ABSTRACT
Numerous distinct pathophysiologic abnormalities have been associated with type 2 diabetes mellitus (T2DM). It is well established that decreased peripheral glucose uptake (mainly muscle) combined with augmented endogenous glucose production are characteristic features of insulin resistance. Increased lipolysis, elevated free fatty acid levels, along with accumulation of intermediary lipid metabolites contributes to further increase glucose output, reduce peripheral glucose utilization, and impair beta-cell function. Adipocyte insulin resistance and inflammation have been identified as important contributors to the development of T2DM. The presence of non-alcoholic fatty liver disease [NAFLD] is now considered an integral part of the insulin resistant state. The traditional concepts of “glucotoxicity” and lipotoxicity, which covers the process of beta cell deterioration in response to chronic elevations of glucose and lipids, has been expanded to encompass all nutrients [‘nutri-toxicity”]. The delayed transport of insulin across the microvascular system is also partially responsible for the development of tissue insulin resistance. Compensatory insulin secretion by the pancreatic beta cells may initially maintain normal plasma glucose levels, but beta cell function is already abnormal at this stage, and progressively worsens over time. Concomitantly, there is inappropriate release of glucagon from the pancreatic alpha-cells, particularly in the post-prandial period. It has been postulated that both impaired insulin and excessive glucagon secretion in T2DM are secondary to an “incretin defect”, defined primarily as inadequate release or response to the gastrointestinal incretin hormones upon meal ingestion. To a certain extent, the gut microbiome appears to play a role in the hormonal and metabolic disturbances seen in T2DM. Moreover, hypothalamic insulin resistance (central nervous system) also impairs the ability of circulating insulin to suppress glucose production, and renal tubular glucose reabsorption capacity may be enhanced, despite hyperglycemia. These pathophysiologic abnormalities should be considered for the treatment of hyperglycemia in patients with T2DM. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
NORMAL GLUCOSE HOMEOSTASIS
In the post-absorptive state (10-12 hour overnight fast), the majority of total body glucose disposal takes place in insulin independent tissues (1). Under basal conditions, approximately 50% of all glucose utilization occurs in the brain, which is insulin independent and becomes saturated at a plasma glucose concentration of about 40 mg/dl (2). Another 25% of glucose uptake occurs in the splanchnic area (liver plus gastrointestinal tissues) and is also insulin independent (3). The remaining 25% of glucose metabolism in the post-absorptive state takes place in insulin-dependent tissues, primarily muscle (4,5). Basal glucose utilization averages ~2.0 mg/kg.min and is precisely matched by the rate of endogenous glucose production (1,3-7). Approximately 85% of endogenous glucose production is derived from the liver, and the remaining amount is produced by the kidney (1,8,9). Approximately one-half of basal hepatic glucose production is derived from glycogenolysis and one-half from gluconeogenesis (9,10).
Following glucose ingestion, the balance between endogenous glucose production and tissue glucose uptake is disrupted. The increase in plasma glucose concentration stimulates insulin release from the pancreatic beta cells, and the resultant hyperinsulinemia and hyperglycemia serve (i) to stimulate glucose uptake by splanchnic (liver and gut) and peripheral (primarily muscle) tissues and (ii) to suppress endogenous glucose production (1,3-7,11-14).
Hyperglycemia, in the absence of hyperinsulinemia, exerts its own independent effect to stimulate muscle glucose uptake and to suppress endogenous glucose production in a dose dependent fashion (14-16). The majority (~80-85%) of glucose that is taken up by peripheral tissues is disposed of in muscle (1,3-7,11-14), with only a small amount (~4-5%) being metabolized by adipocytes (17). Although fat tissue is responsible for only a fraction of total body glucose disposal, it plays a very important role in the maintenance of total body glucose homeostasis (see below). Insulin is a potent inhibitor of lipolysis and even small increments in the plasma insulin concentration exerts a potent anti-lipolytic effect, leading to a marked reduction in the plasma free fatty acid level (18). The decline in plasma FFA concentration results in increased glucose uptake in muscle (19) and contributes to the inhibition of endogenous glucose production (16,20). Thus, changes in the plasma FFA concentration in response to increased plasma levels of insulin and glucose play an important role in the maintenance of normal glucose homeostasis (21,22).
SITE OF INSULIN RESISTANCE IN TYPE 2 DIABETES (T2DM)
The maintenance of whole-body glucose homeostasis is dependent upon a normal insulin secretory response by the pancreatic beta cells and normal tissue sensitivity to the independent effects of hyperinsulinemia and hyperglycemia (i.e., the mass-action effect of glucose) to augment glucose uptake. In turn, the combined effects of insulin and hyperglycemia to promote glucose disposal are dependent on three tightly coupled mechanisms: (i) suppression of endogenous (primarily hepatic) glucose production; (ii) stimulation of glucose uptake by the splanchnic (hepatic plus gastrointestinal) tissues; and (iii) stimulation of glucose uptake by peripheral tissues, primarily muscle (1,4,14). Muscle glucose uptake is regulated by flux through two major metabolic pathways: glycolysis (of which ~90% represents glucose oxidation) and glycogen synthesis.
Hepatic Glucose Production
In the overnight fasted state, the liver of healthy subjects produces glucose at the rate of ~1.8-2.0 mg.kg-1.min-1 (1,3,4,6,18,54). This glucose flux is essential to meet the needs of the brain and other neural tissues, which utilize glucose at a constant rate of ~1-1.2 mg.kg-1.min-1 (2,169). Brain glucose uptake accounts for ~50-60% of glucose disposal during the post-absorptive state and this uptake is insulin independent. Therefore, brain glucose uptake occurs at the same rate during absorptive and post-absorptive periods and is not altered in T2DM (214). Following glucose ingestion, insulin is secreted into the portal vein and glucagon release is inhibited, and this new hormonal ratio is carried to the liver, where it suppresses hepatic glucose output. If the liver does not perceive this insulin signal and continues to produce glucose, there will be two superimposed inputs of glucose into the body, one from the liver and another from the gastrointestinal tract, and marked hyperglycemia will ensue.
In subjects with T2DM and mild to moderate fasting hyperglycemia (140-200 mg/dl, 7.8-11.1 mmol/L) basal endogenous glucose production [EGP] is increased by ~0.5 mg/kg.min. Consequently, during the overnight sleeping hours (i.e., 2200 h to 0800 h), the liver of an 80-kg individual with diabetes and modest fasting hyperglycemia adds an additional 35 g of glucose to the systemic circulation. The increase in basal EGP is closely correlated with the severity of fasting hyperglycemia (1,3,4,6,18,54,157-159,162). Thus, in T2DM with overt fasting hyperglycemia (>140 mg/dl, 7.8 mmol/l), an excessive rate of EGP and glucose output is the major abnormality responsible for the elevated fasting plasma glucose concentration. The close relationship between fasting plasma glucose concentration and EGP has been demonstrated in numerous studies (164-166,170-174).
In the post-absorptive state, the fasting plasma insulin concentration in subjects with T2DM is 2-4-fold greater than in subjects without diabetes. Because hyperinsulinemia is a potent inhibitor of EGP (1,3,4-6,16,18,164,165,175), hepatic resistance to the action of insulin must be present in the post-absorptive state to explain the excessive output of glucose. Hyperglycemia per se also exerts a powerful suppressive action on EGP (15,167,175-177). Therefore, the liver, primarily, also must be glucose resistant with respect to the inhibitory effect of hyperglycemia to suppress glucose output, and this has been well documented (15,167,178,179).
Using the euglycemic insulin clamp technique in combination with tritiated glucose, the dose response relationship between endogenous glucose production and the plasma glucose concentration has been defined by Groop, DeFronzo, et al (18). The following points should be emphasized: (i) first, the dose-response curve relating inhibition of EGP to the plasma insulin concentration is quite steep, with an effective dose for half-maximal insulin concentration (ED50) of ~30-40 µU/ml; (ii) in individuals with T2D the dose response curve is shifted to the right, indicating the presence of hepatic resistance to the inhibitory effect of insulin on hepatic glucose production. However, at plasma insulin concentrations within the high physiologic range (~100 µU/ml), the hepatic insulin resistance can be largely overcome and a near normal suppression of EGP can be achieved; (iii) the severity of the hepatic insulin resistance is related to the severity of the diabetic state. In T2DM with mild fasting hyperglycemia, an increment in plasma insulin concentration of 100 µU/ml causes a complete suppression of EGP. However, in diabetic subjects with more severe fasting hyperglycemia, the ability of the same plasma insulin concentration to suppress EGP is impaired (18). These results suggest that there is an acquired component of hepatic insulin resistance and that this defect becomes progressively worse as the diabetic state decompensates over time.
The glucose released by the liver in the post-absorptive state can be derived from either glycogenolysis or gluconeogenesis (6,16,176). Studies employing the hepatic vein catheter technique have shown that the uptake of gluconeogenic precursors, especially lactate, is increased in subjects with T2DM (180). Consistent with this observation, radioisotope turnover studies, using lactate, alanine, and glycerol have shown that ~90% of the increase in HGP above baseline can be accounted for by accelerated gluconeogenesis (181,182). More recent studies employing 13C-magnetic resonance imaging (183) and D2O (184,185) have confirmed the important contribution of accelerated gluconeogenesis to the increase in HGP. An increased rate of glutamine conversion to glucose also has been shown to contribute to the elevated rate of gluconeogenesis in subjects with T2DM (186), which may be, in part, derived from renal gluconeogenesis (8). The mechanisms responsible for the increase in hepatic gluconeogenesis include hyperglucagonemia (187), increased circulating levels of gluconeogenic precursors (lactate, alanine, glycerol) (181,188), increased FFA oxidation (18,162,189), enhanced sensitivity to glucagon (190) and decreased sensitivity to insulin (1,4.18,164,165). Although the majority of evidence indicates that increased gluconeogenesis is the major cause of the increase in EGP in subjects with T2DM (181- 186), it is likely that accelerated glycogenolysis also contributes to it (181,191).
The presence of both direct and indirect effects of insulin in suppressing EGP and release into the circulation were recently demonstrated in animals using intra-portal and systemic insulin infusions (430). The results provided evidence that, in addition to a direct action of insulin on hepatic enzymes, the inhibition of adipose tissue lipolysis represents an important mechanism by which insulin regulates the rate of gluconeogenesis. This is therefore, accomplished indirectly, by controlling the supply of free fatty acids, which are essential to the process of glucose synthesis de novo. The rate-limiting step in achieving fast and complete inhibition of adipose tissue lipolysis is the transendothelial transport of insulin across tissue capillaries. Additional data obtained during systemic infusions of free fatty acids and in experiments where adipocyte lipolytic factors were manipulated, together with observation in mice lacking hepatic Foxo1 & Akt1/2 signaling have confirmed this indirect action of insulin on gluconeogenesis (430-432). These findings have generated the hypothesis that in patients with T2DM, insulin may be transported slowly across tissue capillaries, which delays the inhibition of lipolysis with subsequent impairment of the suppression of EGP.
On the other hand, animal studies (431), where insulin was infused directly into the portal vein, mimicking normal insulin secretory pattern, showed that there is complete and swift inhibition of EGP. These observations were confirmed when plasma glucagon and fatty acid levels were clamped at basal values, and in conditions where brain insulin action was blocked. Authors conclude that the direct hepatic effect of insulin in the regulation of EGP is more relevant and that, the indirect effect is redundant in physiological conditions. Acute insulin suppression of endogenous gluconeogenesis is largely an indirect effect mediated by the inhibition of adipose tissue lipolysis, which reduces delivery of non-esterified fatty acids and glycerol to the liver. The major direct effect of insulin on hepatic glucose metabolism is the regulation of glycogen metabolism. Hyperglycemia and hyperinsulinemia are required to maximally stimulate net hepatic glycogenesis. In T2DM, lipid-induced hepatic insulin resistance, high rates of adipose tissue lipolysis and hyperglucagonemia impair glucose metabolism in the liver (432).
Because of the inaccessibility of the liver in man, it has been difficult to assess the role of key enzymes involved in the regulation of gluconeogenesis (pyruvate carboxylase, phosphoenol- pyruvate carboxykinase), glycogenolysis (glycogen phosphorylase), and net hepatic glucose output (glucokinase, glucose-6-phosphatase). However, considerable evidence from animal models of T2DM and some evidence in humans have implicated increased activity of PEPCK and G-6-Pase in the accelerated rate of hepatic glucose production (192-194).
Recently, changes in hypothalamic insulin signaling have been shown to affect endogenous glucose production. The activation of the insulin receptor in the third cerebral ventricle is capable of suppressing glucose production, independent of plasma insulin or other counter-regulatory hormones. Conversely, central antagonism to insulin signaling impairs the ability of circulating insulin to inhibit glucose production (6A). These observations have raised the possibility that hypothalamic insulin resistance contributes to hyperglycemia in T2DM.
The Role of the Kidney
The kidney also has been shown to produce glucose and estimates of the renal contribution to total endogenous glucose production have varied from 5% to 20% (8,9,195). These varying estimates of the contribution of renal gluconeogenesis to total glucose production are largely related to the methodology employed to measure glucose production by the kidney (196). One unconfirmed study suggests that the rate of renal gluconeogenesis is increased in T2DM with fasting hyperglycemia (197). Arguing against this possibility are studies employing the hepatic vein catheter technique which have shown that all of the increase in total body EGP (measured with 3-3H-glucose) in T2DM can be accounted for by increased hepatic glucose output (measured by the hepatic vein catheter technique) (3). A more relevant aspect on the role of the kidney in the dysregulation of glucose homeostasis in diabetes is the maintenance of hyperglycemia, which results from a maladaptive enhancement of the tubular glucose transport threshold (9A, 9B). It has been hypothesized that in response to an elevated glucose load presented to the proximal tubular lumen, the sodium glucose co-transporter system increases its reabsorptive capacity by upregulating the SGLT-2 expression and kinetics (9C). However, more recent studies conducted in humans who underwent unilateral nephrectomy were not able to confirm the over-expression of either SGLT-2 or SGLT-1 proteins in proximal renal tubules of patients with T2DM compared to non-diabetic controls (433, 434). The augmented tubular glucose transport described in patients with type 1 and type 2 diabetes may result from a functional enhancement of the activity of these co-transporters. The elevated renal threshold to plasma values between 220-250 mg/dl for the excretion of glucose into the urine in these patients, thus may be secondary to a sustained hyperglycemia. If this is confirmed, the maladaptive process of recycling a substantial amount of glucose back into the peripheral circulation may be attenuated with near-normoglycemia, possibly reversible. In any case, this contribution of the kidney to hyperglycemia in diabetic patients represents one additional pathogenic mechanism that has been underappreciated.
Peripheral (Muscle) Glucose Uptake
Muscle is the major site of glucose disposal in man (1,3-5,14). Under euglycemic hyperinsulinemic conditions, approximately 80% of total body glucose uptake occurs in skeletal muscle (1,3-5). Studies employing the euglycemic insulin clamp in combination with femoral artery/vein catheterization have examined the effect of insulin on leg glucose uptake in subjects with T2DM and control subjects (3). Since bone is metabolically inert with regards to carbohydrate metabolism and adipose tissue takes up less than 5% of an infused glucose load (17,198,199), muscle represents the major tissue responsible for leg glucose uptake.
In response to a physiologic increase in plasma insulin concentration (~80-100 μU/ml), leg (muscle) glucose uptake increases linearly, reaching a plateau value of 10 mg/kg leg wt per minute (3). In contrast, in lean subjects with T2DM, the onset of insulin action is delayed for ~40 min and the ability of the hormone to stimulate leg glucose uptake is markedly blunted, even though the study is carried out for an additional 60 min in the group with T2DMto allow insulin to more fully express its biological effects (3). During the last hour of the insulin clamp study, the rate of glucose uptake was reduced by 50% in the group with T2DM (3). These results provide conclusive evidence that the primary site of insulin resistance during euglycemic insulin clamp studies performed in subjects with T2DM resides in muscle tissue. Using the forearm and leg catheterization techniques (13,153,200,202), a number of investigators have demonstrated a decreased rate of insulin-mediated glucose uptake by peripheral tissues. The use of positron emission tomography (PET) scanning to quantitate leg glucose uptake in subjects with T2DM has provided additional support for the presence of severe muscle resistance to insulin in diabetic subjects (203).
Vascular and Myocardial Insulin Resistance
The first and rate-limiting step in insulin-mediated glucose disposal is the transit of insulin from the plasma to the muscle. Crossing of insulin from the circulation into the muscle interstitium is governed by vascular endothelium. The transendothelial transport depends on the insulin receptor binding to the endothelial cell membrane and requires the activation of the nitric oxide synthase. The transport of insulin across the endothelial cell layer appears to involve a complex vesicular trafficking process, which is saturable. Insulin is known to promote capillary vasodilation particularly in the postprandial period to facilitate entry and distribution of fuel substrates, including glucose. Several studies sampling lymph and interstitial glucose, using dialysis techniques, have suggested that a delay in insulin transfer from the plasma to the tissue may play an important role in the development of insulin resistance (427-429). Thus, impairment of insulin action may be secondary to a decrease in capillary density [chronic situations] or to a defective increase in blood flow or micro-capillary recruitment [acute conditions] (429). These abnormalities have been described in obese insulin-resistant and in the skin flow response of patients with diabetes.
Myocardial insulin resistance translates to abnormal intracellular signaling and reduced glucose oxidation rates in animal models of obesity (435). It adversely affects myocardial mechanical function and tolerance to ischemia and reperfusion. The heart is a dynamic organ that requires continuous energy in the form of ATP in order to meet contractile demands. This is achieved via a constant supply of blood-borne oxidizable substrates. The majority of ATP is derived from fatty acid oxidation [60-70%]. Glucose and lactate extracted from the circulation account for the remainder 30-40%. However, when blood glucose and insulin levels are elevated, such as immediately after a meal, glucose becomes the major fuel for myocardial oxidation and, it may represent up to 70% of the total substrate oxidation by cardio-myocytes. Long–chain fatty acids are taken up by the heart proportionately to circulating levels, via a passive facilitated transport. Once inside the cytosol, they are degraded into acetyl-CoA moieties that enter the mitochondrial oxidative phosphorylation process. The excess fatty acids are re-esterified to form diacyl- and triacyl-glycerides and, these lipid intermediates are stored in the form the myo-cellular lipid pool. Glucose enters the myocardial cells both via GLUT-1 passive and insulin-stimulated GLUT-4 active transport. These are dictated by myocardial contraction demands and circulating insulin levels. Intracellular glucose is phosphorylated and, either stored as muscle glycogen or anaerobically oxidized to pyruvate. Under normal oxygen delivery, pyruvate is converted to acetyl-CoA, which enters mitochondrial oxidation. In conditions of ischemia, low oxygen forces the conversion of pyruvate into lactate (435).
It is believed that myocardial insulin resistance with typical defects in glucose transport and oxidation develops, in part, because of an excess supply of fatty acids. In addition to a direct competition with glucose utilization, there is evidence that the accumulation of intracellular lipid intermediates interferes with insulin signaling. The molecular defects responsible for the insulin resistance in the cardio-myocytes are analogous to the skeletal muscle. The local generation of reactive oxygen species and other elements also participate in obstructing insulin action. Although the cellular and metabolic manifestations may be similar, the consequences of insulin resistance in the heart muscle tends to express with lower tolerance for ischemia and poor mechanical function. Consequently, patients with insulin resistance are susceptible to earlier and more severe cardiovascular complications.
Splanchnic (Hepatic) Glucose Uptake
In humans, it is difficult to catheterize the portal vein, and glucose disposal by the liver has not been examined directly. Using the hepatic vein catheterization technique in combination with the euglycemic insulin clamp, the contribution of the splanchnic (liver plus gastrointestinal) tissues to overall glucose homeostasis has been examined in lean subjects with T2DM with mild to moderate fasting hyperglycemia (3). In the post-absorptive state, there is a net release of glucose from the splanchnic area (i.e., negative balance) in both control and subjects with T2DM, reflecting glucose production by the liver. In response to insulin, splanchnic glucose output is promptly suppressed (reflecting the inhibition of HGP) and, by 20 min, the net glucose balance across the splanchnic region declines to zero (i.e., there was no net uptake or release) (3). After 2 h of sustained hyperinsulinemia, there is a small net uptake of glucose (~0.5 mg.kg- 1.min-1) by the splanchnic area (i.e., positive balance). This uptake is virtually identical to the rate of splanchnic glucose uptake observed in the basal state, indicating that the splanchnic tissues, like the brain, are insensitive to insulin at least with respect to the stimulation of glucose uptake (3,5,6,175). There was no difference between diabetic and control subjects for glucose taken up by the splanchnic tissues at any time during the insulin clamp study (3).
The results of these studies illustrate another important point: namely, that under conditions of euglycemic hyperinsulinemia, very little of the infused glucose is taken up by the splanchnic (and therefore hepatic) tissues (3,5,6,175). During the insulin clamp, the rate of whole-body glucose uptake averaged 7 mg.kg-1.min-1, and of this, only 0.5 mg.kg-1.min-1 or 7%, was disposed of by the splanchnic region. Because the difference in insulin-mediated total body glucose uptake between the T2DM and control groups during the euglycemic insulin clamp study was 2.5 mg.kg-1.min-1, from a purely quantitative standpoint it is obvious that a defect in splanchnic (hepatic) glucose removal never could account for the magnitude of impairment in total body glucose uptake following intravenous glucose/insulin administration. However, after glucose ingestion, the oral route of administration and the resultant hyperglycemia conspire to enhance splanchnic (hepatic) glucose uptake (6,7,11,12,16,26,175) and, under these conditions, diminished hepatic glucose uptake has been shown to contribute to the impairment in glucose tolerance in T2DM (see discussion below) (6,204,205).
Summary: Whole Body Glucose Utilization
Insulin-mediated whole body glucose utilization during the euglycemic insulin clamp represents essentially skeletal muscle glucose utilization. There is a noticeable decrease for glucose taken up in the body in T2DM patients compared with non-diabetic subjects. On the other hand, net splanchnic glucose uptake, quantitated by the hepatic venous catheterization technique, is similar in both groups and averaged 0.5 mg.kg-1.min-1. Adipose tissue glucose uptake accounts for less than 5% of total glucose disposal (17,198,199). Brain glucose uptake, estimated to be 1.0-1.2 mg.kg-1.min-1 in the post-absorptive state (2,169,206), is unaffected by hyperinsulinemia (169). Muscle glucose uptake (extrapolated from leg catheterization data) in control subjects accounts for ~75-80% of the total glucose uptake (1,3,4). In subjects with T2DM, the largest part of the impairment in insulin-mediated glucose uptake is accounted for by a defect in muscle glucose disposal. Even if adipose tissue of subjects with T2DM took up absolutely no glucose, it could, at best, explain only a small fraction of the defect in whole body glucose metabolism.
Glucose Disposal during OGTT
In everyday life, the gastrointestinal tract represents the normal route of glucose entry into the body. However, the assessment of tissue glucose disposal following glucose ingestion presents a challenge because of the difficulties in quantitating the rate of glucose absorption, suppression of hepatic glucose production, and organ (liver and muscle) glucose uptake. Moreover, because the plasma glucose and insulin concentrations are changing simultaneously, it is difficult to draw conclusions about insulin secretion or insulin sensitivity.
To address these issues, Ferrannini, DeFronzo, and colleagues (7,11,12,205) administered oral glucose to healthy control subjects in combination with hepatic vein catheterization to examine splanchnic glucose metabolism. The oral glucose load and endogenous glucose pool were labeled with [1-14C] glucose and [3-3H] glucose, respectively, to quantitate total body glucose disposal (from tritiated glucose turnover) and endogenous HGP (difference between the total rate of glucose appearance, as measured with tritiated glucose, and the rate of oral glucose appearance, as measured with [1-14C] glucose).
During the 3.5 h after glucose (68 g) ingestion: (i) 19 g, or 28%, or the oral load was taken up by splanchnic tissues; (ii) 48 g, or 72%, was disposed of by peripheral (non-splanchnic) tissues; (iii) of the 48 g taken up by peripheral tissues, the brain (an insulin-independent tissue) accounted for ~15 g (~1 mg.kg-1.min-1), or 22%, of the total glucose load (12); (iv) basal HGP declined by 53%. Similar percentages for splanchnic glucose uptake (24%-29%) and suppression of HGP (50%-60%) in normal subjects have been reported by other investigators (13,204,207-209). The contribution of skeletal muscle to the disposal of an oral glucose load has been reported to vary from a low of 26% (207) to a high of 56% (208), with a mean of 45% (11,13,207-209). These results emphasize several important differences between oral and intravenous glucose administration. After glucose ingestion: (i) EGP is less completely suppressed, most likely due to activation of local sympathetic nerves that innervate the liver (210); (ii) peripheral tissue (primarily muscle) glucose uptake is quantitatively less important; (3) splanchnic glucose uptake is quantitatively much more important.
In individuals with T2DM (12,204,205,211,212) the disposition of an oral glucose load is significantly altered. The disturbance in glucose metabolism is accounted for by two factors: (i) decreased tissue glucose uptake and (ii) impaired EGP suppression. Splanchnic glucose uptake is similar in diabetic and control groups. Inappropriate suppression of EGP accounted for nearly one-third of the defect in total-body glucose homeostasis, while reduced peripheral (muscle) glucose uptake accounted for the remaining two-thirds. Since hyperglycemia per se enhances splanchnic (hepatic) glucose uptake in proportion to the increase in plasma glucose concentration (24,175), the splanchnic glucose clearance (SGU/plasma glucose concentration) is markedly reduced in all subjects with T2DM following glucose ingestion. Using a combined insulin clamp/OGTT technique, impairment in glucose uptake by the splanchnic tissues in subjects with T2DM has been demonstrated directly (213).
The gastrointestinal incretin hormones, which are produced in response to nutrient intake and potentiate the stimulus to insulin secretion in the postprandial period have been implicated as additional factors in the pathogenesis of T2DM (4A,28-30). The combined actions of glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) can account for most of the incretin effect in normal subjects (4B). Recent demonstration that in T2DM the incretin effect is impaired, diminished or absent (4B) has rekindled interest in the potential role of these gastrointestinal peptides in the abnormal handling of glucose by splanchnic tissues and perhaps, in the decline in beta-cell insulin secretion.
When viewed in absolute terms, most studies have demonstrated that the total amount of glucose taken up by all tissues of body over the 4-hour period following the ingestion of an oral glucose load is normal (13) or slightly decreased (204,205,211). However, this occurs at the expense of postprandial hyperglycemia. Thus, the efficiency of glucose disposal, i.e., the glucose clearance (tissue glucose uptake/plasma glucose concentration), is severely reduced. It should be emphasized that it is not the absolute glucose disposal rate, but rather the increment in glucose disposal above baseline that determines the rise in plasma glucose concentration above the fasting value. Every published study (13,204,205,211) has demonstrated that the incremental response in whole-body glucose uptake is moderately to severely reduced in individuals with T2DM. Similar results have been reported for forearm muscle glucose uptake (13,201,202,208,209), pointing out the important contribution of diminished muscle glucose disposal to impaired oral glucose tolerance in T2DM.
In summary, results of the OGTT indicate that both impaired suppression of EGP and decreased tissue (muscle) glucose uptake contribute approximately equally to the glucose intolerance of T2DM. The efficiency of the splanchnic (hepatic) tissues to take up glucose (as reflected by the splanchnic glucose clearance) also is impaired in individuals with T2DM.
Summary of Insulin Resistance in T2DM
Insulin resistance involving both muscle and liver are characteristic features of the glucose intolerance in individuals with T2DM. In the basal state, the liver represents a major site of insulin resistance, and this is reflected by overproduction of glucose despite the presence of both fasting hyperinsulinemia and hyperglycemia. This accelerated rate of hepatic glucose output is the primary determinant of the elevated fasting plasma glucose concentration in T2DM. Although tissue (muscle) glucose uptake in the post-absorptive state is increased when viewed in absolute terms, the efficiency with which glucose is taken up (i.e., the glucose clearance) is diminished. After glucose infusion or ingestion (i.e., in the insulin stimulated state) both decreased muscle glucose uptake and impaired suppression of HGP contribute to the insulin resistance. Following glucose ingestion, the defects in insulin-mediated glucose uptake by muscle and the suppression of glucose production by insulin contribute approximately equally to the disturbance in whole-body glucose homeostasis in T2DM. However, under euglycemic hyperinsulinemic conditions, EPG is largely suppressed and impaired muscle glucose uptake is primarily responsible for the insulin resistance.
DYNAMIC INTERACTION BETWEEN INSULIN SENSITIVITY AND INSULIN SECRETION IN T2DM
Subjects with T2DM manifest abnormalities both in tissue (muscle, fat, and liver) sensitivity to insulin and in pancreatic insulin secretion. To understand how these two metabolic disturbances interact to produce the full-blown diabetic condition, it is necessary to quantitate insulin action and insulin secretion in the same individual over a wide range of insulin sensitivity. This dynamic interaction is demonstrated graphically by results obtained in healthy, lean, young normal glucose tolerant women who received a euglycemic insulin clamp (1 mU.kg-1.min-1) and were stratified into quartiles based upon the rate of insulin-mediated glucose disposal (49).
Insulin secretion was measured independently on a separate day with a +125 mg/dl hyperglycemic clamp. Insulin resistance and insulin secretion were strongly and positively correlated (r=0.79, p<0.001). Women who were the most insulin resistant (quartile 1) had the highest fasting plasma insulin concentrations and highest early and late phase plasma insulin responses. Similar results relating the plasma insulin response and the severity of insulin resistance have been reported in normal glucose tolerant subjects with the minimal model technique (46,47) and the insulin suppression test/oral glucose tolerance test (214).
A number of groups have examined the dynamic interaction between insulin secretion and insulin sensitivity in subjects with T2DM (1,4,34,35,38,39,42,46-48,58-61,150,162).
DeFronzo (4) studied lean (ideal body weight < 120%) and obese (ideal body weight > 125%) subjects with varying degrees of glucose tolerance as follows: Group I-obese subjects (n=24) with normal glucose tolerance; Group II-obese subjects (n=23) with impaired glucose tolerance; Group III-obese subjects (n=35) with overt diabetes, subdivided into those with a hyperinsulinemic response and those with a hypoinsulinemic response during a 100-gram OGTT; Group IV-normal weight subjects with T2DM (n=26); Group V-normal weight subjects (n=25) with normal glucose tolerance. All subjects ingested 100 g of glucose to provide a measure of glucose tolerance and insulin secretion. Whole-body insulin sensitivity was quantitated with the euglycemic insulin (~100 µU/ml) clamp technique, which was performed with indirect calorimetry to quantitate rates of glucose oxidation and non-oxidative glucose disposal. The later primarily reflects glycogen synthesis (215).
In normal weight subjects with T2DM, insulin-mediated whole-body glucose uptake was reduced by 40-50% and the impairment in insulin action resulted from defects in both oxidative and non-oxidative glucose metabolism (4). Obese individuals without T2DM were as insulin resistant as the normal-weight subjects with T2DM (4). Defects in both glucose oxidation and glucose storage contributed to the insulin resistance in the obese nondiabetic group. From the metabolic standpoint, therefore, obesity and T2DM closely resemble each other.
Similar results concerning reduced whole-body insulin sensitivity in individuals with obesity and T2DM have been reported by other investigators (160,161,166,216-218). Despite nearly identical degrees of insulin resistance, normal-weight subjects with T2DM manifested fasting hyperglycemia and marked glucose intolerance, whereas the obese individuals without diabetes had normal or only minimally impaired oral glucose tolerance (4). This apparent paradox is explained by the plasma insulin response during the OGTT. Compared with control subjects, the obese group without diabetes secreted more than twice as much insulin, and this was sufficient to offset the insulin resistance. In contrast, in normal-weight subjects with T2DM, the pancreas, when faced with the same challenge, was unable to augment its secretion of insulin sufficiently to compensate for the insulin resistance. This imbalance between insulin supply by the beta-cells and the insulin requirement by tissues resulted in a frankly diabetic state, with fasting hyperglycemia and marked glucose intolerance.
The fact that plasma insulin response to the development of insulin resistance typically is increased during the natural history of T2DM does not mean that the beta cell is functioning normally. To the contrary, recent studies (4C) have demonstrated that the onset of beta-cell failure occurs much earlier and is more severe than previously appreciated.
Recognizing that simply measuring plasma insulin response to a glucose challenge does not provide a valid index of beta cell function, a series of studies were conducted in subjects with normal glucose tolerance (NGT), impaired glucose tolerance (IGT) and T2DM, using an oral glucose tolerance test to evaluate the increment in insulin secretion in response to an increment in plasma glucose. A euglycemic insulin clamp to measure insulin sensitivity was also performed to address the adjustment of the beta cell to the body’s sensitivity to insulin.
Thus, the results yielded a better measure of beta-cell function expressed per increment of plasma glucose and corrected for the degree of insulin resistance, the so-called disposition index [ΔI/ΔG ÷IR]. These data revealed a substantial decrease in beta-cell function, most evident in individuals with IGT who had lost anywhere from 60 to 85% of the total insulin secretory capacity.
When obesity and diabetes coexist in the same individual, the severity of insulin resistance is only slightly greater than that in either the normal-weight diabetic or nondiabetic obese groups (4), and the magnitude of the defects in glucose oxidation and non-oxidative glucose disposal are similar in all obese and diabetic groups. Although hyperinsulinemic and hypoinsulinemic obese diabetic subjects were equally insulin resistant, the severity of glucose intolerance is worse in the hypoinsulinemic group, and this was related entirely to the presence of severe insulin deficiency.
In the obese nondiabetic subjects, tissue sensitivity to insulin is markedly reduced, but glucose tolerance remains perfectly normal because the beta cells are able to augment their insulin secretory capacity appropriately to offset the defect in insulin action. As the obese individual develops impaired intolerance, there is a further reduction in insulin-mediated glucose disposal, which is due primarily to a decrease in glycogen synthesis. However, there is only a small additional impairment in glucose tolerance, because the beta cells are able to further augment their secretion of insulin to counteract the deterioration in insulin sensitivity. The progression of the obese, glucose intolerant person to overt diabetes is heralded by a decline in insulin secretion without any worsening of insulin resistance. The obese diabetic has tipped over the top of Starling's curve of the pancreas and is now on the descending portion. Even though the plasma insulin response is increased compared to nondiabetic control subjects, it is not elevated appropriately for the degree of insulin resistance and there is evidence that there is ~80% of beta-cell functional loss by the time of diagnosis in diabetic subjects. The beta cell insulin response during the OGTT is best represented by the change in plasma insulin over the change in plasma glucose concentration, taking into consideration the degree of insulin resistance for each individual, the so-called Insulin Secretion /Insulin Resistance Index or Disposition Index as shown in Figure 1 below.
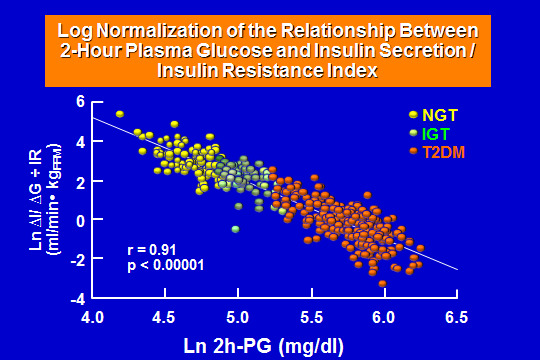
Figure 1
- Log normalization of the relationship between 2-hour plasma glucose and Insulin Secretion/ Insulin Resistance in subjects with normal glucose tolerance (NGT), impaired glucose tolerance (IGT) and patients with type 2 diabetes (T2DM). There is a linear decline in the insulin secretory capacity with the development of the disease, such that by the time clinical diabetes with hyperglycemia become evident, the loss of beta-cell secretion of insulin is below 5% of NGT controls.
The natural history of T2DM described above is consistent with results in humans and monkeys published by other investigators (33-39,42,43,59-61,98,150). In lean subjects with a wide range of glucose tolerance, Reaven et al (42) demonstrated that the progression from normal to impaired glucose tolerance was marked by the development of severe insulin resistance, which was counterbalanced by a compensatory increase in insulin secretion. The onset of T2DM was associated with no (or only slight) further deterioration in tissue sensitivity to insulin. Rather, insulin secretion declined and the impairment in beta cell function was paralleled by a decrease in glucose tolerance. A similar sequence of events has been documented prospectively in Pima Indians (34-39,58,60), in Caucasians (1,4,41,42,44,47,59, 162, 219), Pima Indians (34-39,58,60,219), and Pacific Islanders (33,62,220) and, is consistent with the development of T2DM in the rhesus monkeys (48). As monkeys grow older, they become obese and develop a diabetic condition closely resembling human T2DM. The earliest detectable abnormality in this primate model is a decrease in tissue sensitivity to insulin. Because of a compensatory increase in insulin secretion, the fasting plasma glucose concentration and glucose tolerance remain normal.
The studies detailed above indicate that insulin resistance is an early and characteristic feature of the natural history of T2DM in high-risk populations. Overt diabetes develops only in those individuals whose beta cells are unable to appropriately augment their secretion of insulin to compensate for the defect in insulin action. It should be recognized, however, that there are well-described populations with T2DM in whom insulin sensitivity is normal at the onset of diabetes, whereas insulin secretion is severely impaired (81-83). How frequently this occurs in a typical patient with T2DM remains to be determined. This insulinopenic variety of diabetes appears to be more common in African-Americans, elderly subjects, and in lean Caucasians. In this later group, it is important to exclude type 1 diabetes, since ~10% of Caucasians with older onset diabetes are islet cell antibody and/or GAD positive (220).
Primary Hypersecretion of insulin
An alternative view to explaining the “state of insulin resistance” is the notion that primary beta cell overstimulation results in insulin hypersecretion. This leads to the development of obesity and insulin resistance, and then, to beta cell exhaustion (436). In a model that presupposes beta cell hypersecretion as the initial manifestation of beta cell dysfunction, insulin sensitivity is modulated by insulin secretion. When beta cell hypersecretion occurs, the responsiveness of insulin-sensitive tissues to insulin is downregulated and, these tissues become insulin resistant. The latter becomes necessary to maintain normal glucose tolerance, without the adverse outcome of hypoglycemia. However, considering that beta cell hypersecretion is primary and ‘fixed’, when insulin sensitivity is acutely improved, hypoglycemia would be expected to ensue. In either case, the demonstration of the existence of a feedback loop that regulates glucose metabolism has made it clear that assessment of the adequacy of beta cell function requires knowledge of both the degree of insulin sensitivity and the magnitude of the insulin response.
When considering the feedback loop governing glucose metabolism, in the face of increased insulin secretion, insulin resistance should develop as a protective measure to maintain normal glucose concentrations without hypoglycemia. This is supported by observations in patients with insulinomas, in whom the risk of hypoglycemia is reduced by the downregulation of insulin action with the development of insulin resistance (437). Further support for this downregulation of insulin action comes from studies in healthy individuals with normal glucose tolerance in whom insulin resistance developed during 3–5 days of chronic physiologic hyperinsulinemia, achieved by insulin infusion balanced by glucose infusion to prevent hypoglycemia (438). Higher basal insulin levels have been documented in individuals with obesity and impaired glucose tolerance before the development of T2DM and, identified as a risk factor for diabetes. However, in these studies, OGTT glucose levels were already higher in those who progressed and could be a confounder. Thus, although studies provide some support for the concept of a potential independent pathogenic role of primary hyperinsulinemia in dysglycemia a stronger, more definitive proof is still missing.
Therefore, while it is clear that T2DM is a heterogeneous condition characterized by beta cell failure, whether beta cell dysfunction or primary hyperinsulinemia is the early event in the pathogenesis of dysglycemia is now up for debate. Although there is sufficient evidence in humans (and animal models) to support the principal defect as being early beta cell dysfunction associated with reduced insulin secretion, it is incumbent on the proponents of the primary hyperinsulinemia hypothesis to undertake further studies to make their case more forcefully. Improved understanding of whichever mechanism underlies beta cell dysfunction should allow us to provide better preventative and therapeutic interventions for T2DM.
Delayed Insulin Clearance in Diabetes
The role of delayed (or decreased) insulin clearance as a contributor to insulin resistance and to the development of T2DM has been studied. Insulin availability in the systemic circulation is determined by the rate of beta cell secretion and its rate of hepatic/peripheral/renal clearance. Insulin levels modulate expression and activity of the insulin receptors in target tissues, which ultimately determines insulin action. The main site of insulin clearance is the liver that removes approximately 50% of endogenous insulin with the remainder being cleared by the kidneys and the skeletal muscle. Receptor-mediated insulin endocytosis is the primary mechanism by which insulin is removed from the circulation and inactivated. Upon binding to its receptor, the insulin-receptor complex is internalized through the formation of clathrin-coated vesicles, and is delivered to the endosomes; the acidification of the endosomes then allows the dissociation of the hormone from its receptor and their sorting in different directions. Most of the internalized insulin is next targeted to lysosomes where it is degraded, whereas a smaller fraction remains intact. Both degradation products and intact insulin are segregated in recycling vesicles and released from cell. Defects in the intracellular processing of insulin have been reported in cells from insulin resistant individuals and reduced insulin clearance has been observed in individuals with IGT. More recently, it has also been demonstrated that reduced insulin clearance predicts the development of T2DM independently of confounding factors. There is evidence in animal model of fat-induced insulin resistance supporting the idea that decreased insulin clearance may serve as a compensatory mechanism to alleviate b-cell stress from excessive demand in these conditions of insulin resistance (439). The extent to which delayed insulin clearance is responsible for the advancement of insulin resistance and its role in the pathogenesis of T2DM remains unknown.
Nutrient-induced Stress on Insulin Secretion
There is growing support to the theory that an excess of calorigenic nutrients ingested over time presents the pancreatic islet beta-cells with an overwhelming burden, which might lead to toxic hormonal and metabolic adaptations. It is well recognized that the short-term effects of glucose, lipids and amino acids perfusing the beta cells in the endocrine pancreas include the stimulation of insulin biosynthesis and secretion. Excessive exposure to these nutrients is believed to over-stimulate the beta cells with a constant and uninterrupted demand for insulin release and, possibly induce changes in tissue insulin sensitivity. Chronically, abundant nutritional intake will trigger augmented insulin secretion and insulin resistance, both of which have been shown to contribute to the pathogenesis of T2DM. Eventually, there is altered glucose sensing and depletion of insulin stores. The de-differentiation, with beta cell death that follows is likely to play a role in the progression of the disease. Thus, the traditional concepts of “glucotoxicity” and lipotoxicity”, which defines the process of beta cell deterioration in response to chronic elevation of glucose and lipids in the pericellular milieu, has now been expanded to encompass all nutrients [‘nutri-toxicity”].
The biochemical mechanisms underlying beta cell adaptation and failure associated with “nutri-toxicity” are not entirely clear, but appear to be related to oxidative stress. Various pathways in the cytosol, endoplasmic reticulum [ER] and mitochondria are involved, which tend to affect the insulin secretory capacity of the beta cell. In conditions of mild-to-moderate “nutri-stress”, such as in overweight/obesity, there is exaggerated basal and nutrient stimulated insulin secretion. Slightly elevated blood glucose concentration, hyperinsulinemia and insulin resistance become progressively more evident. Obesity with beta cell failure and T2DM result when there is more advanced and prolonged nutri-stress”. The metabolic machinery of the beta cell is overwhelmed and, there is mitochondrial and ER dysfunction, which result in severe oxidative stress. As a consequence, insulin synthesis and secretion become impaired and there is intra- cellular accumulation of toxic metabolites with beta cell de-differentiation and death (440).
ROLE OF THE ADIPOCYTE IN THE PATHOGENESIS OF T2DM
The majority (>80%) of persons with T2DM in the US are overweight (221). Both lean and especially obese persons with T2DM are characterized by day-long elevations in the plasma free fatty acid concentration, which fail to suppress normally following ingestion of a mixed meal or oral glucose load (222). Free fatty acids (FFA) are stored as triglycerides in adipocytes and serve as an important energy source during conditions of fasting. Insulin is a potent inhibitor of lipolysis, and restrains the release of FFA from the adipocyte by inhibiting the enzyme hormone sensitive lipase. In patients with T2DM the ability of insulin to inhibit lipolysis (as reflected by impaired suppression of radioactive palmitate turnover) and reduce the plasma FFA concentration is markedly reduced (17). It is now recognized that chronically elevated plasma FFA concentrations can lead to insulin resistance in muscle and liver (1,4,19,21,22,51,162,223,224) and impair insulin secretion (22,225,226). Thus, elevated plasma FFA levels can cause/aggravate three major pathogenic disturbances that are responsible for impaired glucose homeostasis in individuals with T2DM and the "triumvirate" (muscle, liver, beta cell) was joined by the "fourth musketeer" (227) to form the "disharmonious quartet". In addition to FFA that circulate in plasma in increased amounts, individuals with T2DM and obese individuals without T2DM have increased stores of triglycerides in muscle (228,229) and liver (230,231) and the increased fat content correlates closely with the presence of insulin resistance in these tissues. Triglycerides in liver and muscle are in a state of constant turnover and the metabolites (i.e., fatty acyl CoAs) of intracellular FFAs have been shown to impair insulin action in both liver and muscle (1,4,92). This sequences of events has been referred to as "lipotoxicity" (1,4,22,93). Evidence also has accumulated to implicate "lipotoxicity" as an important cause of beta cell dysfunction (22,93) (see earlier discussion).
Adipocyte Inflammation and Insulin Resistance
Increased risk of developing T2DM is found in patients who have chronic, low-grade adipocyte inflammation and who are also insulin resistant (441). The mechanisms of adipose tissue inflammation and the related insulin-resistant state are complex. Visceral adiposity is known to be highly active in releasing numerous inflammatory cytokines [Adipokines] that are strongly implicated in the genesis of tissue insulin resistance and T2DM. Adipokines provide an important link between obesity and insulin resistance IR. Adiponectin is a unique adipokine that is inversely related to the metabolic syndrome, T2DM, and atherosclerosis. Adiponectin increases fatty acid oxidation while reducing glucose production in liver, and ablation of the adiponectin gene in mice induces insulin resistance and T2DM. Adiponectin is also anti-inflammatory; it suppresses tumor necrosis factor (TNF) actions in nonalcoholic fatty liver disease and inhibits nuclear factor kappa-beta [NFκB and monocyte adhesion to endothelial cells. Human resistin is an adipokine secreted by infiltrating inflammatory cells in human adiposity and can stimulate synthesis and secretion of other cytokines in adipocytes and endothelial cells. Leptin, a well-known adipokine, normally functions centrally to suppress appetite, but most obese patients are leptin resistant and have increased circulating leptin. In obesity, hyperleptinemia contributes to inflammation through modulation of T-cell and monocyte functions. A role for retinol-binding protein 4 [RBP-4], a more recently described adipokine has been proposed to be linked to inflammation.
Visfatin is a novel adipokine that is increased in obesity, is pro-inflammatory, and has an insulin-mimetic effect via binding to the insulin receptor. A member of the lipocalin family, lipocalin-2, also known as neutrophil gelatinase–associated lipocalin, modulates inflammation and is another adipokine that is elevated in the adipose tissue of obese mouse models and in the plasma of obese and insulin-resistant humans. In vitro studies suggest that lipocalin-2 induces insulin resistance in adipocytes and hepatocytes. The plasma level of another member of the lipocalin family, lipocalin-type prostaglandin D synthase, serves as a biomarker of coronary atherosclerosis. Thus, multiple adipose-secreted factors that are capable of impairing the cellular action of insulin have been suggested to be involved in the development of insulin resistance and facilitate the development of T2DM.
It should be recognized that nutritional fatty acids can modulate the inflammatory response, particularly via NFκB activity, and promote insulin resistance. Further-more, inflammatory modulation of adipocyte differentiation increases free fatty acid release. The mechanisms of free fatty acid-associated insulin resistance include protein kinase C (PKC) activation, endoplasmic reticulum stress, and increased oxidative burden. Free fatty acids also inhibit insulin receptor substrates [IRSs] and induce insulin resistance in skeletal muscle and liver. Increased fatty acid flux from adipose tissue to liver causes hepatic insulin resistance by increasing gluconeogenesis, glycogenolysis, and glucose-6-phosphatase expression and activity, and by enhancing lipogenesis and triglyceride synthesis attributable to activation of the transcription factor sterol-CoA regulatory element binding protein. Finally, free fatty acids cause endothelial insulin resistance and damage by impairing insulin and nitric oxide–dependent signaling, thus contributing to the vascular injury observed in adiposity.
The initial insult in obese individuals that triggers inflammation and systemic insulin resistance may occur through recruitment of macrophages and innate immune antigen activation of inflammatory receptors in the membrane. This can be perpetuated with secretion of chemokines, retention of macrophages in adipose, and secretion of adipokines. The inflammatory milieu induces adipocyte inflammatory cascades, such as the NFκB pathway, via activation of various kinases, and this modulates adipocyte transcription factors, attenuates insulin signaling, and increases the release of pro-inflammatory adipokines and free fatty acids. Inflammatory attenuation of adipocyte differentiation further exacerbates adipose dysfunction. These paracrine and endocrine adipose inflammatory events induce a systemic inflammatory and insulin-resistant state, favoring the development of T2DM.
FFA and Muscle Glucose Metabolism
Four decades ago, Randle (232) proposed that increased FFA oxidation restrains glucose oxidation in muscle by altering the redox potential of the cell and by inhibiting key glycolytic enzymes. The excessive FFA oxidation: (i) leads to the intracellular accumulation of acetyl CoA, a potent inhibitor of pyruvate dehydrogenase (PDH), (ii) increases the NADH/NAD ratio, causing a slowing of the Krebs cycle, and (iii) results in the accumulation of citrate, a powerful inhibitor of phosphofructokinase (PFK). Inhibition of PFK leads to the accumulation of glucose-6-phosphate (G-6-P) which in turn inhibits hexokinase II. The block in glucose phosphorylation causes a buildup of intracellular free glucose which restrains glucose transport into the cell via the GLUT4 transporter. The resultant decrease in glucose transport was postulated to account for the impairment in glycogen synthesis, although a direct inhibitory effect of fatty acyl Co-As on glycogen synthase also has been demonstrated (233). This sequence of events via which accelerated plasma FFA oxidation inhibits muscle glucose transport, glucose oxidation, and glycogen synthesis is referred to as the "Randle Cycle" (232). It should be noted that the same scenario would ensue if the FFA were derived from triglycerides stored in muscle (228,229) or from plasma (222).
Felber and coworkers (59,159,162,234,235) were amongst the first to demonstrate that in obese non-diabetic and diabetic humans, basal plasma FFA levels and lipid oxidation (measured by indirect calorimetry) are increased and fail to suppress normally after glucose ingestion. The elevated basal rate of lipid oxidation was strongly correlated with a decreased basal rate of glucose oxidation, as well as with reduced rates of glucose oxidation and non-oxidative glucose disposal (glycogen synthesis) following ingestion of a glucose load. Further validation of the Randle Cycle in man has come from studies employing the euglycemic insulin clamp. In normal subjects, physiologic hyperinsulinemia (80-100 μU/ml) causes a 60-70% decline in plasma FFA concentration and a parallel decline in plasma FFA and total body lipid oxidation (18). When Intralipid is infused concomitantly with insulin to maintain or increase the plasma FFA concentration/oxidation, both glucose oxidation and non-oxidative glucose disposal are inhibited in a dose dependent fashion (223). Using magnetic resonance imaging, it has been shown that the FFA-induced inhibition of non-oxidative glucose disposal reflects impaired glycogen synthesis (236). The inhibitory effect of elevated plasma FFA levels can be observed at all plasma insulin concentrations, spawning the physiologic and pharmacologic range (223).
The inhibitory effect of an acute elevation in plasma FFA concentration on muscle glucose metabolism is time dependent. Thus, the earliest (within 2 hours) observed abnormality is a defect in glucose oxidation (237), as would be predicted by operation of the Randle cycle (232). This is followed (between 2-3 hours) by defects in glucose transport and phosphorylation and eventually (after 3-4 hours) by impaired glycogen synthesis.
Biochemical/Molecular Basis of FFA-Induced Insulin Resistance
The original description of the Randle cycle was formulated based upon experiments performed in rat diaphragm and heart muscle (232). More recent studies performed in human skeletal muscle suggest that mechanisms in addition to those originally proposed by Randle are involved in the FFA-induced insulin resistance. Thus, several groups (236,238,239) have failed to observe a rise in muscle G-6-P and citrate concentrations when insulin-stimulated glucose metabolism was inhibited by an increase in the plasma FFA concentration. Elevated plasma FFA levels also failed to inhibit muscle phosphofructokinase activity. Thus, while increased FFA/lipid oxidation and decreased glucose oxidation are closely coupled, as originally demonstrated by Randle, mechanisms other than product (i.e., elevated intracellular G-6-P and free glucose concentrations) inhibition of the early steps of glucose metabolism must be invoked to explain the defects in glucose transport, glucose phosphorylation and glycogen synthesis.
Studies in humans and animals have shown a strong inverse correlation between insulin- stimulated glucose metabolism and increased intramuscular lipid pools, including triglyceride (240-242), diacyl-glycerol (DAG) (243,244), and long chain fatty acyl CoAs (FA-CoA) (245). An acute elevation in plasma FFA concentration leads to an increase in muscle fatty acyl CoA and DAG concentrations. Both long chain fatty acyl CoAs and DAG activate PKC theta (243), which increases serine phosphorylation with subsequent inhibition of IRS-1 tyrosine phosphorylation (246,247). Consistent with this observation, two groups have shown that in human muscle elevated plasma FFA levels inhibit insulin-stimulated tyrosine phosphorylation of IRS-1, the association of the p85 subunit of PI-3 kinase with IRS-1, and activation of PI-3-kinase (248,249). Direct effects of long chain fatty acyl CoAs on glucose transport (250), glucose phosphorylation (251), and glycogen synthase (233) also have been demonstrated in muscle. Lastly, increased muscle ceramide levels (secondary to increased long chain fatty acyl CoAs) have been shown to interfere with glucose transport and to inhibit glycogen synthase in muscle via activation of PKB (252). In summary, elevated plasma FFA concentrations can induce insulin resistance in muscle via multiple mechanisms involving alterations in a variety of intracellular lipid signaling molecules which exert their inhibitory effects on multiple steps (insulin signal transduction system, glucose transport, glucose phosphorylation, glycogen synthase, pyruvate dehydrogenase, Krebs cycle) involved in glucose metabolism.
Fatty Liver Disease in T2DM
As the epidemics of obesity increases worldwide in conjunction with T2DM, there is a parallel and proportionate increase in the prevalence of nonalcoholic fatty liver disease (NAFLD). A subtype of NAFLD, which can be characterized as nonalcoholic steato-hepatitis (NASH) is a potentially progressive liver disease that can lead to cirrhosis, hepatocellular carcinoma, liver transplantation, and death. NAFLD is also associated with extrahepatic manifestations such as chronic kidney disease, cardiovascular disease and sleep apnea. Despite this important burden, we are only beginning to understand its pathogenesis and the contribution of environmental and genetic factors to the risk of developing the progressive course of fatty liver disease. Of interest, however, despite the fact that the risk of liver-related mortality and the advancement to liver fibrosis are increased in patients with NAFLD, the leading cause of death is cardiovascular disease (442-443).
NAFLD and NASH are stages of fatty liver disease that are associated with obesity, insulin resistance, T2DM, hypertension, hyperlipidemia, and metabolic syndrome. In these individuals, a net retention of lipids within hepatocytes, mostly in the form of triglycerides, is a prerequisite for the development of fatty liver disease. The primary metabolic abnormality leading to lipid accumulation (steatosis), however, is not well understood, but it could potentially result from insulin resistance and alterations in the uptake, synthesis, degradation or secretory pathways of hepatic lipid metabolism. Insulin resistance represents the most reproducible factor in the development of fatty liver disease. There is also some evidence that lipids synthesis de novo”, a process derived from excess non-utilized carbohydrates accumulated in hepatocytes contributes to the intracellular lipid pool. Once an excessive amount of lipids accumulate inside the hepatocytes, a steatotic liver develops. This makes the cellular architecture of the liver vulnerable to further injury, when challenged by additional insults. There is a presumption that progression from simple, uncomplicated steatosis to steato-hepatitis to advanced fibrosis results from two operating “hits” due to: i) insulin resistance with further accumulation of fat within hepatocytes, and ii) generation of reactive oxygen species due to lipid peroxidation with cytokine production and Fas ligand induction. The oxidative stress and lipid peroxidation are key factors in the development and progression from steatosis to more advanced stages of liver damage. In addition, this sequence of events reflects similar systemic processes, which worsen tissue insulin resistance with impairment of insulin secretion and accelerated atherogenesis, related primarily to the pro-inflammatory state (442).
FFA and Blood Flow
Insulin is a vaso-dilatory hormone and the stimulatory effect of insulin on muscle glucose metabolism has been shown to result from: (i) a direct action of insulin to augment muscle glucose metabolism, and (ii) increased blood flow to muscle (253,254). The vaso-dilatory effect of insulin is mediated via the release of nitric oxide from the vascular endothelium (255). In insulin resistant conditions, such as obesity and T2DM, some investigators have suggested that as much as half of the impairment in insulin-mediated whole body and leg muscle glucose uptake is related to a defect in insulin's vaso-dilatory action (253,254), although the link between insulin-mediated vasodilation and increased blood flow, as well as the underlying mechanisms have been challenged by others (256, 256A). More recent studies employed contrast-enhanced ultrasonography using 1-methyl-xantine to demonstrate that insulin infusion promotes capillary recruitment in healthy individuals. These data have suggested that there is a time-dependent effect of insulin on regional blood flow redistribution with capillary pre-sphincter relaxation preceding vasodilation and consequent increase in skeletal muscle glucose metabolism (256B). These observations also provided a partial explanation for the discrepant findings reported on the topic of insulin, fatty acids and vasodilatation.
Because T2DM and obesity are insulin resistant states characterized by day-long elevation in the plasma FFA concentration (222) and impaired endothelium dependent vasodilation (253), investigators have examined the effect of increased plasma FFA levels on limb blood flow and muscle glucose uptake (257,258). In healthy, non-diabetic subjects an acute physiologic increase in plasma FFA concentration inhibited metha-choline (endothelium dependent) but not nitroprusside (endothelium independent) stimulated blood flow in association with an impairment in insulin-stimulated muscle glucose disposal. In subsequent studies, the inhibitory effect of FFA on insulin-stimulated leg blood flow was shown to be associated with decreased nitric oxide availability (259). FFA elevation also inhibits nitric oxide production in endothelial cell cultures by decreasing nitric oxide synthase activity (259). Since the IRS-1/PI-3 kinase signal transduction pathway is involved in the regulation of nitric oxide synthase activity (260), one could hypothesize that FFA-induced inhibition of the insulin signal transduction pathway is responsible for the blunted vaso-dilatory response to the hormone.
FFA and Hepatic Glucose Metabolism
The liver plays a pivotal role in the regulation of glucose metabolism (1,4,6,11,16,205). Following carbohydrate ingestion, the liver suppresses its basal rate of glucose production and takes up approximately one-third of the glucose in the ingested meal (12,24,25,205).
Collectively, suppression of glucose production and augmentation of hepatic glucose uptake account for the maintenance of nearly one-half of the rise in plasma glucose concentration following ingestion of a carbohydrate meal. Hepatic glucose production is regulated by a number of factors, of which insulin (inhibits) and glucagon and FFA (stimulate) are the most important. In vitro studies have demonstrated that plasma FFA are potent stimulators of endogenous glucose production and do so by increasing the activity of pyruvate carboxylase and phosphoenolpyruvate carboxy-kinase, the rate limiting enzymes for gluconeogenesis (261,262). FFA also enhances the activity of glucose-6- phosphatase, the enzyme that ultimately controls the release of glucose by the liver (263).
In normal subjects, increase plasma FFA levels stimulate gluconeogenesis (264,265), while a decrease in plasma FFA concentration reduces gluconeogenesis (264). It has been shown that a significant portion of the suppressive effect of insulin on hepatic glucose production is mediated via inhibition of lipolysis and a reduction in circulating plasma FFA concentrations (16,266,267). Moreover, FFA infusion in normal humans under conditions that simulate the diabetic state (268) and in obese insulin-resistant subjects (269) enhances hepatic glucose production, most likely secondarily to stimulation of gluconeogenesis. In subjects with T2DM, the fasting plasma FFA concentration and lipid oxidation rate are increased and are strongly correlated with both the elevated fasting plasma glucose concentration and basal rate of hepatic glucose production (18,51,59,162,190,270). The relationship between elevated plasma FFA concentration, FFA oxidation, and hepatic glucose production in obesity and T2DM is explained as follows: (i) increased plasma FFA levels, by mass action, augment FFA uptake by hepatocytes, leading to accelerated lipid oxidation and accumulation of acetyl CoA. The increased concentration of acetyl CoA stimulates pyruvate carboxylase, the rate limiting enzyme in gluconeogenesis (261,262), as well as glucose-6-phosphatase, the rate-controlling enzyme for glucose release from the hepatocyte (263); (ii) the increased rate of FFA oxidation provides a continuing source of energy (in the form of ATP) and reduced nucleotides (NADH) to drive gluconeogenesis; (iii) elevated plasma FFA induce hepatic insulin resistance by inhibiting the insulin signal transduction system (244- 248). In patients with T2DMthese deleterious effects of elevated plasma FFA concentrations occur in concert with increased plasma glucagon levels (181,190,271), increased hepatic sensitivity to glucagon, and increased hepatic uptake of circulating gluconeogenic precursors.
The Role of Gut Microbiome
Recently the potential role of the gut microbiome in metabolic disorders such as obesity and T2DM has been identified (444). Obesity is associated with changes in the composition of the intestinal microbiota, and the obese microbiome seems to be more efficient in harvesting energy from the diet. Lean male donor fecal microbiota transplantation (FMT) in males with the metabolic syndrome resulted in a significant improvement in insulin sensitivity in conjunction with an increased intestinal microbial diversity, including a distinct increase in butyrate-producing bacterial strains. Such differences in gut microbiota composition might function as early diagnostic markers for the development of T2DM in high-risk patients. Products of intestinal microbes such as butyrate may induce beneficial metabolic effects through enhancement of mitochondrial activity, prevention of metabolic endotoxemia, and activation of intestinal gluconeogenesis via different routes of gene expression and hormone regulation. There is currently an enormous effort in trying to better understand, amongst other things, whether bacterial products (like butyrate) have the same effects as the intestinal bacteria that produce it, in order to ultimately pave the way for more successful interventions for obesity and T2DM. Rapid development of the currently available techniques, including the use of fecal transplantations, has already shown promising results, so there is hope for novel therapies based on the microbiota in the future.
Summary: FFA and the Pathogenesis of Obesity and T2DM
n obese individuals and in the majority (>80%) of subjects with T2DM, there is an expanded fat cell mass and the adipocytes are resistant to the anti-lipolytic effects of insulin (18). Most individuals with obesity or T2DM are characterized by visceral adiposity (272) and visceral fat cells have a high lipolytic rate, which is especially refractory to insulin (273). Not surprisingly, both T2DM and obesity are characterized by an elevation in the mean day-long plasma FFA concentration. Elevated plasma FFA levels, as well as increased triglyceride/fatty acyl CoA content in muscle, liver, and beta cell, lead to the development of muscle/hepatic insulin resistance and impaired insulin secretion.
THE OMNIOUS OCTET
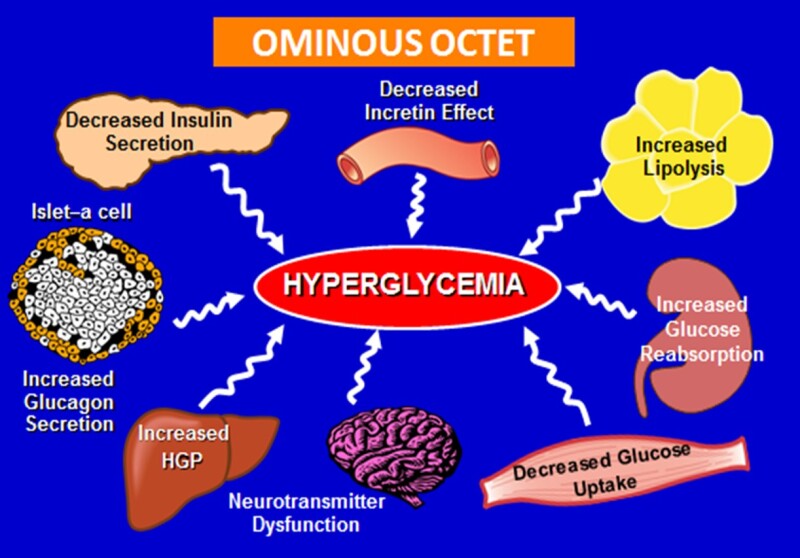
Figure 2.
Summary of the Eight Principal Mechanisms Contributing to Hyperglycemia in Patients with Type 2 Diabetes
The eight principle known causes leading to hyperglycemia through the pathogenesis of T2DM are summarized in Figure 2. It is already established that decreased peripheral glucose uptake combined with augmented endogenous (hepatic) glucose production are characteristic features of insulin resistance. Increased lipolysis with accumulation of intermediary lipid metabolites contributes to further enhance glucose output while reducing peripheral utilization. Compensatory insulin secretion by the pancreatic beta-cells eventually reaches a maximum and, then it progressively deteriorates. Concomitantly, there is inappropriate release of glucagon from the pancreatic alpha-cells, particularly in the post- prandial period. It has been postulated that both impaired insulin and excessive glucagon secretion in T2DM are facilitated by the “incretin defect”, defined primarily as inadequate response of the gastrointestinal “incretin” hormones to meal ingestion in addition to islet-cell resistance to the potentiating action on insulin-secretion by these gastrointestinal peptides. Moreover, considering that hypothalamic insulin resistance (central nervous system) with an elevated sympathetic drive, typically seen in patients with T2DM also impair the ability of circulating insulin to suppress glucose production. The fact that renal tubular glucose reabsorption capacity is enhanced in diabetic patients also contributes to the development and maintenance of chronic hyperglycemia. Thus, the time has arrived to advance the concept from the “triumvirate” to the “omnious octet” (4A). Further, recent observations have recognized that a chronic low-grade inflammation with activation of the immune system are involved in the pathogenesis of obesity-related insulin resistance and T2DM (4D). Adipose tissue, liver, muscle and pancreas are themselves sites of inflammation in presence of obesity. Infiltration of macrophages and other immune cells as well as the presence of pro-inflammatory cytokines in these tissues has been associated with insulin resistance and beta-cell impairment. The possibility that endothelial dysfunction and changes in vascular capillary permeability affect peripheral insulin action has also been raised (4E). These pathogenic mechanisms must be taken into account when deciding for the treatment of hyperglycemia in patients with T2DM.
CELLULAR MECHANISMS OF INSULIN RESISTANCE
The stimulation of glucose metabolism by insulin requires that the hormone must first bind to specific receptors that are present on the cell surface of all insulin target tissues (1,274-277). After insulin has bound to and activated its receptor, "second messengers" are generated and these second messengers initiate a series of events involving a cascade of phosphorylation- de-phosphorylation reactions (1,274-280) that eventually result in the stimulation of intracellular glucose metabolism. The initial step in glucose metabolism involves activation of the glucose transport system, leading to influx of glucose into insulin target tissues, primarily muscle (1,281,282). The free glucose, which has entered the cell, subsequently is metabolized by a series of enzymatic steps that are under the control of insulin. Of these, the most important are glucose phosphorylation (catalyzed by hexokinase), glycogen synthase (which controls glycogen synthesis), and phosphofructokinase (PFK) and PDH (which regulate glycolysis and glucose oxidation, respectively).
Insulin Receptor/Insulin Receptor Tyrosine Kinase
The insulin receptor is a glycoprotein consisting of two alpha subunits and two beta subunits linked by disulfide bonds (1,274-277). The alpha subunit of the insulin receptor is entirely extracellular and contains the insulin-binding domain. The beta subunit has an extracellular domain, a transmembrane domain, and an intracellular domain that expresses insulin- stimulated kinase activity directed towards its own tyrosine residues (1,274-277). Insulin receptor phosphorylation of the beta subunit, with subsequent activation of insulin receptor tyrosine kinase, represents the first step in the action of insulin on glucose metabolism (274- 277). Mutagenesis experiments have shown that insulin receptors devoid of tyrosine kinase activity are completely ineffective in mediating insulin stimulation of cellular metabolism (283,284). Similarly, mutagenesis of any of the three major phosphorylation sites (at residues 1158, 1163, and 1162) impairs insulin receptor kinase activity, resulting in a decrease in the acute metabolic and growth promoting effects of insulin (283,285).
Insulin Receptor Signal Transduction
Following activation, insulin receptor tyrosine kinase phosphorylates specific intracellular proteins, of which at least nine have been identified (282). Four of these belong to the family of insulin-receptor substrate proteins: IRS-1, IRS-2, IRS-3, IRS-4 (the others include Shc, Cbl, Gab-1, p60dok, and APS). In muscle IRS-1 serves as the major docking protein that interacts with the insulin receptor tyrosine kinase and undergoes tyrosine phosphorylation in regions containing amino acid sequence motifs (YXXM or YMXM). When phosphorylated, these serve as recognition sites for proteins containing src-homology 2 (SH2) domains (where y = tyrosine, M = methionine, and x - any amino acid) (274,275). Mutation of these specific tyrosines severely impairs the ability of insulin to stimulate glycogen synthesis and DNA synthesis, establishing the important role of IRS-1 in insulin signal transduction (282). In liver, IRS-2 serves as the primary docking protein that undergoes tyrosine phosphorylation and mediates the effect of insulin on hepatic glucose production, gluconeogenesis and glycogen formation (287). In adipocytes, Cbl represents another substrate which is phosphorylated following its interaction with the insulin receptor tyrosine kinase, which is required for stimulation of GLUT 4 translocation.
Phosphorylation of Cbl occurs when the CAP/Cbl complex associates with flotillin in caveolae, or lipid rafts, containing insulin receptors (288,289).
In muscle, the phosphorylated tyrosine residues on IRS-1 mediate an association between the two SH2 domains of the 85-kDa regulatory subunit of phosphatidylinositol 3-kinase (PI3-kinase), leading to activation of the enzyme (274-284,290,291). PI3-kinase is a heterodimeric enzyme comprised of an 85-kDa regulatory subunit and a 110-kDa catalytic subunit. The latter catalyzes the 3-prime phosphorylation of phosphatidylinositol (PI), PI-4-phosphate, and PI-4,5- diphosphate, resulting in the stimulation of glucose transport (274-277). Activation of PI3-kinase by phosphorylated IRS-1 also leads to activation of glycogen synthase (274,275), via a process that involves activation of PKB/Akt and subsequent inhibition of kinases such as GSK-3 (292) and activation of protein phosphatase 1 (PP1) (293). Inhibitors of PI3-kinase impair glucose transport (274-277,294) by interfering with the translocation of GLUT 4 transporters from their intracellular location (281,282) and block the activation of glycogen synthase (295) and hexokinase (HK)-II expression (296). The action of insulin to increase protein synthesis and inhibit protein degradation also is mediated by PI-3 kinase and involves the activation of mTOR (297,298). mTOR controls translation machinery by phosphorylation and activation of p70 ribosomal S6 kinase (p70rsk) (297) and phosphorylation of initiation factors (299). Insulin also promotes hepatic triglyceride synthesis via increasing the transcription factor steroid regulatory element-binding protein (SREBP)-1c (300), and this lipogenic effect of insulin also appears to be mediated via the PI3-kinase pathway (274).
Other proteins with SH2 domains, including the adapter protein Grb2 and Shc, also interact with IRS-1 and become phosphorylated following exposure to insulin (274-276,301). Grb2 and Shc serve to link IRS-1/IRS-2 to the mitogen-activated protein (MAP) signaling pathway, which plays an important role in the generation of transcription factors (274,275). Following the interaction between IRS-1/IRS-2 and Grb2 and Shc, Ras is activated, leading to the stepwise activation of Raf, MEK, and ERK. Activated ERK then translocates into the nucleus of the cell, where it catalyzes the phosphorylation of transcription factors. These promote cell growth, proliferation, and differentiation (274-276,301-303). Blockade of MAP kinase pathway prevents stimulation of cell growth by insulin but has no effect on the metabolic actions of the hormone (304-306).
Under anabolic conditions insulin stimulates glycogen synthesis by simultaneously activating glycogen synthase and inhibiting glycogen phosphorylase (307-309). The effect of insulin is mediated via the PI3 kinase pathway which inactivates kinases, such as glycogen synthase kinase-3 and activates phosphatases, particularly protein phosphatase 1 (PP1). It is believed that PP1 is the primary regulator of glycogen metabolism (307-310). In skeletal muscle, PP1 associates with a specific glycogen-binding regulatory subunit, causing the activation [de-phosphorylation] of glycogen synthase; PP1 also inactivates [phosphorylates] glycogen phosphorylase. The precise steps that link insulin receptor tyrosine kinase/PI 3-kinase activation to the stimulation of PP1 have yet to be defined. Some evidence suggests that p90 ribosomal S6- kinase may be involved in the activation of glycogen synthase (274). Akt also has been shown to phosphorylate and thus inactivate GSK-3 (292). This decreases glycogen synthase phosphorylation, leading to the enzyme activation (292). A number of studies have convincingly demonstrated that inhibitors of PI3-kinase also inhibit glycogen synthase activity and abolish glycogen synthesis (274,293,310). From the physiological standpoint, it makes sense that activation of glucose transport and glycogen synthase should be linked to the same signaling mechanism to provide a coordinated stimulation of intracellular glucose metabolism.
Insulin Signal Transduction Defects in T2DM
Both receptor and post-receptor defects have been shown to contribute to insulin resistance in individuals with T2DM. Some, but not all studies have demonstrated a modest 20-30% reduction in insulin binding to monocytes and adipocytes from patients with T2DM (1,311- 316). This reduction is due to a decreased number of insulin receptors without change in insulin receptor affinity. In addition to the decreased number of cell-surface receptors, a variety of defects in insulin receptor internalization and processing have been described (314,315).
However, some caution should be employed in interpreting these studies. Muscle and liver, not adipocytes, represent the major tissues responsible for the regulation of glucose homeostasis in vivo and insulin binding to solubilized receptors obtained from skeletal muscle biopsies and liver has been shown to be normal in obese and lean diabetic individuals when expressed per milligram of protein (312,313,316-318). Moreover, a decrease in insulin receptor number cannot be demonstrated in over half of subjects with T2DM (319,320), and it has been difficult to demonstrate a correlation between reduced insulin binding and the severity of insulin resistance (321,322). The insulin receptor gene has been sequenced in a large number of patients with T2DM from diverse ethnic populations using denaturing-gradient gel electrophoresis or single- stranded conformational polymorphism analysis, and, with very rare exceptions (323), physiologically significant mutations in the insulin receptor gene have not been observed (324,325). This excludes a structural gene abnormality in the insulin receptor as a cause of common T2DM.
Insulin receptor tyrosine kinase activity has been examined in a variety of cell types (skeletal muscle, adipocytes, hepatocytes, and erythrocytes) from normal-weight and obese diabetic subjects. Most (278,301,312,313,320,326-328), but not all (317,329) investigators have found reduced tyrosine kinase activity that cannot be explained by alterations in insulin receptor number or insulin receptor binding. However, near-normalization of the fasting plasma glucose concentration, (by weight loss) has been reported to correct the defect in insulin receptor tyrosine kinase activity (330). This observation suggests that the defect in tyrosine kinase is acquired and results from some combination of hyperglycemia, defective intracellular glucose metabolism, hyperinsulinemia, and insulin resistance - all of which improved after weight loss. A glucose-induced reduction in insulin receptor tyrosine kinase activity has been demonstrated in rat fibroblast culture in vitro (331). Insulin receptor tyrosine kinase activity assays are performed in vitro, and the results of these assays could provide misleading information with regard to insulin receptor function in vivo. To circumvent this problem, investigators have employed the euglycemic hyperinsulinemic clamp in combination with muscle biopsies and anti- phospho-tyrosine immunoblot analysis (301). Such analysis yields a "snap shot" of the insulin- stimulated tyrosine phosphorylation state of the receptor in vivo. The results of these studies have demonstrated a substantial decrease in insulin receptor tyrosine phosphorylation in both obese nondiabetic and subjects with T2DM (301,328). When insulin-stimulated insulin receptor tyrosine phosphorylation was examined in normal-glucose-tolerant or impaired- glucose-tolerant individuals at high risk of developing T2DM, a normal increase in tyrosine phosphorylation of the insulin receptor has been observed (332). These observations are consistent with the concept that impaired insulin receptor tyrosine kinase activity in patients with T2DM is acquired secondarily to hyperglycemia or some other metabolic disturbance.
A physiologic increase in the plasma insulin concentration stimulates tyrosine phosphorylation of the insulin receptor and IRS-1 in lean healthy subjects to 150-200% of basal values (280,301,328,332,333). In obese subjects without T2DM, the ability of insulin to activate these two early insulin receptor signaling events in muscle is reduced, while in subjects with T2DM insulin has no significant stimulatory effect on either insulin receptor or IRS-1 tyrosine phosphorylation (301). The association of p85 protein and PI3-kinase activity with IRS-1 also is greatly reduced in obese non-diabetic and subjects with T2DM compared to lean healthy subjects (301,328- 334). Insulin also failed to increase the association of the p85 subunit of PI3-kinase with IRS-2 in muscle, indicating that T2DM is characterized by a combined defect in IRS-1 and IRS-2 function (301,328). The decrease in insulin stimulation of the association of the p85 regulatory subunit of PI3-kinase with IRS-1 is closely correlated with the impairment in muscle glycogen synthase activity and in vivo insulin-stimulated glucose disposal (301). Defective regulation of PI3-kinase gene expression by insulin also has been demonstrated in skeletal muscle and adipose tissue of subjects with T2DM (335). In animal models of diabetes, an 80% decrease in IRS-1 phosphorylation and a greater than 90% reduction in insulin-stimulated PI3-kinase activity have been reported (336).
In the insulin resistant, normal glucose tolerant offspring of two parents with T2DM, IRS-1 tyrosine phosphorylation and the association of p85 protein/PI3-kinase activity with IRS-1 are markedly decreased despite normal tyrosine phosphorylation of the insulin receptor; these insulin signaling defects are correlated closely with the severity of insulin resistance, measured with the euglycemic insulin clamp technique (332). In summary, a defect in the association of PI3-kinase with IRS-1 and its subsequent activation appears to be a characteristic abnormality in T2DM, is closely correlated with in vivo muscle insulin resistance, and is unrelated to a disturbance in insulin receptor tyrosine phosphorylation. Several groups (337,338) have reported that a common mutation in the IRS-1 gene (Gly 972 Arg) is associated with T2DM, insulin resistance, and obesity, but the physiologic significance of this mutation remains to be established (339).
The profound insulin resistance of the PI3-kinase signaling pathway contrasts markedly with the ability of insulin to stimulate MAP kinase pathway activity in insulin-resistant individuals with T2DM and in individuals with obesity without T2DM (301,328). Hyperinsulinemia increases MEK1 activity and ERK1/2 phosphorylation and activity to the same extent in lean healthy individuals as in patients with insulin resistance and obesity without T2DM and patients with T2DM (301,328). This finding of selective insulin resistance is similar to that recently observed in vasculature of Zucker fatty rats (340). Two possible reasons for this difference are alternate insulin signaling pathways and differential signal amplification. With regard to the former, the MAP kinase pathway can be activated either through Grb2/Sos interaction with IRS-1/IRS-2 or with Shc. Because IRS-1 tyrosine phosphorylation is dramatically reduced in the diabetics, it is possible that insulin activation of the MAP kinase pathway in vivo primarily occurs through Shc activation. There is evidence from in vitro studies to support this concept (341). Like ERK and MEK activity, insulin increased Shc phosphorylation to the same extent in lean and obese nondiabetic and subjects with T2DM (301). These results indicate that, in T2DM, insulin induces sufficient activation of the insulin receptor tyrosine kinase to increase Shc phosphorylation normally. It also is possible that differential signal amplification in the PI3-kinase and MAP kinase pathways can explain their differing susceptibilities to the effects of insulin resistance.
Maintenance of insulin stimulation of the MAP kinase pathway in the presence of insulin resistance in the PI3-kinase pathway may be important in the development of insulin resistance. ERKs can phosphorylate IRS-1 on serine residues (342), and serine phosphorylation of IRS-1 and the insulin receptor itself has been implicated in de-sensitization insulin receptor signaling (343). Continued ERK activity, when IRS-1 function already is impaired, could lead to a worsening of insulin resistance. Thus, subjects with T2DM or obesity have inappropriately high MAP kinase activity. One also could postulate that insulin resistance in the metabolic (PI3- kinase) pathway, with its compensatory increase in beta cell function and hyperinsulinemia, leads to excessive stimulation of the MAP kinase pathway in vascular tissue (301,302). This would result in the proliferation of vascular smooth muscle cells, increased collagen formation, and increased production of growth factors and inflammatory cytokines, possibly explaining the accelerated rate of atherosclerosis in individuals with T2DM (340A, 340B).
Glucose Transport
Activation of the insulin signal transduction system in insulin target tissues leads to the stimulation of glucose transport. The effect of insulin is brought about by the translocation of a large intracellular pool of glucose transporters (associated with low-density microsomes) to the plasma membrane (281,282,344). There are five major, different facilitative glucose transporters with distinctive tissue distributions (281,282,345,346) (Table 1). GLUT4, the transporter regulated by insulin is found in insulin-sensitive tissues (muscle and adipocytes), has a Km of ~5 mmol/l, which is close to that of the plasma glucose concentration, and is associated with HK-II (347- 349). In adipocytes and muscle, its concentration in the plasma membrane increases markedly after exposure to insulin, and this increase is associated with a reciprocal decline in the intracellular GLUT4 pool. GLUT1 represents the predominant glucose transporter in the insulin- independent tissues (brain and erythrocytes), but also is found in muscle and adipocytes. It is located primarily in the plasma membrane, where its concentration changes little after the addition of insulin. It has a low Km (~1 mmol/l) and is well suited for its function, which is to mediate basal glucose uptake. It is found in association with HKI (347-349). GLUT2 predominates in the liver and pancreatic beta-cells, where it is found in association with a specific hexokinase, HKIV (347-350). In the beta-cell, HKIV is referred to as gluco-kinase (350,351). GLUT2 has a high Km, (~15-20 mmol/l) and, as a consequence, the glucose concentration in cells expressing this transporter rises in direct proportion to the increase in plasma glucose concentration. This characteristic allows these cells to respond as glucose sensors. In summary, each tissue has a specific glucose transporter and associated hexokinase, which allows it uniquely to carry out its specialized function to maintain whole-body glucose economy.
Table 1.
Classification of Glucose Transport and HK Activity According to their Tissue Distribution and Functional Regulation
Glucose transport activity in patients with T2DM uniformly has been found to be decreased in adipocytes (281,282,320,351,352) and muscle (281,282,354-356). In adipocytes from humans with T2DM and rodent models of diabetes, there is a severe reduction in GLUT4 mRNA and protein, and the ability of insulin to elicit a normal translocation response and to activate the GLUT4 transporter after its insertion into the cell membrane is impaired (281,282,320,353,357). In contrast, muscle tissue obtained from lean and obese subjects with T2DM exhibits normal or increased levels of GLUT4 mRNA expression and normal levels of GLUT4 protein (358-361). Moreover, acute (2- 4-h) physiological hyperinsulinemia does not increase the number of GLUT4 transporters in muscle in either healthy subjects or subjects with T2DM (358-361). Several studies have demonstrated an increase in muscle GLUT4 mRNA levels in response to insulin in control subjects (333,360), but not in subjects with T2DM (360), suggesting insulin resistance at the level of gene transcription. However, the physiological significance of the blunted increase in muscle GLUT4 mRNA levels in subjects with T2DM is unclear, since both basal and insulin- stimulated GLUT4 protein levels are normal. Large populations of subjects with T2DM have been screened for mutations in the GLUT4 gene (362,363). Such mutations are very uncommon and, when detected, have been of questionable physiologic significance.
The results summarized above indicate that the gene (GLUT4) encoding the major insulin- responsive glucose transporter and its transcriptional/translational regulation are not impaired in T2DM. However, in contrast to the normal expression of GLUT4 protein and mRNA in muscle of subjects with T2DM, every study that has examined adipose tissue has reported reduced basal and insulin-stimulated GLUT4 mRNA levels, decreased GLUT4 transporter number in all subcellular fractions, diminished GLUT4 translocation, and impaired intrinsic activity of GLUT4 (281,282,353,361,364). These observations demonstrate that GLUT4 expression in humans is subject to tissue-specific regulation. Although insulin does not increase GLUT4 expression in muscle, it stimulates the translocation of GLUT4 transporters from their intracellular location to the cell membrane (354,365,366). In humans with T2DM, the ability of insulin to stimulate GLUT4 translocation in muscle is impaired (354,367). Using a novel triple- tracer technique, the in vivo dose-response curve for the action of insulin on glucose transport in forearm skeletal muscle has been examined in nondiabetic and subjects with T2DM (368-370). Insulin-stimulated inward muscle glucose transport is severely impaired in subjects with T2DM who are studied under euglycemic conditions. The defect in glucose transport cannot be overcome by repeating the insulin clamp at each subject's normal fasting glucose (hyperglycemia) level. Since the number of GLUT4 transporters in the muscle of subjects with T2DM is normal (358-361), impaired GLUT4 translocation (281,354,367) and decreased intrinsic activity of the glucose transporter (366,371) must be responsible for the defect in muscle glucose transport. Impaired in vivo muscle glucose transport in T2DM also has been demonstrated using MRI (372) and PET (373).
Glucose Phosphorylation
Glucose phosphorylation and glucose transport are tightly coupled phenomena (374). Isozymes of hexokinase (HKI-HKIV) catalyze the first committed intracellular step of glucose metabolism, the conversion of glucose to glucose-6-phosphate (G-6-P) (347-350,375) (Table 1). HKI, HKII, and HKIII are single-chain peptides that have a number of properties in common, including a very high affinity for glucose and product inhibition by G-6-P. HKIV, also called gluco-kinase, has a lower affinity for glucose and is not inhibited by G-6-P. Gluco-kinase (HKIVB) is believed to be the glucose sensor in the beta-cell, while HKIVL plays an important role in the regulation of hepatic glucose metabolism.
In both rat (375-377) and human (333,348,378-380) skeletal muscle, HKII transcription is regulated by insulin. HKI also is present in human skeletal muscle, but it is not regulated by insulin (378). In response to physiological euglycemic hyperinsulinemia, HKII cytosolic activity, protein content, and mRNA levels increase by 50-200% in healthy non-diabetic subjects (378,380) and this is associated with the translocation of hexokinase II from the cytosol to the mitochondria (381). In contrast, insulin has no effect on HK-I activity, protein content, or mRNA levels (378).
In forearm muscle, insulin-stimulated glucose transport (measured with the triple tracer technique) has been shown to be markedly impaired in lean subjects with T2DM (370). However, since the rate of intracellular glucose phosphorylation was impaired to an even greater extent, insulin caused an increase in the intracellular free glucose concentration. By performing the insulin clamp at each subject’s normal level of fasting hyperglycemia, normal rates of whole- body glucose disposal and a normal rate of glucose influx into muscle was elicited. However, the rate of intracellular glucose phosphorylation increased only modestly; consequently, there was a dramatic rise in the free glucose concentration within the intracellular space that is accessible to glucose. These observations indicate that in individuals with T2DM, while both glucose transport and glucose phosphorylation are severely resistant to the action of insulin, impaired glucose phosphorylation (HKII) appears to be the rate-limiting step for insulin action. A similar pattern of impaired muscle glucose phosphorylation and transport is present in the insulin-resistant, normal glucose-tolerant offspring of two diabetic parents (382). These results are consistent with dose-response studies using PET to evaluated glucose phosphorylation and transport in skeletal muscle of subjects with T2DM (373). They also are consistent with 31P-NMR studies (383) which demonstrate that, during hyperinsulinemia, muscle G-6-P concentrations decline in subjects with T2DM versus control subjects. However, subsequent studies using 31P-NMR in combination with 1-14C-glucose suggest that the defect in insulin-stimulated muscle glucose transport exceeds the defect in glucose phosphorylation and is responsible for the decline in muscle glucose-6-P concentration (372). Because of methodologic differences, the results of the triple tracer (370) and MRI (372) studies cannot be reconciled at present. Nonetheless, observations from these studies are consistent in demonstrating that the defects in glucose phosphorylation and glucose transport in muscle are established early in the natural history of T2DM and cannot be explained by glucose toxicity (91). Clear evidence that HKII activity is crucial for glucose uptake derives from studies in transgenic mice who overexpress HKII. In this model, HKII over-expression increased both insulin- and exercise-stimulated muscle glucose uptake (384).
In healthy nondiabetic subjects, physiologic hyperinsulinemia for as little as 2-4 hours increases muscle HKII activity, gene transcription, and translation (333,378). In lean subjects with T2DM insulin-stimulated HKII activity and mRNA levels are markedly reduced compared to controls (383,385). Decreased basal muscle HKII activity and mRNA levels (385) and impaired insulin-stimulated HKII activity (379,380,386,387) in subjects with T2DM have been reported by other investigators. A decrease in insulin-stimulated muscle HKII activity also has been described in individuals with IGT (388). Because of its central role in insulin-mediated muscle glucose metabolism, several groups have looked for point mutations in the HKII gene in individuals with T2DM (388-390). Although several nucleotide substitutions have been found, none have been located close to the glucose and ATP binding sites and none have been associated with insulin resistance. Thus, an abnormality in the HKII gene is unlikely to explain the inherited insulin resistance in common variety T2DM.
Glycogen Synthesis
After glucose is phosphorylated by hexo-kinase II, it either can be converted to glycogen or enter the glycolytic pathway. Of the glucose that enters the glycolytic pathway, ~90% is oxidized. At low physiologic plasma insulin concentrations, glycogen synthesis and glucose oxidation are of approximately equal quantitative importance. With increasing plasma insulin concentrations, glycogen synthesis predominates (18,391). If the rate of glucose oxidation (determined by indirect calorimetry) is subtracted from the rate of whole-body insulin-mediated glucose disposal (determined from the insulin clamp), the difference represents non-oxidative glucose disposal (or glucose storage) (17,360), which primarily reflects glycogen synthesis (1,4,162,216,392). Glucose conversion to lipid accounts for <5% of total body glucose disposal (18,198,199) and, less than 5-10% of the glucose taken up by muscle is released as lactate (5,393,394).
Reduced insulin-stimulated glycogen synthesis is a characteristic finding in all insulin-resistant states, including obesity, diabetes, and the combination of obesity plus diabetes (1,4,18,43,59,159,162,218,219,377,393-395). Impaired glycogen synthesis also represents the major cause of insulin resistance in obese subjects with normal or only slightly impaired glucose tolerance (1,4,162,218,393,395,396). Thus, the inability of insulin to promote glycogen synthesis is a characteristic and early defect in the development of insulin resistance in both obesity and T2DM. The emergence of overt diabetes with fasting hyperglycemia is associated with a major reduction in insulin-mediated non-oxidative glucose disposal (glycogen synthesis) in all ethnic groups (1,4,18,162,377,396). Impaired glycogen synthesis also has been demonstrated in the normal-glucose-tolerant offspring of two diabetic parents (43,397), in the first-degree relatives of people with T2DM (41,398,399), and in a normoglycemic twin of a monozygotic twin pair in which the other has T2DM (101).
Using NMR imaging spectroscopy, a decrease in insulin-stimulated incorporation of [1H, 13C]- glucose into muscle glycogen of subjects with T2DM has been demonstrated directly (215). In T2DM, there was a marked lag in the onset of insulin-stimulated glycogen synthesis that was similar to the delay in insulin-mediated leg muscle glucose uptake. The rate of glycogen synthesis in subjects with T2DM was decreased by ~50%, paralleling the decrease in total glucose uptake by leg muscle (3). Also, impaired muscle glycogen synthesis accounted for essentially all of the defect in whole body glucose disposal.
In summary, an abundance of convincing evidence demonstrates that impaired glycogen synthesis is the major metabolic defect in normal glucose tolerant subjects with obesity, in individuals with IGT, and in patients with overt diabetes. Moreover, numerous studies have documented that the earliest detectable metabolic abnormality responsible for the insulin resistance in normal glucose tolerant individuals who are destined to develop T2DM is impaired glycogen synthesis (4,41,43,101,382,392,399,400).
Glycogen synthase is the key insulin-regulated enzyme which controls the rate of muscle glycogen synthesis (307,308,310,379,401,402). Insulin enhances glycogen synthase activity by stimulating a cascade of phosphorylation/de-phosphorylation reactions (307,308,361-363,403) (see above discussion of insulin receptor signal transduction), which ultimately lead to activate PP1 (also called glycogen synthase phosphatase) (307,308,310,402). The regulatory subunit (G) of PP1 has two serine phosphorylation sites, called site 1 and site 2. Phosphorylation of site 2 by cAMP-dependent kinase (PKA) inactivates PP1, while phosphorylation of site 1 by insulin activates PP1, leading to the stimulation of glycogen synthase (307,308,402,404). Phosphorylation of site 1 of PP1 by insulin in muscle is catalyzed by insulin-stimulated protein kinase 1 (ISPK-1) (309,405), which is part of a family of serine/threonine protein kinases termed ribosomal S6-kinases. Because of their central role in muscle glycogen formation, considerable attention has focused on the three enzymes glycogen synthase, PP1, and ISPK-1 in the pathogenesis of insulin resistance in T2DM.
Glycogen synthase exists in an active (dephosphorylated) and an inactive (phosphorylated) form (307-310). Under fasting conditions, total glycogen synthase activity in subjects with T2DM is reduced and the ability of insulin to activate glycogen synthase is severely impaired (301,384,406-410). An impaired ability of insulin to activate glycogen synthase also has been demonstrated in the normal glucose tolerant relatives of individuals with T2DM (400).
Insulin-mediated activation of glycogen synthase and insulin-stimulated glycogen synthase gene expression has been shown to be impaired in cultured myocytes and fibroblasts from subjects with T2DM (411,412). Studies in insulin-resistant nondiabetic and diabetic Pima Indians have documented that the ability of insulin to activate muscle PP1 (glycogen synthase phosphatase) is severely reduced (413). PP1 dephosphorylates glycogen synthase, leading to its activation. Therefore, a defect in PP1 appears to play an important role in muscle insulin resistance (309).
The effect of insulin on glycogen synthase gene transcription and translation in vivo has been studied extensively. Most studies (378,414,415) have shown that insulin does not increase glycogen synthase mRNA or protein expression in human muscle studied in vivo. However, glycogen synthase mRNA expression is decreased in muscle of patients with T2DM (415,416), explaining in part the decreased glycogen synthase activity in this disease. However, the major abnormality in glycogen synthase regulation in T2DM and other insulin resistant conditions is its lack of de-phosphorylation and activation by insulin as a result of insulin receptor signaling abnormalities (see previous discussion). The glycogen synthase gene (417) has been the subject of intensive investigation. An association between glycogen synthase gene markers and T2DM has been demonstrated in Japanese, French, Finnish, and Pima Indian populations. However, DNA sequencing has revealed either no mutations (418) or rare nucleotide substitutions (419,420) that cannot explain the defect in insulin-stimulated glycogen synthase. Nonetheless, the association between the glycogen synthase gene and T2DM (418) suggests that another gene close to the glycogen synthase gene may be involved in the development of T2DM. The genes encoding the catalytic subunits of PP1 (421) and ISPK-1 (422) have been examined in insulin-resistant Pima Indians and Danes with T2DM. Several silent nucleotide substitutions were found in the PP1 and ISPK-1 genes in the Danish population; the mRNA levels of both genes were normal in skeletal muscle (422). No structural gene abnormalities in the catalytic subunit of PP1 were detected in Pima Indians (422). Thus, neither abnormalities in the PP1 and ISPK-1 genes nor abnormalities in their translation can explain the impaired enzymatic activities of glycogen synthase and PP1 that have been observed in vivo. Similarly, there is no evidence that an alteration in glycogen phosphorylase plays any role in the abnormality in glycogen formation in T2DM (423). In summary, glycogen synthase activity is severely impaired in patients with T2DM and in insulin-resistant normal glucose tolerant individuals who are predisposed to develop T2DM. However, the defect cannot be explained by an abnormality in the genes encoding glycogen synthase or is promoter or by other key genes - PP1 or ISPK-1 - involved in the regulation of glycogen synthase activity.
Glycolysis/Glucose Oxidation
Glucose oxidation accounts for ~90% of total glycolytic flux, while anaerobic glycolysis accounts for the other 10% (393,394). Two enzymes, phosphofructokinase (PFK) and pyruvate dehydrogenase (PDH) play pivotal roles in the regulation of glycolysis and glucose oxidation, respectively. In individuals with T2DM the glycolytic/glucose oxidative pathway has been shown to be impaired in many individuals with T2DM (393,394). Although one study suggested that the activity of PFK is modestly reduced in muscle biopsies from subjects with T2DM (424), the majority of evidence indicates that the activity of PFK is normal (407,412,417). Insulin has no effect on muscle PFK activity, mRNA levels, or protein content in either nondiabetic or diabetic individuals (417). PDH is a key insulin-regulated enzyme whose activity in muscle is acutely stimulated by a physiological increment in the plasma insulin concentration (415). Three previous studies have examined PDH activity in patients with T2DM. Insulin-stimulated PDH activity is decreased in isolated subcutaneous human adipocytes from patients with T2DM (425) and in skeletal muscle from subjects with T2DM undergoing euglycemic hyperinsulinemic clamps (426). However, when patients with T2DM had muscle biopsies during hyperglycemic hyperinsulinemic clamps, activation of PDH by insulin was normal (409), in concert with normalized rates of muscle glucose uptake. These results suggest that insulin stimulation of PDH activity is influenced by glycolytic flux.
Both obesity and T2DM are associated with accelerated FFA turnover and oxidation (1,4,18,162), which would be expected, according to the Randle cycle (232), to inhibit PDH activity and consequently glucose oxidation (see prior discussion). Thus, any observed defect in glucose oxidation or PDH activity could be acquired secondarily to increased FFA oxidation and feedback inhibition of PDH by elevated intracellular levels of acetyl-CoA and reduced availability of NAD. Consistent with this observation, the rates of basal and insulin- stimulated glucose oxidation have been shown to be normal in the normal glucose tolerant offspring of two parents with T2DM (43) and in the first-degree relatives of subjects with T2DM (41,423), while it is decreased in subjects with overt T2DM (1,4,393,394,427). Studies examining PHD activity in muscle tissue from lean diabetic subjects with mild fasting hyperglycemia are needed before the role of this enzyme in the development of insulin resistance in T2DM can be established or excluded.
In summary, post-binding defects in insulin action primarily are responsible for the insulin resistance in T2DM. Diminished insulin binding, when present, is small, occurs in individuals with IGT or very mild diabetes, and results secondarily from downregulation of the insulin receptor by chronic sustained hyperinsulinemia. In patients with T2DM and overt fasting hyperglycemia, post-binding defects are responsible for the insulin resistance. A number of post-binding defects have been documented, including diminished insulin receptor tyrosine kinase activity, insulin signal transduction abnormalities, decreased glucose transport, reduced glucose phosphorylation, and impaired glycogen synthase activity. The glycolytic/glucose oxidative pathway appears to be largely intact and, when defects are observed, they appear to be acquired secondarily to enhanced FFA/lipid oxidation. From the quantitative standpoint, impaired glycogen synthesis represents the major pathway responsible for the insulin resistance in T2DM, and family studies suggests that a defect in the glycogen synthetic pathway represents the earliest detectable abnormality in T2DM. Recent studies link the impairment in glycogen synthase activation to a defect in the ability of insulin to phosphorylate IRS-1, causing a reduced association of the p85 subunit of PI 3-kinase with IRS-1 and decreased activation of the enzyme (PI 3-kinase).
Mitochondrial Dysfunction
In obesity and T2DM, impaired oxidation, reduced mitochondrial contents, lowered rates of oxidative phosphorylation and the production and release of excessive reactive oxygen species (ROS) have been reported. Mitochondrial biogenesis is regulated by various transcription factors such as peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERRs), and nuclear respiratory factors (NRFs). Mitochondrial fusion is promoted by mitofusin 1 (MFN1), mitofusin 2 (MFN2) and optic atrophy 1 (OPA1), while fission is governed by the recruitment of dynamin-related protein 1 (DRP1) by adaptor proteins, such as mitochondrial fission factor (MFF), mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51), and fission 1 (FIS1). Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) and PARKIN promote DRP1-dependent mitochondrial fission, and the outer mitochondrial adaptor MiD51 is required in DRP1 recruitment and PARKIN-dependent mitophagy. Several molecular abnormalities affecting these critical aspects of mitochondrial dynamics have been identified in obese individuals and in patients with T2DM (446).
The generation of new mitochondria, mitochondrial biogenesis, is presumed to be defective in patients with T2DM because the expressions of PGC-1α and its targeted genes are reduced. They are associated with an impaired ability to produce mitochondrial ATP and increased ROS production from the electron transport chain. There is some preliminary evidence that stimulation of mitochondrial biogenesis by pharmacological activation targeting these molecules is beneficial in the treatment of T2DM and obesity. The accumulation of damaged or depolarized mitochondria in pancreatic β cells is associated with oxidative stress and favors subsequent development of diabetes. Mitochondria in pancreatic β cells are continuously recruited in the fusion and fission processes. In a cultured pancreatic β cell line (INS-1), high levels of glucose- and palmitate-induced mitochondrial fusion arrested and reduced respiratory function. In INS1 cells, mitochondria with fission demonstrated reduced Δψ and decreased levels of the fusion protein OPA1. The inhibition of fission machinery proteins using DRP1 and FIS1 RNAi resulted in decreased mitochondrial autophagy, the accumulation of oxidized mitochondrial proteins, reduced respiration, and impaired insulin secretion. All of these suggest that selective fission of damaged mitochondria is followed by their removal by autophagy. In another study, INS-1 cells were treated with palmitate and high glucose, and the fragmentation of mitochondria with reduced fusion activities was observed. The application of FIS1 RNAi that shifts the dynamic balance to favor fusion is able to prevent mitochondrial fragmentation, maintain mitochondrial dynamics, and prevent apoptosis. Thus, although not entirely elucidated, abnormal mitochondrial fusion and fission dynamics in the pancreatic β cells may play an important role in beta cell dysfunction and the progression of T2DM.
Obesity and T2DM are associated with impaired skeletal muscle oxidation, reduced mitochondrial contents, and lowered rates of TCA cycle enzymes and OXPHOS. Patients with T2DM and obesity also demonstrated reduced expression of MFN2, which may be related to the reduced function of mitochondria in skeletal muscle. In 17 subjects with obesity, 12 weeks of exercise improved insulin sensitivity and fat oxidation. Skeletal muscle biopsy in these patients revealed that decreased phosphorylation and reduction of DRP1 at serine 616 were negatively correlated with increases in fat oxidation and insulin sensitivity (447). In this same study, there was a trend towards an increase in the expression of both MFN1 and MFN2. Studies in hepatocytes have recently demonstrated that the role of MAMs in calcium, lipid, and metabolite exchange is altered in obesity and T2DM. Although the ER and mitochondria play distinct cellular roles in the process of intermediary metabolism, obesity is known to lead to a marked reorganization of MAMs, which results in mitochondrial calcium overload, reduced respiratory function, and augmented oxidative stress (448). In contrast, disrupting the integrity of MAMs by knocking out cyclophilin D leads to hepatic insulin resistance through the disruption of inter-organelle Ca2+ transfer, ER stress, mitochondrial dysfunction, lipid accumulation, the activation of c-Jun N-terminal kinase and PKCε. In addition to the beta-cell and skeletal muscle defects described earlier, these altered molecular pathways in the liver represent potential targets for new pharmaceutical intervention to be explored in future studies including individuals with obesity and patients with T2DM.
REFERENCES
- 1.
- DeFronzo RA. Pathogenesis of type 2 diabetes mellitus: metabolic and molecular implications for identifying diabetes genes. Diabetes. 1997;5:117–269.
- 2.
- Grill V. A comparison of brain glucose metabolism in diabetes as measured by positron emission tomography or by arteriovenous techniques. Ann Med. 1990;22:171–175. [PubMed: 2393552]
- 3.
- DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in non-insulin dependent diabetes mellitus. J. Clin Invest. 1985;76:149–155. [PMC free article: PMC423730] [PubMed: 3894418]
- 4.
- DeFronzo RA. Lilly Lecture. The triumvirate: beta cell, muscle, liver. A collusion responsible for NIDDM. Diabetes. 1988;37:667–687. [PubMed: 3289989]
- 4A.
- DeFronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58:773–795. [PMC free article: PMC2661582] [PubMed: 19336687]
- 4B.
- Nauck M, Stockmann F, Ebert R, Creutzfeld W. Reduced incretin effect in type 2 (non- insulin-dependent) diabetes. Diabetologia. 1986;29:46–52. [PubMed: 3514343]
- 4C.
- Gastaldelli A., Ferrannini E, Miyazaki Y, Matsuda M, DeFronzo RA. Beta-cell dysfunction and glucose intolerance: results from the San Antonio Metabolism (SAM) study. Diabetologia. 2004;47:31–39. [PubMed: 14666364]
- 4D.
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J of Clin Invest. 2005;115(5):1111–1119. [PMC free article: PMC1087185] [PubMed: 15864338] [CrossRef]
- 4E.
- Clark MG, Walls MG, Barrett EJ, Vincent MA, Richards SM, Clerk LH, Rattigan, S. Blood flow and muscle metabolism: a focus on insulin action. Am J Physiol: Endocrinol and Metabol 284 (2): E241-E258, 2003 /https://doi.org/ 10.1152/ajpendo.00408.2002. [PubMed: 12531739] [CrossRef]
- 5.
- DeFronzo RA, Jacot E, Jequier E, Maeder E, Wahren J, Felber JP. The effect of insulin on the disposal of intravenous glucose: results from indirect calorimetry. Diabetes. 1981;30:1000–1007. [PubMed: 7030826]
- 6.
- DeFronzo RA, Ferrannini E. Regulation of hepatic glucose metabolism in humans. Diabetes Metab Rev. 1987;3:415–459. [PubMed: 3552529]
- 6A.
- Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nature Medicine. 2002;8(12):1376–1382. [PubMed: 12426561]
- 7.
- Mari A, Wahren J, DeFronzo RA, Ferrannini E. Glucose absorption and production following oral glucose: comparison of compartmental and arteriovenous-difference methods. Metabolism. 1994;43:1419–1425. [PubMed: 7968597]
- 8.
- Cersosimo E, Garlick P, Ferretti J. Insulin regulation of renal glucose metabolism in humans. Am J Physiol. 1999;276(39):E788–E84. [PubMed: 9886953]
- 9.
- Ekberg K, Landau BR, Wajngot A, Chandramouli V, Efendic S, Brunengraber H, Wahren J. Contributions by kidney and liver to glucose production in the post-absorptive state and after 60 h of fasting. Diabetes. 1999;48:292–298. [PubMed: 10334304]
- 9A.
- Farber SJ, Berger EY, Earle DP. Effect of diabetes and insulin on the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest. 1951;30:125–29. [PMC free article: PMC436236] [PubMed: 14814204]
- 9B.
- Morgensen CE. Maximum tubular reabsorption capacity for glucose and renal hemodynamcis during rapid hypertonic glucose infusion in normal and diabetic subjects. Scand J Clin Lab Invest. 1971;28:101–09. [PubMed: 5093515]
- 9C.
- Rahmoune H, Thompson PW, Ward JM, Smith CD, Hong G, Brown J. Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes. 2005;54:3427–34. [PubMed: 16306358]
- 9D.
- Cersosimo E. Renal glucose handling and the kidney as target for anti-diabetic medication. Current Trends in Endocrinology, vol. 7, 2014.
- 10.
- Magnusson I., Katz LD., Shulman RG., Shulman GI. Quantitation of hepatic glycogenolysis and gluconeogenesis in fasting humans with 13C NMR. Science. 1991;254:573–6. [PubMed: 1948033]
- 11.
- Katz LD, Glickman MG, Rapoport S, Ferrannini E, DeFronzo RA. Splanchnic and peripheral disposal of oral glucose in man. Diabetes. 1983;32:675–679. [PubMed: 6862113]
- 12.
- Ferrannini E, Bjorkman O, Reichard GA Jr, Pilo A, Olsson M, Wahren J, DeFronzo RA. The disposal of an oral glucose load in healthy subjects. Diabetes. 1985;34:580–588. [PubMed: 3891471]
- 13.
- Mitrakou A, Kelley D, Veneman T, Jensen T, Pangburn T, Reilly J, Gerich J. Contribution of abnormal muscle and liver glucose metabolism to postprandial hyperglycemia in NIDDM. Diabetes. 1990;39:1381–90. [PubMed: 2121568]
- 14.
- Mandarino L, Bonadonna R, McGuinness O, Wasserman D. Regulation of Muscle Glucose Uptake In Vivo. In: Handbook of Physiology, Section 7, The Endocrine System, Vol. II, The Endocrine Pancreas and Regulation of Metabolism, pp 803-848, L.S. Jefferson and A.D. Cherrington, eds., Oxford University Press, 2001.
- 15.
- Del Prato S., Riccio A. Vigili de Kreutzenberg S. Dorella M. Tiengo A. DeFronzo RA. Basal plasma insulin levels exert a qualitative but not quantitative effect on glucose-mediated glucose uptake. Am J Physiol. 1995;268:E1089–95. [PubMed: 7611383]
- 16.
- Cherrington AD. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48:1198–1214. [PubMed: 10331429]
- 17.
- Jansson P-A, Larsson A, Smith U, Lonroth P. Lactate release from the subcutaneous tissue in lean and obese men. J Clin Invest. 1994;93:240–246. [PMC free article: PMC293758] [PubMed: 8282793]
- 18.
- Groop LC, Bonadonna RC, Del Prato S, Ratheiser K, Zych K, Ferrannini E, DeFronzo RA. Glucose and free fatty acid metabolism in non-insulin dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. J Clin Invest. 1989;84:205–215. [PMC free article: PMC303971] [PubMed: 2661589]
- 19.
- Santomauro A, Boden G, Silva M, Rocha DM, Santos RF, Ursich MJM, Strassman PG, Wajchenberg BL. Overnight lowering of free fatty acids with acipimox improves insulin resistance and glucose tolerance in obese diabetic and non-diabetic subjects. Diabetes. 1999;48:1836–1841. [PubMed: 10480616]
- 20.
- Bergman RN. Non-esterified fatty acids and the liver: why is insulin secreted into the portal vein? Diabetologia. 2000;43:946–52. [PubMed: 10952470]
- 21.
- Boden G. Role of fatty acids in the pathogenesis of insulin resistance and NIDDM. Diabetes. 1997;46:3–10. [PubMed: 8971073]
- 22.
- McGarry JD. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes. 2002;51:7–18. [PubMed: 11756317]
- 23.
- Baron AD., Schaeffer L., Shragg P., Kolterman OG. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes. 1987;36:274–83. [PubMed: 2879757]
- 24.
- DeFronzo RA, Ferrannini E, Hendler R, Wahren J, Felig P. Influence of hyperinsulinemia, hyperglycemia, and the route of glucose administration on splanchnic glucose exchange. Proc Natl Acad Sci. 1978;75:5173–5177. [PMC free article: PMC336287] [PubMed: 283423]
- 25.
- Ferrannini E, Wahren J, Felig P, DeFronzo RA. Role of fractional glucose extraction in the regulation of splanchnic glucose metabolism in normal and diabetic man. Metabolism. 1980;29:28–35. [PubMed: 7351874]
- 26.
- Hsieh P-S, Moore MC, Neal DW, Cherrington AD. Hepatic glucose uptake rapidly decreases after removal of the portal signal in conscious dogs. Am J Physiol. 1998;275:E987–E992. [PubMed: 9843741]
- 27.
- Adkins-Marshall B, Pagliassotti MJ, Asher JR, Connolly CC, Neal DW, Williams PE, Myers SR, Hendrick GK, Adkins RB Jr, Cherrington AD. Role of hepatic nerves in response of liver to intraportal delivery in dogs. Am J Physiol. 1992;262:E679–E686. [PubMed: 1590377]
- 28.
- Creutzfeldt W. The incretin concept today. Diabetologia. 1979;16:75–85. [PubMed: 32119]
- 29.
- Drucker DJ. Glucagon-like peptides. Diabetes. 1998;47:159–169. [PubMed: 9519708]
- 30.
- Holst JJ, Gromada J, Nauck MA. The pathogenesis of NIDDM involves a defective expression of the GIP receptor. Diabetologia. 1997;40:984–986. [PubMed: 9267997]
- 31.
- Polonsky KS, Sturis J, Bell GI. Non-insulin-dependent diabetes mellitus - a genetically programmed failure of the beta cell to compensate for insulin resistance. N Engl J Med. 1996;334:777–783. [PubMed: 8592553]
- 32.
- Cerasi E. Insulin deficiency and insulin resistance in the pathogenesis of NIDDM: is a divorce possible? Diabetologia. 1995;38:992–997. [PubMed: 7589888]
- 33.
- Sicree RA, Zimmet P, King HO, Coventry JO. Plasma insulin response among Nauruans. Prediction of deterioration in glucose tolerance over 6 years. Diabetes. 1987;36:179–186. [PubMed: 3542644]
- 34.
- Saad MF, Knowler WC, Pettitt DJ, Nelson RG, Mott DM, Bennett PH. Sequential changes in serum insulin concentration during development of non-insulin-dependent diabetes. Lancet i. 1989:1356–1359. [PubMed: 2567374]
- 35.
- Saad MF, Knowler WC, Pettitt DJ, Nelson RG, Mott DM, Bennett PH. The natural history of impaired glucose tolerance in the Pima Indians. New Engl J Med. 1988;319:1500–1505. [PubMed: 3054559]
- 36.
- Haffner SM, Miettinen H, Gaskill SP, Stern MP. Decreased insulin secretion and increased insulin resistance are independently related to the 7-year risk of NIDDM in Mexican- Americans. Diabetes. 1995;44:1386–1391. [PubMed: 7589843]
- 37.
- Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance. Evidence for a pathogenic role of relative hyperinsulinemia. Diabetes. 2000;49:2094–2101. [PubMed: 11118012]
- 38.
- Weyer C, Bogardus C, Pratley RE. Metabolic characteristics of individuals with impaired fasting glucose and/or impaired glucose tolerance. Diabetes. 1999;48:2197–2203. [PubMed: 10535454]
- 39.
- Weyer C, Tataranni PA, Bogardus C, Pratley RE. Insulin resistance and insulin secretory dysfunction are independent predictors of worsening of glucose tolerance during each stage of type 2 diabetes development. Diabetes Care. 2000;24:89–94. [PubMed: 11194248]
- 40.
- Pimenta W, Korytkowski M, Mitrakou A, Jenssen T, Yki-Jarvinen H, Evron W, Dailey G, Gerich J. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first degree NIDDM relative. JAMA. 1995;273:1855–1861. [PubMed: 7776502]
- 41.
- Eriksson J, Franssila-Kallunki A, Ekstrand A, Saloranta C, Widen E, Schalin C, Groop L. Early metabolic defects in persons at increased risk for non-insulin-dependent diabetes mellitus. New Engl J Med. 1989;321:337–343. [PubMed: 2664520]
- 42.
- Reaven GM, Hollenbeck CB, Chen YDI. Relationship between glucose tolerance, insulin secretion, and insulin action in non-obese individuals with varying degrees of glucose tolerance. Diabetologia. 1989;32:52–55. [PubMed: 2651188]
- 43.
- Gulli G, Ferrannini E, Stern M, Haffner S, DeFronzo RA. The metabolic profile of NIDDM is fully established in glucose-tolerant offspring of two Mexican-American NIDDM parents. Diabetes. 1992;41:1575–1586. [PubMed: 1446799]
- 44.
- Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn RC. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25- year follow-up study. Lancet. 1992;340:925–929. [PubMed: 1357346]
- 45.
- Lillioja S, Mott DM, Zawadzki JK, Young AA, Abbott WG, Knowler WC, Bennett PH, Moll P, Bogardus C. In vivo insulin action is familial characteristic in nondiabetic Pima Indians. Diabetes. 1987;36:1329–1335. [PubMed: 3311855]
- 46.
- Kahn SE. Clinical Review 135. The importance of ß-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. [PubMed: 11549624]
- 47.
- Bergman RN, Finegood DT, Kahn SE. The evolution of ß-cell dysfunction and insulin resistance in type 2 diabetes. European J Clin Invest. 2002;32:35–45. [PubMed: 12028373]
- 48.
- Hansen BC, Bodkin NH. Heterogeneity of insulin responses: phases leading to type 2 (noninsulin-dependent) diabetes mellitus in the rhesus monkey. Diabetologia. 1986;29:713–719. [PubMed: 3542671]
- 49.
- Diamond MP, Thornton K, Connolly-Diamond M, Sherwin RS, DeFronzo RA. Reciprocal variations in insulin-stimulated glucose uptake and pancreatic insulin secretion in women with normal glucose tolerance. J Soc Gynecol Invest. 1995;2:708–715. [PubMed: 9420879]
- 50.
- Hollenbeck CB, Reaven GM. Variations in insulin-stimulated glucose uptake in healthy individuals with normal glucose tolerance. J Clin Endocrinol Metab. 1987;64:1169–1173. [PubMed: 3553221]
- 51.
- Reaven GM. Banting Lecture. Role of insulin resistance in human disease. Diabetes. 1988;37:595–607. [PubMed: 3056758]
- 52.
- Faber OK, Damsgaard EM. Insulin secretion in type II diabetes. Acta Endocrinol. 1984;262 suppl.:47–50.
- 53.
- Faber OK, Hagen C, Binder C, Markussen J, Naithani VK, Blix PM, Kuzuya H, Horwitz DL, Rubenstein AH, Rossing N. Kinetics of human connecting peptide in normal and diabetic subjects. J Clin Invest. 1978;62:197–203. [PMC free article: PMC371754] [PubMed: 659633]
- 54.
- DeFronzo RA, Ferrannini E, Simonson DC. J. Fasting hyperglycemia in non-insulin- dependent diabetes mellitus: contributions of excessive hepatic glucose production and impaired tissue glucose uptake. Metabolism. 1989;38:387–95. [PubMed: 2657323]
- 55.
- Hales CN. The pathogenesis of NIDDM. Diabetologia. 1994;37:S162–S168. [PubMed: 7821732]
- 56.
- Reaven GM, Hollenbeck C, Jeng C-Y, Wu MS, Chen Y-DI. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 hours in patients with NIDDM. Diabetes. 1988;37:1020–4. [PubMed: 3292322]
- 57.
- Garvey WT, Olefsky JM, Rubenstein AH, Kolterman OG. Day-long integrated urinary C- peptide excretion. Diabetes. 1988;37:590–9. [PubMed: 3282946]
- 58.
- Lillioja S, Mott DM, Howard BV, Bennett PH, Yki-Jarvinen H, Freymond D, Nyomba BL, Zurlo F, Swinburn B, Bogardus C. Impaired glucose tolerance as a disorder of insulin action. Longitudinal and cross-sectional studies in Pima Indians. New Engl J Med. 1988;318:1217–25. [PubMed: 3283552]
- 59.
- Jallut D, Golay A, Munger R, Frascarolo P, Schutz Y, Jequier E, Felber JP. Impaired glucose tolerance and diabetes in obesity: a 6 year follow-up study of glucose metabolism. Metabolism. 1990;39:1068–1075. [PubMed: 2215253]
- 60.
- Lillioja S, Mott DM, Spraul M, Ferraro R, Foley JE, Ravussin E, Knowler WC, Bennett PH, Bogardus C. Insulin resistance and insulin secretory dysfunction as precursors of non- insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:1988–1992. [PubMed: 8247074]
- 61.
- Jensen CC, Cnop M, Hull RL, Fujimoto WY, Kahn SE., American Diabetes Association GENNID Study Group. ß-cell function is a major contributor to oral glucose tolerance in high-risk relatives of our ethnic groups in the U.S. Diabetes. 2002;51:2170–2178.
- 62.
- Dowse GK, Zimmet PZ, Collins VR. Insulin levels and the natural history of glucose intolerance in Nauruans. Diabetes. 1996;45:1367–1372. [PubMed: 8826973]
- 63.
- Haffner SM, Stern MP, Hazuda HP, Pugh JA, Patterson JK. Hyperinsulinemia in a population at high risk for non-insulin-dependent diabetes mellitus. New Engl J Med. 1986;315:220–4. [PubMed: 3523246]
- 64.
- Ho LT, Chang ZY, Wang JT, Li SH, Liu YF, Chen Y-DI, Reaven GM. Insulin insensitivity in offspring of parents with type 2 diabetes mellitus. Diabet Med. 1990;7:31–34. [PubMed: 1967567]
- 65.
- Clement K, Pueyo ME, Vaxillaire M, Rakotoambinina B, Thuillier F, Passa PH, Froguel PH, Robert JJ, Velho G. Assessment of insulin sensitivity in glucokinase-deficient subjects. Diabetologia. 1996;39:82–90. [PubMed: 8720607]
- 66.
- Polonsky KS. Lilly Lecture 1994. The beta cell in diabetes: from molecular genetics to clinical research. Diabetes. 1995;44:705–717. [PubMed: 7789637]
- 67.
- Bell GI, Polonsky KS. Diabetes mellitus and genetically programmed defects in ß-cell function. Nature. 2001;414:788–791. [PubMed: 11742410]
- 68.
- McCarthy MI, Froguel P. Genetic approaches to the molecular understanding of type 2 diabetes. Am J Physiol. 2002;283:E217–E225. [PubMed: 12110525]
- 69.
- Bell GI, Zian K, Newman M, Wu S, Wright L, Fajans S., Spielman RS, Cox NJ. Gene for non-insulin-dependent diabetes mellitus (maturity-onset diabetes of the young subtype) is linked to DNA polymorphism on human chromosome 20q. Proc. Natl. Acad. Sci. 1991;88:1484–1488. [PMC free article: PMC51043] [PubMed: 1899928]
- 70.
- Froguel PH, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, Lesage S, Stoffel M, Takeda J, Passap P. Familial hyperglycaemia due to mutations in glucokinase: definition of a subtype of diabetes mellitus. N Engl J Med. 1993;328:697–702. [PubMed: 8433729]
- 71.
- Menzel S, Yamagata K, Trabb JB, Nerup J, Permutt MA, Fajans SS, Menzel R, Iwasaki N, Omori Y, Cox N, Bell GI. Localization of MODY3 to a 5-cM Region of Human Chromosome 12. Diabetes. 1995;44:1408–1413. [PubMed: 7589847]
- 72.
- Zhang Y, Warren-Perry M, Saker PJ, Hattersley AT, Mackie ADR, Baird JD, Greenwood RH, Stoffel M, Bell GI, Turner RC. Candidate gene studies in pedigrees with maturity-onset diabetes of the young not lined with glucokinase. Diabetologia. 1995;38:1055–1066. [PubMed: 8591819]
- 73.
- Weng J, Macfarlane WM, Lehto M, Gu HF, Shepherd LM, Ivarsson SA, Wibell L, Smith T, Groop LC. Functional consequences of mutations in the MODY4 gene (IPF1) and coexistence with MODY3 mutations. Diabetologia. 2001;44:249–258. [PubMed: 11270685]
- 74.
- Sturis J, Kurland IJ, Byrne MM, Mosekilde E, Froguel P, Pilkis SJ, Bell GI, Polonsky KS. Compensation in pancreatic ß-cell function in subjects with glucokinase mutations. Diabetes. 1994;43:718–723. [PubMed: 8168650]
- 75.
- Beck-Nielsen H, Nielsen OH, Pedersen O, Bak J, Faber O, Schmitz O. Insulin action and insulin secretion in identical twins with MODY: evidence for defects in both insulin action and insulin secretion. Diabetes. 1988;37:730–735. [PubMed: 3289993]
- 76.
- Mohan V, Sharp PS, Aber VR, Mather HM, Kohner EM. Insulin resistance in maturity-onset diabetes of the young. Diabetes Metab. 1988;13:193–197. [PubMed: 3301443]
- 77.
- Elbein SC, Hoffman M, Qin H, Chiu K, Tanizawa Y, Permutt MA. Molecular screening of the glucokinase gene in familial type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia37:182-187, 1994. [PubMed: 8163053]
- 78.
- Efendic S, Grill V, Luft R, Wajngot A. Low insulin response: a marker of pre-diabetes. Adv Exp Med Biol. 1988;246:167–174. [PubMed: 3074647]
- 79.
- Davies MJ, Metcalfe J, Gray IP, Day JL, Hales CN. Insulin deficiency rather than hyperinsulinaemia in newly diagnosed type 2 diabetes mellitus. Diabetic Med. 1993;10:305–312. [PubMed: 8508611]
- 80.
- Chen K-W, Boyko EJ, Bergstrom RW, Leonetti DL, Newell-Morris L, Wahl PW, Fujimoto WY. Earlier appearance of impaired insulin secretion than of visceral adiposity in the pathogenesis of NIDDM. 5-year follow-up of initially nondiabetic Japanese-American men. Diabetes Care. 1995;18:747–753. [PubMed: 7555498]
- 81.
- Arner P, Pollare T, Lithell H. Different etiologies of Type 2 (non-insulin-dependent) diabetes mellitus in obese and non-obese subjects. Diabetologia. 1991;34:483–487. [PubMed: 1916053]
- 82.
- Ferrannini E., Natali A., Bell P., Cavallo-Perin P., Lalic N., Mingrone G. Insulin resistance and hypersecretion in obesity. European Group for the Study of Insulin Resistance (EGIR). Journal of Clinical Investigation. 1997;100(5):1166–73. [Journal Article] [PMC free article: PMC508292] [PubMed: 9303923]
- 83.
- Banjeri MA, Lebovitz HE. Insulin action in black Americans with NIDDM. Diates Care. 1992;15:1295–1302. [PubMed: 1425092]
- 84.
- Mbanya J-CN, Pani LN, Mbanya DNS, Sobngwi E, Ngogang J. Reduced insulin secretion in offspring of African type 2 diabetic patients. Diabetes Care. 2000;23:1761–1765. [PubMed: 11128348]
- 85.
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;6:E214–23. [PubMed: 382871]
- 86.
- Curry DL, Bennett LL, Grodsky GM. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology. 1968;83:572–584. [PubMed: 4877098]
- 87.
- Temple R, Clark PMS, Hales CN. Measurement of insulin secretion in type 2 diabetes: problems and pitfalls. Diab Med. 1992;9:503–512. [PubMed: 1643797]
- 88.
- Brunzell JD, Robertson RP, Lerner RL, Hazzard WR, Ensinck JW, Bierman EL, Porte D. Relationships between fasting plasma glucose levels and insulin secretion during intravenous glucose tolerance tests. J Clin Endocrinol. 1976;46:222–229. [PubMed: 1262429]
- 89.
- Vague P, Moulin J-P. The defective glucose sensitivity of the B cell in insulin dependent diabetes. Improvement after twenty hours of normoglycaemia. Metabolism. 1982;31:139–142. [PubMed: 7043166]
- 90.
- Kosaka K, Kuzuya T, Akanuma Y, Hagura R. Increase in insulin response after treatment of overt maturity onset diabetes mellitus is independent of the mode of treatment. Diabetologia. 1980;18:23–28. [PubMed: 6988263]
- 91.
- Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990;13:610–630. [PubMed: 2192847]
- 92.
- Prentki M, Corkey BE. Are the beta cell signaling molecules malonyl-CoA and cytosolic long-chain acyl-CoA implicated in multiple tissue defects of obesity and NIDDM? Diabetes. 1996;45:273–283. [PubMed: 8593930]
- 93.
- Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes. 1995;44:863–870. [PubMed: 7621989]
- 94.
- Shimabukuro M, Zhou Y-T, Levi M, Unger RH. Fatty acid induced ß cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci. 1998;95:2498–2502. [PMC free article: PMC19389] [PubMed: 9482914]
- 95.
- Bruce DG, Chisholm DJ, Storlien LH, Kraegen EW. Physiological importance of deficiency in early prandial insulin secretion in non-insulin dependent diabetes. Diabetes. 1988;37:736–744. [PubMed: 3289994]
- 96.
- Luzi L. Effect of the loss of first phase insulin secretion on glucose production and disposal in man. Am J Physiol. 1989;257:E241–E246. [PubMed: 2669517]
- 97.
- Bonner-Weir S. ß-cell turnover. Its assessment and implications. Diabetes. 2001;50:S20–S24. [PubMed: 11272192]
- 98.
- Haffner SM, Miettinen H, Stern MP. Insulin secretion and resistance in nondiabetic Mexican Americans and non-Hispanic whites with a parental history of diabetes. J Clin Endocrinol Metab. 1996;81:1846–1851. [PubMed: 8626845]
- 99.
- Gautier J-F, Wilson C, Weyer C, Mott D, Knowler WC, Cavaghan M, Polonsky KS, Bogardus C, Pratley RE. Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes. 2001;50:1828–1833. [PubMed: 11473045]
- 100.
- Vauhkonen N, Niskanen L, Vanninen E, Kainulainen S, Uusitupa M, Laakso M. Defects in insulin secretion and insulin action in non-insulin-dependent diabetes mellitus are inherited. Metabolic studies on offspring of diabetic probands. J Clin Invest. 1997;100:86–96. [PMC free article: PMC508544] [PubMed: 9421470]
- 101.
- Vaag A, Henriksen JE, Madsbad S, Holm N, Beck-Nielsen H. Insulin secretion, insulin action, and hepatic glucose production in identical twins discordant for non-insulin- dependent diabetes mellitus. J Clin Invest. 1995;95:690–698. [PMC free article: PMC295536] [PubMed: 7860750]
- 102.
- Barnett AH, Spilipoulos AJ, Pyke DA, Stubbs WA, Burrin J, Alberti KGMM. Metabolic studies in unaffected co-twins of non-insulin-dependent diabetics. Brit Med J. 1981;282:1656–1658. [PMC free article: PMC1505636] [PubMed: 6786418]
- 103.
- Watanabe RM., Valle T., Hauser ER., Ghosh S., Eriksson J., Kohtamaki K., Ehnholm C., Tuomilehto J., Collins FS., Bergman RN., Boehnke M. Familiality of quantitative metabolic traits in Finnish families with non-insulin-dependent diabetes mellitus. Finland-United States Investigation of NIDDM Genetics (FUSION) Study investigators. Human Heredity. 1999;49(3):159–68. [PubMed: 10364681]
- 104.
- Mahtani MM, Widen E., Lehto M., Thomas J., McCarthy M., Brayer J., Bryant B., Chan G., Daly M., Forsblom C., Kanninen T., Kirby A., Kruglyak L., Munnelly K., Parkkonen M., Reeve-Daly MP., Weaver A., Brettin T., Duyk G., Lander ES., Groop LC. Mapping of a gene for type 2 diabetes associated with an insulin secretion defect by a genome scan in Finnish families. Nature Genetics. 1996;14:90–4. [PubMed: 8782826]
- 105.
- Rossetti L. Shulman Gi, Zawalich W, DeFronzo RA. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J Clin Invest. 1987;80:1037–1044. [PMC free article: PMC442343] [PubMed: 3308956]
- 106.
- Leahy JL, Bonner-Weir S, Weir GC. Abnormal glucose regulation of insulin secretion in models of reduced beta-cell mass. Diabetes. 1984;33:667–73. [PubMed: 6203798]
- 107.
- Leahy JL, Bonner-Weir S, Weir GC. Minimal chronic hyperglycemia is a critical determinant of impaired insulin secretion after an incomplete pancreatectomy. J Clin Invest. 1988;81:1407–14. [PMC free article: PMC442571] [PubMed: 3284912]
- 108.
- Leahy JL, Cooper HE, Weir GC. Impaired insulin secretion associated with near normoglycemia. Diabetes. 1987;36:459–464. [PubMed: 3545946]
- 109.
- Robertson RP, Olson IK, Zhang H-J. Differentiating glucose toxicity from glucose desensitization: a new message from the insulin gene. Diabetes. 1994;43:1085–1089. [PubMed: 8070607]
- 110.
- Newgard CB, McGarry JD. Metabolic coupling factors in pancreatic beta cell signal transduction. Annu Rev Biochem. 1995;64:689–719. [PubMed: 7574498]
- 111.
- Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. [PubMed: 1411571]
- 112.
- Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest. 1994;93:870–876. [PMC free article: PMC293952] [PubMed: 8113418]
- 113.
- Shimabukuro M., Higa M., Zhou YT., Wang MY., Newgard CB., Unger RH. Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J Biol Chem. 1998;273:32487–90. [PubMed: 9829981]
- 114.
- Fehmann HC., Goke R., Goke B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocrine Reviews. 1995;16:390–410. [PubMed: 7671853]
- 115.
- Drucker DJ. Minireview: the glucagon-like peptides. Endocrinology. 2001;142:521–527. [PubMed: 11159819]
- 116.
- Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7- 36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76:912–917. [PubMed: 8473405]
- 117.
- D'Alessio DA., Vogel R., Prigeon R., Laschansky E., Koerker D., Eng J., Ensinck JW. Elimination of the action of glucagon-like peptide 1 causes an impairment of glucose tolerance after nutrient ingestion by healthy baboons. J Clin Invest. 1996;97:133–138. [PMC free article: PMC507071] [PubMed: 8550824]
- 118.
- Schirra J., Sturm K., Leicht P., Arnold R., Goke B., Katschinski M. Exendin(9-39)amide is an antagonist of glucagon-like peptide-1(7-36)amide in humans. J Clin Invest. 1998;101:1421–30. [PMC free article: PMC508720] [PubMed: 9525985]
- 119.
- Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 Amide] but not of synthetic human gastric inhibitory polypeptide in patients with type 2 diabetes mellitus. J Clin Invest. 1993;91:301–307. [PMC free article: PMC330027] [PubMed: 8423228]
- 120.
- Vilsboll T, Krarup T, Deacon CF, Madsbad S, Holst JJ. Reduced postprandial concentrations of intact biologically active glucagon-like peptide 1 in type 2 diabetic patients. Diabetes. 2001;50:609–613. [PubMed: 11246881]
- 121.
- Ahren B, Larsson H, Holst JJ. Effects of glucagon-like peptide-1 on islet function and insulin sensitivity in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1997;82:473–478. [PubMed: 9024239]
- 122.
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7-36 amide) in type 2 (non-insulin- dependent) diabetic patients. Diabetologia. 1993;36:741–744. [PubMed: 8405741]
- 123.
- Xu G., Stoffers DA., Habener JF., Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. 1999;48:2270–2276. [PubMed: 10580413]
- 124.
- Kahn SE, Andrikopoulos S, Verchere CB. Islet amyloid. A long-recognized but underappreciated pathological feature of type 2 diabetes. Diabetes. 1999;48:241–253. [PubMed: 10334297]
- 125.
- Johnson KH, O'Brien TD, Betysholtz C, Westermark P. Islet amyloid, islet-amyloid polypeptide, and diabetes mellitus. New Engl J Med. 1989;321:513–518. [PubMed: 2668761]
- 126.
- Ohsawa H, Kanatsuka A, Yamaguchi T, Makino H, Yoshida S. Islet amyloid polypeptide inhibits glucose-stimulated insulin secretion from isolated rat pancreatic islets. Biochem Biophys Res Comm. 1989;160:961–967. [PubMed: 2655598]
- 127.
- Howard CF. Longitudinal studies on the development of diabetes in individual macaca nigra. Diabetologia. 1986;29:301–306. [PubMed: 3522329]
- 128.
- Hartter E, Svoboda T, Ludvik B, Schuller M, Lell B, Kuenburg E, Brunnbauer M, Woloszczuk W, Prager R. Basal and stimulated plasma levels of pancreatic amylin indicate its co-secretion with insulin in humans. Diabetologia. 1991;34:52–54. [PubMed: 2055340]
- 129.
- Eriksson J, Nakazato M, Miyazato M, Shiomi K, Matsukura S, Groop L. Islet amyloid polypeptide: plasma-concentrations in individuals at increased risk of developing type 2 (non-insulin-dependent) diabetes mellitus. Diabetologia. 1992;35:292–293. [PubMed: 1563587]
- 130.
- Westermark GT, Christmanson L, Terenghi G, Permerth J, Betsoltz C, Larsson J, Polak JM, Westermark P. Islet amyloid polypeptide: demonstration of mRNA in human pancreatic islets by in situ hybridization in islets with and without amyloid deposits. Diabetologia. 1993;36:323–328. [PubMed: 8477877]
- 131.
- Ghatei MA, Datta HK, Zaidi M. BrethertonWatt D, Wimalawansa SJ, Maclntyre 1, Bloom SR. Amylin and amylin-amide lack an acute effect on blood glucose and insulin. J. Endocrinol. 1990;124:R9–R11. [PubMed: 2179452]
- 132.
- Bretherton-Watt D, Gilbey SG, Ghatei MA, Beacham J, Bloom SR. Failure to establish islet amyloid polypeptide (amylin) as a circulating beta cell inhibiting hormone in man. Diabetologia. 1990;33:115–117. [PubMed: 2328845]
- 133.
- Hoppener JWM, Verbeek JS, de Koning EJP, Oosterwijk C, van Hulst KL, Visser-Vernooy HJ, Hofhuis FMA, van Gaalen S, Berends MJH, Hackeng WHL, Jansz HS, Morris JF, Clark A, Capel PJA, Lipis CJM. Chronic overproduction of islet amyloid polypeptide-amylin in transgenic mice: lysosomal localization of human islet amyloid polypeptide and lack of marked hyperglycaemia or hyperinsulinaemia. Diabetologia. 1993;36:1258–1265. [PubMed: 8307253]
- 134.
- Gebre-Medhin S, Olofsson C, Mulder H. Islet amyloid polypeptide in the islets of Langerhans: friend or foe? Diabetologia. 2000;43:687–695. [PubMed: 10907112]
- 135.
- Westermark P, Wilander E. The influence of amyloid deposits on the islet volume in maturity onset diabetes mellitus. Diabetologia. 1978;15:417–421. [PubMed: 367856]
- 136.
- Gepts W, Lecompte PM. The pancreatic islets in diabetes. Am J Med. 1981;70:105–114. [PubMed: 7006384]
- 137.
- Kloppel G, Lohr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type I and type 2 diabetes mellitus revisited. Surv Synth Path Res. 1985;4:110–25. [PubMed: 3901180]
- 138.
- Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, Turner RC. Islet amyloid, increased alpha-cells, reduced beta-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. 1988;9:151–159. [PubMed: 3073901]
- 139.
- Butler AE, Janson J, Bonner-Weir S, Ritzel RA, Butler PC. Decreased beta-cell mass inpatients with type-2 diabetes mellitus. Diabetes. 2002;51 suppl 2:A367.
- 140.
- Stefan Y, Orci L, Malaisse-Lagae F, Perrelet A, Patel Y, Unger R. Quantitation of endocrine cell content in the pancreas of non-diabetic and diabetic humans. Diabetes. 1982;31:694–700. [PubMed: 6131002]
- 141.
- Rahier J, Sempoux C, Moulin P, Guiot Y. No decrease of the B cell mass in type 2 diabetic patients. Diabetologia. 2000;43 Suppl 1:A65. [PubMed: 11272190]
- 142.
- Janson J, Butler AE, Bonner-Weir S, Ritzel RA, Sultana C, Butler PC. Failure of compensatory increase in new islet formation in humans with type-2 diabetes mellitus. Diabetes. 2002;51 suppl 2:A377.
- 143.
- Hales CN, Barker DJP, Clark PM, Cox J, Fall C, Osmond C, Winter PD. Fetal and infant growth and impaired glucose tolerance at age 64 years. BMJ. 1991;303:1019–1022. [PMC free article: PMC1671766] [PubMed: 1954451]
- 144.
- Eriksson UJ. Lifelong consequences of metabolic adaptations in utero? Diabetologia. 1996;39:1123–1125. [PubMed: 8877299]
- 145.
- Hales CN, Barker DJP. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1993;35:595–601. [PubMed: 1644236]
- 146.
- Phillips DIW. Insulin resistance as a programmed response to fetal under nutrition. Diabetologia. 1996;39:1119–1122. [PubMed: 8877298]
- 147.
- Strauss W, Hales C. Plasma insulin in minor abnormalities of glucose tolerance: a 5 year follow-up. Diabetologia. 1974;10:237–243. [PubMed: 4845725]
- 148.
- Hara H, Egusa G, Yamakido M, Kawate R. The high prevalence of diabetes mellitus and hyperinsulinemia among the Japanese-Americans living in Hawaii and Los Angeles. Diabetes Res Clin Pract. 1994;24 Suppl. 1:S37–S42. [PubMed: 7859631]
- 149.
- Berrish TS, Hetherington CS, Alberti KGMM, Walker M. Peripheral and hepatic insulin sensitivity in subjects with impaired glucose tolerance. Diabetologia. 1995;38:699–704. [PubMed: 7672492]
- 150.
- Lillioja S, Nyomba BL, Saad MF, Ferraro R, Castillo C, Bennett PH, Bogardus C. Exaggerated early insulin release and insulin resistance in a diabetes-prone population: a metabolic comparison of Pima Indians and Caucasians. J Clin Endocrinol Metab. 1991;73:866–876. [PubMed: 1890157]
- 151.
- Himsworth HP, Kerr RB. Insulin-sensitive and insulin-insensitive types of diabetes mellitus. Clin Sci. 1939;4:120–152.
- 152.
- Ginsberg H, Kimmerling G, Olefsky JM, Reaven GM. Demonstration of insulin resistance in untreated adult-onset diabetic subjects with fasting hyperglycemia. J Clin Invest. 1975;55:454–461. [PMC free article: PMC301772] [PubMed: 1117064]
- 153.
- Butterfield WJH, Whichelow MJ. Peripheral glucose metabolism in control subjects and diabetic patients during glucose, glucose-insulin, and insulin sensitivity tests. Diabetologia. 1965;1:43–53.
- 154.
- Katz H, Homan M, Jensen M, Caumo A, Cobelli C, Rizza R. Assessment of insulin action in NIDDM in the presence of dynamic changes in insulin and glucose concentration. Diabetes. 1994;43:289–296. [PubMed: 8288053]
- 155.
- Wangot A, Roovete A, Vranic M., Luft R, Efendic S. Insulin resistance and decreased insulin response to glucose in lean type II diabetes. Proc Natl Acad Sci USA. 1982;79:4432–4437. [PMC free article: PMC346686] [PubMed: 6750603]
- 156.
- Bergman RN. Lilly Lecture. Toward physiological understanding of glucose tolerance- minimal-model approach. Diabetes. 1989;38:1512–26. [PubMed: 2684710]
- 157.
- DeFronzo RA, Deibert D, Hendler R, Felig P. Insulin sensitivity and insulin binding to monocytes in maturity-onset diabetes. J Clin Invest. 1982;63:939–946. [PMC free article: PMC372035] [PubMed: 376552]
- 158.
- DeFronzo RA, Simonson D, Ferrannini E. Hepatic and peripheral insulin resistance: a common feature in non-insulin-dependent and insulin-dependent diabetes. Diabetologia. 1982;23:313–319. [PubMed: 6754515]
- 159.
- Golay A, DeFronzo RA, Ferrannini E, Simonson DC, Thorin D, Acheson K, Thiebaud D, Curchod B, Jequier E, Felber JP. Oxidative and non-oxidative glucose metabolism in non- obese Type 2 (non-insulin dependent) diabetic patients. Diabetologia. 1988;31:585–591. [PubMed: 3065112]
- 160.
- Dall'Aglio E, Chang H, Hollenbeck CB, Mondon CE, Sims C, Reaven GM. In vivo and in vitro resistance to maximal insulin-stimulated glucose disposal in insulin deficiency. Am J Physiol. 1985;249:E312–E316. [PubMed: 3898867]
- 161.
- Vuorinen-Markkola H, Koivisto VA. Yki-Järvinen H: Mechanisms of hyperglycemia- induced insulin resistance in whole body and skeletal muscle of type 1 diabetic patients. Diabetes. 1992;41:571–580. [PubMed: 1568526]
- 162.
- Golay A, Felber JP, Jequier E, DeFronzo RA, Ferrannini E. Metabolic basis of obesity and noninsulin-dependent diabetes mellitus. Diabetes Metab Rev. 1988;4:727–747. [PubMed: 3069401]
- 163.
- DeFronzo RA, Sherwin RS, Hendler R, Felig P. Insulin binding to monocytes and insulin action in human obesity, starvation, and refeeding. J Clin Invest. 1978;62:204–213. [PMC free article: PMC371755] [PubMed: 350903]
- 164.
- Firth R, Bell P, Rizza R. Insulin action in non-insulin-dependent diabetes mellitus: the relationship between hepatic and extrahepatic insulin resistance and obesity. Metabolism. 1987;36:1091–1095. [PubMed: 3312938]
- 165.
- Campbell PJ, Mandarino LJ, Gerich JE. Quantification of the relative impairment in actions of insulin on hepatic glucose production and peripheral glucose uptake in non-insulin dependent diabetes mellitus. Metabolism. 1988;37:15–21. [PubMed: 3275857]
- 166.
- Bogardus C, Lillioja S, Howard BV, Reaven G, Mott D. Relationships between insulin secretion, insulin action, and fasting plasma glucose concentration in non-diabetic and noninsulin-dependent subjects. J Clin Invest. 1984;74:1238–46. [PMC free article: PMC425290] [PubMed: 6384267]
- 167.
- Del Prato S, Simonson DC, Sheehan P, Cardi F, DeFronzo RA. Studies on the mass effect of glucose in diabetes. Evidence for glucose resistance. Diabetologia. 1997;40:687–697. [PubMed: 9222649]
- 168.
- Nielsen MF, Basu R, Wise S, Caumo A, Cobelli C, Rizza RA. Normal glucose-induced suppression of glucose production but impaired stimulation of glucose disposal in type 2 diabetes. Evidence for a concentration-dependent defect in uptake. Diabetes. 1998;47:1735–1747. [PubMed: 9792543]
- 169.
- Huang SC, Phelps ME, Hoffman EJ, Sideris K, Selin CJ, Kuhl DE. Non-invasive determination of local cerebral metabolic rate of glucose in man. Am J Physiol. 1980;238:E69–E82. [PubMed: 6965568]
- 170.
- Henry RR, Wallace P, Olefsky JM. Effects of weight loss on mechanisms of hyperglycemia in obese non-insulin dependent diabetes mellitus. Diabetes. 1986;35:990–998. [PubMed: 3527829]
- 171.
- Best JD, Judzewitsch RG, Pfeiffer MA, Beard JC, Halter JB, Porte D. The effect of chronic sulfonylurea therapy on hepatic glucose production in non-insulin-dependent diabetes mellitus. Diabetes. 1982;31:333–338. [PubMed: 6759249]
- 172.
- Tayek JA, Katz J. Glucose production, recycling, and gluconeogenesis in normals and diabetics: a mass isotopomer [U-13C] glucose study. Am J Physiol. 1996;270:E709–E717. [PubMed: 8928779]
- 173.
- Fery F. Role of hepatic glucose production and glucose uptake in the pathogenesis of fasting hyperglycemia in Type 2 diabetes: normalization of glucose kinetics by short-term fasting. J Clin Endocrinol Metab. 1994;78:536–542. [PubMed: 8126123]
- 174.
- Jeng C-Y, Sheu WHH, Fuh MM-T, Chen I, Reaven GM. Relationship between hepatic glucose production and fasting plasma glucose concentration in patients with NIDDM. Diabetes. 1994;43:1440–1444. [PubMed: 7958496]
- 175.
- DeFronzo RA, Ferrannini E, Hendler R, Felig P, Wahren J. Regulation of splanchnic and peripheral glucose uptake by insulin and hyperglycemia. Diabetes. 1983;32:35–45. [PubMed: 6336701]
- 176.
- Cherrington AD, Stevenson RW, Steiner KE, Davis MA, Myers SR, Adkins BA, Abumrad NN, Williams PE. Insulin, glucagon, and glucose as regulators of hepatic glucose uptake and production in vivo. Diabetes Metab Rev. 1987;3:307–332. [PubMed: 3552525]
- 177.
- Bergman RN, Bucolo RJ. Interaction of insulin and glucose in the control of hepatic glucose balance. Am J Physiol. 1974;227:1314–1322. [PubMed: 4440776]
- 178.
- Mevorach M, Giacca A, Aharon Y, Hawkins M, Shamoon H, Rossetti L. Regulation of endogenous glucose production by glucose per se is impaired in type 2 diabetes mellitus. J Clin Invest. 1998;102:744–753. [PMC free article: PMC508937] [PubMed: 9710443]
- 179.
- Basu R, Basu A, Nielsen M, Shah P, Rizza RA. Effect of overnight restoration of euglycemia on glucose effectiveness in type 2 diabetes mellitus. J Cin Endocrinol Metab. 1999;84:2314–2319. [PubMed: 10404795]
- 180.
- Waldhausl W, Bratusch-Marrain P, Gasic S, Korn A, Nowotny P. Insulin production rate, hepatic insulin retention, and splanchnic carbohydrate metabolism after oral glucose ingestion in hyperinsulinemic type II (non-insulin dependent) diabetes mellitus. Diabetologia. 1982;23:6–15. [PubMed: 6749586]
- 181.
- Consoli A, Nurjahn N, Capani F, Gerich J. Predominant role of gluconeogenesis in increased hepatic glucose production in NIDDM. Diabetes. 1989;38:550–556. [PubMed: 2653926]
- 182.
- Nurjhan N, Consoli A, Gerich J. Increased lipolysis and its consequences on gluconeogenesis in noninsulin-dependent diabetes mellitus. J Clin Invest. 1992;89:169–175. [PMC free article: PMC442833] [PubMed: 1729269]
- 183.
- Magnusson I, Rothman D, Katz L, Shulman R, Shulman G. Increased rate of gluconeogenesis in type II diabetes: a 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327. [PMC free article: PMC443176] [PubMed: 1401068]
- 184.
- Gastaldelli A, Baldi S, Pettiti M, Toschi E, Camastra S, Natali A, Landau BR, Ferrannini E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes. 2000;49:1367–1373. [PubMed: 10923639]
- 185.
- Gastaldelli A, Toschi E, Pettiti M, Frascerra S, Qinones-Galvan A, Sironi AM, Natali A, Ferrannini E. Effect of physiological hyperinsulinemia on gluconeogenesis in nondiabetic subjects and in type 2 diabetic patients. Diabetes. 2001;50:1807–1812. [PubMed: 11473042]
- 186.
- Stumvoll M, Perriello G, Nurjhan N, Bucci A, Welle S, Jansson P-A, Dailey G, Bier D, Jenssen T, Gerich J. Glutamine and alanine metabolism in NIDDM. Diabetes. 1996;45:863–868. [PubMed: 8666134]
- 187.
- Baron AD, Schaeffer L, Shragg P, Kolterman OG. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes. 1987;36:274–283. [PubMed: 2879757]
- 188.
- Felig P, Wahren J, Hendler R. Influence of maturity-onset diabetes on splanchnic balance after oral glucose ingestion. Diabetes. 1978;27:121–126. [PubMed: 624441]
- 189.
- Foley JE. Rationale and application of fatty acid oxidation inhibitors in treatment of diabetes mellitus. Diabetes Care. 1992;15:773–784. [PubMed: 1600836]
- 190.
- Matsuda M, DeFronzo RA, Glass L, Consoli A, Giordano M, Bressler P, DelPrato S. Glucagon dose-response curve for hepatic glucose production and glucose disposal in type 2 diabetic patients and normal individuals. Metabolism. 2002;51:1111–1119. [PubMed: 12200754]
- 191.
- Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in NIDDM. J Clin Endocrinol Metab. 1996;81:4059–4067. [PubMed: 8923861]
- 192.
- Clore JN, Stillman J, Sugerman H. Glucose-6-phosphatase flux in vitro is increased in type 2 diabetes. Diabetes. 2000;49:969–974. [PubMed: 10866049]
- 193.
- O'Brien RM, Granner DK. PEPCK gene as model of inhibitory effects of insulin on gene transcription. Diabetes Care. 1990;13:327–339. [PubMed: 2407480]
- 194.
- Sutherland C, O'Brien RM, Granner DK. New connections in the regulation of PEPCK gene expression by insulin. Phil Trans R Soc Long B. 1996;351:191–199. [PubMed: 8650266]
- 195.
- Cersosimo E, Garlick P, Ferretti J. Regulation of splanchnic and renal substrate supply by insulin in humans. Metabolism. 2000;49:676–683. [PubMed: 10831183]
- 196.
- Moller N., Rizza RA., Ford GC., Nair KS. Assessment of postabsorptive renal glucose metabolism in humans with multiple glucose tracers. Diabetes. 2001;50:747–751. [PubMed: 11289038]
- 197.
- Meyer C, Stumvoll M, Nadkarni V, Dostou J, Mitrakou A, Gerich J. Abnormal renal and hepatic glucose metabolism in type 2 diabetes mellitus. J Clin Invest. 1998;102:619–624. [PMC free article: PMC508922] [PubMed: 9691098]
- 198.
- Bjorntorp P, Berchtold P, Holm J. The glucose uptake of human adipose tissue in obesity. Eur J Clin Invest. 1971;1:480–485. [PubMed: 5121738]
- 199.
- Frayn KN, Coppack SW, Humphreys SM, Whyte PL. Metabolic characteristics of human adipose tissue in vivo. Clin Sci. 1989;76:509–516. [PubMed: 2721118]
- 200.
- Campbell P, Mandarino L, Gerich J. Quantification of the relative impairment in actions of insulin on hepatic glucose production and peripheral glucose intake in non-insulin- dependent diabetes mellitus. Metabolism 37:1 5-22, 1988. [PubMed: 3275857]
- 201.
- Capaldo B, Napoli R, Dimarino L, Picardi A, Riccardi G, Sacca L. Quantitation of forearm glucose and free fatty acid (FFA) disposal in normal subjects and type 2 diabetic patients: evidence against an essential role for FFA in the pathogenesis of insulin resistance. J Clin Endocrinol Metab. 1988;67:893–898. [PubMed: 3053749]
- 202.
- Jackson RA, Perry G, Rogers J, Advoni U, Pilkington TRE. Relationship between the basal glucose concentration, glucose tolerance, and forearm glucose uptake in maturity onset diabetes. Diabetes. 1973;22:751–761. [PubMed: 4743470]
- 203.
- Utriainen T., Takala T., Luotolahti M., Ronnemaa T., Laine H., Ruotsalainen U., Haaparanta M., Nuutila P., Yki-Jarvinen H. Insulin resistance characterizes glucose uptake in skeletal muscle but not in the heart in NIDDM. Diabetologia. 1998;41:555–559. [PubMed: 9628273]
- 204.
- Firth RG, Bell PM, Marsh HM, Hansen I, Rizza RA. Postprandial hyperglycemia in patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1986;77:1525–1532. [PMC free article: PMC424555] [PubMed: 3517067]
- 205.
- Ferrannini E, Simonson DC, Katz LD, Reichard G Jr, Bevilacqua S, Barrett EJ, Olsson M, DeFronzo RA. The disposal of an oral glucose load in patients with non-insulin dependent diabetes. Metabolism. 1988;37:79–85. [PubMed: 3275860]
- 206.
- Huang SC, Phelps ME, Hoffman EJ, Sideris K, Selin CJ, Kuhl DE. Non-invasive determination of local cerebral metabolic rate of glucose in man. Am J Physiol. 1980;238:E69–E82. [PubMed: 6965568]
- 207.
- Kelley D, Mitrakou A, Marsh H, Schwenk F, Benn J, Sonnenberg G, Arcangeli M, Aoki T, Sorensen J, Berger M, Sonksen P, Gerich J. Skeletal muscle glycolysis oxidation, and storage of an oral glucose load. J Clin Invest. 1988;81:1563–1571. [PMC free article: PMC442590] [PubMed: 3130396]
- 208.
- Jackson RA, Roshania RD, Hawa MI, Sim BM, DiSilvio L. Impact of glucose ingestion on hepatic and peripheral glucose metabolism in man: an analysis based on simultaneous use of the forearm and double isotope techniques. J Clin Endocrinol Metab. 1986;63:541–549. [PubMed: 3734029]
- 209.
- Jackson RA, Perry G, Rogers J, Advoni U, Pilkington TRE. Relationship between the basal glucose concentration, glucose tolerance, and forearm glucose uptake in maturity onset diabetes. Diabetes. 1973;22:751–761. [PubMed: 4743470]
- 210.
- Shimazu T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes Metab Rev. 1987;3:185–206. [PubMed: 3568978]
- 211.
- McMahon V, Marsh HM, Rizza RA. Effects of basal insulin supplementation on disposition of mixed meal in obese patients with NIDDM. Diabetes. 1989;38:291–303. [PubMed: 2492963]
- 212.
- Basu A, Basu R, Shah P, Vella A, Johnson M, Nair KS, Jensen MD, Schwenk WF, Rizza RA. Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism. Evidence for a defect in hepatic glucokinase activity. Diabetes. 2000;49:272–283. [PubMed: 10868944]
- 213.
- Ludvik B, Nolan JJ, Roberts A, Baloga J, Joyce M, Bell Jo M, Olefsky JM. Evidence for decreased splanchnic glucose uptake after oral glucose administration in non-insulin- dependent diabetes mellitus. J Clin Invest. 1997;100:2354–2361. [PMC free article: PMC508433] [PubMed: 9410915]
- 214.
- Reaven GM, Brand RJ, Ida Chen Y-D, Mathur AK, Goldfine I. Insulin resistance and insulin secretion are determinants of oral glucose tolerance in normal individuals. Diabetes. 1993;42:1324–1332. [PubMed: 8349044]
- 215.
- Shulman GI, Rothman DL, Jue T, Stein P, DeFronzo RA, Shulman RG. Quantitation of muscle glycogen synthesis in normal subjects and subjects with non-insulin-dependent diabetes by 13C nuclear magnetic resonance spectroscopy. New Engl J Med. 1990;322:223–228. [PubMed: 2403659]
- 216.
- Bonadonna RC, Bonora E. Glucose and free fatty acid metabolism in human obesity: relationships to insulin resistance. Diabetes Rev. 1997;5:21–51.
- 217.
- Reaven GM, Chen Y-DI, Donner CC, Fraze E, Hollenbeck CB. How insulin resistant are patients with non-insulin-dependent diabetes mellitus? J Clin Endocrinol Metab. 1985;61:32–36. [PubMed: 3889039]
- 218.
- Lillioja A, Mott DM, Zawadzki JK, Young AA, Abbott WG, Bogardus C. Glucose storage is a major determinant of in vivo 'insulin resistance' in subjects with normal glucose tolerance. J Clin Endocrinol Metab. 1986;62:922–927. [PubMed: 3514652]
- 219.
- Tripathy D, Carlsson M, Almgren P, Osomaa B, Raskinen M-R, Tuomi T, Groop LC. Insulin secretion and insulin sensitivity in relation to glucose tolerance. Lessons from the Botnia Study. Diabetes. 2000;49:975–980. [PubMed: 10866050]
- 220.
- Zimmet PZ. The pathogenesis and prevention of diabetes in adults. Diabetes Care. 1995;18:1050–1064. [PubMed: 7555542]
- 221.
- Mokdad AH, Ford ES, Bowman BA, Nelson DE, Engelgau MM, Vinicor F, Marks JS. Diabetes trends in the United States, 1990-1998. Diabetes Care. 2000;23:1278–1283. [PubMed: 10977060]
- 222.
- Reaven GM, Hollenbeck C, Jeng C-Y, Wu MS, Chen Y-DI. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 hours in patients with NIDDM. Diabetes. 1988;37:1020–1024. [PubMed: 3292322]
- 223.
- Thiebaud D, DeFronzo RA, Jacot E, Golay A, Acheson K, Maeder E, Jequier E, Felber JP. Effect of long-chain triglyceride infusion on glucose metabolism in man. Metabolism. 1982;31:1128–1136. [PubMed: 6752642]
- 224.
- De Kelley, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance. A reexamination. Diabetes. 2000;49:677–683. [PubMed: 10905472]
- 225.
- Kashyap S, Belfort R, Pratipanawatr T, Berria R, Pratipanawatr W, Bajaj M, Mandarino L, DeFronzo R, Cusi K. Chronic elevation in plasma free fatty acids impairs insulin secretion in non-diabetic offspring with a strong family history of T2DM. Diabetes. 2002;51 Suppl 2:A12.
- 226.
- Carpentier A., Mittelman SD., Bergman RN., Giacca A., Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes. 2000;49:399–408. [PubMed: 10868961]
- 227.
- Reaven GM. The fourth Musketeer - from Alexandre Dumas to Calude Bernard. Diabetologia. 1995;38:3–13. [PubMed: 7744226]
- 228.
- Goodpaster BH, Thaete FL, Kelley BE. Thigh adipose tissue distribution is associated with insulin resistance in obesity and in type 2 diabetes mellitus. Am J Clin Nutr. 2000;71:885–892. [PubMed: 10731493]
- 229.
- Greco AV, Mingrone G, Giancaterini A, Manco M, Morroni M, Cinti S, Granzotto M, Vettor R, Camastra S, Ferrannini E. Insulin resistance in morbid obesity. Reversal with intramyocellular fat depletion. Diabetes. 2002;51:144–151. [PubMed: 11756334]
- 230.
- Ryysy L, Hakkinen AM, Goto T, Vehkavaara S, Westerbacka J, Halavaara J, Yki-Jarvinen H. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes. 2000;49:749–758. [PubMed: 10905483]
- 231.
- Miyazaki Y, Mahankali A, Matsuda M, Mahankali S, Hardies J, Cusi K, Mandarino LJ, DeFronzo RA. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab. 2002;87:2784–2791. [PubMed: 12050251]
- 232.
- Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963:1785–789. [PubMed: 13990765]
- 233.
- Wititsuwannakul D, Kim K. Mechanism of palmityl coenzyme A inhibition or liver glycogen synthase. J Biol Chem. 1977;252:7812–7817. [PubMed: 410808]
- 234.
- Golay A, Felber JP, Meyer HU, Curchod B, Maeder E, Jequier E. Study on lipid metabolism in obesity diabetes. Metabolism. 1984;33:111–116. [PubMed: 6694554]
- 235.
- Felber JP, Meyer HU, Curchod B, Iselin HU, Rousselle J, Maeder E, Pahud P, Jequier E. Glucose storage and oxidation in different degrees of human obesity measured by continuous indirect calorimetry. Diabetologia. 1981;20:39–44. [PubMed: 7009283]
- 236.
- Roden M, Krssak M, Stingl H, Gruber S, Hofer A, Furnsinn C, Moser E, Waldhausl W. Rapid impairment of skeletal muscle glucose transport/phosphorylation by free fatty acids in humans. Diabetes. 1999;48:358–364. [PubMed: 10334314]
- 237.
- Bonadonna RC, Groop LC, Zych K, Shank M, DeFronzo RA. Dose dependent effect of insulin on plasma free fatty acid turnover and oxidation in humans. Am J Physiol. 1990;22:736–750. [PubMed: 2240211]
- 238.
- Boden G. Free fatty acids, insulin resistance, and type 2 diabetes mellitus. Proc Assoc Am Physicians. 1999;111:241–248. [PubMed: 10354364]
- 239.
- Boden G, Chen X, Ruiz J, White JV, Rossetti L. Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest. 1994;93:2438–2446. [PMC free article: PMC294452] [PubMed: 8200979]
- 240.
- Krssak M, Falk Petersen K, Dresner A, DiPietro L, Vogel SM, Rothman DL, Roden M, Shulman GI. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1HNMR spectroscopy study. Diabetologia. 1999;42:113–116. [PubMed: 10027589]
- 241.
- Jacobs S, Machann J, Rett K, Brechtel K, Volk A, Renn W, Maerker E, Matthaei S, Schick F, Claussen CD, Hearing H-U. Association of increased intramyocellular lipid content with insulin resistance in lean non-diabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–1119. [PubMed: 10331418]
- 242.
- Kelley DE, Goodpaster BH. Skeletal muscle triglyceride. An aspect of regional adiposity and insulin resistance. Diabetes Care. 2001;24:933–941. [PubMed: 11347757]
- 243.
- Kelley DE, Mandarino LJ. Fuel selection in human skeletal muscle in insulin resistance. Diabetes. 2000;49:677–683. [PubMed: 10905472]
- 244.
- Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkB-a. Diabetes. 2002;51:2005–2011.
- 245.
- Ellis BA, Poynten A, Lowy AJ, Furler SM, Chisholm DJ, Kraegen EW, Cooney GJ. Long- chain acyl-CoA esters as indicators of lipid metabolism and insulin sensitivity in rat and human muscle. Am J Physiol. 2000;279:E554–E560. [PubMed: 10950822]
- 246.
- De Fea K, Roth RA. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry. 1997;36:12939–12947. [PubMed: 9335553]
- 247.
- Ravichandran LV, Esposito DL, Chen J, Quon MJ. PKC-zeta phosphorylates IRS-1 and impairs its ability to activate P-3-kinase in response to insulin. J Biol Chem. 2001;276:3543–3549. [PubMed: 11063744]
- 248.
- Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, Olefsky JM. Fatty acid- induced insulin resistance: decreased muscle PI3K activation but unchanged Akt phosphorylation. J Clin Endocrinol Metab. 2002;87:226–234. [PubMed: 11788651]
- 249.
- Dresner A, Laurent D, Marcucci M, Griffin ME, Dufour S, Cline SW, Slezak LA, Andersen DK, Hundal RS, Rothman DL, Petersen KF, Shulman GI. Effects of free fatty acids on glucose transport and IRS-1 associated phosphatidylinositol 3-kinase activity. J Clin Invest. 1999;103:253–259. [PMC free article: PMC407880] [PubMed: 9916137]
- 250.
- Thompson AL, Cooney GJ. Acyl-CoA inhibition of hexokinase in rat and human skeletal muscle is a potential mechanism of lipid-induced insulin resistance. Diabetes. 2000;49:1761–1764. [PubMed: 11078441]
- 251.
- Tippett PS, Neet KE. An allosteric model for the inhibition of glucokinase by long chain acyl coenzyme A. J Biol Chem. 1982;257:14846–14852. [PubMed: 7130182]
- 252.
- Schmitz-Peiffer C, Craig DL, Biden TJ. Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem. 1999;274:24202–24210. [PubMed: 10446195]
- 253.
- Baron AD. Hemodynamic actions of insulin. Am J Physiol. 1994;267:E187–E202. [PubMed: 8074198]
- 254.
- Mather K, Laakso M, Edelman S, Hook G, Baron A. Evidence for physiological coupling of insulin-mediated glucose metabolism and limb blood flow. Am J Physiol Endocrinol Metab. 2000;279:E1264–E1270. [PubMed: 11093913]
- 255.
- Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–1179. [PMC free article: PMC295191] [PubMed: 8083357]
- 256.
- Utriainen T, Nuutila P, Takala T, Vicini P, Ruotsalainen U, Ronnemaa T, Tolvanen T, Raitakari M, Haaparanta M, Kirvela O, Cobelli C, Yki-Jarvinen H. Intact stimulation of skeletal muscle blood flow, its heterogeneity and redistribution, but not of glucose uptake in non-insulin-dependent diabetes mellitus. J Clin Invest. 1997;100:777–785. [PMC free article: PMC508248] [PubMed: 9259575]
- 256A.
- Cersosimo E, DeFronzo RA. Insulin Reistance and Endothelial Dysfunction: the road map for cardiovscular disease. Diabetes Metab Res Rev. 2006;22:423–436. [PubMed: 16506274]
- 256B.
- Coggins M, Lidner J, Rattigan S, et al. Physiologic hyperinsulinemia enhance human skeletal muscle muscle perfuioson by capillary rectuiment. Diabetes. 2001;50:2682–2690. [PubMed: 11723050]
- 257.
- Steinberg HO, Paradisi G, Hook G, Crowder K, Cronin J, Baron AD. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes. 2000;49:1231–1238. [PubMed: 10909983]
- 258.
- Kreutzenberg SV, Crepaldi C, Marchetto S, Calo L, Tiengo A, Del Prato S, Avogaro A. Plasma free fatty acids and endothelium-dependent vasodilation: effect of chain-length and cyclooxygenase inhibition. J Clin Endocrinol Metab. 2000;85:793–798. [PubMed: 10690892]
- 259.
- Davda RK, Chandler LJ, Guzman NJ. Protein kinase C modulates receptor-independent activation of endothelial nitric oxide synthase. Eur J Pharmacol. 1994;266:237–244. [PubMed: 7513644]
- 260.
- Zeng G, Quon MJ. Insulin stimulated production of nitric oxide is inhibited by wortmannin: direct measurement in vascular endothelial cells. J Clin Invest. 1996;98:894–898. [PMC free article: PMC507502] [PubMed: 8770859]
- 261.
- Exton JH, Corbin JG, Park CR. Control of gluconeogenesis in liver. IV. Differential effects of fatty acids and glucagon on ketogenesis and gluconeogenesis in the perfused rat liver. J Biol Chem. 1969;244:4095–4102. [PubMed: 4308166]
- 262.
- Bahl JJ, Matsuda M, DeFronzo RA, Bressler R. In vitro and in vivo suppression of gluconeogenesis by inhibition of pyruvate carboxylase. Biochem Pharmacol. 1997;53:67–74. [PubMed: 8960065]
- 263.
- Massillon D, Barzailai N, Hawkins M, Prus-Wetheimer D, Rossetti L. Induction of hepatic glucose-6-phosphatase gene expression by lipid infusion. Diabetes. 1997;46:153–157. [PubMed: 8971097]
- 264.
- Chen X, Iqbal N, Boden G. The effect of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest. 1999;103:365–372. [PMC free article: PMC407905] [PubMed: 9927497]
- 265.
- Roden M, Stingl H, Chandramouli V, Schumann WC, Hofer A, Landau BR, Nowotny P, Waldhausl W, Shulman GI. Effects of free fatty acid elevation on postabsorptive endogenous glucose production and gluconeogenesis in humans. Diabetes. 2000;49:701–707. [PubMed: 10905476]
- 266.
- Lewis GF., Vranic M., Harley P., Giacca A. Fatty acids mediate the acute extrahepatic effects of insulin on hepatic glucose production in humans. Diabetes. 1997;46:1111–11119. [PubMed: 9200644]
- 267.
- Rebrin K., Steil GM., Getty L., Bergman RN. Free fatty acid as a link in the regulation of hepatic glucose output by peripheral insulin. Diabetes. 1995;44:1038–45. [PubMed: 7657026]
- 268.
- Ferrannini E, Barrett EJ, Bevilacqua S, DeFronzo RA. Effect of fatty acids on glucose production and utilization in man. J Clin Invest. 1983;72:1737–1747. [PMC free article: PMC370462] [PubMed: 6138367]
- 269.
- Bevilacqua S, Bonadonna R, Buzzigoli G, Boni C, Ciociaro D, Maccari F, Giorico MA, Ferrannini E. Acute elevation of free fatty acid levels leads to hepatic insulin resistance in obese subjects. Metabolism. 1987;36:502–506. [PubMed: 3553852]
- 270.
- Golay A, Swislocki ALM, Chen Y-DI, Reaven GM. Relationships between plasma free fatty acid concentration, endogenous glucose production, and fasting hyperglycemia in normal and non-insulin-dependent diabetic individuals. Metabolism. 1987;36:692–696. [PubMed: 3298937]
- 271.
- Baron AD, Schaeffer L, Shragg P, Kolterman OG. Role of hyperglucagonemia in maintenance of increased rates of hepatic glucose output in type II diabetics. Diabetes. 1987;36:274–283. [PubMed: 2879757]
- 272.
- Bjorntorp P. Metabolic implications of body fat distribution. Diabetes Care. 1991;14:1132–1143. [PubMed: 1773700]
- 273.
- Arner P. Regional adipocity in man. J Endocrinol. 1997;155:191–192. [PubMed: 9415044]
- 274.
- Saltiel AR, Kahn CR. Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. [PubMed: 11742412]
- 275.
- Virkamaki A, Ueki K, Kahn CR. Protein-protein interaction in insulin signaling and the molecular mechanisms of insulin resistance. J Clin Invest. 1999;103:931–943. [PMC free article: PMC408269] [PubMed: 10194465]
- 276.
- Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. [PMC free article: PMC314316] [PubMed: 10903329]
- 277.
- Whitehead JP, Clark SF, Urso B, James DE. Signalling through the insulin receptor. Current Opinion in Cell Biology. 2000;12:222–228. [PubMed: 10712920]
- 278.
- Wilden PA, Kahn CR. The level of insulin receptor tyrosine kinase activity modulates the activities of phosphatidylinositol 3-kinase, microtubule-associated protein, and S6 kinases. Mol Endo. 1994;8:558–567. [PubMed: 8058065]
- 279.
- Haring HU, Mehnert H. Pathogenesis of type 2 (non-insulin-dependent) diabetes mellitus: candidates for a signal transmitter defect causing insulin resistance of the skeletal muscle. Diabetologia. 1993;36:176–182. [PubMed: 8385036]
- 280.
- Wajtaszewski JFP, Hansen BF, Kiens B, Richter EA. Insulin signaling in human skeletal muscle. Time course and effect of exercise. Diabetes. 1997;46:1775–1781. [PubMed: 9356025]
- 281.
- Shepherd PR, Kahn BB. Glucose transporters and insulin action. Implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248–257. [PubMed: 10413738]
- 282.
- Garvey WT. Insulin action and insulin resistance: diseases involving defects in insulin receptors, signal transduction, and the glucose transport effector system. Am J Med. 1998;105:331–345. [PubMed: 9809695]
- 283.
- Chou DK, Dull TJ, Russell DS, Gherzi R, Lebwohl D, Ullrich A, Rosen OM. Human insulin receptors mutated at the ATP-binding site lack protein tyrosine kinase activity and fail to mediate postreceptor effects of insulin. J Biol Chem. 1987;262:1842–1847. [PubMed: 3100537]
- 284.
- Ebina Y, Araki E, Tiara F, Shimada F, Mari M, Craik CS, Siddle K, Pierce SB, Roth RA, Rutter WJ. Replacement of lysine residue 1030 in the putative ATP-binding region of the insulin receptor abolishes insulin and antibody-stimulated glucose uptake and receptor kinase activity. Proc Natl Acad Sci USA. 1987;84:704–708. [PMC free article: PMC304284] [PubMed: 3101064]
- 285.
- Ellis LE, Clauser E, Morgan ME, Roth RA, Rutter WJ. Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-dexoyglucose. Cell. 1986;45:721–732. [PubMed: 3518947]
- 286.
- White MF, Livingston JN, Backer JM, Lauria V, Duli TJ, Ullrich A, Kahn DR. Mutation of the insulin receptor at tyrosine 960 inhibits signal transmission but does not affect its tyrosine kinase activity. Cell. 1992;54:641–649. [PubMed: 2842060]
- 287.
- Kerouz N.J., Horsch D., Pons S., Kahn C.R. Differential regulation of insulin receptor substrates-1 and -2 (IRS-1 and IRS-2) and phosphatidylinositol 3-kinase isoforms in liver and muscle of the obese diabetic (ob/ob) mouse. J Clin Invest. 1997;100:3164–3172. [PMC free article: PMC508530] [PubMed: 9399964]
- 288.
- Baumann CA, Ribon V., Kanzaki M., Thurmond DC., Mora S., Shigematsu S., Bickel PE., Pessin JE. Saltiel AR. CAP defines a second signalling pathway required for insulin- stimulated glucose transport. Nature. 2000;407:202–207. [PubMed: 11001060]
- 289.
- Chiang SH, Baumann CA., Kanzaki M., Thurmond DC., Watson RT., Neudauer CL., Macara IG., Pessin JE., Saltiel AR. Insulin-stimulated GLUT4 translocation requires the CAP- dependent activation of TC10. Nature. 2001;410:944–948. [PubMed: 11309621]
- 290.
- Backer JM, Myers MG Jr, Shoelson SE, Chin DJ, Sun XJ, Miralpeix M., Hu P, Margolis B., Skoinik EY, Schlessinger J, White MF. The phosphatidylinositol 3' kinase is activated by association with IRS-1 during insulin stimulation. EMBO J. 1992;11:3469–3479. [PMC free article: PMC556882] [PubMed: 1380456]
- 291.
- Sun XJ, Miralpeix M, Myers Jr MG, Glasheen EM, Backer JM, Kahn CR, White MF. The expression and function of IRS-1 in insulin signal transmission. J Biol Chem 267-22662- 22672, 1992. [PubMed: 1385403]
- 292.
- Cross DA., Alessi DR., Cohen P., Andjelkovich M., Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. [PubMed: 8524413]
- 293.
- Brady MJ., Nairn AC., Saltiel AR. The regulation of glycogen synthase by protein phosphatase 1 in 3T3-L1 adipocytes. Evidence for a potential role for DARPP-32 in insulin action. J Biol Chem. 1997;272:29698–703. [PubMed: 9368038]
- 294.
- Okada T, Sakuma L, Fukui Y, Hazeki O, Ui M. Essential role of phosphatidylinositol 3- kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. J Biol Chem. 1994;269:3568–3573. [PubMed: 8106400]
- 295.
- Cross D, Alessi D, Vandenheed J, McDowell H, Hundal H, Cohen P. The inhibition of glycogen synthase kinase-3 by insulin or insulin-like growth factor 1 in the rat skeletal muscle cell line L6 is blocked by wortmannin but not rapamycin. Biochem J. 1994;303:21–26. [PMC free article: PMC1137550] [PubMed: 7945242]
- 296.
- Osawa H, Sutherland C, Robey R, Printz R, Granner D. Analysis of the signaling pathway involved in the regulation of hexokinase II gene transcription by insulin. J Biol Chem. 1996;271:16690–16694. [PubMed: 8663315]
- 297.
- Thomas G. Hall MN. TOR signalling and control of cell growth. Current Opinion in Cell Biology. 1997;9:782–7. [PubMed: 9425342]
- 298.
- Nave BT., Ouwens M., Withers DJ., Alessi DR., Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J. 1999;344(Pt 2):427–31. [PMC free article: PMC1220660] [PubMed: 10567225]
- 299.
- Miron M., Verdu J., Lachance PE., Birnbaum MJ., Lasko PF., Sonenberg N. The translational inhibitor 4E-BP is an effector of PI (3)K/Akt signalling and cell growth in Drosophila. Nature Cell Biology. 2001;3:596–601. [PubMed: 11389445]
- 300.
- Shimomura I., Bashmakov Y., Ikemoto S., Horton JD., Brown MS., Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Nat Acad Sci USA. 1999;96:13656–61. [PMC free article: PMC24120] [PubMed: 10570128]
- 301.
- Cusi K, Maezono K, Osman A, Pendergrass M, Patti ME, Pratipanawatr T, DeFronzo RA, Kahn CR, Mandarino LJ. Insulin resistance differentially affects the PI 3-kinase and MAP kinase-mediated signaling in human muscle. J Clin Invest. 2000;105:311–320. [PMC free article: PMC377440] [PubMed: 10675357]
- 302.
- Hsueh WA, Law RE. Insulin signaling in the arterial wall. Am J Cardiol. 1999;84:21J–24J. [PubMed: 10418854]
- 303.
- Xi X-P, Graf K, Goetze S, Hsueh WA, Law RE. Inhibition of MAP kinase blocks insulin- mediated DNA synthesis and transcriptional activation of c-fos by Elk-1 in vascular smooth muscle cells. FEBS Lett. 1997;417:283–286. [PubMed: 9409734]
- 304.
- Boulton TG, Nye SH., Robbins DJ., Ip NY., Radziejewska E., Morgenbesser SD., DePinho RA., Panayotatos N., Cobb MH., Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–75. [PubMed: 2032290]
- 305.
- Lazar DF., Wiese RJ., Brady MJ., Mastick CC., Waters SB., Yamauchi K., Pessin JE., Cuatrecasas P., Saltiel AR. Mitogen-activated protein kinase kinase inhibition does not block the stimulation of glucose utilization by insulin. J Biol Chem. 1995;270:20801–7. [PubMed: 7657664]
- 306.
- Dorrestijn J, Ouwens DM, Van den Berghe N, Box JL, Maassen JA. Expression of dominant-negative Ras mutant does not affect stimulation of glucose uptake and glycogen synthesis by insulin. Diabetologia. 1996;39:558–563. [PubMed: 8739915]
- 307.
- Dent P, Lavoinne A, Nakielny S, Caudwell FB, Watt P, Cohen F. The molecular mechanisms by which insulin stimulates glycogen synthesis in mammalian skeletal muscle. Nature. 1990;348:302–307. [PubMed: 2123524]
- 308.
- Cohen P. The structure and regulation of protein phosphatases. Annu Rev Biochem. 1989;58:453–508. [PubMed: 2549856]
- 309.
- Newgard CB, Brady MJ, O'Doherty RB, Saltiel AR. Organizing glucose disposal. Emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes. 2000;49:1967–1977. [PubMed: 11117996]
- 310.
- Sheperd PR, Nave BT, Siddle K. Insulin stimulation of glycogen synthesis and glycogen synthase activity is blocked by wortmannin and rapamycin in 3T3L1 adipocytes: evidence for the involvement of phosphoinositide 3 kinase and p70 ribosomal protein S6 kinase. Biochem J. 1995;305:25–28. [PMC free article: PMC1136424] [PubMed: 7826337]
- 311.
- Freidenberg GR, Henry RR, Klein HH, Reichart DR, Olefsky JM. Decreased kinase activity of insulin receptors from adipocytes of non-insulin-dependent diabetic studies. J Clin Invest. 1987;79:240–250. [PMC free article: PMC424032] [PubMed: 3540010]
- 312.
- Caro JF, Sinha MK, Raju SM, Ittoop O, Pories WJ, Flickinger EG, Meelheim D, Dohm GL. Insulin receptor kinase in human skeletal muscle from obese subjects with and without non- insulin dependent diabetes. J Clin Invest 79:1330-7.1987. [PMC free article: PMC424378] [PubMed: 3033021]
- 313.
- Caro JF, Ittoop O, Pories WJ, Meelheim D, Flickinger EG, Thomas F, Jenquin M, Silverman JF, Khazanie PG, Sinha MK. Studies on the mechanism of insulin resistance in the liver from humans with non-insulin-dependent diabetes. Insulin action and binding in isolated hepatocytes, insulin receptor structure, and kinase activity. J Clin Invest. 1986;78:249–58. [PMC free article: PMC329556] [PubMed: 3522628]
- 314.
- Molina JM, Ciaraldi TP, Brady D, Olefsky JM. Decreased activation rate of insulin-mediated glucose transport in adipocytes from obese and NIDDM subjects. Diabetes. 1989;38:991–995. [PubMed: 2666204]
- 315.
- Trichitta V, Brunetti A, Chiavetta A, Benzi L, Papa V, Vigneri R. Defects in insulin-receptor internalization and processing in monocytes of obese subjects and obese NIDDM patients. Diabetes. 1989;38:1579–1584. [PubMed: 2684714]
- 316.
- Comi RJ, Grunberger G, Gorden P. Relationship of insulin binding and insulin-stimulated tyrosine kinase activity is altered in type II diabetes. J Clin Invest. 1987;79:453–462. [PMC free article: PMC424100] [PubMed: 3543053]
- 317.
- Klein HH, Vestergaard H, Kotzke G, Pedersen O. Elevation of serum insulin concentration during euglycemic hyperinsulinemic clamp studies leads to similar activation of insulin receptor kinase in skeletal muscle of subjects with and without NIDDM. Diabetes. 1995;344:1310–1317. [PubMed: 7589829]
- 318.
- Obermaier-Kusser B, White MF, Pongratz DE, Su Z, Ermel B, Muhlbacher C, Haring HU. A defective intramolecular autoactivation cascade may cause the reduced kinase activity of the skeletal muscle insulin receptor from patients with non-insulin-dependent diabetes mellitus. J Biol Chem. 1989;264:9497–9503. [PubMed: 2722845]
- 319.
- Arner P, Einarsson K, Ewerth S, Livingston J. Studies on the human liver insulin receptor in non-insulin-dependent diabetes mellitus. J Clin Invest. 1986;77:1716–1718. [PMC free article: PMC424580] [PubMed: 3517068]
- 320.
- Kashiwagi A, Verso MA, Andrews J, Vasquez B, Reaven G, Foley JE. In vitro insulin resistance of human adipocytes isolated from subjects with non-insulin-dependent diabetes mellitus. J Clin Invest. 1983;72:1246–1254. [PMC free article: PMC370408] [PubMed: 6355180]
- 321.
- Lonnroth P, Digirolamo M, Krotkiewski M, Smith U. Insulin binding and responsiveness in fat cells from patients with reduced glucose tolerance and type II diabetes. Diabetes. 1983;32:748–754. [PubMed: 6347772]
- 322.
- Olefsky JM, Reaven GM. Insulin binding in diabetes. Relationships with plasma insulin levels and insulin sensitivity. Diabetes. 1977;26:680–688. [PubMed: 873075]
- 323.
- Cocozza S, Procellini A, Riccardi G, Monticelli A, Condorelli G, Ferrara A, Pianese L, Miele C, Capaldo B, Beguinot F, Varrone S. NIDDM associated with mutation in tyrosine kinase domain of insulin receptor gene. Diabetes. 1992;41:521–526. [PubMed: 1607076]
- 324.
- Moller DE, Yakota A, Flier JS. Normal insulin receptor cDNA sequence in Pima Indians with non-insulin-dependent diabetes mellitus. Diabetes. 1989;38:1496–1500. [PubMed: 2620783]
- 325.
- Kusari J, Verma US, Buse JB, Henry RR, Olefsky JM. Analysis of the gene sequences of the insulin receptor and the insulin-sensitive glucose transporter (GLUT4) in patients with common-type non-insulin-dependent diabetes mellitus. J Clin Invest. 1991;88:1323–1330. [PMC free article: PMC295602] [PubMed: 1918382]
- 326.
- Nyomba BL, Ossowski VM, Bogardus C, Mott DM. Insulin-sensitive tyrosine kinase relationship with in vivo insulin action in humans. Am J Physiol. 1990;258:E964–E974. [PubMed: 2163202]
- 327.
- Nolan JJ, Friedenberg G, Henry R, Reichart D, Olefsky JM. Role of human skeletal muscle insulin receptor kinase in the in vivo insulin resistance of noninsulin-dependent diabetes and obesity. J Clin Endocrinol Metab. 1994;78:471–477. [PubMed: 8106637]
- 328.
- Krook A, Bjornholm M, Galuska D, Jiang XJ, Fahlman R, Myers MG, Wallberg-Henriksson H, Zierath JR. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes. 2000;49:284–292. [PubMed: 10868945]
- 329.
- Bak JF, Moller N, Schmitz O, Saaek A, Pedersen O. In vivo insulin action and muscle glycogen synthase activity in type 2 (non insulin dependent) diabetes mellitus: effects of diet treatment. Diabetologia. 1992;35:777–794. [PubMed: 1511806]
- 330.
- Freidenberg GR, Reichart D, Olefsky JM, Henry RR. Reversibility of defective adipocyte insulin receptor kinase activity in non-insulin dependent diabetes mellitus. Effect of weight loss. J Clin Invest. 1988;82:1398–1406. [PMC free article: PMC442697] [PubMed: 3170749]
- 331.
- Kellerer M, Kroder G, Tippmer S, Berti L, Kiehn L, Mosthaf L, Haring H. Troglitazone prevents glucose-induced insulin resistance of insulin receptor in rat-1 fibroblasts. Diabetes. 1994;43:447–453. [PubMed: 7508875]
- 332.
- Pratipanawatr W, Pratipanawatr T, Cusi K, Berria R, Adams JM, Jenkinson CP, Maezono K, DeFronzo RA, Mandarino LJ. Skeletal muscle insulin resistance in normoglycemic subjects with a strong family history of type 2 diabetes is associated with decreased insulin- stimulated insulin receptor substrate-1 tyrosine phosphorylation. Diabetes. 2001;50:2572–2578. [PubMed: 11679436]
- 333.
- Laville M, Auboeuf D, Khalfallah Y, Vega N, Riou JP, Vidal H. Acute regulation by insulin of phosphatidylinositol-3-kinase, Rad, Glut 4, and lipoprotein lipase in mRNA levels in human muscle. J Clin Invest. 1996;98:43–49. [PMC free article: PMC507399] [PubMed: 8690802]
- 334.
- Kim Y-B, Nikoulina S, Ciaraldi TP, Henry RR, Kahn BB. Normal insulin-dependent activation of Akt/protein kinase B, with diminished activation of phosphoinositide 3-kinase, in muscle in type 2 diabetes. J Clin Invest. 1999;104:733–741. [PMC free article: PMC408433] [PubMed: 10491408]
- 335.
- Andreelli F, Laville M, Ducluzeau P-H, Vega N, Vallier P, Khalfallah Y, Riou J-P, Vidal H. Defective regulation of phosphatidylinositol-3-kinase gene expression in skeletal muscle and adipose tissue of non-insulin-dependent diabetes mellitus patients. Diabetologia. 1999;42:358–364. [PubMed: 10096790]
- 336.
- Folli F, Saad JA, Backer JM, Kahn CR. Regulation of phosphatidylinositol 3-kinase activity in liver and muscle of animal models of insulin-resistant and insulin-deficient diabetes mellitus. J Clin Invest. 1993;92:1787–1794. [PMC free article: PMC288341] [PubMed: 7691886]
- 337.
- Imai Y, Philippe N, Accili D, Taylor SI. Expression of variant forms of insulin receptor substrate-1 identified in patients with noninsulin dependent diabetes mellitus. J Clin Endocrinol Metab. 1997;82:4201–4207. [PubMed: 9398740]
- 338.
- Hitman GA, Hawrammi K, McCarthy MI, Viswanathan M, Snehalatha C, Ramachandran A, Tuomilehto J, Tumilehto-Wolf E, Nissenen A, Pedersen O. Insulin receptor substrate-1 gene mutations in NIDDM: implication for the study of polygenic disease. Diabetologia. 1995;38:481–486. [PubMed: 7796990]
- 339.
- Pratley RE, Thompson DB., Prochazka M., Baier L., Mott D., Ravussin E., Sakul H., Ehm MG., Burns DK., Foroud T., Garvey WT., Hanson RL., Knowler WC., Bennett PH., Bogardus C. An autosomal genomic scan for loci linked to prediabetic phenotypes in Pima Indians. J Clin Invest. 1998;101:1757–64. [PMC free article: PMC508758] [PubMed: 9541507]
- 340.
- Jiang ZY., Lin YW., Clemont A., Feener EP., Hein KD., Igarashi M., Yamauchi T., White MF., King GL. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J Clin Invest. 1999;104:447–57. [PMC free article: PMC408521] [PubMed: 10449437]
- 340A.
- Cersosimo E. XiaoJing X, Musi N. Role of insulin signaling in vascular smooth muscle cell migration, proliferation and inflammation. Am J Physiol: Cell Physiol. 2012 Feb 15th;:C652–C657. [PubMed: 22094332]
- 340B.
- Cersosimo E. XiaoJing X, Upala S, Triplitt C, Musi N. Acute Insulin Resistance Stimulates and Insulin Sensitization Attenuates Vascular Smooth Muscle Cell Migration and Proliferation. Physiological Reports. 2014 August;2(Iss. 8):e12123. Vol. [PMC free article: PMC4246575] [PubMed: 25138792]
- 341.
- Sasaoka T., Draznin B., Leitner JW., Langlois WJ., Olefsky JM. Shc is the predominant signaling molecule coupling insulin receptors to activation of guanine nucleotide releasing factor and p21ras-GTP formation. J Biol Chem. 1994;269:10734–8. [PubMed: 8144662]
- 342.
- De Fea K., Roth RA. Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J Biol Chem. 1997;272:31400–6. [PubMed: 9395471]
- 343.
- Dunaif A., Xia J., Book CB., Schenker E., Tang Z. Excessive insulin receptor serine phosphorylation in cultured fibroblasts and in skeletal muscle. A potential mechanism for insulin resistance in the polycystic ovary syndrome. J Clin Invest. 1995;96:801–10. [PMC free article: PMC185266] [PubMed: 7635975]
- 344.
- Kono T, Robinson FW, Blevins TL, Ezaki O. Evidence that translocation of the glucose transport activity is the major mechanism of insulin action on glucose transport fat cells. J Biol Chem. 1982;257:10942–10947. [PubMed: 7050125]
- 345.
- Bell Gl, Kayano T, Buse JB, Burant CF, Takeda J, Lin D, Fikomoto H, Seino S. Molecular biology of mammalian glucose transporters. Diabetes Care. 1990;13:198–200. [PubMed: 2407475]
- 346.
- Joost H-G, Bell GI, Best JD, Birnbaum MJ, Charron MJ, Chen YT, Doege H, James DE, Lodish HF, Moley KH, Moley JF, Mueckler M, Rogers S, Schurmann A, Seino S, Thorens B. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab. 2002;282:E974–E976. [PubMed: 11882521]
- 347.
- Colowick SP. The hexokinases. In Boyer PD (ed.) The Enzymes, vol. 9. New York: Academic Press, pp 1-48, 1973.
- 348.
- Printz RL, Koch S, Potter LR, O'Doherty RM, Tiesinga JJ, Moritz S, Granner DK. Hexokinase II mRNA and gene structure, regulation by insulin, and evolution. J Biol Chem. 1993;268:5209–5219. [PubMed: 8444897]
- 349.
- Rogers PA, Fisher RA, Harris H. An electrophoretic study of the distribution and properties of human hexokinases. Biochem Genet. 1975;13:857–866. [PubMed: 1239274]
- 350.
- Magnuson MA, Andreone IL, Printz RL, Koch S, Granner DK. The glucokinase gene: structure and regulation by insulin. Proc Natl Acad Sci USA. 1989;86:4838–4842. [PMC free article: PMC297510] [PubMed: 2662183]
- 351.
- Matchinsky FM. Banting Lecture 1995. A lesson in metabolic regulation inspired by the glucokinase glucose sensor paradigm. Diabetes. 1996;45:223–241. [PubMed: 8549869]
- 352.
- Mandarino LJ, Campbell PJ, Gottesman IS, Gerich JE. Abnormal coupling of insulin receptor binding in non-insulin-dependent diabetes. Am J Physiol. 1984;247:E688–E692. [PubMed: 6388357]
- 353.
- Garvey WT, Huecksteadt TP, Mattaei S, Olefsky JM. Role of glucose transporters in the cellular insulin resistance of type II non-insulin dependent diabetes mellitus. J Clin Invest. 1988;81:1528–1536. [PMC free article: PMC442586] [PubMed: 3366906]
- 354.
- Zierath JR, He L, Guma A, Odegoard Wahlstrom E, Klip A, Wallenberg-Henriksson H. Insulin action on glucose transport and plasma membrane GLUT4 content in skeletal muscle from patients with NIDDM. Diabetologia. 1996;39:1180–1189. [PubMed: 8897005]
- 355.
- Krook A, Bjornholm M, Galuska D, Jiang XJ, Fahlman R, Myers MG. Wallenberg- Henriksson H, Zierath JR. Characterization of signal transduction and glucose transport in skeletal muscle from type 2 diabetic patients. Diabetes. 2000;49:284–282. [PubMed: 10868945]
- 356.
- Andreasson K, Galuska D, Thorne T, Sonnenfeld T, Wallberg-Henriksson H. Decreased insulin-stimulated 3-O-methylglucose transport in in vitro incubated muscle strips from type II diabetic subjects. Acta Physiol Scan. 1991;142:255–260. [PubMed: 1877373]
- 357.
- Kahn BB, Shulman GI, DeFronzo RA, Cushman SW, Rossetti L. Normalization of blood glucose in diabetic rats with phlorizin treatment reverses insulin-resistant glucose transport in adipose cells without restoring glucose transporter gene expression. J Clin Invest. 1991;87:561–570. [PMC free article: PMC296344] [PubMed: 1991839]
- 358.
- Pedersen O, Bak J, Andersen P, Lund S, Moller DE, Flier JS, Kahn BB. Evidence against altered expression of GLUT1 or GLUT4 in skeletal muscle of patients with obesity or NIDDM. Diabetes. 1990;39:865–870. [PubMed: 2354749]
- 359.
- Eriksson J, Koranyi L, Bourey R, Schalin-Jantti C, Widen E, Mueckler M, Permutt AM, Groop LC. Insulin resistance in type 2 (non-insulin-dependent) diabetic patients and their relatives is not associated with a defect in the expression of the insulin-responsive glucose transporter (GLUT-4) gene in human skeletal muscle. Diabetologia. 1992;35:143–147. [PubMed: 1547918]
- 360.
- Schalin-Jantti C, Yki-Jarvinen H, Koranyi L, Bourey R, Lindstrom J, Nikula-Ijas P, Franssila-Kallunki A, Groop LC. Effect of insulin on GLUT-4 mRNA and protein concentrations in skeletal muscle of patients with NIDDM and their first-degree relatives. Diabetologia. 1994;37:401–407. [PubMed: 8063042]
- 361.
- Garvey WT, Maianu L, Hancock JA, Golichowski AM, Baron A. Gene expression of glut4 in skeletal muscle from insulin-resistant patients with obesity, IGT, GDM, and NIDDM. Diabetes. 1992;41:465–475. [PubMed: 1535055]
- 362.
- Kusari J, Verma US, Buse JB, Henry RR, Olefsky JM. Analysis of the gene sequences of the insulin receptor and the insulin-sensitive glucose transporter (GLUT 4) in patients with common type non insulin dependent diabetes mellitus. J Clin Invest. 1991;88:1323–1330. [PMC free article: PMC295602] [PubMed: 1918382]
- 363.
- Choi WH, O'Rahilly S, Rees A, Morgan R, Flier JS, Moller DE. Molecular scanning of the insulin-responsive glucose transporter (GLUT 4) gene in patients with non-insulin dependent diabetes mellitus. Diabetes. 1991;40:1712–1718. [PubMed: 1756912]
- 364.
- Sinha M, Raineri-Maldonado C, Buchanan C, Pories W, Carter-Su C, Pilch P, Caro J. Adipose tissue glucose transporters in NIDDM: decreased levels of muscle/fat isoform. Diabetes. 1991;40:474–477. [PubMed: 2010047]
- 365.
- Guma A, Zierath JR, Wallberg-Henriksson H, Klip A. Insulin induces translocation of GLUT-4 glucose transporters in human skeletal muscle. Am J Physiol. 1995;268:E613–E622. [PubMed: 7733259]
- 366.
- Goodyear LJ, Hirschman MF, Napoli R, Calles J, Markuns JF, Ljungqvist O, Horton ES. Glucose ingestion causes GLUT4 translocation in human skeletal muscle. Diabetes. 1996;45:1051–1056. [PubMed: 8690151]
- 367.
- Kelley DE, Mintun MA, Watkins SC, Simoneau J-A, Jadali F, Fredrickson A. The effect of non-insulin-dependent diabetes mellitus and obesity on glucose transport and phosphorylation in skeletal muscle. J Clin Invest. 1996;97:2705–2713. [PMC free article: PMC507362] [PubMed: 8675680]
- 368.
- Bonadonna RC, Saccomani MP, Seely L, Zych KS, Ferrannini E, Cobelli C, DeFronzo RA. Glucose transport in human skeletal muscle. The in vivo response to insulin. Diabetes. 1993;42:191–198. [PubMed: 8093605]
- 369.
- Bonadonna RC, Del Prato S, Saccomani MP, Bonora E, Gulli G, Ferrannini E, Bier D, Cobelli C, DeFronzo RA. Transmembrane glucose transport in skeletal muscle of patients with non-insulin-dependent diabetes. J Clin Invest. 1993;92:486–494. [PMC free article: PMC293636] [PubMed: 8326013]
- 370.
- Bonadonna RC, Del Prato S, Bonora E, Saccomani MP, Gulli G, Natali A, Frascerra S, Pecro N, Ferrannini E, Bier D, Cobelli C, DeFronzo RA. Roles of glucose transport and glucose phosphorylation in muscle insulin resistance of NIDDM. Diabetes. 1996;45:915–925. [PubMed: 8666143]
- 371.
- Napoli R, Hirschman MF, Horton FS. Mechanisms of increased skeletal muscle glucose transport activity after an oral glucose load in rats. Diabetes. 1995;44:1362–1368. [PubMed: 7589839]
- 372.
- Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI. Impaired glucose transport as a cause of decreased insulin stimulated muscle glycogen synthesis in type 2 diabetes. N Engl J Med. 1999;341:240–246. [PubMed: 10413736]
- 373.
- Williams KV, Price JC, Kelley DE. Interactions of impaired glucose transport and phosphorylation in skeletal muscle insulin resistance. A dose-response assessment using positron emission tomography. Diabetes. 2001;50:2069–2079. [PubMed: 11522673]
- 374.
- Perriott LM, Kono T, Whitesell RR, Knobel SM, Piston DW, Granner DK, Powers AC, May JM. Glucose uptake and metabolism by cultured human skeletal muscle cells: rate-limiting steps. Am J Physiol Endocrinol Metab. 2001;281:E72–E80. [PubMed: 11404224]
- 375.
- Printz RL, Ardehali H, Koch S, Granner DK. Human hexokinase II mRNA and gene structure. Diabetes. 1995;44:290–294. [PubMed: 7883116]
- 376.
- Postic CA, Leturque A, Rencurel F, Printz R, Forest C, Granner D, Girard J. The effects of hyperinsulinemia and hyperglycemia on GLUT4 and hexokinase II mRNA and protein in rat skeletal muscle and adipose tissue. Diabetes. 1993;42:922–929. [PubMed: 8495814]
- 377.
- Jones JP, Dohn GL. Regulation of glucose transporter GLUT-4 and hexokinase II gene transcription by insulin and epinephrine. Am J Physiol (Endocrinol Metab 36): E682-E687, 1997. [PubMed: 9357795]
- 378.
- Mandarino LJ, Printz RL, Cusi KA, Kinchington P, O'Doherty RM, Osawa H, Sewell C, Consoli A, Granner DK, DeFronzo RA. Regulation of hexokinase II and glycogen synthase mRNA, protein, and activity in human muscle. Am J Physiol. 1995;269:E701–E708. [PubMed: 7485484]
- 379.
- Kruszynska YT, Mulford MI, Baloga J, Yu JG, Olefsky JM. Regulation of skeletal muscle hexokinase II by insulin in nondiabetic and NIDDM subjects. Diabetes. 1998;47:1107–1113. [PubMed: 9648835]
- 380.
- Vogt C, Ardehali H, Iozzo P, Yki-Jarvinen H, Koval J, Maezono K, Pendergrass M, Printz R, Granner D, DeFronzo R, Mandarino L. Regulation of hexokinase II expression in human skeletal muscle in vivo. Metabolism. 2000;49:814–818. [PubMed: 10877213]
- 381.
- Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, Pendergrass M, Koval J, Ardehali H, Printz R, Granner D, DeFronzo RA, Mandarino L. Effects of insulin on subcellular localization of hexokinase II in human skeletal muscle in vivo. J Clin Endocrinol Metab. 1998;83:230–234. [PubMed: 9435447]
- 382.
- Pendergrass M, Fazioni E, Saccomani MP, Collins D, Bonadonna R, Gulli G. In vivo glucose transport and phosphorylation in skeletal muscle is impaired in insulin resistant, normal glucose tolerant offspring of two NIDDM parents. Diabetes. 1995;44 suppl 1:197A.
- 383.
- Rothman DL, Shulman RG, Shulman GI. 31P nuclear magnetic resonance measurements of muscle glucose-6-phosphate. Evidence for reduced insulin-dependent muscle glucose transport or phosphorylation activity in non-insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:1069–1075. [PMC free article: PMC442962] [PubMed: 1556176]
- 384.
- Halseth AE, Bracy DP, Wasserman DH. Overexpression of hexokinse II increases insulin- and exercise-stimulated muscle glucose uptake in vivo. Am J Physiol. 1999;276:E70–E77. [PubMed: 9886952]
- 385.
- Pendergrass M, Koval J, Vogt C, Yki-Jarvinen H, Iozzo P, Pipek R, Ardehali H, Printz R, Granner D, DeFronzo RA, Mandarino L. Insulin-induced hexokinase II expression is reduced in obesity and NIDDM. Diabetes. 1998;47:387–394. [PubMed: 9519744]
- 386.
- Vestergaard H, Bjorbaek C, Hansen T, Larsen FS, Granner DK, Pedersen O. Impaired activity and gene expression of hexokinase II in muscle from non-insulin-dependent diabetes mellitus patients. J Clin Invest. 1995;96:2639–2645. [PMC free article: PMC185969] [PubMed: 8675629]
- 387.
- Ducluzeau P-H, Perretti N, Laville M, Andreelli F, Vega N, Riou J-P, Vidal H. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes. 2001;50:1134–1142. [PubMed: 11334418]
- 388.
- Lehto M, Huang X, Davis EM, Le Beau MM, Laurila E, Eriksson KF, Bell GI, Groop L. Human hexokinase II gene: exon-intron organization, mutation screening in NIDDM, and its relationship to muscle hexokinase activity. Diabetologia. 1995;38:1466–1474. [PubMed: 8786021]
- 389.
- Laakso M, Malkki M, Kekalainen P, Kuusito J, Deeb SS. Polymorphisms of the human hexokinase II gene: lack of association with NIDDM and insulin resistance. Diabetologia. 1995;38:617–622. [PubMed: 7489847]
- 390.
- Echwald SM, Bjorbaek C, Hansen T, Clausen JO, Vestergaard H, Zierath JR, Printz RL, Granner DK, Pedersen O. Identification of four amino acid substitutions in hexokinase II and studies of relationships to NIDDM, glucose effectiveness, and insulin sensitivity. Diabetes. 1995;44:347–353. [PubMed: 7883123]
- 391.
- Thiebaud D, Jacot E, DeFronzo RA, Maeder E, Jequier E, Felber JP. The effect of graded doses of insulin on total glucose uptake, glucose oxidation, and glucose storage in man. Diabetes. 1982;31:957–963. [PubMed: 6757014]
- 392.
- Young A, Bogardus C, Wolfe-Lopez D, Mott D. Muscle glycogen synthesis and disposition of infused glucose in humans with reduced rates of insulin-mediated carbohydrate storage. Diabetes. 1988;37:303–307. [PubMed: 3286330]
- 393.
- Avogaro A, Toffolo G, Miola M, Valerio A, Tiengo A, Cobelli C, Del Prato S. Intracellular lactate- and pyruvate-interconversion rates are increased in muscle tissue of non-insulin- dependent diabetic individuals. J Clin Invest. 1996;98:108–115. [PMC free article: PMC507406] [PubMed: 8690781]
- 394.
- Del Prato S, Bonadonna RC, Bonora E, Gulli G, Solini A, Shank M, DeFronzo RA. Characterization of cellular defects of insulin action in type 2 (non-insulin-dependent) diabetes mellitus. J Clin Invest. 1993;91:484–494. [PMC free article: PMC287962] [PubMed: 8432857]
- 395.
- Bonadonna RC, Groop L, Kraemer N., Ferrannini E, Del Prato S, DeFronzo RA. Obesity and insulin resistance in man. A dose response study. Metabolism. 1990;39:452–459. [PubMed: 2186255]
- 396.
- Mandarino LJ, Consoli A, Kelley DE, Reilly JP, Nurjhan N. Fasting hyperglycemia normalizes oxidative and nonoxidative pathways of insulin-stimulated glucose metabolism in non-insulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1990;71:1544–1551. [PubMed: 2121778]
- 397.
- Kelley DE, Mandarino LJ. Hyperglycemia normalizes insulin-stimulated skeletal muscle glucose oxidation and storage in noninsulin-dependent diabetes mellitus. J Clin Invest. 1990;86:1999–2007. [PMC free article: PMC329837] [PubMed: 2123890]
- 398.
- Bogardus C, Lillioja A, Stone K, Mott D. Correlation of muscle glycogen synthase activity and in vivo insulin action in man. J. Clin Invest. 1984;73:1185–90. [PMC free article: PMC425132] [PubMed: 6423666]
- 399.
- Rothman DL, Magnusson I, Cline G, Gerard D, Kahn CR, Shulman RG, Shulman GI. Decreased muscle glucose transport/phosphorylation is an early defect in the pathogenesis of non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci USA. 1995;92:983–987. [PMC free article: PMC42621] [PubMed: 7862678]
- 400.
- Vaag A, Henriksen JE. Beck-Nielsen. Decreased insulin activation of glycogen synthase in skeletal muscles in young non-obese Caucasian first-degree relatives of patients with non- insulin-dependent diabetes mellitus. J Clin Invest. 1992;89:782–788. [PMC free article: PMC442922] [PubMed: 1541672]
- 401.
- Yki-Jarvinen H, Mott D, Young AA, Stone K, Bogardus C. Regulation of glycogen synthase and phosphorylase activity by glucose and insulin in human skeletal muscle. J Clin Invest. 1987;80:95–100. [PMC free article: PMC442206] [PubMed: 3110217]
- 402.
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359(Pt 1):1–16. [PMC free article: PMC1222116] [PubMed: 11563964]
- 403.
- Cohen P. The Croonian Lecture 1999. Identification of a protein kinase cascade of major importance in insulin signal transduction. Phil Trans Royal Soc London Series B: Biological Sciences. 1999;354:485–495. [PMC free article: PMC1692513] [PubMed: 10212493]
- 404.
- Stralfors P, Hiraga A, Cohen P. The protein phosphatases involved in cellular regulation: purification and characterization of the glycogen-bound form of protein phosphatase-1 from rabbit skeletal muscle. Eur J Biochem. 1985;149:295–303. [PubMed: 2986973]
- 405.
- Sutherland C, Campbell DG, Cohen P. Identification of insulin-stimulated protein kinase-1 as the rabbit equivalent of rsk-mo-2. Eur J Biochem. 1993;212:581–588. [PubMed: 8444194]
- 406.
- Damsbo P, Vaag A, Hother-Nielsen O, Beck-Nielsen H. Reduced glycogen synthase activity in skeletal muscle from obese patients with and without type 2 (non-insulin- dependent) diabetes mellitus. Diabetologia. 1991;34:239–245. [PubMed: 1906024]
- 407.
- Mandarino LJ, Wright KS, Verity LS, Nichols J, Bell JM, Kolterman OG, Beck-Nielsen H. Effects of insulin infusion on human skeletal muscle pyruvate dehydrogenase, phosphofructokinase, and glycogen synthase. Evidence for their role in oxidative glucose metabolism. J Clin Invest. 1987;80:655–63. [PMC free article: PMC442287] [PubMed: 2957389]
- 408.
- Thorburn AW., Gumbiner B., Bulacan F, Wallace P, Henry RR. Intracellular glucose oxidation and glycogen synthase activity are reduced in non-insulin-dependent (type II) diabetes independent of impaired glucose uptake. J Clin Invest. 1990;85:522–9. [PMC free article: PMC296454] [PubMed: 2105341]
- 409.
- Mandarino LJ, Consoli A, Jain A, Kelley DE. Interaction of carbohdyrate and fat fuels in human skeletal muscle: impact of obesity and NIDDM. Am J Physiol. 1996;270:E463–E470. [PubMed: 8638694]
- 410.
- Nikoulina SE, Ciaraldi TP, Mudaliar S, Mohideen P, Carter L, Henry RR. Potential role of glycogen synthase kinase-3 in skeletal muscle insulin resistance of type 2 diabetes. Diabetes. 2000;49:263–271. [PubMed: 10868943]
- 411.
- Henry RR, Ciaraldi TP, Abrams-Carter L, Mudaliar S, Park KS, Nikoulina SE. Glycogen synthase activity is redued in cultured skeletal muscle cells of non-insulin-dependent diabetes mellitus subjects. J Clin Invest. 1996;98:1231–1236. [PMC free article: PMC507545] [PubMed: 8787686]
- 412.
- Wells AM, Sutcliffe IC, Johnson AB, Taylor R. Abnormal activation of glycogen synthesis in fibroblasts from NIDDM subjects. Evidence for an abnormality specific to glucose metabolism. Diabetes. 1993;42:583–589. [PubMed: 8384133]
- 413.
- Nyomba BL, Freymond D, Raz I, Stone K, Mott DM, Bogardus C. Skeletal muscle glycogen synthase activity in subjects with non-insulin-dependent diabetes mellitus after glyburide therapy. Metabolism. 1990;39:1204–1210. [PubMed: 2122177]
- 414.
- Pratipanawatr T, Cusi K, Ngo P, Pratipanawatr W, Mandarino LJ, DeFronzo RA. Normalization of plasma glucose concentration by insulin therapy improves insulin- stimulated glycogen synthesis in type 2 diabetes. Diabetes. 2002;51:462–468. [PubMed: 11812756]
- 415.
- Vestergaard H, Lund S, Larsen FS, Bjerrum OJ, Pedersen O. Glycogen synthase and phosphofructokinase protein and mRNA levels in skeletal muscle from insulin-resistant patients with non-insulin-dependent diabetes mellitus. J Clin Invest. 1993;91:2342–2350. [PMC free article: PMC443291] [PubMed: 8514849]
- 416.
- Vestergaard H, Bjocbaek C, Andersen PH, Bak JF, Pedersen O. Impaired expression of glycogen synthase mRNA in skeletal muscle of NIDDM patients. Diabetes. 1991;40:1740–1745. [PubMed: 1756915]
- 417.
- Browner MF, Nakano K, Bang AG, Fleffenzk RJ. Human muscle glycogen synthase with DNA sequence: anegatively charged protein with asymmetric charge distribution. Proc Natl Acad Sci USA. 1989;86:1443–1447. [PMC free article: PMC286712] [PubMed: 2493642]
- 418.
- Majer M, Mott DM, Mochizuki H, Rowles JC, Pederson O, Knowler WC, Bogardus C, Prochazka M. Association of the glycogen synthase locus on 19q13 with NIDDM in Pima Indians. Diabetologia. 1996;39:314–321. [PubMed: 8721777]
- 419.
- Orho M, Nikua-Ijas P, Schalin-Jantti C, Permutt M, Groop L. Isolation & characterization of human the muscle glycogen synthase gene. Diabetes. 1995;44:1099–1105. [PubMed: 7657035]
- 420.
- Bjorbaek C, Echward SM, Hubricht P, Vestergaard H, Hansen T, Zierath J, Pedersen O. Genetic variants in promoters and coding regions of the muscle glycogen synthase and the insulin-responsive GLUT4 genes in NIDDM. Diabetes. 1994;43:976–983. [PubMed: 8039605]
- 421.
- Bjorbaek C, Fik TA, Echward SM, Yang P-Y, Vestergaard H, Wang JP, Webb GC, Richmond K, Hansen T, Erikson RL, Miklos GLG, Cohen PTW, Pedersen O. Cloning of human insulin-stimulated protein kinase (ISPK-1) gene and analysis of coding regions and mRNA levels of the ISPK-1 and the protein phosphatase-1 genes in muscle from NIDDM patients. Diabetes. 1995;44:90–97. [PubMed: 7813820]
- 422.
- Procharzka M, Michizuki H, Baier LJ, Cohen PTW, Bogardus C. Molecular and linkage analysis of type-1 protein phosphatase catalytic beta-subunit gene: lack of evidence for its mjaor role in insulin resistance in Pima Indians. Diabetologia. 1995;38:461–466. [PubMed: 7796987]
- 423.
- Schalin-Jantti C, Harkonen M, Groop LC. Impaired activation of glycogen synthase in people at increased risk for developing NIDDM. Diabetes. 1992;41:598–604. [PubMed: 1568529]
- 424.
- Falholt K, Jensen I, Lindkaer Jensen S, Mortensen HB, Volund A, Heding LG, Norskov Petersen P, Falholt W. Carbohydrate and lipid metabolism of skeletal muscle in type 2 diabetic patients. Diab Med. 1988;5:27–31. [PubMed: 2964324]
- 425.
- Mandarino LJ, Madar Z, Kolterman OG, Bell JM, Olefsky JM. Adipocyte glycogen synthase and pyruvate dehydrogenase in obese and type II diabetic patients. Am J Physiol. 1986;251:E489–E496. [PubMed: 3094377]
- 426.
- Kelley D, Mokan M, Mandarino L. Intracellular defects in glucose metabolism in obese patients with noninsulin-dependent diabetes mellitus. Diabetes. 1992;41:698–706. [PubMed: 1587397]
- 427.
- Barrett EJ, Eringa EC. The Vascular Contribution to Insulin Resistance: promise, proof, and pitfalls. Diabetes. 2012;61:3063–65. [PMC free article: PMC3501887] [PubMed: 23172953]
- 428.
- Love KM, et al. Insulin-mediated muscle microvascular perfusion and its phenotypic predictors in humans. Scientific Reports. 2021;11:11433. [doi.org/10.1038/s41598-021 90935-8] [PMC free article: PMC8169863] [PubMed: 34075130]
- 429.
- Clark M, et al. The Microvasculature in Insulin Resistance and Type 2 Diabetes. Semin Vasc Med. 2002;2(1):21–32. [doi:10.1055/s-2002-23506] [PubMed: 16222593]
- 430.
- Bergman RN, Iyer MS. Indirect Regulation of Endogenous Glucose Production by Insulin: The Single Gateway Hypothesis Revisited. Diabetes. 2017;66:1742–47. [doi.org/10.2337/db16-1320] [PubMed: 28637826]
- 431.
- Edgerton DS, Kraft G, Smith M, et al. Insulin direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight. 2017;2(6):e91863. [doi.org/10.1172/jci.insight.91863] [PMC free article: PMC5358484] [PubMed: 28352665]
- 432.
- Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nature Reviews Endocrinology. 2017;13:572–87. [PMC free article: PMC5777172] [PubMed: 28731034]
- 433.
- Solini A, Chiara R, Chiara MM, Proietti A, Koepsell H, Ferrannini E. Sodium glucose co-transporter SGLT-2 and SGLT-1 renal expression in patients with type 2 diabetes. Diabetes, Obesity and Metabolism. 2017;19(9):1289–1294. [doi.org/10.1111/dom12970] [PubMed: 28419670]
- 434.
- Luke N, Shannon C, Fourcaudot M, et al. Sodium-glucose cotransporter and glucose transporter [GLUT] expression in the kidney of type 2 diabetic -subjects. Diabetes, Obesity and Metabolism. 2017;19(9):1322–1326. [doi.org/10.1111/dom13003] [PubMed: 28477418]
- 435.
- du Toit EF and Donner DG. (December 12th 2012). Myocardial Insulin Resistance: An Overview of Its Causes, Effects, and Potential Therapy, Insulin Resistance, Sarika Arora, Intech Open. [DOI: 10.5772/50619] [www
.intechopen.com/chapters/41432] [CrossRef] - 436.
- Esser Nathalie, Utzschneider Kristina M., Kahn Steven E. Early beta-cell dysfunction vs. insulin hypersecretion as the primary event in the pathogenesis of dysglycemia. Diabetologia. 2020;63:2007–2021. [https://doi.org/10.1007/s00125-020-05245-x] [PubMed: 32894311]
- 437.
- Pontiroli AE, Alberetto M, Capra F, Pozza G. The glucose clamp technique for the study of patients with hypoglycemia: insulin resistance as a feature of insulinoma. J Endocrinol Investig. 1990;13(3):241–245. [https://doi.org/10.1007/bf03349549] [PubMed: 2195099]
- 438.
- Del Prato S, Leonetti F, Simonson DC, Sheehan P, Matsuda M, DeFronzo RA. Effect of sustained physiologic hyperinsulinaemia and hyperglycaemia on insulin secretion and insulin sensitivity in man. Diabetologia. 1994;37(10):1025–1035. [https://doi.org/10.1007/bf00400466] [PubMed: 7851681]
- 439.
- Marini MA, Frontoni S, Succurro E, Arturi F, Fiorentino TV, Sciacqua A, Hribal ML, Perticone F, Sesti G. Decreased Insulin Clearance in Individuals with Elevated 1-h Post-Load Plasma Glucose Levels. PLOS ONE. 2013 October;8(Issue 10) Volume. [e77440] [PMC free article: PMC3806727] [PubMed: 24194886]
- 440.
- Prentki M, Peyot ML, Pellegrino M, Madiraju SRM. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic beta cell. Diabetes. 2020;68:279–290. [doi.org/10.2337/dbi19-001] [PubMed: 32079704]
- 441.
- Shah Arti, Mehta Nehal, Reilly Muredach P. Adipose Inflammation, Insulin Resistance, and Cardiovascular Disease. JPEN J Parenter Enteral Nutr. 2008;32(6):638–644. [doi:10.1177/0148607108325251] [PMC free article: PMC3088110] [PubMed: 18974244]
- 442.
- Angulo P, Lindor KD. Non-alcoholic fatty liver disease. Journal of Gastroenterology and Hepatology. 2002;17 Suppl.:S186–S190. [PubMed: 12000605]
- 443.
- Younossi ZM. Non-alcoholic fatty liver disease. Journal of Hepatology. 2019;70:531–544. [PubMed: 30414863]
- 444.
- Hartstra AV, Bouter KEC, Backhed F, Nieuwdorp M. Insights Into the Role of the Microbiome in Obesity and Type 2 Diabetes. Diabetes Care. 2015;38:159–165. [DOI: 10.2337/dc14-0769] [PubMed: 25538312]
- 445.
- Wada J, Nakatsuka A. Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes. Acta Med. Okayama. 2015;70(3):151–158. [http://escholarship.lib.okayama-u.ac.jp/amo/] [PubMed: 27339203]
- 446.
- Fealy CE, Mulya A, Lai N, Kirwan JP. Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle. J Appl Physiol. 2014;1985(117):239–245. [PMC free article: PMC4122691] [PubMed: 24947026]
- 447.
- Sivitz WI, Yorek MA. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. ANTIOXIDANTS & REDOX SIGNALING Volume 12, Number 4, 2010 (by Mary Ann Liebert, Inc.) [DOI: 10.1089/ars.2009.2531] [PMC free article: PMC2824521] [PubMed: 19650713] [CrossRef]
Publication Details
Author Information and Affiliations
Publication History
Last Update: September 27, 2021.
Copyright
This electronic version has been made freely available under a Creative Commons (CC-BY-NC-ND) license. A copy of the license can be viewed at http://creativecommons.org/licenses/by-nc-nd/2.0/.
Publisher
MDText.com, Inc., South Dartmouth (MA)
NLM Citation
Solis-Herrera C, Triplitt C, Cersosimo E, et al. Pathogenesis of Type 2 Diabetes Mellitus. [Updated 2021 Sep 27]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.