121:1553–1562. [PMC free article: PMC3587320] [PubMed: 23297131]
- 39.
- Sinclair D, Webster MJ, Wong J, Weickert CS. Dynamic molecular and anatomical changes in the glucocorticoid receptor in human cortical development. Mol Psychiatry. 2011;16:504–515. [PubMed: 20308989]
- 40.
- Presul E, Schmidt S, Kofler R, Helmberg A. Identification, tissue expression, and glucocorticoid responsiveness of alternative first exons of the human glucocorticoid receptor. J Mol Endocrinol. 2007;38:79–90. [PubMed: 17242171]
- 41.
- Turner JD, Muller CP. Structure of the glucocorticoid receptor (NR3C1) gene 5' untranslated region: identification, and tissue distribution of multiple new human exon 1. J Mol Endocrinol. 2005;35:283–292. [PubMed: 16216909]
- 42.
- Sinclair D, Fullerton JM, Webster MJ, Shannon Weickert C. Glucocorticoid receptor 1B and 1C mRNA transcript alterations in schizophrenia and bipolar disorder, and their possible regulation by GR gene variants. PLoS One. 2012;7:e31720. [PMC free article: PMC3302776] [PubMed: 22427805]
- 43.
- Martin-Blanco A, Ferrer M, Soler J, Salazar J, Vega D, Andion O, Sanchez-Mora C, Arranz MJ, Ribases M, Feliu-Soler A, Perez V, Pascual JC. Association between methylation of the glucocorticoid receptor gene, childhood maltreatment, and clinical severity in borderline personality disorder. J Psychiatr Res. 2014;57:34–40. [PubMed: 25048180]
- 44.
- Na KS, Chang HS, Won E, Han KM, Choi S, Tae WS, Yoon HK, Kim YK, Joe SH, Jung IK, Lee MS, Ham BJ. Association between glucocorticoid receptor methylation and hippocampal subfields in major depressive disorder. PLoS One. 2014;9:e85425. [PMC free article: PMC3897456] [PubMed: 24465557]
- 45.
- Kantake M, Yoshitake H, Ishikawa H, Araki Y, Shimizu T. Postnatal epigenetic modification of glucocorticoid receptor gene in preterm infants: a prospective cohort study. BMJ Open. 2014;4:e005318. [PMC free article: PMC4120337] [PubMed: 25023132]
- 46.
- Bockmuhl Y, Murgatroyd CA, Kuczynska A, Adcock IM, Almeida OF, Spengler D. Differential regulation and function of 5'-untranslated GR-exon 1 transcripts. Mol Endocrinol. 2011;25:1100–1110. [PMC free article: PMC5417247] [PubMed: 21527501]
- 47.
- Denis M, Gustafsson JA, Wikstrom AC. Interaction of the Mr = 90,000 heat shock protein with the steroid-binding domain of the glucocorticoid receptor. J Biol Chem. 1988;263:18520–18523. [PubMed: 3192546]
- 48.
- Czar MJ, Lyons RH, Welsh MJ, Renoir JM, Pratt WB. Evidence that the FK506-binding immunophilin heat shock protein 56 is required for trafficking of the glucocorticoid receptor from the cytoplasm to the nucleus. Mol Endocrinol. 1995;9:1549–1560. [PubMed: 8584032]
- 49.
- Owens-Grillo JK, Hoffmann K, Hutchison KA, Yem AW, Deibel MR Jr, Handschumacher RE, Pratt WB. The cyclosporin A-binding immunophilin CyP-40 and the FK506-binding immunophilin hsp56 bind to a common site on hsp90 and exist in independent cytosolic heterocomplexes with the untransformed glucocorticoid receptor. J Biol Chem. 1995;270:20479–20484. [PubMed: 7657624]
- 50.
- Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Hache RJ, Lefebvre YA. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol. 1999;19:1025–1037. [PMC free article: PMC116033] [PubMed: 9891038]
- 51.
- McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. [PubMed: 10678832]
- 52.
- Voss TC, Schiltz RL, Sung MH, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, Hager GL. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell. 2011;146:544–554. [PMC free article: PMC3210475] [PubMed: 21835447]
- 53.
- Morisaki T, Muller WG, Golob N, Mazza D, McNally JG. Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nat Commun. 2014;5:4456. [PMC free article: PMC4144071] [PubMed: 25034201]
- 54.
- Hache RJ, Tse R, Reich T, Savory JG, Lefebvre YA. Nucleocytoplasmic trafficking of steroid-free glucocorticoid receptor. J Biol Chem. 1999;274:1432–1439. [PubMed: 9880517]
- 55.
- Yang J, Liu J, DeFranco DB. Subnuclear trafficking of glucocorticoid receptors in vitro: chromatin recycling and nuclear export. J Cell Biol. 1997;137:523–538. [PMC free article: PMC2139874] [PubMed: 9151662]
- 56.
- DeFranco DB. Subnuclear trafficking of steroid receptors. Biochem Soc Trans. 1997;25:592–597. [PubMed: 9191162]
- 57.
- Kinyamu HK, Chen J, Archer TK. Linking the ubiquitin-proteasome pathway to chromatin remodeling/modification by nuclear receptors. J Mol Endocrinol. 2005;34:281–297. [PubMed: 15821097]
- 58.
- Kino T, Liou SH, Charmandari E, Chrousos GP. Glucocorticoid receptor mutants demonstrate increased motility inside the nucleus of living cells: time of fluorescence recovery after photobleaching (FRAP) is an integrated measure of receptor function. Mol Med. 2004;10:80–88. [PMC free article: PMC1431369] [PubMed: 16307173]
- 59.
- Kino T, De Martino MU, Charmandari E, Mirani M, Chrousos GP. Tissue glucocorticoid resistance/hypersensitivity syndromes. J Steroid Biochem Mol Biol. 2003;85:457–467. [PubMed: 12943736]
- 60.
- Holaska JM, Black BE, Love DC, Hanover JA, Leszyk J, Paschal BM. Calreticulin is a receptor for nuclear export. J Cell Biol. 2001;152:127–140. [PMC free article: PMC2193655] [PubMed: 11149926]
- 61.
- Black BE, Holaska JM, Rastinejad F, Paschal BM. DNA binding domains in diverse nuclear receptors function as nuclear export signals. Curr Biol. 2001;11:1749–1758. [PubMed: 11719216]
- 62.
- Holaska JM, Black BE, Rastinejad F, Paschal BM. Ca2+-dependent nuclear export mediated by calreticulin. Mol Cell Biol. 2002;22:6286–6297. [PMC free article: PMC133999] [PubMed: 12167720]
- 63.
- Itoh M, Adachi M, Yasui H, Takekawa M, Tanaka H, Imai K. Nuclear export of glucocorticoid receptor is enhanced by c-Jun N-terminal kinase-mediated phosphorylation. Mol Endocrinol. 2002;16:2382–2392. [PubMed: 12351702]
- 64.
- Kino T, Souvatzoglou E, De Martino MU, Tsopanomihalu M, Wan Y, Chrousos GP. Protein 14-3-3s interacts with and favors cytoplasmic subcellular localization of the glucocorticoid receptor, acting as a negative regulator of the glucocorticoid signaling pathway. J Biol Chem. 2003;278:25651–25656. [PubMed: 12730237]
- 65.
- Habib T, Sadoun A, Nader N, Suzuki S, Liu W, Jithesh PV, Kino T. AKT1 has dual actions on the glucocorticoid receptor by cooperating with 14-3-3. Mol Cell Endocrinol. 2017;439:431–443. [PubMed: 27717743]
- 66.
- Psarra AM, Sekeris CE. Glucocorticoids induce mitochondrial gene transcription in HepG2 cells: role of the mitochondrial glucocorticoid receptor. Biochim Biophys Acta. 2011;1813:1814–1821. [PubMed: 21664385]
- 67.
- Hunter RG, Seligsohn M, Rubin TG, Griffiths BB, Ozdemir Y, Pfaff DW, Datson NA, McEwen BS. Stress and corticosteroids regulate rat hippocampal mitochondrial DNA gene expression via the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2016;113:9099–9104. [PMC free article: PMC4987818] [PubMed: 27457949]
- 68.
- He Y, Xu Y, Zhang C, Gao X, Dykema KJ, Martin KR, Ke J, Hudson EA, Khoo SK, Resau JH, Alberts AS, MacKeigan JP, Furge KA, Xu HE. Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci Signal. 2011;4:ra44. [PMC free article: PMC3684214] [PubMed: 21730326]
- 69.
- Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–261. [PubMed: 8771358]
- 70.
- Lieberman BA, Bona BJ, Edwards DP, Nordeen SK. The constitution of a progesterone response element. Mol Endocrinol. 1993;7:515–527. [PubMed: 8388996]
- 71.
- Presman DM, Ogara MF, Stortz M, Alvarez LD, Pooley JR, Schiltz RL, Grontved L, Johnson TA, Mittelstadt PR, Ashwell JD, Ganesan S, Burton G, Levi V, Hager GL, Pecci A. Live cell imaging unveils multiple domain requirements for in vivo dimerization of the glucocorticoid receptor. PLoS Biol. 2014;12:e1001813. [PMC free article: PMC3958349] [PubMed: 24642507]
- 72.
- Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. [PMC free article: PMC2777810] [PubMed: 19372434]
- 73.
- Watson LC, Kuchenbecker KM, Schiller BJ, Gross JD, Pufall MA, Yamamoto KR. The glucocorticoid receptor dimer interface allosterically transmits sequence-specific DNA signals. Nat Struct Mol Biol. 2013;20:876–883. [PMC free article: PMC3702670] [PubMed: 23728292]
- 74.
- Beato M, Sanchez-Pacheco A. Interaction of steroid hormone receptors with the transcription initiation complex. Endocr Rev. 1996;17:587–609. [PubMed: 8969970]
- 75.
- Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. Functional domains of the human glucocorticoid receptor. Cell. 1986;46:645–652. [PubMed: 3742595]
- 76.
- McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344. [PubMed: 10368774]
- 77.
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14:1553–1577. [PubMed: 10887150]
- 78.
- Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani Y. A. p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature. 1996;382:319–324. [PubMed: 8684459]
- 79.
- Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, Evans RM, Nakatani Y, Ozato K. The histone acetylase PCAF is a nuclear receptor coactivator. Genes Dev. 1998;12:1638–1651. [PMC free article: PMC316869] [PubMed: 9620851]
- 80.
- Leo C, Chen JD. The SRC family of nuclear receptor coactivators. Gene. 2000;245:1–11. [PubMed: 10713439]
- 81.
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed: 10652267]
- 82.
- Charmandari E, Kino T, Chrousos GP. Familial/sporadic glucocorticoid resistance: clinical phenotype and molecular mechanisms. Ann N Y Acad Sci. 2004;1024:168–181. [PubMed: 15265781]
- 83.
- Yi P, Wang Z, Feng Q, Pintilie GD, Foulds CE, Lanz RB, Ludtke SJ, Schmid MF, Chiu W, O'Malley BW. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell. 2015;57:1047–1058. [PMC free article: PMC4369429] [PubMed: 25728767]
- 84.
- Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of activity of the transcription factor GATA-1 by acetylation. Nature. 1998;396:594–598. [PubMed: 9859997]
- 85.
- Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. [PubMed: 9288740]
- 86.
- Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell. 1999;98:675–686. [PubMed: 10490106]
- 87.
- Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. [PubMed: 9192902]
- 88.
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR. Structure and specificity of nuclear receptor-coactivator interactions. Genes Dev. 1998;12:3343–3356. [PMC free article: PMC317236] [PubMed: 9808622]
- 89.
- Hurt DE, Suzuki S, Mayama T, Charmandari E, Kino T. Structural Analysis on the Pathologic Mutant Glucocorticoid Receptor Ligand-Binding Domains. Mol Endocrinol. 2016;30:173–188. [PMC free article: PMC4792232] [PubMed: 26745667]
- 90.
- Fry CJ, Peterson CL. Chromatin remodeling enzymes: who's on first? Curr Biol. 2001;11:R185–197. [PubMed: 11267889]
- 91.
- Yoshinaga SK, Peterson CL, Herskowitz I, Yamamoto KR. Roles of SWI1, SWI2, and SWI3 proteins for transcriptional enhancement by steroid receptors. Science. 1992;258:1598–1604. [PubMed: 1360703]
- 92.
- Rachez C, Freedman LP. Mediator complexes and transcription. Curr Opin Cell Biol. 2001;13:274–280. [PubMed: 11343897]
- 93.
- Henriksson A, Almlof T, Ford J, McEwan IJ, Gustafsson JA, Wright AP. Role of the Ada adaptor complex in gene activation by the glucocorticoid receptor. Mol Cell Biol. 1997;17:3065–3073. [PMC free article: PMC232159] [PubMed: 9154805]
- 94.
- Wallberg AE, Neely KE, Hassan AH, Gustafsson JA, Workman JL, Wright AP. Recruitment of the SWI-SNF chromatin remodeling complex as a mechanism of gene activation by the glucocorticoid receptor tau1 activation domain. Mol Cell Biol. 2000;20:2004–2013. [PMC free article: PMC110817] [PubMed: 10688647]
- 95.
- Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, Coetzee GA, Stallcup MR. Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol. 1999;19:6164–6173. [PMC free article: PMC84548] [PubMed: 10454563]
- 96.
- Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, Nguyen MP, Chen D, Huang SM, Subramanian S, McKinerney E, Katzenellenbogen BS, Stallcup MR, Kushner PJ. Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol Endocrinol. 1998;12:1605–1618. [PubMed: 9773983]
- 97.
- Wallberg AE, Neely KE, Gustafsson JA, Workman JL, Wright AP, Grant PA. Histone acetyltransferase complexes can mediate transcriptional activation by the major glucocorticoid receptor activation domain. Mol Cell Biol. 1999;19:5952–5959. [PMC free article: PMC84458] [PubMed: 10454542]
- 98.
- Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 1999;18:5380–5388. [PMC free article: PMC1171607] [PubMed: 10508170]
- 99.
- Lanz RB, McKenna NJ, Onate SA, Albrecht U, Wong J, Tsai SY, Tsai MJ, O'Malley BW. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell. 1999;97:17–27. [PubMed: 10199399]
- 100.
- Benecke A, Chambon P, Gronemeyer H. Synergy between estrogen receptor a activation functions AF1 and AF2 mediated by transcription intermediary factor TIF2. EMBO Rep. 2000;1:151–157. [PMC free article: PMC1084260] [PubMed: 11265755]
- 101.
- Puigserver P, Spiegelman BM. Peroxisome proliferator-activated receptor-g coactivator 1a (PGC-1a): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. [PubMed: 12588810]
- 102.
- Higashida K, Kim SH, Jung SR, Asaka M, Holloszy JO, Han DH. Effects of resveratrol and SIRT1 on PGC-1a activity and mitochondrial biogenesis: a reevaluation. PLoS Biol. 2013;11:e1001603. [PMC free article: PMC3706311] [PubMed: 23874150]
- 103.
- Philp A, Chen A, Lan D, Meyer GA, Murphy AN, Knapp AE, Olfert IM, McCurdy CE, Marcotte GR, Hogan MC, Baar K, Schenk S. Sirtuin 1 (SIRT1) deacetylase activity is not required for mitochondrial biogenesis or peroxisome proliferator-activated receptor-g coactivator-1a (PGC-1a) deacetylation following endurance exercise. J Biol Chem. 2011;286:30561–30570. [PMC free article: PMC3162416] [PubMed: 21757760]
- 104.
- Suzuki S, Iben JR, Coon SL, Kino T. SIRT1 is a transcriptional enhancer of the glucocorticoid receptor acting independently to its deacetylase activity. Mol Cell Endocrinol. 2018;461:178–187. [PMC free article: PMC5756502] [PubMed: 28923345]
- 105.
- Altarejos JY, Montminy M. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol. 2011;12:141–151. [PMC free article: PMC4324555] [PubMed: 21346730]
- 106.
- Hill MJ, Suzuki S, Segars JH, Kino T. CRTC2 Is a Coactivator of GR and Couples GR and CREB in the Regulation of Hepatic Gluconeogenesis. Mol Endocrinol. 2016;30:104–117. [PMC free article: PMC4695631] [PubMed: 26652733]
- 107.
- Wu DY, Ou CY, Chodankar R, Siegmund KD, Stallcup MR. Distinct, genome-wide, gene-specific selectivity patterns of four glucocorticoid receptor coregulators. Nucl Recept Signal. 2014;12:e002. [PMC free article: PMC4242289] [PubMed: 25422592]
- 108.
- Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. [PubMed: 21496643]
- 109.
- Hudson WH, Youn C, Ortlund EA. The structural basis of direct glucocorticoid-mediated transrepression. Nat Struct Mol Biol. 2013;20:53–58. [PMC free article: PMC3539207] [PubMed: 23222642]
- 110.
- Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, Schwalie P, Hubner N, Evans RM. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49:158–171. [PMC free article: PMC3640846] [PubMed: 23159735]
- 111.
- Miner JN, Yamamoto KR. Regulatory crosstalk at composite response elements. Trends Biochem Sci. 1991;16:423–426. [PubMed: 1776172]
- 112.
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, Gass P, Schmid W, Herrlich P, Angel P, Schutz G. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. [PubMed: 9604929]
- 113.
- Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, Herrlich P, Schutz G. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J. 2001;20:7168–7173. [PMC free article: PMC125338] [PubMed: 11742993]
- 114.
- Karin M, Chang L. AP-1-glucocorticoid receptor crosstalk taken to a higher level. J Endocrinol. 2001;169:447–451. [PubMed: 11375114]
- 115.
- Barnes PJ, Karin M. Nuclear factor-kB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. [PubMed: 9091804]
- 116.
- Didonato JA, Saatcioglu F, Karin M. Molecular mechanisms of immunosuppression and anti-inflammatory activities by glucocorticoids. Am J Respir Crit Care Med. 1996;154:S11–15. [PubMed: 8756781]
- 117.
- Liberman AC, Druker J, Garcia FA, Holsboer F, Arzt E. Intracellular molecular signaling. Basis for specificity to glucocorticoid anti-inflammatory actions. Ann N Y Acad Sci. 2009;1153:6–13. [PubMed: 19236322]
- 118.
- Liberman AC, Refojo D, Druker J, Toscano M, Rein T, Holsboer F, Arzt E. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein-protein interaction. FASEB J. 2007;21:1177–1188. [PubMed: 17215482]
- 119.
- Reily MM, Pantoja C, Hu X, Chinenov Y, Rogatsky I. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. 2006;25:108–117. [PMC free article: PMC1356362] [PubMed: 16362036]
- 120.
- Perkins ND. The Rel/NF-kB family: friend and foe. Trends Biochem Sci. 2000;25:434–440. [PubMed: 10973057]
- 121.
- McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kB and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. [PubMed: 10453354]
- 122.
- Caldenhoven E, Liden J, Wissink S, Van de Stolpe A, Raaijmakers J, Koenderman L, Okret S, Gustafsson JA, Van der Saag PT. Negative cross-talk between RelA and the glucocorticoid receptor: a possible mechanism for the antiinflammatory action of glucocorticoids. Mol Endocrinol. 1995;9:401–412. [PubMed: 7659084]
- 123.
- Liden J, Delaunay F, Rafter I, Gustafsson J, Okret S. A new function for the C-terminal zinc finger of the glucocorticoid receptor. Repression of RelA transactivation. J Biol Chem. 1997;272:21467–21472. [PubMed: 9261164]
- 124.
- Wissink S, van Heerde EC, Schmitz ML, Kalkhoven E, van der Burg B, Baeuerle PA, van der Saag PT. Distinct domains of the RelA NF-kB subunit are required for negative cross-talk and direct interaction with the glucocorticoid receptor. J Biol Chem. 1997;272:22278–22284. [PubMed: 9268377]
- 125.
- McKay LI, Cidlowski JA. Cross-talk between nuclear factor-kB and the steroid hormone receptors: mechanisms of mutual antagonism. Mol Endocrinol. 1998;12:45–56. [PubMed: 9440809]
- 126.
- Ito K, Barnes PJ, Adcock IM. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1b-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. [PMC free article: PMC88765] [PubMed: 10958685]
- 127.
- Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000;14:2314–2329. [PMC free article: PMC316928] [PubMed: 10995388]
- 128.
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kB activity through induction of IkB synthesis. Science. 1995;270:286–290. [PubMed: 7569976]
- 129.
- Chinenov Y, Gupte R, Dobrovolna J, Flammer JR, Liu B, Michelassi FE, Rogatsky I. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. 2012;109:11776–11781. [PMC free article: PMC3406827] [PubMed: 22753499]
- 130.
- Herrlich P. Cross-talk between glucocorticoid receptor and AP-1. Oncogene. 2001;20:2465–2475. [PubMed: 11402341]
- 131.
- van Dam H, Castellazzi M. Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene. 2001;20:2453–2464. [PubMed: 11402340]
- 132.
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. [PubMed: 8616895]
- 133.
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. [PubMed: 11483993]
- 134.
- Stauber C, Altschmied J, Akerblom IE, Marron JL, Mellon PL. Mutual cross-interference between glucocorticoid receptor and CREB inhibits transactivation in placental cells. New Biol. 1992;4:527–540. [PubMed: 1387550]
- 135.
- Chatterjee VK, Madison LD, Mayo S, Jameson JL. Repression of the human glycoprotein hormone a-subunit gene by glucocorticoids: evidence for receptor interactions with limiting transcriptional activators. Mol Endocrinol. 1991;5:100–110. [PubMed: 1708098]
- 136.
- Imai E, Miner JN, Mitchell JA, Yamamoto KR, Granner DK. Glucocorticoid receptor-cAMP response element-binding protein interaction and the response of the phosphoenolpyruvate carboxykinase gene to glucocorticoids. J Biol Chem. 1993;268:5353–5356. [PubMed: 8449898]
- 137.
- Liu JL, Papachristou DN, Patel YC. Glucocorticoids activate somatostatin gene transcription through co-operative interaction with the cyclic AMP signalling pathway. Biochem J. 1994;301:863–869. [PMC free article: PMC1137066] [PubMed: 7914402]
- 138.
- ten Dijke P, Miyazono K, Heldin CH. Signaling inputs converge on nuclear effectors in TGF-b signaling. Trends Biochem Sci. 2000;25:64–70. [PubMed: 10664585]
- 139.
- Hata A, Lagna G, Massague J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12:186–197. [PMC free article: PMC316444] [PubMed: 9436979]
- 140.
- Bai S, Shi X, Yang X, Cao X. Smad6 as a transcriptional corepressor. J Biol Chem. 2000;275:8267–8270. [PubMed: 10722652]
- 141.
- Bai S, Cao X. A nuclear antagonistic mechanism of inhibitory Smads in transforming growth factor-b signaling. J Biol Chem. 2002;277:4176–4182. [PubMed: 11711531]
- 142.
- Lin X, Liang YY, Sun B, Liang M, Shi Y, Brunicardi FC, Feng XH. Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol Cell Biol. 2003;23:9081–9093. [PMC free article: PMC309600] [PubMed: 14645520]
- 143.
- Afrakhte M, Moren A, Jossan S, Itoh S, Sampath K, Westermark B, Heldin CH, Heldin NE, ten Dijke P. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-b family members. Biochem Biophys Res Commun. 1998;249:505–511. [PubMed: 9712726]
- 144.
- Miyazono K. Positive and negative regulation of TGF-b signaling. J Cell Sci. 2000;113:1101–1109. [PubMed: 10704361]
- 145.
- Ichijo T, Voutetakis A, Cotrim AP, Bhattachryya N, Fujii M, Chrousos GP, Kino T. The Smad6-histone deacetylase 3 complex silences the transcriptional activity of the glucocorticoid receptor: potential clinical implications. J Biol Chem. 2005;280:42067–42077. [PubMed: 16249187]
- 146.
- Emerson RO, Thomas JH. Adaptive evolution in zinc finger transcription factors. PLoS Genet. 2009;5:e1000325. [PMC free article: PMC2604467] [PubMed: 19119423]
- 147.
- Mackeh R, Marr AK, Fadda A, Kino T. C2H2-type zinc finger proteins: evolutionarily old and new partners of the nuclear hormone receptors. Nucl Recept Signal. 2017. in press. [PMC free article: PMC6348741] [PubMed: 30718982]
- 148.
- Collins T, Stone JR, Williams AJ. All in the family: the BTB/POZ, KRAB, and SCAN domains. Mol Cell Biol. 2001;21:3609–3615. [PMC free article: PMC86980] [PubMed: 11340155]
- 149.
- Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–246. [PMC free article: PMC4610363] [PubMed: 24614316]
- 150.
- Ross-Innes CS, Brown GD, Carroll JS. A co-ordinated interaction between CTCF and ER in breast cancer cells. BMC Genomics. 2011;12:593. [PMC free article: PMC3248577] [PubMed: 22142239]
- 151.
- Awad TA, Bigler J, Ulmer JE, Hu YJ, Moore JM, Lutz M, Neiman PE, Collins SJ, Renkawitz R, Lobanenkov VV, Filippova GN. Negative transcriptional regulation mediated by thyroid hormone response element 144 requires binding of the multivalent factor CTCF to a novel target DNA sequence. J Biol Chem. 1999;274:27092–27098. [PubMed: 10480923]
- 152.
- Kino T, Pavlatou MG, Moraitis AG, Nemery RL, Raygada M, Stratakis CA. ZNF764 haploinsufficiency may explain partial glucocorticoid, androgen, and thyroid hormone resistance associated with 16p11.2 microdeletion. J Clin Endocrinol Metab. 2012;97:E1557–1566. [PMC free article: PMC3410270] [PubMed: 22577170]
- 153.
- Fadda A, Syed N, Mackeh R, Papadopoulou A, Suzuki S, Jithesh PV, Kino T. Genome-wide Regulatory Roles of the C2H2-type Zinc Finger Protein ZNF764 on the Glucocorticoid Receptor. Sci Rep. 2017;7:41598. [PMC free article: PMC5282477] [PubMed: 28139699]
- 154.
- Carter ME, Brunet A. FOXO transcription factors. Curr Biol. 2007;17:R113–114. [PubMed: 17307039]
- 155.
- Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33. [PMC free article: PMC3024537] [PubMed: 21151129]
- 156.
- Ma X, Xu L, Mueller E. Forkhead box A3 mediates glucocorticoid receptor function in adipose tissue. Proc Natl Acad Sci U S A. 2016;113:3377–3382. [PMC free article: PMC4812768] [PubMed: 26957608]
- 157.
- De Martino MU, Bhattachryya N, Alesci S, Ichijo T, Chrousos GP, Kino T. The glucocorticoid receptor and the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II interact with and mutually affect each other's transcriptional activities: implications for intermediary metabolism. Mol Endocrinol. 2004;18:820–833. [PubMed: 14739255]
- 158.
- Pierreux CE, Stafford J, Demonte D, Scott DK, Vandenhaute J, O'Brien RM, Granner DK, Rousseau GG, Lemaigre FP. Antiglucocorticoid activity of hepatocyte nuclear factor-6. Proc Natl Acad Sci U S A. 1999;96:8961–8966. [PMC free article: PMC17715] [PubMed: 10430878]
- 159.
- Philips A, Maira M, Mullick A, Chamberland M, Lesage S, Hugo P, Drouin J. Antagonism between Nur77 and glucocorticoid receptor for control of transcription. Mol Cell Biol. 1997;17:5952–5959. [PMC free article: PMC232443] [PubMed: 9315653]
- 160.
- Nader N, Ng SS, Wang Y, Abel BS, Chrousos GP, Kino T. Liver X receptors regulate the transcriptional activity of the glucocorticoid receptor: implications for the carbohydrate metabolism. PLoS One. 2012;7:e26751. [PMC free article: PMC3310817] [PubMed: 22457708]
- 161.
- Patel R, Patel M, Tsai R, Lin V, Bookout AL, Zhang Y, Magomedova L, Li T, Chan JF, Budd C, Mangelsdorf DJ, Cummins CL. LXRb is required for glucocorticoid-induced hyperglycemia and hepatosteatosis in mice. J Clin Invest. 2011;121:431–441. [PMC free article: PMC3007136] [PubMed: 21123945]
- 162.
- Renga B, Mencarelli A, D'Amore C, Cipriani S, Baldelli F, Zampella A, Distrutti E, Fiorucci S. Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. FASEB J. 2012;26:3021–3031. [PubMed: 22447981]
- 163.
- Sengupta S, Vonesch JL, Waltzinger C, Zheng H, Wasylyk B. Negative cross-talk between p53 and the glucocorticoid receptor and its role in neuroblastoma cells. EMBO J. 2000;19:6051–6064. [PMC free article: PMC305812] [PubMed: 11080152]
- 164.
- Sengupta S, Wasylyk B. Ligand-dependent interaction of the glucocorticoid receptor with p53 enhances their degradation by Hdm2. Genes Dev. 2001;15:2367–2380. [PMC free article: PMC312780] [PubMed: 11562347]
- 165.
- Chandran UR, Warren BS, Baumann CT, Hager GL, DeFranco DB. The glucocorticoid receptor is tethered to DNA-bound Oct-1 at the mouse gonadotropin-releasing hormone distal negative glucocorticoid response element. J Biol Chem. 1999;274:2372–2378. [PubMed: 9891005]
- 166.
- Subramaniam N, Cairns W, Okret S. Glucocorticoids repress transcription from a negative glucocorticoid response element recognized by two homeodomain-containing proteins, Pbx and Oct-1. J Biol Chem. 1998;273:23567–23574. [PubMed: 9722596]
- 167.
- Prefontaine GG, Lemieux ME, Giffin W, Schild-Poulter C, Pope L, LaCasse E, Walker P, Hache RJ. Recruitment of octamer transcription factors to DNA by glucocorticoid receptor. Mol Cell Biol. 1998;18:3416–3430. [PMC free article: PMC108923] [PubMed: 9584182]
- 168.
- Truss M, Bartsch J, Schelbert A, Hache RJ, Beato M. Hormone induces binding of receptors and transcription factors to a rearranged nucleosome on the MMTV promoter in vivo. EMBO J. 1995;14:1737–1751. [PMC free article: PMC398267] [PubMed: 7737125]
- 169.
- Hartig E, Cato AC. In vivo binding of proteins to stably integrated MMTV DNA in murine cell lines: occupancy of NFI and OTF1 binding sites in the absence and presence of glucocorticoids. Cell Mol Biol Res. 1994;40:643–652. [PubMed: 7787882]
- 170.
- Archer TK, Cordingley MG, Wolford RG, Hager GL. Transcription factor access is mediated by accurately positioned nucleosomes on the mouse mammary tumor virus promoter. Mol Cell Biol. 1991;11:688–698. [PMC free article: PMC359719] [PubMed: 1846670]
- 171.
- Chang TJ, Scher BM, Waxman S, Scher W. Inhibition of mouse GATA-1 function by the glucocorticoid receptor: possible mechanism of steroid inhibition of erythroleukemia cell differentiation. Mol Endocrinol. 1993;7:528–542. [PubMed: 8502237]
- 172.
- Lekstrom-Himes J, Xanthopoulos KG. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J Biol Chem. 1998;273:28545–28548. [PubMed: 9786841]
- 173.
- Nishio Y, Isshiki H, Kishimoto T, Akira S. A nuclear factor for interleukin-6 expression (NF-IL6) and the glucocorticoid receptor synergistically activate transcription of the rat a1-acid glycoprotein gene via direct protein-protein interaction. Mol Cell Biol. 1993;13:1854–1862. [PMC free article: PMC359498] [PubMed: 8441418]
- 174.
- Boruk M, Savory JG, Hache RJ. AF-2-dependent potentiation of CCAAT enhancer binding protein b-mediated transcriptional activation by glucocorticoid receptor. Mol Endocrinol. 1998;12:1749–1763. [PubMed: 9817600]
- 175.
- Kulaeva OI, Hsieh FK, Chang HW, Luse DS, Studitsky VM. Mechanism of transcription through a nucleosome by RNA polymerase II. Biochim Biophys Acta. 2013;1829:76–83. [PMC free article: PMC3535581] [PubMed: 22982194]
- 176.
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell. 1999;98:285–294. [PubMed: 10458604]
- 177.
- Kuznetsova T, Wang SY, Rao NA, Mandoli A, Martens JH, Rother N, Aartse A, Groh L, Janssen-Megens EM, Li G, Ruan Y, Logie C, Stunnenberg HG. Glucocorticoid receptor and nuclear factor k-b affect three-dimensional chromatin organization. Genome Biol. 2015;16:264. [PMC free article: PMC4665721] [PubMed: 26619937]
- 178.
- Baker M. Genomics: Genomes in three dimensions. Nature. 2011;470:289–294. [PubMed: 21307943]
- 179.
- Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O'Shaughnessy-Kirwan A, Cramard J, Faure AJ, Ralser M, Blanco E, Morey L, Sanso M, Palayret MG, Lehner B, Di Croce L, Wutz A, Hendrich B, Klenerman D, Laue ED. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017;544:59–64. [PMC free article: PMC5385134] [PubMed: 28289288]
- 180.
- John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. [PMC free article: PMC6386452] [PubMed: 21258342]
- 181.
- Grontved L, John S, Baek S, Liu Y, Buckley JR, Vinson C, Aguilera G, Hager GL. C/EBP maintains chromatin accessibility in liver and facilitates glucocorticoid receptor recruitment to steroid response elements. EMBO J. 2013;32:1568–1583. [PMC free article: PMC3671252] [PubMed: 23665916]
- 182.
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, Vinson C, Stamatoyannopoulos JA, Hager GL. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 2011;43:145–155. [PMC free article: PMC3138120] [PubMed: 21726817]
- 183.
- Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, Janne OA. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013;73:1570–1580. [PubMed: 23269278]
- 184.
- Wiench M, John S, Baek S, Johnson TA, Sung MH, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ, Thurman RE, Stamatoyannopoulos JA, Hager GL. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–3039. [PMC free article: PMC3160184] [PubMed: 21701563]
- 185.
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD, Grimm JB, Morisaki T, Grontved L, Presman DM, Hager GL. Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell. 2016;165:593–605. [PMC free article: PMC4842147] [PubMed: 27062924]
- 186.
- Maranville JC, Luca F, Richards AL, Wen X, Witonsky DB, Baxter S, Stephens M, Di Rienzo A. Interactions between glucocorticoid treatment and cis-regulatory polymorphisms contribute to cellular response phenotypes. PLoS Genet. 2011;7:e1002162. [PMC free article: PMC3131293] [PubMed: 21750684]
- 187.
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. [PMC free article: PMC3771521] [PubMed: 22955828]
- 188.
- Kellendonk C, Tronche F, Reichardt HM, Bauer A, Greiner E, Schmid W, Schutz G. Analysis of glucocorticoid receptor function in the mouse by gene targeting. Ernst Schering Res Found Workshop. 2002:305–318. [PubMed: 12355723]
- 189.
- Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schutz G, Beug H. The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev. 1999;13:2996–3002. [PMC free article: PMC317156] [PubMed: 10580006]
- 190.
- Opherk C, Tronche F, Kellendonk C, Kohlmuller D, Schulze A, Schmid W, Schutz G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–1353. [PubMed: 15031319]
- 191.
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, Muglia LJ. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci U S A. 2005;102:473–478. [PMC free article: PMC544280] [PubMed: 15623560]
- 192.
- Laryea G, Schutz G, Muglia LJ. Disrupting hypothalamic glucocorticoid receptors causes HPA axis hyperactivity and excess adiposity. Mol Endocrinol. 2013;27:1655–1665. [PMC free article: PMC4061381] [PubMed: 23979842]
- 193.
- Chmielarz P, Kusmierczyk J, Parlato R, Schutz G, Nalepa I, Kreiner G. Inactivation of glucocorticoid receptor in noradrenergic system influences anxiety- and depressive-like behavior in mice. PLoS One. 2013;8:e72632. [PMC free article: PMC3748181] [PubMed: 23977333]
- 194.
- Rog-Zielinska EA, Thomson A, Kenyon CJ, Brownstein DG, Moran CM, Szumska D, Michailidou Z, Richardson J, Owen E, Watt A, Morrison H, Forrester LM, Bhattacharya S, Holmes MC, Chapman KE. Glucocorticoid receptor is required for foetal heart maturation. Hum Mol Genet. 2013;22:3269–3282. [PubMed: 23595884]
- 195.
- Goodwin JE, Zhang J, Gonzalez D, Albinsson S, Geller DS. Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J Hypertens. 2011;29:1347–1356. [PMC free article: PMC3439131] [PubMed: 21659825]
- 196.
- Goodwin JE, Feng Y, Velazquez H, Sessa WC. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc Natl Acad Sci U S A. 2013;110:306–311. [PMC free article: PMC3538225] [PubMed: 23248291]
- 197.
- Goodwin JE, Feng Y, Velazquez H, Zhou H, Sessa WC. Loss of the endothelial glucocorticoid receptor prevents the therapeutic protection afforded by dexamethasone after LPS. PLoS One. 2014;9:e108126. [PMC free article: PMC4191990] [PubMed: 25299055]
- 198.
- Kugler DG, Mittelstadt PR, Ashwell JD, Sher A, Jankovic D. CD4+ T cells are trigger and target of the glucocorticoid response that prevents lethal immunopathology in toxoplasma infection. J Exp Med. 2013;210:1919–1927. [PMC free article: PMC3782051] [PubMed: 23980098]
- 199.
- Whirledge SD, Oakley RH, Myers PH, Lydon JP, DeMayo F, Cidlowski JA. Uterine glucocorticoid receptors are critical for fertility in mice through control of embryo implantation and decidualization. Proc Natl Acad Sci U S A. 2015;112:15166–15171. [PMC free article: PMC4679013] [PubMed: 26598666]
- 200.
- Hazra R, Upton D, Jimenez M, Desai R, Handelsman DJ, Allan CM. In vivo actions of the Sertoli cell glucocorticoid receptor. Endocrinology. 2014;155:1120–1130. [PubMed: 24424066]
- 201.
- Adams M, Meijer OC, Wang J, Bhargava A, Pearce D. Homodimerization of the glucocorticoid receptor is not essential for response element binding: activation of the phenylethanolamine N-methyltransferase gene by dimerization-defective mutants. Mol Endocrinol. 2003;17:2583–2592. [PubMed: 12933902]
- 202.
- Rosen J, Miner JN. The search for safer glucocorticoid receptor ligands. Endocr Rev. 2005;26:452–464. [PubMed: 15814846]
- 203.
- Vayssiere BM, Dupont S, Choquart A, Petit F, Garcia T, Marchandeau C, Gronemeyer H, Resche-Rigon M. Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol. 1997;11:1245–1255. [PubMed: 9259316]
- 204.
- Coghlan MJ, Jacobson PB, Lane B, Nakane M, Lin CW, Elmore SW, Kym PR, Luly JR, Carter GW, Turner R, Tyree CM, Hu J, Elgort M, Rosen J, Miner JN. A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol Endocrinol. 2003;17:860–869. [PubMed: 12586843]
- 205.
- Schacke H, Schottelius A, Docke WD, Strehlke P, Jaroch S, Schmees N, Rehwinkel H, Hennekes H, Asadullah K. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci U S A. 2004;101:227–232. [PMC free article: PMC314167] [PubMed: 14694204]
- 206.
- Zalachoras I, Houtman R, Atucha E, Devos R, Tijssen AM, Hu P, Lockey PM, Datson NA, Belanoff JK, Lucassen PJ, Joels M, de Kloet ER, Roozendaal B, Hunt H, Meijer OC. Differential targeting of brain stress circuits with a selective glucocorticoid receptor modulator. Proc Natl Acad Sci U S A. 2013;110:7910–7915. [PMC free article: PMC3651427] [PubMed: 23613579]
- 207.
- Yoshikawa N, Makino Y, Okamoto K, Morimoto C, Makino I, Tanaka H. Distinct interaction of cortivazol with the ligand binding domain confers glucocorticoid receptor specificity: cortivazol is a specific ligand for the glucocorticoid receptor. J Biol Chem. 2002;277:5529–5540. [PubMed: 11741935]
- 208.
- Miner JN, Tyree C, Hu J, Berger E, Marschke K, Nakane M, Coghlan MJ, Clemm D, Lane B, Rosen J. A nonsteroidal glucocorticoid receptor antagonist. Mol Endocrinol. 2003;17:117–127. [PubMed: 12511611]
- 209.
- Clark RD, Ray NC, Williams K, Blaney P, Ward S, Crackett PH, Hurley C, Dyke HJ, Clark DE, Lockey P, Devos R, Wong M, Porres SS, Bright CP, Jenkins RE, Belanoff J. 1H-Pyrazolo[3,4-g]hexahydro-isoquinolines as selective glucocorticoid receptor antagonists with high functional activity. Bioorg Med Chem Lett. 2008;18:1312–1317. [PubMed: 18226897]
- 210.
- Solomon MB, Wulsin AC, Rice T, Wick D, Myers B, McKlveen J, Flak JN, Ulrich-Lai Y, Herman JP. The selective glucocorticoid receptor antagonist CORT 108297 decreases neuroendocrine stress responses and immobility in the forced swim test. Horm Behav. 2014;65:363–371. [PMC free article: PMC4074011] [PubMed: 24530653]
- 211.
- Trebble PJ, Woolven JM, Saunders KA, Simpson KD, Farrow SN, Matthews LC, Ray DW. A ligand-specific kinetic switch regulates glucocorticoid receptor trafficking and function. J Cell Sci. 2013;126:3159–3169. [PMC free article: PMC3711205] [PubMed: 23687373]
- 212.
- Brown AR, Bosies M, Cameron H, Clark J, Cowley A, Craighead M, Elmore MA, Firth A, Goodwin R, Goutcher S, Grant E, Grassie M, Grove SJ, Hamilton NM, Hampson H, Hillier A, Ho KK, Kiczun M, Kingsbury C, Kultgen SG, Littlewood PT, Lusher SJ, Macdonald S, McIntosh L, McIntyre T, Mistry A, Morphy JR, Nimz O, Ohlmeyer M, Pick J, Rankovic Z, Sherborne B, Smith A, Speake M, Spinks G, Thomson F, Watson L, Weston M. Discovery and optimisation of a selective non-steroidal glucocorticoid receptor antagonist. Bioorg Med Chem Lett. 2011;21:137–140. [PubMed: 21129964]
- 213.
- Vollmer TR, Stockhausen A, Zhang JZ. Anti-inflammatory effects of mapracorat, a novel selective glucocorticoid receptor agonist, is partially mediated by MAP kinase phosphatase-1 (MKP-1). J Biol Chem. 2012;287:35212–35221. [PMC free article: PMC3471745] [PubMed: 22898817]
- 214.
- Du J, Cheng B, Zhu X, Ling C. Ginsenoside Rg1, a novel glucocorticoid receptor agonist of plant origin, maintains glucocorticoid efficacy with reduced side effects. J Immunol. 2011;187:942–950. [PubMed: 21666059]
- 215.
- De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A. 2005;102:15827–15832. [PMC free article: PMC1276063] [PubMed: 16243974]
- 216.
- Liberman AC, Antunica-Noguerol M, Ferraz-de-Paula V, Palermo-Neto J, Castro CN, Druker J, Holsboer F, Perone MJ, Gerlo S, De Bosscher K, Haegeman G, Arzt E. Compound A, a dissociated glucocorticoid receptor modulator, inhibits T-bet (Th1) and induces GATA-3 (Th2) activity in immune cells. PLoS One. 2012;7:e35155. [PMC free article: PMC3322149] [PubMed: 22496903]
- 217.
- Reuter KC, Loitsch SM, Dignass AU, Steinhilber D, Stein J. Selective non-steroidal glucocorticoid receptor agonists attenuate inflammation but do not impair intestinal epithelial cell restitution in vitro. PLoS One. 2012;7:e29756. [PMC free article: PMC3266253] [PubMed: 22295067]
- 218.
- De Bosscher K, Beck IM, Dejager L, Bougarne N, Gaigneaux A, Chateauvieux S, Ratman D, Bracke M, Tavernier J, Vanden Berghe W, Libert C, Diederich M, Haegeman G. Selective modulation of the glucocorticoid receptor can distinguish between transrepression of NF-kB and AP-1. Cell Mol Life Sci. 2014;71:143–163. [PMC free article: PMC3889831] [PubMed: 23784308]
- 219.
- Lesovaya E, Yemelyanov A, Kirsanov K, Popa A, Belitsky G, Yakubovskaya M, Gordon LI, Rosen ST, Budunova I. Combination of a selective activator of the glucocorticoid receptor Compound A with a proteasome inhibitor as a novel strategy for chemotherapy of hematologic malignancies. Cell Cycle. 2013;12:133–144. [PMC free article: PMC3570502] [PubMed: 23255118]
- 220.
- Zheng Y, Ishiguro H, Ide H, Inoue S, Kashiwagi E, Kawahara T, Jalalizadeh M, Reis LO, Miyamoto H. Compound A inhibits bladder cancer growth predominantly via glucocorticoid receptor transrepression. Mol Endocrinol. 2015;29:1486–1497. [PMC free article: PMC5414678] [PubMed: 26322830]
- 221.
- Chen Z, Lan X, Wu D, Sunkel B, Ye Z, Huang J, Liu Z, Clinton SK, Jin VX, Wang Q. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat Commun. 2015;6:8323. [PMC free article: PMC4573460] [PubMed: 26374485]
- 222.
- Muir DC, Howard PH. Are there other persistent organic pollutants? A challenge for environmental chemists. Environ Sci Technol. 2006;40:7157–7166. [PubMed: 17180962]
- 223.
- Soto AM, Rubin BS, Sonnenschein C. Interpreting endocrine disruption from an integrative biology perspective. Mol Cell Endocrinol. 2009;304:3–7. [PMC free article: PMC3922629] [PubMed: 19433242]
- 224.
- Kolsek K, Gobec M, Mlinaric Rascan I, Sollner Dolenc M. Screening of bisphenol A, triclosan and paraben analogues as modulators of the glucocorticoid and androgen receptor activities. Toxicol In Vitro. 2015;29:8–15. [PubMed: 25192815]
- 225.
- Sarath Josh MK, Pradeep S, Vijayalekshmy Amma KS, Sudha Devi R, Balachandran S, Sreejith MN, Benjamin S. Human ketosteroid receptors interact with hazardous phthalate plasticizers and their metabolites: an in silico study. J Appl Toxicol. 2016;36:836–843. [PubMed: 26304264]
- 226.
- Lattin CR, Romero LM. Chronic exposure to a low dose of ingested petroleum disrupts corticosterone receptor signalling in a tissue-specific manner in the house sparrow (Passer domesticus). Conserv Physiol. 2014;2:cou058. [PMC free article: PMC4732471] [PubMed: 27293679]
- 227.
- Regnier SM, Kirkley AG, Ye H, El-Hashani E, Zhang X, Neel BA, Kamau W, Thomas CC, Williams AK, Hayes ET, Massad NL, Johnson DN, Huang L, Zhang C, Sargis RM. Dietary exposure to the endocrine disruptor tolylfluanid promotes global metabolic dysfunction in male mice. Endocrinology. 2015;156:896–910. [PMC free article: PMC4330315] [PubMed: 25535829]
- 228.
- Chantigny YA, Murray JC, Kleinman EF, Robinson RP, Plotkin MA, Reese MR, Buckbinder L, McNiff PA, Millham ML, Schaefer JF, Abramov YA, Bordner J. 2-Aryl-3-methyloctahydrophenanthrene-2,3,7-triols as potent dissociated glucocorticoid receptor agonists. J Med Chem. 2015;58:2658–2677. [PubMed: 25706100]
- 229.
- Atucha E, Zalachoras I, van den Heuvel JK, van Weert LT, Melchers D, Mol IM, Belanoff JK, Houtman R, Hunt H, Roozendaal B, Meijer OC. A mixed glucocorticoid/mineralocorticoid selective modulator with dominant antagonism in the male rat brain. Endocrinology. 2015;156:4105–4114. [PubMed: 26305887]
- 230.
- Eda M, Kuroda T, Kaneko S, Aoki Y, Yamashita M, Okumura C, Ikeda Y, Ohbora T, Sakaue M, Koyama N, Aritomo K. Synthesis and biological evaluation of cyclopentaquinoline derivatives as nonsteroidal glucocorticoid receptor antagonists. J Med Chem. 2015;58:4918–4926. [PubMed: 25978072]
- 231.
- Harcken C, Riether D, Kuzmich D, Liu P, Betageri R, Ralph M, Emmanuel M, Reeves JT, Berry A, Souza D, Nelson RM, Kukulka A, Fadra TN, Zuvela-Jelaska L, Dinallo R, Bentzien J, Nabozny GH, Thomson DS. Identification of highly efficacious glucocorticoid receptor agonists with a potential for reduced clinical bone side effects. J Med Chem. 2014;57:1583–1598. [PubMed: 24506830]
- 232.
- Xiao HY, Wu DR, Sheppeck JE 2nd, Habte SF, Cunningham MD, Somerville JE, Barrish JC, Nadler SG, Dhar TG. Heterocyclic glucocorticoid receptor modulators with a 2,2-dimethyl-3-phenyl-N-(thiazol or thiadiazol-2-yl)propanamide core. Bioorg Med Chem Lett. 2013;23:5571–5574. [PubMed: 24011644]
- 233.
- Sindelar DK, Carson MW, Morin M, Shaw J, Barr RJ, Need A, Alexander-Chacko J, Coghlan M, Gehlert DR. LLY-2707, a novel nonsteroidal glucocorticoid antagonist that reduces atypical antipsychotic-associated weight gain in rats. J Pharmacol Exp Ther. 2014;348:192–201. [PubMed: 24163440]
- 234.
- Lin HR. Triterpenes from Alisma orientalis act as androgen receptor agonists, progesterone receptor antagonists, and glucocorticoid receptor antagonists. Bioorg Med Chem Lett. 2014;24:3626–3632. [PubMed: 24915879]
- 235.
- Chirumamilla CS, Palagani A, Kamaraj B, Declerck K, Verbeek MWC, Oksana R, De Bosscher K, Bougarne N, Ruttens B, Gevaert K, Houtman R, De Vos WH, Joossens J, Van Der Veken P, Augustyns K, Van Ostade X, Bogaerts A, De Winter H, Vanden Berghe W. Selective Glucocorticoid Receptor Properties of GSK866 Analogs with Cysteine Reactive Warheads. Front Immunol. 2017;8:1324. [PMC free article: PMC5672024] [PubMed: 29163463]
- 236.
- Ripa L, Edman K, Dearman M, Edenro G, Hendrickx R, Ullah V, Chang HF, Lepistö M, Chapman D, Geschwindner S, Wissler L, Svanberg P, Lawitz K, Malmberg J, Nikitidis A, Olsson RI, Bird J, Llinas A, Hegelund-Myrbäck T, Berger M, Thorne P, Harrison R, Köhler C, Drmota T. Discovery of a Novel Oral Glucocorticoid Receptor Modulator (AZD9567) with Improved Side Effect Profile. J Med Chem. 2018;61(5):1785–1799. [PubMed: 29424542]
- 237.
- Potamitis C, Siakouli D, Papavasileiou KD, Boulaka A, Ganou V, Roussaki M, Calogeropoulou T, Zoumpoulakis P, Alexis MN, Zervou M, Mitsiou DJ. Discovery of new non-steroidal selective glucocorticoid receptor agonists. J Steroid Biochem Mol Biol. 2019;186:142–153. [PubMed: 30321666]
- 238.
- Zhang T, Liang Y, Zuo P, Yan M, Jing S, Li T, Wang Y, Zhang J, Wei Z. Identification of 20(R, S)-protopanaxadiol and 20(R, S)-protopanaxatriol for potential selective modulation of glucocorticoid receptor. Food Chem Toxicol. 2019;131:110642. [PubMed: 31247261]
- 239.
- Leng Y, Sun Y, Lv C, Li Z, Yuan C, Zhang J, Li T, Wang Y. Characterization of β-Sitosterol for Potential Selective GR Modulation. Protein Pept Lett. 2020 Aug 13; doiOnline ahead of print. [PubMed: 32798371] [CrossRef]
- 240.
- Faus H, Haendler B. Post-translational modifications of steroid receptors. Biomed Pharmacother. 2006;60:520–528. [PubMed: 16949786]
- 241.
- Fu M, Liu M, Sauve AA, Jiao X, Zhang X, Wu X, Powell MJ, Yang T, Gu W, Avantaggiati ML, Pattabiraman N, Pestell TG, Wang F, Quong AA, Wang C, Pestell RG. Hormonal control of androgen receptor function through SIRT1. Mol Cell Biol. 2006;26:8122–8135. [PMC free article: PMC1636736] [PubMed: 16923962]
- 242.
- Kim MY, Woo EM, Chong YT, Homenko DR, Kraus WL. Acetylation of estrogen receptor a by p300 at lysines 266 and 268 enhances the deoxyribonucleic acid binding and transactivation activities of the receptor. Mol Endocrinol. 2006;20:1479–1493. [PMC free article: PMC1483068] [PubMed: 16497729]
- 243.
- Ito K, Yamamura S, Essilfie-Quaye S, Cosio B, Ito M, Barnes PJ, Adcock IM. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kB suppression. J Exp Med. 2006;203:7–13. [PMC free article: PMC2118081] [PubMed: 16380507]
- 244.
- Nader N, Chrousos GP, Kino T. Circadian rhythm transcription factor CLOCK regulates the transcriptional activity of the glucocorticoid receptor by acetylating its hinge region lysine cluster: potential physiological implications. FASEB J. 2009;23:1572–1583. [PMC free article: PMC2669420] [PubMed: 19141540]
- 245.
- Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet. 2008;9:764–775. [PMC free article: PMC3758473] [PubMed: 18802415]
- 246.
- Doi M, Hirayama J, Sassone-Corsi P. Circadian regulator CLOCK is a histone acetyltransferase. Cell. 2006;125:497–508. [PubMed: 16678094]
- 247.
- Melvin VS, Harrell C, Adelman JS, Kraus WL, Churchill M, Edwards DP. The role of the C-terminal extension (CTE) of the estrogen receptor a and b DNA binding domain in DNA binding and interaction with HMGB. J Biol Chem. 2004;279:14763–14771. [PubMed: 14739282]
- 248.
- Nader N, Chrousos GP, Kino T. Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab. 2010;21:277–286. [PMC free article: PMC2862789] [PubMed: 20106676]
- 249.
- Charmandari E, Chrousos GP, Lambrou GI, Pavlaki A, Koide H, Ng SS, Kino T. Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man. PLoS One. 2011;6:e25612. [PMC free article: PMC3182238] [PubMed: 21980503]
- 250.
- Pavlatou MG, Vickers KC, Varma S, Malek R, Sampson M, Remaley AT, Gold PW, Skarulis MC, Kino T. Circulating cortisol-associated signature of glucocorticoid-related gene expression in subcutaneous fat of obese subjects. Obesity (Silver Spring). 2013;21:960–967. [PMC free article: PMC4199221] [PubMed: 23784897]
- 251.
- Nicolaides NC, Charmandari E, Kino T, Chrousos GP. Stress-Related and Circadian Secretion and Target Tissue Actions of Glucocorticoids: Impact on Health. Front Endocrinol (Lausanne). 2017;8:70. [PMC free article: PMC5408025] [PubMed: 28503165]
- 252.
- Agorastos A, Nicolaides NC, Bozikas VP, Chrousos GP, Pervanidou P. Multilevel Interactions of Stress and Circadian System: Implications for Traumatic Stress. Front Psychiatry. 2020;10:1003. [PMC free article: PMC6997541] [PubMed: 32047446]
- 253.
- Lamia KA, Papp SJ, Yu RT, Barish GD, Uhlenhaut NH, Jonker JW, Downes M, Evans RM. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. [PMC free article: PMC3245818] [PubMed: 22170608]
- 254.
- Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schutz G, Schibler U. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science. 2000;289:2344–2347. [PubMed: 11009419]
- 255.
- So AY, Bernal TU, Pillsbury ML, Yamamoto KR, Feldman BJ. Glucocorticoid regulation of the circadian clock modulates glucose homeostasis. Proc Natl Acad Sci U S A. 2009;106:17582–17587. [PMC free article: PMC2757402] [PubMed: 19805059]
- 256.
- Leliavski A, Dumbell R, Ott V, Oster H. Adrenal clocks and the role of adrenal hormones in the regulation of circadian physiology. J Biol Rhythms. 2015;30:20–34. [PubMed: 25367898]
- 257.
- Ikeda Y, Kumagai H, Skach A, Sato M, Yanagisawa M. Modulation of circadian glucocorticoid oscillation via adrenal opioid-CXCR7 signaling alters emotional behavior. Cell. 2013;155:1323–1336. [PMC free article: PMC3934808] [PubMed: 24315101]
- 258.
- Kino T, Chrousos GP. Circadian CLOCK-mediated regulation of target-tissue sensitivity to glucocorticoids: implications for cardiometabolic diseases. Endocr Dev. 2011;20:116–126. [PMC free article: PMC3163295] [PubMed: 21164265]
- 259.
- Ismaili N, Garabedian MJ. Modulation of glucocorticoid receptor function via phosphorylation. Ann N Y Acad Sci. 2004;1024:86–101. [PubMed: 15265775]
- 260.
- Orti E, Hu LM, Munck A. Kinetics of glucocorticoid receptor phosphorylation in intact cells. Evidence for hormone-induced hyperphosphorylation after activation and recycling of hyperphosphorylated receptors. J Biol Chem. 1993;268:7779–7784. [PubMed: 8463306]
- 261.
- Kino T. GR-regulating Serine/Threonine Kinases: New Physiologic and Pathologic Implications. Trends Endocrinol Metab. 2018;29(4):260–270. [PubMed: 29501228]
- 262.
- Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol. 1997;17:3947–3954. [PMC free article: PMC232247] [PubMed: 9199329]
- 263.
- Wang Z, Frederick J, Garabedian MJ. Deciphering the phosphorylation "code" of the glucocorticoid receptor in vivo. J Biol Chem. 2002;277:26573–26580. [PubMed: 12000743]
- 264.
- Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19:1569–1583. [PubMed: 15817653]
- 265.
- Carruthers CW, Suh JH, Gustafsson JA, Webb P. Phosphorylation of glucocorticoid receptor tau1c transactivation domain enhances binding to CREB binding protein (CBP) TAZ2. Biochem Biophys Res Commun. 2015;457:119–123. [PubMed: 25511704]
- 266.
- Ismaili N, Blind R, Garabedian MJ. Stabilization of the unliganded glucocorticoid receptor by TSG101. J Biol Chem. 2005;280:11120–11126. [PubMed: 15657031]
- 267.
- Rogatsky I, Waase CL, Garabedian MJ. Phosphorylation and inhibition of rat glucocorticoid receptor transcriptional activation by glycogen synthase kinase-3 (GSK-3). Species-specific differences between human and rat glucocorticoid receptor signaling as revealed through GSK-3 phosphorylation. J Biol Chem. 1998;273:14315–14321. [PubMed: 9603939]
- 268.
- Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3b-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28:7309–7322. [PMC free article: PMC2593428] [PubMed: 18838540]
- 269.
- Wang Z, Chen W, Kono E, Dang T, Garabedian MJ. Modulation of glucocorticoid receptor phosphorylation and transcriptional activity by a C-terminal-associated protein phosphatase. Mol Endocrinol. 2007;21:625–634. [PubMed: 17185395]
- 270.
- Bouazza B, Krytska K, Debba-Pavard M, Amrani Y, Honkanen RE, Tran J, Tliba O. Cytokines alter glucocorticoid receptor phosphorylation in airway cells: role of phosphatases. Am J Respir Cell Mol Biol. 2012;47:464–473. [PMC free article: PMC3488623] [PubMed: 22592921]
- 271.
- Kobayashi Y, Mercado N, Miller-Larsson A, Barnes PJ, Ito K. Increased corticosteroid sensitivity by a long acting b2 agonist formoterol via b2 adrenoceptor independent protein phosphatase 2A activation. Pulm Pharmacol Ther. 2012;25:201–207. [PubMed: 22401993]
- 272.
- Kobayashi Y, Mercado N, Barnes PJ, Ito K. Defects of protein phosphatase 2A causes corticosteroid insensitivity in severe asthma. PLoS One. 2011;6:e27627. [PMC free article: PMC3242752] [PubMed: 22205926]
- 273.
- Kino T, Ichijo T, Amin ND, Kesavapany S, Wang Y, Kim N, Rao S, Player A, Zheng YL, Garabedian MJ, Kawasaki E, Pant HC, Chrousos GP. Cyclin-dependent kinase 5 differentially regulates the transcriptional activity of the glucocorticoid receptor through phosphorylation: clinical implications for the nervous system response to glucocorticoids and stress. Mol Endocrinol. 2007;21:1552–1568. [PubMed: 17440046]
- 274.
- Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol. 2001;2:749–759. [PubMed: 11584302]
- 275.
- Kino T, Jaffe H, Amin ND, Chakrabarti M, Zheng YL, Chrousos GP, Pant HC. Cyclin-dependent kinase 5 modulates the transcriptional activity of the mineralocorticoid receptor and regulates expression of brain-derived neurotrophic factor. Mol Endocrinol. 2010;24:941–952. [PMC free article: PMC2870940] [PubMed: 20357208]
- 276.
- Sousa N, Almeida OF. Corticosteroids: sculptors of the hippocampal formation. Rev Neurosci. 2002;13:59–84. [PubMed: 12013026]
- 277.
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. [PMC free article: PMC2838375] [PubMed: 19198615]
- 278.
- Yulug B, Ozan E, Gonul AS, Kilic E. Brain-derived neurotrophic factor, stress and depression: a minireview. Brain Res Bull. 2009;78:267–269. [PubMed: 19111910]
- 279.
- Papadopoulou A, Siamatras T, Delgado-Morales R, Amin ND, Shukla V, Zheng YL, Pant HC, Almeida OF, Kino T. Acute and chronic stress differentially regulate cyclin-dependent kinase 5 in mouse brain: implications to glucocorticoid actions and major depression. Transl Psychiatry. 2015;5:e578. [PMC free article: PMC4490283] [PubMed: 26057048]
- 280.
- Nader N, Ng SS, Lambrou GI, Pervanidou P, Wang Y, Chrousos GP, Kino T. Adenosine 5'-monophosphate-activated protein kinase regulates metabolic actions of glucocorticoids by phosphorylating the glucocorticoid receptor through p38 mitogen-activated protein kinase. Mol Endocrinol. 2010;24:1748–1764. [PMC free article: PMC2940476] [PubMed: 20660302]
- 281.
- Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, Sanchez-Martin M, Perez-Garcia A, Rigo I, Castillo M, Indraccolo S, Cross JR, de Stanchina E, Paietta E, Racevskis J, Rowe JM, Tallman MS, Basso G, Meijerink JP, Cordon-Cardo C, Califano A, Ferrando AA. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer Cell. 2013;24:766–776. [PMC free article: PMC3878658] [PubMed: 24291004]
- 282.
- Wakui H, Wright AP, Gustafsson J, Zilliacus J. Interaction of the ligand-activated glucocorticoid receptor with the 14-3-3h protein. J Biol Chem. 1997;272:8153–8156. [PubMed: 9079630]
- 283.
- Dennis AP, O'Malley BW. Rush hour at the promoter: how the ubiquitin-proteasome pathway polices the traffic flow of nuclear receptor-dependent transcription. J Steroid Biochem Mol Biol. 2005;93:139–151. [PubMed: 15860256]
- 284.
- Jason LJ, Moore SC, Lewis JD, Lindsey G, Ausio J. Histone ubiquitination: a tagging tail unfolds? Bioessays. 2002;24:166–174. [PubMed: 11835281]
- 285.
- Gillette TG, Gonzalez F, Delahodde A, Johnston SA, Kodadek T. Physical and functional association of RNA polymerase II and the proteasome. Proc Natl Acad Sci U S A. 2004;101:5904–5909. [PMC free article: PMC395896] [PubMed: 15069196]
- 286.
- Wallace AD, Cidlowski JA. Proteasome-mediated glucocorticoid receptor degradation restricts transcriptional signaling by glucocorticoids. J Biol Chem. 2001;276:42714–42721. [PubMed: 11555652]
- 287.
- Deroo BJ, Rentsch C, Sampath S, Young J, DeFranco DB, Archer TK. Proteasomal inhibition enhances glucocorticoid receptor transactivation and alters its subnuclear trafficking. Mol Cell Biol. 2002;22:4113–4123. [PMC free article: PMC133869] [PubMed: 12024025]
- 288.
- Schaaf MJ, Cidlowski JA. Molecular determinants of glucocorticoid receptor mobility in living cells: the importance of ligand affinity. Mol Cell Biol. 2003;23:1922–1934. [PMC free article: PMC149474] [PubMed: 12612067]
- 289.
- Andreou AM, Tavernarakis N. SUMOylation and cell signalling. Biotechnol J. 2009;4:1740–1752. [PubMed: 19946876]
- 290.
- Holmstrom S, Van Antwerp ME, Iniguez-Lluhi JA. Direct and distinguishable inhibitory roles for SUMO isoforms in the control of transcriptional synergy. Proc Natl Acad Sci U S A. 2003;100:15758–15763. [PMC free article: PMC307641] [PubMed: 14663148]
- 291.
- Davies L, Karthikeyan N, Lynch JT, Sial EA, Gkourtsa A, Demonacos C, Krstic-Demonacos M. Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol. 2008;22:1331–1344. [PMC free article: PMC5419542] [PubMed: 18337589]
- 292.
- Tian S, Poukka H, Palvimo JJ, Janne OA. Small ubiquitin-related modifier-1 (SUMO-1) modification of the glucocorticoid receptor. Biochem J. 2002;367:907–911. [PMC free article: PMC1222934] [PubMed: 12144530]
- 293.
- Le Drean Y, Mincheneau N, Le Goff P, Michel D. Potentiation of glucocorticoid receptor transcriptional activity by sumoylation. Endocrinology. 2002;143:3482–3489. [PubMed: 12193561]
- 294.
- Druker J, Liberman AC, Antunica-Noguerol M, Gerez J, Paez-Pereda M, Rein T, Iniguez-Lluhi JA, Holsboer F, Arzt E. RSUME enhances glucocorticoid receptor SUMOylation and transcriptional activity. Mol Cell Biol. 2013;33:2116–2127. [PMC free article: PMC3648064] [PubMed: 23508108]
- 295.
- Lin DY, Huang YS, Jeng JC, Kuo HY, Chang CC, Chao TT, Ho CC, Chen YC, Lin TP, Fang HI, Hung CC, Suen CS, Hwang MJ, Chang KS, Maul GG, Shih HM. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol Cell. 2006;24:341–354. [PubMed: 17081986]
- 296.
- Tirard M, Jasbinsek J, Almeida OF, Michaelidis TM. The manifold actions of the protein inhibitor of activated STAT proteins on the transcriptional activity of mineralocorticoid and glucocorticoid receptors in neural cells. J Mol Endocrinol. 2004;32:825–841. [PubMed: 15171715]
- 297.
- Yang SH, Sharrocks AD. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell. 2004;13:611–617. [PubMed: 14992729]
- 298.
- Holmstrom SR, Chupreta S, So AY, Iniguez-Lluhi JA. SUMO-mediated inhibition of glucocorticoid receptor synergistic activity depends on stable assembly at the promoter but not on DAXX. Mol Endocrinol. 2008;22:2061–2075. [PMC free article: PMC2631372] [PubMed: 18562626]
- 299.
- Hua G, Paulen L, Chambon P. GR. SUMOylation and formation of an SUMO-SMRT/NCoR1-HDAC3 repressing complex is mandatory for GC-induced IR nGRE-mediated transrepression. Proc Natl Acad Sci U S A. 2016;113:E626–634. [PMC free article: PMC4747746] [PubMed: 26712002]
- 300.
- Seckl JR, Walker BR. Minireview: 11b-hydroxysteroid dehydrogenase type 1 - a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. [PubMed: 11250914]
- 301.
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. [PubMed: 11739957]
- 302.
- Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, Shinyama H, Sharp MG, Fleming S, Mullins JJ, Seckl JR, Flier JS. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. [PMC free article: PMC162290] [PubMed: 12840062]
- 303.
- Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275:2–12. [PubMed: 17628337]
- 304.
- Alvira S, Cuellar J, Rohl A, Yamamoto S, Itoh H, Alfonso C, Rivas G, Buchner J, Valpuesta JM. Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat Commun. 2014;5:5484. [PubMed: 25407331]
- 305.
- Paul A, Garcia YA, Zierer B, Patwardhan C, Gutierrez O, Hildenbrand Z, Harris DC, Balsiger HA, Sivils JC, Johnson JL, Buchner J, Chadli A, Cox MB. The cochaperone SGTA (small glutamine-rich tetratricopeptide repeat-containing protein a) demonstrates regulatory specificity for the androgen, glucocorticoid, and progesterone receptors. J Biol Chem. 2014;289:15297–15308. [PMC free article: PMC4140887] [PubMed: 24753260]
- 306.
- Kang KI, Meng X, Devin-Leclerc J, Bouhouche I, Chadli A, Cadepond F, Baulieu EE, Catelli MG. The molecular chaperone Hsp90 can negatively regulate the activity of a glucocorticosteroid-dependent promoter. Proc Natl Acad Sci U S A. 1999;96:1439–1444. [PMC free article: PMC15481] [PubMed: 9990042]
- 307.
- Liu J, DeFranco DB. Chromatin recycling of glucocorticoid receptors: implications for multiple roles of heat shock protein 90. Mol Endocrinol. 1999;13:355–365. [PubMed: 10076993]
- 308.
- Whitesell L, Cook P. Stable and specific binding of heat shock protein 90 by geldanamycin disrupts glucocorticoid receptor function in intact cells. Mol Endocrinol. 1996;10:705–712. [PubMed: 8776730]
- 309.
- Schneikert J, Hubner S, Langer G, Petri T, Jaattela M, Reed J, Cato AC. Hsp70-RAP46 interaction in downregulation of DNA binding by glucocorticoid receptor. EMBO J. 2000;19:6508–6516. [PMC free article: PMC305849] [PubMed: 11101523]
- 310.
- Pratt WB, Morishima Y, Murphy M, Harrell M. Chaperoning of glucocorticoid receptors. Handb Exp Pharmacol. 2006:111–138. [PubMed: 16610357]
- 311.
- Binder EB. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology. 2009;34 Suppl 1:S186–195. [PubMed: 19560279]
- 312.
- Hoeijmakers L, Harbich D, Schmid B, Lucassen PJ, Wagner KV, Schmidt MV, Hartmann J. Depletion of FKBP51 in female mice shapes HPA axis activity. PLoS One. 2014;9:e95796. [PMC free article: PMC3997427] [PubMed: 24759731]
- 313.
- Stohs SJ, Abbott BD, Lin FH, Birnbaum LS. Induction of ethoxyresorufin-O-deethylase and inhibition of glucocorticoid receptor binding in skin and liver of haired and hairless HRS/J mice by topically applied 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology. 1990;65:123–136. [PubMed: 2274963]
- 314.
- Sunahara GI, Lucier GW, McCoy Z, Bresnick EH, Sanchez ER, Nelson KG. Characterization of 2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated decreases in dexamethasone binding to rat hepatic cytosolic glucocorticoid receptor. Mol Pharmacol. 1989;36:239–247. [PubMed: 2770702]
- 315.
- Brake PB, Zhang L, Jefcoate CR. Aryl hydrocarbon receptor regulation of cytochrome P4501B1 in rat mammary fibroblasts: evidence for transcriptional repression by glucocorticoids. Mol Pharmacol. 1998;54:825–833. [PubMed: 9804617]
- 316.
- Czar MJ, Galigniana MD, Silverstein AM, Pratt WB. Geldanamycin, a heat shock protein 90-binding benzoquinone ansamycin, inhibits steroid-dependent translocation of the glucocorticoid receptor from the cytoplasm to the nucleus. Biochemistry. 1997;36:7776–7785. [PubMed: 9201920]
- 317.
- Makino Y, Okamoto K, Yoshikawa N, Aoshima M, Hirota K, Yodoi J, Umesono K, Makino I, Tanaka H. Thioredoxin: a redox-regulating cellular cofactor for glucocorticoid hormone action. Cross talk between endocrine control of stress response and cellular antioxidant defense system. J Clin Invest. 1996;98:2469–2477. [PMC free article: PMC507704] [PubMed: 8958209]
- 318.
- Miura T, Ouchida R, Yoshikawa N, Okamoto K, Makino Y, Nakamura T, Morimoto C, Makino I, Tanaka H. Functional modulation of the glucocorticoid receptor and suppression of NF-kB-dependent transcription by ursodeoxycholic acid. J Biol Chem. 2001;276:47371–47378. [PubMed: 11577102]
- 319.
- Takahashi S, Wakui H, Gustafsson JA, Zilliacus J, Itoh H. Functional interaction of the immunosuppressant mizoribine with the 14-3-3 protein. Biochem Biophys Res Commun. 2000;274:87–92. [PubMed: 10903900]
- 320.
- Mattick JS. The functional genomics of noncoding RNA. Science. 2005;309:1527–1528. [PubMed: 16141063]
- 321.
- Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, Nishida H, Yap CC, Suzuki M, Kawai J, Suzuki H, Carninci P, Hayashizaki Y, Wells C, Frith M, Ravasi T, Pang KC, Hallinan J, Mattick J, Hume DA, Lipovich L, Batalov S, Engstrom PG, Mizuno Y, Faghihi MA, Sandelin A, Chalk AM, Mottagui-Tabar S, Liang Z, Lenhard B, Wahlestedt C. Antisense transcription in the mammalian transcriptome. Science. 2005;309:1564–1566. [PubMed: 16141073]
- 322.
- Zhang R, Zhang L, Yu W. Genome-wide expression of non-coding RNA and global chromatin modification. Acta Biochim Biophys Sin (Shanghai). 2012;44:40–47. [PubMed: 22194012]
- 323.
- Mercer TR, Wilhelm D, Dinger ME, Solda G, Korbie DJ, Glazov EA, Truong V, Schwenke M, Simons C, Matthaei KI, Saint R, Koopman P, Mattick JS. Expression of distinct RNAs from 3' untranslated regions. Nucleic Acids Res. 2011;39:2393–2403. [PMC free article: PMC3064787] [PubMed: 21075793]
- 324.
- Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–1187. [PMC free article: PMC3308137] [PubMed: 22424228]
- 325.
- Guay C, Regazzi R. Circulating microRNAs as novel biomarkers for diabetes mellitus. Nat Rev Endocrinol. 2013;9:513–521. [PubMed: 23629540]
- 326.
- Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, Gorden P, Kahn CR. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017;542:450–455. [PMC free article: PMC5330251] [PubMed: 28199304]
- 327.
- Riester A, Issler O, Spyroglou A, Rodrig SH, Chen A, Beuschlein F. ACTH-dependent regulation of microRNA as endogenous modulators of glucocorticoid receptor expression in the adrenal gland. Endocrinology. 2012;153:212–222. [PubMed: 22128032]
- 328.
- Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, Hong M, Jiang T, Jiang Q, Lu J, Huang X, Huang B. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-a and cAMP/PKA pathways. Leukemia. 2012;26:769–777. [PubMed: 21979877]
- 329.
- Ledderose C, Mohnle P, Limbeck E, Schutz S, Weis F, Rink J, Briegel J, Kreth S. Corticosteroid resistance in sepsis is influenced by microRNA-124-induced downregulation of glucocorticoid receptor-a. Crit Care Med. 2012;40:2745–2753. [PubMed: 22846781]
- 330.
- Uchida S, Nishida A, Hara K, Kamemoto T, Suetsugi M, Fujimoto M, Watanuki T, Wakabayashi Y, Otsuki K, McEwen BS, Watanabe Y. Characterization of the vulnerability to repeated stress in Fischer 344 rats: possible involvement of microRNA-mediated down-regulation of the glucocorticoid receptor. Eur J Neurosci. 2008;27:2250–2261. [PubMed: 18445216]
- 331.
- Vreugdenhil E, Verissimo CS, Mariman R, Kamphorst JT, Barbosa JS, Zweers T, Champagne DL, Schouten T, Meijer OC, de Kloet ER, Fitzsimons CP. MicroRNA 18 and 124a down-regulate the glucocorticoid receptor: implications for glucocorticoid responsiveness in the brain. Endocrinology. 2009;150:2220–2228. [PubMed: 19131573]
- 332.
- Ko JY, Chuang PC, Ke HJ, Chen YS, Sun YC, Wang FS. MicroRNA-29a mitigates glucocorticoid induction of bone loss and fatty marrow by rescuing Runx2 acetylation. Bone. 2015;81:80–88. [PubMed: 26141838]
- 333.
- Davis TE, Kis-Toth K, Szanto A, Tsokos GC. Glucocorticoids suppress T cell function by up-regulating microRNA-98. Arthritis Rheum. 2013;65:1882–1890. [PMC free article: PMC3713788] [PubMed: 23575983]
- 334.
- Zheng Y, Xiong S, Jiang P, Liu R, Liu X, Qian J, Zheng X, Chu Y. Glucocorticoids inhibit lipopolysaccharide-mediated inflammatory response by downregulating microRNA-155: a novel anti-inflammation mechanism. Free Radic Biol Med. 2012;52:1307–1317. [PubMed: 22326887]
- 335.
- Smith LK, Tandon A, Shah RR, Mav D, Scoltock AB, Cidlowski JA. Deep sequencing identification of novel glucocorticoid-responsive miRNAs in apoptotic primary lymphocytes. PLoS One. 2013;8:e78316. [PMC free article: PMC3824063] [PubMed: 24250753]
- 336.
- Palagani A, Op de Beeck K, Naulaerts S, Diddens J, Sekhar Chirumamilla C, Van Camp G, Laukens K, Heyninck K, Gerlo S, Mestdagh P, Vandesompele J, Berghe WV. Ectopic microRNA-150-5p transcription sensitizes glucocorticoid therapy response in MM1S multiple myeloma cells but fails to overcome hormone therapy resistance in MM1R cells. PLoS One. 2014;9:e113842. [PMC free article: PMC4256227] [PubMed: 25474406]
- 337.
- Shi C, Huang P, Kang H, Hu B, Qi J, Jiang M, Zhou H, Guo L, Deng L. Glucocorticoid inhibits cell proliferation in differentiating osteoblasts by microRNA-199a targeting of WNT signaling. J Mol Endocrinol. 2015;54:325–337. [PubMed: 25878056]
- 338.
- Hatchell EC, Colley SM, Beveridge DJ, Epis MR, Stuart LM, Giles KM, Redfern AD, Miles LE, Barker A, MacDonald LM, Arthur PG, Lui JC, Golding JL, McCulloch RK, Metcalf CB, Wilce JA, Wilce MC, Lanz RB, O'Malley BW, Leedman PJ. SLIRP, a small SRA binding protein, is a nuclear receptor corepressor. Mol Cell. 2006;22:657–668. [PubMed: 16762838]
- 339.
- Redfern AD, Colley SM, Beveridge DJ, Ikeda N, Epis MR, Li X, Foulds CE, Stuart LM, Barker A, Russell VJ, Ramsay K, Kobelke SJ, Li X, Hatchell EC, Payne C, Giles KM, Messineo A, Gatignol A, Lanz RB, O'Malley BW, Leedman PJ. RNA-induced silencing complex (RISC) Proteins PACT, TRBP, and Dicer are SRA binding nuclear receptor coregulators. Proc Natl Acad Sci U S A. 2013;110:6536–6541. [PMC free article: PMC3631673] [PubMed: 23550157]
- 340.
- Beato M, Vicent GP. A new role for an old player: steroid receptor RNA Activator (SRA) represses hormone inducible genes. Transcription. 2013;4:167–171. [PMC free article: PMC3977916] [PubMed: 23863201]
- 341.
- Kino T, Marr AK. A lovely leap toward the development of breast cancer therapy with long non-coding RNAs. Transl Cancer Res. 2016;5:S400–404.
- 342.
- Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8. [PMC free article: PMC2819218] [PubMed: 20124551]
- 343.
- Mayama T, Marr AK, Kino T. Differential expression of glucocorticoid receptor noncoding RNA repressor Gas5 in autoimmune and inflammatory diseases. Horm Metab Res. 2016;48:550–557. [PubMed: 27214311]
- 344.
- Lucafò M, Di Silvestre A, Romano M, Avian A, Antonelli R, Martelossi S, Naviglio S, Tommasini A, Stocco G, Ventura A, Decorti G, De Ludicibus S. Role of the Long Non-Coding RNA Growth Arrest-Specific 5 in Glucocorticoid Response in Children with Inflammatory Bowel Disease. Basic Clin Pharmacol Toxicol. 2018;122(1):87–93. [PubMed: 28722800]
- 345.
- Gharesouran J, Taheri M, Sayad A, Ghafouri-Fard S, Mazdeh M, Omrani MD. The Growth Arrest-Specific Transcript 5 (GAS5) and Nuclear Receptor Subfamily 3 Group C Member 1 (NR3C1): Novel Markers Involved in Multiple Sclerosis. Int J Mol Cell Med. 2018 Spring;7(2):102–110. [PMC free article: PMC6148504] [PubMed: 30276165]
- 346.
- Mahdi Eftekharian M, Noroozi R, Komaki A, Mazdeh M, Taheri M, Ghafouri-Fard S. GAS5 genomic variants and risk of multiple sclerosis. Neurosci Lett. 2019;701:54–57. [PubMed: 30790644]
- 347.
- Moradi M, Gharesouran J, Ghafouri-Fard S, Noroozi R, Talebian S, Taheri M, Rezazadeh M. Role of NR3C1 and GAS5 genes polymorphisms in multiple sclerosis. Int J Neurosci. 2020;130(4):407–412. [PubMed: 31724909]
- 348.
- Esguerra JLS, Ofori JK, Nagao M, Shuto Y, Karagiannopoulos A, Fadista J, Sugihara H, Groop L, Eliasson L. Glucocorticoid induces human beta cell dysfunction by involving riborepressor GAS5 LincRNA. Mol Metab. 2020;32:160–167. [PMC free article: PMC6976904] [PubMed: 32029226]
- 349.
- Gasic V, Stankovic B, Zukic B, Janic D, Dokmanovic L, Krstovski N, Lazic J, Milosevic G, Lucafò M, Stocco G, Decorti G, Pavlovic S, Kotur N. Expression Pattern of Long Non-coding RNA Growth Arrest-specific 5 in the Remission Induction Therapy in Childhood Acute Lymphoblastic Leukemia. J Med Biochem. 2019;38(3):292–298. [PMC free article: PMC6534956] [PubMed: 31156339]
- 350.
- Ketab FNG, Gharesouran J, Ghafouri-Fard S, Dastar S, Mazraeh SA, Hosseinzadeh H, Moradi M, Javadlar M, Hiradfar A, Rezamand A, Taheri M, Rezazadeh M. Dual biomarkers long non-coding RNA GAS5 and its target, NR3C1, contribute to acute myeloid leukemia. Exp Mol Pathol. 2020;114:104399. [PubMed: 32032633]
- 351.
- Yang L, Lin C, Jin C, Yang JC, Tanasa B, Li W, Merkurjev D, Ohgi KA, Meng D, Zhang J, Evans CP, Rosenfeld MG. lncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 2013;500:598–602. [PMC free article: PMC4034386] [PubMed: 23945587]
- 352.
- Kino T, Su YA, Chrousos GP. Human glucocorticoid receptor isoform b: recent understanding of its potential implications in physiology and pathophysiology. Cell Mol Life Sci. 2009;66:3435–3448. [PMC free article: PMC2796272] [PubMed: 19633971]
- 353.
- Schaaf MJ, Champagne D, van Laanen IH, van Wijk DC, Meijer AH, Meijer OC, Spaink HP, Richardson MK. Discovery of a functional glucocorticoid receptor b-isoform in zebrafish. Endocrinology. 2008;149:1591–1599. [PubMed: 18096659]
- 354.
- Otto C, Reichardt HM, Schutz G. Absence of glucocorticoid receptor-b in mice. J Biol Chem. 1997;272:26665–26668. [PubMed: 9334249]
- 355.
- DuBois DC, Sukumaran S, Jusko WJ, Almon RR. Evidence for a glucocorticoid receptor b splice variant in the rat and its physiological regulation in liver. Steroids. 2013;78:312–320. [PMC free article: PMC3552070] [PubMed: 23257260]
- 356.
- Kino T, Chrousos GP. Glucocorticoid and mineralocorticoid receptors and associated diseases. Essays Biochem. 2004;40:137–155. [PubMed: 15242344]
- 357.
- Charmandari E, Chrousos GP, Ichijo T, Bhattacharyya N, Vottero A, Souvatzoglou E, Kino T. The human glucocorticoid receptor (hGR) b isoform suppresses the transcriptional activity of hGRa by interfering with formation of active coactivator complexes. Mol Endocrinol. 2005;19:52–64. [PubMed: 15459252]
- 358.
- Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor b isoform. Specificity and mechanisms of action. J Biol Chem. 1999;274:27857–27866. [PubMed: 10488132]
- 359.
- Li LB, Leung DY, Hall CF, Goleva E. Divergent expression and function of glucocorticoid receptor b in human monocytes and T cells. J Leukoc Biol. 2006;79:818–827. [PubMed: 16461744]
- 360.
- Zhang X, Clark AF, Yorio T. Regulation of glucocorticoid responsiveness in glaucomatous trabecular meshwork cells by glucocorticoid receptor-b. Invest Ophthalmol Vis Sci. 2005;46:4607–4616. [PubMed: 16303956]
- 361.
- Kino T, Manoli I, Kelkar S, Wang Y, Su YA, Chrousos GP. Glucocorticoid receptor (GR) b has intrinsic, GRa-independent transcriptional activity. Biochem Biophys Res Commun. 2009;381:671–675. [PMC free article: PMC2796800] [PubMed: 19248771]
- 362.
- Lewis-Tuffin LJ, Jewell CM, Bienstock RJ, Collins JB, Cidlowski JA. Human glucocorticoid receptor b binds RU-486 and is transcriptionally active. Mol Cell Biol. 2007;27:2266–2282. [PMC free article: PMC1820503] [PubMed: 17242213]
- 363.
- de Castro M, Elliot S, Kino T, Bamberger C, Karl M, Webster E, Chrousos GP. The non-ligand binding b-isoform of the human glucocorticoid receptor (hGRb): tissue levels, mechanism of action, and potential physiologic role. Mol Med. 1996;2:597–607. [PMC free article: PMC2230188] [PubMed: 8898375]
- 364.
- Oakley RH, Sar M, Cidlowski JA. The human glucocorticoid receptor b isoform. Expression, biochemical properties, and putative function. J Biol Chem. 1996;271:9550–9559. [PubMed: 8621628]
- 365.
- Hinds TD Jr, Ramakrishnan S, Cash HA, Stechschulte LA, Heinrich G, Najjar SM, Sanchez ER. Discovery of glucocorticoid receptor-b in mice with a role in metabolism. Mol Endocrinol. 2010;24:1715–1727. [PMC free article: PMC2940475] [PubMed: 20660300]
- 366.
- van der Vaart M, Schaaf MJ. Naturally occurring C-terminal splice variants of nuclear receptors. Nucl Recept Signal. 2009;7:e007. [PMC free article: PMC2716050] [PubMed: 19636396]
- 367.
- Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, Muramatsu M. Molecular cloning and characterization of human estrogen receptor bcx: a potential inhibitor of estrogen action in human. Nucleic Acids Res. 1998;26:3505–3512. [PMC free article: PMC147730] [PubMed: 9671811]
- 368.
- Benbrook D, Pfahl M. A novel thyroid hormone receptor encoded by a cDNA clone from a human testis library. Science. 1987;238:788–791. [PubMed: 3672126]
- 369.
- Ebihara K, Masuhiro Y, Kitamoto T, Suzawa M, Uematsu Y, Yoshizawa T, Ono T, Harada H, Matsuda K, Hasegawa T, Masushige S, Kato S. Intron retention generates a novel isoform of the murine vitamin D receptor that acts in a dominant negative way on the vitamin D signaling pathway. Mol Cell Biol. 1996;16:3393–3400. [PMC free article: PMC231333] [PubMed: 8668154]
- 370.
- Arnold KA, Eichelbaum M, Burk O. Alternative splicing affects the function and tissue-specific expression of the human constitutive androstane receptor. Nucl Recept. 2004;2:1. [PMC free article: PMC406421] [PubMed: 15043764]
- 371.
- Hossain A, Li C, Saunders GF. Generation of two distinct functional isoforms of dosage-sensitive sex reversal-adrenal hypoplasia congenita-critical region on the X chromosome gene 1 (DAX-1) by alternative splicing. Mol Endocrinol. 2004;18:1428–1437. [PubMed: 15044589]
- 372.
- Ohkura N, Hosono T, Maruyama K, Tsukada T, Yamaguchi K. An isoform of Nurr1 functions as a negative inhibitor of the NGFI-B family signaling. Biochim Biophys Acta. 1999;1444:69–79. [PubMed: 9931442]
- 373.
- Petropoulos I, Part D, Ochoa A, Zakin MM, Lamas E. NOR-2 (neuron-derived orphan receptor), a brain zinc finger protein, is highly induced during liver regeneration. FEBS Lett. 1995;372:273–278. [PubMed: 7556683]
- 374.
- Gervois P, Torra IP, Chinetti G, Grotzinger T, Dubois G, Fruchart JC, Fruchart-Najib J, Leitersdorf E, Staels B. A truncated human peroxisome proliferator-activated receptor a splice variant with dominant negative activity. Mol Endocrinol. 1999;13:1535–1549. [PubMed: 10478844]
- 375.
- Sabatino L, Casamassimi A, Peluso G, Barone MV, Capaccio D, Migliore C, Bonelli P, Pedicini A, Febbraro A, Ciccodicola A, Colantuoni V. A novel peroxisome proliferator-activated receptor g isoform with dominant negative activity generated by alternative splicing. J Biol Chem. 2005;280:26517–26525. [PubMed: 15857827]
- 376.
- He B, Cruz-Topete D, Oakley RH, Xiao X, Cidlowski JA. Human glucocorticoid receptor b regulates gluconeogenesis and inflammation in mouse liver. Mol Cell Biol. 2015;36:714–730. [PMC free article: PMC4760220] [PubMed: 26711253]
- 377.
- McBeth L, Nwaneri AC, Grabnar M, Demeter J, Nestor-Kalinoski A, Hinds TD Jr. Glucocorticoid receptor b increases migration of human bladder cancer cells. Oncotarget. 2016;7:27313–27324. [PMC free article: PMC5053652] [PubMed: 27036026]
- 378.
- Hinds TD, Peck B, Shek E, Stroup S, Hinson J, Arthur S, Marino JS. Overexpression of glucocorticoid receptor b enhances myogenesis and reduces catabolic gene expression. Int J Mol Sci. 2016;17:232. [PMC free article: PMC4783964] [PubMed: 26875982]
- 379.
- Stechschulte LA, Wuescher L, Marino JS, Hill JW, Eng C, Hinds TD Jr. Glucocorticoid receptor b stimulates Akt1 growth pathway by attenuation of PTEN. J Biol Chem. 2014;289:17885–17894. [PMC free article: PMC4067219] [PubMed: 24817119]
- 380.
- Wang Q, Lu PH, Shi ZF, Xu YJ, Xiang J, Wang YX, Deng LX, Xie P, Yin Y, Zhang B, Mu HJ, Qiao WZ, Cui H, Zou J. Glucocorticoid receptor b acts as a co-activator of T-cell factor 4 and enhances glioma cell proliferation. Mol Neurobiol. 2015;52:1106–1118. [PubMed: 25301232]
- 381.
- Goleva E, Li LB, Eves PT, Strand MJ, Martin RJ, Leung DY. Increased glucocorticoid receptor b alters steroid response in glucocorticoid-insensitive asthma. Am J Respir Crit Care Med. 2006;173:607–616. [PMC free article: PMC2662945] [PubMed: 16387802]
- 382.
- Derijk RH, Schaaf MJ, Turner G, Datson NA, Vreugdenhil E, Cidlowski J, de Kloet ER, Emery P, Sternberg EM, Detera-Wadleigh SD. A human glucocorticoid receptor gene variant that increases the stability of the glucocorticoid receptor b-isoform mRNA is associated with rheumatoid arthritis. J Rheumatol. 2001;28:2383–2388. [PubMed: 11708406]
- 383.
- Lee CK, Lee EY, Cho YS, Moon KA, Yoo B, Moon HB. Increased expression of glucocorticoid receptor b messenger RNA in patients with ankylosing spondylitis. Korean J Intern Med. 2005;20:146–151. [PMC free article: PMC3891384] [PubMed: 16134770]
- 384.
- Longui CA, Vottero A, Adamson PC, Cole DE, Kino T, Monte O, Chrousos GP. Low glucocorticoid receptor a/b ratio in T-cell lymphoblastic leukemia. Horm Metab Res. 2000;32:401–406. [PubMed: 11069204]
- 385.
- Pujols L, Mullol J, Benitez P, Torrego A, Xaubet A, de Haro J, Picado C. Expression of the glucocorticoid receptor a and b isoforms in human nasal mucosa and polyp epithelial cells. Respir Med. 2003;97:90–96. [PubMed: 12556018]
- 386.
- Shahidi H, Vottero A, Stratakis CA, Taymans SE, Karl M, Longui CA, Chrousos GP, Daughaday WH, Gregory SA, Plate JM. Imbalanced expression of the glucocorticoid receptor isoforms in cultured lymphocytes from a patient with systemic glucocorticoid resistance and chronic lymphocytic leukemia. Biochem Biophys Res Commun. 1999;254:559–565. [PubMed: 9920778]
- 387.
- Piotrowski P, Burzynski M, Lianeri M, Mostowska M, Wudarski M, Chwalinska-Sadowska H, Jagodzinski PP. Glucocorticoid receptor b splice variant expression in patients with high and low activity of systemic lupus erythematosus. Folia Histochem Cytobiol. 2007;45:339–342. [PubMed: 18165172]
- 388.
- Diaz PV, Pinto RA, Mamani R, Uasapud PA, Bono MR, Gaggero AA, Guerrero J, Goecke A. Increased expression of the glucocorticoid receptor b in infants with RSV bronchiolitis. Pediatrics. 2012;130:e804–811. [PubMed: 23008453]
- 389.
- Leung DY, Hamid Q, Vottero A, Szefler SJ, Surs W, Minshall E, Chrousos GP, Klemm DJ. Association of glucocorticoid insensitivity with increased expression of glucocorticoid receptor b. J Exp Med. 1997;186:1567–1574. [PMC free article: PMC2199113] [PubMed: 9348314]
- 390.
- Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative b isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A. 2001;98:6865–6870. [PMC free article: PMC34444] [PubMed: 11381138]
- 391.
- Xu Q, Leung DY, Kisich KO. Serine-arginine-rich protein p30 directs alternative splicing of glucocorticoid receptor pre-mRNA to glucocorticoid receptor b in neutrophils. J Biol Chem. 2003;278:27112–27118. [PubMed: 12738786]
- 392.
- Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, Leung DY. High constitutive glucocorticoid receptor b in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med. 2001;193:585–593. [PMC free article: PMC2193396] [PubMed: 11238589]
- 393.
- Orii F, Ashida T, Nomura M, Maemoto A, Fujiki T, Ayabe T, Imai S, Saitoh Y, Kohgo Y. Quantitative analysis for human glucocorticoid receptor a/b mRNA in IBD. Biochem Biophys Res Commun. 2002;296:1286–1294. [PubMed: 12207914]
- 394.
- Tliba O, Damera G, Banerjee A, Gu S, Baidouri H, Keslacy S, Amrani Y. Cytokines induce an early steroid resistance in airway smooth muscle cells: novel role of interferon regulatory factor-1. Am J Respir Cell Mol Biol. 2008;38:463–472. [PMC free article: PMC2274949] [PubMed: 17947510]
- 395.
- Chung CC, Shimmin L, Natarajan S, Hanis CL, Boerwinkle E, Hixson JE. Glucocorticoid receptor gene variant in the 3' untranslated region is associated with multiple measures of blood pressure. J Clin Endocrinol Metab. 2009;94:268–276. [PMC free article: PMC2630865] [PubMed: 18854398]
- 396.
- van den Akker EL, Koper JW, van Rossum EF, Dekker MJ, Russcher H, de Jong FH, Uitterlinden AG, Hofman A, Pols HA, Witteman JC, Lamberts SW. Glucocorticoid receptor gene and risk of cardiovascular disease. Arch Intern Med. 2008;168:33–39. [PubMed: 18195193]
- 397.
- van den Akker EL, Nouwen JL, Melles DC, van Rossum EF, Koper JW, Uitterlinden AG, Hofman A, Verbrugh HA, Pols HA, Lamberts SW, van Belkum A. Staphylococcus aureus nasal carriage is associated with glucocorticoid receptor gene polymorphisms. J Infect Dis. 2006;194:814–818. [PubMed: 16941349]
- 398.
- Charmandari E, Kino T. Chrousos syndrome: a seminal report, a phylogenetic enigma and the clinical implications of glucocorticoid signalling changes. Eur J Clin Invest. 2010;40:932–942. [PMC free article: PMC2948853] [PubMed: 20649902]
- 399.
- Charmandari E, Kino T, Ichijo T, Chrousos GP. Generalized glucocorticoid resistance: clinical aspects, molecular mechanisms, and implications of a rare genetic disorder. J Clin Endocrinol Metab. 2008;93:1563–1572. [PMC free article: PMC2386273] [PubMed: 18319312]
- 400.
- Nicolaides N, Lamprokostopoulou A, Sertedaki A, Charmandari E. Recent advances in the molecular mechanisms causing primary generalized glucocorticoid resistance. Hormones (Athens). 2016;15(1):23–34. [PubMed: 27086682]
- 401.
- Nicolaides NC, Charmandari E. Novel insights into the molecular mechanisms underlying generalized glucocorticoid resistance and hypersensitivity syndromes. Hormones (Athens). 2017;16(2):124–138. [PubMed: 28742501]
- 402.
- Nicolaides NC, Charmandari E. Glucocorticoid Resistance. Exp Suppl. 2019;111:85–102. [PubMed: 31588529]
- 403.
- Chrousos GP, Vingerhoeds A, Brandon D, Eil C, Pugeat M, DeVroede M, Loriaux DL, Lipsett MB. Primary cortisol resistance in man. A glucocorticoid receptor-mediated disease. J Clin Invest. 1982;69:1261–1269. [PMC free article: PMC370198] [PubMed: 6282933]
- 404.
- Nicolaides NC, Charmandari E. Chrousos syndrome: from molecular pathogenesis to therapeutic management. Eur J Clin Invest. 2015;45(5):504–14. [PubMed: 25715669]
- 405.
- Vingerhoeds ACM, Thijssen JHH, Schwarts F. Spontaneous hypercortisolism without Cushing's syndrome. J Clin Endocrinol Metab. 1976;43:1128–1133. [PubMed: 186477]
- 406.
- Lamberts SW, Poldermans D, Zweens M, de Jong FH. Familial cortisol resistance: differential diagnostic and therapeutic aspects. J Clin Endocrinol Metab. 1986;63:1328–1333. [PubMed: 3782421]
- 407.
- Nawata H, Sekiya K, Higuchi K, Kato K, Ibayashi H. Decreased deoxyribonucleic acid binding of glucocorticoid-receptor complex in cultured skin fibroblasts from a patient with the glucocorticoid resistance syndrome. J Clin Endocrinol Metab. 1987;65:219–226. [PubMed: 3597702]
- 408.
- Iida S, Gomi M, Moriwaki K, Itoh Y, Hirobe K, Matsuzawa Y, Katagiri S, Yonezawa T, Tarui S. Primary cortisol resistance accompanied by a reduction in glucocorticoid receptors in two members of the same family. J Clin Endocrinol Metab. 1985;60:967–971. [PubMed: 3980675]
- 409.
- Vecsei P, Frank K, Haack D, Heinze V, Ho AD, Honour JW, Lewicka S, Schoosch M, Ziegler R. Primary glucocorticoid receptor defect with likely familial involvement. Cancer Res. 1989;49:2220s–2221s. [PubMed: 2539257]
- 410.
- Lamberts SW, Koper JW, Biemond P, den Holder FH, de Jong FH. Cortisol receptor resistance: the variability of its clinical presentation and response to treatment. J Clin Endocrinol Metab. 1992;74:313–321. [PubMed: 1309833]
- 411.
- Hurley DM, Accili D, Stratakis CA, Karl M, Vamvakopoulos N, Rorer E, Constantine K, Taylor SI, Chrousos GP. Point mutation causing a single amino acid substitution in the hormone binding domain of the glucocorticoid receptor in familial glucocorticoid resistance. J Clin Invest. 1991;87:680–686. [PMC free article: PMC296359] [PubMed: 1704018]
- 412.
- Karl M, Lamberts SW, Detera-Wadleigh SD, Encio IJ, Stratakis CA, Hurley DM, Accili D, Chrousos GP. Familial glucocorticoid resistance caused by a splice site deletion in the human glucocorticoid receptor gene. J Clin Endocrinol Metab. 1993;76:683–689. [PubMed: 8445027]
- 413.
- Karl M, Lamberts SW, Koper JW, Katz DA, Huizenga NE, Kino T, Haddad BR, Hughes MR, Chrousos GP. Cushing's disease preceded by generalized glucocorticoid resistance: clinical consequences of a novel, dominant-negative glucocorticoid receptor mutation. Proc Assoc Am Physicians. 1996;108:296–307. [PubMed: 8863343]
- 414.
- Malchoff DM, Brufsky A, Reardon G, McDermott P, Javier EC, Bergh CH, Rowe D, Malchoff CD. A mutation of the glucocorticoid receptor in primary cortisol resistance. J Clin Invest. 1993;91:1918–1925. [PMC free article: PMC288186] [PubMed: 7683692]
- 415.
- Michailidou Z, Carter RN, Marshall E, Sutherland HG, Brownstein DG, Owen E, Cockett K, Kelly V, Ramage L, Al-Dujaili EA, Ross M, Maraki I, Newton K, Holmes MC, Seckl JR, Morton NM, Kenyon CJ, Chapman KE. Glucocorticoid receptor haploinsufficiency causes hypertension and attenuates hypothalamic-pituitary-adrenal axis and blood pressure adaptions to high-fat diet. FASEB J. 2008;22:3896–3907. [PMC free article: PMC2749453] [PubMed: 18697839]
- 416.
- Kino T, Stauber RH, Resau JH, Pavlakis GN, Chrousos GP. Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: Importance of the ligand-binding domain for intracellular GR trafficking. J Clin Endocrinol Metab. 2001;86:5600–5608. [PubMed: 11701741]
- 417.
- Mendonca BB, Leite MV, de Castro M, Kino T, Elias LL, Bachega TA, Arnhold IJ, Chrousos GP, Latronico AC. Female pseudohermaphroditism caused by a novel homozygous missense mutation of the GR gene. J Clin Endocrinol Metab. 2002;87:1805–1809. [PubMed: 11932321]
- 418.
- Ruiz M, Lind U, Gafvels M, Eggertsen G, Carlstedt-Duke J, Nilsson L, Holtmann M, Stierna P, Wikstrom AC, Werner S. Characterization of two novel mutations in the glucocorticoid receptor gene in patients with primary cortisol resistance. Clin Endocrinol (Oxf). 2001;55:363–371. [PubMed: 11589680]
- 419.
- Charmandari E, Kino T, Ichijo T, Zachman K, Alatsatianos A, Chrousos GP. Functional characterization of the natural human glucocorticoid receptor (hGR) mutants hGRaR477H and hGRaG679S associated with generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2006;91:1535–1543. [PubMed: 16449337]
- 420.
- Vottero A, Kino T, Combe H, Lecomte P, Chrousos GP. A novel, C-terminal dominant negative mutation of the GR causes familial glucocorticoid resistance through abnormal interactions with p160 steroid receptor coactivators. J Clin Endocrinol Metab. 2002;87:2658–2667. [PubMed: 12050230]
- 421.
- Charmandari E, Raji A, Kino T, Ichijo T, Tiulpakov A, Zachman K, Chrousos GP. A novel point mutation in the ligand-binding domain (LBD) of the human glucocorticoid receptor (hGR) causing generalized glucocorticoid resistance: the importance of the C terminus of hGR LBD in conferring transactivational activity. J Clin Endocrinol Metab. 2005;90:3696–3705. [PubMed: 15769988]
- 422.
- Nader N, Bachrach BE, Hurt DE, Gajula S, Pittman A, Lescher R, Kino T. A novel point mutation in helix 10 of the human glucocorticoid receptor causes generalized glucocorticoid resistance by disrupting the structure of the ligand-binding domain. J Clin Endocrinol Metab. 2010;95:2281–2285. [PMC free article: PMC2869551] [PubMed: 20335448]
- 423.
- Zhu HJ, Dai YF, Wang O, Li M, Lu L, Zhao WG, Xing XP, Pan H, Li NS, Gong FY. Generalized glucocorticoid resistance accompanied with an adrenocortical adenoma and caused by a novel point mutation of human glucocorticoid receptor gene. Chin Med J (Engl). 2011;124:551–555. [PubMed: 21362280]
- 424.
- Nicolaides NC, Skyrla E, Vlachakis D, Psarra AM, Moutsatsou P, Sertedaki A, Kossida S, Charmandari E. Functional characterization of the hGRaT556I causing Chrousos syndrome. Eur J Clin Invest. 2016;46:42–49. [PubMed: 26541474]
- 425.
- Nicolaides NC, Geer EB, Vlachakis D, Roberts ML, Psarra AM, Moutsatsou P, Sertedaki A, Kossida S, Charmandari E. A novel mutation of the hGR gene causing Chrousos syndrome. Eur J Clin Invest. 2015;45:782–791. [PubMed: 26031419]
- 426.
- McMahon SK, Pretorius CJ, Ungerer JP, Salmon NJ, Conwell LS, Pearen MA, Batch JA. Neonatal complete generalized glucocorticoid resistance and growth hormone deficiency caused by a novel homozygous mutation in helix 12 of the ligand binding domain of the glucocorticoid receptor gene (NR3C1). J Clin Endocrinol Metab. 2010;95:297–302. [PubMed: 19933394]
- 427.
- Charmandari E, Kino T, Souvatzoglou E, Vottero A, Bhattacharyya N, Chrousos GP. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab. 2004;89:1939–1949. [PubMed: 15070967]
- 428.
- Roberts ML, Kino T, Nicolaides NC, Hurt DE, Katsantoni E, Sertedaki A, Komianou F, Kassiou K, Chrousos GP, Charmandari E. A novel point mutation in the DNA-binding domain (DBD) of the human glucocorticoid receptor causes primary generalized glucocorticoid resistance by disrupting the hydrophobic structure of its DBD. J Clin Endocrinol Metab. 2013;98:E790–795. [PMC free article: PMC3615201] [PubMed: 23426617]
- 429.
- Bouligand J, Delemer B, Hecart AC, Meduri G, Viengchareun S, Amazit L, Trabado S, Feve B, Guiochon-Mantel A, Young J, Lombes M. Familial glucocorticoid receptor haploinsufficiency by non-sense mediated mRNA decay, adrenal hyperplasia and apparent mineralocorticoid excess. PLoS One. 2010;5:e13563. [PMC free article: PMC2962642] [PubMed: 21042587]
- 430.
- Charmandari E, Ichijo T, Jubiz W, Baid S, Zachman K, Chrousos GP, Kino T. A novel point mutation in the amino terminal domain of the human glucocorticoid receptor (hGR) gene enhancing hGR-mediated gene expression. J Clin Endocrinol Metab. 2008;93:4963–4968. [PMC free article: PMC2626453] [PubMed: 18827003]
- 431.
- Charmandari E, Kino T, Ichijo T, Jubiz W, Mejia L, Zachman K, Chrousos GP. A novel point mutation in helix 11 of the ligand-binding domain of the human glucocorticoid receptor gene causing generalized glucocorticoid resistance. J Clin Endocrinol Metab. 2007;92:3986–3990. [PubMed: 17635946]
- 432.
- Lin L, Wu X, Hou Y, Zheng F, Xu R. A novel mutation in the glucocorticoid receptor gene causing resistant hypertension: a case report. Am J Hypertens. 2019;32:1126–1128. [PubMed: 31414133]
- 433.
- Paragliola RM, Costella A, Corsello A, Urbani A, Concolino P. A Novel Pathogenic Variant in the N-Terminal Domain of the Glucocorticoid Receptor, Causing Glucocorticoid Resistance. Mol Diagn Ther. 2020;24(4):473–485. [PubMed: 32607951]
- 434.
- Tatsi C, Xekouki P, Nioti O, Bachrach B, Belyavskaya E, Lyssikatos C, Stratakis CA. A novel mutation in the glucocorticoid receptor gene as a cause of severe glucocorticoid resistance complicated by hypertensive encephalopathy. J Hypertens. 2019;37:1475–1481. [PMC free article: PMC10913058] [PubMed: 31145715]
- 435.
- Al Argan R, Saskin A, Yang JW, D’Agostino MD, Rivera J. Glucocorticoid resistance syndrome caused by a novel NR3C1 point mutation. Endocr J. 2018;65:1139–1146. [PubMed: 30158362]
- 436.
- Vitellius G, Fagart J, Delemer B, Amazit L, Ramos N, Bouligand J, Le Billan F, Castinetti F, Guiochon-Mantel A, Trabado S, Lombès M. Three novel heterozygous point mutations of NR3C1 causing glucocorticoid resistance. Hum Mutat. 2016;37:794–803. [PubMed: 27120390]
- 437.
- Velayos T, Grau G, Rica I, Pérez-Nanclares G, Gaztambide S. Glucocorticoid resistance syndrome caused by two novel mutations in the NR3C1 gene. Endocrinol Nutr. 2016;63:369–371. [PubMed: 27211791]
- 438.
- Vitellius G, Trabado S, Hoeffel C, Bouligand J, Bennet A, Castinetti F, Decoudier B, Guiochon-Mantel A, Lombes M, Delemer B. investigators of the MUTA-GR Study. Significant prevalence of NR3C1 mutations in incidentally discovered bilateral adrenal hyperplasia: results of the French MUTA-GR Study. Eur J Endocrinol. 2018;178:411–423. [PubMed: 29444898]
- 439.
- Ma L, Tan X, Li J, Long Y, Xiao Z, De J, Ren Y, Tian H, Md TC. A novel glucocorticoid receptor mutation in primary generalized glucocorticoid resistance disease. Endocr Pract. 2020;11 doiOnline ahead of print. [PubMed: 32045292] [CrossRef]
- 440.
- Cannavò S, Benvenga S, Messina E, Moleti M, Ferraù F. Comment to ’Glucocorticoid resistance syndrome caused by a novel NR3C1 point mutation’ by Al Argan et al. Endocr J. 2019;66:657. [PubMed: 31189748]
- 441.
- Trebble P, Matthews L, Blaikley J, Wayte AW, Black GC, Wilton A, Ray DW. Familial glucocorticoid resistance caused by a novel frameshift glucocorticoid receptor mutation. J Clin Endocrinol Metab. 2010;95:E490–E499. [PMC free article: PMC4110505] [PubMed: 20861124]
- 442.
- Vitellius G, Delemer B, Caron P, Chabre O, Bouligand J, Pussard E, Trabado S, Lombes M. Impaired 11β-hydroxysteroid dehydrogenase type 2 in glucocorticoid-resistant patients. J Clin Endocrinol Metab. 2019;104:5205–5216. [PubMed: 31225872]
- 443.
- Molnár Á, Patócs A, Likó I, Nyírő G, Rácz K, Tóth M, Sármán B. An unexpected, mild phenotype of glucocorticoid resistance associated with glucocorticoid receptor gene mutation case report and review of the literature. BMC Med Genet. 2018;19:37. [PMC free article: PMC5840839] [PubMed: 29510671]
- 444.
- Donner KM, Hiltunen TP, Jänne OA, Sane T, Kontula K. Generalized glucocorticoid resistance caused by a novel two-nucleotide deletion in the hormone-binding domain of the glucocorticoid receptor gene NR3C1. Eur J Endocrinol. 2012;168:K9–K18. [PubMed: 23076843]
- 445.
- Murani E, Reyer H, Ponsuksili S, Fritschka S, Wimmers K. A substitution in the ligand binding domain of the porcine glucocorticoid receptor affects activity of the adrenal gland. PLoS One. 2012;7:e45518. [PMC free article: PMC3445511] [PubMed: 23029068]
- 446.
- Kino T. Single Nucleotide Variations of the Human GR Gene Manifested as Pathologic Mutations or Polymorphisms. Endocrinology. 2018;159(7):2506–2519. [PubMed: 29762667]
- 447.
- Rosmond R, Bouchard C, Bjorntorp P. Tsp509I polymorphism in exon 2 of the glucocorticoid receptor gene in relation to obesity and cortisol secretion: cohort study. BMJ. 2001;322:652–653. [PMC free article: PMC26546] [PubMed: 11250851]
- 448.
- Huizenga NA, Koper JW, De Lange P, Pols HA, Stolk RP, Burger H, Grobbee DE, Brinkmann AO, De Jong FH, Lamberts SW. A polymorphism in the glucocorticoid receptor gene may be associated with and increased sensitivity to glucocorticoids in vivo. J Clin Endocrinol Metab. 1998;83:144–151. [PubMed: 9435432]
- 449.
- Dobson MG, Redfern CP, Unwin N, Weaver JU. The N363S polymorphism of the glucocorticoid receptor: potential contribution to central obesity in men and lack of association with other risk factors for coronary heart disease and diabetes mellitus. J Clin Endocrinol Metab. 2001;86:2270–2274. [PubMed: 11344238]
- 450.
- Russcher H, van Rossum EF, de Jong FH, Brinkmann AO, Lamberts SW, Koper JW. Increased expression of the glucocorticoid receptor-A translational isoform as a result of the ER22/23EK polymorphism. Mol Endocrinol. 2005;19:1687–1696. [PubMed: 15746190]
- 451.
- van Rossum EF, Voorhoeve PG, te Velde SJ, Koper JW, Delemarre-van de Waal HA, Kemper HC, Lamberts SW. The ER22/23EK polymorphism in the glucocorticoid receptor gene is associated with a beneficial body composition and muscle strength in young adults. J Clin Endocrinol Metab. 2004;89:4004–4009. [PubMed: 15292341]
- 452.
- van Rossum EF, Koper JW, Huizenga NA, Uitterlinden AG, Janssen JA, Brinkmann AO, Grobbee DE, de Jong FH, van Duyn CM, Pols HA, Lamberts SW. A polymorphism in the glucocorticoid receptor gene, which decreases sensitivity to glucocorticoids in vivo, is associated with low insulin and cholesterol levels. Diabetes. 2002;51:3128–3134. [PubMed: 12351458]
- 453.
- van der Voorn B, Wit JM, van der Pal SM, Rotteveel J, Finken MJ. Antenatal glucocorticoid treatment and polymorphisms of the glucocorticoid and mineralocorticoid receptors are associated with IQ and behavior in young adults born very preterm. J Clin Endocrinol Metab. 2015;100:500–507. [PubMed: 25406795]
- 454.
- van Rossum EF, Koper JW, van den Beld AW, Uitterlinden AG, Arp P, Ester W, Janssen JA, Brinkmann AO, de Jong FH, Grobbee DE, Pols HA, Lamberts SW. Identification of the BclI polymorphism in the glucocorticoid receptor gene: association with sensitivity to glucocorticoids in vivo and body mass index. Clin Endocrinol (Oxf). 2003;59:585–592. [PubMed: 14616881]
- 455.
- Manenschijn L, van den Akker EL, Lamberts SW, van Rossum EF. Clinical features associated with glucocorticoid receptor polymorphisms. An overview. Ann N Y Acad Sci. 2009;1179:179–198. [PubMed: 19906240]
- 456.
- van Moorsel D, van Greevenbroek MM, Schaper NC, Henry RM, Geelen CC, van Rossum EF, Nijpels G. t Hart LM, Schalkwijk CG, van der Kallen CJ, Sauerwein HP, Dekker JM, Stehouwer CD, Havekes B. BclI glucocorticoid receptor polymorphism in relation to cardiovascular variables: the Hoorn and CODAM studies. Eur J Endocrinol. 2015;173:455–464. [PubMed: 26139210]
- 457.
- Koetz KR, van Rossum EF, Ventz M, Diederich S, Quinkler M. BclI polymorphism of the glucocorticoid receptor gene is associated with increased bone resorption in patients on glucocorticoid replacement therapy. Clin Endocrinol (Oxf). 2013;78:831–837. [PubMed: 23134110]
- 458.
- Bouma EM, Riese H, Nolte IM, Oosterom E, Verhulst FC, Ormel J, Oldehinkel AJ. No associations between single nucleotide polymorphisms in corticoid receptor genes and heart rate and cortisol responses to a standardized social stress test in adolescents: the TRAILS study. Behav Genet. 2011;41:253–261. [PMC free article: PMC3044823] [PubMed: 20680430]
- 459.
- Lian Y, Xiao J, Wang Q, Ning L, Guan S, Ge H, Li F, Liu J. The relationship between glucocorticoid receptor polymorphisms, stressful life events, social support, and post-traumatic stress disorder. BMC Psychiatry. 2014;14:232. [PMC free article: PMC4149199] [PubMed: 25113244]
- 460.
- Maciel GA, Moreira RP, Bugano DD, Hayashida SA, Marcondes JA, Gomes LG, Mendonca BB, Bachega TA, Baracat EC. Association of glucocorticoid receptor polymorphisms with clinical and metabolic profiles in polycystic ovary syndrome. Clinics (Sao Paulo). 2014;69:179–184. [PMC free article: PMC3935131] [PubMed: 24626943]
- 461.
- Syed AA, Irving JA, Redfern CP, Hall AG, Unwin NC, White M, Bhopal RS, Weaver JU. Association of glucocorticoid receptor polymorphism A3669G in exon 9b with reduced central adiposity in women. Obesity (Silver Spring). 2006;14:759–764. [PubMed: 16855182]
- 462.
- Simpson DM, Bender AN. Human immunodeficiency virus-associated myopathy: analysis of 11 patients. Ann Neurol. 1988;24:79–84. [PubMed: 2843080]
- 463.
- Kotler DP, Rosenbaum K, Wang J, Pierson RN. Studies of body composition and fat distribution in HIV-infected and control subjects. J Acquir Immune Defic Syndr Hum Retrovirol. 1999;20:228–237. [PubMed: 10077170]
- 464.
- Yanovski JA, Miller KD, Kino T, Friedman TC, Chrousos GP, Tsigos C, Falloon J. Endocrine and metabolic evaluation of human immunodeficiency virus-infected patients with evidence of protease inhibitor-associated lipodystrophy. J Clin Endocrinol Metab. 1999;84:1925–1931. [PubMed: 10372688]
- 465.
- Hadigan C, Miller K, Corcoran C, Anderson E, Basgoz N, Grinspoon S. Fasting hyperinsulinemia and changes in regional body composition in human immunodeficiency virus-infected women. J Clin Endocrinol Metab. 1999;84:1932–1937. [PubMed: 10372689]
- 466.
- Dube MP. Disorders of Glucose Metabolism in Patients Infected with Human Immunodeficiency Virus. Clin Infect Dis. 2000;31:1467–1475. [PubMed: 11096014]
- 467.
- Pavlakis GN. The molecular biology of HIV-1. In: DeVita VT, Hellman S, Rosenberg SA, eds. AIDS: Diagnosis, Treatment and Prevention. 4 ed. Philadelphia: Lippincott Raven; 1996:45-74.
- 468.
- Emerman M. HIV-1, Vpr and the cell cycle. Curr Biol. 1996;6:1096–1103. [PubMed: 8805364]
- 469.
- Kino T, Gragerov A, Kopp JB, Stauber RH, Pavlakis GN, Chrousos GP. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J Exp Med. 1999;189:51–62. [PMC free article: PMC1887689] [PubMed: 9874563]
- 470.
- Kino T, Gragerov A, Slobodskaya O, Tsopanomichalou M, Chrousos GP, Pavlakis GN. Human immunodeficiency virus type-1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J Virol. 2002;76:9724–9734. [PMC free article: PMC136530] [PubMed: 12208951]
- 471.
- Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1994;91:10873–10877. [PMC free article: PMC45128] [PubMed: 7971975]
- 472.
- Henklein P, Bruns K, Sherman MP, Tessmer U, Licha K, Kopp J, de Noronha CM, Greene WC, Wray V, Schubert U. Functional and structural characterization of synthetic HIV-1 vpr that transduces cells, localizes to the nucleus, and induces G2 cell cycle arrest. J Biol Chem. 2000;275:32016–32026. [PubMed: 10903315]
- 473.
- Mirani M, Elenkov I, Volpi S, Hiroi N, Chrousos GP, Kino T. HIV-1 protein Vpr suppresses IL-12 production from human monocytes by enhancing glucocorticoid action: potential implications of Vpr coactivator activity for the innate and cellular immunity deficits observed in HIV-1 infection. J Immunol. 2002;169:6361–6368. [PubMed: 12444143]
- 474.
- Shrivastav S, Kino T, Cunningham T, Ichijo T, Schubert U, Heinklein P, Chrousos GP, Kopp JB. Human immunodeficiency virus (HIV)-1 viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor g and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy. Mol Endocrinol. 2008;22:234–247. [PMC free article: PMC2234580] [PubMed: 17932108]
- 475.
- Shrivastav S, Zhang L, Okamoto K, Lee H, Lagranha C, Abe Y, Balasubramanyam A, Lopaschuk GD, Kino T, Kopp JB. HIV-1 Vpr enhances PPARb/d-mediated transcription, increases PDK4 expression, and reduces PDC activity. Mol Endocrinol. 2013;27:1564–1576. [PMC free article: PMC3753422] [PubMed: 23842279]
- 476.
- Balasubramanyam A, Mersmann H, Jahoor F, Phillips TM, Sekhar RV, Schubert U, Brar B, Iyer D, Smith EO, Takahashi H, Lu H, Anderson P, Kino T, Henklein P, Kopp JB. Effects of transgenic expression of HIV-1 Vpr on lipid and energy metabolism in mice. Am J Physiol Endocrinol Metab. 2007;292:E40–48. [PubMed: 16882932]
- 477.
- Agarwal N, Iyer D, Patel SG, Sekhar RV, Phillips TM, Schubert U, Oplt T, Buras ED, Samson SL, Couturier J, Lewis DE, Rodriguez-Barradas MC, Jahoor F, Kino T, Kopp JB, Balasubramanyam A. HIV-1 Vpr induces adipose dysfunction in vivo through reciprocal effects on PPAR/GR co-regulation. Sci Transl Med. 2013;5:ra164. [PMC free article: PMC4009012] [PubMed: 24285483]
- 478.
- Kino T, Chrousos GP. Virus-mediated modulation of the host endocrine signaling systems: Clinical implications. Trends Endocrinol Metab. 2007;18:159–166. [PMC free article: PMC7128651] [PubMed: 17400471]
- 479.
- Jeang KT, Xiao H, Rich EA. Multifaceted activities of the HIV-1 transactivator of transcription, Tat. J Biol Chem. 1999;274:28837–28840. [PubMed: 10506122]
- 480.
- Kino T, Chrousos GP. Glucocorticoid and mineralocorticoid resistance/hypersensitivity syndromes. J Endocrinol. 2001;169:437–445. [PubMed: 11375113]
- 481.
- Kino T, Slobodskaya O, Pavlakis GN, Chrousos GP. Nuclear receptor coactivator p160 proteins enhance the HIV-1 long terminal repeat promoter by bridging promoter-bound factors and the Tat-P-TEFb complex. J Biol Chem. 2002;277:2396–2405. [PubMed: 11704662]
- 482.
- Price DH. P-TEFb, a cyclin-dependent kinase controlling elongation by RNA polymerase II. Mol Cell Biol. 2000;20:2629–2634. [PMC free article: PMC85478] [PubMed: 10733565]
- 483.
- Fawell S, Seery J, Daikh Y, Moore C, Chen LL, Pepinsky B, Barsoum J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci U S A. 1994;91:664–668. [PMC free article: PMC43009] [PubMed: 8290579]
- 484.
- Kino T, Mirani M, Alesci S, Chrousos GP. AIDS-related lipodystrophy/insulin resistance syndrome. Horm Metab Res. 2003;35:129–136. [PubMed: 12734771]
- 485.
- Liu C. Adenoviruses. In: Belshe RB, ed. Textbook of human virology. 2nd ed. St. Louis: Mosby-Year Book, Inc.; 1991.
- 486.
- Brockmann D, Esche H. The multifunctional role of E1A in the transcriptional regulation of CREB/CBP-dependent target genes. Curr Top Microbiol Immunol. 2003;272:97–129. [PubMed: 12747548]
- 487.
- Chinnadurai G. CtBP, an unconventional transcriptional corepressor in development and oncogenesis. Mol Cell. 2002;9:213–224. [PubMed: 11864595]
- 488.
- Ng SS, Li A, Pavlakis GN, Ozato K, Kino T. Viral infection increases glucocorticoid-induced interleukin-10 production through ERK-mediated phosphorylation of the glucocorticoid receptor in dendritic cells: potential clinical implications. PLoS One. 2013;8:e63587. [PMC free article: PMC3648469] [PubMed: 23667643]
- 489.
- Hinzey A, Alexander J, Corry J, Adams KM, Claggett AM, Traylor ZP, Davis IC, Webster Marketon JI. Respiratory syncytial virus represses glucocorticoid receptor-mediated gene activation. Endocrinology. 2011;152:483–494. [PMC free article: PMC3037158] [PubMed: 21190962]
- 490.
- Webster Marketon JI, Corry J, Teng MN. The respiratory syncytial virus (RSV) nonstructural proteins mediate RSV suppression of glucocorticoid receptor transactivation. Virology. 2014;449:62–69. [PMC free article: PMC3904736] [PubMed: 24418538]
- 491.
- Xie J, Long X, Gao L, Chen S, Zhao K, Li W, Zhou N, Zang N, Deng Y, Ren L, Wang L, Luo Z, Tu W, Zhao X, Fu Z, Xie X, Liu E. Respiratory Syncytial Virus Nonstructural Protein 1 Blocks Glucocorticoid Receptor Nuclear Translocation by Targeting IPO13 and May Account for Glucocorticoid Insensitivity. J Infect Dis. 2017;217(1):35–46. [PubMed: 28968829]
- ABSTRACT
- INTRODUCTION
- EVOLUTION OF GR
- STRUCTURE OF THE HUMAN GR GENE AND PROTEIN
- TRANSCRIPTIONAL AND TRANSLATIONAL REGULATION OF GR ISOFORMS
- ACTIONS OF GR
- GENOME-WIDE TRANSCRIPTIONAL REGULATION BY GRa
- FACTORS THAT MODULATE GR ACTIONS
- EPIGENETIC MODULATION OF GRa
- NON-CODING RNAS
- THE SPLICING VARIANT GRbeta ISOFORM
- PATHOLOGIC MODULATION OF GR ACTIVITY
- ACKNOWLEDGEMENTS
- REFERENCES
- Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: importance of the ligand-binding domain for intracellular GR trafficking.[J Clin Endocrinol Metab. 2001]Pathologic human GR mutant has a transdominant negative effect on the wild-type GR by inhibiting its translocation into the nucleus: importance of the ligand-binding domain for intracellular GR trafficking.Kino T, Stauber RH, Resau JH, Pavlakis GN, Chrousos GP. J Clin Endocrinol Metab. 2001 Nov; 86(11):5600-8.
- Review Glucocorticoid Therapy and Adrenal Suppression.[Endotext. 2000]Review Glucocorticoid Therapy and Adrenal Suppression.Nicolaides NC, Pavlaki AN, Maria Alexandra MA, Chrousos GP. Endotext. 2000
- Review GR-regulating Serine/Threonine Kinases: New Physiologic and Pathologic Implications.[Trends Endocrinol Metab. 2018]Review GR-regulating Serine/Threonine Kinases: New Physiologic and Pathologic Implications.Kino T. Trends Endocrinol Metab. 2018 Apr; 29(4):260-270. Epub 2018 Feb 28.
- Review New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation.[Endocrinology. 2013]Review New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation.Vandevyver S, Dejager L, Tuckermann J, Libert C. Endocrinology. 2013 Mar; 154(3):993-1007. Epub 2013 Feb 5.
- Review The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease.[J Allergy Clin Immunol. 2013]Review The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease.Oakley RH, Cidlowski JA. J Allergy Clin Immunol. 2013 Nov; 132(5):1033-44. Epub 2013 Sep 29.
- Glucocorticoid Receptor - EndotextGlucocorticoid Receptor - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...