

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Lipodystrophy syndromes are a heterogeneous group of diseases, characterized by selective absence of adipose tissue. In one sense, these diseases are lipid-partitioning disorders, where the primary defect is the loss of functional adipocytes, leading to ectopic steatosis, severe dyslipidemia, and insulin resistance. These syndromes have attracted significant attention since the mid-1990s as the understanding of adipose tissue biology grew, initially spurred by the discovery of the pathways leading to adipocyte differentiation and maturation, and then by the discovery of leptin. Although lipodystrophy syndromes are known since the beginning of the 20th century, significant progress in understanding these syndromes were made in the last two decades, placing these syndromes at the forefront of the translational metabolism field. Currently, more than 20 distinctive molecular etiologies have been attributed to cause human diseases most of which map to adipocyte differentiation or lipid droplet pathways. Seemingly acquired syndromes are recently reported to have a genetic basis, suggesting that our “pre-genome” understanding of the syndromes was inadequate and that we need to likely change our classification schemes. Regardless of the etiology, it is the selective absence of adipose tissue and its function, leading to the reduced ability to store long-term energy that perturbs insulin sensitivity and lipid metabolism. The treatment of these syndromes has also attracted considerable interest. The most successful example of the treatment of these syndromes came from the demonstration that leptin replacement strategy improved insulin resistance and dyslipidemia in the most severely affected forms of the disease, leading to an FDA approved therapy for generalized lipodystrophy syndromes. In the partial forms of the disease, the phenotypes are more complex, and the efficacy of leptin is not as uniform. Currently, standard treatment-resistant partial lipodystrophy is an EMA-approved indication, and numerous trials are in progress, either evaluating the efficacy of leptin in familial partial lipodystrophy or aiming to develop potential new treatments for the partial forms of the disease. These rare metabolic diseases are likely to continue to fuel novel breakthroughs in the field of metabolism in the foreseeable future. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Lipodystrophy syndromes comprise a heterogeneous group of disorders characterized by either generalized or partial lack of adipose tissue depending on the type of lipodystrophy (1, 2). Lipodystrophy classically has been classified as congenital or acquired. Patients with partial lipodystrophy may exhibit excess adipose tissue accumulation in preserved areas of the body. Lipodystrophy syndromes usually manifest with several metabolic abnormalities associated with severe insulin resistance that include diabetes mellitus, hypertriglyceridemia, and hepatic steatosis which can progress to steatohepatitis. Other common manifestations are acanthosis nigricans, polycystic ovarian syndrome (PCOS), and eruptive xanthomas (due to severe hypertriglyceridemia) (3, 4). Metabolic derangements are mostly responsible for the serious comorbidities associated with lipodystrophy; some of which are chronic complications of poorly controlled diabetes, acute pancreatitis, hepatic cirrhosis, proteinuria and renal failure, and premature cardiovascular disease (Fig.1) (1, 2). Typically, standard treatments fail to achieve good glycemic control in most patients with lipodystrophy, although episodes of diabetic ketoacidosis have rarely been reported (5). The severity of the comorbidities depends on the subtype, extent of fat loss, and other clinical characteristics such as gender and age. Major causes of mortality are cardiovascular diseases (6-9), liver diseases (2, 10), acute pancreatitis (2), renal failure (10), and sepsis (3). In certain areas of the world, infectious etiology also rises to the surface suggesting that perturbed immune function may be at play (11). Clinical features of lipodystrophy are shown in Table 1. It is important to note that there are additional components of the disease that may be specific to each molecular etiology. In addition, we are beginning to recognize that patients often report reduced quality of life with increased overall pain (requiring frequent use of pain medications), sleep disturbances and sleep apnea, gastrointestinal dysmotility, mood disturbances such as depression and anxiety and psychiatric diseases (12, 13).

Figure 1.
Consequences of Lipodystrophy.
Lipodystrophy is an intriguing rare disease that helps us obtain a better understanding of the pathophysiology of metabolic abnormalities associated with insulin resistance. The main cause of insulin resistance in lipodystrophy is the fact that the excess energy cannot be stored in adipose tissue, which is secondary to either the near total lack of adipocyte storage in patients with generalized lipodystrophy or a limited capacity to store in partial lipodystrophy. Limited lipid storage capacity causes the failure of buffering for postprandial lipids and secreting substantial adipokines, which in turn results in excessive levels of triglycerides and lipid intermediates in circulation. The body stores fat at ectopic sites such as the liver because of inability to store energy in the subcutaneous adipose depots. Levels of adipokines and hormones secreted from the adipose tissue, most characteristically leptin, are decreased in these patients especially if fat loss is extensive (1, 2, 9, 14, 15). Leptin has a fundamental role in glucose and lipid homeostasis, but more importantly, leptin is the key hormone responsible for regulating food intake (16). Low levels of leptin in lipodystrophy trigger hyperphagia, which is often extreme (17-19). In addition, leptin protects pancreatic beta cells from lipotoxicity at least in rodent models (20, 21). Leptin improves insulin sensitivity by increasing glucose uptake in peripheral tissues such as muscle via sympathetic nervous system activation. Leptin also decreases hepatic gluconeogenesis (22-24).
DIAGNOSIS
The diagnosis of lipodystrophy is usually made clinically based on history, body distribution of adipose tissue, physical examination, and metabolic profile. Lipodystrophy should be suspected in any person with partial or complete lack of subcutaneous adipose tissue. However, the diagnosis of lipodystrophy is often delayed because of the rarity of these syndromes and the failure of the physicians to recognize this disease. Although patients with congenital generalized lipodystrophy lack subcutaneous adipose tissue from birth, specific diagnosis is usually made during childhood or even adulthood when they start developing metabolic abnormalities. This is at least partly because the awareness of lipodystrophy is still low among physicians. The problem of recognition is much more common for partial lipodystrophy. The distribution of fat loss varies in different types of partial lipodystrophy. At first glance, certain types of partial lipodystrophy cannot be clearly distinguished from other common metabolic diseases (e.g., poorly controlled diabetes mellitus with truncal obesity) based on phenotype unless the physician is suspicious for lipodystrophy and checks carefully for certain characteristic such as the appearance of the limbs which look thinner than in a normal person. Also, the onset of fat loss may be gradual and delay the diagnosis both in genetic and acquired forms (3, 4). Lipodystrophy syndromes should be considered in the differential diagnosis in patients with relatively early onset insulin resistant diabetes mellitus, persistent hypertriglyceridemia, hepatic steatosis, PCOS, and hepatosplenomegaly. Other diseases that should be considered in the differential diagnosis of lipodystrophy are listed in Table 2.
Table 2.
Differential Diagnosis of Lipodystrophy Syndromes
| Generalized Lipodystrophy Syndromes | Constitutional thinness |
| Uncontrolled type 1 diabetes mellitus | |
| HIV-associated wasting | |
| Anorexia nervosa, cachexia and starvation | |
| Chronic infections | |
| Adrenocortical insufficiency | |
| Thyrotoxicosis | |
| Acromegaly | |
| Diencephalic syndrome | |
| Partial Lipodystrophy Syndromes | Cushing’s syndrome |
| Truncal obesity | |
| Type 2 diabetes (lipodystrophy like phenotype) | |
| HIV associated lipodystrophy | |
| Multiple symmetric lipomatosis | |
| Progeroid syndromes | |
| Acromegaly |
A thorough physical examination is required for clinical diagnosis of lipodystrophy. Clinicians should pay specific attention to evaluating the extremities and the gluteal region for leanness and muscularity. In addition, other body parts should be examined for accumulation of excessive amounts of fat. Due to marked abdominal obesity and excessive fat accumulation in the neck, patients with familial partial lipodystrophy (FPLD) may be misdiagnosed as Cushing’s syndrome (2).
The absence of subcutaneous fat can be quantified by using conventional anthropometric measurements, dual energy x-ray absorptiometry (DXA) scan, whole-body magnetic resonance imaging (MRI), and computed tomography (CT) scan (4). Anthropometry including skinfold thickness and limb circumference measurements are easy and affordable ways to estimate the fat loss and redistribution (9). Use of skin calipers may aid, but when unavailable, simple inspection and palpation of skin thickness may be very useful. We have used a cut-off of 11 mm in men and 22 mm in women in mid-thigh thickness as a screening point to suspect clinical presence of lipodystrophy that warrants further detailed work-up. For facial fat loss, serial photography may be used to evaluate the gradual loss of facial fat. DXA, MRI, and CT scans are non-invasive modalities that may be used for quantification of fat on a tissue-specific basis, but at least in the United States, none are covered for this purpose by insurance companies (3, 4, 25).
New Diagnostic Strategies and Technological Tools
Due to the complex nature, heterogeneity, and rarity of lipodystrophy syndromes, the need for accurate and objective diagnostic tools is increasing. Imaging techniques play an important role in visualizing fat distribution and assisting in the diagnosis of lipodystrophy. While measuring subcutaneous fat tissue thickness with high-resolution ultrasound is a practical and non-invasive method, results vary significantly depending on age, gender, and ethnicity (26).
Body fat distribution can be quantitatively determined using whole body Dual Energy X-ray absorptiometry (DXA scans). Work from our group indicates that images obtained with the "Fat Shadows" method, based on highlighting only the fat tissue in DXA images, can support the diagnosis of FPLD and generalized lipodystrophy (GL). Using this approach, FPLD was distinguished from control subjects with 85% sensitivity and 96% specificity, while GL was distinguished from nonobese control subjects with 100% sensitivity and specificity (27). In addition, Garg and colleagues reported that the DXA-derived lower limb fat (%) is diagnostic of FPLD2 with 99% specificity and 100% sensitivity in adult females below the 1st percentile (28).
A study by our group also suggests that the combined use of measurements performed on pelvic MRI images can be promising for the reliable diagnosis of FPLD. The combination of gluteal fat thickness ≤13 mm and pubic/gluteal fat ratio ≥2.5 was found to have 97% sensitivity and 91% specificity in the overall cohort and 100% sensitivity and 90% specificity in females for the diagnosis of FPLD (29).
Technological applications are also being developed to increase awareness of lipodystrophy and provide practical solutions for non-experts. LipoDDx, designed as a new mobile app, stands out with its 80% effectiveness in recognizing lipodystrophy subtypes when screening patients with adipose tissue loss, but more research is still needed in this area (30).
In addition, integrating artificial intelligence (AI) into medical diagnosis may hold promise for diagnosing lipodystrophy syndromes in the future. In this regard, a study by da Cunha Olegario NB et al. (31) was designed to enable the identification of the CGL phenotype from patient images with a deep learning model, analyzing more than 330 images, including individuals of different ages with phenotypically confusing features. After a fourfold cross-validation technique, the CGL phenotype could be identified with a mean accuracy, sensitivity, and specificity of over 90% (31).
These developments in diagnostic methods and technological tools for lipodystrophy syndromes not only increase diagnostic accuracy but also pave the way for creating a more personalized and effective treatment algorithms.
Laboratory Testing
Laboratory testing is a valuable tool for physicians to support the diagnosis. If the physical phenotype is not recognized, hyperglycemia, insulin resistance, and severe hypertriglyceridemia that is non-responsive to therapy may provide important clues for the diagnosis. When fat loss is not confirmed by the physical examination or by an imaging modality, hyperglycemia and hypertriglyceridemia that are resistant or unresponsive to conventional treatment may serve as surrogate indicators to the clinician that a patient may have lipodystrophy. Lipodystrophy should be suspected in patients with uncontrolled diabetes (e.g., requiring ≥200 units/day (≥2 units/kg/day) of insulin) or triglyceride levels that remain persistently elevated (e.g., ≥500 mg/dL) despite fully optimized therapy and diet modifications. All patients except those with localized lipodystrophy, should be tested for blood glucose levels, glycated hemoglobin (HbA1c), serum lipids (especially triglyceride levels), and liver function tests on the initial evaluation and during subsequent encounters. In addition to these laboratory evaluations, leptin levels may be used in support of the diagnosis. However, it should be noted that leptin assays are not standardized, and low leptin levels may be observed in other conditions such as hypothalamic amenorrhea and malnutrition. Thus, low leptin level is not specific for the diagnosis lipodystrophy (4, 32). Circulating adiponectin though not a clinically available test, may be helpful in differentiating patients with generalized lipodystrophy from those who have constitutional leanness, fat loss due to calorie imbalance or excessive exercise as well as poorly controlled diabetes mellitus with insulin deficiency. In all of the cases except lipodystrophy, adiponectin levels will be normal or even higher than normal whereas in lipodystrophy including familial partial lipodystrophy, serum adiponectin levels are usually low.
Genetic Testing
In the genetic forms of lipodystrophy, parental consanguinity and the mode of inheritance should be questioned (2). Genetic testing is available for known genetic forms of lipodystrophy. In our earlier version of this review, we had mentioned that genetic tests to be available only in certain clinical and research laboratories; however, there has been incredible growth in the availability of genetic testing through commercial or certified clinical labs in the US since 2016. Because additional loci for genetic lipodystrophy syndromes are presumed to be present, negative genetic tests do not rule out a genetic condition. When commercial panels are not positive, an attempt for whole exome or even whole genome testing with mitochondrial gene evaluation has evolved as viable and increasingly more available strategies. If these latter tests cannot be undertaken commercially, ongoing research in specialty centers can be considered where pipelines to analyze VUS (variant of uncertain significance) and evaluate transcriptomic profiles from PBMCs or tissues may be considered.
Gauging Disease Severity in Lipodystrophy: Roadmap for Clinical Follow Up
Given that lipodystrophy syndromes are complex and may impact multiple organs and systems, it is important to start a follow-up schedule while paying attention to all aspects of the conditions. To help assess the status and disease progression of patients with lipodystrophy, the LD Severity Score study group has developed the 'LD Severity Score (LDS)', an online tool easily accessible to all clinicians. This online application generates an overall score using multisystem assessments across eight domains (diabetes and its complications, lipid status, cardiovascular conditions, liver and kidney function, reproductive system status, and other conditions) to capture the various clinical manifestations of the disease holistically. The Clinical Global Impression (CGI) and the global improvement (GI) scores were generated based on the subjective assessments of all these categories by a group of experts during a representative patient visit as part of the app's validation. The LDS demonstrated high content validity and feasibility, along with high reliability indicated by interclass correlation coefficients greater than 0.95 (33). The results of the lipodystrophy severity scores calculated for each patient in the app are shown in a figure, compared to the Clinical Global Impression scores generated by the experts. The LD Scoring tool, developed to predict the clinical outcomes and/or treatment effects of lipodystrophy, can be accessed at https://ldscoring.com/.
CLASSIFICATION OF LIPODYSTROPHY SYNDROMES
Lipodystrophy syndromes can be classified as genetic or acquired. However, they are simply classified as generalized and partial in clinical practice most of the time (Table 3).
Table 3.
Classification of Lipodystrophy Syndromes
Type | Lipodystrophy Phenotype | Subtype (Genes Involved) | Key Clinical Features |
|---|---|---|---|
| Generalized lipodystrophy syndromes | |||
| Congenital Generalized Lipodystrophy (CGL) | Near total absence of the body fat starting at birth or shortly after, generalized muscularity, metabolic abnormalities | CGL1 (AGPAT2) Autosomal recessive | Loss of metabolically active fat with sparing of mechanically functioning fat |
| CGL2 (BSCL2) Autosomal recessive | Generalized absence of adipose tissue | ||
| CGL3 (CAV1) Autosomal recessive | Short stature, vitamin D resistance, hypocalcemia, hypomagnesemia, achalasia | ||
| CGL4 (PTRF) Autosomal recessive | Myopathy, skeletal abnormalities, pyloric stenosis and gastrointestinal motility problems, cardiac arrhythmias | ||
| Other genes associated with GL phenotype | LMNA (e.g., T10I, biallelic lamin A specific variants), PPARG (biallelic variants), PCYT1A, PLAAT3 | ||
| Acquired Generalized Lipodystrophy (AGL) | Near total absence of the body fat commonly develops during childhood or adolescence, metabolic abnormalities | Autoimmune | AGL follows an autoimmune disease, e.g. JDM |
| Panniculitis-associated | Tender subcutaneous nodules that herald the onset of AGL | ||
| Idiopathic | No history of auto-immune disease or panniculitis | ||
| Partial lipodystrophy syndromes | |||
| Familial Partial Lipodystrophy (FPLD) | Loss of fat from the limbs, metabolic abnormalities | FPLD1, Kobberling (Unknown) | Loss of subcutaneous fat from the limbs, although they usually have truncal obesity. Palpable “ledge” formation between the normal and lipodystrophic areas |
| FPLD2, Dunnigan (LMNA) Autosomal dominant | Increased muscularity and loss of fat in the limbs, excess fat accumulation in the face and neck | ||
| FPLD3 (PPARG) Autosomal dominant | Loss of subcutaneous fat from the limbs, specifically distally | ||
| FPLD4 (PLIN1) Autosomal dominant | Loss of subcutaneous fat from the limbs, histologically; small adipocytes, macrophage infiltration and fibrosis of adipose tissue | ||
| FPLD5 (CIDEC) Autosomal recessive | Loss of subcutaneous fat from the limbs, small, multilocular lipid droplets in adipocytes | ||
| FPLD6 (LIPE) Autosomal recessive | Increased visceral fat, dyslipidemia, hepatosteatosis, insulin resistance, and diabetes, some may present with muscular dystrophy and elevated serum creatine phosphokinase | ||
| Acquired Partial Lipodystrophy (APL) | Loss of subcutaneous fat starts from the face, neck, upper extremities, and progresses to the trunk. Lower limbs are typically spared, some patients have excess fat over the gluteal region, thighs and calves | Autoimmune | Coinciding autoimmune disorders; dermatomyositis/polymyositis and SLE are most associated disorders |
| MPGN-associated | Low serum complement 3, glomerulonephritis, hematuria, urinary casts, proteinuria, nephritic syndrome, renal failure | ||
| Idiopathic | No history of auto-immune disease or MPGN | ||
| Progeria associated lipodystrophy | LMNA, ZMPSTE24, POLD1, WRN, MTX2, FBN1, BANF1, KCNJ6, SPRTN, ALDH18A1, ERCC8, ERCC6 ( 34 ) BUD13 ( 35 ) , EPHX1 ( 36 ), OPA3 ( 37 ), PDGFRB ( 38 ), SLC25A24 ( 39 ), SUPT7L ( 40 ) | Progeroid features: most present with partial lipodystrophy, though in rare cases, fat loss can occur in a more generalized fashion | |
| Other genes associated with lipodystrophy | AKT2, PCYT1A, PIK3R1, MFN2, PSMB8, ADRA2A | Various presentations of lipodystrophy | |
GENERALIZED LIPODYSTROPHY SYNDROMES
Generalized lipodystrophy syndromes are rare disorders that are either inherited (Berardinelli-Seip Syndrome) (41-43) or acquired (Lawrence Syndrome) (9).
Congenital Generalized Lipodystrophy
Congenital Generalized Lipodystrophy (CGL) or Berardinelli-Seip syndrome is a rare syndrome which manifests with near total absence of adipose tissue. It is inherited in an autosomal recessive manner. Fat loss is usually recognized shortly after birth or in the first years of life, although patients may be diagnosed later during teenage years or adulthood. There have been over 300 reported cases to date (14, 44, 45).
In addition to lack of subcutaneous fat, patients may present with hepatomegaly and umbilical protuberance during infancy. Extensive acanthosis nigricans and prominent musculature may also contribute to the striking phenotype of these patients (46). Affected females may have irregular menstrual cycles, oligomenorrhea, clitoromegaly, and hirsutism. Premature menarche and pubarche are also rarely seen. Most males were reported to be fertile whereas only a few females had successful pregnancies (47). Sperm abnormalities have been reported in CGL. Other clinical manifestations include advanced bone age, bone cysts which may progress over time, mild mental retardation, cardiomyopathy, and cardiac rhythm disturbances (48-50). A significant association has been found between diabetic foot ulcers and, specifically, generalized lipodystrophy; foot ulcer complications may arise earlier in GL than in partial lipodystrophy (PL) (51).
Children with CGL usually have a voracious appetite and accelerated growth. Basal metabolic rate may be increased. Patients also report heat intolerance, especially after meals and sometimes gustatory sweating. Hypertriglyceridemia usually presents with high levels of chylomicrons and very low-density lipoproteins (VLDL) and reduced levels of high-density lipoproteins (HDL). Low HDL cholesterol levels are the most common lipid abnormality (49). Severe hypertriglyceridemia usually results in recurrent acute pancreatitis. Insulin resistance commonly results in diabetes in adolescence or later. Diabetes is rarely responsive to insulin therapy. Serum leptin levels are very low (32). Metabolic dysfunction–associated liver disease (MASLD) and steatohepatitis are common in individuals with CGL and have the potential to advance to cirrhosis at relatively early stages of life (49, 52). A study conducted in Brazil determined that the average age of death of 20 CGL patients who died between 1997-2017 was 27.1±12.4 years. In this patient group, most of whom were CGL2, the most common causes of death were infectious causes (35%), such as pneumonia and liver complications (35%), such as cirrhosis (11).
The genetic defect can be determined in the majority of patients with CGL. There are at least four molecularly distinct types of CGL. However, it is noteworthy that there are some cases of CGL reported without any pathogenic variants in any of the four genes described below.
CONGENITAL GENERALIZED LIPODYSTROPHY TYPE 1 (CGL1)
1-acylglycerol-3-phophate O-acyltransferase 2 (AGPAT2), a key enzyme in triglyceride synthesis, is deficient in CGL1. AGPAT2 gene is located on chromosome 9q34. AGPAT2 catalyzes the acylation of lysophosphaditic acid to form phosphaditic acid, a key intermediate in the biosynthesis of triglyceride and glycerophospholipids (53). Precisely how AGPAT2 deficiency causes lipodystrophy remains unsolved, but possible mechanisms include impaired lipogenesis, altered differentiation of preadipocytes to adipocytes, altering normal activation of phosphatidylinositol 3-kinase (PI3K)/Akt and PPARG pathways in the early stages of adipogenesis, and apoptosis/necrosis of adipocytes (2, 54, 55). More recent findings suggest that lysophosphatidic acid (LPA) could potentially trigger inflammation and fibrosis in the adipose tissue, leading to eventual loss of adipose tissue. In addition, LPA accumulation in the liver can also trigger the progress of metabolic dysfunction-associated fatty liver disease (MAFLD) to MASH (56).
Adiposity is preserved in certain body parts such as orbits, palms and soles, which constitute the mechanical adipose tissue (32, 57-59) (Fig.2). AGPAT2 pathogenic variants along with BSCL2 pathogenic variants are responsible for the majority of the CGL cases.

Figure 2.
CGL1. Near total absence of adipose tissue in CGL1 (2A, 2C, 2D). Magnetic resonance images document the lack of subcutaneous fat (2B). Liver biopsy reveals severe hepatic steatosis with both micro and macrovesicular steatosis (Hematoxylin and eosin staining; magnification 200X), 2E).
CONGENITAL GENERALIZED LIPODYSTROPHY TYPE 2 (CGL2)
CGL2 is caused by pathogenic variants in the BSCL2 gene which have been mapped to chromosome 11q13. This gene encodes a 398-amino acid integral endoplasmic reticulum membrane protein called seipin (60). This protein is assumed to take part in lipid droplet formation and adipocyte differentiation (61, 62). Patients with BSCL2 pathogenic variants have the most severe disease and are born without any adipose tissue. Hypertriglyceridemia and hepatic steatosis can be detected in early childhood; and hepatic involvement can be more severe in CGL2 than other subtypes (49, 63). Intellectual disability and cardiomyopathy are more common than in CGL1. CGL2 patients are also distinguished from the CGL1 patients with the loss of mechanical adipose tissue (64) (Fig.3). Although the mechanism is not clear, adiponectin levels are relatively higher in patients with CGL2 despite severely suppressed leptin levels which can help in the differential diagnosis (65).
In addition, rare specific variants of the BSCL2 gene that lead to the skipping of exon 7 are associated with Celia’s encephalopathy (Progressive Encephalopathy with/without Lipodystrophy, PELD), a severe progressive neurodegenerative disorder that also presents with a GL phenotype. (66-68).

Figure 3.
CGL2. Near total absence of adipose tissue in a patient with CGL2 (3A, 3B). Also note that the patient shown now deceased was only 29 years old at the time the picture was taken, suggesting the possibility of accelerated aging.
CONGENITAL GENERALIZED LIPODYSTROPHY TYPE 3 (CGL3)
CGL3 is caused by pathogenic variants in the CAV1 gene which are located on chromosome 7q31 (9, 15, 69). This gene encodes the protein caveolin-1, which is an integral part of caveolae found in plasma membranes. Caveolin 1 binds fatty acids on the plasma membranes and translocates them into lipid droplets. Mutated caveolin 1 disrupts lipid droplet formation and adipocyte differentiation (70). CGL3 is distinguished from other CGLs by the presence of unique features such as preserved bone marrow fat, vitamin D resistance, hypocalcemia, hypomagnesemia, and decreased bone density (48). In addition to this classical presentation, whole exome sequencing has identified de novo heterozygous null CAV1 pathogenic variants in two patients of European origin with generalized fat loss, thin mottled skin, and progeroid features at birth; however, no differences in the number and morphology of caveolae have been found in dermal fibroblasts (71), which suggests that this observation needs to be confirmed in further pedigrees. Heterozygous CAV1 frameshift mutations have also been reported to be associated with partial lipodystrophy (Fig.4) (72). Several features such as congenital cataracts and cerebellar progressive ataxia were also present (73). Apart from this, a novel p.(His79Glnfs*3) CAV1 variant was identified in four consanguineous patients diagnosed with CGL3. In addition to typical findings, two patients had esophageal achalasia, while the other had atypical retinitis pigmentosa findings (74).

Figure 4.
Partial LD with Heterozygous CAV1 Pathogenic Variant.
CONGENITAL GENERALIZED LIPODYSTROPHY TYPE 4 (CGL4)
Type 4 CGL (CGL4) is caused by pathogenic variants in the PTRF gene. The product of this gene, CAVIN, is a polymerase 1 and transcript release factor which regulates caveolae 1 and 3 (75). CGL4 can be recognized by distinct clinical characteristics. The majority of CGL4 patients that have been documented so far have had null mutations in the CAVIN1/PTRF gene. However, a novel homozygous mutation (c.21T>A; p.Tyr7Ter) was described in this gene in two pediatric siblings who exhibited slight variations in their phenotypical presentation, and whose clinical manifestations were compatible with CGL4 (76).
This rare subtype of CGL is associated with myopathy, pyloric stenosis, gastrointestinal dysmotility, arrhythmias that include exercise-induced ventricular tachycardia and sudden death, and skeletal abnormalities such as atlantoaxial instability and scoliosis (77-79) (Fig.5). Regardless of metabolic illness, patients with CGL4 are more prone to suffering life-threatening arrhythmias and cardiac problems throughout childhood (49).

Figure 5.
CGL4. Lack of subcutaneous fat (5A), scoliosis (5A), gastrointestinal dysmotility (5B), and exercise-induced ventricular arrhythmia (5C) in CGL4.
OTHER GENES ASSOCIATED WITH GENERALIZED LIPODYSTROPHY
Biallellic loss-of-function pathogenic variants in phosphate cytidylyltransferase 1 alpha (PCYT1A), the rate-limiting enzyme in the Kennedy pathway of de novo phosphatidylcholine synthesis, have been reported to be associated with generalized lipodystrophy, severe hepatic steatosis and low HDL cholesterol levels (80). Although widely involved in the familial partial lipodystrophy pathogenesis, several pathogenic variants in the LMNA and PPARG genes have been associated with generalized lipodystrophy. Heterozygous LMNA p.T10I pathogenic variant was reported to be associated with generalized lipodystrophy, diabetes mellitus, acanthosis nigricans, hypertriglyceridemia, and hepatomegaly (Fig.6) (81). Biallelic pathogenic variants in PPARG has also been reported to cause generalized lipodystrophy (82).
Furthermore, research has shown that a deficiency of phospholipase A/acyltransferase 3 (PLAAT3), an enzyme that modifies phospholipids and is predominantly found in neural and white adipose tissue (WAT), leads to monogenic lipodystrophy syndrome. Patients with biallelic loss-of-function variants in PLAAT3 show varying degrees of fat loss, from partial to generalized, insulin resistance, diabetes, hypertriglyceridemia, fatty liver, and polycystic ovary syndrome. Additionally, these patients exhibit numerous neurogenic symptoms, including demyelinating neuropathy, migraines, and intellectual disability, along with musculoskeletal dysmorphisms (83).

Figure 6.
Heterozygous LMNA p.T10I Pathogenic Variant. Generalized lack of subcutaneous fat (6A), eruptive xanthomata (6B), and lipemia retinalis (6C) secondary to severe hypertriglyceridemia in a patient with heterozygous LMNA p.T10I pathogenic variant.
Acquired Generalized Lipodystrophy
Acquired generalized lipodystrophy (AGL), also known as Lawrence Syndrome, is very rare. Generalized fat loss is not present at birth but develops later in life. It occurs over a variable period, ranging from a few weeks to years (Fig.7) (9).

Figure 7.
AGL. Generalized loss of subcutaneous fat in two patients with AGL (7A-D). Note the distal fat loss around the feet as opposed to patients with CGL phenotypes.
Although the pathogenesis of AGL has been elusive previously, it has always been hypothesized to be linked to autoimmune destruction of adipocytes. Autoantibodies against adipocyte membranes have been reported (84-86). Recently, the presence of antibodies against the lipid droplet surface protein perilipin-1 (PLIN1), an essential regulator of the lipolytic pathway, has been demonstrated in some AGL patients (87-90). Anti-PLIN1 autoantibodies in AGL patients were first detected in 2018 in three out of five patients (90). Subsequently, in a study focused on anti-PLIN1 antibodies, 50% of the 40 AGL patients examined exhibited the antibodies, whereas in another investigation, 37% of the 46 patients were found to have these antibodies (88, 89). Interestingly, one of the AGL patients with perilipin-1 antibody also had a mutation in the AIRE gene that causes autoimmune polyendocrine syndrome type 1 (APS1) (88). Considering these studies, whether perilipin-1 antibodies can be a potential biomarker in AGL patients and the relationship between APS1 and lipodystrophy are still curious.
AGL is associated with panniculitis in approximately 25% of the patients. This type may manifest with subcutaneous inflammatory nodules (panniculitis), which heal by localized loss of fat and eventually results in complete loss of subcutaneous fat (9). Another one fourth of the AGL patients present with an autoimmune disease that include juvenile dermatomyositis (JDM), Sjogren’s syndrome, rheumatoid arthritis, systemic sclerosis, and systemic lupus erythematosus (9, 85). Of these, JDM particularly correlates with AGL. 8-40% of patients with JDM develop AGL (Fig.8) (86, 91, 92). In the remaining 50% of the cases, AGL is not associated with any autoimmune or inflammatory condition (9). Some patients with AGL exhibit low serum complement 4 levels and auto-immune hepatitis, sometimes together with type 1 diabetes, which suggests the involvement of classical complement pathway in AGL pathogenesis (93). Recently, with the groundbreaking and increasing use of immune checkpoint inhibitors in cancer treatments, it has been reported in the literature that acquired generalized lipodystrophy developed after anti-PD-1 treatment (nivolumab and pembrolizumab) in four patients (94-96).
As mentioned above, it is of note that some of the patients with AGL are recently recognized to have additional progeroid features and may harbor a specific pathogenic of LMNA gene at position 10 (p.T10I). We have reported clinical presentations of these patients recently in a case series report. One of these patients also had biopsy proven juvenile dermatomyositis suggesting that the long-recognized association between AGL and JDM may be linked through distinctive molecular mechanisms (81).
In patients with AGL, metabolic abnormalities associated with severe insulin resistance that include hypertriglyceridemia, diabetes mellitus, hepatic steatosis, acanthosis nigricans, menstrual irregularities and PCOS may develop soon after the recognition of fat loss. Patients have suppressed levels of leptin and adiponectin (9, 32).

Figure 8.
Juvenile Dermatomyositis and AGL. Generalized loss of subcutaneous fat in a patient with juvenile dermatomyositis associated AGL (8A, 8B). Note the absence of muscle tissue as well in this severely affected patient.
PARTIAL LIPODYSTROPHY
Fat loss affects only part of the body in partial lipodystrophy. Partial lipodystrophy is categorized into inherited (familial partial lipodystrophy, FPLD) and acquired forms (acquired partial lipodystrophy, APL). Both patients with FPLD and APL start losing fat at some point during their life. Lower limbs are most frequently affected in FPLD. There might be accumulation of adipose tissue in the face and neck. On the other hand, APL is characterized by fat loss that spreads through a cephalocaudal distribution from the face, neck, shoulders, arms, and forearms and that extends to the thoracic region and upper abdomen. There are numerous genes associated with FPLD. Despite the growing number of proven genetic markers, about half of the patients do not have a discernible single gene variation.
Inherited Partial Lipodystrophy Syndromes
Patients with these syndromes usually notice partial fat loss around puberty. Fat loss pattern is very heterogeneous in patients with FPLD. Even among patients with pathogenic variants of the same gene, fat loss patterns may vary.
FAMILIAL PARTIAL LIPODYSTROPHY TYPE 1 (FPLD1)
The loss of adipose tissue is mainly limited to the extremities in patients with FPLD1 or Kobberling-type lipodystrophy (97). There is a normal or slightly increased fat in the face and neck. Truncal obesity is a common finding. The hallmark of this syndrome is the formation of a palpable “ledge” between the normal and lipodystrophic areas (98). It is believed that women are diagnosed more easily as they usually present with a more severe disease. Metabolic complications usually develop in early adulthood. Insulin resistant diabetes and metabolic syndrome are common and may cause premature coronary artery disease. Hypertriglyceridemia may trigger episodes of acute pancreatitis. Acanthosis nigricans is commonly seen. Leptin levels are variable and correlate with body mass index (BMI), which suggests that the levels of leptin are appropriate for the fat content in FPLD1 (98). The Cambridge group recently reported that this form of lipodystrophy may have a polygenic etiology (99). There is a remarkable phenotypical heterogeneity among patients with FPLD1. In this spectrum of FPLD1, patients with significant central obesity are likely polygenic. This type of presentation is relatively more common, and it is sometimes difficult to make a distinction between FPLD1 and truncal obesity complicated with metabolic syndrome (100). The use of radiological methods such as DXA, CT, or MRI can help in this population to further define body fat distribution in addition to physical examination and skinfold measurements. On the other hand, some FPLD patients with no increase in truncal fat are classified as FPLD1, if no gene is identified. These patients can still have a monogenic form of FPLD that has not been discovered. Two different presentations of FPLD1 are shown in Fig.9.

Figure 9.
FPLD1. Heterogeneity in FPLD1. Patient in A to D presented with decrease in peripheral fat depots and preservation of abdominal fat. Patient in E to H has increased abdominal adiposity. The formation of a palpable “ledge” between the normal and lipodystrophic areas is shown (9C and 9E). (Images E-H used with permission by Dr. Jonathan Q. Purnell from publication Diabetes Care 2003;26(6):1819-24).
FAMILIAL PARTIAL LIPODYSTROPHY TYPE 2 (FPLD2)
FPLD2 or Dunnigan Variety lipodystrophy is an autosomal dominant syndrome which is characterized by gradual onset of subcutaneous fat loss from the extremities during puberty. Affected individuals have prominent muscularity in their extremities. Excess fat accumulates in the neck causing a buffalo hump (Fig.10). This phenotype sometimes can be misdiagnosed as Cushing’s syndrome at first glance (15). Pathogenic variants in the LMNA gene, which is located on chromosome 1q21-22, cause FPLD2. The LMNA gene codes nuclear lamina proteins, lamin A and C. Pathogenic variants in the LMNA gene can be scattered across many exons of the gene and are missense mutations (101). Mutant lamins disrupt the interaction between nuclear lamina and chromatin and may result in apoptosis, which may be followed by premature adipocyte death (102).

Figure 10.
FPLD2. Subcutaneous adipose tissue loss from the extremities, excess fat accumulation in the face and neck, and Cushingoid appearance in FPLD2 (10A-D; Note that one of the patients (10A) previously underwent liposuction for removal of unwanted excess fat from the neck).
Females have a more recognizable phenotype and more severe metabolic complications (103). Most patients with FPLD2 develop diabetes in their twenties and thirties. Other components of insulin resistance are usually present. Patients with FPLD2 are at high risk for cardiovascular diseases that usually develop at relatively younger ages (104). Arrhythmias such as atrial fibrillation or flutter are more common in patients with LMNA pathogenic variants and may occur at an earlier age. Detailed cardiac analyses among patients with LMNA pathogenic variants showed that individuals with non-482 LMNA variants exhibited a high possibility of suffering from vigorous cardiac complications such as myocardial infarction, atrial fibrillation/flutter, cardiomyopathy, and congestive heart failure (105). Our retrospective analysis of 494 patients with LMNA-associated lipodystrophy revealed that the most prevalent LMNA variants were R482Q and R482W. This paper also highlights that patients with the R482W variant are diagnosed with diabetes at a significantly younger age compared to those with the R482Q variant (27 years vs. 40 years, respectively) (106).
There is a phenotypic heterogeneity among patients with FPLD2. For instance, less severe loss of fat has been reported in patients with exon 11 LMNA pathogenic variants which affects only lamin A protein (107). LMNA R349W pathogenic variant (exon 6) is associated with facial fat loss which is uncommon in FPLD2 (104, 108, 109). Exon 1 variants are associated with severe cardiac disease that require cardiac transplant at an early age and may be coupled with arrhythmias and conduction system abnormalities. Variants across exon 4 through 8 have been noted to cause muscular dystrophy related symptoms together with fat distribution abnormalities. In a retrospective evaluation of 12 pediatric FPLD2 patients, although all patients had the same LMNA variant p.(R482W), there were marked differences in the severity of the phenotype. Despite the absence of comorbidities in patients under the age of ten, the earliest age of onset of diabetes in the cohort was 12, and the earliest age of onset of hepatic steatosis was observed to be 10 (110). LMNA gene pathogenic variants are also involved in the pathogenesis of progeroid disorders including Hutchinson-Gilford progeria syndrome (HGPS), mandibuloacral dysplasia, and atypical progeroid syndrome (APS).
FAMILIAL PARTIAL LIPODYSTROPHY TYPE 3 (FPLD3)
FPLD3 is caused by pathogenic variants in the PPARG gene, a key regulator of adipocyte differentiation. Patients with FPLD3 usually show milder fat loss; and there is no accumulation of adipose tissue in the face and neck (Fig.11); however, they manifest metabolic complications at a similar rate and severity to those with FPLD2 (104, 111-115). Even more, in a retrospective analysis, FPLD3 patients exhibited a notably greater prevalence of hypertriglyceridemia and diabetes, along with elevated levels of median serum triglycerides and mean HbA1c, compared to FPLD2 patients (116).

Figure 11.
FPLD3. Moderate partial subcutaneous adipose tissue loss in a patient with FPLD3 (11A-C).
FAMILIAL PARTIAL LIPODYSTROPHY TYPE 4 (FPLD4)
FPLD4 is caused by pathogenic variants in the PLIN1 gene encoding perilipin 1, which is an essential lipid droplet coat protein (117). Although frameshift mutations in PLIN1 are known to cause partial lipodystrophy, a recent comprehensive study suggests that null variants in the PLIN1 gene are not associated with the formation of lipodystrophy (118).
Perilipin plays a key role in coordinating access of lipases to the core triacylglycerol. It is characterized by the loss of adipose tissue which is most striking in the lower limbs and femorogluteal depot, severe insulin resistance, diabetes, hypertriglyceridemia, and hepatic steatosis (119-121).
FAMILIAL PARTIAL LIPODYSTROPHY TYPE 5 (FPLD5)
FPLD5 is an autosomal recessive syndrome caused by pathogenic variants in the CIDEC gene. It is characterized by partial lipodystrophy, acanthosis nigricans, severe insulin resistance leading to diabetes, and hepatic steatosis. The CIDEC gene is located on chromosome 3 (3p25.3) and encodes the CIDEC protein, which is expressed in the lipid droplets. Pathogenic variants of the CIDEC gene are postulated to result in the loss of ability of lipid droplets to store fat (122).
FAMILIAL PARTIAL LIPODYSTROPHY TYPE 6 (FPLD6)
FPLD type 6 is caused by pathogenic variants in the LIPE (lipase E, hormone sensitive type) gene which has an autosomal recessive inheritance (123). This FPLD subtype is characterized by late-onset partial fat loss from the lower extremities and also multiple symmetric lipomatosis and progressive distal symmetric myopathy (123, 124). Hormone sensitive lipase is the predominant regulator of lipolysis from adipocytes. Pathogenic variants in the LIPE gene appear to result in impaired lipolysis which may induce lipomatosis and partial fat loss at the same time that is associated with hypertriglyceridemia, hepatic steatosis, and insulin resistant diabetes (124).
NEWER AND EMERGING GENES ASSOCIATED WITH FAMILIAL PARTIAL LIPODYSTROPHY
FPLD has also been reported to be caused by pathogenic variants in the AKT2 gene (125). AKT is a serine/threonine protein kinase, which is involved in cell signaling/growth, glycogen synthesis, and insulin-stimulated glucose transport. Lipodystrophy in patients with AKT2 mutations is thought to be due to defective adipocyte differentiation and post-receptor insulin signaling (126). Additional variants may be found through extensive sequencing platforms of ongoing studies such as UK Biobank (127), RADIANT (128) or All of Us (129) studies. Exome sequencing has identified a heterozygous variant in the adrenoceptor α 2A (ADRA2A) gene, which encodes the main presynaptic inhibitory feedback G protein–coupled receptor regulating norepinephrine release, in an African-American pedigree with atypical FPLD (130), which needs to be confirmed in additional pedigrees and to date, no additional cases have been reported with similar phenotypes.
Progeroid Syndromes and Lipodystrophy
Mandibuloacral Dysplasia (MAD) is a rare progeroid syndrome which manifests with craniofacial, skeletal and cutaneous abnormalities and lipodystrophy (Fig.12) (131). The clinical manifestations present gradually over time, most commonly during childhood. There are two types of MAD currently recognized. Mandibuloacral dysplasia type A (MADA) is characterized by the loss of subcutaneous fat from the extremities along with normal or excessive fat in the face and the neck. Mandibuloacral dysplasia type B manifests with a more generalized loss of subcutaneous fat (131-134).

Figure 12.
Mandibuloacral Dysplasia. Hypoplasia of the mandible in a patient with Mandibuloacral Dysplasia.
MADA is caused by mutations in the LMNA gene which results in the accumulation of prelamin A protein (135). This, in return disrupts the interaction between nuclear lamina and chromatin (134-136). Compound heterozygous pathogenic variants in the zinc metalloproteinase (ZMPSTE24) gene have been reported to cause MADB associated lipodystrophy (137, 138). ZMPSTE24 is essential in the post-translational proteolytic cleavage of carboxy terminal residues of farnesylated prelamin A to form mature lamin A and vimentin processing (137, 139, 140).
In addition, homozygous mutation in the MTX2 gene causes mandibuloacral dysplasia progeroid syndrome (MDPS), an autosomal recessive severe laminopathy-like disorder characterized by mandibular recession, clavicular hypoplasia and acroosteolysis, progeroid appearance and loss of subcutaneous fat (141).
MDP (mandibular hypoplasia, deafness and progeroid features syndrome) has been reported to be caused by pathogenic variants of the POLD1 gene that encodes catalytic subunit of DNA polymerase δ which play an essential role in the lagging-strand DNA synthesis during DNA replication (142). In addition to progressive lipodystrophy and severe insulin resistance, patients with MDP suffer from mandibular hypoplasia, sensorineural deafness, progeroid features, scleroderma and skin telangiectasia, ligament contractures, reduced mass of limb muscles, hypogonadism and undescended testes in males (142-145). We recently observed a mother daughter pair with a different POLD1 variant near the carboxyl terminal of the protein at a very highly conserved residue (Fig.13).

Figure 13.
Partial Lipodystrophy in a Patient with POLD1 Variant.
Biallelic WRN null mutations linked to partial lipodystrophy with severe insulin resistance in adult progeria Werner syndrome (Fig.14) (146). The WRN gene encodes a RecQ DNA helicase which plays a critical role in repairing damaged DNA (147). An unusual Werner syndrome with the absence of progeroid findings, early-onset diabetes, severe dyslipidemia, and hepatic fibrosis has been reported in a patient with partial lipodystrophy who had a novel variant in the WRN gene (148).

Figure 14.
Werner Syndrome.
Fibrillin-1 (FBN1) gene pathogenic variants are found in more than 90% of patients with Marfan syndrome (149). Pathogenic variants in the penultimate exon of FBN1 have been reported to be associated with a distinct phenotype of generalized lipodystrophy that share some clinical features with neonatal progeroid syndrome (Wiedemann–Rautenstrauch syndrome), a very severe disorder with only a few patients described who could reach their late childhood (150-152). Although these patients have marfanoid/progeroid appearance, skeletal features, dilated aortic bulb, bilateral subluxation of the lens, myopia in addition to the severe generalized lipodystrophy, no significant metabolic abnormality caused by the lack of adipose tissue has been reported (150, 151, 153).
Pathogenic variants in BANF1 have been reported to be associated with progeroid features, growth retardation, decreased subcutaneous fat, thin limbs, and stiff joints. This disease is also called Néstor-Guillermo progeria syndrome (NGPS) (154).
Heterozygous pathogenic variants in KCNJ6 (GIRK2), which encodes an inwardly rectifying potassium channel, cause Keppen-Lubinsky syndrome that is characterized by severe developmental delay and intellectual disability, microcephaly, large prominent eyes, an open mouth, progeroid appearance, and generalized lipodystrophy (155).
Pathogenic variants of the Spartan (SPRTN) gene, which encodes a protein that is essential in the maintenance of genomic stability, have reported to be associated progeroid features, lipodystrophy and hepatocellular carcinoma (156).
Pathogenic variants in the ALDH18A1 gene, which encodes pyrroline-5-carboxylate-synthetase, a mitochondrial enzyme important in ornithine biosynthesis, cause Cutis Laxa Autosomal Dominant 3 syndrome. This syndrome is characterized by intellectual disability, hypotonia, retinal abnormalities, craniofacial dysmorphism, joint laxity, and abnormal fat distribution (34, 157).
Several other genes associated with progeroid lipodystrophy are listed in Table 3.
Complex Syndromes and Their Genes Associated with Lipodystrophy
Pathogenic variants in the phosphatidylinositol 3-kinase, regulatory subunit 1 (PIK3R1), which mediates insulin’s metabolic actions, have been reported in patients with SHORT syndrome (short stature, joint hyperextensibility, ocular depression, Rieger anomaly, and teething delay) that is associated with lipodystrophy in many patients (158, 159). It has also been reported that patients with C-terminal PIK3R1 pathogenic variants exhibit severe insulin resistance but normolipidemia and no hepatic steatosis (160).
Pathogenic variants in the proteasome subunit, beta-type, 8 (PSMB8) gene, which encodes a catalytic subunit of the 20S immunoproteasomes called β5i, has been linked to an autosomal-recessive autoinflammatory syndrome characterized by joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy (JMP syndrome) (161-163).
CANDLE syndrome is another rare autoinflammatory syndrome characterized by chronic atypical neutrophilic dermatitis, recurrent fever, and partial loss of adipose tissue from the upper limbs and face (164). An eponym for the syndrome was proposed as Nakajo–Nishimura syndrome (165, 166). Homozygous or compound heterozygous mutations in the gene PSMB8 have been reported in patients with CANDLE syndrome (167, 168).
Two families with AREDYLD syndrome that is characterized by acrorenal field defect, ectodermal dysplasia, generalized lipodystrophy, and multiple abnormalities have been reported (169, 170). The genetic basis of this very rare syndrome is still unknown.
Lipomatosis Syndromes
In studies conducted in 2016-2018, a pathogenic variant in the MFN2 gene that encodes mitofusin 2, a membrane-bound mediator of mitochondrial membrane fusion and inter-organelle communication, have been reported to be associated with partial lipodystrophy, upper body adipose hyperplasia, and suppression of leptin expression (171) (Fig.15). MFN2 gene-related Multiple Symmetric Lipomatosis (MSL) is an unusual type of lipodystrophy that includes both lipomatous masses and lipoatrophy (172). In these cases, one may observe various indicators, including insulin resistance, diabetes, non-alcoholic fatty liver disease, dyslipidemia, peripheral neuropathy, autonomic neuropathy, and extremely low leptin and adiponectin levels. Fibroblast growth factor 21 (FGF21) serum levels were found to be remarkably higher in these cases (173). Of note, there are additional etiologies linked to the development of lipomatosis, with or without lipodystrophy.

Figure 15.
Disease progression in a patient with a pathogenic variant in the MFN2 gene (15A-D).
Acquired Partial Lipodystrophy
Acquired partial lipodystrophy (APL) is characterized by fat loss typically starting in childhood or early adulthood. Loss of adipose tissue first manifests in the face and gradually progresses to the upper extremities, thorax, and upper abdomen symmetrically. It typically proceeds in a cephalocaudal fashion but spares the lower extremities (Fig.16). There might be accumulation of fat in the lower abdomen, gluteal region, and lower extremities.
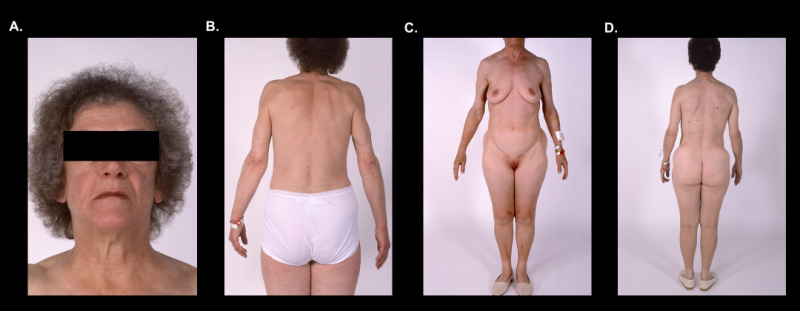
Figure 16.
APL. Typical cephalocaudal adipose tissue loss pattern in two patients with APL (16A-D). Note preservation of fat depots below the waist line.
Although the etiology of APL is still unknown, some patients may have coinciding autoimmune conditions. Systemic lupus erythematosus and dermatomyositis/polymyositis are among the most frequently associated auto-immune diseases (174). In recent years, cases of APL associated with hematopoietic stem cell transplantation and total body irradiation have also been described. These patients are thought to constitute a subset of APL (175, 176). APL has been associated with abnormalities of the alternative complement pathway that may cause membranoproliferative glomerulonephritis (MPGN) (177). Subsequent chronic renal disease constitutes the major cause of morbidity in these patients. It has been suggested that C3-nephritic factor might be the cause for the lysis of adipocytes expressing factor D, although there is no solid evidence supporting this hypothesis (178). A comprehensive review of renal complications in lipodystrophy by the Turkish Lipodystrophy Study Group verified low complement C3 levels in more than 45% of APL patients (179, 180).
Rare variants in LMNB2 were previously reported in five patients with APL, but two of four variants were also present in normal controls (181). In addition, subcutaneous loss of fat from the legs and the gluteal region, presence of diabetes, type IV and V hyperlipoproteinemias were atypical presentations in these patients (181).
Metabolic complications are less common compared to other types of lipodystrophy syndromes (4). Not all patients develop insulin resistance, diabetes, or hypertriglyceridemia. Leptin levels vary from hypoleptinemia to normal range (32, 174). However, patients may develop metabolic abnormalities such as diabetes, hypertriglyceridemia, low HDL cholesterol levels, and hepatic steatosis in later stages of the disorder. In addition, several patients with APL have been reported to develop diabetes or other metabolic abnormalities at a relatively young age, which are apparently associated with insulin resistance (182). It is also known that metabolic complications such as hepatic steatosis, poorly controlled diabetes, and pancreatitis are severe in a group of APL patients who are thought to have advanced fat loss (180). Thus, patients with APL should also be followed for metabolic abnormalities, as is done for other subtypes of lipodystrophy.
ANIMAL MODELS OF LIPODYSTROPHY
Numerous animal models of lipodystrophy have shown that adipose tissue dysfunction triggers the development of severe insulin resistance, which is associated with metabolic abnormalities and end-organ complications as mentioned above and shown in Fig.1. Extensive and authoritative reviews of these studies can be found in articles by Drs. David B. Savage (183) and by Xavier Prieur (121). The introduction of these animal models has allowed researchers to explore the fundamental characteristics of lipodystrophy and insulin resistance and allowed studies of the effects of different treatment approaches. Regardless of the strategy used, ablation of white adipose tissue led to the development of insulin resistance, hypertriglyceridemia, and hepatic steatosis (sometimes 6-fold elevation in total liver weight). In now classical experiments of Reitman and colleagues, fat transplantation from littermates rescued metabolic derangements in the famous A-ZIP mice (184-186). Dr. Beutler’s group identified kelch repeat and BTB (POZ) domain containing 2 (KBTBD2) deficiency as a cause of lipodystrophy associated with insulin resistance and diabetes and they also showed that transplantation of wild-type adipose tissue rescued diabetes and the hepatic steatosis phenotypes of Kbtbd2−/− mice (187). The administration of leptin into aP2–SREBP-1c transgenic mice from the Brown and Goldstein laboratory resulted in dramatic benefits in glycemic parameters, insulin action, and hepatic steatosis, which could not be explained by its effect on food intake alone, providing the premise to undertake leptin replacement in human patients (188). What was also striking was that if fat from the leptin deficient obese mice was transplanted into littermates of the A-ZIP mice, the metabolic rescue was far less effective, suggesting that leptin played an important role in the regulation of metabolism in lipodystrophy in rodents (189). The replacement of deficient leptin in a small but severely affected cohort of human patients with lipodystrophy with recombinant human leptin (metreleptin) was first reported in 2002 and brought further attention to lipodystrophy research (190). Longer-term studies subsequently confirmed the role of metreleptin therapy in lipodystrophy syndromes especially in the most severe forms (32, 41, 42).
Recent Animal Models Advancing Our Understanding of Lipodystrophy and Fat Dysfunction
While we are not intending to provide a comprehensive review of all animal models generated recently, we selected a few to highlight recent advances in this field. A study by Tapia et al.'s (191) showed that a generalized lipodystrophy gene and a critical enzyme in triglyceride synthesis AGPAT2 is essential for the expression of critical mitochondrial proteins. In this study, genes involved in the type-1 interferon response were overexpressed in differentiated Agpat2−/− adipocytes, and this condition could be associated with their defective mitochondria. They also showed that differentiated Agpat2−/− brown adipocytes have a lower proportion of lipid-laden cells, Adiponectin and Perilipin1 synthesis, indicating that these cells were able to initiate brown adipogenesis but could not carry it to further stages (191). Interestingly, another study of Agpat2 null mice, demonstrated the absence of caveolae in adipocytes lacking AGPAT2, which hints at a possible mechanistic connection between different Congenital Generalized Lipodystrophy (CGL) types (121, 192, 193).
The Macdougald lab and our group collaboratively developed adipocyte specific knock out of LMNA gene which recapitulates most of the features of human FPLD2 (194). Loss of adipocyte-specific lamin A/C in mice (LmnaADKO) caused a significant reduction in the weight of posterior subcutaneous white adipose tissue (WAT), gonadal WAT, pericardial WAT, renal WAT, and retroperitoneal WAT, as well as markedly reduced circulating adiponectin and leptin levels in both sexes. Hyperglycemia, hyperinsulinemia, liver enlargement, and ectopic fat accumulation in the liver were also noted in these mice under a high-fat diet, consistent with the clinical presentation of FPLD2 (194).
A novel mouse model was generated to explore whether maintaining intact BSCL2 expression in the liver prevents the onset of metabolic disorders in mice with specific BSCL2 deficiency in adipose tissue. BSCL2 was simultaneously targeted for ablation in adipose tissue and hepatocytes. It was observed that liver-specific seipin deficiency did not result in hepatosteatosis and insulin resistance. These results suggest that most of the metabolic pathophysiology is driven by the function of BSCL2 outside of hepatocytes, and likely in adipose tissue (195).
In an animal model created to elucidate the molecular mechanisms of lipomatosis and lipodystrophy associated with the MFN2 gene, which encodes an outer membrane GTPase required for mitochondrial fusion (196), no change in mitochondrial oxidative capacity was observed in homozygous Mfn2R707W/R707W mice (197). Although the triggering of a cellular integrated stress response and selective impairment in mitochondrial morphology and function were detected in adipose tissues, no significant changes in glucose and lipid metabolism were observed in homozygous mice, unlike humans. Interestingly, leptin and adiponectin levels were low in these mice (197).
A new animal model study showed that a novel R133L heterozygous mutation in the LMNA gene causes signs of aging, such as impaired mitochondrial functions, decreased lipid storage capacity in subcutaneous adipose tissue, as well as metabolic disorders such as insulin resistance and ectopic lipid accumulation. LmnaR133L/+mice exhibited findings consistent with lipodystrophy, such as a reduction in epididymal white adipose tissue (eWAT) mass, as well as smaller adipocytes and upregulated inflammation genes in the inguinal subcutaneous white adipose tissue (iWAT) (198).
Furthermore, the importance of the lipid kinase phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3) in the function of white adipose tissues (WATs) and brown adipose tissues (BATs) has been demonstrated using adipocyte-specific Pik3c3 knock-out model. Deletion of PIK3C3 has been linked to lipodystrophy due to impaired adipocyte differentiation, autophagy, and thermogenesis mechanisms (199).
We have also covered recent animal models of novel gene therapies under a new heading: Emerging Therapeutic Technologies and Gene Replacement Therapy Approaches.
TREATMENT
Currently, treatment modalities are restricted to ameliorating or preventing the comorbidities of the lipodystrophic syndromes. There is no cure for these syndromes. For the metabolic disturbances, lifestyle modification (diet and exercise as needed), metformin, and fibrates (and/or statins) are generally required. Insulin or other antidiabetics (e.g., metformin, thiazolidinediones) can also be used if needed. Metreleptin, a leptin analog, is indicated as an adjunct to diet as replacement therapy to treat the complications of leptin deficiency in patients with generalized lipodystrophy.
Lifestyle Modification
There is limited knowledge on the effectiveness of diet and exercise in the management of metabolic disturbances in patients with lipodystrophy. In general, a balanced macronutrient composition is recommended. In patients with severe hypertriglyceridemia, a balanced low- fat diet (<15% of daily caloric intake) is appropriate. To control diabetes, increased physical activity and carbohydrate restriction are advised. Dietary fiber intake and foods that are rich in omega-3 fatty acids are suggested (3).
Most patients with lipodystrophy are encouraged to be physically active. In patients with cardiomyopathy and cardiac arrhythmias strenuous exercise should be avoided. Patients with CGL4 should avoid exercise as they may develop exercise-induced ventricular arrhythmias (75, 79). Contact sports are not advised to patients with severe hepatosplenomegaly and CGL patients presenting with lytic bone lesions.
Patients should abstain from drinking alcohol due to the risk of developing acute pancreatitis and metabolic dysfunction-associated steatohepatitis (MASH). Patients should also be advised to avoid smoking and maintain an optimal blood pressure to decrease the risk of cardiovascular disease.
Insulin Resistance
In patients presenting with lipodystrophy and diabetes, both metformin and thiazolidinediones are somewhat effective to treat hyperglycemia and hyperlipidemia (200-204). Metformin is used as the first-line agent in insulin resistant diabetes. Thiazolidinediones may improve the metabolic profile in partial lipodystrophy syndromes (204). The very first thiazolidinedione to be approved in the United States troglitazone actually worked remarkably well in lowering both HbA1c and triglyceride levels in a cohort of patients with predominantly partial lipodystrophy syndromes. However, data on the currently approved thiazolidinediones are limited and contradictory (202, 205, 206). Thiazolidinediones should be considered in the management of diabetes in patients with partial lipodystrophy, however they should not be routinely used in generalized lipodystrophy as their efficacy has not been studied (3, 204). Insulin is usually needed in very high doses and concentrated forms, such as U-500. Patients with extreme insulin resistance, however, may not respond to concentrated insulin. Administration of insulin-like growth factor-1 (IGF-1) has been shown to be effective in maintaining glycemic control and insulin resistance in short-term studies, as well as in type 2 diabetes (207-209). A retrospective study found that administration of sodium-glucose cotransporter 2 (SGLT2) inhibitors resulted in a 0.8% drop in HbA1c levels after one year in individuals with partial lipodystrophy. Additionally, SGLT-2 inhibitors led to a significant decrease in both systolic and diastolic blood pressure (210). In clinical practice, it should be considered that combination therapy with metreleptin and SGLT2 inhibitors may contribute to prognosis by improving insulin resistance in adipose tissue and reducing the risk of cardiovascular events (211). Many other hypoglycemic agents have been used in lipodystrophy, but their efficacy has not been studied (3). Recently we published a retrospective review of GLP-1 agonist use in FPLD syndromes showing substantial improvement in glucose control and body weight (212). Dual incretin therapeutic tirzepatide is postulated to have even more efficacy due to the potential impact of GIP agonism and potentially improving inflammation.
Dyslipidemia
Statins are normally used as first-line agents to treat hypercholesterolemia but patients with FPLD have low tolerance to statins. Rosuvastatin and pravastatin have been proven to reduce total LDL cholesterol levels (213, 214). Statins are used with caution to prevent side effects such as myopathy and hepatotoxicity. Along with diet, fibrates and fish oil rich in omega-3 fatty acids, should be prescribed for serum triglyceride levels >500 mg/dL and may be considered for triglycerides >200 mg/dL. Combining fibrates with statins has proved to be effective in dyslipidemia; however, there is an increased risk for muscle toxicity. Therapeutic apheresis can be used in extreme hypertriglyceridemia to prevent recurrent episodes of acute pancreatitis in acutely life-threatening situations (190).
Cosmetic Treatment
Cosmetic correction of lipoatrophy and fat excess is associated with improved quality of life in patients with lipodystrophy. Autologous adipose tissue transplantation, facial reconstruction with free flaps and silicone or other implants have been used in lipoatrophic areas. In addition, liposuction or surgical excision is used for removal of unwanted excess fat from body parts such as the chin, buffalo hump and vulvar region.
Bariatric Surgery
Roux-en-Y Gastric Bypass Surgery (RYGB) is associated with effective weight loss and resolution of metabolic comorbidities in patients with obesity (215). RYGB was used with success in several patients with FPLD1 and with FPLD2 (216-218). RYGB resulted in weight loss and significant improvements in metabolic parameters in patients with FPLD1 that allowed patients to stop using insulin (216). FPLD2 patients also benefited from RYBG. Substantial improvements in metabolic parameters and a significant weight loss were reported after the surgery (218, 219).
Leptin
A large group of lipodystrophy patients present with low leptin levels. Metreleptin (r-metHuLeptin) is an analog of human leptin made through recombinant DNA technology. It has been tested in congenital and acquired forms of lipodystrophy and has been shown to ameliorate the metabolic derangements (43, 190).
Leptin replacement therapy is approved in Japan as a therapy indicated specifically for the treatment of diabetes and/or hypertriglyceridemia in patients with congenital or acquired lipodystrophy. In the United States, metreleptin, now called MYALEPT, has been approved by the FDA in 2014 for use in patients with congenital generalized or acquired generalized lipodystrophy for the treatment of complications of leptin deficiency as an adjunct to diet and lifestyle modifications. There is no lower age limit for initiation of Myalept nor a specific degree of metabolic abnormality so long as the diagnosis of generalized lipodystrophy can be substantiated. However, it is not approved for use in human immunodeficiency virus (HIV)-related lipodystrophy, or in patients with metabolic diseases such as diabetes and hypertriglyceridemia, or partial lipodystrophy in the US. Metreleptin is an approved treatment for generalized lipodystrophy (GL) and partial lipodystrophy (PL) in the European Union (EU), United Kingdom (UK), Canada, and Brazil. However, there is an age limit of ≥ 2 years for GL. The approval for PL states that patients with confirmed PL can initiate treatment if they are aged ≥ 12 years and only when standard treatments have not achieved adequate metabolic control. Based on this indication, in the EU, UK, Canada, Brazil, and Japan, patients with PL who do not respond to available diabetes and lipid-lowering agents can be treated with recombinant leptin therapy. It is still controversial whether basal endogenous leptin levels can be used to predict response to metreleptin treatment in lipodystrophy patients. However, a recent study revealed that baseline endogenous leptin levels are poor predictors for response to metreleptin therapy among individuals with partial lipodystrophy (220). The clinical effects of leptin treatment in patients with lipodystrophy are summarized below.
APPETITE
Metreleptin decreases hyperphagia, leading to weight loss that usually stabilize with long-term treatment (190, 221-223). This effect can be noted by the patients right after the treatment with metreleptin. Functional MRI studies combined with behavioral assessments showed that metreleptin treatment is associated with long-term improvements of hedonic and homeostatic central nervous networks regulating appetite and food intake (224-226). Food related neural activity and development of satiety were effectively restored by leptin replacement in lipodystrophy (227).
METABOLIC PARAMETERS
Metabolic changes become evident quickly within days to weeks of treatment with metreleptin. Metreleptin therapy has been shown to improve fasting plasma glucose levels starting from the first week. In the first set of patients with lipodystrophy treated with metreleptin, four months of therapy with metreleptin decreased average triglyceride levels by 60%. The absolute decrease in HbA1c was 1.9% among patients with diabetes. Liver volume reduced by an average of 28% and led to the discontinuation of or a large reduction in antidiabetic therapy (190). In the long term, more than three-fourths of patients with GL treated with metreleptin discontinued concomitant treatments, including insulin and oral antidiabetics (228).
In clinical studies, metreleptin led to significant improvements in patients with GL. After 1 year of treatment initiation, a mean change of -2.2% in HbA1c and a mean percent change of -32.1% in triglycerides were observed. Significant reductions in alanine aminotransferase levels occurred. Mean liver volume decreased by 33.8% at month 12. Nearly 80% of patients with GL achieved a ≥1% actual decrease in HbA1c or a ≥30% decrease in triglycerides after 1 year of treatment with 66% achieving decreases of ≥2% in HbA1c or a ≥40% in triglycerides. Among patients with baseline HbA1c of 7% or greater, the mean reduction at Month 12 was 2.8%. In subjects with baseline TG level 500 mg/dL or greater, the mean percent reduction in triglycerides at month 12 was 72%. Patients with GL overall sustained clinically significant reductions in HbA1c and triglycerides in the longer term follow up.
In patients with PL, metreleptin treatment led to statistically significant reductions in HbA1c (−0.6%), fasting TGs (−20.8%), and liver volume (−13.4%) after 1 year treatment. In a subgroup of patients with baseline HbA1c ≥ 6.5% or triglycerides ≥ 5.65 mmol/L, more prominent reductions were observed in HbA1c (−0.9%) and fasting TGs (−37.4%). In this subgroup, 68% of patients had a ≥ 1% decrease in HbA1c or ≥ 30% decrease in fasting TGs, and 43% had a ≥ 2% decrease in HbA1c or ≥ 40% decrease in fasting triglycerides. Longer-term treatment in the PL subgroup led to significant reductions at months 12, 24, and 36 in HbA1c and fasting triglycerides (229).
12 months of long-term metreleptin therapy has been shown to reduce mean fasting plasma glucose levels by 2.8 mmol/L in the generalized lipodystrophy group and 1.2 mmol/L in the partial lipodystrophy group (228, 229). In a subset of patients undergoing hyperinsulinemic-euglycemic clamp studies, leptin replacement therapy improved peripheral glucose disposal and decreased both hepatic glucose output and hepatic steatosis (230). It is generally recommended to lower the insulin doses by 50% on initiation of metreleptin therapy (especially in GL) to avoid hypoglycemia in well-controlled patients with diabetes. Metreleptin treatment has no suppressive effect on beta cell function in patients with lipodystrophy (231). On the contrary, it has been reported that metreleptin therapy improves insulin secretion in the setting of diabetes (232). A prospective study observed that metreleptin administration for two weeks suppressed basal gluconeogenesis (GNG) by reducing carbon sources for GNG and increasing insulin-mediated suppression of GNG. Peripheral insulin sensitivity increased significantly throughout the 6-month follow-up of these patients (233).
Apolipoprotein C-III and angiopoietin-like protein 8 (ANGPTL8), recognized as inhibitors of lipoprotein lipase, play a role in modulating hypertriglyceridemia. A notable reduction of these hepatokine plasma concentrations is observed, especially after six months of metreleptin treatment (234). While about a 60% decrease in TG values was detected in 1-year follow-up in GL patients using concomitant lipid-lowering medications, a 30% decrease was observed in patients with partial lipodystrophy. These reductions were less in individuals who did not take concomitant medication (235). In a real world study from France, during a follow-up period of more than 15 months, TG levels decreased by approximately 150 mg/dL in patients with generalized lipodystrophy (236). It should be noted that acute withdrawal of metreleptin therapy might result in acute pancreatitis episodes (237, 238). Metreleptin also decreased total cholesterol and LDL-cholesterol levels (237, 239). But did not alter HDL cholesterol levels (236, 237, 239).
As we have noted, the beneficial effect of metreleptin on glycemic and lipid measures in generalized lipodystrophy are clear and usually quite remarkable. Although the response is variable in patients with partial lipodystrophy and it is not approved for this indication in the US, studies have shown that some patients can benefit from metreleptin treatment. A selected cohort of partial lipodystrophy patients with moderately to severely low leptin and significant baseline metabolic abnormalities is more likely to benefit from metreleptin therapy (13, 235, 236, 240-242). Recent studies confirm that most partial lipodystrophy patients with inadequate metabolic control also respond to metreleptin treatment (235, 236). Furthermore, when comparing the metabolic responses to metreleptin treatment of patients with PPARG and LMNA pathogenic variants in the FPLD group, both groups reported remarkable and similar reductions in HbA1c, insulin dose, and triglyceride levels after 12 months of treatment (243). However, the real-life study from France showed that leptin treatment did not significantly change HbA1c levels (7.7 [7.1‐9.1]% at baseline vs. 7.7 [7.4‐9.5]% at one year) in 19 patients with partial lipodystrophy. In the same group of patients, a significant decrease in the median value of fasting triglycerides from 3.3 mmol/L (1.9-9.9) to 2.5 mmol/L (1.6-5.3) was observed with short-term metreleptin treatment (p<0.01), whereas the median serum triglyceride levels were 5.2 mmol/L (2.2-11.3) at the last evaluation after long-term leptin treatment (p=0.94) (236). When a responder analysis is performed, however, 61% of the patients with PL showed improvements in glucose homeostasis, with responders exhibiting lower leptin levels compared to non-responders. Similarly, 61% of the PL patients in the French real-world study cohort were responders regarding hypertriglyceridemia.
LIVER
Starting from the earliest studies, leptin replacement therapy was observed to improve hepatic steatosis and lower serum transaminases within 6-12 months (Fig.17) (223, 230, 244).The liver volume decreases in parallel (222, 244). Metabolic associated steatohepatitis (MASH) score has been reported to improve after metreleptin treatment and no progression in hepatic fibrosis has been reported (245, 246). When treated at least for a year, the majority of patients showed improved liver histology, steatosis and hepatocyte ballooning, and only 33% of patients continued fulfilling the criteria for MASH after 1 year of treatment with metreleptin (247, 248). A significant improvement in the metabolic dysfunction-associated steatotic liver disease (MASLD) score has been reported after metreleptin treatment in pediatric patients who underwent liver biopsies (249). Also, our study showed that metreleptin can induce improvements in hepatic steatosis and injury in patients with MASH associated partial lipodystrophy (246). In a mechanistic study, leptin therapy resulted in significant increase in insulin suppression of hepatic glucose production (230). This improvement in insulin action helps reverse hepatic steatosis by decreasing triglyceride content (230). Other mechanisms associated with reducing hepatic steatosis may be leptin effectively increases hepatic VLDL-TG secretion via possibly vagal effect (250) and decreases de novo lipogenesis (251). These observations suggest that the effect of metreleptin on hepatic steatosis can be somewhat blunted when the autonomic innervation of the liver is not intact. However, it should be noted that the liver disease can still benefit from improving metabolic profile and hepatic steatosis can diminish as a result of controlled de novo lipogenesis after improved insulin sensitivity with leptin replacement. To support this view, metreleptin has also been reported to result in rapid clearance of fat from the liver and normalization of liver histology in an AGL patient with recurrence of MASLD in the first few months of liver transplantation (252).

Figure 17.
Liver Histology Before and After Metreleptin. Liver histology shows regression of hepatic steatosis and ballooning injury after metreleptin treatment (left before metreleptin and right 4 months on metreleptin treatment, Hematoxylin and eosin staining; magnification 200X).
KIDNEYS
Patients with lipodystrophy may develop proteinuric kidney disease. Metreleptin decreases proteinuria in most patients (223, 253). The reduction in proteinuria coincided with improvement in hyperfiltration in 11 of 15 patients treated with metreleptin. However, four patients had worsening renal function. Hence, renal functions should be closely monitored during metreleptin therapy (253). In further studies, metreleptin significantly reduced proteinuria in patients with GL (254).
CARDIOVASCULAR SYSTEM AND OVERALL SURVIVAL
A study conducted in lipodystrophic mice showed that metreleptin treatment inhibited proatherosclerotic cytokine growth/differentiation factor 15 (GDF15) and reduced macrophage accumulation in atherosclerotic lesions via endothelial to mesenchymal transition. Thus, the positive effect of leptin on the endothelium by reducing inflammation has been reported (255). Metreleptin treatment provides a significant improvement in serum levels of atherogenic lipoproteins, which are known to be associated with increased cardiovascular disease risk (256). A potential increase in cardiovascular risk was detected in a prospective coronary calcium score (CCS) evaluation in 19 individuals diagnosed with CGL, although not sufficiently powered, this study did not report a significant change after metreleptin in CCS score (257). Another recent study, on the other hand, reported an approximately 30% regression in left ventricular hypertrophy and an improvement in septal e' velocity in the GL patients after leptin treatment (258). Cardiovascular problems are a significant risk of mortality in lipodystrophy patients. Our large retrospective evaluation from a large multicenter dataset, found that after adjusting covariates such as lipodystrophy diagnosis, triglyceride levels, elevated HbA1c, ≥1 episode of pancreatitis, and abnormalities in the heart or kidneys, metreleptin treatment resulted in a mortality reduction of over 60% (259).
CENTRAL NERVOUS SYSTEM AND OTHER EFFECTS
A functional MRI study revealed that leptin may induce alterations in brain activity in the subgenual area (Brodmann area 25), a region not strictly linked to eating behavior, known for its connections to the reward network. This suggests that leptin may also affect brain regions other than eating behavior (260). Another observational study of ten individuals with partial lipodystrophy who were naïve to metreleptin found a rapid decline in Beck's depression inventory score one week after metreleptin therapy. In the first 3-month follow-up of the individuals, a significant improvement was observed compared to their initial scores. Although this shows that metreleptin treatment may have an antidepressant effect, the findings need to be supported by long-term clinical studies (261). Metreleptin treatment also significantly contributes to improving indirect findings related to quality of life, such as hyperphagia, inability to go to work/school, and deterioration in physical appearance (262).
REPRODUCTIVE SYSTEM
In females, metreleptin was found to normalize gonadotropin secretion. It led to normal progression of puberty, normalized menstrual cycles, and improved fertility (223, 263-265). Leptin replacement improved low estradiol levels and corrected the attenuated luteinizing hormone (LH) response to luteinizing hormone-releasing hormone (LHRH) in young women with lipodystrophy and leptin deficiency (263). One-year treatment with metreleptin resulted a significant decrease in testosterone and sex hormone binding globulin (SHBG) levels in lipodystrophic women with PCOS (266). Several pregnancies have occurred in patients with lipodystrophy while they were on metreleptin without any evidence for teratogenicity (47, 267), although it has not been approved for use in pregnancy. Leptin replacement was associated with a small increase (clinically non-significant) in serum testosterone and SHBG in males. No change was observed in serum LH response to LHRH (264). No impact of leptin therapy on bone mineral density and content and bone metabolism has been reported in both sexes (244, 268, 269) .
ADVERSE EFFECTS
Majority of patients treated with metreleptin in clinical studies experienced ≥1 treatment-emergent adverse event, which mainly were mild-to-moderate in severity (228, 229). The most common side effects were hypoglycemia, nausea, decreased appetite and injection site reactions e.g. erythema and urticaria. Headache, fatigue, weight loss, and abdominal pain were also seen (228). During metreleptin treatment, iron parameters might be affected, and a decrease in ferritin levels might be observed (270). There may also be a need to make dose adjustments or cessation of concomitant treatments such as insulin, oral antidiabetics, and lipid lowering drugs after metreleptin therapy. In some cases, in vivo neutralizing antibodies to metreleptin have been reported (271, 272) and is the main reason underlying the FDA's restriction of metreleptin use. Anti-metreleptin antibodies developed in most patients with lipodystrophy; however, neutralizing activity concurrent with worsened metabolic control has been reported only in a small number of patients treated with metreleptin (267, 271). In addition, in the few patients who presented with neutralizing antibody formation, occurrence of severe infections such as sepsis has been reported. Two of these patients developed multiple sepsis episodes around the time of detection of neutralizing antibody (271). T-cell lymphoma has been reported in three patients with acquired generalized lipodystrophy receiving metreleptin (273). In acquired lipodystrophy patients with autoimmunity and immunodeficiency before metreleptin therapy, T-cell lymphoma development was also described (273, 274), suggesting that lymphoma development in acquired lipodystrophy is more likely to be associated with the disease itself rather than being related to metreleptin treatment.
In other cases with acquired generalized lipodystrophy, progression of kidney disease and liver disease have been observed while receiving metreleptin therapy (275). Since patients with AGL with distinct autoimmune conditions clearly benefit from metreleptin, treatment for their metabolic abnormalities should be considered in patients with AGL with close clinical follow up considering the cautionary preclinical data (276, 277).
More recently attention has been devoted to emergence of new cancers while on metreleptin therapy. Data on these parameters has been reported with the prospective, non-randomized, open-label clinical trial of metreleptin in lipodystrophy. Ten patients with general lipodystrophy had neoplasm development throughout the research period. These included peripheral T-cell lymphoma, granular cell tumor, breast cancer, ovarian neoplasm, papillary thyroid cancer, and basal cell carcinoma. However, only the anaplastic large cell lymphoma case was thought to be drug-related (228). During the study period, neoplasms (e.g., adrenal adenoma, benign ovarian germ cell teratoma, schwannoma, and a nervous system neoplasm) were observed to develop in five partial lipodystrophy patients. Still, none of them were considered drug-related (229).
Additionally, MEASuRE (Metreleptin Efficacy and Safety Registry), a voluntary registry that collects long-term safety and effectiveness data on metreleptin, was developed as a post-authorization requirement by the FDA and EMA. This database contains comprehensive data on long-term adverse effects and drug efficacy in lipodystrophy patients using commercially available metreleptin (278).
Investigational Treatments for Lipodystrophy
Since in the United States partial lipodystrophy has been without medical therapy, several companies with interesting compounds have initiated development of their products for this indication. Current and recently completed investigational treatments with registered studies in ClinicalTrials.gov are presented in Table 4. Data on these trials will be updated as results become available.
The BROADEN study which was the largest global double blinded placebo controlled study published in 2022 revealed that treatment with volanesorsen, an antisense oligonucleotide to apolipoprotein C-III, significantly reduced serum triglyceride levels by over 88% and improved hepatic steatosis in patients with FPLD by 53% from original level (279, 280). While the exact mechanisms of how volanesorsen led to the effects observed in the FPLD population are unclear, a small scale study showed that hepatic insulin resistance concurrently improved in parallel to an increase in lipoprotein lipase (LPL) activity in a few FPLD patients treated with this drug (281).While Volanesorsen is not approved in the US due to the adverse events of systemic inflammation and thrombocytopenia, this drug has been approved for use in FPLD in Brazil under the name Waylivra, marketed by PTC Therapeutics. The same drug is also available in Canada and EU for treatment of familial hyperchylomicronemia syndrome and can be an off-label option for patients with recurrent pancreatitis and severe hypertriglyceridemia. Currently, second generation anti-sense inhibitors of ApoCIII are under development in the US for severe hypertriglyceridemia and patients with FPLD are not excluded from these studies if they meet the trial population criteria (282-284). We are aware of a few patients with FPLD who are in ongoing studies.
A therapeutic compassionate use trial with the melanocortin-4 receptor agonist setmelanotide was conducted in a patient with atypical partial lipodystrophy, whose metabolic status continued to worsen despite the presence of neutralizing antibodies to metreleptin. The reported findings indicated that the treatment had no improving effect on the lipid profile, insulin resistance, and liver fat percentage (285). The same patient was successfully treated with Mibavademab a monoclonal leptin receptor agonist antibody and remains on long term treatment (286). The promising results obtained from this patient experience led to the development of this therapeutic as an option for patients with generalized lipodystrophy. A small-scale proof of concept study in FPLD patients was also started but very recently discontinued and results are not yet available. Treatment of 16 enrolled patients with generalized lipodystrophy over a year resulted in meaningful changes in metabolic parameters which are similar to metreleptin effects; but final completion of the study and the definitive dosing schema has not been published so far (287).
Table 4.
Ongoing or Recently Completed Investigational Therapies for Lipodystrophy
Investigational agent | Status | Type of lipodystrophy | Primary outcome | Results |
|---|---|---|---|---|
| Volanesorsen (anti-sense oligonucleotide to apoC-III) | Completed | Familial partial lipodystrophy | Change in fasting triglycerides | 88% reduction in triglycerides, 53% reduction in hepatic steatosis |
| Vupanorsen (Targeting angiopoietin-like 3 “ANGPTL3”) (288) | (Pfizer and Ionis announced discontinuation of vupanorsen clinical development program due to concern for increase in liver fat | Familial partial lipodystrophy | Reduction in fasting triglycerides and free fatty acids | 59.9% reduction in triglycerides, 54.7 % reduction in ANGPTL3, 53.5% reduction in VLDL cholesterol, 41.7% reduction in free fatty acids (FFA) |
| Obeticholic Acid (farnesoid X receptor agonist) | Completed | Familial partial lipodystrophy | Change in liver triglycerides | 6.8% reduction in liver triglycerides median value, 16.9 mg/dL reduction in serum triglycerides, 0.5 U/L reduction in serum alanine aminotransferase (ALT) median value |
| Cholic Acid (primary bile acid) | Completed | Various forms of lipodystrophy | Reduction in liver triglyceride content | 7.9% reduction in hepatic triglycerides median value (Fat/Fat+Water) |
| Setmelanotide (melanocortin-4 receptor agonist) (285) | Expanded access in a single patient | Partial lipodystrophy associated with leptin deficiency | Treatment of refractory hypertriglyceridemia leading to recurrent bouts of pancreatitis | No sustained improvement in glycemic control or hypertriglyceridemia, slight decrease in visceral fat |
| Gemcabene (monocalcium salt of a dialkyl ether dicarboxylic acid) | Completed | Familial partial lipodystrophy | Change in fasting triglycerides, hepatic steatosis | The mean percentage change in fasting serum triglycerides between weeks 12 and 24 in group 1 (receiving 300 mg Gemcabene daily) was -0.44 (-41.27 to 40.38), and in group 2 (receiving 600 mg Gemcabene daily) was -20.27 (-53.98 to 12.77) |
| Baricitinib (inhibitor of Janus kinases 1 and 2 “JAK1/2”) | Expanded access available | Autoinflammatory syndromes, familial partial lipodystrophy type 2, Hutchinson-Gilford Progeria Syndrome, and MAD (289) | Clinical benefit from JAK 1/2 inhibition, improves adipogenesis and lipid droplet formation | Increased number of adipocytes and formation of lipid droplets, higher adipocyte differentiation ability, increased lipid droplet size (~76 µm2) in the FPLD2 group |
| Evinacumab (Anti-ANGPTL3) | Completed | Patients with severe hypertriglyceridemia | Percent lowering of triglycerides | 59.9% reduction in mean fasting triglycerides levels from baseline at the end of the treatment (week 27) |
| Mibavademab | Active, not recruiting | Generalized lipodystrophy | Meaningful improvements in metabolic parameters | Reduction in HbA1c (mean: -1.9%, SD: 2.0) and triglycerides (median: -102.1 mg/dL, IQR: 1355.5 mg/dL, -49.3% [57.2%]) (290) With longer treatment there was a further 0.7% reduction in HbA1c and 42% reduction in fasting triglycerides in patients receiving high dose mibavademab (287) |
Emerging Therapeutic Technologies and Gene Replacement Therapy Approaches
Research on novel and emerging technologies is ongoing to broaden therapy options and better understand diseases of complex nature like lipodystrophy. LipocyteProfiler, a sophisticated image-based tool for deep phenotypic analysis, facilitates assessing numerous morphological and cellular profiles that can be methodically associated with genes and genetic variations that have significance in cardiometabolic diseases. LipocyteProfiler is algorithmically generated from over 3,000 morphological and cellular features that map to the cell, cytoplasm, and nucleus across channels differentiating the organelles, namely DNA, mitochondria, AGP (actin, Golgi, plasma membrane), and Lipid. It was observed that LipocyteProfiler was able to detect significant changes in cell profiles during white and brown adipocyte differentiation after genetic and pharmacological manipulations and transcriptional states in adipocytes. Also, profiles associated with the polygenic risk of lipodystrophy confirmed increased mitochondrial activity, reduced actin cytoskeleton remodeling, and reduced lipid accumulation capacity in subcutaneous adipocytes. This tool may shed light on a deeper insight into both known and novel mechanisms by generating cellular profiles that are specific to the processes occurring in response to polygenic metabolic events, particularly in hepatocytes and adipocytes. Analyzing deep phenotypic profiles, especially in lipocytes, can enhance our understanding of lipodystrophy pathophysiology and allow us to develop better therapies (291).
Gene therapy studies conducted in recent years are an emerging and promising approach to treating various genetic lipodystrophy syndromes. A novel study shows that the in vivo embryonic re-expression of human AGPAT2 (hAGPAT2) induces adipocyte differentiation and regenerates 30%–50% of the WAT and BAT depots in Agpat2−/− mice when compared to age-matched wild-type mice. Due to this genetic reorganization in Agpat2-null mice, it was observed that the differentiation of adipocytes, specifically the metabolically active depots (SubQ, Gonadal, and BAT) and dWAT (dermal) was stimulated (292).
An in vivo gene therapy study demonstrated that delivering the human BSCL2 gene using an adeno-associated virus (AAV) in seipin knockout mice resulted in the restoration of adipose tissue, and this led to a significant improvement in metabolic disease (293). Following AAV-mediated gene therapy, it has been documented that there was a reduction in serum triglyceride levels, an improvement in insulin sensitivity and glucose tolerance, and a partial enhancement in the development of visceral WAT depots in seipin knockout mice. However, a dramatic increase in circulating leptin and adiponectin levels could not be achieved. Nevertheless, this study suggests a potential therapeutic approach for treating metabolic conditions related to CGL2 and other lipodystrophy syndromes (293).
Introducing a functional copy of the defective gene associated with the condition or modulating the expression of related genes to restore normal adipose tissue function holds promise for alleviating severe metabolic complications in patients with lipodystrophy. CrispR-cas editing of single nucleotide variants is also rapidly approaching clinical use in all rare diseases. However, further studies are required to move gene therapies from the experimental stage to clinical applications.
CONCLUSION
Lipodystrophy syndromes are a group of fascinating diseases that are caused by mechanisms that disrupt predominantly adipocyte differentiation or lipid droplet formation. LMNA gene defects, the most common single gene defects leading to the development of lipodystrophy syndromes, leads to lipodystrophy possibly due to inducing adipocyte apoptosis or death, but more work is needed on this front. Regardless of the mechanism and whether the diseases present with generalized or partial fat loss, common metabolic complications include severe insulin resistance, hypertriglyceridemia, and ectopic fat deposition, especially hepatic steatosis. This common theme is recapitulated in numerous animal models as well. The diseases are typically progressive and lead to multi-organ involvement and increased mortality. Molecular advances in the understanding of disease mechanisms may lead to better and specific treatments for lipodystrophy syndromes. So far, the most exciting therapeutic development for the treatment of lipodystrophy syndromes has been the approval of leptin replacement therapy for generalized lipodystrophy in the form of metreleptin which started a new era in lipodystrophy research; leading to the launch of registries and natural history studies (e.g. the LD Lync Study-Natural History Study of Lipodystrophy Syndromes (NCT03087253) (294-296), organization of research consortia around the world (e.g., European Consortium of Lipodystrophies (ECLip) (297) and country specific patient advocacy foundations or organizations. All these efforts will contribute to discovery of new disease forms and disease mechanisms, understanding of the natural course of lipodystrophy diseases, development of improved treatment options and possibly cures.
REFERENCES
- 1.
- Chan JL, Oral EA. Clinical classification and treatment of congenital and acquired lipodystrophy. Endocr Pract. 2010;16(2):310-23. [PubMed: 20061300]
- 2.
- Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J Clin Endocrinol Metab. 2011;96(11):3313-25. [PMC free article: PMC7673254] [PubMed: 21865368]
- 3.
- Brown RJ, Araujo-Vilar D, Cheung PT, Dunger D, Garg A, Jack M, et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J Clin Endocrinol Metab. 2016;101(12):4500-11. [PMC free article: PMC5155679] [PubMed: 27710244]
- 4.
- Handelsman Y, Oral EA, Bloomgarden ZT, Brown RJ, Chan JL, Einhorn D, et al. The clinical approach to the detection of lipodystrophy - an AACE consensus statement. Endocr Pract. 2013;19(1):107-16. [PMC free article: PMC4108221] [PubMed: 23435042]
- 5.
- Robbins DC, Sims EA. Recurrent ketoacidosis in acquired, total lipodystrophy (lipoatrophic diabetes). Diabetes Care. 1984;7(4):381-5. [PubMed: 6432502]
- 6.
- Jackson SN, Howlett TA, McNally PG, O'Rahilly S, Trembath RC. Dunnigan-Kobberling syndrome: an autosomal dominant form of partial lipodystrophy. QJM. 1997;90(1):27-36. [PubMed: 9093586]
- 7.
- Lupsa BC, Sachdev V, Lungu AO, Rosing DR, Gorden P. Cardiomyopathy in congenital and acquired generalized lipodystrophy: a clinical assessment. Medicine (Baltimore). 2010;89(4):245-50. [PMC free article: PMC3090142] [PubMed: 20616664]
- 8.
- Bjornstad PG, Semb BK, Trygstad O, Seip M. Echocardiographic assessment of cardiac function and morphology in patients with generalised lipodystrophy. Eur J Pediatr. 1985;144(4):355-9. [PubMed: 2934255]
- 9.
- Misra A, Garg A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: case reports and review of the literature. Medicine (Baltimore). 2003;82(2):129-46. [PubMed: 12640189]
- 10.
- Seip M. Generalized lipodystrophy. Ergeb Inn Med Kinderheilkd. 1971;31:59-95. [PubMed: 4933543]
- 11.
- Lima JG, Nobrega LHC, Lima NN, Dos Santos MCF, Silva PHD, Baracho MdFP, et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PLoS One. 2018;13(6):e0199052. [PMC free article: PMC5993255] [PubMed: 29883474]
- 12.
- Ajluni N, Meral R, Neidert AH, Brady GF, Buras E, McKenna B, et al. Spectrum of disease associated with partial lipodystrophy: lessons from a trial cohort. Clin Endocrinol (Oxf). 2017;86(5):698-707. [PMC free article: PMC5395301] [PubMed: 28199729]
- 13.
- Ajluni N, Dar M, Xu J, Neidert AH, Oral EA. Efficacy and Safety of Metreleptin in Patients with Partial Lipodystrophy: Lessons from an Expanded Access Program. J Diabetes Metab. 2016;7(3). [PMC free article: PMC5026130] [PubMed: 27642538]
- 14.
- Garg A, Misra A. Lipodystrophies: rare disorders causing metabolic syndrome. Endocrinol Metab Clin North Am. 2004;33(2):305-31. [PubMed: 15158521]
- 15.
- Garg A. Acquired and inherited lipodystrophies. N Engl J Med. 2004;350(12):1220-34. [PubMed: 15028826]
- 16.
- Caron A, Lee S, Elmquist JK, Gautron L. Leptin and brain-adipose crosstalks. Nat Rev Neurosci. 2018;19(3):153-65. [PMC free article: PMC5962962] [PubMed: 29449715]
- 17.
- Musso C, Cochran E, Moran SA, Skarulis MC, Oral EA, Taylor S, et al. Clinical course of genetic diseases of the insulin receptor (type A and Rabson-Mendenhall syndromes): a 30-year prospective. Medicine (Baltimore). 2004;83(4):209-22. [PubMed: 15232309]
- 18.
- Oral EA, Chan JL. Rationale for leptin-replacement therapy for severe lipodystrophy. Endocr Pract. 2010;16(2):324-33. [PubMed: 20061299]
- 19.
- Melvin A, O'Rahilly S, Savage DB. Genetic syndromes of severe insulin resistance. Curr Opin Genet Dev. 2018;50:60-7. [PubMed: 29477938]
- 20.
- Lee Y, Ravazzola M, Park B-H, Bashmakov YK, Orci L, Unger RH. Metabolic mechanisms of failure of intraportally transplanted pancreatic β-cells in rats: role of lipotoxicity and prevention by leptin. Diabetes. 2007;56(9):2295-301. [PubMed: 17563069]
- 21.
- Morioka T, Asilmaz E, Hu J, Dishinger JF, Kurpad AJ, Elias CF, et al. Disruption of leptin receptor expression in the pancreas directly affects β cell growth and function in mice. The Journal of clinical investigation. 2007;117(10):2860-8. [PMC free article: PMC1994606] [PubMed: 17909627]
- 22.
- D'Souza A M, Neumann UH, Glavas MM, Kieffer TJ. The glucoregulatory actions of leptin. Mol Metab. 2017;6(9):1052-65. [PMC free article: PMC5605734] [PubMed: 28951828]
- 23.
- Triantafyllou GA, Paschou SA, Mantzoros CS. Leptin and Hormones: Energy Homeostasis. Endocrinol Metab Clin North Am. 2016;45(3):633-45. [PubMed: 27519135]
- 24.
- Meek TH, Morton GJ. The role of leptin in diabetes: metabolic effects. Diabetologia. 2016;59(5):928-32. [PubMed: 26969486]
- 25.
- Goodpaster BH. Measuring body fat distribution and content in humans. Curr Opin Clin Nutr Metab Care. 2002;5(5):481-7. [PubMed: 12172470]
- 26.
- Anvery N, Wan HT, Dirr MA, Christensen RE, Weil A, Raja S, et al. Utility of high‐resolution ultrasound in measuring subcutaneous fat thickness. Lasers in Surgery and Medicine. 2022;54(9):1189-97. [PubMed: 36183386]
- 27.
- Meral R, Ryan BJ, Malandrino N, Jalal A, Neidert AH, Muniyappa R, et al. “Fat shadows” from DXA for the qualitative assessment of lipodystrophy: when a picture is worth a thousand numbers. Diabetes Care. 2018;41(10):2255-8. [PMC free article: PMC6150431] [PubMed: 30237235]
- 28.
- Vasandani C, Li X, Sekizkardes H, Adams-Huet B, Brown RJ, Garg A. Diagnostic value of anthropometric measurements for familial partial lipodystrophy, Dunnigan variety. The Journal of Clinical Endocrinology & Metabolism. 2020;105(7):2132-41. [PMC free article: PMC7202860] [PubMed: 32193531]
- 29.
- Adiyaman SC, Altay C, Kamisli BY, Avci ER, Basara I, Simsir IY, et al. Pelvis magnetic resonance imaging to diagnose familial partial lipodystrophy. The Journal of Clinical Endocrinology & Metabolism. 2023:dgad063. [PubMed: 36808247]
- 30.
- Araújo-Vilar D, Fernández-Pombo A, Rodríguez-Carnero G, Martínez-Olmos MÁ, Cantón A, Villar-Taibo R, et al. LipoDDx: A mobile application for identification of rare lipodystrophy syndromes. Orphanet Journal of Rare Diseases. 2020;15(1):1-9. [PMC free article: PMC7118879] [PubMed: 32241282]
- 31.
- da Cunha Olegario NB, da Cunha Neto JS, Barbosa PCS, Pinheiro PR, Landim PLA, Montenegro APDR, et al. Identifying congenital generalized lipodystrophy using deep learning-DEEPLIPO. Scientific Reports. 2023;13(1):2176. [PMC free article: PMC9905595] [PubMed: 36750605]
- 32.
- Haque WA, Shimomura I, Matsuzawa Y, Garg A. Serum adiponectin and leptin levels in patients with lipodystrophies. J Clin Endocrinol Metab. 2002;87(5):2395. [PubMed: 11994394]
- 33.
- Rebecca J. Brown M, Baris Akinci, MD, Matheos Yosef, PhD, Robert Alexander Hegele, MD, FACP, FRCPC, David Araujo-Vilar, MD, PhD, Martin JF Wabitsch, MD, PhD, Helen Phillips, MB ChB, FFPM, Shokoufeh Khalatbari, PhD, Ekaterina Sorkina, MD, Ferruccio Santini, MD, Corinne Vigouroux, MD, Lipodystrophy Severity Score Study Group, Elif A. Oral, MD. SUN-787 - Lipodystrophy Severity Score: Validation Of A Tool To Assess Disease Burden In Lipodystrophy. [Abstract]. In press 2024.
- 34.
- Araújo-Vilar D, Fernández-Pombo A, Cobelo-Gómez S, Castro AI, Sánchez-Iglesias S. Lipodystrophy-associated progeroid syndromes. Hormones. 2022;21(4):555-71. [PubMed: 35835948]
- 35.
- Kornak U, Saha N, Keren B, Neumann A, Taylor Tavares AL, Piard J, et al. Alternative splicing of BUD13 determines the severity of a developmental disorder with lipodystrophy and progeroid features. Genet Med. 2022;24(9):1927-40. [PubMed: 35670808]
- 36.
- Gautheron J, Morisseau C, Chung WK, Zammouri J, Auclair M, Baujat G, et al. EPHX1 mutations cause a lipoatrophic diabetes syndrome due to impaired epoxide hydrolysis and increased cellular senescence. Elife. 2021;10. [PMC free article: PMC8331186] [PubMed: 34342583]
- 37.
- Bourne SC, Townsend KN, Shyr C, Matthews A, Lear SA, Attariwala R, et al. Optic atrophy, cataracts, lipodystrophy/lipoatrophy, and peripheral neuropathy caused by a de novo OPA3 mutation. Cold Spring Harb Mol Case Stud. 2017;3(1):a001156. [PMC free article: PMC5171695] [PubMed: 28050599]
- 38.
- Bredrup C, Stokowy T, McGaughran J, Lee S, Sapkota D, Cristea I, et al. A tyrosine kinase-activating variant Asn666Ser in PDGFRB causes a progeria-like condition in the severe end of Penttinen syndrome. European Journal of Human Genetics. 2019;27(4):574-81. [PMC free article: PMC6460636] [PubMed: 30573803]
- 39.
- Writzl K, Maver A, Kovačič L, Martinez-Valero P, Contreras L, Satrustegui J, et al. De novo mutations in SLC25A24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. The American Journal of Human Genetics. 2017;101(5):844-55. [PMC free article: PMC5673633] [PubMed: 29100094]
- 40.
- Kopp J, Koch LA, Lyubenova H, Küchler O, Holtgrewe M, Ivanov A, et al. Loss-of-function variants affecting the STAGA complex component SUPT7L cause a developmental disorder with generalized lipodystrophy. Human Genetics. 2024:1-12. [PMC free article: PMC11098864] [PubMed: 38592547]
- 41.
- Seip M. Lipodystrophy and gigantism with associated endocrine manifestations. A new diencephalic syndrome? Acta Paediatr. 1959;48:555-74. [PubMed: 14444642]
- 42.
- Berardinelli W. An undiagnosed endocrinometabolic syndrome: report of 2 cases. J Clin Endocrinol Metab. 1954;14(2):193-204. [PubMed: 13130666]
- 43.
- Tsoukas MA MC. Endocrinology Adult and Pediatric. In: Jameson JL DL, editor. 7 ed: Saunders, In Press.
- 44.
- Agarwal AK, Simha V, Oral EA, Moran SA, Gorden P, O'Rahilly S, et al. Phenotypic and genetic heterogeneity in congenital generalized lipodystrophy. J Clin Endocrinol Metab. 2003;88(10):4840-7. [PubMed: 14557463]
- 45.
- Van Maldergem L, Magre J, Khallouf TE, Gedde-Dahl T, Jr., Delepine M, Trygstad O, et al. Genotype-phenotype relationships in Berardinelli-Seip congenital lipodystrophy. J Med Genet. 2002;39(10):722-33. [PMC free article: PMC1734991] [PubMed: 12362029]
- 46.
- Capeau J, Magre J, Caron-Debarle M, Lagathu C, Antoine B, Bereziat V, et al. Human lipodystrophies: genetic and acquired diseases of adipose tissue. Endocr Dev. 2010;19:1-20. [PMC free article: PMC3892722] [PubMed: 20551664]
- 47.
- Maguire M, Lungu A, Gorden P, Cochran E, Stratton P. Pregnancy in a woman with congenital generalized lipodystrophy: leptin's vital role in reproduction. Obstet Gynecol. 2012;119(2 Pt 2):452-5. [PMC free article: PMC3470464] [PubMed: 22270436]
- 48.
- Patni N, Garg A. Congenital generalized lipodystrophies--new insights into metabolic dysfunction. Nat Rev Endocrinol. 2015;11(9):522-34. [PMC free article: PMC7605893] [PubMed: 26239609]
- 49.
- Yildirim Simsir I, Tuysuz B, Ozbek MN, Tanrikulu S, Celik Guler M, Karhan AN, et al. Clinical features of generalized lipodystrophy in Turkey: A cohort analysis. Diabetes, Obesity and Metabolism. 2023;25(7):1950-63. [PubMed: 36946378]
- 50.
- Saydam O, Ozgen Saydam B, Adiyaman CS, Sonmez Ince M, Eren MA, Keskin FE, et al. Diabetic foot ulcers: a neglected complication of lipodystrophy. Diabetes care. 2020;43(10):e149-e51. [PubMed: 32801128]
- 51.
- Saydam O, Ozgen Saydam B, Adiyaman SC, Sonmez Ince M, Eren MA, Keskin FE, et al. Risk factors for diabetic foot ulcers in metreleptin naïve patients with lipodystrophy. Clinical Diabetes and Endocrinology. 2021;7(1):1-10. [PMC free article: PMC8485489] [PubMed: 34593051]
- 52.
- Akinci B, Oral EA, Neidert A, Rus D, Cheng WY, Thompson-Leduc P, et al. Comorbidities and survival in patients with lipodystrophy: an international chart review study. The Journal of Clinical Endocrinology & Metabolism. 2019;104(11):5120-35. [PMC free article: PMC6760298] [PubMed: 31314093]
- 53.
- Garg A, Wilson R, Barnes R, Arioglu E, Zaidi Z, Gurakan F, et al. A gene for congenital generalized lipodystrophy maps to human chromosome 9q34. J Clin Endocrinol Metab. 1999;84(9):3390-4. [PubMed: 10487716]
- 54.
- Fernandez-Galilea M, Tapia P, Cautivo K, Morselli E, Cortes VA. AGPAT2 deficiency impairs adipogenic differentiation in primary cultured preadipocytes in a non-autophagy or apoptosis dependent mechanism. Biochem Biophys Res Commun. 2015;467(1):39-45. [PubMed: 26417690]
- 55.
- Subauste AR, Das AK, Li X, Elliott BG, Evans C, El Azzouny M, et al. Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes. 2012;61(11):2922-31. [PMC free article: PMC3478532] [PubMed: 22872237]
- 56.
- Sakuma I, Gaspar RC, Luukkonen PK, Kahn M, Zhang D, Zhang X, et al. Lysophosphatidic acid triggers inflammation in the liver and white adipose tissue in rat models of 1-acyl-sn-glycerol-3-phosphate acyltransferase 2 deficiency and overnutrition. Proceedings of the National Academy of Sciences. 2023;120(52):e2312666120. [PMC free article: PMC10756285] [PubMed: 38127985]
- 57.
- Simha V, Agarwal AK, Aronin PA, Iannaccone ST, Garg A. Novel subtype of congenital generalized lipodystrophy associated with muscular weakness and cervical spine instability. Am J Med Genet A. 2008;146A(18):2318-26. [PMC free article: PMC2716114] [PubMed: 18698612]
- 58.
- Garg A, Fleckenstein JL, Peshock RM, Grundy SM. Peculiar distribution of adipose tissue in patients with congenital generalized lipodystrophy. J Clin Endocrinol Metab. 1992;75(2):358-61. [PubMed: 1639935]
- 59.
- Simha V, Garg A. Phenotypic heterogeneity in body fat distribution in patients with congenital generalized lipodystrophy caused by mutations in the AGPAT2 or seipin genes. J Clin Endocrinol Metab. 2003;88(11):5433-7. [PubMed: 14602785]
- 60.
- Magre J, Delepine M, Khallouf E, Gedde-Dahl T, Jr., Van Maldergem L, Sobel E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat Genet. 2001;28(4):365-70. [PubMed: 11479539]
- 61.
- Cartwright BR, Goodman JM. Seipin: from human disease to molecular mechanism. J Lipid Res. 2012;53(6):1042-55. [PMC free article: PMC3351812] [PubMed: 22474068]
- 62.
- Cartwright BR, Binns DD, Hilton CL, Han S, Gao Q, Goodman JM. Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology. Mol Biol Cell. 2015;26(4):726-39. [PMC free article: PMC4325842] [PubMed: 25540432]
- 63.
- Akinci B, Onay H, Demir T, Ozen S, Kayserili H, Akinci G, et al. Natural History of Congenital Generalized Lipodystrophy: A Nationwide Study From Turkey. J Clin Endocrinol Metab. 2016;101(7):2759-67. [PMC free article: PMC7958923] [PubMed: 27144933]
- 64.
- Altay C, Secil M, Demir T, Atik T, Akinci G, Kutbay NO, et al. Determining residual adipose tissue characteristics with MRI in patients with various subtypes of lipodystrophy. Diagn Interv Radiol. 2017;23(6):428-34. [PMC free article: PMC5669542] [PubMed: 29044029]
- 65.
- Antuna-Puente B, Boutet E, Vigouroux C, Lascols O, Slama L, Caron-Debarle M, et al. Higher Adiponectin Levels in Patients with Berardinelli-Seip Congenital Lipodystrophy due to Seipin as compared with 1-Acylglycerol-3-Phosphate-O-Acyltransferase-2 Deficiency. J Clin Endocr Metab. 2010;95(3):1463-8. [PubMed: 20097706]
- 66.
- Ruiz-Riquelme A, Sanchez-Iglesias S, Rabano A, Guillen-Navarro E, Domingo-Jimenez R, Ramos A, et al. Larger aggregates of mutant seipin in Celia's Encephalopathy, a new protein misfolding neurodegenerative disease. Neurobiol Dis. 2015;83:44-53. [PubMed: 26282322]
- 67.
- Sanchez-Iglesias S, Unruh-Pinheiro A, Guillin-Amarelle C, Gonzalez-Mendez B, Ruiz-Riquelme A, Rodriguez-Canete BL, et al. Skipped BSCL2 Transcript in Celia's Encephalopathy (PELD): New Insights on Fatty Acids Involvement, Senescence and Adipogenesis. PLoS One. 2016;11(7):e0158874. [PMC free article: PMC4938205] [PubMed: 27391332]
- 68.
- Hsu RH, Lin WD, Chao MC, Hsiao HP, Wong SL, Chiu PC, et al. Congenital generalized lipodystrophy in Taiwan. J Formos Med Assoc. 2019;118(1 Pt 1):142-7. [PubMed: 29478747]
- 69.
- Kim CA, Delepine M, Boutet E, El Mourabit H, Le Lay S, Meier M, et al. Association of a homozygous nonsense caveolin-1 mutation with Berardinelli-Seip congenital lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1129-34. [PubMed: 18211975]
- 70.
- Garg A, Agarwal AK. Caveolin-1: a new locus for human lipodystrophy. J Clin Endocrinol Metab. 2008;93(4):1183-5. [PMC free article: PMC2291498] [PubMed: 18390817]
- 71.
- Garg A, Kircher M, Del Campo M, Amato RS, Agarwal AK, University of Washington Center for Mendelian G. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A. 2015;167A(8):1796-806. [PMC free article: PMC5086082] [PubMed: 25898808]
- 72.
- Lim K, Haider A, Adams C, Sleigh A, Savage DB. Lipodistrophy: a paradigm for understanding the consequences of “overloading” adipose tissue. Physiological Reviews. 2021;101(3):907-93. [PubMed: 33356916]
- 73.
- Cao H, Alston L, Ruschman J, Hegele RA. Heterozygous CAV1 frameshift mutations (MIM 601047) in patients with atypical partial lipodystrophy and hypertriglyceridemia. Lipids Health Dis. 2008;7:3. [PMC free article: PMC2276215] [PubMed: 18237401]
- 74.
- Karhan AN, Zammouri J, Auclair M, Capel E, Apaydin FD, Ates F, et al. Biallelic CAV1 null variants induce congenital generalized lipodystrophy with achalasia. European Journal of Endocrinology. 2021;185(6):841-54. [PubMed: 34643546]
- 75.
- Hayashi YK, Matsuda C, Ogawa M, Goto K, Tominaga K, Mitsuhashi S, et al. Human PTRF mutations cause secondary deficiency of caveolins resulting in muscular dystrophy with generalized lipodystrophy. J Clin Invest. 2009;119(9):2623-33. [PMC free article: PMC2735915] [PubMed: 19726876]
- 76.
- Mancioppi V, Daffara T, Romanisio M, Ceccarini G, Pelosini C, Santini F, et al. A new mutation in the CAVIN1/PTRF gene in two siblings with congenital generalized lipodystrophy type 4: case reports and review of the literature. Frontiers in Endocrinology. 2023;14. [PMC free article: PMC10369054] [PubMed: 37501786]
- 77.
- Rajab A, Straub V, McCann LJ, Seelow D, Varon R, Barresi R, et al. Fatal cardiac arrhythmia and long-QT syndrome in a new form of congenital generalized lipodystrophy with muscle rippling (CGL4) due to PTRF-CAVIN mutations. PLoS Genet. 2010;6(3):e1000874. [PMC free article: PMC2837386] [PubMed: 20300641]
- 78.
- Shastry S, Delgado MR, Dirik E, Turkmen M, Agarwal AK, Garg A. Congenital generalized lipodystrophy, type 4 (CGL4) associated with myopathy due to novel PTRF mutations. Am J Med Genet A. 2010;152A(9):2245-53. [PMC free article: PMC2930069] [PubMed: 20684003]
- 79.
- Akinci G, Topaloglu H, Akinci B, Onay H, Karadeniz C, Ergul Y, et al. Spectrum of clinical manifestations in two young Turkish patients with congenital generalized lipodystrophy type 4. Eur J Med Genet. 2016;59(6-7):320-4. [PMC free article: PMC7605892] [PubMed: 27167729]
- 80.
- Payne F, Lim K, Girousse A, Brown RJ, Kory N, Robbins A, et al. Mutations disrupting the Kennedy phosphatidylcholine pathway in humans with congenital lipodystrophy and fatty liver disease. Proc Natl Acad Sci U S A. 2014;111(24):8901-6. [PMC free article: PMC4066527] [PubMed: 24889630]
- 81.
- Hussain I, Patni N, Ueda M, Sorkina E, Valerio CM, Cochran E, et al. A Novel Generalized Lipodystrophy-associated Progeroid Syndrome due to recurrent heterozygous LMNA p.T10I Mutation. J Clin Endocrinol Metab. 2017. [PMC free article: PMC6283411] [PubMed: 29267953]
- 82.
- Dyment DA, Gibson WT, Huang L, Bassyouni H, Hegele RA, Innes AM. Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli-Seip syndrome. Eur J Med Genet. 2014;57(9):524-6. [PubMed: 24980513]
- 83.
- Schuermans N, El Chehadeh S, Hemelsoet D, Gautheron J, Vantyghem M-C, Nouioua S, et al. Loss of phospholipase PLAAT3 causes a mixed lipodystrophic and neurological syndrome due to impaired PPARγ signaling. Nature genetics. 2023;55(11):1929-40. [PubMed: 37919452]
- 84.
- Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical course of the syndrome of autoantibodies to the insulin receptor (type B insulin resistance): a 28-year perspective. Medicine (Baltimore). 2002;81(2):87-100. [PubMed: 11889410]
- 85.
- Pope E, Janson A, Khambalia A, Feldman B. Childhood acquired lipodystrophy: a retrospective study. J Am Acad Dermatol. 2006;55(6):947-50. [PubMed: 17097389]
- 86.
- Bingham A, Mamyrova G, Rother KI, Oral E, Cochran E, Premkumar A, et al. Predictors of acquired lipodystrophy in juvenile-onset dermatomyositis and a gradient of severity. Medicine (Baltimore). 2008;87(2):70-86. [PMC free article: PMC2674585] [PubMed: 18344805]
- 87.
- Savage DB. Perilipin 1 Antibodies in Patients With Acquired Generalized Lipodystrophy. Diabetes. 2023;72(1):16-8. [PubMed: 36538601]
- 88.
- Mandel-Brehm C, Vazquez SE, Liverman C, Cheng M, Quandt Z, Kung AF, et al. Autoantibodies to perilipin-1 define a subset of acquired generalized lipodystrophy. Diabetes. 2023;72(1):59-70. [PMC free article: PMC9797316] [PubMed: 35709010]
- 89.
- Corvillo F, Abel BS, López-Lera A, Ceccarini G, Magno S, Santini F, et al. Characterization and clinical association of autoantibodies against perilipin 1 in patients with acquired generalized lipodystrophy. Diabetes. 2023;72(1):71-84. [PMC free article: PMC9797321] [PubMed: 35771980]
- 90.
- Corvillo F, Aparicio V, López-Lera A, Garrido S, Araújo-Vilar D, De Miguel MP, et al. Autoantibodies against perilipin 1 as a cause of acquired generalized lipodystrophy. Frontiers in immunology. 2018;9:2142. [PMC free article: PMC6156147] [PubMed: 30283460]
- 91.
- Verma S, Singh S, Bhalla AK, Khullar M. Study of subcutaneous fat in children with juvenile dermatomyositis. Arthritis Rheum. 2006;55(4):564-8. [PubMed: 16874777]
- 92.
- Billings JK, Milgraum SS, Gupta AK, Headington JT, Rasmussen JE. Lipoatrophic panniculitis: a possible autoimmune inflammatory disease of fat. Report of three cases. Arch Dermatol. 1987;123(12):1662-6. [PubMed: 3688906]
- 93.
- Eren E, Ozkan TB, Cakir ED, Saglam H, Tarim O. Acquired generalized lipodystrophy associated with autoimmune hepatitis and low serum C4 level. J Clin Res Pediatr Endocrinol. 2010;2(1):39-42. [PMC free article: PMC3005665] [PubMed: 21274335]
- 94.
- Gnanendran SS, Miller JA, Archer CA, Jain SV, Hwang SJ, Peters G, et al. Acquired lipodystrophy associated with immune checkpoint inhibitors. Melanoma research. 2020;30(6):599-602. [PubMed: 32141964]
- 95.
- Jehl A, Cugnet-Anceau C, Vigouroux C, Legeay AL, Dalle S, Harou O, et al. Acquired generalized lipodystrophy: a new cause of anti-PD-1 immune-related diabetes. Diabetes care. 2019;42(10):2008-10. [PubMed: 31434650]
- 96.
- Unal MC, Semiz GG, Ozdogan O, Altay C, Yildirim EC, Semiz HS, et al. Nivolumab Associated Endocrine Abnormalities: Challenging Cases from a Reference Clinic. Acta Endocrinol (Buchar). 2022;18(4):516-22. [PMC free article: PMC10162826] [PubMed: 37152877]
- 97.
- Guillin-Amarelle C, Sanchez-Iglesias S, Castro-Pais A, Rodriguez-Canete L, Ordonez-Mayan L, Pazos M, et al. Type 1 familial partial lipodystrophy: understanding the Kobberling syndrome. Endocrine. 2016;54(2):411-21. [PubMed: 27473102]
- 98.
- Herbst KL, Tannock LR, Deeb SS, Purnell JQ, Brunzell JD, Chait A. Kobberling type of familial partial lipodystrophy: an underrecognized syndrome. Diabetes Care. 2003;26(6):1819-24. [PubMed: 12766116]
- 99.
- Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17-26. [PMC free article: PMC5774584] [PubMed: 27841877]
- 100.
- Akinci B, von Schnurbein J, Araujo-Vilar D, Wabitsch M, Oral EA. Lipodystrophy Prevalence, "Lipodystrophy-Like Phenotypes," and Diagnostic Challenges. Diabetes. 2024;73(7):1039-42. [PubMed: 38900954]
- 101.
- Hegele RA, Joy TR, Al-Attar SA, Rutt BK. Thematic review series: Adipocyte Biology. Lipodystrophies: windows on adipose biology and metabolism. J Lipid Res. 2007;48(7):1433-44. [PubMed: 17374881]
- 102.
- Agarwal AK, Barnes RI, Garg A. Genetic basis of congenital generalized lipodystrophy. Int J Obes Relat Metab Disord. 2004;28(2):336-9. [PubMed: 14557833]
- 103.
- Garg A. Gender differences in the prevalence of metabolic complications in familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. 2000;85(5):1776-82. [PubMed: 10843151]
- 104.
- Akinci B, Onay H, Demir T, Savas-Erdeve S, Gen R, Simsir IY, et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism. 2017;72:109-19. [PubMed: 28641778]
- 105.
- Eldin AJ, Akinci B, da Rocha AM, Meral R, Simsir IY, Adiyaman SC, et al. Cardiac phenotype in familial partial lipodystrophy. Clinical endocrinology. 2021;94(6):1043-53. [PMC free article: PMC9003538] [PubMed: 33502018]
- 106.
- Besci O, Foss de Freitas MC, Guidorizzi NR, Guler MC, Gilio D, Maung JN, et al. Deciphering the clinical presentations in LMNA-related lipodystrophy: report of 115 cases and a systematic review. The Journal of Clinical Endocrinology & Metabolism. 2024;109(3):e1204-e24. [PMC free article: PMC10876415] [PubMed: 37843397]
- 107.
- Garg A, Vinaitheerthan M, Weatherall PT, Bowcock AM. Phenotypic heterogeneity in patients with familial partial lipodystrophy (dunnigan variety) related to the site of missense mutations in lamin a/c gene. J Clin Endocrinol Metab. 2001;86(1):59-65. [PubMed: 11231979]
- 108.
- Fountas A, Giotaki Z, Dounousi E, Liapis G, Bargiota A, Tsatsoulis A, et al. Familial partial lipodystrophy and proteinuric renal disease due to a missense c.1045C > T LMNA mutation. Endocrinol Diabetes Metab Case Rep. 2017;2017. [PMC free article: PMC5467650] [PubMed: 28620495]
- 109.
- Mory PB, Crispim F, Freire MB, Salles JE, Valerio CM, Godoy-Matos AF, et al. Phenotypic diversity in patients with lipodystrophy associated with LMNA mutations. Eur J Endocrinol. 2012;167(3):423-31. [PubMed: 22700598]
- 110.
- Zhong ZX, Harris J, Wilber E, Gorman S, Savage DB, O'Rahilly S, et al. Describing the natural history of clinical, biochemical and radiological outcomes of children with familial partial lipodystrophy type 2 (FPLD2) from the United Kingdom: A retrospective case series. Clinical Endocrinology. 2022;97(6):755-62. [PMC free article: PMC9804585] [PubMed: 35920656]
- 111.
- Monajemi H, Zhang L, Li G, Jeninga EH, Cao H, Maas M, et al. Familial partial lipodystrophy phenotype resulting from a single-base mutation in deoxyribonucleic acid-binding domain of peroxisome proliferator-activated receptor-gamma. J Clin Endocrinol Metab. 2007;92(5):1606-12. [PubMed: 17299075]
- 112.
- Ludtke A, Buettner J, Wu W, Muchir A, Schroeter A, Zinn-Justin S, et al. Peroxisome proliferator-activated receptor-gamma C190S mutation causes partial lipodystrophy. J Clin Endocrinol Metab. 2007;92(6):2248-55. [PubMed: 17356052]
- 113.
- Al-Shali K, Cao H, Knoers N, Hermus AR, Tack CJ, Hegele RA. A single-base mutation in the peroxisome proliferator-activated receptor gamma4 promoter associated with altered in vitro expression and partial lipodystrophy. J Clin Endocrinol Metab. 2004;89(11):5655-60. [PubMed: 15531525]
- 114.
- Agarwal AK, Garg A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab. 2002;87(1):408-11. [PubMed: 11788685]
- 115.
- Demir T, Onay H, Savage DB, Temeloglu E, Uzum AK, Kadioglu P, et al. Familial partial lipodystrophy linked to a novel peroxisome proliferator activator receptor -gamma (PPARG) mutation, H449L: a comparison of people with this mutation and those with classic codon 482 Lamin A/C (LMNA) mutations. Diabet Med. 2016;33(10):1445-50. [PubMed: 26756202]
- 116.
- Vasandani C, Li X, Sekizkardes H, Brown RJ, Garg A. Phenotypic differences among familial partial lipodystrophy due to LMNA or PPARG variants. Journal of the Endocrine Society. 2022;6(12):bvac155. [PMC free article: PMC9664976] [PubMed: 36397776]
- 117.
- Gandotra S, Lim K, Girousse A, Saudek V, O'Rahilly S, Savage DB. Human frame shift mutations affecting the carboxyl terminus of perilipin increase lipolysis by failing to sequester the adipose triglyceride lipase (ATGL) coactivator AB-hydrolase-containing 5 (ABHD5). J Biol Chem. 2011;286(40):34998-5006. [PMC free article: PMC3186430] [PubMed: 21757733]
- 118.
- Laver TW, Patel KA, Colclough K, Curran J, Dale J, Davis N, et al. PLIN1 haploinsufficiency is not associated with lipodystrophy. The Journal of Clinical Endocrinology & Metabolism. 2018;103(9):3225-30. [PMC free article: PMC6126890] [PubMed: 30020498]
- 119.
- Gandotra S, Le Dour C, Bottomley W, Cervera P, Giral P, Reznik Y, et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N Engl J Med. 2011;364(8):740-8. [PMC free article: PMC3773916] [PubMed: 21345103]
- 120.
- Kozusko K, Tsang V, Bottomley W, Cho YH, Gandotra S, Mimmack ML, et al. Clinical and molecular characterization of a novel PLIN1 frameshift mutation identified in patients with familial partial lipodystrophy. Diabetes. 2015;64(1):299-310. [PMC free article: PMC4361744] [PubMed: 25114292]
- 121.
- Le Lay S, Magré J, Prieur X. Not enough fat: mouse models of inherited lipodystrophy. Frontiers in Endocrinology. 2022;13:785819. [PMC free article: PMC8895270] [PubMed: 35250856]
- 122.
- Rubio-Cabezas O, Puri V, Murano I, Saudek V, Semple RK, Dash S, et al. Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1(5):280-7. [PMC free article: PMC2891108] [PubMed: 20049731]
- 123.
- Farhan SM, Robinson JF, McIntyre AD, Marrosu MG, Ticca AF, Loddo S, et al. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol. 2014;30(12):1649-54. [PubMed: 25475467]
- 124.
- Zolotov S, Xing C, Mahamid R, Shalata A, Sheikh-Ahmad M, Garg A. Homozygous LIPE mutation in siblings with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy. Am J Med Genet A. 2017;173(1):190-4. [PMC free article: PMC5788284] [PubMed: 27862896]
- 125.
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science. 2004;304(5675):1325-8. [PMC free article: PMC2258004] [PubMed: 15166380]
- 126.
- Tan K, Kimber WA, Luan J, Soos MA, Semple RK, Wareham NJ, et al. Analysis of genetic variation in Akt2/PKB-beta in severe insulin resistance, lipodystrophy, type 2 diabetes, and related metabolic phenotypes. Diabetes. 2007;56(3):714-9. [PMC free article: PMC2672155] [PubMed: 17327441]
- 127.
- 128.
- The rare and atypical diabetes network (RADIANT) study: design and early results. Diabetes Care. 2023;46(6):1265-70. [PMC free article: PMC10234756] [PubMed: 37104866]
- 129.
- 130.
- Garg A, Sankella S, Xing C, Agarwal AK. Whole-exome sequencing identifies ADRA2A mutation in atypical familial partial lipodystrophy. JCI Insight. 2016;1(9). [PMC free article: PMC4927009] [PubMed: 27376152]
- 131.
- Simha V, Garg A. Body fat distribution and metabolic derangements in patients with familial partial lipodystrophy associated with mandibuloacral dysplasia. J Clin Endocrinol Metab. 2002;87(2):776-85. [PubMed: 11836320]
- 132.
- Garavelli L, D'Apice MR, Rivieri F, Bertoli M, Wischmeijer A, Gelmini C, et al. Mandibuloacral dysplasia type A in childhood. Am J Med Genet A. 2009;149A(10):2258-64. [PubMed: 19764019]
- 133.
- Lombardi F, Gullotta F, Columbaro M, Filareto A, D'Adamo M, Vielle A, et al. Compound heterozygosity for mutations in LMNA in a patient with a myopathic and lipodystrophic mandibuloacral dysplasia type A phenotype. J Clin Endocrinol Metab. 2007;92(11):4467-71. [PubMed: 17848409]
- 134.
- Agarwal AK, Zhou XJ, Hall RK, Nicholls K, Bankier A, Van Esch H, et al. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J Investig Med. 2006;54(4):208-13. [PubMed: 17152860]
- 135.
- Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. 2002;71(2):426-31. [PMC free article: PMC379176] [PubMed: 12075506]
- 136.
- Garg A, Cogulu O, Ozkinay F, Onay H, Agarwal AK. A novel homozygous Ala529Val LMNA mutation in Turkish patients with mandibuloacral dysplasia. J Clin Endocrinol Metab. 2005;90(9):5259-64. [PubMed: 15998779]
- 137.
- Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. 2003;12(16):1995-2001. [PubMed: 12913070]
- 138.
- Miyoshi Y, Akagi M, Agarwal AK, Namba N, Kato-Nishimura K, Mohri I, et al. Severe mandibuloacral dysplasia caused by novel compound heterozygous ZMPSTE24 mutations in two Japanese siblings. Clin Genet. 2008;73(6):535-44. [PMC free article: PMC2732118] [PubMed: 18435794]
- 139.
- Peinado JR, Quiros PM, Pulido MR, Marino G, Martinez-Chantar ML, Vazquez-Martinez R, et al. Proteomic profiling of adipose tissue from Zmpste24-/- mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol Cell Proteomics. 2011;10(11):M111 008094. [PMC free article: PMC3226399] [PubMed: 21828285]
- 140.
- Akinci B, Sankella S, Gilpin C, Ozono K, Garg A, Agarwal AK. Progeroid syndrome patients with ZMPSTE24 deficiency could benefit when treated with rapamycin and dimethylsulfoxide. Cold Spring Harb Mol Case Stud. 2017;3(1):a001339. [PMC free article: PMC5171694] [PubMed: 28050601]
- 141.
- Elouej S, Harhouri K, Le Mao M, Baujat G, Nampoothiri S, Kayserili H, et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat Commun. 2020;11(1):4589. [PMC free article: PMC7486921] [PubMed: 32917887]
- 142.
- Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. 2013;45(8):947-50. [PMC free article: PMC3785143] [PubMed: 23770608]
- 143.
- Shastry S, Simha V, Godbole K, Sbraccia P, Melancon S, Yajnik CS, et al. A novel syndrome of mandibular hypoplasia, deafness, and progeroid features associated with lipodystrophy, undescended testes, and male hypogonadism. J Clin Endocrinol Metab. 2010;95(10):E192-7. [PMC free article: PMC3050107] [PubMed: 20631028]
- 144.
- Pelosini C, Martinelli S, Ceccarini G, Magno S, Barone I, Basolo A, et al. Identification of a novel mutation in the polymerase delta 1 (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism. 2014;63(11):1385-9. [PubMed: 25131834]
- 145.
- Elouej S, Beleza-Meireles A, Caswell R, Colclough K, Ellard S, Desvignes JP, et al. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (MDPL). Metabolism. 2017;71:213-25. [PubMed: 28521875]
- 146.
- Donadille B, D'Anella P, Auclair M, Uhrhammer N, Sorel M, Grigorescu R, et al. Partial lipodystrophy with severe insulin resistance and adult progeria Werner syndrome. Orphanet J Rare Dis. 2013;8:106. [PMC free article: PMC3720184] [PubMed: 23849162]
- 147.
- Sidorova JM. Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair (Amst). 2008;7(11):1776-86. [PMC free article: PMC2659608] [PubMed: 18722555]
- 148.
- Atallah I, McCormick D, Good J-M, Barigou M, Fraga M, Sempoux C, et al. Partial lipodystrophy, severe dyslipidaemia and insulin resistant diabetes as early signs of Werner syndrome. Journal of clinical lipidology. 2022;16(5):583-90. [PubMed: 35780059]
- 149.
- Becerra-Munoz VM, Gomez-Doblas JJ, Porras-Martin C, Such-Martinez M, Crespo-Leiro MG, Barriales-Villa R, et al. The importance of genotype-phenotype correlation in the clinical management of Marfan syndrome. Orphanet J Rare Dis. 2018;13(1):16. [PMC free article: PMC5778633] [PubMed: 29357934]
- 150.
- Graul-Neumann LM, Kienitz T, Robinson PN, Baasanjav S, Karow B, Gillessen-Kaesbach G, et al. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3' terminus of the FBN1-gene. Am J Med Genet A. 2010;152A(11):2749-55. [PubMed: 20979188]
- 151.
- Takenouchi T, Hida M, Sakamoto Y, Torii C, Kosaki R, Takahashi T, et al. Severe congenital lipodystrophy and a progeroid appearance: Mutation in the penultimate exon of FBN1 causing a recognizable phenotype. Am J Med Genet A. 2013;161A(12):3057-62. [PubMed: 24039054]
- 152.
- Rautenstrauch T, Snigula F, Wiedemann HR. [Neonatal progeroid syndrome (Wiedemann-Rautenstrauch). A follow-up study]. Klin Padiatr. 1994;206(6):440-3. [PubMed: 7823529]
- 153.
- Davis MR, Arner E, Duffy CR, De Sousa PA, Dahlman I, Arner P, et al. Expression of FBN1 during adipogenesis: Relevance to the lipodystrophy phenotype in Marfan syndrome and related conditions. Mol Genet Metab. 2016;119(1-2):174-85. [PMC free article: PMC5044862] [PubMed: 27386756]
- 154.
- Cabanillas R, Cadinanos J, Villameytide JA, Perez M, Longo J, Richard JM, et al. Nestor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A(11):2617-25. [PubMed: 21932319]
- 155.
- Masotti A, Uva P, Davis-Keppen L, Basel-Vanagaite L, Cohen L, Pisaneschi E, et al. Keppen-Lubinsky syndrome is caused by mutations in the inwardly rectifying K+ channel encoded by KCNJ6. Am J Hum Genet. 2015;96(2):295-300. [PMC free article: PMC4320262] [PubMed: 25620207]
- 156.
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, et al. Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet. 2014;46(11):1239-44. [PMC free article: PMC4343211] [PubMed: 25261934]
- 157.
- Wolthuis DF, Van Asbeck E, Mohamed M, Gardeitchik T, Lim-Melia ER, Wevers RA, et al. Cutis laxa, fat pads and retinopathy due to ALDH18A1 mutation and review of the literature. European journal of paediatric neurology. 2014;18(4):511-5. [PubMed: 24767728]
- 158.
- Chudasama KK, Winnay J, Johansson S, Claudi T, Konig R, Haldorsen I, et al. SHORT syndrome with partial lipodystrophy due to impaired phosphatidylinositol 3 kinase signaling. Am J Hum Genet. 2013;93(1):150-7. [PMC free article: PMC3710758] [PubMed: 23810379]
- 159.
- Thauvin-Robinet C, Auclair M, Duplomb L, Caron-Debarle M, Avila M, St-Onge J, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Genet. 2013;93(1):141-9. [PMC free article: PMC3710759] [PubMed: 23810378]
- 160.
- Huang-Doran I, Tomlinson P, Payne F, Gast A, Sleigh A, Bottomley W, et al. Insulin resistance uncoupled from dyslipidemia due to C-terminal PIK3R1 mutations. JCI Insight. 2016;1(17):e88766. [PMC free article: PMC5070960] [PubMed: 27766312]
- 161.
- Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87(6):866-72. [PMC free article: PMC2997366] [PubMed: 21129723]
- 162.
- Garg A, Hernandez MD, Sousa AB, Subramanyam L, Martinez de Villarreal L, dos Santos HG, et al. An autosomal recessive syndrome of joint contractures, muscular atrophy, microcytic anemia, and panniculitis-associated lipodystrophy. J Clin Endocrinol Metab. 2010;95(9):E58-63. [PMC free article: PMC2936059] [PubMed: 20534754]
- 163.
- Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011;121(10):4150-60. [PMC free article: PMC3195477] [PubMed: 21881205]
- 164.
- Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendinez F, Hernandez A, et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol. 2010;62(3):489-95. [PubMed: 20159315]
- 165.
- Kanazawa N. Nakajo-Nishimura syndrome: an autoinflammatory disorder showing pernio-like rashes and progressive partial lipodystrophy. Allergol Int. 2012;61(2):197-206. [PubMed: 22441638]
- 166.
- Torrelo A. CANDLE Syndrome As a Paradigm of Proteasome-Related Autoinflammation. Front Immunol. 2017;8:927. [PMC free article: PMC5552674] [PubMed: 28848544]
- 167.
- Cavalcante MP, Brunelli JB, Miranda CC, Novak GV, Malle L, Aikawa NE, et al. CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature-a rare case with a novel mutation. Eur J Pediatr. 2016;175(5):735-40. [PubMed: 26567544]
- 168.
- Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A. 2011;108(36):14914-9. [PMC free article: PMC3169106] [PubMed: 21852578]
- 169.
- Pinheiro M, Freire-Maia N, Chautard-Freire-Maia EA, Araujo LM, Liberman B. AREDYLD: a syndrome combining an acrorenal field defect, ectodermal dysplasia, lipoatrophic diabetes, and other manifestations. Am J Med Genet. 1983;16(1):29-33. [PubMed: 6638067]
- 170.
- Breslau-Siderius EJ, Toonstra J, Baart JA, Koppeschaar HP, Maassen JA, Beemer FA. Ectodermal dysplasia, lipoatrophy, diabetes mellitus, and amastia: a second case of the AREDYLD syndrome. Am J Med Genet. 1992;44(3):374-7. [PubMed: 1488989]
- 171.
- Rocha N, Bulger DA, Frontini A, Titheradge H, Gribsholt SB, Knox R, et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife. 2017;6. [PMC free article: PMC5422073] [PubMed: 28414270]
- 172.
- Zammouri J, Vatier C, Capel E, Auclair M, Storey-London C, Bismuth E, et al. Molecular and cellular bases of lipodystrophy syndromes. Frontiers in Endocrinology. 2022;12:803189. [PMC free article: PMC8763341] [PubMed: 35046902]
- 173.
- Capel E, Vatier C, Cervera P, Stojkovic T, Disse E, Cottereau A-S, et al. MFN2-associated lipomatosis: clinical spectrum and impact on adipose tissue. Journal of clinical lipidology. 2018;12(6):1420-35. [PubMed: 30158064]
- 174.
- Misra A, Peethambaram A, Garg A. Clinical features and metabolic and autoimmune derangements in acquired partial lipodystrophy: report of 35 cases and review of the literature. Medicine (Baltimore). 2004;83(1):18-34. [PubMed: 14747765]
- 175.
- Tews D, Schulz A, Denzer C, von Schnurbein J, Ceccarini G, Debatin K-M, et al. Lipodystrophy as a late effect after stem cell transplantation. Journal of Clinical Medicine. 2021;10(8):1559. [PMC free article: PMC8068033] [PubMed: 33917653]
- 176.
- Lorenc A, Hamilton-Shield J, Perry R, Stevens M, Adipose CH, Roche MLEWGSWMFLODMDSSAMH. Body composition after allogeneic haematopoietic cell transplantation/total body irradiation in children and young people: a restricted systematic review. Journal of Cancer Survivorship. 2020;14:624-42. [PMC free article: PMC7473918] [PubMed: 32388841]
- 177.
- Savage DB, Semple RK, Clatworthy MR, Lyons PA, Morgan BP, Cochran EK, et al. Complement abnormalities in acquired lipodystrophy revisited. J Clin Endocrinol Metab. 2009;94(1):10-6. [PubMed: 18854390]
- 178.
- Mathieson PW, Wurzner R, Oliveria DB, Lachmann PJ, Peters DK. Complement-mediated adipocyte lysis by nephritic factor sera. J Exp Med. 1993;177(6):1827-31. [PMC free article: PMC2191037] [PubMed: 8496694]
- 179.
- Akinci B, Unlu SM, Celik A, Simsir IY, Sen S, Nur B, et al. Renal complications of lipodystrophy: A closer look at the natural history of kidney disease. Clinical endocrinology. 2018;89(1):65-75. [PMC free article: PMC5999575] [PubMed: 29722904]
- 180.
- Ozgen Saydam B, Sonmez M, Simsir IY, Erturk MS, Kulaksizoglu M, Arkan T, et al. A subset of patients with acquired partial lipodystrophy developing severe metabolic abnormalities. Endocrine Research. 2019;44(1-2):46-54. [PubMed: 30182761]
- 181.
- Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79(2):383-9. [PMC free article: PMC1559499] [PubMed: 16826530]
- 182.
- Akinci B, Koseoglu FD, Onay H, Yavuz S, Altay C, Simsir IY, et al. Acquired partial lipodystrophy is associated with increased risk for developing metabolic abnormalities. Metabolism. 2015;64(9):1086-95. [PubMed: 26139569]
- 183.
- Savage DB. Mouse models of inherited lipodystrophy. Dis Model Mech. 2009;2(11-12):554-62. [PubMed: 19892886]
- 184.
- Moitra J, Mason MM, Olive M, Krylov D, Gavrilova O, Marcus-Samuels B, et al. Life without white fat: a transgenic mouse. Genes Dev. 1998;12(20):3168-81. [PMC free article: PMC317213] [PubMed: 9784492]
- 185.
- Reitman ML, Gavrilova O. A-ZIP/F-1 mice lacking white fat: a model for understanding lipoatrophic diabetes. Int J Obes Relat Metab Disord. 2000;24 Suppl 4:S11-4. [PubMed: 11126232]
- 186.
- Gavrilova O, Marcus-Samuels B, Graham D, Kim JK, Shulman GI, Castle AL, et al. Surgical implantation of adipose tissue reverses diabetes in lipoatrophic mice. J Clin Invest. 2000;105(3):271-8. [PMC free article: PMC377444] [PubMed: 10675352]
- 187.
- Zhang Z, Turer E, Li X, Zhan X, Choi M, Tang M, et al. Insulin resistance and diabetes caused by genetic or diet-induced KBTBD2 deficiency in mice. Proc Natl Acad Sci U S A. 2016;113(42):E6418-E26. [PMC free article: PMC5081616] [PubMed: 27708159]
- 188.
- Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature. 1999;401(6748):73-6. [PubMed: 10485707]
- 189.
- Colombo C, Cutson JJ, Yamauchi T, Vinson C, Kadowaki T, Gavrilova O, et al. Transplantation of adipose tissue lacking leptin is unable to reverse the metabolic abnormalities associated with lipoatrophy. Diabetes. 2002;51(9):2727-33. [PubMed: 12196465]
- 190.
- Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346(8):570-8. [PubMed: 11856796]
- 191.
- Tapia PJ, Figueroa A-M, Eisner V, González-Hódar L, Robledo F, Agarwal AK, et al. Absence of AGPAT2 impairs brown adipogenesis, increases IFN stimulated gene expression and alters mitochondrial morphology. Metabolism. 2020;111:154341. [PubMed: 32810486]
- 192.
- Rochford JJ. When Adipose Tissue Lets You Down: Understanding the Functions of Genes Disrupted in Lipodystrophy. Diabetes. 2022;71(4):589-98. [PubMed: 35316838]
- 193.
- Gonzalez-Hodar L, McDonald JG, Vale G, Thompson BM, Figueroa A-M, Tapia PJ, et al. Decreased caveolae in AGPAT2 lacking adipocytes is independent of changes in cholesterol or sphingolipid levels: a whole cell and plasma membrane lipidomic analysis of adipogenesis. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2021;1867(9):166167. [PubMed: 33989739]
- 194.
- Corsa CAS, Walsh CM, Bagchi DP, Foss Freitas MC, Li Z, Hardij J, et al. Adipocyte-Specific Deletion of Lamin A/C Largely Models Human Familial Partial Lipodystrophy Type 2. Diabetes. 2021;70(9):1970-84. [PMC free article: PMC8576431] [PubMed: 34088712]
- 195.
- Mcilroy GD, Mitchell SE, Han W, Delibegović M, Rochford JJ. Ablation of Bscl2/seipin in hepatocytes does not cause metabolic dysfunction in congenital generalised lipodystrophy. Disease Models & Mechanisms. 2020;13(1):dmm042655. [PMC free article: PMC6994952] [PubMed: 31848133]
- 196.
- Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nature reviews Molecular cell biology. 2020;21(4):204-24. [PubMed: 32071438]
- 197.
- Mann JP, Duan X, Patel S, Tábara LC, Scurria F, Alvarez-Guaita A, et al. A mouse model of human mitofusin-2-related lipodystrophy exhibits adipose-specific mitochondrial stress and reduced leptin secretion. Elife. 2023;12:e82283. [PMC free article: PMC9937658] [PubMed: 36722855]
- 198.
- Qiu R, Wang S, Lin D, He Y, Huang S, Wu B, et al. Mice harboring a R133L heterozygous mutation in LMNA exhibited ectopic lipid accumulation, aging, and mitochondrial dysfunction in adipose tissue. The FASEB Journal. 2023;37(2):e22730. [PubMed: 36583724]
- 199.
- Song W, Postoak JL, Yang G, Guo X, Pua HH, Bader J, et al. Lipid kinase PIK3C3 maintains healthy brown and white adipose tissues to prevent metabolic diseases. Proceedings of the National Academy of Sciences. 2023;120(1):e2214874120. [PMC free article: PMC9910429] [PubMed: 36574710]
- 200.
- Vantyghem MC, Vigouroux C, Magre J, Desbois-Mouthon C, Pattou F, Fontaine P, et al. Late-onset lipoatrophic diabetes. Phenotypic and genotypic familial studies and effect of treatment with metformin and lispro insulin analog. Diabetes Care. 1999;22(8):1374-6. [PubMed: 10480788]
- 201.
- Luedtke A, Boschmann M, Colpe C, Engeli S, Adams F, Birkenfeld AL, et al. Thiazolidinedione response in familial lipodystrophy patients with LMNA mutations: a case series. Horm Metab Res. 2012;44(4):306-11. [PubMed: 22274718]
- 202.
- Moreau F, Boullu-Sanchis S, Vigouroux C, Lucescu C, Lascols O, Sapin R, et al. Efficacy of pioglitazone in familial partial lipodystrophy of the Dunnigan type: a case report. Diabetes Metab. 2007;33(5):385-9. [PubMed: 17936664]
- 203.
- McLaughlin PD, Ryan J, Hodnett PA, O'Halloran D, Maher MM. Quantitative whole-body MRI in familial partial lipodystrophy type 2: changes in adipose tissue distribution coincide with biochemical improvement. AJR Am J Roentgenol. 2012;199(5):W602-6. [PubMed: 23096204]
- 204.
- Arioglu E, Duncan-Morin J, Sebring N, Rother KI, Gottlieb N, Lieberman J, et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann Intern Med. 2000;133(4):263-74. [PubMed: 10929166]
- 205.
- Sleilati GG, Leff T, Bonnett JW, Hegele RA. Efficacy and safety of pioglitazone in treatment of a patient with an atypical partial lipodystrophy syndrome. Endocr Pract. 2007;13(6):656-61. [PubMed: 17954424]
- 206.
- Iwanishi M, Ebihara K, Kusakabe T, Chen W, Ito J, Masuzaki H, et al. Clinical characteristics and efficacy of pioglitazone in a Japanese diabetic patient with an unusual type of familial partial lipodystrophy. Metabolism. 2009;58(12):1681-7. [PubMed: 19793595]
- 207.
- Kuzuya H, Matsuura N, Sakamoto M, Makino H, Sakamoto Y, Kadowaki T, et al. Trial of insulinlike growth factor I therapy for patients with extreme insulin resistance syndromes. Diabetes. 1993;42(5):696-705. [PubMed: 8482426]
- 208.
- Moses AC, Morrow LA, O'Brien M, Moller DE, Flier JS. Insulin-like growth factor I (rhIGF-I) as a therapeutic agent for hyperinsulinemic insulin-resistant diabetes mellitus. Diabetes Res Clin Pract. 1995;28 Suppl:S185-94. [PubMed: 8529513]
- 209.
- Satoh M, Yoshizawa A, Takesue M, Saji T, Yokoya S. Long-term effects of recombinant human insulin-like growth factor I treatment on glucose and lipid metabolism and the growth of a patient with congenital generalized lipodystrophy. Endocr J. 2006;53(5):639-45. [PubMed: 16902264]
- 210.
- Bansal R, Cochran E, Startzell M, Brown RJ. Clinical Effects of Sodium-Glucose Transporter Type 2 Inhibitors in Patients With Partial Lipodystrophy. Endocrine Practice. 2022;28(6):610-4. [PMC free article: PMC9177708] [PubMed: 35301125]
- 211.
- Nagayama A, Ashida K, Watanabe M, Moritaka K, Sonezaki A, Kitajima Y, et al. Case report: metreleptin and SGLT2 inhibitor combination therapy is effective for acquired incomplete lipodystrophy. Frontiers in Endocrinology. 2021;12:690996. [PMC free article: PMC8201990] [PubMed: 34135866]
- 212.
- Foss-Freitas MC, Imam S, Neidert A, Gomes AD, Broome DT, Oral EA. Efficacy and Safety of Glucagon-Like Peptide 1 Agonists in a Retrospective Study of Patients With Familial Partial Lipodystrophy. Diabetes Care. 2024;47(4):653. [PMC free article: PMC10973902] [PubMed: 38300898]
- 213.
- Johns KW, Bennett MT, Bondy GP. Are HIV positive patients resistant to statin therapy? Lipids Health Dis. 2007;6:27. [PMC free article: PMC2186312] [PubMed: 17958912]
- 214.
- Macallan DC, Baldwin C, Mandalia S, Pandol-Kaljevic V, Higgins N, Grundy A, et al. Treatment of altered body composition in HIV-associated lipodystrophy: comparison of rosiglitazone, pravastatin, and recombinant human growth hormone. HIV Clin Trials. 2008;9(4):254-68. [PubMed: 18753120]
- 215.
- Lager CJ, Esfandiari NH, Subauste AR, Kraftson AT, Brown MB, Cassidy RB, et al. Roux-En-Y Gastric Bypass Vs. Sleeve Gastrectomy: Balancing the Risks of Surgery with the Benefits of Weight Loss. Obes Surg. 2017;27(1):154-61. [PMC free article: PMC5187368] [PubMed: 27342739]
- 216.
- Melvin A, Adams C, Flanagan C, Gaff L, Gratton B, Gribble F, et al. Roux-en-Y Gastric Bypass Surgery in the Management of Familial Partial Lipodystrophy Type 1. J Clin Endocrinol Metab. 2017;102(10):3616-20. [PMC free article: PMC5630252] [PubMed: 28973478]
- 217.
- Utzschneider KM, Trence DL. Effectiveness of gastric bypass surgery in a patient with familial partial lipodystrophy. Diabetes Care. 2006;29(6):1380-2. [PubMed: 16732025]
- 218.
- Ciudin A, Baena-Fustegueras JA, Fort JM, Encabo G, Mesa J, Lecube A. Successful treatment for the Dunnigan-type familial partial lipodystrophy with Roux-en-Y gastric bypass. Clin Endocrinol (Oxf). 2011;75(3):403-4. [PubMed: 21521325]
- 219.
- Grundfest-Broniatowski S, Yan J, Kroh M, Kilim H, Stephenson A. Successful Treatment of an Unusual Case of FPLD2: The Role of Roux-en-Y Gastric Bypass-Case Report and Literature Review. J Gastrointest Surg. 2017;21(4):739-43. [PubMed: 27778252]
- 220.
- Meral R, Malandrino N, Walter M, Neidert AH, Muniyappa R, Oral EA, et al. Endogenous leptin concentrations poorly predict metreleptin response in patients with partial lipodystrophy. The Journal of Clinical Endocrinology & Metabolism. 2022;107(4):e1739-e51. [PMC free article: PMC8947785] [PubMed: 34677608]
- 221.
- McDuffie JR, Riggs PA, Calis KA, Freedman RJ, Oral EA, DePaoli AM, et al. Effects of exogenous leptin on satiety and satiation in patients with lipodystrophy and leptin insufficiency. J Clin Endocrinol Metab. 2004;89(9):4258-63. [PMC free article: PMC2266890] [PubMed: 15356018]
- 222.
- Moran SA, Patten N, Young JR, Cochran E, Sebring N, Reynolds J, et al. Changes in body composition in patients with severe lipodystrophy after leptin replacement therapy. Metabolism. 2004;53(4):513-9. [PubMed: 15045701]
- 223.
- Ebihara K, Kusakabe T, Hirata M, Masuzaki H, Miyanaga F, Kobayashi N, et al. Efficacy and safety of leptin-replacement therapy and possible mechanisms of leptin actions in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2007;92(2):532-41. [PubMed: 17118991]
- 224.
- Schlogl H, Muller K, Horstmann A, Miehle K, Puschel J, Villringer A, et al. Leptin Substitution in Patients With Lipodystrophy: Neural Correlates for Long-term Success in the Normalization of Eating Behavior. Diabetes. 2016;65(8):2179-86. [PubMed: 27207511]
- 225.
- Schlogl H, Muller K, Horstmann A, Pleger B, Miehle K, Moller H, et al. Leptin-substitution in patients with congenital lipodystrophy increases connectivity in reward-related brain structures: an fMRI study. Exp Clin Endocr Diab. 2014;122(3).
- 226.
- Schlogl H, Muller K, Horstmann A, Miehle K, Pleger B, Moller H, et al. Leptin-substitution increases connectivity in reward-related brain areas in patients with congenital lipodystrophy. Diabetologia. 2015;58:S71-S.
- 227.
- Aotani D, Ebihara K, Sawamoto N, Kusakabe T, Aizawa-Abe M, Kataoka S, et al. Functional magnetic resonance imaging analysis of food-related brain activity in patients with lipodystrophy undergoing leptin replacement therapy. J Clin Endocrinol Metab. 2012;97(10):3663-71. [PMC free article: PMC3462942] [PubMed: 22872692]
- 228.
- Brown RJ, Oral EA, Cochran E, Araújo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with generalized lipodystrophy. Endocrine. 2018;60:479-89. [PMC free article: PMC5936645] [PubMed: 29644599]
- 229.
- Oral EA, Gorden P, Cochran E, Araújo-Vilar D, Savage DB, Long A, et al. Long-term effectiveness and safety of metreleptin in the treatment of patients with partial lipodystrophy. Endocrine. 2019;64:500-11. [PMC free article: PMC7340120] [PubMed: 30805888]
- 230.
- Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109(10):1345-50. [PMC free article: PMC150981] [PubMed: 12021250]
- 231.
- Muniyappa R, Brown RJ, Mari A, Joseph J, Warren MA, Cochran EK, et al. Effects of leptin replacement therapy on pancreatic beta-cell function in patients with lipodystrophy. Diabetes Care. 2014;37(4):1101-7. [PMC free article: PMC3964492] [PubMed: 24496806]
- 232.
- Vatier C, Fetita S, Boudou P, Tchankou C, Deville L, Riveline J, et al. One-year metreleptin improves insulin secretion in patients with diabetes linked to genetic lipodystrophic syndromes. Diabetes Obes Metab. 2016;18(7):693-7. [PubMed: 26584826]
- 233.
- Quaye E, Chacko S, Startzell M, Brown RJ. Leptin decreases gluconeogenesis and gluconeogenic substrate availability in patients with lipodystrophy. The Journal of Clinical Endocrinology & Metabolism. 2023:dgad445. [PMC free article: PMC10735288] [PubMed: 37515588]
- 234.
- Lightbourne M, Wolska A, Abel BS, Rother KI, Walter M, Kushchayeva Y, et al. Apolipoprotein CIII and angiopoietin-like protein 8 are elevated in lipodystrophy and decrease after metreleptin. Journal of the Endocrine Society. 2021;5(2):bvaa191. [PMC free article: PMC7787426] [PubMed: 33442570]
- 235.
- Adamski K, Cook K, Gupta D, Morris E, Tuttle E, Carr E, et al. Effects of metreleptin in patients with lipodystrophy with and without baseline concomitant medication use. Current Medical Research and Opinion. 2021;37(11):1881-9. [PubMed: 34490811]
- 236.
- Mosbah H, Vantyghem MC, Nobécourt E, Andreelli F, Archambeaud F, Bismuth E, et al. Therapeutic indications and metabolic effects of metreleptin in patients with lipodystrophy syndromes: Real‐life experience from a national reference network. Diabetes, Obesity and Metabolism. 2022;24(8):1565-77. [PMC free article: PMC9541305] [PubMed: 35445532]
- 237.
- Chan JL, Lutz K, Cochran E, Huang W, Peters Y, Weyer C, et al. Clinical effects of long-term metreleptin treatment in patients with lipodystrophy. Endocr Pract. 2011;17(6):922-32. [PMC free article: PMC3498767] [PubMed: 22068254]
- 238.
- Kamran F, Rother KI, Cochran E, Safar Zadeh E, Gorden P, Brown RJ. Consequences of stopping and restarting leptin in an adolescent with lipodystrophy. Hormone research in paediatrics. 2012;78(5-6):320-5. [PMC free article: PMC3590018] [PubMed: 22965160]
- 239.
- Chong AY, Lupsa BC, Cochran EK, Gorden P. Efficacy of leptin therapy in the different forms of human lipodystrophy. Diabetologia. 2010;53(1):27-35. [PubMed: 19727665]
- 240.
- Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and generalized lipodystrophy: comparison of baseline characteristics and response to metreleptin. J Clin Endocrinol Metab. 2015;100(5):1802-10. [PMC free article: PMC4422900] [PubMed: 25734254]
- 241.
- Simha V, Subramanyam L, Szczepaniak L, Quittner C, Adams-Huet B, Snell P, et al. Comparison of efficacy and safety of leptin replacement therapy in moderately and severely hypoleptinemic patients with familial partial lipodystrophy of the Dunnigan variety. J Clin Endocrinol Metab. 2012;97(3):785-92. [PMC free article: PMC3319219] [PubMed: 22170723]
- 242.
- Park JY, Javor ED, Cochran EK, DePaoli AM, Gorden P. Long-term efficacy of leptin replacement in patients with Dunnigan-type familial partial lipodystrophy. Metabolism. 2007;56(4):508-16. [PMC free article: PMC2595136] [PubMed: 17379009]
- 243.
- Sekizkardes H, Cochran E, Malandrino N, Garg A, Brown RJ. Efficacy of metreleptin treatment in familial partial lipodystrophy due to PPARG vs LMNA pathogenic variants. The Journal of Clinical Endocrinology & Metabolism. 2019;104(8):3068-76. [PMC free article: PMC6563798] [PubMed: 31194872]
- 244.
- Simha V, Szczepaniak LS, Wagner AJ, DePaoli AM, Garg A. Effect of leptin replacement on intrahepatic and intramyocellular lipid content in patients with generalized lipodystrophy. Diabetes Care. 2003;26(1):30-5. [PubMed: 12502655]
- 245.
- Javor ED, Ghany MG, Cochran EK, Oral EA, DePaoli AM, Premkumar A, et al. Leptin reverses nonalcoholic steatohepatitis in patients with severe lipodystrophy. Hepatology. 2005;41(4):753-60. [PubMed: 15791619]
- 246.
- Akinci B, Subauste A, Ajluni N, Esfandiari NH, Meral R, Neidert AH, et al. Metreleptin therapy for nonalcoholic steatohepatitis: Open-label therapy interventions in two different clinical settings. Med. 2021;2(7):814-35. e6. [PMC free article: PMC8920072] [PubMed: 35291351]
- 247.
- Safar Zadeh E, Lungu AO, Cochran EK, Brown RJ, Ghany MG, Heller T, et al. The liver diseases of lipodystrophy: the long-term effect of leptin treatment. Journal of hepatology. 2013;59(1):131-7. [PMC free article: PMC3924897] [PubMed: 23439261]
- 248.
- Machado MV, Cortez-Pinto H. Leptin in the treatment of lipodystrophy-associated nonalcoholic fatty liver disease: are we there already? Expert Rev Gastroenterol Hepatol. 2013;7(6):513-5. [PubMed: 23985000]
- 249.
- Brown RJ, Meehan CA, Cochran E, Rother KI, Kleiner DE, Walter M, et al. Effects of Metreleptin in Pediatric Patients With Lipodystrophy. J Clin Endocrinol Metab. 2017;102(5):1511-9. [PMC free article: PMC5443330] [PubMed: 28324110]
- 250.
- Metz M, Beghini M, Wolf P, Pfleger L, Hackl M, Bastian M, et al. Leptin increases hepatic triglyceride export via a vagal mechanism in humans. Cell Metabolism. 2022;34(11):1719-31. e5. [PubMed: 36220067]
- 251.
- Baykal AP, Parks EJ, Shamburek R, Syed-Abdul MM, Chacko S, Cochran E, et al. Leptin decreases de novo lipogenesis in patients with lipodystrophy. JCI insight. 2020;5(14). [PMC free article: PMC7453896] [PubMed: 32573497]
- 252.
- Casey SP, Lokan J, Testro A, Farquharson S, Connelly A, Proietto J, et al. Post-liver transplant leptin results in resolution of severe recurrence of lipodystrophy-associated nonalcoholic steatohepatitis. Am J Transplant. 2013;13(11):3031-4. [PubMed: 24007548]
- 253.
- Javor ED, Moran SA, Young JR, Cochran EK, DePaoli AM, Oral EA, et al. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. J Clin Endocrinol Metab. 2004;89(7):3199-207. [PubMed: 15240593]
- 254.
- Javor ED, Moran SA, Young JR, Cochran EK, DePaoli AM, Oral EA, et al. Proteinuric nephropathy in acquired and congenital generalized lipodystrophy: baseline characteristics and course during recombinant leptin therapy. The Journal of Clinical Endocrinology & Metabolism. 2004;89(7):3199-207. [PubMed: 15240593]
- 255.
- Stürzebecher PE, Kralisch S, Schubert MR, Filipova V, Hoffmann A, Oliveira F, et al. Leptin treatment has vasculo-protective effects in lipodystrophic mice. Proceedings of the National Academy of Sciences. 2022;119(40):e2110374119. [PMC free article: PMC9546548] [PubMed: 36161905]
- 256.
- Kinzer AB, Shamburek RD, Lightbourne M, Muniyappa R, Brown RJ. Advanced lipoprotein analysis shows atherogenic lipid profile that improves after metreleptin in patients with lipodystrophy. Journal of the Endocrine Society. 2019;3(8):1503-17. [PMC free article: PMC6676079] [PubMed: 31620670]
- 257.
- Rebouças BMXCR, Mendonça RM, Egito EST, Lima DN, de Melo Campos JTA, Lima JG. Coronary arterial calcification in patients with congenital generalised lipodystrophy: A case series. Clinical endocrinology. 2022;97(6):863-6. [PubMed: 35864565]
- 258.
- Nguyen M-L, Sachdev V, Burklow TR, Li W, Startzell M, Auh S, et al. Leptin attenuates cardiac hypertrophy in patients with generalized lipodystrophy. The Journal of Clinical Endocrinology & Metabolism. 2021;106(11):e4327-e39. [PMC free article: PMC8530723] [PubMed: 34223895]
- 259.
- Cook K, Ali O, Akinci B, Foss de Freitas MC, Montenegro Jr RM, Fernandes VO, et al. Effect of leptin therapy on survival in generalized and partial lipodystrophy: a matched cohort analysis. The Journal of Clinical Endocrinology & Metabolism. 2021;106(8):e2953-e67. [PMC free article: PMC8277211] [PubMed: 33822100]
- 260.
- Schlögl H, Janssen L, Fasshauer M, Miehle K, Villringer A, Stumvoll M, et al. Reward Processing During Monetary Incentive Delay Task After Leptin Substitution in Lipodystrophy—an fMRI Case Series. Journal of the Endocrine Society. 2023;7(6):bvad052. [PMC free article: PMC10174197] [PubMed: 37180211]
- 261.
- Vieira DB, Antel J, Peters T, Miehle K, Stumvoll M, Hebebrand J, et al. Suggestive evidence for an antidepressant effect of metreleptin treatment in patients with lipodystrophy. Obesity Facts. 2022;15(5):685-93. [PMC free article: PMC9669995] [PubMed: 36037795]
- 262.
- Cook K, Adamski K, Gomes A, Tuttle E, Kalden H, Cochran E, et al. Effects of metreleptin on patient outcomes and quality of life in generalized and partial lipodystrophy. Journal of the Endocrine Society. 2021;5(4):bvab019. [PMC free article: PMC7993583] [PubMed: 33817539]
- 263.
- Oral EA, Ruiz E, Andewelt A, Sebring N, Wagner AJ, Depaoli AM, et al. Effect of leptin replacement on pituitary hormone regulation in patients with severe lipodystrophy. J Clin Endocrinol Metab. 2002;87(7):3110-7. [PubMed: 12107209]
- 264.
- Musso C, Cochran E, Javor E, Young J, Depaoli AM, Gorden P. The long-term effect of recombinant methionyl human leptin therapy on hyperandrogenism and menstrual function in female and pituitary function in male and female hypoleptinemic lipodystrophic patients. Metabolism. 2005;54(2):255-63. [PubMed: 15690321]
- 265.
- Abel BS, Muniyappa R, Stratton P, Skarulis MC, Gorden P, Brown RJ. Effects of Recombinant Human Leptin (Metreleptin) on Nocturnal Luteinizing Hormone Secretion in Lipodystrophy Patients. Neuroendocrinology. 2016;103(3-4):402-7. [PMC free article: PMC7269156] [PubMed: 26336809]
- 266.
- Lungu AO, Zadeh ES, Goodling A, Cochran E, Gorden P. Insulin resistance is a sufficient basis for hyperandrogenism in lipodystrophic women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 2012;97(2):563-7. [PMC free article: PMC3275358] [PubMed: 22090274]
- 267.
- Meehan CA, Cochran E, Kassai A, Brown RJ, Gorden P. Metreleptin for injection to treat the complications of leptin deficiency in patients with congenital or acquired generalized lipodystrophy. Expert Rev Clin Pharmacol. 2016;9(1):59-68. [PMC free article: PMC4931926] [PubMed: 26465174]
- 268.
- Christensen JD, Lungu AO, Cochran E, Collins MT, Gafni RI, Reynolds JC, et al. Bone mineral content in patients with congenital generalized lipodystrophy is unaffected by metreleptin replacement therapy. J Clin Endocrinol Metab. 2014;99(8):E1493-500. [PMC free article: PMC4121033] [PubMed: 25070319]
- 269.
- Simha V, Zerwekh JE, Sakhaee K, Garg A. Effect of subcutaneous leptin replacement therapy on bone metabolism in patients with generalized lipodystrophy. J Clin Endocrinol Metab. 2002;87(11):4942-5. [PubMed: 12414854]
- 270.
- Akinci EY, Boutros S, Ryan BJ, Sargin P, Akinci B, Neidert AH, et al. Iron parameters in patients with partial lipodystrophy and impact of exogenous leptin therapy. BMJ Open Diabetes Research and Care. 2021;9(1):e002385. [PMC free article: PMC8354259] [PubMed: 34373262]
- 271.
- Chan JL, Koda J, Heilig JS, Cochran EK, Gorden P, Oral EA, et al. Immunogenicity associated with metreleptin treatment in patients with obesity or lipodystrophy. Clin Endocrinol (Oxf). 2016;85(1):137-49. [PMC free article: PMC4875885] [PubMed: 26589105]
- 272.
- Beltrand J, Lahlou N, Le Charpentier T, Sebag G, Leka S, Polak M, et al. Resistance to leptin-replacement therapy in Berardinelli-Seip congenital lipodystrophy: an immunological origin. Eur J Endocrinol. 2010;162(6):1083-91. [PubMed: 20236991]
- 273.
- Brown RJ, Chan JL, Jaffe ES, Cochran E, DePaoli AM, Gautier JF, et al. Lymphoma in acquired generalized lipodystrophy. Leuk Lymphoma. 2016;57(1):45-50. [PMC free article: PMC4755279] [PubMed: 25864863]
- 274.
- Aslam A, Savage DB, Coulson IH. Acquired generalized lipodystrophy associated with peripheral T cell lymphoma with cutaneous infiltration. Int J Dermatol. 2015;54(7):827-9. [PubMed: 24168394]
- 275.
- MYALEPT PRESCRIBING INFORMATION. https://www
.accessdata .fda.gov/drugsatfda_docs /label/2014/125390s000lbl.pdf [ - 276.
- Lebastchi J, Ajluni N, Neidert A, Oral EA. A Report of Three Cases With Acquired Generalized Lipodystrophy With Distinct Autoimmune Conditions Treated With Metreleptin. J Clin Endocrinol Metab. 2015;100(11):3967-70. [PMC free article: PMC4702455] [PubMed: 26390101]
- 277.
- Park JY, Chong AY, Cochran EK, Kleiner DE, Haller MJ, Schatz DA, et al. Type 1 diabetes associated with acquired generalized lipodystrophy and insulin resistance: the effect of long-term leptin therapy. J Clin Endocrinol Metab. 2008;93(1):26-31. [PMC free article: PMC2729152] [PubMed: 17940115]
- 278.
- Haymond MW, Araújo-Vilar D, Balser J, Lewis JH, Louzado R, Musso C, et al. The Metreleptin Effectiveness and Safety Registry (MEASuRE): concept, design and challenges. Orphanet Journal of Rare Diseases. 2023;18(1):127. [PMC free article: PMC10214317] [PubMed: 37237416]
- 279.
- Oral EA, Garg A, Tami J, Huang EA, O'Dea LSL, Schmidt H, et al. Assessment of efficacy and safety of volanesorsen for treatment of metabolic complications in patients with familial partial lipodystrophy: Results of the BROADEN study: Volanesorsen in FPLD; The BROADEN Study. Journal of Clinical Lipidology. 2022;16(6):833-49. [PubMed: 36402670]
- 280.
- Prohaska TA, Alexander VJ, Karwatowska-Prokopczuk E, Tami J, Xia S, Witztum JL, et al. APOC3 inhibition with volanesorsen reduces hepatic steatosis in patients with severe hypertriglyceridemia. Journal of Clinical Lipidology. 2023. [PubMed: 37164837]
- 281.
- Lightbourne M, Startzell M, Bruce KD, Brite B, Muniyappa R, Skarulis M, et al. Volanesorsen, an antisense oligonucleotide to apolipoprotein C-III, increases lipoprotein lipase activity and lowers triglycerides in partial lipodystrophy. Journal of Clinical Lipidology. 2022;16(6):850-62. [PMC free article: PMC9771980] [PubMed: 36195542]
- 282.
- Karwatowska-Prokopczuk E, Tardif J-C, Gaudet D, Ballantyne CM, Shapiro MD, Moriarty PM, et al. Effect of olezarsen targeting APOC-III on lipoprotein size and particle number measured by NMR in patients with hypertriglyceridemia. Journal of Clinical Lipidology. 2022;16(5):617-25. [PubMed: 35902351]
- 283.
- Clifton P, Sullivan D, Baker J, Schwabe C, Thackwray S, Scott R, et al. Pharmacodynamic effect of aro-apoc3, an investigational hepatocyte-targeted rna interference therapeutic targeting apolipoprotein c3, in patients with hypertriglyceridemia and multifactorial chylomicronemia. Circulation. 2020;142(Suppl_3):A12594-A.
- 284.
- Clifton P, Sullivan D, Baker J, Schwabe C, Thackwray S, Scott R, et al. ARO-APOC3, an Investigational RNAi Therapeutic, Shows Similar Efficacy and Safety in Genetically Confirmed FCS and Non-FCS Participants with Severe Hypertriglyceridemia. Circulation. 2021;144(Suppl_1):A10357-A.
- 285.
- Akinci B, Meral R, Rus D, Hench R, Neidert AH, DiPaola F, et al. The complicated clinical course in a case of atypical lipodystrophy after development of neutralizing antibody to metreleptin: treatment with setmelanotide. Endocrinology, Diabetes & Metabolism Case Reports. 2020;2020(1). [PMC free article: PMC7159256] [PubMed: 32213649]
- 286.
- Foss de Freitas MC, DillGomes A, Neidert A, Podgrabinska S, Olenchock B, SinhaRoy R, et al. FRI059 Long-term Follow-up On Partial Lipodystrophy Treated With Leptin Receptor Agonist Mibavademab (REGN4461). Journal of the Endocrine Society. 2023;7(Supplement_1):bvad114. 070.
- 287.
- Odelya Pagovich M, Simona Podgrabinska, PhD, Jian Zhao, Bret Musser, Jeanne Mendell, PhD, MPH, Robert J. Sanchez, Kathryn Miller, Prodromos Parasoglou, Stefanie Hectors, Satyajit Karnik, PhD, Baris Akinci, MD, Ilgin Yildirim Simsir, Samim Ozen, MD, Ekaterina Shestakova, Ekaterina Mishina, Abhimanyu Garg, MD, Rolando Jesus Vargas Gonzalez, Gary Herman, Elif A. Oral, Rebecca J. Brown, Benjamin A. Olenchock. MON-789 - Changes In Metabolic Parameters For Mibavademab, A Novel Leptin Receptor Agonist, In Individuals With Generalized Lipodystrophy. ENDO 2024. [Abstract]. In press 2024.
- 288.
- Foss-Freitas MC, Akinci B, Neidert A, Bartlett VJ, Hurh E, Karwatowska-Prokopczuk E, et al. Selective targeting of angiopoietin-like 3 (ANGPTL3) with vupanorsen for the treatment of patients with familial partial lipodystrophy (FPLD): results of a proof-of-concept study. Lipids in Health and Disease. 2021;20(1):1-12. [PMC free article: PMC8647384] [PubMed: 34865644]
- 289.
- Hartinger R, Lederer E-M, Schena E, Lattanzi G, Djabali K. Impact of Combined Baricitinib and FTI Treatment on Adipogenesis in Hutchinson–Gilford Progeria Syndrome and Other Lipodystrophic Laminopathies. Cells. 2023;12(10):1350. [PMC free article: PMC10216179] [PubMed: 37408186]
- 290.
- Olenchock B, Podgrabinska S, Hou C, Musser B, Mendell J, Harman A, et al. FRI058 Treatment With Mibavademab, A Novel Leptin Receptor Agonist, Is Associated With Improvement In Metabolic Parameters In Individuals With Generalized Lipodystrophy. Journal of the Endocrine Society. 2023;7(Supplement_1):bvad114. 069.
- 291.
- Laber S, Strobel S, Mercader JM, Dashti H, Dos Santos FR, Kubitz P, et al. Discovering cellular programs of intrinsic and extrinsic drivers of metabolic traits using LipocyteProfiler. Cell Genomics. 2023;3(7). [PMC free article: PMC10363917] [PubMed: 37492099]
- 292.
- Agarwal AK, Tunison K, Vale G, McDonald JG, Li X, Scherer PE, et al. Regulated adipose tissue-specific expression of human AGPAT2 in lipodystrophic Agpat2-null mice results in regeneration of adipose tissue. Iscience. 2023;26(10). [PMC free article: PMC10518674] [PubMed: 37752957]
- 293.
- Sommer N, Roumane A, Han W, Delibegović M, Rochford JJ, Mcilroy GD. Gene therapy restores adipose tissue and metabolic health in a pre-clinical mouse model of lipodystrophy. Molecular Therapy-Methods & Clinical Development. 2022;27:206-16. [PMC free article: PMC9589143] [PubMed: 36320417]
- 294.
- Foss de Freitas MC, Rosenberg DS, Guler MC, Yosef M, Khalatbari S, Carman R, et al. FRI055 LYNC-LD: Prospective Multicenter Natural History Study Of Lipodystrophy Syndromes To Determine Prevalence, Incidence And Predictors Of Diabetes And Severe Hypertriglyceridemia, And Their Complications. Journal of the Endocrine Society. 2023;7(Supplement_1):bvad114. 066.
- 295.
- Merve Celik Guler M, Maria Cristina Foss de Freitas, MD PhD, Matheos Yosef, Shokoufeh Khalatbari, Diarratou Kaba, Michelle Ashmus, Demircan Guler, MD, Donatella Gilio, MD, Anabela Dill Gomes, B.S. Psychology, Drake Stanton Rosenberg, BS candidate, Trinity Neal, Becca Tuska, Maiah Brush, B.S., Michael Hwang, BA, Britney Tracey, Marinna Okawa, Brianna Brite, Bachelors of Science, Carman Richison, Ilgin Yildirim Simsir, MD, Baris Akinci, MD, Rebecca J. Brown, MD, Elif A. Oral, MD. SAT-614-Unraveling the Natural History of Lipodystrophy Syndromes with LD LYNC: Time to Development of Important Comorbidities. 2024.
- 296.
- Merve Celik Guler M, Maria Cristina Foss de Freitas, MD PhD, Matheos Yosef, Shokoufeh Khalatbari, Diarratou Kaba, Michelle Ashmus, Demircan Guler, MD, Donatella Gilio, MD, Anabela Dill Gomes, B.S. Psychology, Drake Stanton Rosenberg, BS candidate, Trinity Neal, Becca Tuska, Maiah Brush, B.S., Michael Hwang, BA, Britney Tracey, Marinna Okawa, Brianna Brite, Bachelors of Science, Carman Richison, Ilgin Yildirim Simsir, MD, Baris Akinci, MD, Rebecca J. Brown, MD, Elif A. Oral, MD. SAT-605-Predictors of Psychosocial Burden and Pain in Lipodystrophy Syndromes. 2024.
- 297.
- Von Schnurbein J, Adams C, Akinci B, Ceccarini G, D’apice MR, Gambineri A, et al. European lipodystrophy registry: background and structure. Orphanet journal of rare diseases. 2020;15:1-11. [PMC free article: PMC6964101] [PubMed: 31941540]
- Review Molecular and Cellular Bases of Lipodystrophy Syndromes.[Front Endocrinol (Lausanne). 2...]Review Molecular and Cellular Bases of Lipodystrophy Syndromes.Zammouri J, Vatier C, Capel E, Auclair M, Storey-London C, Bismuth E, Mosbah H, Donadille B, Janmaat S, Fève B, et al. Front Endocrinol (Lausanne). 2021; 12:803189. Epub 2022 Jan 3.
- Review Partial lipodystrophy: Clinical presentation and treatment.[Ann Endocrinol (Paris). 2024]Review Partial lipodystrophy: Clinical presentation and treatment.Mosbah H, Vatier C, Vigouroux C. Ann Endocrinol (Paris). 2024 Jun; 85(3):197-200. Epub 2024 Jun 12.
- Review Leptin replacement therapy in the management of lipodystrophy syndromes.[Ann Endocrinol (Paris). 2024]Review Leptin replacement therapy in the management of lipodystrophy syndromes.Vigouroux C, Mosbah H, Vatier C. Ann Endocrinol (Paris). 2024 Jun; 85(3):201-204. Epub 2024 Jun 12.
- Review How to diagnose a lipodystrophy syndrome.[Ann Endocrinol (Paris). 2012]Review How to diagnose a lipodystrophy syndrome.Vantyghem MC, Balavoine AS, Douillard C, Defrance F, Dieudonne L, Mouton F, Lemaire C, Bertrand-Escouflaire N, Bourdelle-Hego MF, Devemy F, et al. Ann Endocrinol (Paris). 2012 Jun; 73(3):170-89. Epub 2012 Jun 28.
- Review Treatment Options for Lipodystrophy in Children.[Front Endocrinol (Lausanne). 2...]Review Treatment Options for Lipodystrophy in Children.Mainieri F, Tagi VM, Chiarelli F. Front Endocrinol (Lausanne). 2022; 13:879979. Epub 2022 May 4.
- Lipodystrophy Syndromes: Presentation and Treatment - EndotextLipodystrophy Syndromes: Presentation and Treatment - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...