

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Congenital hypopituitarism refers to a deficiency of one or more pituitary hormones resulting from issues in fetal development. Embryological pituitary development involves a complex interplay of transcription factors, extrinsic and intrinsic to the oral ectoderm and neuroectoderm which develop to form the mature pituitary during early embryogenesis. Disruption of this process can result in isolated pituitary dysfunction, or effect nearby structures, such as the eye, olfactory bulbs, midline structures, and forebrain. Genetic causes make up an important portion of cases and have varying phenotypes even within identical mutations. This chapter provides an overview of pituitary development, and various causes of hypopituitarism including septo-optic dysplasia, syndromic and non-syndromic causes, isolated pituitary deficiencies, and syndromes associated with hypopituitarism. It also provides guidance on the investigation of genetic causes of hypopituitarism in the current genetic landscape. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
The pituitary gland can be thought of as the “hormonal control center” from which most endocrine organs are regulated. It is located in the sella turcica, posterior to the sphenoid sinus, inferior to the optic chiasm and the hypothalamus. The pituitary comprises 3 lobes, the anterior, intermediate and posterior.
The posterior lobe or neurohypophysis contains antidiuretic hormone (ADH or AVP) and oxytocin, contained in vesicles, as part of axonal projections from hypothalamic cells, known as the hypothalamic-hypophyseal tract. The intermediate lobe contains melanotrophs which produce alpha-melanocyte stimulating hormone (αMSH), although in humans is typically an embryological remnant and may not be present.
The anterior lobe or adenohypophysis contains 5 cell lines, producing 6 different hormones regulated by hypothalamic hormones via the hypophyseal portal system. Somatotrophs produce growth hormone (GH) while lactotrophs produce prolactin making up the somatomammotroph class of hormones. The glycoprotein hormone class consists of hormones with identical alpha subunits and different beta subunits, with thyroid stimulating hormone (TSH) produced by thyrotroph cells, and gonadotroph cells producing follicle stimulating hormone (FSH) and luteinizing hormone (LH). Corticotroph cells produce adrenocorticotrophic hormone (ACTH).
Growth hormone mediates its effect through induction of insulin like growth factor 1 (IGF-1) from the liver and is critical for linear growth in the child. Prolactin stimulates development of the mammary gland and lactation and is typically quiescent outside of pregnancy or childrearing. TSH stimulates production of thyroid hormone from the thyroid gland, which regulates metabolic rate and is critical for growth and cognitive development in early childhood. FSH and LH support gonadal development, production of testosterone or estrogen, and maturation of gamete cells. ACTH stimulates production of cortisol which is an essential stress hormone and ADH regulates plasma osmolality by inducing resorption of water in the collecting duct of the kidney.
Congenital hypopituitarism refers to a deficiency of one or more pituitary hormones resulting from events during fetal development. This may be the result of genetic mutation, antenatal insult, or as is commonly the case, be idiopathic. Pituitary deficiencies may be detected during the neonatal period; however, in some individuals’ pituitary deficiencies may not manifest until later in childhood. Furthermore, the severity can be variable.
Hypopituitarism may be isolated, affecting one cell line (isolated hormone deficiency), or multiple, termed combined or multiple pituitary hormone deficiency (CPHD, MPHD). Pan-hypopituitarism is typically used to describe deficiency of all anterior pituitary hormones; however, in some cases this term may also include the presence of AVP deficiency. AVP (or ADH) deficiency is termed as central diabetes insipidus (DI) but is undergoing a formal process to change the name to AVP deficiency. The new name is preferred as it highlights both etiology and treatment, while avoiding confusion with diabetes mellitus. AVP deficiency may coexist with other deficiency’s or be isolated.
The incidence of isolated growth hormone deficiency (IGHD) is reportedly 1 in 4000 live births (3), with CPHD also seen in 1 in 4000 live births (4), thus presenting an uncommon but important chronic condition.
Embryological pituitary development involves a complex interplay of transcription factors, extrinsic and intrinsic to the oral ectoderm and neuroectoderm which develop to form the mature pituitary. Disorders occurring during early development may affect nearby structures commonly the eye, olfactory bulbs, midline structures, and forebrain, whereas later events are typically isolated to pituitary abnormalities. Transcription factor gene defects have been demonstrated to cause different biochemical and structural forms of congenital hypopituitarism, although the majority of cases remain idiopathic. There is considerable phenotypic variability within known genetic causes of hypopituitarism, with some forms having incomplete penetrance, and presentation ranging from asymptomatic, to severe neonatal onset forms. This review discusses the clinical phenotypes of various genetic anomalies associated with hypopituitarism. Clinical manifestations and investigation of hypopituitarism are discussed in the “Hypopituitarism” chapter of Endotext.
This chapter will present an overview of the embryology of pituitary development before discussing the genetic causes of hypopituitarism. It will firstly discuss genes that cause septo-optic dysplasia, before discussing genes that cause other clinical phenotypes and hypopituitarism. After describing the genes associated with non-syndromic hypopituitarism, and isolated hormone deficiencies we will discuss other syndromes where hypopituitarism may be part of the phenotype. Each gene is described within the subheading that they most typically present; however, due to phenotypic variability some genes can present in multiple categories. For example, PROKR2 mutation classically causes isolated hypogonadotrophic hypogonadism, but can result in non-syndromic CPHD or septo-optic dysplasia in some cases (5-8). The information within this review is up to date as of May 2022; however, understanding of these genes continues to progress.

Figure 1.
Stages of rodent pituitary development. (a) Oral ectoderm. (b) Rudimentary pouch. (c) Definitive pouch. (d) Adult pituitary gland. I infundibulum; NP neural plate; N notochord; PP pituitary placode; OM oral membrane; H heart; F forebrain; MB midbrain; HB hindbrain; RP Rathke's pouch; AN anterior neural pore; O oral cavity; PL posterior lobe; OC optic chiasm; P pontine flexure; PO pons; IL intermediate lobe; AL anterior lobe; DI diencephalon; SC sphenoid cartilage. Adapted from Sheng and Westphal, Trends in Genetics 1999;15:236-240, with permission (licence number: 5420490065848” (2).
EMBRYOLOGY
Pituitary gland development is classically expressed based on timing with murine development. The pituitary gland forms from the hypophyseal placode on the anterior neural ridge of the neural plate (evident embryonic (E) day 7.5 in the mouse model). Ventral displacement leads to the formation of the oral ectoderm and the roof of the mouth. A portion of this ectoderm thickens (E8.5) before invaginating rostrally at E9. At E9.5, corresponding with 4 weeks gestation, this migrates dorsally resulting in a cone shape, known as the rudimentary Rathke’s pouch. This continues to thicken with increased rostral mitosis spreading into the mesenchyme. Following ongoing proliferation, it separates from the oral ectoderm at E12.5 (approximately 6 weeks gestation). Hormonal cell progenitors arise from ventral proliferation forming the anterior pituitary. (Figure 1) The somatotrophs arise caudomedially, gonadotrophs rostroventrally and corticotrophs ventrally. The dorsal aspect later adjoins the descending infundibulum and remains thin (Intermediate lobe) (9).
Throughout this process the tissue is in contact with the neural plate neuroectoderm, at the base of the developing diencephalon. This neuroectoderm evaginates and progress dorsally to form the posterior pituitary. The tissue immediately ventral to this evagination forms the optic chiasm. The diencephalon develops throughout this process to form the hypothalamus (9).
The olfactory placode forms immediately lateral to the hypophyseal placode on the neural plate. These cells form the olfactory bulb but also the GnRH secreting cells which migrate to the forebrain from 6 weeks gestation, progressing to the hypothalamus by week 15. Axons from these cells extend into the hypothalamic portal system allowing regulation of gonadotroph cells (10-12). The optic nerve origin and optic chiasm develop from the neuroectoderm immediately anterior to the posterior pituitary, which has a complex interplay with some causes of hypopituitarism (13).
Correlation of pituitary development in mice to humans is difficult. Whilst pluripotent stem cells have been induced to form hypothalamic and anterior pituitary cells, the complex interplay of transcription factors, as well as interaction from other anatomical structures, mean these methods cannot give us a clear timeline in humans (14). Historical embryo samples demonstrate the formation of Rathke’s pouch by week 4, with interruption of connection to the oral cavity by week 6 with the anterior pituitary fully differentiated by 16 weeks (15).
Using hormone expression to correlate these two embryonic timelines, humans start producing GH, FSH, LH and ACTH at 8 weeks gestation, with TSH and prolactin at 13 weeks (9). Somatotrophs make up 50% of the cellular composition of the anterior pituitary, with 15-20% of cells being lactotrophs, 5% thyrotropes, 10-15% gonadotrophs and 15-20% corticotrophs (16) (Figure 2).

Figure 2.
Structural development of pituitary. Lateral representation of process of oral and neuroectoderm folding. Cross-sectional representation of hormone producing pituitary cell groups. Adapted from Larkin S. et al, Endotext (1).
Various transcription factors are involved in coordinating these processes and gene mutation can result in underdevelopment or arrest of pituitary development. Figure 3 summarizes the temporal expression of various transcription factors within the pituitary. Abnormalities in genes expressed early in development such as SOX2, HESX1 and GLI2 more frequently effect other nearby structures, whereas those expressed later such as PROP1 and POU1F1 typically have localized effects. Transcription factors may be induced, upregulated or downregulated by multiple other transcription factors and the degree and site of expression can vary across the embryological development. For a diagrammatic overview of the role each transcription factor plays in pituitary development please see Figure 1 of the 2020 JCEM review by Gregory L and Dattani M (4).

Figure 3.
Timing of gene expression of transcription factors implicated in combined hypopituitarism.
Disruptions to this cascade of transcription factors causes a range of structural phenotypes, varying from a normal pituitary appearance, to anterior pituitary hypoplasia or agenesis. The infundibulum/pituitary stalk may be thin or absent and the posterior pituitary may be hypoplastic, absent, or ectopic. The combination of anterior pituitary hypoplasia (APH), infundibular thinning/absence, and ectopic posterior pituitary (EPP), is known as pituitary stalk interruption syndrome (PSIS) (Figure 4).

Figure 4.
Lateral appearance of structural pituitary phenotypes. Adapted from Larkin et al, Endotext (1).
SEPTO-OPTIC DYSPLASIA
Septo-optic dysplasia (SOD) is defined by the presence of two or more of: optic nerve hypoplasia, hypopituitarism, and midline defects (agenesis of the corpus callosum or septum pellucidum) forming a clinical triad. SOD is a rare condition with incidence estimates between 1 in 9,000 to 1 in 40,000 (17,18). The association between hypopituitarism and optic nerve hypoplasia relates to the embryological origin of the optic chiasm being immediately anterior to the infundibular tissue, thus an insult at this location can interrupt both pathways. The phenotype is highly variable even in familial SOD (13). Although several genetic causes have been identified, these make up less than 10% of cases of SOD, and are summarized in Table 1 (19). Septo-optic dysplasia is commonly seen in young primiparous mothers (20) and while antenatal exposure to alcohol and drug use have been suggested, there is a lack of evidence to support causality (21).
Optic nerve hypoplasia (ONH) is typically bilateral (80-90%) with midline defects typically being septum pellucidum (85%) or corpus callosum (30%) hypoplasia or aplasia (21). 55-80% of cases have pituitary hormone deficiency, with growth hormone (GHD) and TSH deficiency (TSHD) being the most common deficiencies, followed by ACTH. Diabetes insipidus and hypogonadotrophic hypogonadism (HH) are found in less than 30% of hormone deficient cases (22-25). Approximately half of these hormone deficiencies are detected in the first 2 years of life and can present with severe neonatal hypoglycemia and cardiovascular instability, with the remainder of cases presenting across childhood to adolescence. The degree of deficiency is also noted to progress over time (22). Children with SOD typically have at least some degree of visual impairment, with 44- 80% being legally blind (22,26), while epilepsy and behavioral issues can be seen in approximately a quarter of patients, and cognitive impairment in 42.5% (22). Recently, 31% of cases were found to be obese despite therapy, suggesting the contribution of hypothalamic obesity (22).
HESX1
HESX1 is a member of the paired-like class of homeobox genes, located on chromosome 3p14.3 with autosomal dominant and recessive mutations described, making up approximately 1% of septo-optic dysplasia cases (19,27).
It appears from E8.5 through to E13.5 in the ventral diencephalon and oral ectoderm (28). HESX1 regulates the structural organization of the pituitary and interacts with the infundibulum and surrounding tissues through repressing transcription. Murine studies suggest absence typically does not prevent Rathke’s pouch/anterior pituitary formation (5%) but can result in polypituitary. It is suggested HESX1 regulates the regional expression of LHX3, PROP1, FGF8 and FGF10, preventing excessive expansion of the pituitary (19). HESX1 is upregulated by OTX2 and LHX3 transcription factors while being repressed by FGF8 and PROP1 (28,29).
Dattani et al. described a sibling pair with consanguineous parents, with a homozygous (R160C) HESX1 mutation in the late 1990’s. The SOD phenotype comprised optic nerve hypoplasia, midline defects (absent corpus callosum (CC) and septum pellucidum (SP)), ectopic posterior pituitary and anterior pituitary hypoplasia with hypopituitarism (30). Subsequent reports indicate that HESX1 mutation results in the complete SOD triad in approximately 30% of cases with ophthalmic changes including anophthalmia, microphthalmia, optic nerve hypoplasia and optic nerve aplasia (28,31). The majority of HESX1 pathogenic variants manifest GHD, with variability in deficiency of the other anterior hormones and ADH deficiency being uncommon. MRI appearance varies from normal to posterior pituitary ectopia and anterior pituitary hypoplasia in affected individuals (19). It is also associated with hypoplastic nasal cavities/olfactory bulbs and underdevelopment of the forebrain (28).
Autosomal dominant forms typically cause a milder phenotype, with undescended posterior pituitary and GHD, but phenotype-genotype correlation is poor (19). Penetrance is variable with asymptomatic parents and siblings being reported and midline defects and optic nerve anomalies absent in some cases (31,32). Testing of 217 patients with Kallmann syndrome (hypogonadotrophic hypogonadism (HH) and anosmia) revealed 1.4% had a mutation of HESX1 and normal pituitary on imaging (33).
SOX2
SOX2 and SOX3 are from the SOX (SRY-related high mobility group HMG Box) family of transcription factors and are both single exon genes that are expressed early in development and decline as cells differentiate. SOX 2 is located on chromosome 3q26.33 with autosomal dominant expression and cause 10-15% of microphthalmia and anophthalmia cases (19,34).
SOX2 is expressed during the morula stage, E2.5 but from gastrulation is restricted to the presumptive neuroectoderm, and E9.5 is found in the brain, CNS, sensory placodes, in the branchial arches, esophagus and trachea (28). At E11.5 it is expressed throughout Rathke’s pouch but from E18.5 is restricted to the intermediate lobe (35). In humans it is expressed within the developing anterior pituitary from weeks 3.5-9 and in the diencephalon but not in the neurohypophysis or infundibulum (19).
In 2006 Williamson et al. described heterozygous de novo mutations of SOX2 causing bilateral anophthalmia or severe microphthalmia associated with developmental delay, esophageal atresia and genital anomalies (36). Later that year, Kelberman et al. reported the association of SOX2 with HH, GHD and anterior pituitary hypoplasia (37). A cohort of 18 participants with SOX2 mutation showed 33% (6/18) had short stature or GHD, and 44% (8/18) had genital anomalies or hypogonadism (38).
Alongside hypoplastic anterior pituitary, hypothalamic hamartomas and corpus callosum abnormalities are often seen, as are hippocampus anomalies, sensorineural hearing loss, learning difficulties and cognitive impairment. The vast majority have anophthalmia in at least one eye, but milder cases have microphthalmia, coloboma and optic nerve hypoplasia, with normal ocular structures seen very rarely (34). SOX2 mutations are associated with esophageal/tracheal abnormalities and underdeveloped genitalia known as Anophthalmia-Esophageal-Genital (AEG) syndrome. Micropenis and cryptorchidism are commonly seen, and while genital anomalies are noted less commonly in females, they can be severe, with some cases having vaginal agenesis (28).
Anophthalmia or microphthalmia with esophageal abnormalities, sensorineural hearing loss or GHD and HH should raise the question of a SOX2 mutation for a clinician. Given the autosomal dominant inheritance, detection would be critical for family planning for both the patient and their parents.
SOX3
SOX3 gene is found at Xq27 causing X-linked recessive inheritance found to cause 0.2% of all GH deficient cases (28,39). SOX3 is expressed throughout the central nervous system during embryonic development.
It is first expressed at E6.5 but is rapidly restricted to the anterior ectoderm, and to the presumptive neuroectoderm. It is highly expressed in the infundibulum and presumptive hypothalamus but not in Rathke’s pouch (28).
SOX3 supports development of the hypothalamus and infundibulum as well as the development of the AP. Absence results in thinning of the infundibulum, which is thought to reduce expression of other genes involved with supporting pituitary proliferation. As a result, Rathke’s pouch is initially expanded but eventually the anterior pituitary becomes hypoplastic. It is important in confining the expression of FGF8 and BMP4, preventing excessive activity. Interestingly, duplications of SOX3 are also associated with similar phenotypes to those with non-functional mutations (typically polyalanine expansions). This suggests that correct dose of SOX3 is required for optimal embryonic development (28).
In 2002, Laumonnier et al. described a boy with IGHD and learning difficulties associated with a SOX3 mutation (40). Subsequently, GHD has been found in affected males and is associated with developmental delay or cognitive impairment of varying severity. One family of 5 affected females with short stature and language/ hearing impairment has been described (28). Other concurrent pituitary hormone deficiencies are present with variable frequency with panhypopituitarism described in some cases (28).
MRI findings include absent corpus callosum and absent or hypoplastic infundibulum and ectopic posterior pituitary, with cognitive impairment and learning difficulties also reported. SOX3 is not associated with optic nerve hypoplasia and has not been reported in patients with the complete SOD triad. Craniofacial abnormalities were reported frequently in murine models but are rare in human populations and 1 case of spina bifida has been reported (28).
Patients with an x-linked pattern of inheritance, and isolated GHD or CPHD should raise clinical suspicion for SOX3 mutation.
OTX2
Orthodentic Homeobox 2 (OTX2) is critical for anterior structure development and forebrain maintenance (28). It is located on chromosome 14q22.3 with an autosomal dominant inheritance that has been detected in 2-3% of cases of anophthalmia/microphthalmia and can be associated with hypopituitarism (29,38,41).
From E10.5 OTX2 is strongly expressed in the neurohypophysis and hypothalamus but the expression in Rathke’s pouch is minimal. Expression continues until E16.5 but is silenced by E12.5. Knockout within Rathke’s pouch caused no abnormalities in pituitary development or function (42). OTX2 deficiency results in decreased posterior lobe and pituitary stalk development and secondary anterior lobe hypoplasia. This is mediated through failure of FGF10 production, resulting in the hypoplastic anterior pituitary, but with normal cell differentiation (42). It is also important for upregulating HESX1 and POU1F1 (29).
Isolated GHD is classical for OTX2 mutations but CPHD, typically TSHD or HH and panhypopituitarism have been described. Imaging typically demonstrates ectopic or absent posterior pituitary with hypoplastic or normal anterior pituitary but may be normal (28,41). Ocular manifestations vary from anophthalmia/microphthalmia, to optic nerve hypoplasia or retinal dystrophy, with normal ocular phenotype also described. Additional anomalies are rarely reported (41).
OTX2 abnormalities should be considered in cases of anophthalmia/microphthalmia and hypopituitarism. Posterior pituitary anomalies and absence of extracranial features may differentiate from SOX2 mutations (28,41).
PAX6
Other genes known to be associated with anophthalmia/microphthalmia have been shown to causes septo-optic dysplasia and hypopituitarism. PAX6 is located on chromosome 11p13 and is inherited in autosomal dominant pattern with incomplete penetrance (43). In murine pituitary development, PAX6 is expressed in the dorsal side of Rathke’s pouch from E9-12.5 and establishes the ventral-dorsal cell boundaries (44). It has a major role in early eye development, classically causing aniridia, with optic nerve anomalies including hypoplasia or microphthalmia or anophthalmia also described (43). Over 500 cases of PAX6 mutation have been described with only four cases of hypopituitarism, including isolated ACTH deficiency in one, GH deficiency in the remainder, two of which had plausible HH. Two were noted to have a hypoplastic pituitary (45-47). Thus, PAX6 is a rare cause of hypopituitarism in patients with eye abnormalities and may present with SOD.
RAX
The RAX gene found on chromosome 18q21.32 is involved in forebrain and eye development and is inherited in an autosomal recessive nature. In humans, it is associated with anophthalmia, microphthalmia and palatal anomalies and is present in approximately 3% of anophthalmia/microphthalmia cases (38). One patient to date with a severe mutation had anterior and posterior pituitary agenesis on MRI resulting in panhypopituitarism, but other patients have not been found to have pituitary anomalies (48).
TCF7L1
The transcription factor 7-like 1 (TCF7L1) gene on chromosome 2p11.2 is a transcription regulator gene, important for brain development through WNT/β-catenin signaling pathway. Heterozygous mutations of TCF7L1 have been identified in 2 SOD patients, 1 of whom had GHD and ACTHD and MRI demonstrating ONH, anterior pituitary hypoplasia and absent posterior pituitary (49).
Other Genes
GLI2, BMP4, TBC1D32, PROKR2, FGF8, FGFR1 mutations are also associated with SOD but are described later as this is not their typical phenotype. Genes causing SOD are summarized in Table 1.
Table 1.
Genes Associated with Septo-Optic Dysplasia
| Gene | Inheritance/ prevalence | Ocular Phenotype | Pituitary appearance | Pituitary deficiencies | Other Features |
|---|---|---|---|---|---|
| HESX1 Chr 3p14.3 | AD/AR (incomplete penetrance) 1% of SOD | ONH | EPP, APH | GH, CPHD, ACTH + ADH less common | CC/SP changes, hypoplastic olfactory, underdeveloped forebrain |
| SOX2 Chr 3q26.33 | AD 10-15% of AO/MO | AO/MO | APH, hypothalamic hamartoma | GH, LH/FSH | CC changes, SNHL, ID, esophageal anomalies, genital anomalies |
| SOX3 Chr Xq27 | XR 0.4% of IGHD | Nil | Thin infundibulum, EPP, APH, PSIS | GH, CPHD uncommon | CC changes, ID |
| OTX2 Chr 14q22.3 | AD 3% of AO/MO | AO/MO, ONH, retinal dystrophy | EPP, APH, PSIS | GH, LH/FSH, TSH. ACTH less common | |
| PAX6 Chr 11p13 | AD Rare | Aniridia, ONH, AO/MO | APH | Rare GH, ACTH, FSH/LH | ASD, ADHD, obesity, diabetes mellitus |
| RAX 18q21.32 | AR 3% of AO/MO | AO/MO | Rare CPHD | Palate changes | |
| TCF7L1 Chr 2p11.2 | AD Rare | ONH | Normal, APH, absent posterior pituitary | GH, ACTH | CC/SP changes |
| GLI2 2q14.2 | AD 1-13% of CPHD | ONH | APH, EPP, PSIS | GH, ACTH, FSH/LH, TSH | SP/CC changes, polydactyly, midface hypoplasia, cleft palate/lip, HPE |
| BMP4 Chr 14q22-23 | AD Rare | AO/MO | Normal | GH, TSH | Myopia, cleft palate/lip, polydactyly |
| TBC1D32 Chr 6q22.31 | AR Rare | ONH | APH, EPP or absent pituitary | GH, CPHD | Oro-facial-digital syndrome, CC agenesis |
| PROKR2 Chr 20p12.3 | AD/AR 5-23% of HH | ONH | PSIS | HH, CPHD | |
| FGF8 Chr 10q24 | AD/AR 1% of HH | ONH | HH, CPHD | HPE, VACTERL | |
| FGFR1 Chr 8p11.2p12 | AD 5-11% of HH | ONH | PSIS | HH, CPHD | Split hand/foot malformation |
AD – Autosomal Dominant, ADHD – Attention Deficit Hyperactivity Disorder, AO – Anophthalmia, APH – Anterior Pituitary Hypoplasia, AR – Autosomal Recessive, ASD – Autism Spectrum Disorder, CC– Corpus Callosum, CPHD - Combined Pituitary Hormone Deficiency, EPP – Ectopic Posterior Pituitary, HH – Hypogonadotrophic Hypogonadism, HPE – Holoprosencephaly, ID – Intellectual Disability, MO – Microphthalmia, ONH – Optic Nerve Hypoplasia, PSIS – Pituitary Stalk Interruption Syndrome, SP – Septum Pellucidum, SNHL – Sensorineural Hearing Loss, VACTERL – Vertebral defects, Anal atresia, Cardiac defects, Tracheo-esophageal fistula, Renal anomalies, and Limb abnormalities.
SYNDROMIC CAUSES OF HYPOPITUITARISM
Septo-optic dysplasia is the most common syndromic presentation of hypopituitarism. Other genes causing hypopituitarism, which do not typically cause optic nerve hypoplasia are discussed below, although GLI2 and BMP4 may present with SOD. Syndromic causes of hypopituitarism are summarized in Table 3.
LHX3
The LIM homeobox proteins LHX3 and LHX4 are expressed in early development and have a co-dependent role in supporting pituitary development. The LHX3 gene located on chromosome 9q34.3 has autosomal recessive expression and is seen in 0.5-1.2% of hypopituitarism cases (39,50,51).
LHX3 is first expressed in Rathke’s pouch at E9.5 following induction by FGF8 and by E12.5 is predominantly expressed in the dorsal aspect of the pouch. LHX3 is also expressed in the ventral hindbrain, inner ear and spinal cord, and is upregulated by LHX4 in the pituitary and SOX2 in the inner ear. LHX3 is expressed in the anterior and intermediate pituitary throughout development, persisting into adulthood and is thus thought to have a role in maintaining some of the cell types. In LHX3 null mice, Rathke’s pouch is initially formed, but regresses resulting in aplasia of the anterior and intermediate lobes, which is at least in part due to failure to support HESX1 and POU1F1 production. LHX3 is critical for maturation of Rathke’s pouch into the anterior pituitary and regulates differentiation of all anterior cell lines, although corticotrophs are somewhat preserved (52).
As a result, LHX3 mutations commonly cause CPHD or panhypopituitarism, with ACTH production normal in approximately half. Children typically present with CPHD at birth but milder phenotypes of developing hypopituitarism throughout childhood are described. A variable appearance is seen on imaging, ranging from normal (10%) to aplasia or hyperplasia, with one case having a microadenoma type appearance (53). Sensorineural hearing loss is seen in up to 50% of cases as LHX3 is present in a restricted region of the inner ear in tandem with SOX2. Intellectual or learning difficulties are reported in 38% of cases (51). A short stiff neck with limited rotation is seen in approximately 70% of cases, with other vertebral anomalies such as scoliosis seen in approximately 50% of cases. Respiratory distress has occasionally been described (54,55).
LHX3 should be suspected in patients with early-onset CPHD with a stiff neck and/or hearing impairment. Neonatal respiratory distress is a rare but important association that may also point to LHX3 mutation.
LHX4
The LHX4 gene is found at chromosome 1q25.2 with autosomal dominant expression causing 0.9-1.4% of hypopituitarism cases (50,56).
LHX4 is expressed in Rathke’s pouch from E9.5 like LHX3; however, by E12.5 it is restricted to the anterior lobe. It also expressed in hindbrain, cerebral cortex and spinal cord tissues. LHX4 and LHX3 work in tandem to support specialization of pituitary cells and LHX4 is important for upregulating LHX3 and POU1F1 and ensuring correct timing of this induction. Thus, LHX4 deficiency may result in an element of LHX3 deficiency and causes a hypoplastic anterior pituitary with appropriate cell differentiation but reduced numbers of hormone producing cells (2,57-59).
Like in LHX3, LHX4 also typically results in CPHD, but milder presentations, including transient isolated GHD, have been reported (56). One family of recessive lethal LHX4 has been reported with severe combined hypopituitarism and fatal respiratory distress thought to be secondary to their hypopituitarism (60). Appearances are again variable on imaging; ectopic posterior pituitary is common with the anterior pituitary ranging from normal to aplasia. Chiari 1 malformation is also associated but likely represents a minority of cases (56). Cardiac defects have been reported in 2 cases and respiratory distress in 4 cases (56,60-62). Penetrance is variable with asymptomatic effected parents being a common occurrence (56).
LHX4 mutation should be considered in patients with Chiari malformation or respiratory/cardiac defects in a patient with hypopituitarism.
GLI2
The GLI2 gene is a zinc finger transcription factor found on chromosome 2q14.2 and has an autosomal dominant inheritance with incomplete penetrance (63). Whilst prevalence has been reported in as many of 13.6% of cases of hypopituitarism, other cohorts have found it to be rare (<1%) (39,63).
GLI2 is expressed in the ventral diencephalon, where it induces BMP4 and FGF8 and is present in the oral ectoderm and Rathke’s pouch from E8.5, where it is important for mediating SHH (Sonic Hedgehog) response. GLI2 is important for supporting pituitary progenitor proliferation and its absence results in reduced pituitary cell numbers, particularly corticotrophs, somatotrophs and lactotrophs, but appropriate differentiation of cells in general (64).
GLI2 has been shown to mediate SHH effect and causes of holoprosencephaly (HPE) are commonly known to affect SHH. In 2003, Roessler et al. identified GLI2 gene mutations in 7 out of 390 patients meeting HPE criteria (65); however, this has since been shown to be an uncommon manifestation GLI2 (66). A study of 112 cases with GLI2 mutations found only three children with holoprosencephaly, and one of whom had an alternative causative gene detected. This study showed that of those with a truncated GLI2 mutation, 62% had pituitary abnormalities and polydactyly and only 15% did not have either anomaly. 58% of those with non-truncated variants had hypopituitarism. 13% had midface hypoplasia with 16% having cleft palate/lip (66). Unlike other causes of holoprosencephaly, GLI2 typically manifests with anterior pituitary anomalies, particularly GH and ACTH deficiency (63).
MRI findings vary, including optic nerve hypoplasia, septum pellucidum or corpus callosum hypoplasia/agenesis, anterior pituitary hypoplasia, ectopic posterior pituitary or PSIS. Thus SOD, PSIS, and rarely HPE are all plausible manifestations of GLI2 mutation (67).
The presence of anterior pituitary deficiency and polydactyly or midface abnormalities/central incisor should raise suspicion for GLI2 and trigger further investigation.
GLI3
Heterozygous mutation of another zinc finger transcription factor GLI3 found on chromosome 7p14.1, results in Pallister-Hall syndrome which presents with mesoaxial polydactyly and hypothalamic hamartoma (68). In mouse studies, GLI3 is not critical for pituitary or hypothalamic development or differentiation, but if combined with GLI2 mutation, the phenotype is more severe than GLI2 anomalies alone (64,69). Mutation in the amino-terminal third of the GLI3 gene results in Greig Cephalosyndactyly syndrome, associated with polydactyly, macrocephaly and hypotelorism, without hypothalamic abnormalities, with Pallister Hall being caused by mutations in the middle third of the gene (70).
In 1980 Hall and Pallister et al. described 6 cases with hypothalamic hamartoma, panhypopituitarism, postaxial polydactyly and imperforate anus, with lethality in all cases (71). Since this time, milder/non-lethal familial forms have been denoted, with the pituitary phenotype varying from normal pituitary function to isolated GHD and panhypopituitarism, thus all cases should be assessed for pituitary dysfunction (72). As with other causes of hypothalamic hamartoma, central precocious puberty can also be associated and should be monitored for (68).
Hypopituitarism Associated With Holoprosencephaly
The prosencephalon cleaves into right and left hemispheres between day 18 and 28 gestation in humans. Failure of this process results in holoprosencephaly (HPE) of varying degrees with lobar holoprosencephaly having separated 3rd ventricles with a connection within the frontal cortex, semilobarholoprosencephaly being partially separated and alobarholoprosencephaly having a continuous single anterior ventricle. (Figure 5) Facial development is affected by the same process and can cause hypotelorism or cleft palate, but also severe cases may have anophthalmia or cyclopia with a superiorly displaced proboscis. Holoprosencephaly is present in 1 in 250 embryos’ but there is a high rate of fetal demise. It is estimated to occur in 1 in 8,000-16,000 live births (73).
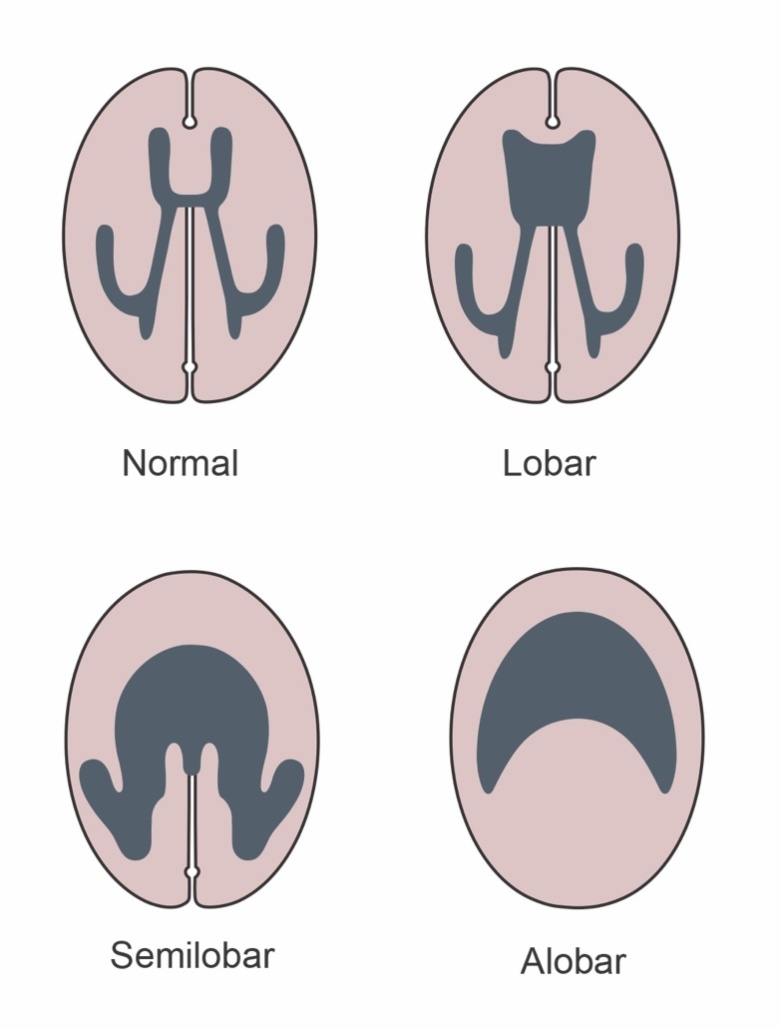
Figure 5.
Types of holoprosencephaly.
Other associated midline defects include underdevelopment of olfactory bulbs and corpus callosum, single midline maxillary incisor and hypoplastic pituitary or ectopic posterior pituitary. Holoprosencephaly is associated with anterior hypopituitarism in 5-10% of cases, with 70% having diabetes insipidus (DI) (74).
Many cases are caused by trisomy 13 making up 40-60% of all causes with a genetic anomaly, trisomy 18 and triploidy are also associated. Sonic hedgehog and ZIC2 mutations make up 5% of non-syndromic cases each. Importantly, structural chromosomal anomalies in almost all chromosomes have been shown to cause holoprosencephaly (75,76).
The SHH gene on chromosome 7q36 is critical for early development of the CNS, particularly the forebrain and is present in the notochord, neural tube and posterior limb buds of the gut. It is a major gene implicated in holoprosencephaly and its effect is mediated through other transcription factors, many of which have been demonstrated to cause HPE shown in Table 2 (73).
There is limited data regarding the rates of anterior pituitary dysfunction with each gene type although cases in SHH and ZIC2 have been reported (67,77).
Table 2.
Genes Associated with Holoprosencephaly and Hypopituitarism
| Gene | Chromosome | % of HPE | Other Features |
|---|---|---|---|
| SHH | 7q36 | 5.4-5.9% | Renal-urinary anomalies |
| ZIC2 | 13q32 | 4.8-5.2% | Renal-urinary anomalies |
| SIX3 | 2p21 | 3% | Typically, severe HPE phenotype |
| TGIF | 18p11.3 | <1% | Wide spectrum of severity |
| CDON | 11q24.2 | rare | CHD, renal dysplasia, radial defects, gallbladder agenesis |
CHD – Congenital Heart Disease, HPE – Holoprosencephaly. Adapted from Tekendo-Ngongang C, et al. Holoprosencephaly Overview. GeneReviews. 2020 (76).
Pituitary Stalk Interruption Syndrome
Pituitary stalk interruption syndrome (PSIS) is a triad of a thin/absent pituitary stalk, ectopic posterior pituitary and aplasia/hypoplasia of the anterior pituitary (Figure 6). SHH and other downstream genes of the SHH signaling pathway, including SIX3, TGIF, CDON and GPR161 have all been reported in 1-2 cases with PSIS and CPHD, without holoprosencephaly (76,78-82). Other causes of hypopituitarism that have been associated include POU1F1, PROP1, LHX3, LHX4, HESX1, SOX3, OTX2, PROKR2 and FOXA2. 100% of patients have been GHD in large case series with gonadotrophin, ACTH and TSH deficiencies in the majority and hyperprolactinemia in a minority (83).

Figure 6.
Lateral appearance of pituitary stalk interruption syndrome (PSIS). Adapted from Larkin S. et al, Endotext (1).
ROBO1
Heterozygous ROBO1 gene mutations (found on chromosome 3p12.3) has been described in 5 patients with PSIS, with a 6th case of homozygous mutation (84,85). Ocular anomalies, including hypermetropia and ptosis were also seen, but penetrance was incomplete with unaffected parents in some cases. Combined GHD and TSHD, and a case of panhypopituitarism have been described (84).
FOXA2
The forkhead box A2 (FOXA2) gene is found on chromosome 20p11.21 with dominant or de novo inheritance (86). FOXA2 regulates expression of GLI2 and SHH and mutations of the FOXA2 gene and 20p11 deletions have been reported to cause hypopituitarism (87-89). Imaging demonstrates anterior pituitary hypoplasia, absent or ectopic posterior pituitary and absent or thin pituitary stalk (86,90). FOXA2 is also involved in regulation of the ABCC8 and KCNJ11 genes, critical for the K-ATP channels in pancreatic beta-cells, and mutation has been shown to cause congenital hyperinsulinism (91). Other phenotypic features include craniofacial dysmorphism, choroidal coloboma and malformations of the liver, lung and heart (90,92,93).
EIF2S3
Mutations in the EIF2S3 gene found on chromosome Xp22.11, result in the MEHMO syndrome (Mental retardation, Epileptic seizures, Hypogenitalism, Microcephaly and Obesity). EIF2S3 is expressed in the pituitary and hypothalamus during embryological development (94). Mutation typically results in GHD, and panhypopituitarism has been described (94,95). Imaging typically demonstrates corpus callosum thinning and either normal, or hypoplastic anterior pituitary (94).
BMP4
Bone morphogenetic protein 4 (BMP4) gene is found on chromosome 14q22-q23 with dominant inheritance and incomplete penetrance (70). BMP4 is expressed in the diencephalon and infundibulum from E8.5, and absence results in failure of pituitary placode and Rathke’s pouch development in mouse studies (96). Mutations are frequently associated with anophthalmia/microphthalmia, cleft lip/palate and polydactyly/syndactyly. In 2018 Rodríguez-Contreras et al. reported a pathogenic BMP4 mutation in a boy with GH and TSH deficiency and myopia, but further cases have not yet been described (97).
ARNT2
Chromosome 15q25.1 is the locus for the aryl hydrocarbon receptor nuclear translocator 2 (ARNT2) gene and has autosomal recessive inheritance. ARNT2 is expressed in the developing anterior and posterior pituitary and hypothalamus and mutations result in CPHD with diabetes insipidus. Pituitary imaging demonstrates absent posterior pituitary bright spot with stalk thinning and a hypoplastic anterior pituitary. It is also associated with postnatal microcephaly, hypoplastic frontal and temporal lobes, renal anomalies and vision impairment (98).
TBC1D32
The TBC1 domain family member 32 (TBC1D32) gene is found at Chromosome 6q22.31 and results in disease with an autosomal recessive inheritance. TBC2D32 is expressed in the developing hypothalamus and Rathke’s pouch and is involved in SHH signaling and ciliary functioning. Isolated GHD or panhypopituitarism were found in the limited cases reported to date. Anterior pituitary hypoplasia or aplasia is described with ectopic or absent posterior pituitary as is partial agenesis of the corpus callosum and optic chiasm hypoplasia. Other phenotypic features include oral anomalies (cleft palate), facial dysmorphism (hypertelorism/cleft lip) and limb anomalies (polydactyly/syndactyly), consistent with oro-facial-digital syndrome (99).
Table 3.
Syndromic Causes of Hypopituitarism
| Gene | Inheritance/ prevalence | SOD/ PSIS/ HPE | Pituitary appearance | Pituitary deficiencies | Other Features |
|---|---|---|---|---|---|
| LHX3 Chr 9q34.3 | AR 0.5-1.2% | No | APH, Normal, hyperplasia | CPHD, ACTH in 50% | SNHL, ID, short stiff neck, scoliosis, rarely RDS |
| LHX4 1q25.2 | AD 0.9-1.4% | No | APH, EPP | CPHD | CC hypoplasia, Chiari 1 malformation, rarely CHD/RDS |
| GLI2 2q14.2 | AD 1-13% | SOD, HPE, PSIS | APH, EPP, PSIS | GH, ACTH, FSH/LH, TSH | ONH, SP/CC changes, polydactyly, midface hypoplasia, cleft palate/lip |
| GLI3 Chr 7p14.1 | AD Rare | No | Hypothalamic hamartoma | GH, ACTH, FSH/LH, TSH | Postaxial polydactyly, imperforate anus. CPP |
| ROBO1 Chr 3p12.3 | AD/AR Rare | PSIS | PSIS | GH, TSH Pan-hypopituitarism | Hypermetropia, ptosis |
| EIF2S3 Chr Xp22.11 | XR | No | Normal, APH | GH, Pan-hypopituitarism | CC hypoplasia, ID, epilepsy, gonadal failure, microcephaly, obesity |
| BMP4 Chr 14q22-23 | AD Rare | SOD | Normal | GH, TSH | AO/MO, myopia, cleft palate/lip, polydactyly |
| FOXA2 Chr 20p11.21 | AD/De novo | PSIS | APH, EPP, PSIS | Pan-hypopituitarism, | Hyperinsulinism, craniofacial dysmorphism, choroidal coloboma, and liver, lung and heart malformations |
| ARNT2 Chr 15q25.1 | AR | No | APH, thin stalk, absent PP | Pan-hypopituitarism, ADH | Microcephaly, fronto-temporal hypoplasia, renal anomalies and vision impairment. |
| TBC1D32 Chr 6q22.31 | AR | SOD | APH, EPP or absent pituitary | GH, pan-hypopituitarism | Oro-facial-digital syndrome, CC agenesis |
| HPE Genes SHH ZIC2 SIX3 TGIF CDON | Rare | HPE, PSIS | Normal Rarely PSIS | ADH Rarely CPHD |
AO – Anophthalmia, APH – Anterior Pituitary Hypoplasia, CC – Corpus Callosum, CHD – Congenital Heart Disease, CPP – Central Precocious Puberty, EPP – Ectopic Posterior Pituitary, FTT – Failure To Thrive, HPE – Holoprosencephaly, ID – Intellectual Disability, MO – Microphthalmia, PP – Posterior Pituitary, PSIS – Pituitary Stalk Interruption Syndrome, RDS - Respiratory Distress, SOD – Septo-optic Dysplasia, SNHL – sensorineural hearing loss, SP – Septum Pellucidum, XR – X-linked Recessive
NON-SYNDROMIC CAUSES OF HYPOPITUITARISM
POU1F1

Figure 7.
Cross-sectional representation of pituitary cell groups in POU1F1. Adapted from Larkin S. et al, Endotext (1).
POU1F1 was the first genetic cause of CPHD detected, initially described in mice (known as PIT-1) and 2 years later in humans. It is a member of the POU family of transcription factors, and the gene is located at 3p11.2. Approximately 65% of cases are autosomal recessive (homozygous/compound heterozygous) with 35% having an autosomal dominant form, the most common being the R271W mutation. In a review of 15 cases, the majority had affected parents, with only 3 cases of hormonally intact parents (100). POU1F1 is present in 2.8% of CPHD, with up to 21% of familial CPHD (50). There is considerable variability between countries, with one cohort of 23 CPHD patients in India detected 6 POU1F1 mutations and many other studies having low numbers of detected mutations (50,100,101).
Expression of POU1F1 is triggered by PROP1 and is seen within the pituitary from E13.5, persisting through to adulthood. It is critical for supporting the differentiation and proliferation of thyrotroph, lactotroph and somatotroph cells, and stimulates the GH1, prolactin and TSHB genes, required for hormone synthesis (102). LHX3, LHX4 and OTX2 upregulate POU1F1 but PROP1 is essential for its function (52,57,59).
Tatsumi et al. detected the mutation in a case of “cretinism” with cognitive impairment and short stature, with GH, TSH and prolactin deficiency, reported in 1992 (103). 100% of cases in the literature have GH deficiency with 87.5% having TSH deficiency and 95.6% having prolactin deficiency (demonstrated in Figure 7). TSH deficiency is typically detected in infancy, either prior to, or simultaneously with GH and prolactin deficiency, with ~13.5% detected after GHD. 70% have anterior pituitary hypoplasia, but other midline and peripheral defects are rarely seen. Autosomal dominant forms are less likely to have an absent pituitary (63.6%) and typically have less severe GHD with 20% having an undetectable GH peak on stimulation testing, compared to 48%. Apart from hypopituitarism, there are no other phenotypic features known to be associated with POU1F1 (100).
Patients with severe short stature, GHD, prolactin deficiency and TSH deficiency only and isolated anterior pituitary findings on MRI should raise suspicion of POU1F1 mutation, especially if there is a known family history.
PROP1
The prophet of POU1F1 (PROP1) is a paired like homeodomain transcription factor on chromosome 5q35 with autosomal recessive inheritance (104). It has been shown to cause 11% of all CPHD patients, with up to 50% of familial forms and approximately 7% of sporadic forms (50). This is highly variable across different populations and ethnicities with no cases found in studies in Japan, Germany, the United Kingdom, Spain or Australia and numbers in Lithuania being as high as 65% of CPHD cases and >90% of familial cases (50).
Prop1 is initially expressed in Rathke’s pouch at E10, with levels rising until E12 and decreasing to E15.5 when it ceases. It stimulates stem cell transformation to mesenchymal cells and supports cell migration and differentiation, without which cells remain static. It is essential for POU1F1 initiation and failure of this results in further issues with cell differentiation. It is involved in the stimulation and differentiation of all cell lines including gonadotrophins and to a lesser extent, corticotrophs (104).
Most cases have CPHD, and is typically progressive, with the number of diagnosed deficiencies and severity of deficiency increasing over time. Onset of GHD, TSHD and HH can occur antenatally with diagnosis at birth, but often present later, and in rare cases, some cell lines are normal. ACTH deficiency has been described from the age of 6 and typically develops across late childhood and adolescence. Imaging demonstrates a normal or hypoplastic anterior pituitary (occasionally hyperplastic) with normal posterior pituitary structures, and aside from features secondary to hypogonadism (micropenis and undescended testis) no extracranial phenotype has been described (105).
The presentation is typically milder than POU1F1 mutation and the presence of features of hypogonadism and adrenal insufficiency in a non-syndromic child with CPHD should raise suspicion for PROP1 over POU1F1.
ISOLATED HORMONE DEFICIENCIES
The presence of any pituitary hormone deficiency raises the question of other hormone deficiencies. While isolated forms of pituitary deficiency occur, there is the potential for other deficiencies to develop over time as is seen in POU1F1 or LHX, and routine monitoring is required (53,100). It is also true that genes classically causing isolated hormone deficiencies, like GH1, can cause CPHD, and thus screening may be required (106,107).
Isolated Growth Hormone Deficiency

Figure 8.
Cross-sectional representation of pituitary cell groups in isolated growth hormone deficiency. Adapted from Larkin S. et al, Endotext (1).
Isolated growth hormone deficiency (IGHD) is reported to have a prevalence between 1 in 4,000-10,000. Whilst most cases are sporadic, suggesting an in-utero insult to development of a specific cell-line, 3-30% have a first degree relative, supporting the possibility of genetic causes (108). Approximately 6.5% of patients have a genetic cause currently detectable (39) with rates as high as 38% in those with a family history (109). Growth Hormone 1 (GH1) and Growth Hormone Releasing Hormone Receptor (GHRHR) genes are the most commonly implicated, although genes implicated in CPHD, such as HESX1, SOX3, OTX2, LHX4 and GLI2 can also present as isolated GHD, (appearance shown in Figure 8). Genetic causes are classified into 4 categories, listed in table 4. Type 1A which has undetectable GH levels caused by autosomal recessive GH1 gene deletions, Type 1B caused by GH1 gene splice site mutation or other genes (such as listed above), Type 2 caused by exon skipping mutations in GH1 cause an autosomal dominant phenotype, and Type 3 is X-linked recessive, and is associated with agammaglobulinemia (108).
The GH1 gene located on 17q22-24consists of 5 exons and 4 introns and encodes for a 191 amino acid peptide with a molecular weight of 22kDa. Transcription of GH1 is triggered by cAMP release in response to GHRH binding to the GHRHR, and is down-regulated by somatostatin via inhibiting cAMP release, in response to receptor binding (108). This pathway is demonstrated in Figure 9. GH1 mutations cause 22.7% of familial isolated GHD, the majority being type 2 autosomal dominant mutations (109).

Figure 9.
Cellular function of somatotroph cell. cAMP- Cyclic Adenosine Monophosphate, GH – Growth Hormone, GHRH – Growth Hormone Releasing Hormone, GHRHR – Growth Hormone Releasing Hormone Receptor.
TYPE 1A
In 1981 Phillips et al. described GH1 mutation in Swiss children with a homozygous deletion causing Type 1A GHD (110). Type 1A GHD is caused by a GH1 gene deletion or less commonly, by a frameshift or nonsense mutation and makes up <15% of GH1 mutation related disease (109). The resulting phenotype is of severe GHD, with undetectable levels and can present with hypoglycemia neonatally as well as growth failure in the first 6 months. Complicating therapy is the frequent occurrence of anti-GH antibodies requiring therapy with recombinant insulin-like growth factor 1 (IGF1) (108).
TYPE 1B
Type 1B GHD caused by GH1 mutations are typically the result of gene splice site mutations and are again autosomal recessive. They have a variable phenotype ranging from an IGHD Type 1A phenotype to mid-childhood growth failure (108).
GHRHR anomalies also cause a Type 1B phenotype. GHRHR is found on chromosome 7p14.3 and was reported in 1996 by Wajnrajch et al. (111). Patients have normal GHRH levels but undetectable GH and low IGF1 levels. The phenotype is of proportional short stature, with anterior pituitary hypoplasia (108). GHRHR mutations are responsible for 16-19% of familial IGHD but are rarely the cause of sporadic IGHD (109). No GHRH mutations causing short stature have been reported to date (108).
TYPE 2
Type 2 GHD originates from mutations that result in loss of exon 3 from the final protein. This causes a dominant negative effect on secretion of normal GH from the other allele, through retention in the endoplasmic reticulum, impairing Golgi apparatus activity (108). Type 2 GHD has varying degrees of severity as well as of pituitary hypoplasia and represents approximately 70% of GH1 mutation related disease (109).
TYPE 3
IGHD has been reported in 10 cases of X-linked agammaglobulinemia, known as IGHD type 3; however, an individual gene causing both associations has not been well characterized (112).
Sporadic IGHD does not typically have a genetic basis; however, familial forms have a high likelihood of detecting a mutation (38%) (107). It is important in patients with IGHD to always consider the possibility of a developing CPHD, with this occurring in 5.5% without structural anomalies, and 20.7% with structural anomalies(106,107(113)). This should be considered even in patients with GH1 mutation’s, in particular Type 2 mutations have been found to develop other deficiencies, in particular TSH and ACTH deficiency (106,107).
Table 4.
Summary of Types of Isolated Growth Hormone Deficiency.
| IGHD type | Genetic abnormalities | Inheritance | Pituitary imaging | Percentage of familial cases | Comments |
|---|---|---|---|---|---|
| 1A | GH1 gene deletions and nonsense mutations | AR | Normal/ hypoplastic | 2-4% | GH levels undetectable, anti-GH antibodies, transient response to GH therapy, |
| 1B | GH1 gene splice site mutations | AR | Normal/ hypoplastic | 2-4% | |
| GHRHR gene mutations | AR | Normal/ hypoplastic | 16-19% | ||
| 2 | GH1 gene splice site and missense mutations and intronic deletions | AD | Normal/ hypoplastic | 15-22% | Variable height, CPHD may occur |
| 3 | Unknown | XR | EPP | Unknown | Agammaglobulinemia |
AD – Autosomal Dominant, AR – Autosomal Recessive, CPHD – Combined Pituitary Hormone Deficiency, EPP – Ectopic Posterior Pituitary, GHRHR – Growth Hormone Releasing Hormone Receptor, IGHD – Isolated Growth Hormone Deficiency, XR – X-li linked Recessive. Adapted from Alatzoglou K. et al, JCEM;94:3191-3199 and Casteras A. et al, Endocrinology, Diabetes and Metabolic Case Reports (109,114).
Central Hypothyroidism
Neonatal screening studies using T4 testing determined neonatal central hypothyroidism to occur in 1 in 16000-30000 infants (115,116). Up to 40% of these cases have isolated central hypothyroidism, with neonatal rates between 1 in 40,000 to 75,000, while some cases of isolated central hypothyroidism develop after the neonatal period (117). Thyroid hormone receptors (THR) are present on both the hypothalamus and pituitary and downregulate hormone secretion. Thyrotropin releasing hormone (TRH) is released from the hypothalamus and binds the TRH receptor (TRHR) upregulating TSH production (pathway demonstrated in Figure 10). 5 different genes have been implicated but there is limited data on the frequency of genetic mutations in patient with isolated central hypothyroidism (118).
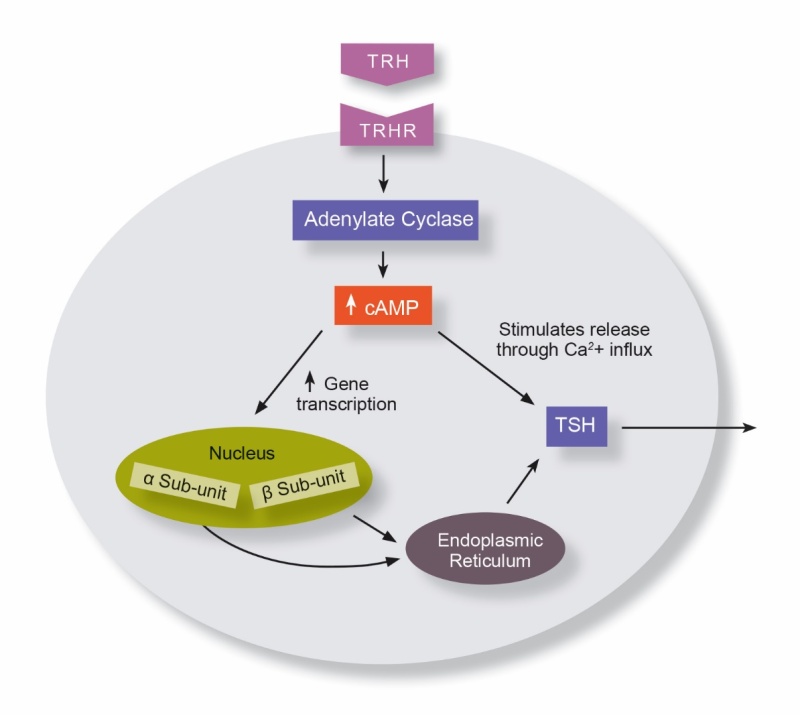
Figure 10.
Cellular function of thyrotroph cell. cAMP- Cyclic Adenosine Monophosphate, TRH – Thyrotropin Releasing Hormone, TRHR – Thyrotropin Releasing Hormone Receptor, TSH – Thyroid Stimulating Hormone.
TSHB
Thyroid stimulating hormone beta subunit (TSHB) gene is found on chromosome 1p13.1 and is associated with autosomal recessive inheritance (118). Hayashizaki et al. described the mutation in 1989 in a patient with severe neonatal onset hypothyroidism associated with prolonged jaundice, failure to thrive and developmental delay (119). Subsequent cases have also had severe deficiencies presenting neonatally (118).
TRHR
The thyrotropin releasing hormone receptor (TRHR) gene is found on chromosome 8q23.1 and again has autosomal recessive inheritance. The phenotype is milder than for TSHB with detection ranging from neonatal period to late childhood, and other patients being asymptomatic in adulthood. This is likely due to the presence of thyroid hormone receptors on the thyrotroph cells, upregulating TSH release independently of TRH (118).
IGSF1
Immunoglobulin superfamily 1 (IGSF1) gene is found on chromosome Xq26.1 and is associated with X-linked recessive central hypothyroidism. Hypothyroidism is seen in all cases, but the degree of deficiency can be variable, with many presenting in the neonatal period with jaundice and failure to thrive, others are detected asymptomatically on family screening (118).
IGSF1 is expressed in Rathke’s pouch and the adult pituitary gland and mutation results in low TSHB and TRHR mRNA, thus it is proposed IGSF1 has a role in regulating expression of these genes. 60% of patients have prolactin deficiency, supporting the involvement of decreased TRHR activity. GHD has been reported in a few cases, and 1 patient has been reported to have a hypoplastic anterior pituitary and ectopic posterior pituitary (118).
Other known associations include macroorchidism in 80% with low adulthood testosterone levels but preserved fertility. Follow up shows that despite thyroxine replacement, 25% of children and 75% of adults are overweight and 65% of cases have ADHD (attention deficit hyperactivity disorder) (118).
OTHER X-LINKED CAUSES
Transducing b-like protein X-linked (TBL1X) gene is found at chromosome Xp22.2 and mutations are thought to prevent upregulation of TSHB and TRHR transcription, causing central hypothyroidism. It has a mild hypothyroid phenotype and is associated with hearing loss, and ADHD (118).
Insulin receptor substrate 4 (IRS4) is found on chromosome Xq22.3 is often described as a cause of TSHD; however, mutations to date have resulted in low T4 but normal T3 and TSH, which does not suggest true central hypothyroidism (118).
Central hypothyroidism is rare and while genetic testing may be appropriate, the prevalence of individual causative genes is unknown. If genetics are not performed, siblings of patients should have formal thyroid function tests including a T4 arranged in the newborn period.
Table 5.
Genetic Causes of Isolated Central Hypothyroidism
| Gene | Inheritance | Degree of hypothyroidism | Other pituitary deficiency | Other features |
|---|---|---|---|---|
| TSHB 1p13.1 | AR | Severe | Nil | Jaundice, FTT, developmental delay |
| TRHR 8q23.1 | AR | Mild | Prolactin | |
| IGSF1 Xq26.1 | XR | Variable (mild to severe) | Prolactin, (rarely GH) | Macroorchidism, low testosterone, obesity, ADHD |
| TBL1X Xp22.2 | XR | Mild | Nil | Hearing loss, ADHD |
AD – Autosomal Dominant, ADHD – Attention Deficit Hyperactivity Disorder, AR – Autosomal Recessive, XR – X-linked Recessive. Adapted from Tajima T. et al, Clin Pediatr Endocrinol;28(3):69-79 (118).
Isolated ACTH Deficiency
Isolated ACTH deficiency (IAD) is an extremely rare but serious condition with mortality rates of up to 25% in severe neonatal-onset forms (120).
Corticotrophin releasing hormone (CRH) is produced by the hypothalamus in response to low cortisol levels and binds to corticotroph cells. CRH stimulates cAMP release triggering protein kinase A (PKA) which binds CRH response element-binding protein (CREB) which in turn stimulates TBX19 which stimulates production of pro-opiomelanocortin (POMC). This is converted by prohormone convertase 1/3 (PCSK1 gene) to ACTH, which stimulates glucocorticoid production in the adrenal gland (Figure 11) (121). Abnormalities in TBX19, POMC and PCSK1 genes have all been shown to cause IAD, and NFKB2 mutation causes IAD and immunodeficiency. Genetic causes are summarized in Table 6.

Figure 11.
Cellular function of the corticotroph cell. ACTH – Adrenocorticotrophic Hormone, cAMP- Cyclic Adenosine Monophosphate, CRH – Corticotropin Releasing Hormone, CRHR – Corticotropin Releasing Hormone Receptor, PC 1/3 – Prohormone Convertase 1/3, POMC – Pro-opiomelanocortin, TBX19 – T-Box Transcription Factor 19.
TBX19
T-box transcription factor 19 (TBX19) previously known as TPIT is located on chromosome 1q24.2 and causes severe neonatal onset IAD. It has an autosomal recessive inheritance and has been detected in 65% of cases of severe IAD, with 100% penetrance reported to date (120). As reported above, TBX19 is an important part of the pathway from CRH stimulation to ACTH release but is also critical for terminal differentiation of corticotrophs and can result in absence or severely hypoplastic cells (120).
All patients present with severe neonatal hypoglycemia and >50% have hypoglycemic seizures, with a mortality rate of 25%. >60% have prolonged cholestatic/ conjugated jaundice and if diagnosis is delayed, intellectual impairment and developmental delay have been reported (120). Other abnormalities have occasionally been reported including Chiari type 1 malformation, and subtle dysmorphic features (120,122).
Cortisol assessment should be performed in any neonate with recurrent hypoglycemia and cholestatic jaundice, and consideration of TBX19 gene assessment on confirmation of the diagnosis. Low levels of maternal estriol on antenatal testing has been shown to herald adrenal steroidogenesis issues and seen in a case of IAD caused by TBX19, but estriol is no longer routinely used in antenatal screening (123).
PCSK1
Proprotein convertase of Subtisilin/Kexin 1 gene (PCSK1) found on chromosome 5q15-21, codes for prohormone convertase 1/3 (PC 1/3) and has an autosomal recessive pattern of inheritance. It is found throughout the body including the pituitary, hypothalamus and endocrine pancreas. It plays a critical role in processing POMC, proinsulin and proglucagon but multiple other hormones as well (124).
PC 1/3 is required for conversion of POMC to ACTH in corticotroph cells. Deficiency thus results in low levels or absence of ACTH production. Despite this, there is typically a mild clinical phenotype, only 75% of patients have ACTH deficiency and not all are associated with hypocortisolemia. It is unclear whether this is due to partial ACTH binding capacity of POMC or other ACTH precursors, or whether an alternative enzyme compensates (124).
PC1/3 deficiency can also cause CPHD. HH, TSHD, and GHD are seen in approximately 50%, 70% and 35% of cases respectively and have been demonstrated or proposed to be secondary to failure of hypothalamic conversion of pro-releasing hormones (proGnRH to GnRH (suspected), proTRH to TRH, proGHRH to GHRH). Like with the corresponding adrenal insufficiency, thyroid and GH deficiency are often phenotypically mild. DI, presumed due to ineffective provasopressin conversion to vasopressin is also reported in a subset of patients (124).
Other associated features include obesity, occurring in nearly all patients, associated with hyperphagia and develops from approximately 2 years of age, the cause of which is multifactorial. Like in Prader-Willi syndrome, initially children present with failure to thrive; however, this is likely due to the presence of malabsorptive diarrhea. All cases to date have had severe neonatal diarrhea with demonstrated fat, amino acid and monosaccharide malabsorption, with the majority requiring hospitalization and parenteral nutrition. The underlying mechanism is not understood, and the intestinal architecture is typically normal on biopsy. The malabsorptive state typically displays partial improvement after the first year of life (124).
PC1/3 is also critical for cleavage of proinsulin to insulin, resulting in undetectable insulin levels. To compensate, much higher quantities of proinsulin are produced which typically prevents diabetes mellitus as it is maintains a 2-5% activity at the insulin receptor. The most common symptom is in fact postprandial hypoglycemia, resulting from proinsulins four to six times longer half-life, causing a relative hyperinsulinism after meals. Later in life some patients proceed to diabetes mellitus likely due to b-cell exhaustion (124).
PCSK1 mutation should be considered in patients with early onset malabsorptive diarrhea and later onset obesity. Other features may include post-prandial hypoglycemia, gonadal abnormalities and reduced linear growth. CPHD and DI should be screened for but are typically mild (124).
POMC
Pro-opiomelanocortin gene (POMC) is found on chromosome 2p23.3 and deficiency manifests in an autosomal recessive inheritance pattern (125). It causes a classic triad of adrenal insufficiency, hypopigmented skin with red hair, and obesity associated with hyperphagia, that can commence within infancy (125,126).
Children typically present with neonatal hypoglycemia (~70%) and adrenal insufficiency is detected at this point. Convulsions, hyperbilirubinemia and hyponatremia were present in over 30% of presenting cases. All known cases to date have been reported to have adrenal insufficiency and obesity, with 75% having red or Reddish-brown hair (125).
Other endocrinopathies have been reported, TSHD being the most common. Melanocyte stimulating hormones (MSH) bind the melanocortin 4 receptor which has been shown to regulate TRH release and it is hypothesized that the limited production of MSH’s drives the hypothyroidism. TSHD is reported in 35% of cases to date and is often subclinical. GHD and HH have been reported in 10-20% of patients with mechanisms not well understood. Type 1 diabetes has also been seen in 10-20% of cases (double antibody positive) it is thought this may be secondary to the increased inflammatory state in the absence of melanocortin which have been demonstrated to have an anti-inflammatory effect (125).
DAVID SYNDROME
DAVID syndrome (Deficient Anterior pituitary with Variable Immune Deficiency) was first described in 2011 and results from a mutation of NFKB2 gene on chromosome 10q24.32. Classically it presents with common variable immunodeficiency (CVID) due to hypogammaglobulinemia, typically resulting in recurrent sinopulmonary infections. IAD typically develops years or decades later; however, initial presentation with IAD has been reported in 1 case (127). GHD has been reported in 2 cases and TSHD in a single case, and imaging may demonstrate a normal or hypoplastic anterior pituitary (127,128).
Neonatal onset isolated adrenal deficiency should be screened for TBX19 mutations, and if absent, especially in a child with red hair, POMC gene testing should be considered, similarly the possibility of PCSK1 mutation in children with malabsorptive diarrhea and adrenal insufficiency.
Table 6.
Summary of Genetic Causes of Isolated ACTH Deficiency (IAD).
| Gene | Inheritance | Onset of IAD | Presentation | Other pituitary deficiencies | Other phenotypes |
|---|---|---|---|---|---|
| TBX19 Chr 1q24.2 | AR, 100% penetrance | 65% of severe neonatal onset IAD | Neonatal hypoglycemia, cholestatic jaundice | Nil | Intellectual impairment if delayed diagnosis, Chiari type 1 malformation |
| PCSK1 Chr 5q15-21 | AR | Variable | Infantile malabsorptive diarrhea, | FSH, LH, GH, TSH, Rarely ADH | Obesity in early childhood |
| POMC Chr 2p23.3 | AR | Neonatal | Neonatal hypoglycemia, jaundice | TSH (35%), GH, LH/FSH (10%) | Obesity, reddish brown hair. Type 1 diabetes |
| NFKB2 Chr 10q24.32 | AD | Variable | Recurrent sinopulmonary infections | GH, TSH (rare) | Combined variable immunodeficiency |
AD -- Autosomal Dominant, AR – Autosomal Recessive.
Central Hypogonadism
Congenital hypogonadotrophic hypogonadism (HH), also known as Kallmann syndrome when seen in association with anosmia, occurs in 1 in 10,000 – 50,000 people, with a 4 to 1 male predominance (12,129).
Gonadotrophin releasing hormone (GnRH) is secreted in a pulsatile manner from the hypothalamus and has a complex pattern of regulation by estradiol and other factors. It binds GnRH receptors on gonadotroph cells leading to secretion of LH and FSH. In fetal development, the cells responsible for GnRH release migrate to the hypothalamus from the olfactory placode, and thus mutations triggering HH are often associated with olfactory dysgenesis and anosmia (12).
There are over 25 gene defects implicated in HH, and a mutation is found in up to 50% of cases. No gene is implicated in more than 10% of familial cases. Oligogenicity is also described in multiple genes which further complicates diagnosis. Broadly speaking, HH is a result of one of four mechanisms, defects in GnRH fate specification, GnRH neuron migration, abnormal neuroendocrine secretion/homeostasis or gonadotroph cell defects (130).
55% of cases have anosmia/hyposmia (129). The majority of anosmic cases have an autosomal dominant inheritance, except for FEZF1, PROK2 and PROKR2, while norosmic cases may be autosomal dominant or recessive. However, incomplete penetrance, oligogenicity and variability of anosmia and norosmia of some genes makes delineating likely causative genes difficult. X-linked recessive inheritance is seen in KAL1 mutations only to date (12,130).
While individual genes have specific associations, larger studies demonstrate between 5 and 15% of cases have hearing loss, palate anomalies or unilateral renal agenesis. Up to 30% have synkinesia or eye movement disorders and one study found 5-15% of patients had dental agenesis, scoliosis or finger anomalies (129).
In the absence of anosmia or congenital anomalies, HH can be difficult to separate from constitutional delay (normal late onset puberty). Adolescents require pubertal induction to support bone, metabolic and sexual health and to limit the effect on mental health, and input from fertility specialists may be required when starting a family (129).
Multiple genes associated with HH have been implicated in combined hypopituitarism including PROKR2, FGF8, FGFR1 and CHD7 and summarized in Table 7.
CHARGE SYNDROME (CHD7)
CHARGE syndrome results from mutation of the CHD7 gene on chromosome 8q12.2 with autosomal dominant inheritance, although typically resulting from de novo mutation and occurs in 1 in 15,000 to 17,000 live births (131). The acronym CHARGE is of Coloboma of the eye, Heart defects, Atresia of the choanae, Retardation of growth and development, Genital hypoplasia and Ear and hearing abnormalities, and was coined by Pagon et al. in 1981 (132).
HH, typically presenting with cryptorchidism and micropenis in males is present in 60-88% of patients, most of whom will not enter puberty spontaneously (131,133). Growth failure is present in 60-72% of cases, with approximately 10% being due to GHD (131,134-136). Other pituitary abnormalities are rare with one study reporting 2 out of 25 patients with TSHD, and while other studies report hypothyroidism, these are typically primary, or the etiology is not reported. Imaging typically demonstrates olfactory bulb anomalies, but the pituitary gland is typically normal (134,136). Thus, while hypogonadism is a well-known consequence of CHARGE syndrome, GHD and TSHD must also be considered.
PROKR2
Prokineticin-2 Receptor (PROKR2) gene is found on chromosome 20p13 and typically has dominant inheritance with incomplete penetrance, but recessive mutations are also described (70). PROKR2 prevalence within HH varies greatly, ranging from 5.1-23.3% of mutations in different populations(137). PROKR2 is highly expressed in the olfactory placode and GnRH neurons in the mouse model but is not seen in the pituitary or infundibulum. It typically causes HH and individuals often harbor mutations in other HH genes, demonstrating it sometimes has an oligogenic effect (70). CPHD has been described in 20 patients, 13 of whom had SOD and 4 with pituitary stalk interruption syndrome (PSIS) (5-8). The pathogenicity of the mutations in these cases is uncertain (6,70).
FGF8
The fibroblast growth factor 8 (FGF8) gene is found on chromosome 10q24 and mutations can have either autosomal dominant or recessive inheritance, making up approximately 1% of HH cases (70,137). FGF8 is first expressed at E9.25 in the diencephalon, prospective hypothalamus and infundibulum, and activates LHX3 (70,96,138,139). FGF8 plays a role in coordinating anterior pituitary development, with excess in mice resulting in a dysplastic, hyperplastic pituitary and absence results in pituitary hypoplasia. Most mutations are associated with Kallmann syndrome; however, multiple cases of CPHD have been identified as well as a case of Holoprosencephaly, and SOD with GH deficiency. VACTERL (Vertebral defects, Anal atresia, Cardiac defects, Tracheo-esophageal fistula, Renal anomalies, and Limb abnormalities) or VACTERL like phenotype has also been described (70).
FGFR1
The Fibroblast Growth Factor Receptor 1 (FGFR1) gene is found on chromosome 8p11.2-p12 with autosomal dominant inheritance making up 5-11.7% of mutations associated with HH (138,140). It is expressed in the olfactory placode where is ensures fate specification, proliferation and migration of GnRH neurons (140). It typically causes HH, with or without anosmia but also been reported in CPHD. 9 cases have been described, 3 associated with SOD, 3 with PSIS and 3 without (5,7,141,142). While some variants have demonstrable pathogenicity for this phenotype, it is unclear in others. Split hand/foot malformation is frequently part of the FGFR1 phenotype and the mutation is also seen in Pfeiffer syndrome, Hartsfield syndrome and Jackson-Weiss syndrome (142).
Table 7.
Causes of Hypogonadotrophic Hypogonadism Associated with Combined Pituitary Hormone Deficiency.
| Gene | Inheritance/ prevalence | SOD/ PSIS/HPE | Other pituitary deficiencies | Other Features |
|---|---|---|---|---|
| PROKR2 Chr 20p12.3 | AD/AR 5-23% of HH | SOD/ PSIS | CPHD | |
| FGF8 Chr 10q24 | AD/AR 1% of HH | SOD/HPE | GH/CPHD | VACTERL |
| FGFR1 Chr 8p11.2-p12 | AD 5-11% of HH | SOD/PSIS | GH/CPHD | Split hand/foot malformation |
| CHARGE CHD7 Chr 8q12.2 | AD/de novo 6% of HH | Nil | GH/TSH | Coloboma, CHD, choanal atresia, short stature, ID, genital hypoplasia, ear abnormalities |
AD – Autosomal Dominant, AR – Autosomal Recessive, CHD – Congenital Heart Disease HPE – Holoprosencephaly, ID – Intellectual Disability, PSIS – Pituitary Stalk Interruption Syndrome, SOD – Septo-optic Dysplasia, VACTERL- Vertebral defects, Anal atresia, Cardiac defects, Tracheo-esophageal fistula, Renal anomalies, and Limb abnormalities.
Central Diabetes Insipidus
Isolated diabetes insipidus (DI) is rare, with a reported prevalence of 1 in 25000, including both nephrogenic and central forms (143). Arginine vasopressin (AVP) or antidiuretic hormone (ADH) is produced from pre-pro-AVP in the supraoptic and paraventricular hypothalamic nuclei, and their axonal projections extend into the posterior pituitary. AVP mediates its affect through the AVPR2 receptor which increases urine concentration in the collecting duct. Apart from cases related to holoprosencephaly, genetic causes are rare (144).
AVP
The AVP gene is located on chromosome 20p13 and has an autosomal dominant inheritance except for a few recessive mutations. The AVP mutant proteins are thought to accumulate in the endoplasmic reticulum resulting in cell death. As a result, it usually manifests between 1 to 6 years of age with gradual progression of deficiency but there is wide variation even within affected families. Along with polyuria and polydipsia classically seen in DI, failure to thrive is commonly seen and resolves with AVP replacement (144).
WOLFRAM SYNDROME
The WFS1 gene, located on chromosome 4p16.1 encodes for wolframin which is an endoplasmic reticulum channel and has an autosomal recessive inheritance (144). Prevalence is estimated between 1 in 500,000 – 770,000 children with rates higher in areas of increased consanguinity (1 in 68,000 in Lebanon) (145). Wolfram syndrome causes diabetes mellitus, usually diagnosed at 6 years of age, optic nerve atrophy at around age 11 and sensorineural hearing loss typically progressing over time. DI is seen in approximately 70% of cases and is typically diagnosed in the 2nd or third decade of life (143). A case DI without other manifestations of Wolfram syndrome has been reported (146). The mechanism of DI in Wolfram syndrome is not well understood (144).
SYNDROMES ASSOCIATED WITH HYPOPITUITARISM
Hypopituitarism has been reported in a wide variety of syndromes as an associated feature. These syndromes are reported separately as hypopituitarism is a less common association.
Prader-Willi Syndrome
Prader Willi Syndrome (PWS) is a complex condition with major neurological and endocrine challenges. It results from a lack of the paternally imprinted genes on chromosome 15q11.2-q13 which can result from paternal gene deletion, maternal uniparental disomy, or imprinting defects (147). Children typically have hypotonia and failure to thrive in infancy, but later progress to hyperphagia and obesity. Obesity and pituitary hormone deficiencies result from hypothalamic dysfunction. Growth failure and GHD are commonly seen but CPHD can also occur, and the anterior pituitary can be hypoplastic in approximately 70% of patients (148,149).
Growth failure in Prader-Willi Syndrome occurs due to failure within the GH-IGF-1 axis, with GHD reported in 40-100% of patients and IGF-1 deficiency in nearly 100% of cases. Growth hormone replacement early in life (<1 year of age) is recommended regardless of GHD and is shown to improve lean muscle mass, motor development and cardiovascular health (150-153).
Primary gonadal failure is present in most cases, with underdeveloped gonads typical in both genders. HH may compound this in some cases (154). TSHD has been reported in up to 30% cases, but the true prevalence is unclear. High rates were reported in the population younger than 2 years of age but in older children, rates did not exceed the general population, suggesting CNS maturation may result in this early childhood phenotype being transient. It is unclear if there is an association between PWS and ACTH deficiency with reported rates ranging from 0-60%, with rates likely being 5-14%. Routine screening of 8 AM cortisol is recommended (147).
The mechanism for hypopituitarism in PWS is not well understood. The MAGEL2 gene, located within the PWS locus causes Schaaf-Yang Syndrome and presents similarly to PWS but with arthrogryposis. MAGEL2 is highly expressed in the developing hypothalamus and pituitary between 7-8 weeks gestation and mutations can result in IGHD or CPHD (155).
Genetic testing is indicated in children with a history of neonatal hypotonia and failure to thrive that progress to obesity, with or without short stature.
Axenfield-Rieger Syndrome (PITX2)
Axenfield-Rieger syndrome involves dysgenesis of the anterior segment of the eye, typically resulting in glaucoma. The cause of many cases is unknown; however, 40% are due to either the PITX2 or FOXC2 genes. PITX2 is a pituitary homeobox gene located on chromosome 4q25 with autosomal dominant inheritance and variable penetrance (156). PITX2 is expressed early in the oral ectoderm (E8.5), and later in the anterior pituitary where it induces POU1F1 amongst other factors (156,157). Some cases of PITX2 mutation present with GHD, with dental and cardiac issues also associated (156).
Johanson-Blizzard Syndrome (UBR1)
Mutation of the UBR1 gene on 15q15-21 results in Johanson-Blizzard syndrome (JBS) and is inherited autosomal recessively, occurring in 1 in 250,000 live births, with 70 patients reported in the literature (158). It is characterized by exocrine pancreatic insufficiency, nasal alae hypoplasia/aplasia and cutis aplasia of the scalp. Along with developmental delay, failure to thrive and hearing impairment, hypothyroidism, genitourinary abnormalities and hypopituitarism are sometimes reported (159).
In 2007, Hoffman et al. reported a case of a 4-year-old deceased boy with ACTH, GH and TSH deficiency, with undescended testicles and hypoplastic pituitary gland with an absent stalk (160). 3 cases of IGHD and isolated TSHD have also been reported, with microgenitalia reported in 4 patients, who have not been evaluated for HH. Other known endocrine associations include diabetes mellitus and primary hypothyroidism (158,159).
Oliver-McFarlane Syndrome (PNPLA6)
Oliver McFarlane syndrome is caused by a recessive mutation of chromosome 19p13 encoding for the PNPLA6 gene. This encodes for neuropathy target esterase (NTE) and is found in the human eye, brain and pituitary, which also results in other syndromes including Gordon-Holmes, Lawrence-Moon and Boucher–Neuhäuser syndrome, which are associated with HH and occasionally hypopituitarism (161). A 2021 systematic review found of the 31 cases in the literature, 90% had hypopituitarism, with 67% having GHD or HH and 50% having TSHD (162). Other features include chorioretinal atrophy, trichomegaly, alopecia, and spinocerebellar involvement (161,162).
Wiedemann-Steiner Syndrome (KMT2A)
Wiedemann-Steiner syndrome results from heterozygous mutation of the KMT2A gene on chromosome 11q23.3. It classically presents with intellectual disability, facial dysmorphism, hypertrichosis of the elbow and short stature. A study of 104 individuals found short stature in 57.8% (163) and of the 12 patients tested within a cohort of 33, 6 were found to have GHD. The mechanism for hypopituitarism is not well understood (164).
Kabuki Syndrome (KMT2D)
Mutations of KMT2D gene on chromosome 12q13.12 are responsible for 55-80% of cases of Kabuki syndrome and result in autosomal dominant inheritance (165). Kabuki syndrome is characterized by distinctive facial features, intellectual disability, short stature and cardiac disease. 10 cases of hypopituitarism have been reported in the literature, 6 with GHD, 2 with DI and precocious puberty, 1 with ACTH deficiency and 1 with GHD and DI. Imaging showed 2 had PSIS and 1 had an absent posterior pituitary bright spot (166).
Williams Syndrome
Deletions of the 7q11.23 region result in Williams or Williams-Beuren syndrome, which has autosomal dominant inheritance. Williams syndrome occurs in between 1 in 7,500 and 20,000 live births and is characterized by congenital heart disease (supravalvular aortic stenosis), intellectual impairment, short stature and facial dysmorphism (167). 40% of patients have short stature, with hypercalcemia, precocious puberty and primary hypothyroidism commonly reported (168). Growth hormone deficiency is less frequently reported (168-170), and a case of combined ACTH and TSH deficiency has also been described (167). MRI imaging is typically normal, with one patient having an absent posterior-pituitary bright spot but normal ADH production (170), and another with ONH and normal pituitary function has been described (171).
PHACES
PHACES syndrome is a rare constellation of signs including Posterior fossa brain malformations, cervicofacial infantile Hemangiomas, Arterial anomalies, Cardiac defects, Eye anomalies and midline/ ventral defects. It is typically diagnosed following the detection of large, segmental facial hemangiomas and no gene defect has been identified. Over 300 cases have been reported to date (172). Goddard et al. reported a child with partially empty sella and GHD and TSHD in 2006 (173). Since then, 11 cases with hypopituitarism have been reported, 7 with abnormal pituitary MRI, 2 unreported, and 2 with normal pituitary appearance. The majority had GH (9/11) or TSH deficiency (7/11) with adrenal insufficiency and HH occurring in 2 out of 11 each. There is also an association with thyroid development issues and primary hypothyroidism (172). Thus, patients with PHACES syndrome should be monitored for growth and development with consideration of endocrine assessment if there are concerns. Abnormal pituitary appearance on MRI requires routine endocrine follow up (174).
Table 8.
Syndromes Associated with Hypopituitarism.
| Gene | Inheritance/ prevalence | Pituitary appearance | Pituitary deficiencies | Other Features |
|---|---|---|---|---|
| Prader Willi 15q11.2-q13 | Imprinted | Normal | GH, TSH, FSH/LH, ACTH | Infantile FTT, obesity, hyperphagia, ID, typically primary hypogonadism, dysmorphic features |
| Axenfield-Rieger PITX2 Chr 4q25 | AD | Normal | GH | Eye changes, glaucoma, dental issues, CHD |
| Johansson Blizzard UBR1 15q15-21 | AR | APH, absent stalk | GH, TSH, ACTH ?FSH/LH | Exocrine pancreas insufficiency, nasal anomalies, cutis aplasia, ID, genitourinary anomalies |
| Oliver-McFarlane PNPLA6 Chr 19p13 | AR | Normal | GH, FSH/LH, TSH | Chorioretinal atrophy, trichomegaly, alopecia, spinocerebellar involvement. |
| Wiedemann-Steiner KMT2A Chr 11q23.3 | AR | Normal | GH | ID, facial dysmorphism, elbow hypertrichosis, short stature |
| Kabuki KMT2D Chr 12q13.12 | AD | Normal, PSIS / absent PP | GH, ADH, ACTH | Facial dysmorphism, ID, short stature, CHD |
| Williams Chr7q11.23 | AD | Normal | GH, TSH, ACTH | Facial dysmorphism, ID, short stature, CHD, hypercalcemia, precocious puberty, primary hypothyroidism |
| PHACES (gene unknown) | Unknown | APH | GH, TSH, ACTH, FSH/LH | Posterior fossa brain malformations, hemangiomas, arterial anomalies, CHD, eye anomalies and midline/ ventral defects. |
ACTH – Adrenocorticotrophic Hormone, AD – Autosomal Dominant, APH – Anterior Pituitary Hypoplasia, CHD – Congenital Heart Disease, FTT – Failure To Thrive, ID – Intellectual Disability, PP – Posterior Pituitary, PSIS – Pituitary Stalk Interruption Syndrome.
INDICATIONS FOR GENETIC TESTING
Known genetic causes make up only a small proportion of the current hypopituitarism population, thus routine broad screening will not be appropriate at most centers and currently remains in the realms of research. Testing should be targeted to patients with consistent features, such as anophthalmia and SOX2 or a short stiff neck in LHX4. Individual genes can have a wide range of presentations and thus presence of a family history of hypopituitarism, whether similar in presentation or varied, should raise a high index of suspicion and screening should be strongly considered. Autosomal recessive causes are frequently seen, and dominant causes often have incomplete penetrance thus even relatively distant family associations should cause consideration for testing. Finally, presence of hypopituitarism in the context of a consanguineous parents should again raise suspicion of a likely genetic cause. Genetic testing typically is through next generation sequencing (NGS), although some mutations may be detectable on chromosomal microarray. Deletions on chromosomal microarray may include associated genes causing hypopituitarism. All the genes described above are summarized in table 9, in order of gene location, to assist in determining which mutations may be associated in such cases.
Septo-Optic Dysplasia
Current evidence suggests genetic causes of SOD make up <10% of cases thus routine screening will not be appropriate in most centers (19). In the absence of family history, the main indication for testing is in patients with microphthalmia or anophthalmia, where testing for SOX2 and OTX2 would be appropriate, and if negative, testing could be extended to PAX6, RAX and BMP4 (38). In patients with facial midline defects or polydactyly, GLI2 testing should also be considered (66).
Syndromic Causes
Syndromic causes should be tested for based on clinical suspicion, with family history and consanguinity again increasing the likelihood. Respiratory distress syndrome and a short stiff neck or Chiari I malformation should raise suspicion of LHX3 and LHX4 mutations respectively (53,56). Polydactyly and either midline facial changes or hypothalamic hamartoma suggest GLI2 and GLI3 mutations respectively (66,68). In cases of holoprosencephaly with anterior pituitary dysfunction, outside of GLI2, there are only case reports of other known genes causing hypopituitarism (66).
Non-Syndromic
POU1F1 testing may be appropriate in patients with severe short stature, GHD, TSHD and prolactin deficiency alone, especially in Western-Indian populations where rates of up to 25% of the CPHD cohort have been reported (100). PROP1 typically has progressive hypopituitarism with late onset ACTH deficiency and routine screening in European countries, particularly Lithuania, may be appropriate (50).
Isolated Hormone Deficiencies
Unless familial, isolated GH deficiency rarely has a genetic basis thus genetic testing is not currently indicated with the exception of those with absent GH levels on stimulation testing as seen in type 1a GH1 mutations (109). The frequency of genetic causes within the TSH deficient cohort is unknown. Macroorchidism points to IGSF1 mutation but otherwise family history is the main indicator for genetic testing (118). Cases with isolated ACTH deficiency in the neonatal period should be tested for TBX19 mutations, and if absent, especially in a child with red hair, POMC gene testing should be performed (120,124). Children with malabsorptive diarrhea and adrenal insufficiency should be screened for PCSK1 mutation, and NFKB2 if there is known immunodeficiency (125,127). Hypogonadotrophic hypogonadism testing is well described and typically involves panel testing, but targeted testing based on inheritance pattern may be appropriate (12,130). Isolated diabetes insipidus is rare and testing should be based on a family history or presence of other features consistent with Wolfram syndrome, such as childhood onset optic atrophy or diabetes mellitus (144).
Syndromes Associated with Hypopituitarism
Testing for Prader-Willi, Williams, PITX2, UBR1, PNPLA6, KMT2A and KMT2D gene mutations should be considered based on their clinical features as listed in table 8, and in patients known to have these mutations, pituitary screening may be necessary, especially in the context of growth failure.
Table 9.
Summary of Genes Associated with Hypopituitarism
| Gene | Inheritance | Pituitary deficiencies | Phenotype |
|---|---|---|---|
| 1p13.1 - TSHB | AR | TSH | Jaundice, FTT, developmental delay |
| 1q24.2 - TBX19 | AR, | ACTH | Intellectual impairment if delayed diagnosis, Chiari type 1 malformation |
| 1q25.2 - LHX4 | AD | CPHD | CC hypoplasia, Chiari 1 malformation, rarely CHD/RDS |
| 2p11.2 - TCF7L1 | AD | GH, ACTH | SOD, CC/SP changes |
| 2p21 - SIX3 | AD | CPHD | HPE, PSIS |
| 2p23.3 - POMC | AR | ACTH, TSH (35%), GH, LH/FSH (10%) | Obesity, reddish brown hair. Type 1 diabetes |
| 2q14.2 - GLI2 | AD | GH, CPHD | SOD, PSIS, SP/CC changes, polydactyly, midface hypoplasia, cleft palate/lip, HPE |
| 3p11.2 - POU1F1 | AR/AD | GH, TSH, prolactin (typically CPHD) | |
| 3p12.3 - ROBO1 | AD/AR | GH, TSH, CPHD | PSIS, hypermetropia, ptosis |
| 3p14.3 - HESX1 | AD/AR | GH, CPHD, (ACTH, ADH uncommon) | SOD, CC/SP changes, hypoplastic olfactory, underdeveloped forebrain |
| 3q26.33 - SOX2 | AD | GH, LH/FSH | SOD, AO/MO, hypothalamic hamartoma, CC changes, SNHL, ID, esophageal anomalies, genital anomalies |
| 4p16.1 - WFS1 Wolfram | AR | ADH | Diabetes mellitus, optic nerve atrophy, SNHL |
| 4q25 - PITX2 Axenfield-Rieger | AD | GH | Eye changes, glaucoma, dental issues, CHD |
| 5q15-21 - PCSK1 | AR | ACTH, FSH, LH, GH, TSH, Rarely ADH | Obesity in early childhood |
| 5q35 - PROP1 | AR | CPHD | |
| 6q22.31 - TBC1D32 | AR | GH, CPHD | SOD, Oro-facial-digital syndrome, CC agenesis |
| 7p14.1 - GLI3 Pallister-Hall | AD | CPHD | Hypothalamic hamartoma, Postaxial polydactyly, imperforate anus. CPP |
| 7p14.3 - GHRHR | AR | GH | |
| 7q11.23 - Williams | AD | GH, (TSH, ACTH uncommon) | Facial dysmorphism, ID, short stature, CHD, hypercalcemia, precocious puberty, primary hypothyroidism |
| 7q36 - SHH | AD | CPHD | HPE, PSIS, renal-urinary anomalies |
| 8p11.2-p12 - FGFR1 | AD | FSH/LH, (GH/CPHD uncommon) | SOD, PSIS, split hand/foot malformation |
| 8q12.2 – CHD7 CHARGE | AD/de novo | FSH/LH, (GH/TSH uncommon) | Coloboma, CHD, choanal atresia, short stature, ID, genital hypoplasia, ear abnormalities |
| 8q23.1 - TRHR | AR | TSH, Prolactin | |
| 9q34.3 - LHX3 | AR | CPHD, ACTH in 50% | SNHL, ID, short stiff neck, scoliosis, rarely RDS |
| 10q24.32 - NFKB2 | AD | ACTH, (GH, TSH uncommon) | Combined variable immunodeficiency |
| 10q24.32 - FGF8 | AD/AR | FSH/LH, (GH/CPHD uncommon) | SOD, HPE, VACTERL |
| 11p13 - PAX6 | AD | GH, ACTH, FSH/LH | SOD, AO/MO, ASD, ADHD, obesity, diabetes mellitus |
| 11q23.3 - KMT2A Wiedemann-Steiner | AR | GH | ID, facial dysmorphism, elbow hypertrichosis, short stature |
| 11q24.2 - CDON | AD | CPHD | HPE, PSIS, CHD, renal dysplasia, radial defects, gallbladder agenesis |
| 12q13.12 - KMT2D Kabuki | AD | GH, ADH, ACTH | Facial dysmorphism, ID, short stature, CHD |
| 13q32 - ZIC2 | AD | CPHD | HPE, PSIS, renal-urinary anomalies |
| 14q22.3 - OTX2 | AD | GH, CPHD, (ACTH uncommon) | SOD, PSIS, AO/MO |
| 14q22-23 - BMP4 | AD | GH, TSH | myopia, cleft palate/lip, polydactyly |
| 15q11.2-q13 - Prader Willi | Imprinted | GH, TSH, FSH/LH, ACTH | Infantile FTT, obesity, hyperphagia, ID, primary hypogonadism, dysmorphic features |
| 15q15-21 - UBR1 Johansson Blizzard | AR | GH, TSH, (ACTH uncommon) | Exocrine pancreas insufficiency, nasal anomalies, cutis aplasia, ID, genitourinary anomalies |
| 15q25.1 - ARNT2 | AR | CPHD + ADH | Microcephaly, fronto-temporal hypoplasia, renal anomalies and vision impairment. |
| 17q22-24 - GH1 | AR/AD | GH, (CPHD uncommon) | Anti-GH antibodies in some forms |
| 18p11.3 - TGIF | AD | CPHD | HPE, PSIS |
| 18q21.32 - RAX | AR | CPHD | SOD, AO/MO, palate changes |
| 19p13 - PNPLA6 Oliver-McFarlane | AR | GH, FSH/LH, TSH | Chorioretinal atrophy, trichomegaly, alopecia, spinocerebellar involvement. |
| 20p11.21 - FOXA2 | AD/de novo | CPHD | PSIS, hyperinsulinism, craniofacial dysmorphism, choroidal coloboma, and liver, lung and heart malformations |
| 20p12.3 - PROKR2 | AD/AR | FSH/LH, (CPHD uncommon) | SOD, PSIS |
| 20p13 - AVP | AD/AR | ADH | FTT, polyuria |
| Xp22.11 - EIF2S3 | XR | GH, CPHD | CC hypoplasia, ID, epilepsy, gonadal failure, microcephaly, obesity |
| Xp22.2 - TBL1X | XR | TSH | Hearing loss, ADHD |
| Xq26.1 - IGSF1 | XR | TSH, Prolactin (GH uncommon) | Macroorchidism, low testosterone, obesity, ADHD |
| Xq27 - SOX3 | XR | GH (CPHD uncommon) | PSIS, CC changes, ID |
AD – Autosomal Dominant, ADHD – Attention Deficit Hyperactivity Disorder, AO – Anophthalmia, AR – Autosomal Recessive, ASD – Autism Spectrum Disorder, CC– Corpus Callosum, CHD – Congenital Heart Disease, CPHD – Combined Pituitary Hormone Deficiency, CPP – Central Precocious Puberty, FTT – Failure To Thrive, HH – Hypogonadotrophic Hypogonadism, HPE – Holoprosencephaly, ID – Intellectual Disability, MO – Microphthalmia, PSIS – Pituitary Stalk Interruption Syndrome, RDS – Respiratory Distress Syndrome, SOD – Septo-optic dysplasia, SP – Septum Pellucidum, SNHL – Sensorineural Hearing Loss, VACTERL- Vertebral defects, Anal atresia, Cardiac defects, Tracheo-esophageal fistula, Renal anomalies, and Limb abnormalities, XR – X-linked Recessive.
ABBREVIATION LIST
ACTH – Adrenocorticotrophic Hormone
AD – Autosomal Dominant
ADH – Antidiuretic Hormone (also known as AVP)
ADHD – Attention Deficit Hyperactivity Disorder
APH – Anterior Pituitary Hypoplasia
αMSH – Alpha-Melanocyte Stimulating Hormone
AR – Autosomal Recessive
AVP – Arginine Vasopressin (also known as ADH)
CC – Corpus Callosum
CHD – Congenital Heart Disease
CPHD – Combined Pituitary Hormone Deficiency
CRH – Corticotrophin Releasing Hormone
CVID – Common Variable Immunodeficiency
DI – Diabetes Insipidus
EPP – Ectopic Posterior Pituitary
FSH – Follicle Stimulating Hormone
GH – Growth Hormone
GHD – Growth Hormone Deficiency
HH – Hypogonadotrophic Hypogonadism
HPE – Holoprosencephaly
IAD – Isolated ACTH Deficiency
ID – Intellectual Disability
IGF1 – Insulin-like Growth Factor 1
IGHD – Isolated Growth Hormone Deficiency
LH – Luteinizing Hormone
MPHD – Multiple Pituitary Hormone Deficiency
NGS – Next Generation Sequencing
ONH – Optic Nerve Hypoplasia
PSIS – Pituitary Stalk Interruption Syndrome
SOD – Septo-Optic Dysplasia
SP – Septum Pellucidum
THR – Thyroid Hormone Receptor
TRH – Thyrotropin Releasing Hormone
TRHR – Thyrotropin Releasing Hormone Receptor
TSH – Thyroid Stimulating Hormone
TSHD – Thyroid Stimulating Hormone Deficiency
XR – X-linked Recessive
REFERENCES
- 1.
- Larkin S, Ansorge O. Development And Microscopic Anatomy Of The Pituitary Gland. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA)2000.
- 2.
- Sheng HZ, Westphal H. Early steps in pituitary organogenesis. Trends Genet. 1999;15(6):236–240. [PubMed: 10354584]
- 3.
- Vimpani GV, Vimpani AF, Lidgard GP, Cameron EH, Farquhar JW. Prevalence of severe growth hormone deficiency. Br Med J. 1977;2(6084):427–430. [PMC free article: PMC1631237] [PubMed: 890325]
- 4.
- Gregory LC, Dattani MT. The Molecular Basis of Congenital Hypopituitarism and Related Disorders. J Clin Endocrinol Metab. 2020;105(6):2103–2120. [PubMed: 31702014]
- 5.
- Correa FA, Trarbach EB, Tusset C, Latronico AC, Montenegro LR, Carvalho LR, Franca MM, Otto AP, Costalonga EF, Brito VN, Abreu AP, Nishi MY, Jorge AAL, Arnhold IJP, Sidis Y, Pitteloud N, Mendonca BB. FGFR1 and PROKR2 rare variants found in patients with combined pituitary hormone deficiencies. Endocrine Connections. 2015;4:100–107. [PMC free article: PMC4401104] [PubMed: 25759380]
- 6.
- McCabe MJ, Gaston-Massuet C, Gregory LC, Alatzoglou KS, Tziaferi V, Sbai O, Rondard P, Masumoto K-h, Nagano M, Shigeyoshi Y, Pfeifer M, Hulse T, Buchanan CR, Pitteloud N, Martinez-Barbera J-P, Dattani MT. Variations in PROKR2, But Not PROK2, Are Associated With Hypopituitarism and Septo-optic Dysplasia. J Clin Endocrinol Metab. 2013;98(3):547–557. [PMC free article: PMC3612801] [PubMed: 23386640]
- 7.
- Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, Sykiotis GP, Gregory LC, Diaczok D, Tziaferi V, Elting MW, Padidela R, Plummer L, Martin C, Feng B, Zhang C, Zhou QY, Chen H, Mohammadi M, Quinton R, Sidis Y, Radovick S, Dattani MT, Pitteloud N. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012;97(4):E694–699. [PMC free article: PMC3319178] [PubMed: 22319038]
- 8.
- Reynaud R, Jayakody SA, Monnier C, Saveanu A, Bouligand J, Guedj AM, Simonin G, Lecomte P, Barlier A, Rondard P, Martinez-Barbera JP, Guiochon-Mantel A, Brue T. PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J Clin Endocrinol Metab. 2012;97(6):E1068–1073. [PubMed: 22466334]
- 9.
- Skowronska-Krawczyk D, Rosenfield M, eds. Development of the Pituitary. 6 ed. Philadelphia: Saunders Elsevier; 2010. Jameson JL, Groot LD, eds. Endocrinology; No. 1.
- 10.
- Zhu X, Gleiberman AS, Rosenfeld MG. Molecular physiology of pituitary development: signaling and transcriptional networks. Physiol Rev. 2007;87(3):933–963. [PubMed: 17615393]
- 11.
- Rizzoti K, Lovell-Badge R. Early development of the pituitary gland: induction and shaping of Rathke's pouch. Rev Endocr Metab Disord. 2005;6(3):161–172. [PubMed: 16151620]
- 12.
- Millar AC, Faghfoury H, Bieniek JM. Genetics of hypogonadotropic hypogonadism. Transl Androl Urol. 2021;10(3):1401–1409. [PMC free article: PMC8039576] [PubMed: 33850776]
- 13.
- Ludwig P, Lopez MJ, Czyz CN. Embryology, Eye Malformations. StatPearls [Internet].2021. [PubMed: 29494102]
- 14.
- Matsumoto R, Takahashi Y. Human pituitary development and application of iPSCs for pituitary disease. Cell Mol Life Sci. 2021;78(5):2069–2079. [PMC free article: PMC11071979] [PubMed: 33206204]
- 15.
- Hill M. Embryology Endocrine - Pituitary Development. 2022.
- 16.
- Mitrofanova LB, Konovalov PV, Krylova JS, Polyakova VO, Kvetnoy IM. Plurihormonal cells of normal anterior pituitary: Facts and conclusions. Oncotarget. 2017;8(17):29282–29299. [PMC free article: PMC5438730] [PubMed: 28418929]
- 17.
- Patel L, McNally RJ, Harrison E, Lloyd IC, Clayton PE. Geographical distribution of optic nerve hypoplasia and septo-optic dysplasia in Northwest England. J Pediatr. 2006;148(1):85–88. [PubMed: 16423603]
- 18.
- Garne E, Rissmann A, Addor MC, Barisic I, Bergman J, Braz P, Cavero-Carbonell C, Draper ES, Gatt M, Haeusler M, Klungsoyr K, Kurinczuk JJ, Lelong N, Luyt K, Lynch C, O'Mahony MT, Mokoroa O, Nelen V, Neville AJ, Pierini A, Randrianaivo H, Rankin J, Rouget F, Schaub B, Tucker D, Verellen-Dumoulin C, Wellesley D, Wiesel A, Zymak-Zakutnia N, Lanzoni M, Morris JK. Epidemiology of septo-optic dysplasia with focus on prevalence and maternal age - A EUROCAT study. Eur J Med Genet. 2018;61(9):483–488. [PubMed: 29753093]
- 19.
- McCabe MJ, Alatzoglou KS, Dattani MT. Septo-optic dysplasia and other midline defects: the role of transcription factors: HESX1 and beyond. Best Pract Res Clin Endocrinol Metab. 2011;25(1):115–124. [PubMed: 21396578]
- 20.
- Atapattu N, Ainsworth J, Willshaw H, Parulekar M, MacPherson L, Miller C, Davies P, Kirk JM. Septo-optic dysplasia: antenatal risk factors and clinical features in a regional study. Horm Res Paediatr. 2012;78(2):81–87. [PubMed: 22907285]
- 21.
- Garcia-Filion P, Borchert M. Prenatal determinants of optic nerve hypoplasia: review of suggested correlates and future focus. Surv Ophthalmol. 2013;58(6):610–619. [PMC free article: PMC3879141] [PubMed: 24160732]
- 22.
- Cerbone M, Guemes M., Wade A, Improda N, Dattani M. Endocrine morbidity in midline brain defects: Differences between septo-optic dysplasia and related disorders. EClinicalMedicine. 2020;19:1–17. [PMC free article: PMC7046495] [PubMed: 32140665]
- 23.
- Cemeroglu AP, Coulas T, Kleis L. Spectrum of clinical presentations and endocrinological findings of patients with septo-optic dysplasia: a retrospective study. J Pediatr Endocrinol Metab. 2015;28(9-10):1057–1063. [PubMed: 25879316]
- 24.
- Oatman OJ, McClellan DR, Olson ML, Garcia-Filion P. Endocrine and pubertal disturbances in optic nerve hypoplasia, from infancy to adolescence. Int J Pediatr Endocrinol. 2015;2015(1):8. [PMC free article: PMC4397734] [PubMed: 25878671]
- 25.
- Ahmad T, Garcia-Filion P, Borchert M, Kaufman F, Burkett L, Geffner M. Endocrinological and auxological abnormalities in young children with optic nerve hypoplasia: a prospective study. J Pediatr. 2006;148(1):78–84. [PubMed: 16423602]
- 26.
- Siatkowski RM, Sanchez JC, Andrade R, Alvarez A. The clinical, neuroradiographic, and endocrinologic profile of patients with bilateral optic nerve hypoplasia. Ophthalmology. 1997;104(3):493–496. [PubMed: 9082278]
- 27.
- McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou A, Keller E, Keller A, Haufs N, Krude H, Shalet SM, Dattani MT. HESX1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol Metab. 2007;92(2):691–697. [PubMed: 17148560]
- 28.
- Kelberman D, Dattani MT. Genetics of septo-optic dysplasia. Pituitary. 2007;10(4):393–407. [PubMed: 17587179]
- 29.
- Tajima T, Ohtake A, Hoshino M, Amemiya S, Sasaki N, Ishizu K, Fujieda K. OTX2 loss of function mutation causes anophthalmia and combined pituitary hormone deficiency with a small anterior and ectopic posterior pituitary. J Clin Endocrinol Metab. 2009;94(1):314–319. [PubMed: 18854396]
- 30.
- Dattani MT, Martinez-Barbera JP, Thomas PQ, Brickman JM, Gupta R, Martensson IL, Toresson H, Fox M, Wales JK, Hindmarsh PC, Krauss S, Beddington RS, Robinson IC. Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998;19(2):125–133. [PubMed: 9620767]
- 31.
- Durmaz B, Cogulu O, Dizdarer C, Stobbe H, Pfaeffle R, Ozkinay F. A novel homozygous HESX1 mutation causes panhypopituitarism without midline defects and optic nerve anomalies J Pediatr Endocr Met.2011;24(9-10):779-782. [PubMed: 22145475]
- 32.
- Thomas PQ, Dattani MT, Brickman JM, McNay D, Warne G, Zacharin M, Cameron F, Hurst J, Woods K, Dunger D, Stanhope R, Forrest S, Robinson IC, Beddington RS. Heterozygous HESX1 mutations associated with isolated congenital pituitary hypoplasia and septo-optic dysplasia. Hum Mol Genet. 2001;10(1):39–45. [PubMed: 11136712]
- 33.
- Newbern K, Natrajan N, Kim HG, Chorich LP, Halvorson LM, Cameron RS, Layman LC. Identification of HESX1 mutations in Kallmann syndrome. Fertil Steril. 2013;99(7):1831–1837. [PMC free article: PMC3888813] [PubMed: 23465708]
- 34.
- Williamson KA, Yates TM, FitzPatrick DR. SOX2 Disorder. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993. [PMC free article: PMC1300] [PubMed: 20301477]
- 35.
- Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson ICAF. SOX2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci U S A. 2008;105(8):2907–2912. [PMC free article: PMC2268558] [PubMed: 18287078]
- 36.
- Williamson KA, Hever AM, Rainger J, Rogers RC, Magee A, Fiedler Z, Keng WT, Sharkey FH, McGill N, Hill CJ, Schneider A, Messina M, Turnpenny PD, Fantes JA, van Heyningen V, FitzPatrick DR. Mutations in SOX2 cause anophthalmia-esophageal-genital (AEG) syndrome. Hum Mol Genet. 2006;15(9):1413–1422. [PubMed: 16543359]
- 37.
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, Chong WK, Kirk JMW, Achermann JC, Ross R, Carmignac D, Lovell-Badge R, Robinson ICAF, Dattani MT. Mutations within SOX2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. . The Journal of Clinical Investigation. 2006;116(9):2442–2455. [PMC free article: PMC1551933] [PubMed: 16932809]
- 38.
- Chassaing N., et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/ microphthalmia. Clin Genet. 2013 [PubMed: 24033328]
- 39.
- Blum WF, Klammt J, Amselem S, Pfaffle HM, Legendre M, Sobrier ML, Luton MP, Child CJ, Jones C, Zimmermann AG, Quigley CA, Cutler GB Jr, Deal CL, Lebl J, Rosenfeld RG, Parks JS, Pfaffle RW. Screening a large pediatric cohort with GH deficiency for mutations in genes regulating pituitary development and GH secretion: Frequencies, phenotypes and growth outcomes. EBioMedicine. 2018;36:390–400. [PMC free article: PMC6197701] [PubMed: 30266296]
- 40.
- Laumonnier F, Ronce N, Hamel BCJ, Thomas P, Lespinasse J, Raynaud M, Paringaux C. Bokhoven Hv, Kalscheuer V, Fryns J-P, Chelly J, Moraine C, Briault S. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am J Hum Genet. 2002;71(6):1450–1455. [PMC free article: PMC420004] [PubMed: 12428212]
- 41.
- Gregory LC, Gergics P, Nakaguma M, Bando H, Patti G, McCabe MJ, Fang Q, Ma Q, Ozel AB, Li JZ, Poina MM, Jorge AAL, Benedetti AFF, Lerario AM, Arnhold IJP, Mendonca BB, Maghnie M, Camper SA, Carvalho LRS, Dattani MT. The phenotypic spectrum associated with OTX2 mutations in humans. Eur J Endocrinol. 2021;185(1):121–135. [PMC free article: PMC8437083] [PubMed: 33950863]
- 42.
- Mortensen AH, Schade V, Lamonerie T, Camper SA. Deletion of OTX2 in neural ectoderm delays anterior pituitary development. Hum Mol Genet. 2015;24(4):939–953. [PMC free article: PMC4834879] [PubMed: 25315894]
- 43.
- Cunha D, Arno G, Corton M, Moosajee M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes. 2019;10:1050–1072. [PMC free article: PMC6947179] [PubMed: 31861090]
- 44.
- Kioussi C, O’Connell S, St-Onge L, Treier M, Gleiberman AS, Gruss P, Rosenfeld MG. <i>Pax6</i> is essential for establishing ventral-dorsal cell boundaries in pituitary gland development. Proceedings of the National Academy of Sciences. 1999;96(25):14378–14382. [PMC free article: PMC24444] [PubMed: 10588713]
- 45.
- Hergott-Faure L, Borot S, Kleinclauss C, Abitbol M, Penfornis A. Pituitary function and glucose tolerance in a family with a PAX6 mutation. Ann Endocrinol (Paris). 2012;73(6):510–514. [PubMed: 23146210]
- 46.
- Takagi M, Nagasaki K, Fujiwara I, Ishii T, Amano N, Asakura Y, Muroya K, Hasegawa Y, Adachi M, Hasegawa T. Heterozygous defects in PAX6 gene and congenital hypopituitarism. Eur J Endocrinol. 2015;172(1):37–45. [PubMed: 25342853]
- 47.
- Shimo N, Yasuda T, Kitamura T, Matsushita K, Osawa S, Yamamoto Y, Kozawa J, Otsuki M, Funahashi T, Imagawa A, Kaneto H, Nishida K, Shimomura I. Aniridia with a heterozygous PAX6 mutation in which the pituitary function was partially impaired. Intern Med. 2014;53(1):39–42. [PubMed: 24390526]
- 48.
- Brachet C, Kozhemyakina EA, Boros E, Heinrichs C, Balikova I, Soblet J, Smits G, Vilain C, Mathers PH. Truncating RAX Mutations: Anophthalmia, Hypopituitarism, Diabetes Insipidus, and Cleft Palate in Mice and Men. J Clin Endocrinol Metab. 2019;104(7):2925–2930. [PMC free article: PMC6543774] [PubMed: 30811539]
- 49.
- Gaston-Massuet C, McCabe MJ, Scagliotti V, Young RM, Carreno G, Gregory LC, Jayakody SA, Pozzi S, Gualtieri A, Basu B, Koniordou M, Wu C-I, Bancalari RE, Rahikkala E, Veijola R, Lopponen T, Graziola F, Turton J, Signore M, Gharavy SNM, Charolidi N, Sokol SY, Andoniadou CL, Wilson SW, Merrill BJ, Dattani MT, Martinez-Barberaa JP. Transcription factor 7-like 1 is involved in hypothalamo–pituitary axis development in mice and humans. PNAS. 2016;113(5):E548–E557. [PMC free article: PMC4747739] [PubMed: 26764381]
- 50.
- De Rienzo F, Mellone S, Bellone S, Babu D, Fusco I, Prodam F, Petri A, Muniswamy R, De Luca F, Salerno M, Momigliano-Richardi P, Bona G, Giordano M. Italian Study Group on Genetics of C. Frequency of genetic defects in combined pituitary hormone deficiency: a systematic review and analysis of a multicentre Italian cohort. Clin Endocrinol (Oxf). 2015;83(6):849–860. [PubMed: 26147833]
- 51.
- Rajab A, Kelberman D, de Castro SC, Biebermann H, Shaikh H, Pearce K, Hall CM, Shaikh G, Gerrelli D, Grueters A, Krude H, Dattani MT. Novel mutations in LHX3 are associated with hypopituitarism and sensorineural hearing loss. Hum Mol Genet. 2008;17(14):2150–2159. [PubMed: 18407919]
- 52.
- Ellsworth BS, Butts DL, Camper SA. Mechanisms underlying pituitary hypoplasia and failed cell specification in Lhx3-deficient mice. Dev Biol. 2008;313(1):118–129. [PMC free article: PMC2768753] [PubMed: 18037398]
- 53.
- Xatzipsalti M, Voutetakis A, Stamoyannou L, Chrousos GP, Kanaka-Gantenbein C. Congenital Hypopituitarism: Various Genes, Various Phenotypes. Horm Metab Res. 2019;51:81–90. [PubMed: 30759489]
- 54.
- Bechtold-Dalla Pozza S, Hiedl S, Roeb J, Lohse P, Malik RE, Park S, Duran-Prado M, Rhodes SJ. A recessive mutation resulting in a disabling amino acid substitution (T194R) in the LHX3 homeodomain causes combined pituitary hormone deficiency. Horm Res Paediatr. 2012;77(1):41–51. [PMC free article: PMC3355643] [PubMed: 22286346]
- 55.
- Sobrier ML, Brachet C, Vie-Luton MP, Perez C, Copin B, Legendre M, Heinrichs C, Amselem S. Symptomatic heterozygotes and prenatal diagnoses in a nonconsanguineous family with syndromic combined pituitary hormone deficiency resulting from two novel LHX3 mutations. J Clin Endocrinol Metab. 2012;97(3):E503–509. [PubMed: 22238406]
- 56.
- Cohen E, Maghnie M, Collot N, Leger J, Dastot F, Polak M, Rose S, Touraine P, Duquesnoy P, Tauber M, Copin B, Bertrand AM, Brioude F, Larizza D, Edouard T, Gonzalez Briceno L, Netchine I, Oliver-Petit I, Sobrier ML, Amselem S, Legendre M. Contribution of LHX4 Mutations to Pituitary Deficits in a Cohort of 417 Unrelated Patients. J Clin Endocrinol Metab. 2017;102(1):290–301. [PubMed: 27820671]
- 57.
- Machinis K, Amselem S. Functional Relationship between LHX4 and POU1F1 in Light of the LHX4 Mutation Identified in Patients with Pituitary Defects. J Clin Endocrinol Metab. 2005;90(9):5456–5462. [PubMed: 15998782]
- 58.
- Liu Y, Fan M, Yu S, Zhou Y, Wang J, Yuan J, Qiang B. cDNA cloning, chromosomal localization and expression pattern analysis of human LIM-homeobox gene LHX4. Brain Res. 2002;928(1-2):147–155. [PubMed: 11844481]
- 59.
- Colvin SC, Mullen RD, Pfaeffle RW, Rhodes SJ. LHX3 and LHX4 transcription factors in pituitary development and disease. Pediatr Endocrinol Rev. 2009;6 Suppl 2:283–290. [PubMed: 19337183]
- 60.
- Gregory LC, Humayun KN, Turton JP, McCabe MJ, Rhodes SJ, Dattani MT. Novel Lethal Form of Congenital Hypopituitarism Associated With the First Recessive LHX4 Mutation. J Clin Endocrinol Metab. 2015;100(6):2158–2164. [PMC free article: PMC4454798] [PubMed: 25871839]
- 61.
- Tajima T, Hattori T, Nakajima T, Okuhara K, Tsubaki J, Fujieda K. A novel missense mutation (P366T) of the LHX4 gene causes severe combined pituitary hormone deficiency with pituitary hypoplasia, ectopic posterior lobe and a poorly developed sella turcica. Endocr J. 2007;54(4):637–641. [PubMed: 17527005]
- 62.
- Filges I, Bischof-Renner A, Rothlisberger B, Potthoff C, Glanzmann R, Gunthard J, Schneider J, Huber AR, Zumsteg U, Miny P, Szinnai G. Panhypopituitarism presenting as life-threatening heart failure caused by an inherited microdeletion in 1q25 including LHX4. Pediatrics. 2012;129(2):e529–534. [PubMed: 22232309]
- 63.
- Franca MM, Jorge AA, Carvalho LR, Costalonga EF, Otto AP, Correa FA, Mendonca BB, Arnhold IJ. Relatively high frequency of non-synonymous GLI2 variants in patients with congenital hypopituitarism without holoprosencephaly. Clin Endocrinol (Oxf). 2013;78(4):551–557. [PubMed: 22967285]
- 64.
- Wang Y, Martin JF, Bai CB. Direct and indirect requirements of Shh/Gli signaling in early pituitary development. Dev Biol. 2010;348(2):199–209. [PMC free article: PMC3200223] [PubMed: 20934421]
- 65.
- Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci U S A. 2003;100(23):13424–13429. [PMC free article: PMC263830] [PubMed: 14581620]
- 66.
- Bear K, Solomon BD, Antonini S, Arnhold IJP, França MM, Gerkes EH, Grange DK, Hadley DW, Jääskeläinen J, Paulo SS, Rump P, Stratakis CA, Thompson EM, Willis M, Winder TL, Jorge AAL, Roessler E, Muenke M. Pathogenic mutations in GLI2 cause a specific phenotype that is distinct from holoprosencephaly. J Med Genet. 2014;51(6):413–418. [PMC free article: PMC6417429] [PubMed: 24744436]
- 67.
- Paulo S, Fernandes-Rosa FL, Turatti W, Coeli-Lacchini FB, Martinelli CE, Nakiri GS, Moreira AC, Santos AC. Castro Md, Antonini SR. Sonic Hedgehog mutations are not a common cause of congenital hypopituitarism in the absence of complex midline cerebral defects. Clinical Endocrinology. 2015;82:562–569. [PubMed: 25056824]
- 68.
- Biesecker LG. GLI3-Related Pallister-Hall Syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993. [PMC free article: PMC1465] [PubMed: 20301638]
- 69.
- Haddad-Tóvolli R, Paul F, Zhang Y, Zhou X, Theil T, Puelles L, Blaess S, Alvarez-Bolado G. Differential requirements for Gli2 and Gli3 in the regional specification of the mouse hypothalamus. Front Neuroanat. 2015:9. [PMC free article: PMC4373379] [PubMed: 25859185]
- 70.
- Fang Q, George A, Brinkmeier M, Mortensen A, Gergics P, Cheung L, Daly AZ, Ajmal A, Millán MIP, Ozel A, Kitzman J, Mills R, Li J, Camper S. Genetics of Combined Pituitary Hormone Deficiency: Roadmap into the Genome Era. Endocrine Reviews. 2016;37(6):636–675. [PMC free article: PMC5155665] [PubMed: 27828722]
- 71.
- Hall JG, Pallister PD, Clarren SK, Beckwith JB, Wiglesworth FW, Fraser FC, Cho S, Benke PJ, Reed SD. Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly--a new syndrome? Part I: clinical, causal, and pathogenetic considerations. Am J Med Genet. 1980;7(1):47–74. [PubMed: 7211952]
- 72.
- Biesecker L, Graham J. Pallister-Hall Syndrome. J Med Genet. 1996;33:585–589. [PMC free article: PMC1050667] [PubMed: 8818945]
- 73.
- Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis. 2007;2(8):8. [PMC free article: PMC1802747] [PubMed: 17274816]
- 74.
- Hahn JS, Hahn SM, Kammann H, Barkovich AJ, Clegg NJ, Delgado MR, Levey E. Endocrine disorders associated with holoprosencephaly. J Pediatr Endocrinol Metab. 2005;18(10):935–941. [PubMed: 16355806]
- 75.
- Dubourg C, Kim A, Watrin E, de Tayrac M, Odent S, David V, Dupe V. Recent advances in understanding inheritance of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2018;178(2):258–269. [PubMed: 29785796]
- 76.
- Tekendo-Ngongang C, Muenke M, Kruszka P. Holoprosencephaly Overview. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993. [PMC free article: PMC1530] [PubMed: 20301702]
- 77.
- Tasdemir S, Sahin I, Cayir A, Doneray H, Solomon BD, Muenke M, Yuce I, Tatar A. Holoprosencephaly: ZIC2 mutation in a case with panhypopituitarism. J Pediatr Endocrinol Metab. 2014;27(7-8):777–781. [PubMed: 24706429]
- 78.
- Bando H, Brinkmeier M, Gergics P, Fang Q, Mortensen AH, Ozel AB, Ma Q, Li J, Reynaud R, Castinetti F, Brue TC, Camper SA. SIX3 Variant Causes Pituitary Stalk Interruption Syndrome and Combined Pituitary Hormone Deficiency. J Endocrine Soc. 2021;5(1):A530.
- 79.
- Tatsi C, Sertedaki A, Voutetakis A, Valavani E, Magiakou M-A, Kanaka-Gantenbein C, Chrousos GP, Dacou-Voutetakis C. Pituitary Stalk Interruption Syndrome and Isolated Pituitary Hypoplasia May Be Caused by Mutations in Holoprosencephaly-Related Genes. J Clin Endocrinol Metab. 2013;98(4):779–784. [PubMed: 23476075]
- 80.
- Obara‑Moszyńska M, Budny B, Kałużna M, Zawadzka K, Jamsheer A, Rohde A, Ruchała M, Ziemnicka K, Niedziela M. CDON gene contributes to pituitary stalk interruption syndrome associated with unilateral facial and abducens nerve palsy. Journal of Applied Genetics. 2021;62:621–629. [PMC free article: PMC8571149] [PubMed: 34235642]
- 81.
- Karaca E, Buyukkaya R, Pehlivan D, Charng WL, Yaykasli KO, Bayram Y, Gambin T, Withers M, Atik MM, Arslanoglu I, Bolu S, Erdin S, Buyukkaya A, Yaykasli E, Jhangiani SN, Muzny DM, Gibbs RA, Lupski JR. Whole-exome sequencing identifies homozygous GPR161 mutation in a family with pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2015;100(1):E140–147. [PMC free article: PMC4283017] [PubMed: 25322266]
- 82.
- Bashamboo A, Bignon-Topalovic J, Rouba H, McElreavey K, Brauner R. A Nonsense Mutation in the Hedgehog Receptor CDON Associated With Pituitary Stalk Interruption Syndrome. J Clin Endocrinol Metab. 2016;101(1):12–15. [PubMed: 26529631]
- 83.
- Wang CZ, Guo LL, Han BY, Su X, Guo QH, Mu YM. Pituitary Stalk Interruption Syndrome: From Clinical Findings to Pathogenesis. J Neuroendocrinol. 2017;29(1) [PubMed: 27917547]
- 84.
- Bashamboo A, Bignon-Topalovic J, Moussi N, McElreavey K, Brauner R. Mutations in the Human ROBO1 Gene in Pituitary Stalk Interruption Syndrome. J Clin Endocrinol Metab. 2017;102(7):2401–2406. [PubMed: 28402530]
- 85.
- Dateki S, Watanabe S, Mishima H, Shirakawa T, Morikawa M, Kinoshita E, Yoshiura KI, Moriuchi H. A homozygous splice site ROBO1 mutation in a patient with a novel syndrome with combined pituitary hormone deficiency. J Hum Genet. 2019;64(4):341–346. [PubMed: 30692597]
- 86.
- Vajravelu ME, Chai J, Krock B, Baker S, Langdon D, Alter C, De Leon DD. Congenital Hyperinsulinism and Hypopituitarism Attributable to a Mutation in FOXA2. J Clin Endocrinol Metab. 2018;103(3):1042–1047. [PMC free article: PMC6276717] [PubMed: 29329447]
- 87.
- Dayem-Quere M, Giuliano F, Wagner-Mahler K, Massol C, Crouzet-Ozenda L, Lambert JC, Karmous-Benailly H. Delineation of a region responsible for panhypopituitarism in 20p11.2. Am J Med Genet A. 2013;161A(7):1547–1554. [PubMed: 23657910]
- 88.
- Williams PG, Wetherbee JJ, Rosenfeld JA, Hersh JH. 20p11 deletion in a female child with panhypopituitarism, cleft lip and palate, dysmorphic facial features, global developmental delay and seizure disorder. Am J Med Genet A. 2011;155A(1):186–191. [PubMed: 21204230]
- 89.
- Garcia-Heras J, Kilani RA, Martin RA, Lamp S. A deletion of proximal 20p inherited from a normal mosaic carrier mother in a newborn with panhypopituitarism and craniofacial dysmorphism. Clin Dysmorphol. 2005;14(3):137–140. [PubMed: 15930903]
- 90.
- Giri D, Vignola ML, Gualtieri A, Scagliotti V, McNamara P, Peak M, Didi M, Gaston-Massuet C, Senniappan S. Novel FOXA2 mutation causes Hyperinsulinism, Hypopituitarism with Craniofacial and Endoderm-derived organ abnormalities. Hum Mol Genet. 2017;26(22):4315–4326. [PubMed: 28973288]
- 91.
- Heddad Masson M, Poisson C, Guerardel A, Mamin A, Philippe J, Gosmain Y. Foxa1 and Foxa2 regulate alpha-cell differentiation, glucagon biosynthesis, and secretion. Endocrinology. 2014;155(10):3781–3792. [PubMed: 25057789]
- 92.
- Tsai EA, Grochowski CM, Falsey AM, Rajagopalan R, Wendel D, Devoto M, Krantz ID, Loomes KM, Spinner NB. Heterozygous deletion of FOXA2 segregates with disease in a family with heterotaxy, panhypopituitarism, and biliary atresia. Hum Mutat. 2015;36(6):631–637. [PMC free article: PMC4477513] [PubMed: 25765999]
- 93.
- Stekelenburg C, Gerster K, Blouin JL, Lang-Muritano M, Guipponi M, Santoni F, Schwitzgebel VM. Exome sequencing identifies a de novo FOXA2 variant in a patient with syndromic diabetes. Pediatr Diabetes. 2019;20(3):366–369. [PubMed: 30684292]
- 94.
- Gregory LC, Ferreira CB, Young-Baird SK, Williams HJ, Harakalova M. Haaften Gv, Rahman SA, Gaston-Massuet C, Kelberman D, Qasim W, Camper SA, Dever TE, Shah P, Robinson ICAF, Dattani MT. Impaired EIF2S3 function associated with a novel phenotype of X-linked hypopituitarism with glucose dysregulatio. EBioMedicine. 2019;42:470–480. [PMC free article: PMC6492072] [PubMed: 30878599]
- 95.
- Skopkova M, Hennig F, Shin BS, Turner CE, Stanikova D, Brennerova K, Stanik J, Fischer U, Henden L, Muller U, Steinberger D, Leshinsky-Silver E, Bottani A, Kurdiova T, Ukropec J, Nyitrayova O, Kolnikova M, Klimes I, Borck G, Bahlo M, Haas SA, Kim JR, Lotspeich-Cole LE, Gasperikova D, Dever TE, Kalscheuer VM. EIF2S3 Mutations Associated with Severe X-Linked Intellectual Disability Syndrome MEHMO. Hum Mutat. 2017;38(4):409–425. [PMC free article: PMC6267786] [PubMed: 28055140]
- 96.
- Takuma N, Sheng HZ, Furuta Y, Ward JM, Sharma K, Hogan BL, Pfaff SL, Westphal H, Kimura S, Mahon KA. Formation of Rathke's pouch requires dual induction from the diencephalon. Development. 1998;125(23):4835–4840. [PubMed: 9806931]
- 97.
- Rodriguez-Contreras FJ, Marban-Calzon M, Vallespin E, Del Pozo A, Solis-Lopez M, Lobato-Vidal N, Fernandez-Elvira M, Del Valle Rex-Romero M, Heath KE, Gonzalez-Casado I, Campos-Barros A. Loss of function BMP4 mutation supports the implication of the BMP/TGF-beta pathway in the etiology of combined pituitary hormone deficiency. Am J Med Genet A. 2019;179(8):1591–1597. [PubMed: 31120642]
- 98.
- Hietamäki Johanna. Gregory LC, Ayoub S, Iivonen A-P, Vaaralahti K, Liu X, Brandstack N, Buckton AJ, Laine T, Känsäkoski J, Hero M, Miettinen PJ, Varjosalo M, Wakeling E, Dattani MT, Raivio T. Loss-of-Function Variants in TBC1D32 Underlie Syndromic Hypopituitarism. J Clin Endocrinol Metab. 2020;105(6):1748–1758. [PMC free article: PMC7138537] [PubMed: 32060556]
- 99.
- Webb EA, AlMutair A, Kelberman D, Bacchelli C, Chanudet E, Lescai F, Andoniadou CL, Banyan A, Alsawaid A, Alrifai MT, Alahmesh MA, Balwi M, Mousavy-Gharavy SN, Lukovic B, Burke D, McCabe MJ, Kasia T, Kleta R, Stupka E, Beales PL, Thompson DA, Chong WK, Alkuraya FS, Martinez-Barbera JP, Sowden JC, Dattani MT. ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain. 2013;136(Pt 10):3096–3105. [PMC free article: PMC3784281] [PubMed: 24022475]
- 100.
- Jadhav S, Diwaker C, Lila A, Gada J, Kale S, Sarathi V, Thadani P, Arya S, Patil V, Shah N, Bandgar T, Ajmal A. POU1F1 mutations in combined pituitary hormone deficiency: differing spectrum of mutations in a Western‑Indian cohort and systematic analysis of world literature. Pituitary. 2021;24:657–669. [PubMed: 33742319]
- 101.
- McLennan K, Jeske Y, Cotterill A, Cowley D, Penfold J, Jones T, Howard N, Thomsett M, Choong C. Combined pituitary hormone deficiency in Australian children: clinical and genetic correlates. Clin Endocrinol (Oxf). 2003;58(6):785–794. [PubMed: 12780757]
- 102.
- Anderson B, Rosenfeld M. Pit-1 Determines Cell types during Development of the Anterior Pituitary Gland The Journal of Biological Chemistry.1994;269(47):29335-29338. [PubMed: 7961905]
- 103.
- Tatsumi K, Miyai K, Notomi T, Kaibe K, Amino N, Mizuno Y, Kohno H. Cretinism with combined hormone deficiency caused by a mutation in the PIT1 gene. Nat Genet. 1992;1(1):56–58. [PubMed: 1302000]
- 104.
- Ward RD, Raetzman LT, Suh H, Stone BM, Nasonkin IO, Camper SA. Role of PROP1 in pituitary gland growth. Mol Endocrinol. 2005;19(3):698–710. [PubMed: 15591534]
- 105.
- Bottner A, Keller E, Kratzsch J, Stobbe H, Weigel JF, Keller A, Hirsch W, Kiess W, Blum WF, Pfaffle RW. PROP1 mutations cause progressive deterioration of anterior pituitary function including adrenal insufficiency: a longitudinal analysis. J Clin Endocrinol Metab. 2004;89(10):5256–5265. [PubMed: 15472232]
- 106.
- Mullis PE, Robinson ICAF, Salemi S, Eble A, Besson A, Vuissoz J-M, Deladoey J, Simon D, Czernichow P, Binder G. Isolated Autosomal Dominant Growth Hormone Deficiency: An Evolving Pituitary Deficit? A Multicentre Follow-Up Study. J Clin Endocrinol Metab. 2005;90(4):2089–2096. [PubMed: 15671105]
- 107.
- Cerbone M, Dattani MT. Progression from isolated growth hormone deficiency to combined pituitary hormone deficiency. Growth Horm IGF Res. 2017;37:19–25. [PubMed: 29107171]
- 108.
- Mullis PE. Genetics of isolated growth hormone deficiency. J Clin Res Pediatr Endocrinol. 2010;2(2):52–62. [PMC free article: PMC3014602] [PubMed: 21274339]
- 109.
- Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, Aylwin S, Crowne EC, Christesen HT, Hertel NT, Trainer PJ, Savage MO, Raza J, Banerjee K, Sinha SK, Ten S, Mushtaq T, Brauner R, Cheetham TD, Hindmarsh PC, Mullis PE, Dattani MT. Expanding the spectrum of mutations in GH1 and GHRHR: genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab. 2009;94(9):3191–3199. [PubMed: 19567534]
- 110.
- Phillips JA 3rd, Hjelle BL, Seeburg PH, Zachmann M. Molecular basis for familial isolated growth hormone deficiency. Proc Natl Acad Sci U S A. 1981;78(10):6372–6375. [PMC free article: PMC349041] [PubMed: 6273867]
- 111.
- Wajnrajch MP, Gertner JM, Harbison MD, Chua SC Jr, Leibel RL. Nonsense mutation in the human growth hormone-releasing hormone receptor causes growth failure analogous to the little (lit) mouse. Nat Genet. 1996;12(1):88–90. [PubMed: 8528260]
- 112.
- Mazhar M, Waseem M. Agammaglobulinaemia. StatPearls [Internet].2022.
- 113.
- Child CJ, Blum WF, Deal C, Zimmermann AG, Quigley CA, Drop SLS, Cutler GB, Rosenfeld RG. Development of additional pituitary hormone deficiencies in pediatric patients originally diagnosed with isolated growth hormone deficiency due to organic causes. Eur J Endocrinol. 2016;174(5):669–679. [PubMed: 26888628]
- 114.
- Casteras A, Kratzsch J, Ferrandez A, Zafon C, Carrascosa A, Mesa J. Clinical challenges in the management of isolated GH deficiency type IA in adulthood. Endocrinol Diabetes Metab Case Rep. 2014;2014:130057. [PMC free article: PMC3965272] [PubMed: 24683479]
- 115.
- Lanting CI. Tijn DAv, Loeber JG, Vulsma T, Vijlder JJMd, Verkerk PH. Clinical Effectiveness and Cost-Effectiveness of the Use of the Thyroxine/Thyroxine-Binding Globulin Ratio to Detect Congenital Hypothyroidism of Thyroidal and Central Origin in a Neonatal Screening Program. Pediatrics. 2005;116(1):168–173. [PubMed: 15995048]
- 116.
- Fisher DA. Second International Conference on Neonatal Thyroid Screening: progress report. J Pediatr. 1983;102(5):653–654. [PubMed: 6842320]
- 117.
- Zwaveling-Soonawala N, Naafs JC, Verkerk PH, van Trotsenburg ASP. Mortality in Children With Early-Detected Congenital Central Hypothyroidism. J Clin Endocrinol Metab. 2018;103(8):3078–3082. [PubMed: 29889220]
- 118.
- Tajima T, Nakamura A, Oguma M, Yamazaki M. Recent advances in research on isolated congenital central hypothyroidism. Clin Pediatr Endocrinol. 2019;28(3):69–79. [PMC free article: PMC6646241] [PubMed: 31384098]
- 119.
- Hayashizaki Y, Hiraoka Y, Endo Y, Miyai K, Matsubara K. Thyroid-stimulating hormone (TSH) deficiency caused by a single base substitution in the CAGYC region of the beta-subunit. EMBO J. 1989;8(8):2291–2296. [PMC free article: PMC401161] [PubMed: 2792087]
- 120.
- Couture C, Saveanu A, Barlier A, Carel JC, Fassnacht M, Fluck CE, Houang M, Maes M, Phan-Hug F, Enjalbert A, Drouin J, Brue T, Vallette S. Phenotypic homogeneity and genotypic variability in a large series of congenital isolated ACTH-deficiency patients with TPIT gene mutations. J Clin Endocrinol Metab. 2012;97(3):E486–495. [PubMed: 22170728]
- 121.
- Stevens A, White A, eds. Adrenocorticotrophic Hormone. 6 ed. Philadelphia: Saunders Elsevier; 2010. Jameson JL, Groot LD, eds. Endocrinology; No. 1.
- 122.
- Derya-Kardelen-Al A, Poyrazoğlu Ş, Aslanger A, Yeşil G, Ceylaner S, Baş F, Darendeliler F. A Rare Cause of Adrenal Insufficiency – Isolated ACTH Deficiency Due to TBX19 Mutation: Long-Term Follow-Up of Two Cases and Review of the Literature. HORMONE RESEARCH IN PÆDIATRICS. 2019;92:395–403. [PubMed: 32344415]
- 123.
- Weintrob N, Drouin J, Vallette-Kasic S, Taub E, Marom D, Lebenthal Y, Klinger G, Bron-Harlev E, Shohat M. Low Estriol Levels in the Maternal Triple-Marker Screen as a Predictor of Isolated AdrenocorticotropicHormone Deficiency Caused by a New Mutation in the TPIT Gene. Pediatrics. 2006;117(2):322–327. [PubMed: 16390921]
- 124.
- Stijnen P, Ramos-Molina B, O'Rahilly S, Creemers JW. PCSK1 Mutations and Human Endocrinopathies: From Obesity to Gastrointestinal Disorders. Endocr Rev. 2016;37(4):347–371. [PubMed: 27187081]
- 125.
- Gregoric N, Groselj U, Bratina N, Debeljak M, Zerjav Tansek M, Suput Omladic J, Kovac J, Battelino T, Kotnik P, Avbelj Stefanija M. Two Cases With an Early Presented Proopiomelanocortin Deficiency-A Long-Term Follow-Up and Systematic Literature Review. Front Endocrinol (Lausanne). 2021;12:689387. [PMC free article: PMC8220084] [PubMed: 34177811]
- 126.
- Mendiratta MS, Yang Y, Balazs AE, Willis AS, Eng CM, Karaviti LP, Potocki L. Early onset obesity and adrenal insufficiency associated with a homozygous POMC mutation. Int J Pediatr Endocrinol. 2011;2011(1):5. [PMC free article: PMC3159139] [PubMed: 21860632]
- 127.
- Brue T, Quentien M, Khetchoumian K, Bensa M, Capo-Chichi J, Delemer B, Balsalobre A, Nassif C, Papadimitriou D, Pagnier A, Hasselmann C, Patry L, Schwartzentruber J, Souchon P, Takayasu S, Enjalbert A, Vliet GV, Majewski J, Drouin J, Samuels M. Mutations in NFKB2 and potential genetic heterogeneity in patients with DAVID syndrome, having variable endocrine and immune deficiencies. BMC Medical Genetics. 2014;15(139) [PMC free article: PMC4411703] [PubMed: 25524009]
- 128.
- Lal RA, Bachrach LK, Hoffman AR, Inlora J, Rego S, Snyder MP, Lewis DB. A Case Report of Hypoglycemia and Hypogammaglobulinemia: DAVID Syndrome in a Patient With a Novel NFKB2 Mutation. J Clin Endocrinol Metab. 2017;102(7):2127–2130. [PubMed: 28472507]
- 129.
- Young J, Xu C, Hietamaki J, Papadakis GE, Acierno JS, Maione L, Raivio T, Pitteloud N. Clinical Mangement of Congenital Hypogonadotrophic Hypogonadism. Endocrine Reviews. 2019;40(2):669–710. [PubMed: 30698671]
- 130.
- Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, Maghnie M, Pitteloud N, Prevot V, Raivio T, Tena-Sempere M, Quinton R, Young J. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015;11(9):547–564. [PubMed: 26194704]
- 131.
- Dijk DR, Bocca G, van Ravenswaaij-Arts CM. Growth in CHARGE syndrome: optimizing care with a multidisciplinary approach. J Multidiscip Healthc. 2019;12:607–620. [PMC free article: PMC6682174] [PubMed: 31534343]
- 132.
- Pagon RA, Graham JM Jr, Zonana J, Yong SL. Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99(2):223–227. [PubMed: 6166737]
- 133.
- Aramaki M, Udaka T, Kosaki R, Makita Y, Okamoto N, Yoshihashi H, Oki H, Nanao K, Moriyama N, Oku S, Hasegawa T, Takahashi T, Fukushima Y, Kawame H, Kosaki K. Phenotypic spectrum of CHARGE syndrome with CHD7 mutations. J Pediatr. 2006;148(3):410–414. [PubMed: 16615981]
- 134.
- Asakura Y, Toyota Y, Muroya K, Kurosawa K, Fujita K, Aida N, Kawame H, Kosaki K, Adachi M. Endocrine and radiological studies in patients with molecularly confirmed CHARGE syndrome. J Clin Endocrinol Metab. 2008;93(3):920–924. [PubMed: 18089695]
- 135.
- Pinto G, Abadie V, Mesnage R, Blustajn J, Cabrol S, Amiel J, Hertz-Pannier L, Bertrand AM, Lyonnet S, Rappaport R, Netchine I. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J Clin Endocrinol Metab. 2005;90(10):5621–5626. [PubMed: 16030162]
- 136.
- Shoji Y, Ida S, Etani Y, Yamada H, Kayatani F, Suzuki Y, Kosaki K, Okamoto N. Endocrinological Characteristics of 25 Japanese Patients with CHARGE Syndrome. Clin Pediatr Endocrinol. 2014;23(2):45–51. [PMC free article: PMC4004997] [PubMed: 24790386]
- 137.
- Sarfati J, Fouveaut C, Leroy C, Jeanpierre M, Hardelin J-P, Dode C. Greater prevalence of PROKR2 mutations in Kallmann syndrome patients from the Maghreb than in European patients. Eur J Endocrinol. 2013;169:805–809. [PubMed: 24031091]
- 138.
- Ericson J, Norlin S, Jessell TM, Edlund T. Integrated FGF and BMP signaling controls the progression of progenitor cell differentiation and the emergence of pattern in the embryonic anterior pituitary. Development. 1198(125):1005–1015. [PubMed: 9463347]
- 139.
- Davis SW, Ellsworth BS, Perez Millan MI, Gergics P, Schade V, Foyouzi N, Brinkmeier ML, Mortensen AH, Camper SA. Pituitary gland development and disease: from stem cell to hormone production. Curr Top Dev Biol. 2013;106:1–47. [PMC free article: PMC4039019] [PubMed: 24290346]
- 140.
- Tsai PS, Moenter SM, Postigo HR, El Majdoubi M, Pak TR, Gill JC, Paruthiyil S, Werner S, Weiner RI. Targeted expression of a dominant-negative fibroblast growth factor (FGF) receptor in gonadotropin-releasing hormone (GnRH) neurons reduces FGF responsiveness and the size of GnRH neuronal population. Mol Endocrinol. 2005;19(1):225–236. [PubMed: 15459253]
- 141.
- Erbas IM, Paketci A, Acar S, Kotan LD, Demir K, Abaci A, Bober E. A nonsense variant in FGFR1: a rare cause of combined pituitary hormone deficiency. J Pediatr Endocrinol Metab. 2020;33(12):1613–1615. [PubMed: 32853167]
- 142.
- Fukami M, Iso M, Sato N, Igarashi M, Seo M, Kazukawa I, Kinoshita E, Dateki S, Ogata T. Submicroscopic deletion involving the fibroblast growth factor receptor 1 gene in a patient with combined pituitary hormone deficiency. Endocr J. 2013;60(8):1013–1020. [PubMed: 23657145]
- 143.
- Di Iorgi N, Napoli F, Allegri AE, Olivieri I, Bertelli E, Gallizia A, Rossi A, Maghnie M. Diabetes insipidus--diagnosis and management. Horm Res Paediatr. 2012;77(2):69–84. [PubMed: 22433947]
- 144.
- Schernthaner-Reiter MH, Stratakis CA, Luger A. Genetics of Diabetes Insipidus. Endocrinol Metab Clin North Am. 2017;46(2):305–334. [PubMed: 28476225]
- 145.
- Boutzios G, Livadas S, Marinakis E, Opie N, Economou F, Diamanti-Kandarakis E. Endocrine and metabolic aspects of the Wolfram syndrome. Endocrine. 2011;40(1):10–13. [PubMed: 21725703]
- 146.
- Perrotta S, Di Iorgi N, Della Ragione F, Scianguetta S, Borriello A, Allegri AEM, Ferraro M, Santoro C, Napoli F, Calcagno A, Giaccardi M, Cappa M, Salerno MC, Cozzolino D, Maghnie M. Early-onset central diabetes insipidus is associated with de novo arginine vasopressin-neurophysin II or Wolfram syndrome 1 gene mutations. European Journal of Endocrinology. 2015;172(4):461–472. [PubMed: 25740874]
- 147.
- Heksch R, Kamboj M, Anglin K, Obrynba K. Review of Prader-Willi syndrome: the endocrine approach. Transl Pediatr. 2017;6(4):274–285. [PMC free article: PMC5682385] [PubMed: 29184809]
- 148.
- Miller JL, Goldstone AP, Couch JA, Shuster J, He G, Driscoll DJ, Liu Y, Schmalfuss IM. Pituitary abnormalities in Prader-Willi syndrome and early onset morbid obesity. Am J Med Genet A. 2008;146A(5):570–577. [PubMed: 17431897]
- 149.
- Tauber M, Barbeau C, Jouret B, Pienkowski C, Malzac P, Moncla A, Rochiccioli P. Auxological and endocrine evolution of 28 children with Prader-Willi syndrome: effect of GH therapy in 14 children. Horm Res. 2000;53(6):279–287. [PubMed: 11146368]
- 150.
- Carrel AL, Myers SE, Whitman BY, Allen DB. Benefits of long-term GH therapy in Prader-Willi syndrome: a 4-year study. J Clin Endocrinol Metab. 2002;87(4):1581–1585. [PubMed: 11932286]
- 151.
- Festen DA, de Lind van Wijngaarden R, van Eekelen M, Otten BJ, Wit JM, Duivenvoorden HJ, Hokken-Koelega AC. Randomized controlled GH trial: effects on anthropometry, body composition and body proportions in a large group of children with Prader-Willi syndrome. Clin Endocrinol (Oxf). 2008;69(3):443–451. [PubMed: 18363884]
- 152.
- Haqq AM, Stadler DD, Jackson RH, Rosenfeld RG, Purnell JQ, LaFranchi SH. Effects of growth hormone on pulmonary function, sleep quality, behavior, cognition, growth velocity, body composition, and resting energy expenditure in Prader-Willi syndrome. J Clin Endocrinol Metab. 2003;88(5):2206–2212. [PubMed: 12727976]
- 153.
- Myers SE, Whitman BY, Carrel AL, Moerchen V, Bekx MT, Allen DB. Two years of growth hormone therapy in young children with Prader-Willi syndrome: physical and neurodevelopmental benefits. Am J Med Genet A. 2007;143A(5):443–448. [PubMed: 17103437]
- 154.
- Radicioni AF, Di Giorgio G, Grugni G, Cuttini M, Losacco V, Anzuini A, Spera S, Marzano C, Lenzi A, Cappa M, Crino A. Multiple forms of hypogonadism of central, peripheral or combined origin in males with Prader-Willi syndrome. Clin Endocrinol (Oxf). 2012;76(1):72–77. [PubMed: 21718342]
- 155.
- Gregory LC, Shah P, Sanner JRF, Arancibia M, Hurst J, Jones WD, Spoudeas H, Le Quesne Stabej P, Williams HJ, Ocaka LA, Loureiro C, Martinez-Aguayo A, Dattani MT. Mutations in MAGEL2 and L1CAM Are Associated With Congenital Hypopituitarism and Arthrogryposis. J Clin Endocrinol Metab. 2019;104(12):5737–5750. [PMC free article: PMC6916815] [PubMed: 31504653]
- 156.
- F. Castinetti, Brue. T, Deficiency. CPH, Cohen IL, eds. Growth Hormone Deficiency: Physiology and Clinical Management Boston Children's Hospital: Springer International; 2016.
- 157.
- Suh H, Gage PJ, Drouin J, Camper SA. Pitx2 is required at multiple stages of pituitary organogenesis: pituitary primordium formation and cell specification. Development. 2002;129(2):329–337. [PubMed: 11807026]
- 158.
- Liu S, Wang Z, Jiang J, Luo X, Hong Q, Zhang Y, OuYang H, Wei S, Liang J, Chen N, Zeng W. Severe forms of Johanson-Blizzard syndrome caused by two novel compound heterozygous variants in UBR1: Clinical manifestations, imaging findings and molecular genetics. Pancreatology. 2020;20(3):562–568. [PubMed: 31980351]
- 159.
- Almashraki N, Abdulnabee MZ, Sukalo M, Alrajoudi A, Sharafadeen I, Zenker M. Johanson-Blizzard syndrome. World J Gastroenterol. 2011;17(37):4247–4250. [PMC free article: PMC3208372] [PubMed: 22072859]
- 160.
- Hoffman WH, Lee JR, Kovacs K, Chen H, Yaghmai F. Johanson-Blizzard syndrome: autopsy findings with special emphasis on hypopituitarism and review of the literature. Pediatr Dev Pathol. 2007;10(1):55–60. [PubMed: 17378628]
- 161.
- Hufnagel RB, Arno G, Hein ND, Hersheson J, Prasad M, Anderson Y, Krueger LA, Gregory LC, Stoetzel C, Jaworek TJ, Hull S, Li A, Plagnol V, Willen CM, Morgan TM, Prows CA, Hegde RS, Riazuddin S, Grabowski GA, Richardson RJ, Dieterich K, Huang T, Revesz T, Martinez-Barbera JP, Sisk RA, Jefferies C, Houlden H, Dattani MT, Fink JK, Dollfus H, Moore AT, Ahmed ZM. Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet. 2015;52(2):85–94. [PMC free article: PMC8108008] [PubMed: 25480986]
- 162.
- Lisbjerg K, Andersen MKG, Bertelsen M, Brost AG, Buchvald FF, Jensen RB, Bisgaard AM, Rosenberg T, Tumer Z, Kessel L. Oliver McFarlane syndrome: two new cases and a review of the literature. Ophthalmic Genet. 2021;42(4):464–473. [PubMed: 33818269]
- 163.
- Baer S, Afenja A, Smol T, Piton A, Gérard B, Alembik Y, Bienvenu T, Boursier G, Boute O, Colson C, Cordier M-P, Cormier-Daire V, Delobel B, Doco-Fenzy M, Duban-Bedu B, Fradin M, Geneviève D, Goldenberg A, Grelet M, Haye D, Heron D, Isidor B, Keren B, Lacombe D, Lèbre A-S, Lesca G, Masurel A, Mathieu-Dramard M, Nava C, Pasquier L, Petit A, Philip N, Piard J, Sukno S, Thevenon J, Van-Gils J, Vincent-Delorme C, Willems M, Schaefer E, Morin G. Wiedemann-Steiner syndrome as a major cause of syndromic intellectual disability: A study of 33 French cases. Clinical Genetics. 2018;94:141–152. [PubMed: 29574747]
- 164.
- Sheppard SE, Campbell IM, Harr MH, Gold N, Li D, Bjornsson HT, Cohen JS, Fahrner JA, Fatemi A, Harris JR, Nowak C, Stevens CA, Grand K, Au M, Graham JM Jr, Sanchez-Lara PA, Campo MD, Jones MC, Abdul-Rahman O, Alkuraya FS, Bassetti JA, Bergstrom K, Bhoj E, Dugan S, Kaplan JD, Derar N, Gripp KW, Hauser N, Innes AM, Keena B, Kodra N, Miller R, Nelson B, Nowaczyk MJ, Rahbeeni Z, Ben-Shachar S, Shieh JT, Slavotinek A, Sobering AK, Abbott MA, Allain DC, Amlie-Wolf L, Au PYB, Bedoukian E, Beek G, Barry J, Berg J, Bernstein JA, Cytrynbaum C, Chung BH, Donoghue S, Dorrani N, Eaton A, Flores-Daboub JA, Dubbs H, Felix CA, Fong CT, Fung JLF, Gangaram B, Goldstein A, Greenberg R, Ha TK, Hersh J, Izumi K, Kallish S, Kravets E, Kwok PY, Jobling RK, Knight Johnson AE, Kushner J, Lee BH, Levin B, Lindstrom K, Manickam K, Mardach R, McCormick E, McLeod DR, Mentch FD, Minks K, Muraresku C, Nelson SF, Porazzi P, Pichurin PN, Powell-Hamilton NN, Powis Z, Ritter A, Rogers C, Rohena L, Ronspies C, Schroeder A, Stark Z, Starr L, Stoler J, Suwannarat P, Velinov M, Weksberg R, Wilnai Y, Zadeh N, Zand DJ, Falk MJ, Hakonarson H, Zackai EH, Quintero-Rivera F. Expanding the genotypic and phenotypic spectrum in a diverse cohort of 104 individuals with Wiedemann-Steiner syndrome. Am J Med Genet A. 2021;185(6):1649–1665. [PMC free article: PMC8631250] [PubMed: 33783954]
- 165.
- Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, eds. GeneReviews((R)). Seattle (WA)1993. [PMC free article: PMC62111] [PubMed: 21882399]
- 166.
- Ito N, Ihara K, Miyake N, Hara T, Tsutsumi Y, Matsumoto N. Hypothalamic pituitary complications in Kabuki syndrome. Pituitary. 2019;16:133–138. [PubMed: 22434255]
- 167.
- Dayal D, Giri D, Senniappan S. A rare association of central hypothyroidism and adrenal insufficiency in a boy with Williams-Beuren syndrome. Ann Pediatr Endocrinol Metab. 2017;22(1):65–67. [PMC free article: PMC5401826] [PubMed: 28443262]
- 168.
- Levy-Shraga Y, Gothelf D, Pinchevski-Kadir S, Katz U, Modan-Moses D. Endocrine manifestations in children with Williams-Beuren syndrome. Acta Paediatr. 2018;107(4):678–684. [PubMed: 29266477]
- 169.
- Güven A. Seven cases with Williams-Beuren syndrome: endocrine evaluation and long-term follow-up. J Pediatr Endocr Met. 2016;30(2):159–165. [PubMed: 28085672]
- 170.
- Xekouki P, Fryssira H, Maniati-Christidi M, Amenta S, Karavitakis EM, Kanaka-Gantenbein C, Dacou-Voutetakis C. Growth hormone deficiency in a child with Williams-Beuren syndrome. The response to growth hormone therapy. J Pediatr Endocrinol Metab. 2005;18(2):205–207. [PubMed: 15751610]
- 171.
- Garcia ML, Ty EB, Taban M, David Rothner A, Rogers D, Traboulsi EI. Systemic and ocular findings in 100 patients with optic nerve hypoplasia. J Child Neurol. 2006;21(11):949–956. [PubMed: 17092460]
- 172.
- Bongsebandhu-Phubhakdi C, Tempark T, Supornsilchai V. Endocrine manifestations of PHACE syndrome. J Pediatr Endocrinol Metab. 2019;32(8):797–802. [PubMed: 31323007]
- 173.
- Goddard DS, Liang MG, Chamlin SL, Svoren BM, Spack NP, Mulliken JB. Hypopituitarism in PHACES Association. Pediatr Dermatol. 2006;23(5):476–480. [PubMed: 17014646]
- 174.
- Garzon MC, Epstein LG, Heyer GL, Frommelt PC, Orbach DB, Baylis AL, Blei F, Burrows PE, Chamlin SL, Chun RH, Hess CP, Joachim S, Johnson K, Kim W, Liang MG, Maheshwari M, McCoy GN, Metry DW, Monrad PA, Pope E, Powell J, Shwayder TA, Siegel DH, Tollefson MM, Vadivelu S, Lew SM, Frieden IJ, Drolet BA. PHACE Syndrome: Consensus-Derived Diagnosis and Care Recommendations. J Pediatr. 2016;178:24–33 e22. [PMC free article: PMC6599593] [PubMed: 27659028]
- Review Hypopituitarism oddities: congenital causes.[Horm Res. 2007]Review Hypopituitarism oddities: congenital causes.Kelberman D, Dattani MT. Horm Res. 2007; 68 Suppl 5:138-44. Epub 2007 Dec 10.
- Review Genetic regulation of pituitary gland development in human and mouse.[Endocr Rev. 2009]Review Genetic regulation of pituitary gland development in human and mouse.Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Endocr Rev. 2009 Dec; 30(7):790-829. Epub 2009 Oct 16.
- Review [Clinical and genetic aspects of combined pituitary hormone deficiencies].[Ann Endocrinol (Paris). 2008]Review [Clinical and genetic aspects of combined pituitary hormone deficiencies].Castinetti F, Reynaud R, Saveanu A, Quentien MH, Albarel F, Barlier A, Enjalbert A, Brue T. Ann Endocrinol (Paris). 2008 Feb; 69(1):7-17. Epub 2008 Mar 4.
- Review Genetic forms of hypopituitarism and their manifestation in the neonatal period.[Early Hum Dev. 2009]Review Genetic forms of hypopituitarism and their manifestation in the neonatal period.Alatzoglou KS, Dattani MT. Early Hum Dev. 2009 Nov; 85(11):705-12. Epub 2009 Sep 16.
- Review Hypopituitarism.[Endotext. 2000]Review Hypopituitarism.Chung TT, Monson JP. Endotext. 2000
- Genetic Etiology of Congenital Hypopituitarism - EndotextGenetic Etiology of Congenital Hypopituitarism - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...