

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
The neurohypophysis is the structural foundation of a neuro-humoral system coordinating fluid balance and reproductive function through the action of two peptide hormones: vasopressin and oxytocin. Vasopressin is the principle endocrine regulator of renal water excretion, facilitating adaptive physiological responses to maintain plasma volume and plasma osmolality. Oxytocin is important in parturition and lactation. Data support a wider role for both peptides in the neuro-regulation of complex behavior. Clinically, deficits in the production or action of vasopressin manifest as diabetes insipidus. An understanding of the physiology and pathophysiology of vasopressin is also critical in approaching the diagnosis and management of hyponatremia, the most common electrolyte disturbance in clinical practice. This chapter explores the anatomy, physiology, and pathophysiology of the neurohypophysis, vasopressin and oxytocin: highlighting developments in the neural basis of osmo-sensing; the mechanism of action of vasopressin and oxytocin; together with a description of the cell and molecular biology underpinning some of the disease processes in which both the structure and functions of the two hormones are involved. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
The neurohypophysis consists of three parts: the supraoptic and paraventricular nuclei of the hypothalamus; the supraoptico-hypophyseal tract; and the posterior pituitary. The neurohypophysis is one component of a complex neurohumoral system coordinating physiological responses to changes in both the internal and external environment. This chapter will concentrate on the physiology and pathophysiology of two hormones made by the hypothalamus and posterior pituitary, vasopressin (AVP) and oxytocin (OT). These hormones have key roles in water balance and reproductive function.
ANATOMY, CELL BIOLOGY AND PHYSIOLOGY OF THE OF THE HYPOTHALAMO-POSTERIOR PITUITARY AXIS
Anatomy Of the Neurohypophysis
The posterior pituitary is derived from the forebrain during development and is composed predominantly of neural tissue. It lies below the hypothalamus, with which it forms a structural and functional unit: the neurohypophysis. The supraoptic nucleus (SON) is situated along the proximal part of the optic tract. It consists of the cell bodies of discrete vasopressinergic and oxytotic magnocellular neurons projecting to the posterior pituitary along the supraoptico-hypophyseal tract. The paraventricular nucleus (PVN) also contains discrete vasopressinergic and oxytotic magnocellular neurons, also projecting to the posterior pituitary along the supraoptico-hypophyseal tract. The PVN contains additional, smaller parvocellular neurons that project to the median eminence and additional extra-hypothalamic areas including forebrain, brain stem, and spinal cord. Some of these parvocellular neurons are vasopressinergic. A group of those projecting via the median eminence co-secrete VP and corticotrophin releasing hormone (CRH), and terminate in the hypophyseal-portal bed of the anterior pituitary. These and other vasopressinergic parvocellular neurons terminating in the hypophyseal-portal bed have a role in the regulation of adrenocorticotrophin (ACTH) release from the anterior pituitary gland, acting synergistically with CRH produced by other hypothalamic neurons. A schematic overview of the anatomy of the neurohypophysis and its major connections is shown in Figure 1.

Figure 1.
Schematic representation of the anatomy of the neurohypophysis, and its major afferent and efferent connections.
The posterior pituitary receives an arterial blood supply from the inferior hypophyseal artery and the artery of the trabecula (a branch of the superior hypophyseal artery), derivatives of the internal carotid artery and its branches. The SON and PVN receive an arterial supply from the supra-hypophyseal, anterior communicating, anterior cerebral, posterior communicating and posterior cerebral arteries, all derived from the circle of Willis. Venous drainage of the neurohypophysis is via the dural, cavernous, and inferior petrosal sinuses.
Molecular-Cell Biology of Vasopressin and Oxytocin
AVP is a 9 amino acid peptide with a disulphide bridge between the cysteine residues at positions 1 and 6 (Figure 2). Most mammals have the amino-acid arginine at position 8, though in the Pig family arginine is substituted by lysine. The structure of OT differs from that of AVP by only 2 amino acids: isoleucine for phenylalanine at position 3; and leucine for arginine at position 8. Non-mammalian species have a variety of peptides very similar to AVP and OT, suggesting they derive from a common ancestral gene.

Figure 2.
The structural and chemical characteristics of Vasopressin and Oxytocin. The cyclical peptides differ in only 2 amino acid positions. Both contain disulphide bridges between Cysteine residues at positions 1 and 6.
THE VASOPRESSIN-NEUROPHYSIN AND OXYTOCIN-EUROPHYSIN GENES
The genes encoding AVP and OT are in a head-to-head tandem array on chromosome 20p13 in Man, separated by 12 Kb of DNA. Each has 3 exons, and encodes a polypeptide precursor with a modular structure: an amino-terminal signal peptide; the AVP or OT peptide; a hormone-specific mid-molecule peptide termed a neurophysin (NPI and NPII for OT and AVP respectively); and a carboxyl-terminal peptide known as copeptin (Figure 3). There is considerable homology between the NP sequences of the AVP-NP and OT-NP genes, positions 10-74 of the NP sequences being highly conserved at the amino acid level.

Figure 3.
Structural organization of the Vasopressin-neurophysin II gene, and processing of its product. The AVP-NPII gene has 3 exons. Translation of the mRNA yields a larger preprohormone precursor, subsequently modified through substantial post-translational modification. The OT gene has a similar structure, and its product undergoes similar processing and post-translational modification. AVP: Vasopressin. NPII: Neurophysin II. OsRE: osmo-sensitive response element. GRE: glucocorticoid response element. ERE: estrogen response element. AP1-RE: AP1 response element.
Regulatory control of AVP gene expression is mediated through positive and negative elements in the proximal promoter. Several transcription factors bind to these elements. AP1, AP2 and CREB stimulate AVP gene expression. The glucocorticoid receptor (GR) represses expression (1, 2). The human, rat and mouse OT promoters contain half estrogen-response elements, and IL-6 response elements (3). The inter-genic region between the AVP and OT genes contains regulatory elements responsible for selective expression. The region -288 to -116 5′ upstream of the AVP gene promoter confers cell-type (magnocellular neuron) specific expression of the AVP gene (4). AVP gene expression can also be regulated at a post-transcriptional level. The length of the poly (A) tail of AVP mRNA increases in response to water deprivation, influencing mRNA stability (4). AVP mRNA also contains a dendritic localization sequence (DLS). Interaction of the DLS with a multifunctional poly(A) binding protein (PABP) may play key role in RNA stabilization, initiation of translation and translational silencing (5).
Synthesis And Release of Vasopressin and Oxytocin
Synthesis of the AVP and OT precursors occurs in the cell bodies of discrete vasopressinergic and oxytotic magnocellular neurosecretory neurons within the supraoptic (SON) and paraventricular (PVN) nuclei of the hypothalamus. Generation of the mature hormone entails post-translational modification of the large primary precursor (Figure 4). Following ribosomal translation of the respective mRNA, the carboxyl terminal domain of the precursor is glycosylated, and the product packaged in vesicles of the regulated secretory pathway. These migrate along the axons of the magnocellular neurons, during which the precursor is cleaved by basic endopeptidases into the mature hormone and the associated NP and copeptin. These are stored in secretory granules within the terminals of the magnocellular neurons in the posterior pituitary. Increased firing frequency of vasopressinergic and oxytotic neurons opens voltage-gated Ca2+ channels in these nerve terminals. This, in turn, leads to transient Ca2+ influx, fusion of the neurosecretory granules with the nerve terminal membrane, and release of the hormone and its NP and copeptin into the systemic circulation in equimolar quantities. NPs act as carrier proteins for AVP and OT during axonal migration, and appear to serve no other function. The function of copeptin remains unclear to date, it has been postulated to play a role as a prolactin-releasing factor, but never confirmed (6, 7). Another role as a chaperone-like molecule involved in the folding of the AVP precursor has been speculated (8) .In addition, it was reported to interact with the calnexin–calreticulin system (9), which prevents the export of misfolded, glucose-tagged proteins from the endoplasmic reticulum.

Figure 4.
Schematic overview of the post-translational processing of the AVP-NP II gene product. Sequential modification of 164 amino-acid AVP-NPII preprohormone in endoplasmic reticulum and golgi lead to trafficking through the regulated secretory pathway and ultimately release from neurosecretory vesicles in the posterior pituitary. AVP-NPII precursor is complexed as tetramers or high oligomers during processing. A small amount of partially processed precursor is released through the constitutive secretory pathway. OT is processed in a similar manner.
AVP and OT circulate unbound to plasma proteins, though AVP does bind to specific receptors on platelets. AVP concentrations in platelet-rich plasma are 5-fold higher than in platelet-depleted plasma (10). AVP and OT have short circulating half-lives of 5-15 minutes. Several endothelial and circulating endo- and amino-peptidases degrade the peptides. A specific placental cysteine amino-peptidase degrades AVP and OT rapidly during pregnancy and the peri-partum period.
The Physiology of The Secretion of Vasopressin and Thirst
AVP is a key component in the regulation of fluid and electrolyte balance, through direct effects on renal water handling. However, the physiology of AVP has a wider context, encompassing roles in the integrated response to changes in cardiovascular status.
VASOPRESSIN RECEPTORS
There are three distinct AVP receptor (V-R) subtypes (Table 1). All have seven transmembrane spanning domains, and all are G protein coupled. They are encoded by different genes and differ in tissue distribution, down-stream signal transduction and function. The human V2-R gene maps to Xq28. Interestingly, the V2-R is up regulated by its ligand (11).
Table 1.
Vasopressin Receptor Subtypes
| Vasopressin receptor | |||
|---|---|---|---|
| V1a | V1b | V2 | |
| Expression | Vascular smooth muscle Liver Platelets CNS | Pituitary corticotroph | Basolateral membrane of distal nephron |
| Amino acid structure | 418 amino acids (human) | 424 amino acids (human) | 370 amino acids (human) |
| Second messenger system | Gq/11mediated phospholipase C activation: Ca2+, inositol triphosphate & diacyl glycerol mobilization | As V1a | Gs mediated adenylate cyclase activation: cAMP production & protein kinase A stimulation |
| Physiological effects | ●Smooth muscle contraction ●Stimulation of glycogenolysis. ●Enhanced platelet adhesion ●Neurotransmitter & neuromodulatory function | Enhanced ACTH release | Increased synthesis & assembly of aquaporin-2 |
VASOPRESSIN AND RENAL WATER HANDLING
Although AVP has multiple actions, its principle physiological effect is in the regulation of water resorption in the distal nephron, the structure and transport processes of which allow the kidney to both concentrate and dilute urine in response to the prevailing circulating AVP concentration. Active transport of solute out of the thick ascending loop of Henle generates an osmolar gradient in the renal interstitium, which increases from renal cortex to inner medulla, a gradient through which distal parts of the nephron pass end route to the collecting system. AVP stimulates the expression of a specific water channel protein (aquaporin) on the luminal surface of the interstitial cells lining the collecting duct. The presence of aquaporin (AQP) in the wall of the distal nephron allows resorption of water from the duct lumen along an osmotic gradient, and excretion of concentrated urine. To date, 13 different AQPs have been identified in Man, seven of which (AQP1-4, AQP6-8) are found in the kidney. AQPs act as passive pores for small substrates and are divided into 2 families: the water only channels; and the aquaglyceroporins that can conduct other small molecules such as glycerol and urea. Most substrates are neutral. However, this is not always the case. For example, AQP6 is a gated ion channel. AQPs are involved in a variety of cell processes: small molecule permeation; gas conduction and cell-cell interaction. As with other membrane channels, specific structural arrangements within the primary, secondary, and tertiary structure convey the three functional characteristics of permeation, selectivity, and gating. The structure of AQPs involves 2 tandem repeats, each formed from 3 transmembrane domains, together with 2 highly conserved loops containing the signature motif asparagine-proline-alanine (NPA). All AQPs form homotetramers in the membrane, providing 4 functionally independent pores with an additional central pore formed between the 4 monomers. Water can pass through all the 4 independent channels of water-permeable AQPs. There are data to suggest that the central pore may act as independent channel in some AQPs (12-14). AQP1 is constitutively expressed in the apical and basolateral membranes of the proximal tubule and descending loop of Henle, where it facilitates isotonic fluid movement. Loss of function mutations of AQP1 in man lead to defective renal water conservation (15). AQP2 is expressed on the luminal surface of collecting duct cells and is the water channel responsible for AVP-dependent water transport from the lumen of the nephron into the collecting duct cells. V2-R activation in collecting duct cells produces a biphasic increase in expression of AQP2. Ligand-receptor binding triggers an intracellular phosphorylation cascade ultimately resulting in phosphorylation of the nuclear transcription factor CREB and expression of c-Fos. In turn, these transcription factors stimulate AQP2 gene expression through CRE and AP-1 elements in the AQP2 gene promoter. In addition, AVP stimulates an immediate increase in AQP2 expression by accelerating trafficking and assembly of pre-synthesized protein into functional, homo-tetrameric water channels.
Maximum diuresis occurs at plasma AVP concentrations of 0.5 pmol/l or less. As AVP levels rise, there is a sigmoid relationship between plasma AVP concentration and urine osmolality, with maximum urine concentration achieved at plasma AVP concentrations of 3-4 pmol/L (Figure 5). Following persistent AVP secretion, antidiuresis may diminish. Down-regulation of both V2-R function and AQP2 expression may be responsible for this escape phenomenon.

Figure 5.
The relationship of plasma AVP concentration to urine osmolality. Shaded area represents range of normal; single line indicates representative individual. AVP has additional effects at other sites in the nephron: decreasing medullary blood flow; stimulating active urea transport in the distal collecting duct; and stimulating active sodium transport into the renal interstitium. AVP up-regulates the bumetanide-sensitive sodium-potassium-chloride cotransporter (SLC12A1) in the thick ascending loop of Henle through both a rapid acceleration of post-translational processing/trafficking and an increase in SCLC12A1 gene expression. Together, these contribute to the generation and maintenance of a hypertonic medullary interstitium, and augment AVP-dependent water resorption (16).
REGULATION OF VASOPRESSIN RELEASE
Osmoregulation of Vasopressin
Plasma osmolality is the most important determinant of AVP secretion. The osmoregulatory systems for thirst and AVP secretion, and in turn the actions of AVP on renal water excretion, maintain plasma osmolality within narrow limits of 284 to 295 mOsmol/kg. The relationship between plasma osmolality and plasma AVP concentration has 3 characteristics.
- The osmotic threshold or 'set point' for AVP release.
- The shape of the line describing changes in plasma AVP concentration with changing plasma osmolality
- The sensitivity of the osmoregulatory mechanism coupling plasma osmolality and AVP release.
Increases in plasma osmolality increase plasma AVP concentrations in a linear manner (Figure 6). The abscissal intercept of this line indicates the mean 'osmotic threshold' for AVP release (284 mOsmol/kg): the mean plasma osmolality above which plasma AVP increases in response to increases in plasma osmolality. AVP levels increase from a basal rate through activation of stimulatory osmoreceptor afferents, and decrease to minimal values when this drive is removed and synergistic inhibitory afferents are activated. The slope of the line relating plasma osmolality to plasma AVP concentration reflects the sensitivity of osmoregulated AVP release. There are considerable inter-individual variations in both the threshold and sensitivity of AVP release. However, they are remarkably reproducible within an individual over time (17).
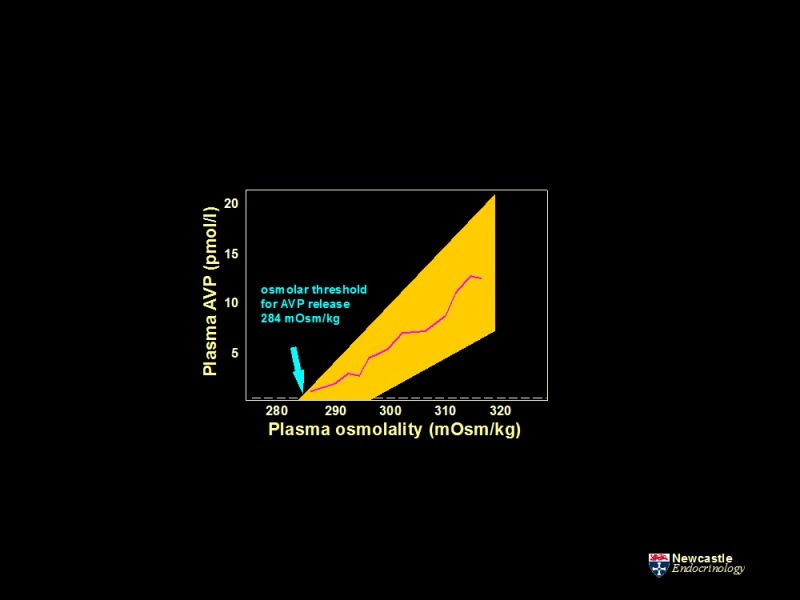
Figure 6.
The relationship of plasma AVP concentration to changes in plasma osmolality during controlled hypertonic stimulation. AVP concentration determined during progressive hypertonicity induced by infusion of 855 mmol/l saline in a group of healthy adults. Increases in plasma osmolality increase plasma AVP concentrations in a linear manner, defined by the function, plasma AVP = 0.43 (plasma osmolality - 284), r = +0.96. The abscissal intercept of this regression line indicates the mean 'osmotic threshold' for AVP release: the mean plasma osmolality above which plasma AVP starts to increase. The shaded area represents the range of normal response. LD represents the limit of detection of the assay, 0.3 pmol/l.
There are situations where the normal relationship between plasma osmolality and AVP concentration breaks down:
- Rapid changes of plasma osmolality: rapid increases in plasma osmolality result in exaggerated AVP release.
- During the act of drinking: drinking rapidly suppresses AVP release, through afferent pathways originating in the oropharynx.
- Pregnancy: the osmotic threshold for AVP release is lowered in pregnancy.
- Whether aging is accompanied by changes in AVP concentrations is controversial.
As befits its major function and physiological role, AVP production by the neurohypophysis is influenced by sensory signals reflecting osmotic status and blood pressure/circulating volume. The relationships of the SON and PVN with the autonomic afferents and central nervous system nuclei responsible for osmo- and baroregulation are key to the physiological regulation of AVP. Functional osmoreceptors are situated in anterior circumventricular structures: the subfornicular organ (SFO), and the organum vasculosum of the lamina terminalis (OVLT). Local fenestrations in the blood brain barrier at these sites allow neural tissue direct contact with the circulation. Subsequent sensory input to the SON and PVN is via glutaminergic afferents. Moreover, these neurons integrate osmolar status with additional endocrine signals reflecting circulating volume status through the action of angiotensin II (A-II), relaxin, and atrial natriuretic peptide (ANP). A-II and relaxin excite both OT and AVP magnocellular neurons. In contrast, ANP inhibits AVP neuron activity. AVP neurons themselves have independent osmo-sensing properties and V-Rs are present on vasopressinergic neurons of both the PVN and SON, highlighting the potential for auto-control of AVP release through direct osmoregulation and short loop feedback (18). AVP magnocellular neurons in the SON and PVN co-express the peptide Apelin and its G-protein coupled receptor. A ‘yin and yang’ relationship has been proposed between AVP and these 36 amino-acid peptides (and indeed it’s shorter active derivatives Apelin-17 and Apelin-13). Intra-cerebroventricular injection of Apelin-17 inhibits the phasic firing of AVP magnocellular neurons, reducing AVP release and stimulating aquaresis. Hypertonic stress and water loading have reciprocal effects on plasma AVP and Apelin concentrations. Apelin receptors are also co-expressed in AVP target cells in the renal collecting duct. AVP and Apelin are thus regulated in opposite directions to maintain volume and osmolar homeostasis (19-22).
Changes in the osmotic environment of osmo-sensitive neurons in the OVLT, SFO and vasopressinergic neurons of the SON and PVN result in altered cell volume. These physical changes alter the activity of the stretch-sensitive cationic channel TRPV1, expressed on the cell surface of these neurons. TRPVI thus acts as the transduction mechanism linking changes in osmolality to altered membrane potential and firing frequency. Osmoregulatory function is not lost in Trpv1−/−mice, indicating additional osmo-sensing pathways must be in operation (23).
A related, but distinct osmo-sensory input feeds additional data on peripheral osmolar status to the neurohypophysis. Hepatic portal blood vessels contain sensory neurons responsive to changes in the osmolality of peripheral blood. In contrast to central mechanisms, the key transducing element of the peripheral process is the stretch-sensitive ion channel, TRPV4. Plasma osmolality is frequently elevated in patients after liver transplant in which the donor organ is denervated, demonstrating the function of this peripheral pathway (24).
Osmosensitivity of AVP release is influenced by circadian rhythms. AVP release increases during sleep. This effect is mediated by clock neurons projecting from the suprachismatic nucleus, increasing the activity of osmosensory afferent input to the SON (25).
Baroregulation of Vasopressin
Reductions in circulating volume stimulate AVP release. Falls in arterial blood pressure of 5 to 10 per cent are necessary to increase circulating AVP concentrations in man. Progressive reduction in blood pressure produces an exponential increase in plasma AVP, in contrast to the linear increases of osmoregulated AVP release. Baroregulatory influences on neurohypophyseal AVP release derive from aortic arch, carotid sinus, cardiac atrial, and great vein stretch-sensitive afferents via cranial nerves IX and X. Ascending projections are via the nucleus tractus solitarius (NTS) in the brain stem. From the NTS, further afferents project to the SON and PVN, which also receive additional adrenergic afferents from other brain stem nuclei involved in cardiovascular control, such as the locus coeruleus. These nuclei integrate a number of afferent inputs that reflect volume status. Ascending baroregulatory pathways must affect some tonic inhibition of AVP release, as interruption increases plasma AVP levels (26, 27). Osmoregulated AVP responses can be modified by factors triggered as part of the coordinated neurohumoral response to changes in circulating volume and blood pressure. A-II amplifies the proportional relationship between osmolality and action potential firing in the SON. The peptide produces this effect through polymerization of intracellular actin filaments, resulting in altered cell shape, a mechanism that is synergistic with those mediating responses to changes in extracellular osmolality. A-II thus enhances osmosensitivity. This mechanism underpins the changes in osmo-regulated AVP release in hypo- and hypervolemia: the osmotic threshold and sensitivity of AVP release is lowered by hypovolemia; while the converse is found in hypervolemia and hypertension (19).
Additional Mechanisms Regulating Vasopressin Release
A number of other stimuli influence AVP release independent of osmotic and hemodynamic status.
- Nausea and emesis
- Unspecific stress
- Pain
- Manipulation of abdominal contents
- Immune-response mediators and inflammatory triggers
These stimuli contribute to high plasma AVP values observed in acute illness and after surgery.
ADDITIONAL EFFECTS OF VASOPRESSIN
Cardiovascular Effects
AVP is a potent pressor agent; its effects mediated through a specific receptor (V1-R) expressed by vascular smooth muscle cells. Though systemic effects on arterial blood pressure are only apparent at high concentrations, AVP is important in maintaining blood pressure in mild volume depletion. The most striking vascular effects of AVP are in the regulation of regional blood flow. The sensitivity of vascular smooth muscle to the pressor effects of AVP varies according to the vascular bed. Vasoconstriction of splanchnic, hepatic and renal vessels occur at AVP concentrations close to the physiological range. Furthermore, there are differential pressor responses within a given vascular bed. Selective effects on intrarenal vessels lead to redistribution of renal blood flow from medulla to cortex. Baroregulated AVP release thus constitutes one of the key physiological mediators of an integrated hemodynamic response to volume depletion.4.2.
Effects on the Pituitary
AVP is an ACTH secretagogue, acting through pituitary corticotroph-specific V1b-Rs. Though the effect is weak in isolation, AVP and CRF act synergistically. AVP and CRF co-localize in neurohypophyseal parvocellular neurons projecting to the median eminence and the neurohypophyseal portal blood supply of the anterior pituitary. Levels of both AVP and CRF in these neurons are inversely related to glucocorticoid levels, consistent with a role in feedback regulation.
Effects of AVP on Regulation of Bone Mass
AVP exerts its action both on osteoblasts and osteoclasts thru AVPR1 and AVPR2 receptors (28). Mice studies showed that AVP upregulates osteoclast differentiation genes and inhibits osteoblast formation. Mice rendered deficient in AVPr1a (Avpr1a-/-) have a high bone mass, and they show an increase in osteoblastogenesis after additional inhibition of AVPR2 (29) .
The AVP effect is opposed by oxytocin, which stimulates osteoblast formation and inhibits mature osteoclast activation. Mice studies showed that both haploinsufficiency for oxytocin and deletion of the oxytocin receptor (Oxr-/-) result in osteopenia (30). The role of both hormones in the regulation of skeletal physiology remains to be further explored (31).
Behavioral Effects of Vasopressin
Vasopressinergic fibers and V-Rs are present in CNS neural networks anatomically and functionally independent of the neurohypophysis, including the cerebral cortex and limbic system. An increasing amount of data highlight the role of central vasopressinergic systems in mediating complex social behavior. Data in Man link V1a-R gene sequence variation with a range of normal and abnormal behavior patterns, including gender dimorphic behavior. Dysregulation of central AVP action may be a distal end point in complex conditions characterized by altered social and emotional behavior (32, 33).
Thirst
Renal free water clearance can be reduced to a minimum by the antidiuretic actions of vasopressin, but water loss is not completely eliminated, and insensible water loss from respiration and sweating is a continuous process. To maintain water homeostasis, water must also be consumed to replace the obligate urinary and insensible fluid losses. This is regulated by thirst. Thirst and drinking are key processes in the maintenance of fluid and electrolyte balance. Thirst perception and the regulation of water ingestion involve complex, integrated neural and neurohumoral pathways. As with those mediating AVP release, the osmoreceptors regulating thirst are situated in the OVLT, effectively outside the blood-brain barrier and distinct from those mediating AVP release. Neural activity in and around the OVLT remains active in hyperosmolar states following immediate satiety of thirst, indicating that other centers must be involved in thirst perception. The anterior cingulate cortex and insular cortex receive input from osmo-sensitive afferents and have been implicated as key higher centers in thirst pathways (20). There is a linear relationship between thirst and plasma osmolalities in the physiological range. The mean osmotic threshold for thirst perception is 281 mOsm/kg, similar to that for AVP release. Thirst occurs when plasma osmolality rises above this threshold. As with osmoregulated AVP release, the characteristics of osmoregulated thirst remain consistent within an individual on repeated testing, despite wide inter-individual variation.
As with AVP release, there are also specific physiological situations in which the relationship between plasma osmolality and thirst breaks down.
- The act of drinking: reduces osmotically stimulated thirst.
- Extracellular volume depletion: this stimulates thirst through volume-sensitive cardiac afferents and the generation of circulating and intra-cerebral A-II, a powerful dipsogen.
- Pregnancy, the luteal phase of the menstrual cycle and super ovulation syndrome: these states reduce the osmolar threshold for thirst.
- Aging: both thirst appreciation and fluid intake can be blunted in the elderly
The act of drinking reduces thirst perception before any change in plasma osmolality. This effect is produced through three mechanisms: oropharyngeal sensory afferents; gastro-intestinal stretch-sensitive afferents; and peripheral osmoreceptors in the hepatic portal vein. Recent data have highlighted how thirst-promoting neurons in the SFO integrate sensory inputs from the oropharynx (drinking and food composition) with central osmolar status to influence thirst perception. This complex mechanism effectively explains anticipatory changes in water consumption that precede changes in plasma osmolality (34). As with AVP release, hypovolemia resets the relationship between plasma osmolality and thirst. A-II is one of the key mediators of this physiological response. Peripheral A-II generation can act on central osmoreceptors, to increase both thirst and AVP release. An independent, intra-cerebral A-II system is activated in parallel. A-II is a powerful central dipsogen.
The Physiology of Oxytocin
OT binds to specific G-protein coupled cell surface receptors (OT-Rs) on target cells to mediate a variety of physiological effects, largely concerned with reproductive function. The classical physiological roles of OT are the regulation of lactation, parturition and reproductive behavior. Data from transgenic animals with targeted disruption of the oxytocin gene (and thus lacking OT) have forced a review of this dogma (35).
OXYTOCIN AND LACTATION
In the rat, stimulation of vagal sensory afferents in the nipple by the act of suckling triggers reflex synchronized firing of oxytotic magnocellular neurons in the neurohypophysis, and corresponding pulsatile OT release. OT acts on OT-Rs on smooth muscle cells lining the milk ducts of the breast, initiating milk ejection. OT is essential for completion of this milk ejection reflex in rodent. Mice lacking OT fail to transfer milk to their suckling young. This deficit is corrected by injection of OT. In contrast, women lacking posterior pituitary function can breast-feed normally, illustrating that OT is not necessary for lactation in man. Pituitary lactotrophs express OT-R mRNA, and OT released into the hypophyseal portal blood supply from the median eminence can stimulate prolactin release. However, the role of OT in the physiology of prolactin release remains unclear.
OXYTOCIN AND PARTURITION
OT is a uterotonic agent. In many mammals, there is both an increase in OT secretion and an increase in uterine responsiveness to OT during parturition (3). These data suggest a key role for the hormone in the initiation and progression of labor. Falling progesterone concentrations toward the end of pregnancy lead to up-regulation of uterine myometrial OT-Rs, enhanced contractility, and increased sensitivity to circulating OT. Stretching of the 'birth canal' during parturition leads to the stimulation of specific autonomic afferents, reflex firing of oxytotic neurons and OT release. A positive feedback loop is formed, OT stimulating uterine contraction further and enhancing the production of additional local uterotonic mediators such as prostaglandins. The difficulties of analyzing pulsatile release, and the short circulating half-life of the hormone (due to placental cysteine aminopeptidase), have made it difficult to demonstrate increased circulating OT levels in women during labor. Mice lacking OT have normal parturition. Moreover, women with absent posterior pituitary function can have a normal labor. However, the importance of OT in the birth process is highlighted by the effectiveness of OT antagonists in the management of pre-term labor (36). The role of OT in parturition is not limited to maternal responses. Maternal OT produces a switch to inhibitory GABAergic signaling in the fetal CNS. This, in turn, increases fetal neuronal resistance to damage that may occur during delivery. OT therefore mediates direct adaptive mother-fetal signaling during parturition in line with a wider-ranging role in maternal-fetal physiology (37).
OXYTOCIN AND BEHAVIOR
OT-R expression is widespread in the CNS of many species, and OT has widespread roles as a neurotransmitter. The central oxytocinergic system and related limbic networks affect complex neural circuits of socio-emotional behavior and promote pro-social effects such as in-group favoritism and protection against social threats, interpersonal trust and attachment, empathy, and emotion recognition (38-41). In humans, brain regions such as the amygdala, hippocampus, cingulate cortex, and nucleus accumbens, which play a key role in human socio-emotional behavior, show a high density of OT-R expression. In some cases, these overlap those involving AVP (32, 33).
OT knockout (OT-KO) models have been used to identify aspects of dysfunctions in social behavior. Generally, an impairment in forming social memories and higher anxiety-related behaviors were observed (42-44). These results were substantiated by the fact that central administration of OT rescued OT-KO mice from these changes. In the context of behavior; OT facilitates both lordosis and the development of maternal behavior patterns in rat (3). However, mice lacking OT exhibit normal sexual and maternal behavior, suggesting the behavioral effects to be species-specific or the potential for considerable redundancy in neural pathways. In humans, lower endogenous OT or impaired signaling has been linked with mental disorders associated with social deficits, such as autism spectrum disorder (ASD), anxiety and depression disorder or borderline personality disorder (45-50). In these disorders, no OT deficiency per se has been proven; the evidence is largely based on observational studies with individual variations in social behavior associated with alteration in peripheral OT levels, in genes involved in OT signaling or OT-R polymorphisms (51-53). In ASD, recent data have highlighted associations with the single nucleotide polymorphisms (SNPs) rs7632287, rs237887, rs2268491 and rs2254298 (54-56). Intranasal OT has been investigated to ameliorate symptoms of these conditions. However, overall, the effect size is inconsistent, and the results of these studies are controversial. Central oxytocinergic transmission reduces anxiety behavior and hypothalamo-pituitary-adrenal stress responses in female rats. It may be that OT has a complex role in the stress response, with context-dependent differential effects. It buffers responses to social stress, reduces cortisol levels during conflict situations, improves self-representations in patients with anxiety disorder, and reduces amygdala response to emotional stimuli, consequently reducing fear reactivity and anxiety (57-63).
Only limited research has been devoted to the role of OT in patients with hypothalamic-pituitary dysfunction and focused primarily on patients with craniopharyngioma (CP). These data assume direct tumor-induced or post-surgical damage to the SON/PVN and consequent disruption of the oxytocinergic system (64-68). Results demonstrate personality changes and increased psycho-social comorbidities – including anxiety, depression, and social withdrawal (64, 69-71). This was confirmed by a systematic review showing behavioral dysfunctions in 57%, and social impairment and difficulties holding relationships in 40% (72). Interestingly, the age of onset appears to influence the type of socio-behavioral dysfunction: while adult-onset had higher levels of anxiety and depression, younger patients had more impact in the domains of social isolation. Research in patients with confirmed posterior pituitary dysfunction, i.e., CDI, demonstrated higher depression and anxiety levels, self-reported autistic traits, lower joy when socializing and worse scores on an emotional recognition task than healthy adults (73, 74).
Similar to other hormones, single basal OT levels are unreliable and insufficient in identifying a deficiency (75) with inconclusive results in patients with hypothalamic-pituitary dysfunction. Research still disagrees regarding the precise relationship between peripheral OT and centrally generated behavior. Cerebrospinal fluid (CSF) concentrations of OT are more likely to mirror the behavioral effects than plasma concentrations, and whether plasma levels may serve as a surrogate for CSF OT is controversially discussed (76, 77). Accordingly, peripheral levels of OT correlate to central levels only after stimulation but not at baseline (78). Established pituitary provocation tests do not show a consistent strong OT increase to test for a suspected deficiency (79). OT-R expression is widespread in the CNS of many species and OT has widespread roles as a neurotransmitter, including neural networks that mediate a range of complex behaviors. In some cases, these overlap those involving AVP (32, 33).
CLINICAL PROBLEMS SECONDARY TO DEFECTS IN THE HYPOTHALAMO-POSTERIOR PITUITARY AXIS
Defects in the production or action of AVP manifest as clinical problems in maintaining plasma sodium concentration and fluid balance, reflecting the key role of the hormone in these processes. A further group of related clinical conditions reflect primary defects in thirst. In some cases, the two may coincide, reflecting the close anatomical and functional relationship of both processes.
Diabetes Insipidus
CLASSIFICATION (see Figure 7)
Diabetes insipidus (DI) is characterized by production of dilute urine in excess of >50 ml/kg/24 hours in adults. DI arises through one of four mechanisms (Figure 7 and Table 2).
- Deficiency of AVP: central diabetes insipidus (CDI). Also called Arginine Vasopressin Deficiency
- Inappropriate, excessive water drinking: primary polydipsia.
- Renal resistance to the antidiuretic action of AVP: nephrogenic diabetes insipidus (NDI). Also called Arginine Vasopressin Resistance
- increased vasopressinase expression in pregnancy: Gestational diabetes insipidus

Table 2.
Classification of Hypotonic Polyuria (80)
| Type of hypotonic polyuria | Basic defect | Causes |
|---|---|---|
| Central DI | Deficiency in AVP synthesis or secretion | Acquired Trauma (surgery, deceleration injury) Neoplastic (craniopharyngioma, meningioma, germinoma, metastases) Vascular (cerebral/ hypothalamic hemorrhage, infarction or ligation of anterior communicating artery aneurysm) Granulomatous (histiocytosis, sarcoidosis) Infectious (meningitis, encephalitis, tuberculosis) Inflammatory/autoimmune (lymphocytic infundibuloneurohypophysitis, IgG4 neurohypophysitis) Drug/toxin-induced Osmoreceptor dysfunction (adipsic DI) Others (hydrocephalus, ventricular/suprasellar cyst, trauma, degenerative disease) Idiopathic Congenital Autosomal dominant: AVP gene mutation Autosomal recessive: Wolfram Syndrome (DIDMOAD) X-linked recessive |
| Primary Polydipsia | Excessive osmotically unregulated fluid intake | Dipsogenic (downward resetting of the thirst threshold; idiopathic or similar lesions as with central DI) Psychosis intermittent hyponatremia polydipsia (PIP syndrome) Compulsive water drinking Health enthusiasts |
| Nephrogenic DI | Reduced renal sensitivity to antidiuretic effect of physiological AVP levels | Acquired Drug exposure (lithium, demeclocycline, cisplatin etc.) Hypercalcemia, hypokalemia Infiltrating lesions (sarcoidosis, amyloidosis, multiple myeloma etc.) Vascular disorders (sickle cell anemia) Mechanical (polycystic kidney disease, ureteral obstruction) Congenital X-linked AVPR2 gene mutations autosomal recessive or dominant AQP2 gene mutations |
| Gestational DI | Exaggerated enzymatic metabolism of circulating AVP hormone | Increased AVP metabolism Pregnancy |
Central Diabetes Insipidus (CDI) (Arginine Vasopressin Deficiency)
CDI (also known as neurogenic or cranial DI) is the result of partial or complete lack of osmoregulated AVP secretion. Plasma AVP concentrations are inappropriately low with respect to prevailing plasma osmolalities. Presentation with CDI implies destruction or loss of function of more than 80% of vasopressinergic magnocellular neurons. It is rare (estimated prevalence of 1: 25000), with an equal gender distribution. Though persistent polyuria can lead to dehydration, most patients can maintain water balance through appropriate polydipsia if given free access to water.
Most cases of CDI are acquired. Trauma (head injury or surgery) can produce CDI through damage to the hypothalamus, pituitary stalk, or posterior pituitary. Pituitary stalk trauma may lead to a triphasic disturbance in water balance, an immediate polyuric phase followed within days by a more prolonged period (up to several weeks) of antidiuresis suggestive of AVP excess. This second phase can be followed by reversion to CDI, or recovery. This characteristic 'triple response' reflects initial axonal damage; the subsequent unregulated release of large amounts of pre-synthesized AVP; and either recovery or development of permanent CDI (as determined by the magnitude of initial damage to vasopressinergic neurons). All phases of the response are not apparent in all cases.
Hypothalamic tumors (e.g., craniopharyngioma) or pituitary metastases (e.g., breast or bronchus) can present with CDI. However, primary pituitary tumors rarely cause CDI. In childhood, craniopharyngioma and germinoma/teratoma are a relatively common cause (81).
When obvious causes are not present, most cases of CDI will be “idiopathic”. However, the possibility of an autoimmune process should be considered, as many idiopathic cases are considered to be autoimmune in origin (82). A well-recognized cause of autoimmune CDI is lymphocytic infundibulohypophysitis (83).
Familial forms are rare, but are a recognized cause of CDI in childhood. Most reported cases are expressed as autosomal dominant and the genetic defect is usually in the biologically inactive neurophysin or in the signal peptide of the pre-prohormone. Lack of normal cleavage of the signal peptide from the prohormone and abnormal folding of the vasopressin/neurophysin precursor are thought to produce fibrillar aggregations in the endoplasmic reticulum, which is cytotoxic to the neuron, explaining the dominant phenotype (84, 85). Wolfram syndrome is a rare autosomal recessive disease with diabetes insipidus, diabetes mellitus, optic atrophy, and deafness (DIDMOAD). The genetic defect is for the protein wolframin that is found in the endoplasmic reticulum and is important for folding proteins (86). Wolframin is localized to chromosome 4. It is involved in beta-cell proliferation and intracellular protein processing and calcium homeostasis, producing a wide spectrum of endocrine and central nervous system (CNS) disorders. Diabetes insipidus is usually a late manifestation and is associated with decreased magnocellular neurons in the paraventricular and supraoptic nuclei (87, 88).
Primary Polydipsia (PP)
PP is a polyuric syndrome secondary to excess fluid intake. PP can be associated with organic structural brain lesions, e.g. sarcoidosis of the hypothalamus (89) and craniopharyngioma (90). It can also be produced by drugs that cause a dry mouth or by any peripheral disorder causing an elevation of renin and/or angiotensin. However, mostly there is no identifiable pathologic etiology; in this circumstance the disorder is often associated with psychiatric syndromes. Series of patients in psychiatric hospitals have shown that as many as 42% have some form of polydipsia and for over half of those there was no obvious explanation for the polydipsia (91). It also seems to be increasingly prevalent in health conscious people who voluntarily change their drinking habits with the aim to improve their well-being, in which case it is often called habitual polydipsia (92).
Nephrogenic Diabetes Insipidus (NDI)
NDI is due to renal resistance to the antidiuretic effects of AVP. Genetic variants of NDI usually present in infancy (93). In these forms, NDI can occur as a result of mutations in the V2 receptor and mutations of the aquaporin 2 water channels. Over 90% of cases are X-linked recessive in males, and over 200 different mutations of the V2 receptor have been reported affecting all aspects of receptor function: expression; ligand binding; and G-protein coupling. Most lead to complete loss of function, though a few are associated with a mild phenotype. 10% of kindreds with familial NDI have an autosomal recessive form, with normal V2-R function. Affected individuals harbor loss of function mutations of the AQP2 gene. Most mutations occur in the region coding for the transmembrane domain of the protein. Additional rare kindreds have been described harboring a mutation in the portion of the gene encoding the carboxyl-terminal intracellular tail of AQP2. The NDI of these kindreds is inherited as an autosomal dominant trait, mutant protein sequestering the product of the wild type AQP2 allele within mixed tetramers in a dominant-negative manner.
The development of NDI in an adult is less likely to reflect a genetic cause. The commonest cause of acquired NDI in clinical practice is lithium therapy, with other causes including hypokalemia, hypercalcemia, and release of bilateral urinary tract obstruction associated with downregulation of aquaporin 2 and decreased function of vasopressin (94, 95). NDI secondary to lithium is characterized by dysregulated AQP2 expression and trafficking along the whole collecting duct as well as dysregulated expression of the amiloride-sensitive epithelial sodium channel (ENaC) in the cortical collecting duct. Lithium enters collecting duct cells through ENaC expressed on the apical cell membrane and leads to inhibition of glycogen synthase kinase type 3 (GSK-3) signaling pathways. NDI secondary to lithium toxicity can persist after drug withdrawal, and may be irreversible. Demeclocycline is another commonly recognized drug to causes NDI and is sometimes used clinically to treat SIAD. The final common pathway producing NDI in many of these cases is down-regulation of AQP2 expression (96).
Gestational Diabetes Insipidus
In normal pregnancy, physiologic adaptations include expansion of blood volume and decreased plasma osmolality and serum sodium. Thirst and increased fluid intake are commonly reported in pregnancy, but in some patients the increased thirst is driven by marked polyuria, which may point to the presence of diabetes insipidus. Two types of transient diabetes insipidus must be differentiated in pregnancy, both caused by the placental enzyme cysteine aminopeptidase, named oxytocinase, which enzymatically degrades oxytocin (97). Because of the close structural homology between AVP and oxytocin, this enzyme also metabolizes AVP. In the first type of pregnancy-associated DI, the activity of oxytocinase is abnormally elevated. This syndrome has been referred to as vasopressin resistant diabetes insipidus of pregnancy (98) and has been reported to be associated with preeclampsia, acute fatty liver, and coagulopathies. Symptoms usually develop at the end of the second or early third trimester and it is more common during multiple pregnancy (99). In the second type of pregnancy-associated DI, the accelerated metabolic clearance of vasopressin produces DI in a patient with borderline pre-existing vasopressin function from a mild nephrogenic diabetes insipidus or partial central diabetes insipidus. Vasopressin is rapidly destroyed and the neurohypophysis is unable to keep up with the increased demand. Symptoms in this second type usually appear early in pregnancy (100).
INVESTIGATIONS FOR DIAGNOSIS
Investigations have the following three aims:
- To confirm DI
- To classify the DI into central or nephrogenic DI or PP (or gestational DI in case of pregnancy)
- To establish the etiology of the specific form of DI
After establishing polyuria and polydipsia, and excluding hyperglycemia, hypokalemia, hypercalcemia, and significant renal insufficiency, attention should be focused on the AVP axis.
For many years, the indirect water deprivation test was the gold standard for differential diagnosis of diabetes insipidus. This test indirectly assesses AVP activity by measurement of the urine concentration capacity during a prolonged period of dehydration, and again after a subsequent injection of an exogenous synthetic AVP variant, desmopressin. Interpretation of the test results goes back to the publication of Miller et al (101). If upon thirsting, urinary osmolality remains <300mOsm/kg and does not increase >50% after desmopressin injection, complete nephrogenic DI is diagnosed. If the urinary osmolality increase after desmopressin injection is >50%, complete central DI is diagnosed. In partial central DI and primary polydipsia, urinary concentration increases to 300–800mosm/kg, with an increase of >9% (in partial central DI) and <9% (in primary polydipsia), respectively, after desmopressin injection. However, these published criteria are based on post hoc data from only 36 patients, who had a wide overlap in their urinary osmolalities. Furthermore, the diagnostic criteria for this test are derived from a single study with post-hoc assessment (101) and have not been prospectively validated. Consequently, the indirect water deprivation test has been shown to have considerable diagnostic limitations with an overall diagnostic accuracy of 70%, and an accuracy of only 41% in patients with primary polydipsia (102).
Several reasons exist for this limited diagnostic outcome of the indirect water deprivation test. First, chronic polyuria itself can affect renal concentration capacity, through renal washout (103) (104, 105) or downregulation of AQP2 expression in the kidneys (106). This may lead to a reduced renal response to osmotic stimulation or exogenous desmopressin in different forms of chronic polyuria. Second, in patients with AVP deficiency, urine concentration can be higher than expected (107, 108), especially in those patients with impaired glomerular function, or can result from a compensatory increase in AVPR2 expression in patients with chronic central DI (109). Finally, patients with acquired nephrogenic DI are often only partially resistant to AVP, resulting in a clinical presentation that is similar to partial central DI.
To overcome these limitations, direct measurement of AVP levels has been proposed. In a study published in 1981, patients with central DI were reported to have AVP levels below a calculated normal area (defining the normal relationship between plasma osmolality and AVP levels), whereas AVP levels were above the normal area in patients with nephrogenic DI and within the normal area in patients with primary polydipsia (110). However, despite these promising initial results, direct measurement of AVP levels failed to enter routine clinical use for various reasons. First, several technical limitations of the AVP assay result in a high preanalytical instability of AVP in samples (111). Second, the accuracy of diagnoses using commercially available AVP assays has been disappointing, with correct diagnoses in only 38% of patients with DI, and particularly poor differentiation between partial central DI and primary polydipsia (102, 112). Third, an accurate definition of the normal physiological area defining the relationship between plasma AVP levels and osmolality is still lacking, especially for commercially available assays (113, 114), which is a crucial prerequisite for the identification of AVP secretion outside the normal range in patients suspected of having DI (102).
Copeptin, the C-terminal segment of the AVP prohormone, is an easy-to- measure AVP surrogate that is very stable ex vivo (111), mirroring AVP concentrations. Two studies have shown that a basal copeptin level >21.4pmol/l without prior thirsting unequivocally identifies nephrogenic DI, rendering a further water deprivation test unnecessary in these patients (102, 115). For the differentiation between patients with primary polydipsia and central DI, results of a study including 144 patients (116) showed that an osmotically stimulated copeptin level >4.9pmol/l after infusion of 3% saline (aiming at a sodium level >150mmol/l) had an overall diagnostic accuracy of 96.5% (93.2% sensitivity and 100% specificity) in distinguishing between patients with primary polydipsia and those with central DI. The classic water deprivation test had an overall diagnostic accuracy of only 76.5%. Importantly, the hypertonic saline infusion test requires close monitoring of sodium levels to ascertain a diagnostically meaningful increase of plasma sodium within the hyperosmotic range while preventing a marked increase. Addition of copeptin measurement did not improve the diagnostic performance of the indirect water deprivation test, most likely due to the lack of osmotic stimulus by thirsting alone. Another study suggests that non-osmotically stimulated copeptin levels upon arginine infusion also provide a high diagnostic accuracy in differentiating central DI from PP (117). These data indicate that plasma copeptin is a promising biomarker to distinguish between different forms of polyuria–polydipsia syndrome. A possible diagnostic algorithm with and without the availability of copeptin measurement is shown in Figure 8.

Figure 8.
Algorithm for the differential diagnosis of diabetes insipidus (Rev Christ-Crain, Nat Rev Primer).
In establishing the underlying mechanisms of central DI once the diagnosis is confirmed, imaging of the hypothalamus, pituitary and surrounding structures with MRI is essential. If no mass lesion is identified, imaging should be repeated after 6-12 months so that slow growing germ cell tumors are not missed. Idiopathic and familial central DI are often associated with loss of the normal hyper-intense signal of the posterior pituitary on T1-weighted images (Figure 9). Signal intensity is correlated strongly with AVP content of the gland (118). In the absence of appropriate history and diagnostic testing, the loss of a posterior pituitary bright spot does not make the diagnosis of central DI. Importantly, presence of an appropriate bright spot does not exclude the diagnosis of central DI.

Figure 9.
Loss of the posterior pituitary 'bright spot' on T1 weighted MRI in hypothalamic diabetes insipidus. The normal posterior pituitary can be demonstrated as a 'bright spot' within the sella turcica on T1-weighted MRI (a). This increased signal intensity can be lost in HDI (b). An ectopic posterior pituitary 'bright-spot' can be seen some cases of childhood onset hypopituitarism, implying failure to complete normal developmental migration. Function can be normal despite the aberrant position.
Evidence of anterior pituitary dysfunction should be looked for in central DI, though it is relatively uncommon in the adult population. Interestingly, evidence of organ-specific autoimmune disease is relatively common in adult patients with isolated central DI, consistent with an autoimmune basis for the condition (119).
TREATMENT OF DIABETES INSIPIDUS
Treatment of Central DI
The primary aim of treatment in patients with diagnosed central DI should be to reduce polyuria and polydipsia to levels that allow maintenance of a normal lifestyle. The treatment of choice for those with significant symptoms is the synthetic, long-acting AVP analogue DDAVP. The long half-life, selectivity for AVPR2 and availability of multiple preparations renders desmopressin an ideal treatment for central DI. Optimal dosage and dosing intervals should be determined for each patient. The available treatment options are the intranasal spray, oral tablets, sublingual tablets or a parenteral injection, in divided doses. Oral preparations provide greater convenience and are usually preferred by patients. However, starting with a nasal spray initially is preferable because of greater consistency of absorption and physiological effect, after which the patient can be switched to an oral preparation. After trying both preparations, the patient can then choose which they prefer for long-term treatment. A satisfactory schedule can generally be determined using modest doses of desmopressin. The maximum dose of desmopressin required rarely exceeds 0.2 mg orally, 120 µg sublingually or 10 µg (one nasal spray) given 2–3 times daily. These doses usually produce plasma desmopressin levels higher than those required to cause maximum antidiuresis but reduce the need for more frequent treatment (120).
Hyponatremia is the major complication of desmopressin therapy — a 27% incidence of mild hyponatremia (serum sodium 131–134 mmol/l) and a 15% incidence of more severe hyponatremia (serum sodium ≤130 mmol/lm) have been reported after long-term follow-up of patients with chronic central DI (121). Hyponatremia usually occurs if the patient is antidiuretic while continuing normal fluid intake. Severe hyponatremia in patients with central DI who are treated with desmopressin can be avoided first by monitoring serum electrolyte levels frequently during initiation of therapy and second, patients should be instructed to delay a scheduled dose of desmopressin once or twice weekly until polyuria recurs, thereby allowing excess retained fluid to be excreted. A recent publication showed that patients using this approach had a significantly lower the risk of hyponataemia compared to those who did not follow this approach (OR 0.4, 95%CI 0.3-0.7, p<0.01) (122).
Mostly, Desmopressin is a lifelong treatment. An exception is treatment of postsurgical central DI. If the patient is awake and responds to thirst, thirst is a sufficient guide for water replacement. If the duration of diabetes insipidus is transient, it is therefore acceptable to treat simply with fluid replacement, parenterally or orally. However, most patients who develop diabetes insipidus require desmopressin 0.5 to 2 μg subcutaneously, intramuscularly, or intravenously. Urine output will be reduced in 1 to 2 hours and the duration of effect is 6 to 24 hours. Care should be taken that hypotonic intravenous fluids are not given excessively after administering desmopressin, as the combination can lead to profound hyponatremia. Because there is always the possibility of developing the triphasic response due to pituitary stalk damage, it is recommended that polyuria should be recurrent before a decision to administer subsequent doses of desmopressin is made (123).
Patients with hypernatremia due to osmoreceptor dysfunction (adipsic central DI) should be treated acutely with the same treatment as any hyperosmolar patient. The long-term management of osmoreceptor dysfunction syndromes requires a thorough search for potentially treatable causes, combined with measures to prevent dehydration. Because hypodipsia cannot be cured, the focus of management is based on education of the patient and family about the importance of regulating their fluid intake according to their hydration status (124). This can be accomplished most efficaciously by establishing a daily schedule of fluid intake regardless of the patient's thirst, which can be adjusted in response to changes in body weight (125). As these patients will not drink spontaneously, daily fluid intake must be fixed and prescribed. If the patient has polyuria, desmopressin should also be prescribed, as in any patient with central DI. The success of the fluid prescription should be monitored periodically by measuring serum Na+ concentration. In addition, periodic recalculation of the target weight (at which hydration status and serum Na+ concentration are normal) might be required.
Treatment of NDI
Treatment of acquired nephrogenic DI should target the underlying cause (e.g., correction of hypercalcemia or hypokalemia), if possible. If not, different approaches are possible.
- Low salt diet to minimize the osmotic load
- For patients on long-term lithium therapy, amiloride prevents uptake of lithium in the collecting duct epithelial cells and thus the inhibitory effects of intracellular lithium on water transport (126).
- Hydrochlorothiazide has been shown to reduce urine output in both central and nephrogenic DI (127, 128). Thiazides decrease salt reabsorption by inhibiting the thiazide-sensitive co-transporter SLC12A3 in the distal tubule. The loss of sodium reduces plasma volume, so that less water is presented to the collecting duct and lost in the urine.
- In an animal model of nephrogenic DI, use of the NSAID indomethacin reduced water diuresis independently of AVP (129). A similar effect of prostaglandin synthesis inhibitors was later reported in patients with nephrogenic DI (130). Since these early studies, prostaglandin synthesis inhibitors have become an essential component of the treatment of nephrogenic DI
- High dose DDAVP can produce a response in partial NDI
Treatment of PP
Treatment of primary polydipsia entails reduction of excessive fluid intakes, best done in a graded fashion to allow patients to slowly achieve a level of intake that reduced urine volume below polyuric levels (50 mL/kg BW). Measures to reduce mouth dryness (e.g., ice chips, hard candy to stimulate salivary flow) are useful adjuncts to reduce thirst. Pharmacologic therapies have been tried but without consistent evidence of success. A recent study suggests that GLP-1 analogues reduce fluid intake, urine output, and thirst perception(131).
Treatment of Gestational DI
Desmopressin is the only therapy recommended for treatment of diabetes insipidus during pregnancy. Desmopressin is reported to be safe for both the mother and the child (132, 133). During delivery, patients can maintain adequate oral intake and are therefore safe to continue administration of desmopressin. Physicians should be cautious about over administration of fluid parenterally during delivery because these patients will not be able to excrete the fluid and can develop water intoxication and hyponatremia. After delivery, plasma oxytocinase decreases and patients can recover completely or be asymptomatic with regard to fluid intake and urine excretion.
Syndrome Of Inappropriate Antidiuresis
Hyponatremia
Hyponatremia (serum sodium <135 mmol/l) is a clinical feature in some 15–20% of non-selected emergency admissions to hospital. It is associated with increased morbidity and mortality across a range of conditions. Moreover, data support the association of hyponatremia correction with improvements in clinical outcome. The relationship of serum sodium and outcome is not straightforward. Co-morbidity and disease severity, rather than hyponatremia per se, may make a significant contribution to adverse outcome in these patients. Further data are needed to clarify whether the relationship between sodium levels and outcome is causal or the association of two variables linked with disease severity (134-137).
Hyponatremia is not invariably associated with a low serum osmolality; high concentrations of other circulating osmolytes (e.g., glucose) can lead to a fall in plasma sodium that is appropriate to maintain normal osmolar status. A reduced plasma aqueous phase secondary to dyslipidemia can result in artefactual hyponatremia with normal plasma osmolality, even when using an ion-specific electrode. This is consequent to the use of a standard dilution step in most clinical biochemistry laboratories. This type of artefactual hyponatremia is not seen when a direct potentiometric method is used, such as when using a blood-gas analyzer. In many clinical situations, hyponatremia is multifactorial (Table 3).
Table 3.
Causes of Hyponatremia
| Pseudohyponatremia | Reduced renal free water clearance | ||
|---|---|---|---|
| Hyperglycemia, Hyperlipidemia, Non-physiological osmolyte, Elevated paraprotein | Hypovolemia Cardiac failure Nephrotic syndrome Hypothyroidism Hypoadrenalism SIAD Nephrogenic syndrome of antidiuresis | Drugs Renal failure Portal hypertension & ascites Hypoalbuminemia Sepsis Fluid sequestration | |
| Sodium depletion | |||
| Renal loss | Diuretics Salt wasting nephropathy Hypoadrenalism Central salt wasting | ||
| Extra-renal loss | Gut loss | ||
| Excess water intake | |||
| Dipsogenic DI Sodium-free, hypo-osmolar irrigant solutions Dilute infant feeding formula Exercise-associated hyponatremia | |||
AVP plays a key role in many pathophysiological situations of which hyponatremia is a feature. Importantly however, even when AVP plays a role in the development of hyponatremia, AVP production may not be inappropriate. Hyponatremia may reflect an appropriate physiological response to volume depletion. To maintain circulating volume in hypovolemia, baroregulated AVP release may proceed despite plasma osmolalities below the osmotic threshold for AVP release. This can result in hyponatremia, which can become persistent. Though clinical assessment can identify the extracellular volume status of some patients, it is unreliable and has poor sensitivity and specificity (138).
Pathophysiology and diagnosis of SIAD
An individual with hypoosmolar plasma, a normal circulating volume, and a plasma AVP concentration high for the prevailing osmolality, has the syndrome of inappropriate antidiuresis (SIAD). The clinical criteria to diagnose SIAD go back to Schwartz and Bartter (139) in 1967 and are summarized in Table 4.
Excretion of urine that is not maximally dilute in the context of dilute plasma (i.e., urine concentration greater than 100mOsm/Kg) indicates the action of AVP on renal water resorption. Importantly however, it does not define whether this action is appropriate (for instance in the context of hypovolemia and baro-stimulated AVP release) or inappropriate. Measurement of urinary sodium concentration is key in the differential diagnosis of SIAD from hypovolemia. Renal sodium excretion should be above 30mol/L to make a diagnosis of SIAD. Below this value, volume depletion needs to be considered more likely and below 20 mmol/L, hypovolemia is the likely cause of hyponatremia. SIAD is often associated with urine sodium concentrations of 60 mmol/L or more. SIAD is a volume-expanded state and there is evidence of mild sodium loss as other homeostatic regulators of volume homeostasis attempt to minimize volume expansion. The utility of urinary sodium concentration in defining the etiology of hyponatremia is limited by concurrent use of drugs that produce a natriuresis: diuretics, angiotensin converting enzyme inhibitors, and angiotensin II antagonists. In this situation, a serum urate <4 mg/dl, or a fractional urate excretion >12% can help differentiate SIAD from mild hypovolemia (140, 141).
Cortisol deficiency is a key differential diagnosis as secondary adrenal deficiency can present a biochemical picture identical to SIAD. Therefore, formal biochemical exclusion of adrenal insufficiency is mandatory before diagnosing SIAD.
Four patterns of abnormal AVP secretion have been identified (table 5). The same pattern has also been shown for copeptin (142). Absolute plasma AVP or copeptin concentrations may not be strikingly high and in fact AVP and copeptin measurement is not helpful in establishing the diagnosis (143). The key feature is that they are inappropriate for the prevailing plasma osmolality (10).
Table 4.
Criteria for Diagnosis of SIAD
| Low serum osmolality <275 mOsmol/kg H20 Elevated urine osmolality >100 mOsmol/kg H20 (inappropriately concentrated) Clinical euvolemia (absence of signs of hypovolemia or hypervolemia) Elevated urinary sodium excretion >30mEq/L with normal salt and water intake Absence of other potential causes of euvolemic hypo-osmolality (glucocorticoid insufficiency, severe hypothyroidism) Normal renal function and absence of diuretic use |
Table 5.
Classification of SIAD
| Characteristics | Prevalence | |
|---|---|---|
| SIAD Type A | Wide fluctuations in plasma AVP concentration independent of plasma osmolality | 35% |
| SIAD Type B | Osmotic threshold for AVP release subnormal Osmoregulation around subnormal osmolar set point | 30% |
| SIAD Type C | Failure to suppress AVP release at low plasma osmolality Normal response to osmotic stimulation | |
| SIAD Type D | Normal osmoregulated AVP release Unable to excrete water load. | <10% |
Etiology of SIAD
Many conditions have been reported to cause SIAD, though the mechanism(s) of inappropriate AVP release are not clear in many cases (Table 6). SIAD is a non-metastatic manifestation of small cell lung cancer and other malignancies. Some tumors express AVP ectopically. However, excessive posterior pituitary AVP secretion also occurs in association with malignancy. The normal osmoregulated AVP release found in the Type D syndrome suggests an increase in renal sensitivity to AVP, or the action of an additional antidiuretic factor.
Table 6.
Causes of SIAD
| Neoplastic disease | Chest disorders |
| Carcinoma (bronchus, duodenum, pancreas, bladder, ureter, prostate) Thymoma Mesothelioma Lymphoma, leukemia Ewing's sarcoma Carcinoid Bronchial adenoma | Pneumonia Tuberculosis Empyema Cystic fibrosis Pneumothorax Aspergillosis |
| Neurological disorders | Drugs |
| Head injury, neurosurgery Brain abscess or tumor Meningitis, encephalitis Guillain-Barré syndrome Cerebral hemorrhage Cavernous sinus thrombosis Hydrocephalus Cerebellar and cerebral atrophy Shy-Drager syndrome Peripheral neuropathy Seizures Subdural hematoma Alcohol withdrawal | Sulphonylureas Alkylating agents & Vinca alkaloids Thiazide diuretics Dopamine antagonists Tricyclic antidepressants MAOIs SSRIs 3,4-MDMA ("Ecstasy") Anti-convulsants |
| Miscellaneous | |
| Idiopathic Psychosis Porphyria Abdominal surgery Diverse non-osmotic stimuli (e.g., nausea, stress, pain) | |
SIAD is a common mechanism of drug-induced hyponatremia, and can reflect direct stimulation of AVP release from the hypothalamus; indirect action on the hypothalamus; or aberrant resetting of the hypothalamic osmostat (table 7). The prevalence of hyponatremia in patients taking high dose dopamine antagonists is greater than 25%, and is not restricted to one class of these drugs. Hyponatremia secondary to antidepressants is well recognized, occurring with most SSRIs, and the related drug Venlafaxine. It can arise in the first few weeks of treatment. Anticonvulsants are another common cause of SIAD and hyponatremia. The frequency in patients treated with carbamazepine (CBZ) ranges from 4.8 to 40%. Increased sensitivity of central osmoreceptors and increased renal responses to AVP have both been described with CBZ.
Table 7.
Mechanisms of Drug Induced Hyponatremia
| Reduction in free water clearance | Sodium depletion | ||
|---|---|---|---|
| SIAD | Dopamine antagonists Tricyclic antidepressants MAOIs SSRIs Venlafaxine Carbamazepine Oxcarbamazepine Sodium valproate 3,4-MDMA ('ecstasy') Clofibrate Cyclophosphamide Sulphonylureas | Diuretics | Spironolactone Thiazides Loop diuretics |
| AVP-like activity | DDAVP Oxytocin | ACE inhibitors/Angiotensin II receptor antagonists | |
| Potentiation of AVP action | NSAIDS Carbamazepine Sulphonylureas Cyclophosphamide | Direct renal toxicity | Cyclophosphamide Ifosfamide Cisplatin Carboplatin Vincristine Vinblastine |
Exercise Associated Hyponatremia
Extreme endurance exercise is a profound physiological stressor. While the magnitude of the physiological stress is likely to reflect a number of factors, duration of the event and the effort entailed are likely to be major contributors. Non-osmoregulated AVP release is a feature of extreme endurance exercise: a reflection of the stressed state. When combined with reduced renal blood flow, another feature of extreme endurance exercise, this can lead to a marked antidiuretic state (figure 10). If endurance athletes maintain a fluid intake in excess of water loss, hyponatremia will ensue. This can be further complicated if there is aggressive fluid resuscitation in the event of collapse. There is a positive correlation between the odds ratio for developing hyponatremia during extreme endurance exercise and the length of time taken to complete the event. Athletes developing hyponatremia also demonstrate weight gain over the course of the event, clearly implicating water intake in excess of water and electrolyte loss as the cause. Occasional runners should be advised to follow their thirst as they run and avoid rigid, time-based fluid intake. Health professionals attending endurance events need to be aware of the problem of exercise-associated hyponatremia. In addition, they should avoid attempting resuscitation with large volumes of hypotonic fluid in the absence of appropriate indications and without biochemical monitoring (144).

Figure 10.
Exercise associated hyponatremia in triathletes. 1089 triathletes were studied. The mean plasma sodium level at the finish was 140.5±4.2 mmol/L (range 111-152). Among athletes completing the study events, 10.6% had documented hyponatremia: 8.7% mild; 1.6% severe; and 0.3% critical (3 athletes in total with plasma sodium levels 120, 119, and 111 mmol/L respectively). Multivariate analysis showed a significant association between development of hyponatremia and the following factors: female gender; longer times to complete a race. Critical hyponatremia occurred in participants who finished in the 12th and 14th hours of the race (145).
Nephrogenic Syndrome of Inappropriate Antidiuresis
The G-protein-coupled V2-R mediates the action of AVP on renal water excretion. Rare kindreds have been found that harbor constitutively activating mutations in the V2-R that led to AVP-independent, V2-R mediated, antidiuresis associated with persistent hyponatremia (Figure 11). This nephrogenic syndrome of inappropriate antidiuresis (NSIAD) was initially described in male infants with persistent hyponatremia in keeping with the haploinsufficiency associated with the V2-Rgene being on the X chromosome. However, subsequent studies have found the condition is not limited to males, expression of the condition being clearly identified in heterozygous females. The true prevalence of NSIAD is not known. However, as some 10% of patients with SIAD have been described as having undetectable AVP, it seems likely that at least some of these cases may be due to activating mutations of the V2-R (146, 147).

Figure 11.
In vitro bioactivity of different V2-R constructs relative to wild type (WT) in a cAMP-dependent luciferase reporter system in the absence of AVP. The R137H construct is the V2-R found in X-linked NDI. R137C and R137L are receptor variants found in NSIAD. Constructs differ only by the amino acid at position 137. R137C and R137L found in NSIAD demonstrate constitutive, AVP-independent activity. R: arginine. H: histidine. C: cysteine. L: leucine.
Cerebral Salt Wasting
This acquired, primary natriuretic state remains a subject of controversy. It has been characterized as a combination of hyponatremia with hypovolemia associated with neurological or (more often) neurosurgical pathologies. The underlying mechanism(s) remain unclear, but increased release of natriuretic peptides and/or reduced sympathetic drive have been proposed. The diagnosis of cerebral salt wasting (CSW) hinges on the natural history: the development of hyponatremia being preceded by natriuresis and diuresis with ensuing clinical and biochemical features of hypovolemia. In contrast to SIAD, urea and creatinine are elevated and there may be postural hypotension. The simple observation of weight loss over the period in question can be helpful.
SIAD can occur in the same group of patients in whom CSW has been reported. Both CSW and SIAD are associated with urine sodium concentrations greater than 40mmols/L. However, the natriuresis of CSW is much more profound than that of SIAD and precedes the development of hyponatremia. CSW is a particular concern for the neurosurgical patient in whom autoregulation of cerebral blood flow is disturbed and in whom small reductions in circulating volume can reduce cerebral perfusion. The management of CSW is volume replacement with 0.9% saline; while the cause-directed approach to SIAD would often involve restriction of fluid. The clinical and practice context, together with the opposed cause-directed management approaches to CSW and SIAD can lead to significant tension. A cause-independent approach to the management of the neurosurgical patient with hyponatremia is often the practical and pragmatic approach to take: balancing management of hyponatremia with the need to avoid threatening cerebral perfusion and avoidable vasospasm (148). Importantly, in a prospective, single center study of 100 patients developing hyponatremia after subarachnoid hemorrhage not a single case of CSW was identified: hyponatremia was attributable to SIAD, glucocorticoid deficiency, or inappropriate fluid administration (Figure 12). Therefore, CSW is probably a very rare condition.

Figure 12.
Hyponatremia after subarachnoid hemorrhage. Prospective study of 100 patients. Demographics, outcomes and etiology of hyponatremia noted. SAH: subarachnoid hemorrhage (149).
Management of hyponatremia secondary to SIAD
The morbidity and mortality of hyponatremia secondary to SIAD are the result of disturbance in central nervous system (CNS) function: the combined impact of cerebral oedema and direct neuronal dysfunction (Table 8). While it is intuitive that the greater the derangement in serum sodium should be associated with the most profound neurological disturbance, the relationship between serum sodium and neurological function is not simple. Patients with marked biochemical disturbance may have mild symptoms if hyponatremia develops over a prolonged period. This reflects CNS adaptation: brain edema being limited by efflux of organic solutes. This adaptation can complicate the management of hyponatremia. Rapid correction of serum sodium following the gradual development of hyponatremia (which has resulted in a degree of CNS adaptation) can lead to significant changes in brain volume as the osmolar gradient across the blood-brain barrier alters. This can trigger CNS demyelination (osmotic demyelination syndrome, ODS). This is a rare but serious complication of hyponatremia and its treatment. ODS develops within 1-4 days of rapid correction of serum sodium, irrespective of the method employed to achieve it. It can even occur when sodium levels are corrected slowly. Risk factors for ODS are severe liver disease, potassium depletion, malnutrition and profound hyponatremia, especially if <105mmol/L. Neurological manifestations include quadriplegia, opthalmoplegia, pseudo-bulbar palsy and coma.
Table 8.
Clinical Features of Hyponatremia Secondary to SIAD
|
Management of Hyponatremia Associated with Severe or Moderately Severe Symptoms
Hyponatremia associated with severe or moderately severe symptoms requires urgent management as an emergency. Treatment is cause-independent. Treatment should be given priority over establishing the etiology of hyponatremia. The aim of treatment is to reduce immediate risk through increasing serum sodium to a level that decreases morbidity and mortality. Importantly, the rate of increase in serum sodium must be at a rate that does not result in harm through precipitating ODS (Figure 13). The immediate target should not be to normalize serum sodium. Once the patient is stable, investigations to establish the cause of hyponatremia can begin, the outcome of which subsequently guiding cause-directed therapy (140, 150, 151).

Figure 13.
Immediate, cause-independent management of hyponatremia associated with severe or moderately severe symptoms.
Previous strategies for the emergency management of hyponatremia have stratified patients based on presumed duration of the electrolyte disturbance. Furthermore, some have advocated hypertonic fluid administration volume and rates based on calculated deficits in serum sodium using a standard formula (152). A more pragmatic and reliable approach to patient stratification is to base the decision on use of hypertonic fluid on patient symptoms and signs; while the determining hypertonic fluid prescription by calculated serum sodium deficit is associated with a significant risk of over-correction (153). Over-correction of hyponatremia (increase in serum sodium above the recommended rate) should prompt review of the fluid regimen and consideration of active management with hypotonic fluid, with or without concurrent use of DDAVP (140, 154).
Two recent studies have compared bolus versus infusion hypertonic saline, both in favor of a bolus regimen. An Irish observational study compared outcomes for 22 hyponatremic patients treated with bolus 100ml 3% NaCl, compared to a historical cohort of 28 patients managed with 20ml/hour 3% NaCl infusion (155). Bolus hypertonic saline was associated with a faster elevation of serum sodium at 6 hours and significantly greater improvement in Glasgow Coma Scale at 6 hours. There were no cases of ODS in either group. A larger randomized trial from Korea included 178 patients with hyponatremia who were randomized to either bolus or infusion NaCl 3% (156). The results showed that both therapies were effective and safe, with no significant difference in overcorrection risk, but there was less need for therapeutic relowering in the bolus group compared to the slow continuous infusion. Bolus therapy is therefore currently recommended in guidelines.
Management of SIAD Associated with Mild to Moderate Symptoms
Fluid restriction: Fluid restriction of 0.5–1L/day is the first-line treatment recommended by EU and US guidelines (157). It is a reasonable initial intervention when the clinical condition is not critical. All fluids need to be included in the restriction. As SIAD is associated with a degree of natriuresis, sodium intake should be maintained. Fluid restriction may need to be maintained for several days before sodium levels normalize and it is important that a negative fluid balance is confirmed during this period. As cause-directed therapy progresses (e.g., treatment of underlying infection or removal of drug causing SIAD), fluid restriction may be relaxed. A recent randomized controlled trial evaluated the efficacy of fluid restriction in comparison to no treatment showing a modest early rise in serum sodium of 3mmol/L in fluid restricted patients compared to 1mmol/L with no treatment. Similar results were seen in other recent trials (158).
Of note, fluid restriction is not always effective, and a clinically useful response is only seen in around half of hyponatremic patients. In a large hyponatremia registry fluid restriction was unsuccessful in 55% of cases (159). Factors predicting a poor response to fluid restriction include high urine osmolality (>500 mosm/L), and high urine sodium concentration (or high urine/serum electrolyte ratio i.e. (UNa + UK)/pNa > 1, i.e., ‘Furst equation’) (150, 160, 161).
Drug treatment in SIAD: If patients do not respond to fluid restriction, a second-line treatment has to be initiated. There are several different possibilities, which are discussed in the following.
Urea is a product of hepatic nitrogen metabolism which is renally excreted and has an osmotic effect to promote free water excretion. It is recommended as a second line treatment in the European and US guidelines(140, 150). The evidence base for the use of urea is limited due to the retrospective observational nature of most current studies. In fact, there are several observational studies of urea, but no randomized trials to date. A retrospective observational trial reported a mean increase in sodium levels of 7 mmol/L over 4 days, with no instances of rapid correction (162). Another retrospective study reported that the majority of patients normalized sodium levels within 48 hours despite fluid intake >2 L/d (163). Finally, in patients with SIAD in the context of subarachnoid hemorrhage urea treatment led to sodium normalization within a mean time of 3 days (164).
Although overcorrection has been described, there have been no published cases of ODS due to urea therapy. The main limitations are the difficult access and poor palatability due to bitter taste, which can be ameliorated by combining it with a small volume of orange juice. Urea therapy can cause an increase in serum urea concentration and blood urea nitrogen, but this does not necessarily represent deterioration in renal function or hypovolemia (162).
Tolvaptan is an oral vasopressin V2-receptor antagonist that blocks AVP action in the kidney, inducing water diuresis and thereby raising sodium levels. An intravenous alternative conivaptan is also approved, but its use is limited to a maximum duration of 4 days because of drug interaction effects with other agents metabolized by the CYP3A4 hepatic isoenzyme. The efficacy of tolvaptan versus placebo was investigated in the randomized placebo-controlled SALT-1 and SALT-2 trials in patients with chronic eu- or hypervolemic hyponatremia (165). The results showed that tolvaptan increased sodium levels within 4 days by 4 mmol/L compared to only 1 mmol/L in the placebo group and an even higher increase within 30 days. The American expert panel recommends its use as a second-line treatment, whereas the European clinical practice guidelines recommend against the use of tolvaptan in moderate to profound hyponatremia, mainly due to lack of proven outcome benefit aside from increase in sodium levels and the concern of overcorrection. Importantly, discontinuing fluid restriction when vaptans are started, initiation of treatment in the hospital and regular sodium monitoring limit the risk for overcorrection. A lower initial dose of tolvaptan, 7.5 mg, which has been associated with lower rates of overcorrection, should also be considered (166). Common side effects of tolvaptan include dry mouth, thirst, and urinary frequency.
Sodium-glucose co-transporter 2 inhibitors are approved for treatment of diabetes and heart failure, but may also be beneficial in SIAD. The sodium-glucose co-transporter 2 in the proximal renal tubule is responsible for 90% of renal glucose resorption (167). Inhibition of SGLT2 leads to glycosuria, accompanied by an osmotic diuresis. A randomized trial of inpatients with SIAD compared 4 days of empagliflozin 25mg or placebo, in addition to fluid restriction (168). Plasma sodium increased by 10mmol/L over 4 days in the empagliflozin group, compared to 7mmol/L with placebo (p = 0.04). There was no hypotension or hypoglycemia observed. The relatively high sodium increment in the placebo group suggests that at least in some patients, reversible stimuli to AVP secretion may have been present. A second, randomized controlled cross-over study in 14 patients’ outpatients with chronic SIAD compared a 4-week treatment with empagliflozin 25mg/day to placebo, without concomitant fluid restriction. Empagliflozin treatment resulted in a serum sodium increase of 4.1mmol/L (p=0.004) under empagliflozin as compared to placebo. Treatment with empagliflozin was generally well tolerated with no events of sodium overcorrection, hypoglycemia, hypotension, urinary tract or genital infection occurring during the observation period (169). Future larger studies are needed to validate these findings.
Loop diuretics block the Na+-K+-2CL- cotransporter in the thick ascending limb of the loop of Henle and decrease delivery of sodium and chloride to the kidney medulla, decreasing the medullary gradient necessary for water reabsorption in the collecting duct. Salt tablets are added to replace loop diuretic-induced sodium losses. They are listed in the European practice guidelines as a second line therapy for SIAD in combination with salt tablets. However, a recent randomized controlled trial comparing the effect of FR alone versus FR plus loop diuretics versus FR plus loop diuretics plus salt tablets did not show a significant additive effect of loop diuretics or salt tablets (158). Acute kidney injury and hypokalemia were more common in patients receiving furosemide, therefore caution should be taken when adopting this strategy.
Demeclocycline has been used for many years in management of hyponatremia of SIAD. It produces a form of NDI and so increases renal water loss even in the presence of AVP. There is a lag time of some 3–4 days in onset of action and the effect is unpredictable occurring in 33-60% of patients. Photosensitive skin reactions are a significant additional adverse effect. There are very limited data to support long-term efficacy and a systematic review did not find good quality evidence for its use (170).
Lithium also induces nephrogenic diabetes insipidus and has therefore been tried as treatment for SIAD, however, due to adverse effects and questionable efficacy it is not recommended in European guidelines.
Adipsic And Hypodipsic Syndromes
Adipsic and hypodipsic disorders are characterized by inadequate spontaneous fluid intake due to defects in osmoregulated thirst. Patients deny thirst and do not drink, despite dehydration and hypovolemia. If the defect is mild, the resultant hypernatremia is often well tolerated. Severe disorders can lead to somnolence, seizures, and coma. Because of the close anatomical relationship of the osmoregulatory centers for thirst and AVP release, adipsic syndromes are often associated with defects in osmoregulated AVP release and HDI.
Classification and etiology
Four patterns of adipsic/hypodipsic syndrome are recognized (Table 9, Figure 14). Causes are outlined in Table 10. Patients with the Type A syndrome osmoregulate around a supra-normal osmolar set point and are protected from extreme hypernatremia, as are those with the Type B syndrome. In Type C adipsia, osmoregulated thirst and AVP release are absent. Patients present with adipsic diabetes insipidus. Precipitants include rupture and repair of anterior communicating artery (ACA) aneurysm, as the osmoreceptors mediating both thirst and AVP release receive a blood supply from perforating branches of the anterior cerebral artery and ACA. Some patients with the Type C syndrome have constitutive low level AVP release, and are at risk of dilutional hyponatremia.
Table 9.
Classification of Adipsic and Hypodipsic Syndromes
| Adipsia/hypodipsia Syndrome | Osmoregulated Thirst | Osmoregulated AVP release |
|---|---|---|
| Type A (essential hypernatremia) | Osmotic threshold increased Normal sensitivity | Osmotic threshold increased Normal sensitivity Normal non-osmotic stimulation |
| Type B | Normal osmotic threshold Reduced sensitivity | Normal osmotic threshold Reduced sensitivity Normal non-osmotic stimulation |
| Type C | No response to osmotic stimulation | Persistent low level AVP release No response to osmotic stimulation Normal non-osmotic stimulation |
| Type D | No response to osmotic stimulation | Normal |
Table 10.
Causes of Adipsic and Hypodipsic Syndromes
| Neoplastic (50%) | |
| Primary | Craniopharyngioma Germ cell tumor Meningioma |
| Secondary | Pituitary tumor Bronchial carcinoma Breast carcinoma |
| Granulomatous (20%) Histiocytosis Sarcoidosis | |
| Miscellaneous (15%) Hydrocephalus Ventricular cyst Trauma Toluene poisoning | |
| Vascular (15%) Internal carotid artery ligation Anterior communicating artery aneurysm Intra-hypothalamic hemorrhage | |

Figure 14.
Patterns of plasma AVP and thirst responses to hypertonic stress in patients with adipsic syndromes. Normal range responses to osmolar stimulation are shown by the shaded areas. The 4 types of adipsic syndrome are demonstrated. Patients with the Type A syndrome osmoregulate around a higher osmolar set point, while those with the Type B syndrome mount AVP and thirst responses but with reduced sensitivity. Patients with the Type C syndrome have much reduced or absent AVP and thirst responses to osmolar stimulation. Those with the Type D syndrome demonstrate normal AVP responses to osmolar stimulation but much reduced thirst responses.
Management
As those with Type A and Type B adipsia are protected from extreme hypernatremia, treatment is to recommend an obligate fluid intake of about 2L/24 hours with appropriate adjustment for climate and season. If fluid balance cannot be maintained during intercurrent illness, hospital in-patient management may be required. The adipsic central diabetes insipidus of the Type C syndrome can be difficult to manage. The structural and vascular problems producing the syndrome often led to associated defects in short term memory and task organization, complicating long-term management. A pragmatic approach is to effectively dictate an acceptable urine output (1-2L/24 hours) with regular DDAVP (producing a fixed obligate antidiuresis). Together with an estimation of standard insensible fluid loss (approximately 0.4L), this can be set to create a fixed net fluid loss of some 2Ls. In turn, this can be balanced by a daily fluid intake that varies in response to depending on day-to-day fluctuations from a target weight at which the patient is euvolemic and normonatremic.
Daily fluid intake = 2L + (daily weight in Kg- target weight in Kg).
Plasma sodium should be checked weekly, to avoid the creeping development of hyper- and hyponatremia as dry weight changes. This approach can result in stable fluid balance and successful independent living (171).
REFERENCES
- 1.
- Waller SJ, Ratty A, Burbach JP, Murphy D. Transgenic and transcriptional studies on neurosecretory cell gene expression. Cell Mol Neurobiol. 1998;18(2):149–71. [PubMed: 9535288]
- 2.
- Iwasaki Y, Oiso Y, Saito H, Majzoub JA. Positive and negative regulation of the rat vasopressin gene promoter. Endocrinology. 1997;138(12):5266–74. [PubMed: 9389510]
- 3.
- Russell JA, Leng G. Sex, parturition and motherhood without oxytocin? J Endocrinol. 1998;157(3):343–59. [PubMed: 9691969]
- 4.
- Ponzio TA, Fields RL, Rashid OM, Salinas YD, Lubelski D, Gainer H. Cell-type specific expression of the vasopressin gene analyzed by AAV mediated gene delivery of promoter deletion constructs into the rat SON in vivo. PLoS One. 2012;7(11):e48860. [PMC free article: PMC3498266] [PubMed: 23155418]
- 5.
- Mohr E, Kächele I, Mullin C, Richter D. Rat vasopressin mRNA: a model system to characterize cis-acting elements and trans-acting factors involved in dendritic mRNA sorting. Prog Brain Res. 2002;139:211–24. [PubMed: 12436937]
- 6.
- Nagy G, Mulchahey JJ, Smyth DG, Neill JD. The glycopeptide moiety of vasopressin-neurophysin precursor is neurohypophysial prolactin releasing factor. Biochem Biophys Res Commun. 1988;151(1):524–9. [PubMed: 3126738]
- 7.
- Hyde JF, Ben-Jonathan N. The posterior pituitary contains a potent prolactin-releasing factor: in vivo studies. Endocrinology. 1989;125(2):736–41. [PubMed: 2526728]
- 8.
- Barat C, Simpson L, Breslow E. Properties of human vasopressin precursor constructs: inefficient monomer folding in the absence of copeptin as a potential contributor to diabetes insipidus. Biochemistry. 2004;43(25):8191–203. [PubMed: 15209516]
- 9.
- Schrag JD, Procopio DO, Cygler M, Thomas DY, Bergeron JJ. Lectin control of protein folding and sorting in the secretory pathway. Trends Biochem Sci. 2003;28(1):49–57. [PubMed: 12517452]
- 10.
- Bichet DG, Arthus MF, Barjon JN, Lonergan M, Kortas C. Human platelet fraction arginine-vasopressin. Potential physiological role. J Clin Invest. 1987;79(3):881–7. [PMC free article: PMC424228] [PubMed: 2950136]
- 11.
- Laycock JF, Hanoune J. From vasopressin receptor to water channel: intracellular traffic, constraint and by-pass. J Endocrinol. 1998;159(3):361–72. [PubMed: 9834453]
- 12.
- King LS, Yasui M. Aquaporins and disease: lessons from mice to humans. Trends Endocrinol Metab. 2002;13(8):355–60. [PubMed: 12217493]
- 13.
- Nielsen S, Kwon TH, Frøkiaer J, Agre P. Regulation and dysregulation of aquaporins in water balance disorders. J Intern Med. 2007;261(1):53–64. [PubMed: 17222168]
- 14.
- Verkman AS. Aquaporins at a glance. J Cell Sci. 2011;124(Pt 13):2107–12. [PubMed: 21670197]
- 15.
- King LS, Choi M, Fernandez PC, Cartron JP, Agre P. Defective urinary concentrating ability due to a complete deficiency of aquaporin-1. N Engl J Med. 2001;345(3):175–9. [PubMed: 11463012]
- 16.
- Knepper MA, Kwon TH, Nielsen S. Molecular physiology of water balance. N Engl J Med. 2015;372(14):1349–58. [PMC free article: PMC6444926] [PubMed: 25830425]
- 17.
- McKenna K, Thompson C. Osmoregulation in clinical disorders of thirst appreciation. Clin Endocrinol (Oxf). 1998;49(2):139–52. [PubMed: 9828896]
- 18.
- Prager-Khoutorsky M, Bourque CW. Osmosensation in vasopressin neurons: changing actin density to optimize function. Trends Neurosci. 2010;33(2):76–83. [PubMed: 19963290]
- 19.
- Roberts EM, Newson MJ, Pope GR, Landgraf R, Lolait SJ, O'Carroll AM. Abnormal fluid homeostasis in apelin receptor knockout mice. J Endocrinol. 2009;202(3):453–62. [PMC free article: PMC2729781] [PubMed: 19578099]
- 20.
- Azizi M, Iturrioz X, Blanchard A, Peyrard S, De Mota N, Chartrel N, et al. Reciprocal regulation of plasma apelin and vasopressin by osmotic stimuli. J Am Soc Nephrol. 2008;19(5):1015–24. [PMC free article: PMC2386722] [PubMed: 18272843]
- 21.
- Llorens-Cortes C, Moos F. Apelin and vasopressin: two work better than one. J Neuroendocrinol. 2012;24(7):1085–6. [PubMed: 22712789]
- 22.
- Blanchard A, Steichen O, De Mota N, Curis E, Gauci C, Frank M, et al. An abnormal apelin/vasopressin balance may contribute to water retention in patients with the syndrome of inappropriate antidiuretic hormone (SIADH) and heart failure. J Clin Endocrinol Metab. 2013;98(5):2084–9. [PubMed: 23515451]
- 23.
- Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9(7):519–31. [PubMed: 18509340]
- 24.
- Lechner SG, Markworth S, Poole K, Smith ES, Lapatsina L, Frahm S, et al. The molecular and cellular identity of peripheral osmoreceptors. Neuron. 2011;69(2):332–44. [PubMed: 21262470]
- 25.
- Trudel E, Bourque CW. Circadian modulation of osmoregulated firing in rat supraoptic nucleus neurones. J Neuroendocrinol. 2012;24(4):577–86. [PubMed: 22330181]
- 26.
- Cunningham JT, Bruno SB, Grindstaff RR, Grindstaff RJ, Higgs KH, Mazzella D, et al. Cardiovascular regulation of supraoptic vasopressin neurons. Prog Brain Res. 2002;139:257–73. [PubMed: 12436941]
- 27.
- Ishikawa SE, Schrier RW. Pathophysiological roles of arginine vasopressin and aquaporin-2 in impaired water excretion. Clin Endocrinol (Oxf). 2003;58(1):1–17. [PubMed: 12519405]
- 28.
- Verbalis J, Usala R. Disorders of water and sodium homeostasis and bone. Current Opinion in Endocrine and Metabolic Research. 2018;3:83–92.
- 29.
- Tamma R, Sun L, Cuscito C, Lu P, Corcelli M, Li J, et al. Regulation of bone remodeling by vasopressin explains the bone loss in hyponatremia. Proc Natl Acad Sci U S A. 2013;110(46):18644–9. [PMC free article: PMC3831977] [PubMed: 24167258]
- 30.
- Tamma R, Colaianni G, Zhu LL, DiBenedetto A, Greco G, Montemurro G, et al. Oxytocin is an anabolic bone hormone. Proc Natl Acad Sci U S A. 2009;106(17):7149–54. [PMC free article: PMC2678458] [PubMed: 19369205]
- 31.
- Sun L, Tamma R, Yuen T, Colaianni G, Ji Y, Cuscito C, et al. Functions of vasopressin and oxytocin in bone mass regulation. Proc Natl Acad Sci U S A. 2016;113(1):164–9. [PMC free article: PMC4711832] [PubMed: 26699482]
- 32.
- Aspé-Sánchez M, Moreno M, Rivera MI, Rossi A, Ewer J. Oxytocin and Vasopressin Receptor Gene Polymorphisms: Role in Social and Psychiatric Traits. Front Neurosci. 2015;9:510. [PMC free article: PMC4729929] [PubMed: 26858594]
- 33.
- Dumais KM, Veenema AH. Vasopressin and oxytocin receptor systems in the brain: Sex differences and sex-specific regulation of social behavior. Front Neuroendocrinol. 2016;40:1–23. [PMC free article: PMC4633405] [PubMed: 25951955]
- 34.
- Zimmerman CA, Lin YC, Leib DE, Guo L, Huey EL, Daly GE, et al. Thirst neurons anticipate the homeostatic consequences of eating and drinking. Nature. 2016;537(7622):680–4. [PMC free article: PMC5161740] [PubMed: 27487211]
- 35.
- Nishimori K, Young LJ, Guo Q, Wang Z, Insel TR, Matzuk MM. Oxytocin is required for nursing but is not essential for parturition or reproductive behavior. Proc Natl Acad Sci U S A. 1996;93(21):11699–704. [PMC free article: PMC38121] [PubMed: 8876199]
- 36.
- Goodwin TM, Valenzuela GJ, Silver H, Creasy G. Dose ranging study of the oxytocin antagonist atosiban in the treatment of preterm labor. Atosiban Study Group. Obstet Gynecol. 1996;88(3):331–6. [PubMed: 8752234]
- 37.
- Tyzio R, Cossart R, Khalilov I, Minlebaev M, Hübner CA, Represa A, et al. Maternal oxytocin triggers a transient inhibitory switch in GABA signaling in the fetal brain during delivery. Science. 2006;314(5806):1788–92. [PubMed: 17170309]
- 38.
- Schulze L, Lischke A, Greif J, Herpertz SC, Heinrichs M, Domes G. Oxytocin increases recognition of masked emotional faces. Psychoneuroendocrinology. 2011;36(9):1378–82. [PubMed: 21477929]
- 39.
- De Dreu CK, Greer LL, Van Kleef GA, Shalvi S, Handgraaf MJ. Oxytocin promotes human ethnocentrism. Proc Natl Acad Sci U S A. 2011;108(4):1262–6. [PMC free article: PMC3029708] [PubMed: 21220339]
- 40.
- Kosfeld M, Heinrichs M, Zak PJ, Fischbacher U, Fehr E. Oxytocin increases trust in humans. Nature. 2005;435(7042):673–6. [PubMed: 15931222]
- 41.
- Hurlemann R, Patin A, Onur OA, Cohen MX, Baumgartner T, Metzler S, et al. Oxytocin enhances amygdala-dependent, socially reinforced learning and emotional empathy in humans. J Neurosci. 2010;30(14):4999–5007. [PMC free article: PMC6632777] [PubMed: 20371820]
- 42.
- Mantella RC, Vollmer RR, Li X, Amico JA. Female oxytocin-deficient mice display enhanced anxiety-related behavior. Endocrinology. 2003;144(6):2291–6. [PubMed: 12746288]
- 43.
- Ferguson JN, Young LJ, Hearn EF, Matzuk MM, Insel TR, Winslow JT. Social amnesia in mice lacking the oxytocin gene. Nat Genet. 2000;25(3):284–8. [PubMed: 10888874]
- 44.
- Winslow JT, Insel TR. The social deficits of the oxytocin knockout mouse. Neuropeptides. 2002;36(2-3):221–9. [PubMed: 12359512]
- 45.
- Leppanen J, Ng KW, Kim YR, Tchanturia K, Treasure J. Meta-analytic review of the effects of a single dose of intranasal oxytocin on threat processing in humans. J Affect Disord. 2018;225:167–79. [PubMed: 28837950]
- 46.
- Kiani Z, Farkhondeh T, Aramjoo H, Aschner M, Beydokhti H, Esmaeili A, et al. Oxytocin effect in adult patients with autism: An updated systematic review and meta-analysis of randomized controlled trials. CNS Neurol Disord Drug Targets. 2022 [PubMed: 35585805]
- 47.
- Yoon S, Kim YK. The Role of the Oxytocin System in Anxiety Disorders. Adv Exp Med Biol. 2020;1191:103–20. [PubMed: 32002925]
- 48.
- De Cagna F, Fusar-Poli L, Damiani S, Rocchetti M, Giovanna G, Mori A, et al. The Role of Intranasal Oxytocin in Anxiety and Depressive Disorders: A Systematic Review of Randomized Controlled Trials. Clin Psychopharmacol Neurosci. 2019;17(1):1–11. [PMC free article: PMC6361048] [PubMed: 30690935]
- 49.
- Jawad MY, Ahmad B, Hashmi AM. Role of Oxytocin in the Pathogenesis and Modulation of Borderline Personality Disorder: A Review. Cureus. 2021;13(2):e13190. [PMC free article: PMC7942026] [PubMed: 33717733]
- 50.
- Carson DS, Berquist SW, Trujillo TH, Garner JP, Hannah SL, Hyde SA, et al. Cerebrospinal fluid and plasma oxytocin concentrations are positively correlated and negatively predict anxiety in children. Mol Psychiatry. 2015;20(9):1085–90. [PubMed: 25349162]
- 51.
- Parker KJ, Garner JP, Libove RA, Hyde SA, Hornbeak KB, Carson DS, et al. Plasma oxytocin concentrations and OXTR polymorphisms predict social impairments in children with and without autism spectrum disorder. Proc Natl Acad Sci U S A. 2014;111(33):12258–63. [PMC free article: PMC4143031] [PubMed: 25092315]
- 52.
- Kranz TM, Kopp M, Waltes R, Sachse M, Duketis E, Jarczok TA, et al. Meta-analysis and association of two common polymorphisms of the human oxytocin receptor gene in autism spectrum disorder. Autism Res. 2016;9(10):1036–45. [PubMed: 26788924]
- 53.
- Ebstein RP, Knafo A, Mankuta D, Chew SH, Lai PS. The contributions of oxytocin and vasopressin pathway genes to human behavior. Horm Behav. 2012;61(3):359–79. [PubMed: 22245314]
- 54.
- Hammock EA, Young LJ. Oxytocin, vasopressin and pair bonding: implications for autism. Philos Trans R Soc Lond B Biol Sci. 2006;361(1476):2187–98. [PMC free article: PMC1764849] [PubMed: 17118932]
- 55.
- LoParo D, Waldman ID. The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: a meta-analysis. Mol Psychiatry. 2015;20(5):640–6. [PubMed: 25092245]
- 56.
- Lefevre A, Sirigu A. The two fold role of oxytocin in social developmental disorders: A cause and a remedy? Neurosci Biobehav Rev. 2016;63:168–76. [PubMed: 26828138]
- 57.
- Heinrichs M, Baumgartner T, Kirschbaum C, Ehlert U. Social support and oxytocin interact to suppress cortisol and subjective responses to psychosocial stress. Biol Psychiatry. 2003;54(12):1389–98. [PubMed: 14675803]
- 58.
- Sobota R, Mihara T, Forrest A, Featherstone RE, Siegel SJ. Oxytocin reduces amygdala activity, increases social interactions, and reduces anxiety-like behavior irrespective of NMDAR antagonism. Behav Neurosci. 2015;129(4):389–98. [PMC free article: PMC4518468] [PubMed: 26214213]
- 59.
- Labuschagne I, Phan KL, Wood A, Angstadt M, Chua P, Heinrichs M, et al. Oxytocin attenuates amygdala reactivity to fear in generalized social anxiety disorder. Neuropsychopharmacology. 2010;35(12):2403–13. [PMC free article: PMC3055328] [PubMed: 20720535]
- 60.
- Ditzen B, Schaer M, Gabriel B, Bodenmann G, Ehlert U, Heinrichs M. Intranasal oxytocin increases positive communication and reduces cortisol levels during couple conflict. Biol Psychiatry. 2009;65(9):728–31. [PubMed: 19027101]
- 61.
- Guastella AJ, Howard AL, Dadds MR, Mitchell P, Carson DS. A randomized controlled trial of intranasal oxytocin as an adjunct to exposure therapy for social anxiety disorder. Psychoneuroendocrinology. 2009;34(6):917–23. [PubMed: 19246160]
- 62.
- Domes G, Heinrichs M, Gläscher J, Büchel C, Braus DF, Herpertz SC. Oxytocin attenuates amygdala responses to emotional faces regardless of valence. Biol Psychiatry. 2007;62(10):1187–90. [PubMed: 17617382]
- 63.
- Kirsch P, Esslinger C, Chen Q, Mier D, Lis S, Siddhanti S, et al. Oxytocin modulates neural circuitry for social cognition and fear in humans. J Neurosci. 2005;25(49):11489–93. [PMC free article: PMC6725903] [PubMed: 16339042]
- 64.
- Karavitaki N, Brufani C, Warner JT, Adams CB, Richards P, Ansorge O, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf). 2005;62(4):397–409. [PubMed: 15807869]
- 65.
- Brandi ML, Gebert D, Kopczak A, Auer MK, Schilbach L. Oxytocin release deficit and social cognition in craniopharyngioma patients. J Neuroendocrinol. 2020;32(5):e12842. [PubMed: 32294805]
- 66.
- Daubenbüchel AM, Hoffmann A, Eveslage M, Özyurt J, Lohle K, Reichel J, et al. Oxytocin in survivors of childhood-onset craniopharyngioma. Endocrine. 2016;54(2):524–31. [PubMed: 27585663]
- 67.
- Gebert D, Auer MK, Stieg MR, Freitag MT, Lahne M, Fuss J, et al. De-masking oxytocin-deficiency in craniopharyngioma and assessing its link with affective function. Psychoneuroendocrinology. 2018;88:61–9. [PubMed: 29175721]
- 68.
- Daughters K, Manstead ASR, Rees DA. Hypopituitarism is associated with lower oxytocin concentrations and reduced empathic ability. Endocrine. 2017;57(1):166–74. [PMC free article: PMC5486505] [PubMed: 28597171]
- 69.
- Pereira AM, Schmid EM, Schutte PJ, Voormolen JH, Biermasz NR, van Thiel SW, et al. High prevalence of long-term cardiovascular, neurological and psychosocial morbidity after treatment for craniopharyngioma. Clin Endocrinol (Oxf). 2005;62(2):197–204. [PubMed: 15670196]
- 70.
- Karavitaki N, Cudlip S, Adams CB, Wass JA. Craniopharyngiomas. Endocr Rev. 2006;27(4):371–97. [PubMed: 16543382]
- 71.
- Wijnen M, van den Heuvel-Eibrink MM, Janssen J, Catsman-Berrevoets CE, Michiels EMC, van Veelen-Vincent MC, et al. Very long-term sequelae of craniopharyngioma. Eur J Endocrinol. 2017;176(6):755–67. [PubMed: 28325825]
- 72.
- Zada G, Kintz N, Pulido M, Amezcua L. Prevalence of neurobehavioral, social, and emotional dysfunction in patients treated for childhood craniopharyngioma: a systematic literature review. PLoS One. 2013;8(11):e76562. [PMC free article: PMC3818366] [PubMed: 24223703]
- 73.
- Eisenberg Y, Murad S, Casagrande A, McArthur M, Dugas LR, Barengolts E, et al. Oxytocin alterations and neurocognitive domains in patients with hypopituitarism. Pituitary. 2019;22(2):105–12. [PubMed: 30656597]
- 74.
- Aulinas A, Plessow F, Asanza E, Silva L, Marengi DA, Fan W, et al. Low Plasma Oxytocin Levels and Increased Psychopathology in Hypopituitary Men With Diabetes Insipidus. J Clin Endocrinol Metab. 2019;104(8):3181–91. [PMC free article: PMC6570634] [PubMed: 30882859]
- 75.
- McCullough ME, Churchland PS, Mendez AJ. Problems with measuring peripheral oxytocin: can the data on oxytocin and human behavior be trusted? Neurosci Biobehav Rev. 2013;37(8):1485–92. [PubMed: 23665533]
- 76.
- Landgraf R, Neumann ID. Vasopressin and oxytocin release within the brain: a dynamic concept of multiple and variable modes of neuropeptide communication. Front Neuroendocrinol. 2004;25(3-4):150–76. [PubMed: 15589267]
- 77.
- Ludwig M, Leng G. Dendritic peptide release and peptide-dependent behaviours. Nat Rev Neurosci. 2006;7(2):126–36. [PubMed: 16429122]
- 78.
- Valstad M, Alvares GA, Egknud M, Matziorinis AM, Andreassen OA, Westlye LT, et al. The correlation between central and peripheral oxytocin concentrations: A systematic review and meta-analysis. Neurosci Biobehav Rev. 2017;78:117–24. [PubMed: 28442403]
- 79.
- Sailer CO, Winzeler B, Urwyler SA, Schnyder I, Refardt J, Eckert A, et al. Oxytocin levels in response to pituitary provocation tests in healthy volunteers. Eur J Endocrinol. 2021;185(3):355–64. [PMC free article: PMC8650762] [PubMed: 34181566]
- 80.
- Christ-Crain M, Bichet DG, Fenske WK, Goldman MB, Rittig S, Verbalis JG, et al. Diabetes insipidus. Nat Rev Dis Primers. 2019;5(1):54. [PubMed: 31395885]
- 81.
- Baylis PH, Cheetham T. Diabetes insipidus. Arch Dis Child. 1998;79(1):84–9. [PMC free article: PMC1717616] [PubMed: 9771260]
- 82.
- Tomkins M, Lawless S, Martin-Grace J, Sherlock M, Thompson CJ. Diagnosis and Management of Central Diabetes Insipidus in Adults. J Clin Endocrinol Metab. 2022;107(10):2701–15. [PMC free article: PMC9516129] [PubMed: 35771962]
- 83.
- Pivonello R, De Bellis A, Faggiano A, Di Salle F, Petretta M, Di Somma C, et al. Central diabetes insipidus and autoimmunity: relationship between the occurrence of antibodies to arginine vasopressin-secreting cells and clinical, immunological, and radiological features in a large cohort of patients with central diabetes insipidus of known and unknown etiology. J Clin Endocrinol Metab. 2003;88(4):1629–36. [PubMed: 12679449]
- 84.
- Birk J, Friberg MA, Prescianotto-Baschong C, Spiess M, Rutishauser J. Dominant pro-vasopressin mutants that cause diabetes insipidus form disulfide-linked fibrillar aggregates in the endoplasmic reticulum. J Cell Sci. 2009;122(Pt 21):3994–4002. [PubMed: 19825939]
- 85.
- Brachet C, Birk J, Christophe C, Tenoutasse S, Velkeniers B, Heinrichs C, et al. Growth retardation in untreated autosomal dominant familial neurohypophyseal diabetes insipidus caused by one recurring and two novel mutations in the vasopressin-neurophysin II gene. Eur J Endocrinol. 2011;164(2):179–87. [PubMed: 21088058]
- 86.
- Rohayem J, Ehlers C, Wiedemann B, Holl R, Oexle K, Kordonouri O, et al. Diabetes and neurodegeneration in Wolfram syndrome: a multicenter study of phenotype and genotype. Diabetes Care. 2011;34(7):1503–10. [PMC free article: PMC3120194] [PubMed: 21602428]
- 87.
- Ghirardello S, Garrè ML, Rossi A, Maghnie M. The diagnosis of children with central diabetes insipidus. J Pediatr Endocrinol Metab. 2007;20(3):359–75. [PubMed: 17451074]
- 88.
- Hilson JB, Merchant SN, Adams JC, Joseph JT. Wolfram syndrome: a clinicopathologic correlation. Acta Neuropathol. 2009;118(3):415–28. [PMC free article: PMC2758421] [PubMed: 19449020]
- 89.
- Bell NH. Endocrine complications of sarcoidosis. Endocrinol Metab Clin North Am. 1991;20(3):645–54. [PubMed: 1935922]
- 90.
- Smith D, Finucane F, Phillips J, Baylis PH, Finucane J, Tormey W, et al. Abnormal regulation of thirst and vasopressin secretion following surgery for craniopharyngioma. Clin Endocrinol (Oxf). 2004;61(2):273–9. [PubMed: 15272926]
- 91.
- de Leon J, Dadvand M, Canuso C, Odom-White A, Stanilla J, Simpson GM. Polydipsia and water intoxication in a long-term psychiatric hospital. Biol Psychiatry. 1996;40(1):28–34. [PubMed: 8780852]
- 92.
- Sailer C, Winzeler B, Christ-Crain M. Primary polydipsia in the medical and psychiatric patient: characteristics, complications and therapy. Swiss Med Wkly. 2017;147:w14514. [PubMed: 29120013]
- 93.
- Bichet DG. Nephrogenic diabetes insipidus. Am J Med. 1998;105(5):431–42. [PubMed: 9831428]
- 94.
- Sands JM, Bichet DG. Nephrogenic diabetes insipidus. Ann Intern Med. 2006;144(3):186–94. [PubMed: 16461963]
- 95.
- Robben JH, Knoers NV, Deen PM. Cell biological aspects of the vasopressin type-2 receptor and aquaporin 2 water channel in nephrogenic diabetes insipidus. Am J Physiol Renal Physiol. 2006;291(2):F257–70. [PubMed: 16825342]
- 96.
- Grünfeld JP, Rossier BC. Lithium nephrotoxicity revisited. Nat Rev Nephrol. 2009;5(5):270–6. [PubMed: 19384328]
- 97.
- Oiso Y. Transient diabetes insipidus during pregnancy. Intern Med. 2003;42(6):459–60. [PubMed: 12857039]
- 98.
- Barron WM, Cohen LH, Ulland LA, Lassiter WE, Fulghum EM, Emmanouel D, et al. Transient vasopressin-resistant diabetes insipidus of pregnancy. N Engl J Med. 1984;310(7):442–4. [PubMed: 6694683]
- 99.
- Davison JM, Sheills EA, Philips PR, Barron WM, Lindheimer MD. Metabolic clearance of vasopressin and an analogue resistant to vasopressinase in human pregnancy. Am J Physiol. 1993;264(2 Pt 2):F348–53. [PubMed: 8447444]
- 100.
- Refardt J, Christ-Crain M. Diabetes insipidus in pregnancy: how to advice the patient? Minerva Endocrinol. 2018;43(4):458–64. [PubMed: 29463074]
- 101.
- Miller M, Dalakos T, Moses AM, Fellerman H, Streeten DH. Recognition of partial defects in antidiuretic hormone secretion. Ann Intern Med. 1970;73(5):721–9. [PubMed: 5476203]
- 102.
- Fenske W, Quinkler M, Lorenz D, Zopf K, Haagen U, Papassotiriou J, et al. Copeptin in the differential diagnosis of the polydipsia-polyuria syndrome--revisiting the direct and indirect water deprivation tests. J Clin Endocrinol Metab. 2011;96(5):1506–15. [PubMed: 21367924]
- 103.
- Van de Heijning BJ, Koekkoek-van den Herik I, Iványi T, Van Wimersma Greidanus TB. Solid-phase extraction of plasma vasopressin: evaluation, validation and application. J Chromatogr. 1991;565(1-2):159–71. [PubMed: 1874864]
- 104.
- Wun T. Vasopressin and platelets: a concise review. Platelets. 1997;8(1):15–22. [PubMed: 16793628]
- 105.
- Preibisz JJ, Sealey JE, Laragh JH, Cody RJ, Weksler BB. Plasma and platelet vasopressin in essential hypertension and congestive heart failure. Hypertension. 1983;5(2 Pt 2):I129–38. [PubMed: 6826223]
- 106.
- Cadnapaphornchai MA, Summer SN, Falk S, Thurman JM, Knepper MA, Schrier RW. Effect of primary polydipsia on aquaporin and sodium transporter abundance. Am J Physiol Renal Physiol. 2003;285(5):F965–71. [PubMed: 12876065]
- 107.
- Berliner RW, Davidson DG. Production of hypertonic urine in the absence of pituitary antidiuretic hormone. J Clin Invest. 1957;36(10):1416–27. [PMC free article: PMC1072744] [PubMed: 13475482]
- 108.
- Robertson RP, Porte D Jr. The glucose receptor. A defective mechanism in diabetes mellitus distinct from the beta adrenergic receptor. J Clin Invest. 1973;52(4):870–6. [PMC free article: PMC302334] [PubMed: 4693651]
- 109.
- Block LH, Furrer J, Locher RA, Siegenthaler W, Vetter W. Changes in tissue sensitivity to vasopressin in hereditary hypothalamic diabetes insipidus. Klin Wochenschr. 1981;59(15):831–6. [PubMed: 6267361]
- 110.
- Zerbe RL, Robertson GL. A comparison of plasma vasopressin measurements with a standard indirect test in the differential diagnosis of polyuria. N Engl J Med. 1981;305(26):1539–46. [PubMed: 7311993]
- 111.
- Morgenthaler NG, Struck J, Alonso C, Bergmann A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem. 2006;52(1):112–9. [PubMed: 16269513]
- 112.
- Fenske W, Allolio B. Clinical review: Current state and future perspectives in the diagnosis of diabetes insipidus: a clinical review. J Clin Endocrinol Metab. 2012;97(10):3426–37. [PubMed: 22855338]
- 113.
- Baylis PH. Diabetes insipidus. J R Coll Physicians Lond. 1998;32(2):108–11. [PMC free article: PMC9663009] [PubMed: 9597622]
- 114.
- Baylis PH, Gaskill MB, Robertson GL. Vasopressin secretion in primary polydipsia and cranial diabetes insipidus. Q J Med. 1981;50(199):345–58. [PubMed: 7036200]
- 115.
- Timper K, Fenske W, Kühn F, Frech N, Arici B, Rutishauser J, et al. Diagnostic Accuracy of Copeptin in the Differential Diagnosis of the Polyuria-polydipsia Syndrome: A Prospective Multicenter Study. J Clin Endocrinol Metab. 2015;100(6):2268–74. [PubMed: 25768671]
- 116.
- Fenske W, Refardt J, Chifu I, Schnyder I, Winzeler B, Drummond J, et al. A Copeptin-Based Approach in the Diagnosis of Diabetes Insipidus. N Engl J Med. 2018;379(5):428–39. [PubMed: 30067922]
- 117.
- Winzeler B, Cesana-Nigro N, Refardt J, Vogt DR, Imber C, Morin B, et al. Arginine-stimulated copeptin measurements in the differential diagnosis of diabetes insipidus: a prospective diagnostic study. Lancet. 2019;394(10198):587–95. [PubMed: 31303316]
- 118.
- Kurokawa H, Fujisawa I, Nakano Y, Kimura H, Akagi K, Ikeda K, et al. Posterior lobe of the pituitary gland: correlation between signal intensity on T1-weighted MR images and vasopressin concentration. Radiology. 1998;207(1):79–83. [PubMed: 9530302]
- 119.
- Hannon MJ, Orr C, Moran C, Behan LA, Agha A, Ball SG, et al. Anterior hypopituitarism is rare and autoimmune disease is common in adults with idiopathic central diabetes insipidus. Clin Endocrinol (Oxf). 2012;76(5):725–8. [PubMed: 22026638]
- 120.
- Lam KS, Wat MS, Choi KL, Ip TP, Pang RW, Kumana CR. Pharmacokinetics, pharmacodynamics, long-term efficacy and safety of oral 1-deamino-8-D-arginine vasopressin in adult patients with central diabetes insipidus. Br J Clin Pharmacol. 1996;42(3):379–85. [PMC free article: PMC2042683] [PubMed: 8877030]
- 121.
- Behan LA, Sherlock M, Moyles P, Renshaw O, Thompson CJ, Orr C, et al. Abnormal plasma sodium concentrations in patients treated with desmopressin for cranial diabetes insipidus: results of a long-term retrospective study. Eur J Endocrinol. 2015;172(3):243–50. [PubMed: 25430399]
- 122.
- Atila C, Loughrey PB, Garrahy A, Winzeler B, Refardt J, Gildroy P, et al. Central diabetes insipidus from a patient's perspective: management, psychological co-morbidities, and renaming of the condition: results from an international web-based survey. Lancet Diabetes Endocrinol. 2022;10(10):700–9. [PubMed: 36007536]
- 123.
- Loh JA, Verbalis JG. Disorders of water and salt metabolism associated with pituitary disease. Endocrinol Metab Clin North Am. 2008;37(1):213–34. x. [PubMed: 18226738]
- 124.
- Eisenberg Y, Frohman LA. ADIPSIC DIABETES INSIPIDUS: A REVIEW. Endocr Pract. 2016;22(1):76–83. [PubMed: 26401579]
- 125.
- Cuesta M, Hannon MJ, Thompson CJ. Adipsic diabetes insipidus in adult patients. Pituitary. 2017;20(3):372–80. [PubMed: 28074401]
- 126.
- Batlle DC, von Riotte AB, Gaviria M, Grupp M. Amelioration of polyuria by amiloride in patients receiving long-term lithium therapy. N Engl J Med. 1985;312(7):408–14. [PubMed: 3969096]
- 127.
- Crawford JD, Kennedy GC. Chlorothiazid in diabetes insipidus. Nature. 1959;183(4665):891–2. [PubMed: 13644230]
- 128.
- Earley LE, Orloff J. THE MECHANISM OF ANTIDIURESIS ASSOCIATED WITH THE ADMINISTRATION OF HYDROCHLOROTHIAZIDE TO PATIENTS WITH VASOPRESSIN-RESISTANT DIABETES INSIPIDUS. J Clin Invest. 1962;41(11):1988–97. [PMC free article: PMC291129] [PubMed: 16695887]
- 129.
- Rosa RM, Bierer BE, Thomas R, Stoff JS, Kruskall M, Robinson S, et al. A study of induced hyponatremia in the prevention and treatment of sickle-cell crisis. N Engl J Med. 1980;303(20):1138–43. [PubMed: 6999348]
- 130.
- Usberti M, Dechaux M, Guillot M, Seligmann R, Pavlovitch H, Loirat C, et al. Renal prostaglandin E2 in nephrogenic diabetes insipidus: effects of inhibition of prostaglandin synthesis by indomethacin. J Pediatr. 1980;97(3):476–8. [PubMed: 6251193]
- 131.
- Winzeler B, Sailer CO, Coynel D, Zanchi D, Vogt DR, Urwyler SA, et al. A randomized controlled trial of the GLP-1 receptor agonist dulaglutide in primary polydipsia. J Clin Invest. 2021;131(20) [PMC free article: PMC8516458] [PubMed: 34473645]
- 132.
- Källén BA, Carlsson SS, Bengtsson BK. Diabetes insipidus and use of desmopressin (Minirin) during pregnancy. Eur J Endocrinol. 1995;132(2):144–6. [PubMed: 7858730]
- 133.
- Ray JG. DDAVP use during pregnancy: an analysis of its safety for mother and child. Obstet Gynecol Surv. 1998;53(7):450–5. [PubMed: 9662731]
- 134.
- Waikar SS, Mount DB, Curhan GC. Mortality after hospitalization with mild, moderate, and severe hyponatremia. Am J Med. 2009;122(9):857–65. [PMC free article: PMC3033702] [PubMed: 19699382]
- 135.
- Chawla A, Sterns RH, Nigwekar SU, Cappuccio JD. Mortality and serum sodium: do patients die from or with hyponatremia? Clin J Am Soc Nephrol. 2011;6(5):960–5. [PMC free article: PMC3087791] [PubMed: 21441132]
- 136.
- Corona G, Giuliani C, Verbalis JG, Forti G, Maggi M, Peri A. Hyponatremia improvement is associated with a reduced risk of mortality: evidence from a meta-analysis. PLoS One. 2015;10(4):e0124105. [PMC free article: PMC4408113] [PubMed: 25905459]
- 137.
- Refardt J, Pelouto A, Potasso L, Hoorn EJ, Christ-Crain M. Hyponatremia Intervention Trial (HIT): Study Protocol of a Randomized, Controlled, Parallel-Group Trial With Blinded Outcome Assessment. Front Med (Lausanne). 2021;8:729545. [PMC free article: PMC8450416] [PubMed: 34552947]
- 138.
- Ball SG, Iqbal Z. Diagnosis and treatment of hyponatraemia. Best Pract Res Clin Endocrinol Metab. 2016;30(2):161–73. [PubMed: 27156756]
- 139.
- Bartter FC, Schwartz WB. The syndrome of inappropriate secretion of antidiuretic hormone. Am J Med. 1967;42(5):790–806. [PubMed: 5337379]
- 140.
- Spasovski G, Vanholder R, Allolio B, Annane D, Ball S, Bichet D, et al. Clinical practice guideline on diagnosis and treatment of hyponatraemia. Eur J Endocrinol. 2014;170(3):G1–47. [PubMed: 24569125]
- 141.
- Fenske W, Störk S, Koschker AC, Blechschmidt A, Lorenz D, Wortmann S, et al. Value of fractional uric acid excretion in differential diagnosis of hyponatremic patients on diuretics. J Clin Endocrinol Metab. 2008;93(8):2991–7. [PubMed: 18477658]
- 142.
- Fenske WK, Christ-Crain M, Hörning A, Simet J, Szinnai G, Fassnacht M, et al. A copeptin-based classification of the osmoregulatory defects in the syndrome of inappropriate antidiuresis. J Am Soc Nephrol. 2014;25(10):2376–83. [PMC free article: PMC4178436] [PubMed: 24722436]
- 143.
- Nigro N, Winzeler B, Suter-Widmer I, Schuetz P, Arici B, Bally M, et al. Evaluation of copeptin and commonly used laboratory parameters for the differential diagnosis of profound hyponatraemia in hospitalized patients: 'The Co-MED Study'. Clin Endocrinol (Oxf). 2017;86(3):456–62. [PubMed: 27658031]
- 144.
- Rosner MH. Exercise-associated hyponatremia. Semin Nephrol. 2009;29(3):271–81. [PubMed: 19523574]
- 145.
- Danz M, Pöttgen K, Tönjes PM, Hinkelbein J, Braunecker S. Hyponatremia among Triathletes in the Ironman European Championship. N Engl J Med. 2016;374(10):997–8. [PubMed: 26962745]
- 146.
- Feldman BJ, Rosenthal SM, Vargas GA, Fenwick RG, Huang EA, Matsuda-Abedini M, et al. Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med. 2005;352(18):1884–90. [PMC free article: PMC5340184] [PubMed: 15872203]
- 147.
- Decaux G, Vandergheynst F, Bouko Y, Parma J, Vassart G, Vilain C. Nephrogenic syndrome of inappropriate antidiuresis in adults: high phenotypic variability in men and women from a large pedigree. J Am Soc Nephrol. 2007;18(2):606–12. [PubMed: 17229917]
- 148.
- Yee AH, Burns JD, Wijdicks EF. Cerebral salt wasting: pathophysiology, diagnosis, and treatment. Neurosurg Clin N Am. 2010;21(2):339–52. [PubMed: 20380974]
- 149.
- Hannon MJ, Behan LA, O'Brien MM, Tormey W, Ball SG, Javadpour M, et al. Hyponatremia following mild/moderate subarachnoid hemorrhage is due to SIAD and glucocorticoid deficiency and not cerebral salt wasting. J Clin Endocrinol Metab. 2014;99(1):291–8. [PubMed: 24248182]
- 150.
- Verbalis JG, Goldsmith SR, Greenberg A, Korzelius C, Schrier RW, Sterns RH, et al. Diagnosis, evaluation, and treatment of hyponatremia: expert panel recommendations. Am J Med. 2013;126(10) Suppl 1:S1–42. [PubMed: 24074529]
- 151.
- Ball S, Barth J, Levy M. SOCIETY FOR ENDOCRINOLOGY ENDOCRINE EMERGENCY GUIDANCE: Emergency management of severe symptomatic hyponatraemia in adult patients. Endocr Connect. 2016;5(5):G4–g6. [PMC free article: PMC5314809] [PubMed: 27935814]
- 152.
- Adrogué HJ, Madias NE. Hyponatremia. N Engl J Med. 2000;342(21):1581–9. [PubMed: 10824078]
- 153.
- Mohmand HK, Issa D, Ahmad Z, Cappuccio JD, Kouides RW, Sterns RH. Hypertonic saline for hyponatremia: risk of inadvertent overcorrection. Clin J Am Soc Nephrol. 2007;2(6):1110–7. [PubMed: 17913972]
- 154.
- Sterns RH, Hix JK, Silver S. Treating profound hyponatremia: a strategy for controlled correction. Am J Kidney Dis. 2010;56(4):774–9. [PubMed: 20709440]
- 155.
- Garrahy A, Dineen R, Hannon AM, Cuesta M, Tormey W, Sherlock M, et al. Continuous Versus Bolus Infusion of Hypertonic Saline in the Treatment of Symptomatic Hyponatremia Caused by SIAD. J Clin Endocrinol Metab. 2019;104(9):3595–602. [PubMed: 30882872]
- 156.
- Baek SH, Jo YH, Ahn S, Medina-Liabres K, Oh YK, Lee JB, et al. Risk of Overcorrection in Rapid Intermittent Bolus vs Slow Continuous Infusion Therapies of Hypertonic Saline for Patients With Symptomatic Hyponatremia: The SALSA Randomized Clinical Trial. JAMA Intern Med. 2021;181(1):81–92. [PMC free article: PMC7589081] [PubMed: 33104189]
- 157.
- Garrahy A, Galloway I, Hannon AM, Dineen R, O'Kelly P, Tormey WP, et al. Fluid Restriction Therapy for Chronic SIAD; Results of a Prospective Randomized Controlled Trial. J Clin Endocrinol Metab. 2020;105(12) [PubMed: 32879954]
- 158.
- Krisanapan P, Vongsanim S, Pin-On P, Ruengorn C, Noppakun K. Efficacy of Furosemide, Oral Sodium Chloride, and Fluid Restriction for Treatment of Syndrome of Inappropriate Antidiuresis (SIAD): An Open-label Randomized Controlled Study (The EFFUSE-FLUID Trial). Am J Kidney Dis. 2020;76(2):203–12. [PubMed: 32199708]
- 159.
- Verbalis JG, Greenberg A, Burst V, Haymann JP, Johannsson G, Peri A, et al. Diagnosing and Treating the Syndrome of Inappropriate Antidiuretic Hormone Secretion. Am J Med. 2016;129(5):537.e9-.e23. [PubMed: 26584969]
- 160.
- Winzeler B, Lengsfeld S, Nigro N, Suter-Widmer I, Schütz P, Arici B, et al. Predictors of nonresponse to fluid restriction in hyponatraemia due to the syndrome of inappropriate antidiuresis. J Intern Med. 2016;280(6):609–17. [PubMed: 27481546]
- 161.
- Furst H, Hallows KR, Post J, Chen S, Kotzker W, Goldfarb S, et al. The urine/plasma electrolyte ratio: a predictive guide to water restriction. Am J Med Sci. 2000;319(4):240–4. [PubMed: 10768609]
- 162.
- Rondon-Berrios H, Tandukar S, Mor MK, Ray EC, Bender FH, Kleyman TR, et al. Urea for the Treatment of Hyponatremia. Clin J Am Soc Nephrol. 2018;13(11):1627–32. [PMC free article: PMC6237061] [PubMed: 30181129]
- 163.
- Decaux G, Andres C, Gankam Kengne F, Soupart A. Treatment of euvolemic hyponatremia in the intensive care unit by urea. Crit Care. 2010;14(5):R184. [PMC free article: PMC3219290] [PubMed: 20946646]
- 164.
- Pierrakos C, Taccone FS, Decaux G, Vincent JL, Brimioulle S. Urea for treatment of acute SIADH in patients with subarachnoid hemorrhage: a single-center experience. Ann Intensive Care. 2012;2(1):13. [PMC free article: PMC3488535] [PubMed: 22647340]
- 165.
- Schrier RW, Gross P, Gheorghiade M, Berl T, Verbalis JG, Czerwiec FS, et al. Tolvaptan, a selective oral vasopressin V2-receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099–112. [PubMed: 17105757]
- 166.
- Sterns RH. Tolvaptan for the Syndrome of Inappropriate Secretion of Antidiuretic Hormone: Is the Dose Too High? Am J Kidney Dis. 2018;71(6):763–5. [PubMed: 29801549]
- 167.
- Braunwald E. Gliflozins in the Management of Cardiovascular Disease. N Engl J Med. 2022;386(21):2024–34. [PubMed: 35613023]
- 168.
- Refardt J, Imber C, Sailer CO, Jeanloz N, Potasso L, Kutz A, et al. A Randomized Trial of Empagliflozin to Increase Plasma Sodium Levels in Patients with the Syndrome of Inappropriate Antidiuresis. J Am Soc Nephrol. 2020;31(3):615–24. [PMC free article: PMC7062212] [PubMed: 32019783]
- 169.
- Refardt J, Imber C, Nobbenhuis R, Sailer C, Haslbauer A, Monnerat S, et al. Treatment Effect of the SGLT2-inhibitor Empagliflozin on chronic SIAD –results of a double-blind, randomized, crossover, placebo-controlled trial. J Am Soc Nephrol. 2022 in press. [PMC free article: PMC10103093] [PubMed: 36396331]
- 170.
- Miell J, Dhanjal P, Jamookeeah C. Evidence for the use of demeclocycline in the treatment of hyponatraemia secondary to SIADH: a systematic review. Int J Clin Pract. 2015;69(12):1396–417. [PMC free article: PMC5042094] [PubMed: 26289137]
- 171.
- Ball SG, Vaidja B, Baylis PH. Hypothalamic adipsic syndrome: diagnosis and management. Clin Endocrinol (Oxf). 1997;47(4):405–9. [PubMed: 9404436]
- Review Neurochemical Circuits Subserving Fluid Balance and Baroreflex: A Role for Serotonin, Oxytocin, and Gonadal Steroids.[Neurobiology of Body Fluid Hom...]Review Neurochemical Circuits Subserving Fluid Balance and Baroreflex: A Role for Serotonin, Oxytocin, and Gonadal Steroids.Vivas L, Godino A, Dalmasso C, Caeiro XE, Macchione AF, Cambiasso MJ. Neurobiology of Body Fluid Homeostasis: Transduction and Integration. 2014
- Review The neurohypophysis: recent developments.[J Lab Clin Med. 1987]Review The neurohypophysis: recent developments.Robinson AG. J Lab Clin Med. 1987 Mar; 109(3):336-45.
- Specific immunoelectronmicroscopic localization of vasopressin and oxytocin in the neurohypophysis of the rat.[Cell Tissue Res. 1977]Specific immunoelectronmicroscopic localization of vasopressin and oxytocin in the neurohypophysis of the rat.van Leeuwen FW, Swaab DF. Cell Tissue Res. 1977 Feb 14; 177(4):493-501.
- Enkephalins co-exist with oxytocin and vasopressin in nerve terminals of rat neurohypophysis.[Nature. 1981]Enkephalins co-exist with oxytocin and vasopressin in nerve terminals of rat neurohypophysis.Martin R, Voigt KH. Nature. 1981 Feb 5; 289(5797):502-4.
- The vasopressor and oxytocic activities of the hypothalamus and neurohypophysis are influenced by inhibited alpha-adrenergic transmission during dehydration and subsequent rehydration in the white rat.[Acta Physiol Pol. 1981]The vasopressor and oxytocic activities of the hypothalamus and neurohypophysis are influenced by inhibited alpha-adrenergic transmission during dehydration and subsequent rehydration in the white rat.Guzek JW, Janus J, Ciosek J. Acta Physiol Pol. 1981 Nov-Dec; 32(6):667-79.
- The Neurohypophysis: Endocrinology of Vasopressin and Oxytocin - EndotextThe Neurohypophysis: Endocrinology of Vasopressin and Oxytocin - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...