

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
Central nervous system (CNS) tumors are the second commonest childhood malignancy, with 10% of these affecting the suprasellar and/or intrasellar regions. Survival has increased significantly over the last decade as a result of improved multimodality cancer therapies and better supportive care. Measurements of serum prolactin, α-fetoprotein, and β-hCG as well as baseline pituitary function tests are essential at diagnosis prior to commencement of any therapy. Craniopharyngiomas and low-grade gliomas account for most of these tumors, whilst other histological subtypes such as pituitary adenomas, germinomas, and hamartomas are rare. Non-neoplastic masses include pituitary hyperplasia and Rathke’s cleft cysts. Neurological syndromes and endocrine dysfunction are often present at diagnosis, and may be missed if not sought for. Post-diagnosis, endocrinopathies can evolve over decades secondary to tumor and/or treatment, necessitating long-term follow-up of such patients. Treatment of endocrine dysfunction is crucial not just to avoid the fatal consequences of untreated secondary adrenal insufficiency and/or diabetes insipidus, but also to improve quality of survival, and should be closely supervised by a pediatric endocrinologist with experience in the management of such patients. Growth hormone therapy in replacement doses in particular has not been shown to increase the risk of tumor recurrence. The “hypothalamic syndrome”, including variable hypothalamic dysfunction (e.g., sleep-wake cycle disturbances, temperature dysregulation, adipsia, and behavioral disorders) and hypothalamic obesity, is a common and as yet untreatable sequela of both tumor and treatment. The latter is caused by dysregulation of a network anorexigenic and orexigenic hormone signals which is only beginning to be elucidated. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Central nervous system (CNS) tumors are the second commonest childhood malignancy after leukemias, accounting for 25% of cancers in children <15 years of age with an annual incidence rate of 35 cases/million/year (1–4). As with all childhood cancers, their incidence is gradually increasing worldwide (1,2,5), an effect largely attributed to improvements in diagnosis and tumor registration (6–8), and more recently campaigns such as the UK HeadSmart project aimed at increasing awareness of pediatric brain tumor symptoms (http://www.headsmart.org.uk/) (9). Concurrently, 5-year survival for CNS tumors has increased much more steeply from 57% to 65% in the last decade (~95% in low-grade gliomas) as a result of improved multimodality cancer therapies and better supportive care (10–12).
However, while survival is high, increasingly intensive treatment strategies aimed at improving cure in a small minority can conversely cause a higher toxicity burden in the larger majority, with a rapidly accruing cohort of survivors faced with reduced quality of life due to late and evolving multi-organ toxicities (13–15). Over 40% of these chronic morbidities (“late effects”) are severe, disabling or life-threatening (16), and more than 80% of CNS tumor survivors develop at least one endocrinopathy, most frequently growth hormone deficiency (17). Indeed, suprasellar tumors have been found to be the commonest cause of hypothalamo-pituitary dysfunction in adult cohort studies (18,19). However, when compared with adult CNS tumors, pediatric tumors tend to be more curable, and the early presentation of some tumors (e.g., craniopharyngiomas, primitive neuroectodermal tumors (PNET)), and their association with mutations in neural development genes blur the delineation between congenital malformations and neoplasia (20–22).
Tumor location and histology is distinctly age-dependent: 30% of tumors under the age of 14 years are infratentorial (medulloblastomas, posterior fossa juvenile pilocytic astrocytomas, and ependymomas), whilst 26% and 16% of tumors diagnosed in young adulthood (15 to 24 years) are supratentorial or suprasellar respectively (non-pilocytic astrocytomas, other gliomas, pituitary adenomas, and germinomas) (4,23). Supra- and intrasellar tumors constitute 10% of all pediatric CNS tumors (23,24) and their close proximity to the vital hypothalamo-pituitary axis (HPA) increases the risk of important endocrine dysfunction. This may occur secondary to tumor mass effect and/or treatment, and can therefore be manifest at presentation or evolve subsequently during or after completion of oncological therapies. Dissecting the effect of tumor from treatment on endocrinopathies diagnosed after commencement of therapy is particularly complicated. We aim here to (1) outline the epidemiology, clinical features, and management of common pediatric suprasellar tumors not readily addressed in other chapters, (2) examine the common clinical neuroendocrine presenting features and (3) summarize common themes in the neuroendocrine late effects observed at follow-up of these patients.
THE DIFFERENTIAL DIAGNOSIS OF PEDIATRIC SUPRA- AND INTRASELLAR MASSES
The definitive diagnosis of pediatric suprasellar and intrasellar masses is crucial, as therapeutic strategies differ markedly depending on histological subtype. However, a tissue diagnosis may not always be possible due to their location, as even minor procedures such as biopsies can lead to life-threatening endocrinopathies such as diabetes insipidus (DI) (25). Biochemical measurements of serum prolactin (PRL), α-fetoprotein (AFP), and β-human chorionic gonadotrophin (β-hCG) to aid the diagnosis of prolactinomas and secreting germinomas respectively are therefore absolutely essential prior to commencement of any therapy.
Table 1.
The Differential Diagnosis of Pediatric Suprasellar Tumors and Other Disorders
| Neoplastic |
| Craniopharyngioma Low-grade glioma (mainly pilocytic astrocytoma) Pituitary adenoma Germ cell tumor (mainly germinoma) Hamartoma Meningeal metastases |
| Non-neoplastic |
| Pituitary hyperplasia Pituitary stalk thickening Langerhans cell histiocytosis* Tuberculosis Sarcoidosis Rathke cleft cyst Arachnoid cyst Epidermoid/dermoid cyst Meningioma |
- *
The classification of Langerhans cell histiocytosis as a non-neoplastic disease is debatable.
Craniopharyngiomas

Figure 1.
T1-weighted MRI images of a craniopharyngioma demonstrating the coexistence of solid, cystic and calcified components with the tendency for multiple progressions over seven years. (a) After initial endoscopic cyst fenestration and ventriculoperitoneal shunt insertion, (b) after first transcranial debulking, (c) first cystic progression, (d) after first cyst drainage via reservoir, (e) second cystic progression, (f) after second transcranial debulking, (g) after adjuvant radiotherapy and third cystic progression, (h) after second cyst drainage via reservoir, (a) after fourth cystic & solid progression, (j) after complete resection.
Craniopharyngiomas are by far the commonest suprasellar tumor of childhood, accounting for up to 50-80% of masses in this region (24,26–28) and 1.5-11.6% of all pediatric CNS tumors (3,24,26,29,30). There is a bimodal age distribution in incidence, with the peak incidence in childhood occurring between the ages of 5-14 years at 1.4 cases/million/year (29,31). They are benign tumors originating from the embryonal epithelium lining Rathke’s pouch and are almost invariably adamantinomatous in childhood, characterized by the presence of intratumoral calcifications (32). Over-activation of the Sonic hedgehog (SHH) and Wnt/β-catenin pathways, both important in both pituitary stem cell development and carcinogenesis, have been shown to be key to their formation (20,21), but they occur typically sporadically, with only one case report of familial adamantinomatous craniopharyngiomas occurring in a consanguineous pedigree reported in the English literature (33). Contrastingly, papillary craniopharyngiomas are found almost exclusively in adults and harbor the BRAF V600E mutation instead (34).
Symptoms related to hypothalamo-pituitary dysfunction, such as weight gain, growth failure, prolonged recovery from infections, and abnormalities of puberty are often under-recognized but in fact constitute the third commonest group of clinical findings at diagnosis, after symptoms related to raised intracranial pressure (e.g., headaches, vomiting) and visual deterioration (22,35–47). Radiologically, 65-93% of these tumors are calcified but a plain X-ray or computerized tomography (CT) scan may be required to demonstrate this. The coexistence of solid, cystic, and calcified structures on neuroimaging, as well as the characteristic cholesterol crystals seen under microscopy of the “engine fluid” aspirated surgically from cystic components are so highly suggestive of the diagnosis that histological confirmation from biopsies of solid components may be unnecessary, particularly as this may further compromise hypothalamo-pituitary function (32,48). Anatomically, 75% of craniopharyngiomas are suprasellar with an intrasellar extension, 20% are exclusively suprasellar, and 5% are exclusively intrasellar, with over 50% involving the hypothalamus and nearly one-third invading the floor of the third ventricle (26,37,44).
Due to their location, a significant proportion of these tumors are not completely resectable, but their relative rarity, high rates of survival, and benign histology have precluded them from pan-European randomized trials, resulting in a lack of agreement on the optimal treatment strategy. Most recently, the first evidence- and consensus-based national UK guideline for the management of craniopharyngiomas in children and young people has been published by the UK Children’s Cancer and Leukemia Group (CCLG), with endorsement from the Royal College of Pediatrics and Child Health (RCPCH) and British Society of Pediatric Endocrinology & Diabetes (BSPED) (49).Importantly, these guidelines advocate a more conservative approach to the degree of surgical resection in the presence of significant hypothalamic involvement in order to minimize further damage to the hypothalamo-pituitary axis (39,50,51), balanced against the need to relieve symptoms of raised intracranial pressure, preserve vision, and provide long-term control and reduced recurrence rates (49,52,53). The use of adjuvant radiotherapy in combination with subtotal tumor resection has been shown to achieve survival rates which are on par with complete tumor resection (5-year progression-free survival 73-100% vs 73-82%), with the potential for less neuroendocrine dysfunction (54–56). More recently, the use of proton beam therapy has increased, with equivalent survival outcomes to conventional radiotherapy, but there remains the issue of insufficient follow-up data to ascertain its long-term toxicity profile (57,58). Experience with systemic or intracystic chemotherapy, intracystic interferon, and radioisotope instillation of 32P or 90Y have been met with conflicting success and cannot therefore be currently recommended as primary treatment approaches in children (59–62). Ultimately, despite high long-term overall survival (80% at 30 years), (37) up to 98% of survivors experience dysfunction in at least one hypothalamo-pituitary axis with high rates of morbid obesity (45,63).
Low-grade Gliomas (LGGs)
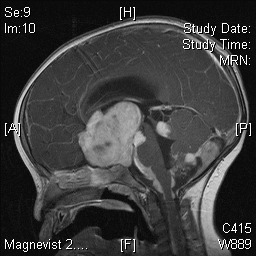
Figure 2.
T1-weighted MRI image demonstrating appearances of a large, lobulated optic pathway astrocytoma with hydrocephalus and widespread leptomeningeal dissemination affecting the brainstem, cerebellum, and spinal cord.
LGGs account for >40% of all CNS tumors and are thus the commonest pediatric intracranial tumor (3,8). The optic pathway, hypothalamus, and suprasellar midline are the second most frequent location for LGGs (30-50%) after the cerebellum, cerebral hemispheres, and brainstem (12,64). Even in the suprasellar region they are the second commonest pediatric tumor after craniopharyngiomas, and are similarly regarded as benign (grade I or II), the vast majority being juvenile pilocytic astrocytomas (65). The genetic tumor predisposition syndrome neurofibromatosis type 1 (NF-1) is present in 10-16% of cases, whilst 15% of asymptomatic NF-1 children will have LGGs on neuroimaging. NF-1-associated tumors more often originate from the optic nerves (70%) than from the hypothalamochiasmatic area (27-40%) and tend to a more indolent course (11,12,64,66–69). Mutations involving KIAA1549, BRAF and Ras proto-oncogenes are associated with pilocytic astrocytomas and disruptors targeted at these pathways form the basis of current clinical therapeutic trials (70–72). Similar to craniopharyngiomas, the commonest symptoms at diagnosis are related to visual changes or raised intracranial pressure, with disorders of the LH/ FSH axis being the most prevalent endocrinopathy at presentation (25,66,73–75). In infancy, hypothalamic LGGs can present with diencephalic syndrome (see below) (11,76–78), which significantly increases the risk of future neuroendocrine dysfunction (79).
Complete tumor resection has been shown to be a favorable risk factor for survival (12,64) but suprasellar and/or optic pathway tumors cannot be completely resected without causing major visual and neuroendocrine morbidity. Treatment trials have thus focused on medical strategies, with radiotherapy being delayed in favor of chemotherapy in young children due to concerns of cognitive dysfunction (80), subsequent primary cancers (SPCs) (81,82) and radiation-induced vasculopathies (83), despite showing superior 5-year progression-free survival rates (65% vs. 47%) (11). However, to date none of the previous international treatment trials – LGG1 (1997-2004) or LGG2 (2005-2010) – were randomized, these being purely observational studies aimed at improving visual outcomes but with little reported success (11,12,84). At the time of writing, the first randomized interventional study of chemotherapeutic strategies (LGG3) is being designed with careful long-term prospective measurements of visual and neuroendocrine outcomes. More recently, tumors harboring BRAF mutations have been the target of MAPK/ERK kinase (MEK) and BRAF inhibitors such as trametinib and dabrafenib (72,85–87), although these can still lead to various side effects including endocrinopathies(88).
A 30-year survival analysis has revealed the extent of long-term neuroendocrine dysfunction affecting these patients with new endocrine deficits appearing up to 15 years post-diagnosis, and 20-year endocrine event-free survival approaching 20% (25). Hypothalamic tumor location is a more important independent risk factor for long-term anterior hypothalamo-pituitary deficits than radiotherapy exposure; however only surgical intervention (regardless of extent) has been shown to be independently associated with posterior pituitary dysfunction and life-threatening salt and water imbalances (25,64). Similar to craniopharyngiomas, overall survival is high (85% at 25 years), but ~80% of survivors experience at least one endocrinopathy (25,79).
Pituitary Adenomas

Figure 3.
T1-weighted MRI image demonstrating appearances of a giant prolactinoma. There is obscuration of normal pituitary morphology due to the tumor arising from the pituitary gland itself.
Pituitary adenomas are rare in childhood, accounting for just 3% of all supratentorial tumors with an estimated annual incidence of 0.1 cases/million/year in children (89). The vast majority are functioning, with prolactinomas alone accounting for 50% of adenomas and 2% of all pediatric and adolescent intracranial tumors. Therefore, the measurement of plasma prolactin (PRL) may be diagnostic and is absolutely mandatory prior to planning surgery for any pituitary mass, as medical treatment alone may be entirely curative (90,91). ACTH- and GH-secreting adenomas are the next commonest, whilst TSH-secreting, gonadotrophin-secreting, and non-functioning adenomas are vanishingly rare (91–93).
A child with a pituitary adenoma may be the index case for a genetic tumor predisposition syndrome (up to 22%), particularly given their rarity, and therefore careful documentation of their family history and testing for multiple endocrine neoplasia type 1 (MEN1) and aryl-hydrocarbon receptor interacting protein (AIP) gene mutations are therefore paramount in all cases (94–96). Other genetic syndromes associated with pituitary adenomas that need to be considered are multiple endocrine neoplasia type 4 (CDKN1B), Carney complex (PRKAR1A), McCune-Albright syndrome (GNAS), SDH-related pituitary adenoma syndrome (SDHB, SDHC, SDHD), and DICER1 syndrome (97).
Investigation and management of pituitary adenomas depends on whether they are functioning or non-functioning, and in the case of the former, which hormones are being secreted in excess. Similar to craniopharyngiomas, an evidence- and consensus-based national UK guideline is being written for the management of pituitary adenomas in children and young people as a collaborative effort between the CCLG, RCPCH and BSPED.
PROLACTINOMA
Pituitary adenomas are classified as microadenomas (<1 cm), macroadenomas (>1 cm), and giant adenomas (>4 cm). In prolactinomas plasma PRL levels generally, but not exclusively, increase with tumor size. Hyperprolactinemia may also result from stalk compression by tumor mass (interrupting hypothalamic dopaminergic inhibition of PRL secretion) and antipsychotic medication but PRL concentrations are usually <2000 mU/l and patients rarely symptomatic (98). Laboratories should always screen for artefactual hyperprolactinemia due to macroprolactin, but levels >5000 mU/l are usually diagnostic and symptomatic. Occasionally, falsely low results can be due to interference by extreme hyperprolactinemia on antibody-antigen sandwich complex formation, a phenomenon known as the hook effect. In cases of large tumors, samples should therefore be diluted 100-fold and repeated for confirmation (99). Clinical presentation varies according to the size of tumor, gender, and pubertal status, with girls usually experiencing galactorrhea, pubertal delay, or amenorrhea and boys presenting later with larger, more aggressive tumors with raised intracranial pressure (90).
Given the paucity of good quality outcome data in children, treatment guidelines follow those for adults (53,91), recommending dopamine agonists (DAs) as first line, ideally cabergoline due to its high efficacy and tolerability (98). Starting doses, dose escalation and duration of therapy in children remain undefined and are critical questions given the potential for more aggressive disease and cardiac valve abnormalities with long-term cumulative exposure (100). Surgery should be reserved for those cases resistant to DAs or for neurosurgical emergencies (e.g., neuro-ophthalmic deficits, pituitary apoplexy) and both trans-sphenoidal and transcranial approaches should be considered by an experienced pediatric neurosurgeon. Radiotherapy has usually been reserved for treatment failures in view of the presumed risk of post-treatment endocrine morbidity and second primary cancers. However, the former may have been overestimated in view of the high incidence of endocrinopathies already present at diagnosis (101), and therefore this treatment modality should be considered earlier and prior to other more experimental treatments such as temozolomide chemotherapy (98). As with other hypothalamo-pituitary tumors, long-term neuroendocrine and secondary cardiovascular morbidity is significant (102).
CORTICOTROPHINOMAS
The age distribution for corticotrophinomas is younger than that of prolactinomas (where the incidence increases in adolescence and young adulthood), with Cushing disease accounting for the vast proportion of Cushing syndrome in children aged >5 years, and >70% of pituitary adenomas in the prepubertal age group (103,104). These tumors are nearly always microadenomas. Common presenting features include weight gain with linear growth arrest or short stature, change in facial appearance, fatigue, striae, hirsutism, emotional lability, hypertension, acne, headaches, or psychosis (104–106). Diagnosis is achieved by firstly screening for Cushing syndrome indicated by a raised urine free cortisol (sensitivity 89%) or midnight cortisol concentration of >50 nmol/l (sensitivity 99%, specificity 20%). This is then followed by a low-dose (sensitivity 100%, specificity 80%) then high-dose (sensitivity 94%, specificity 70%) dexamethasone suppression test (104,107–111). CRH-stimulated bilateral inferior petrosal sinus sampling (BIPSS) may help successfully localize the position of the microadenoma (104,105). Transsphenoidal resection is the first-line treatment of choice, superseding bilateral adrenalectomy which carries a risk of post-operative Nelson syndrome (112). Cure rates are 45-78% with nearly 40% requiring adjuvant radiotherapy (113–115).
SOMATOTROPHINOMAS
8-15% of all pituitary adenomas in patients <20 years of age secrete GH, with a significant proportion co-secreting PRL and TSH (103,116). Genetic syndromes associated with somatotrophinomas include MEN-1 (MEN1), Carney complex (PRKAR1A), McCune-Albright syndrome (GNAS), and familial isolated pituitary adenoma (FIPA, AIP) syndrome (97). Due to the absence of complete epiphyseal fusion, in childhood and adolescence, somatotrophinomas present with pituitary gigantism rather than acromegaly. Tall stature and increased growth velocity however can still be associated with other acromegalic features such as mild obesity, macrocephaly, acral enlargement, frontal bossing, and macrognathia (93,117). Investigations reveal high random GH and IGF-1 concentrations, loss of GH pulsatility, and failure of GH suppression to an oral glucose tolerance test (87). Like corticotrophinomas, transsphendoidal resection is the treatment of choice but a significant proportion of patients require adjuvant medical therapy with somatostatin analogues (octreotide, lanreotide), dopamine agonists (cabergoline, bromocriptine), or the GH receptor antagonist pegvisomant (118). Radiotherapy has been used with limited effect (119).
Germ Cell Tumors

Figure 4.
T1-weighted MRI image demonstrating the appearance of a contrast-enhancing suprasellar β-hCG-secreting germinoma in a patient who presented with central diabetes insipidus.
Germ cell tumors (GCTs) are tumors arising from primordial germ cells normally sited in the testes and ovaries and can be subclassified into germinomatous (GGCT, usually non-secreting but can occasionally produce βhCG) and non-germinomatous germ cell tumors (NGGCT). NGGCTs and can be further classified into yolk sac tumors (secreting α-fetoprotein (AFP)), choriocarcinomas (secreting βhCG), and embryonal carcinomas. In contrast to craniopharyngiomas and LGGs, intracranial GCTs account for just 3-4% of all primary pediatric and young adult CNS tumors <24 years (23,120). There is a clear peak in incidence in adolescence and young adulthood, with age-adjusted incidence rates rising from 0.9 cases/million/year in patients <10 years to 1.3-2.1 cases/million/year in patients aged 15-24 years (23,120). Boys are affected nearly three times as often as girls, and this sex distribution is magnified in adolescence (male: female ratio of >8:1) (23). GCTs are also the commonest CNS tumor in Klinefelter and Down syndromes (121). Diabetes Insipidus (DI) and gonadotrophin-independent precocious puberty (due to βhCG acting on the LH receptor) are common findings at diagnosis and present in 30-50% and 11-12% of patients respectively. Unlike NGGCTs, GGCTs can grow indolently (if at all), meaning that both clinical and radiological features can often be subtle at onset, and delays in diagnosis up to 21 years have been reported (122–124).
Histologically, intracranial GCTs resemble their gonadal counterparts (ovarian teratoma or testicular seminoma) and account for 34% of all such tumors (125). They have a particular predilection for the pineal gland (37-66%) and suprasellar region (23-35%), such that synchronous (bifocal) pineal and suprasellar tumors are pathognomonic. Both GGCTs and NGGCTs are extremely chemo- and radiosensitive, and their propensity to metastasize throughout the cerebrospinal fluid (26,121,126) has meant that whole neuraxial (craniospinal) irradiation has been standard therapy for decades, with overall and progression-free survival rates approaching 100% (119). Chemotherapy alone has been shown to result in inferior survival (127), and more recent attempts to reduce the irradiation field with adjuvant chemotherapy in an effort to preserve cognitive function have shown little reduction in overall survival (121,128,129). The latest SIOP CNS GCTII however aims to reduce the radiation dose and field by stratifying treatment strategies between NGGCT and GGCTs, and based on the absence or presence of metastatic disease (https://www.skion.nl/workspace/uploads/2_siop_cns_gct_ii_final_version_2_15062011_unterschrift_hoppenheit.pdf). As for other suprasellar tumors, the rate of post-treatment endocrine morbidity is significant, with 50-60% of patients having at least one endocrinopathy (122).
Hypothalamic Hamartomas

Figure 5.
T1-weighted MRI image demonstrating the appearances of a pedunculated hypothalamic hamartoma (arrowheads) arising from the floor of the third ventricle in a patient who presented with central precocious puberty. The pituitary morphology is otherwise normal.
Hypothalamic hamartomas are extremely rare congenital (rather than neoplastic) malformations consisting of grey matter heterotopia in the tuber cinereum and inferior hypothalamus (24,26,130). Their true prevalence is unknown but is estimated to occur in between 1 in 50,000 – 1 million individuals (131–133). Symptom onset occurs in infancy to early childhood, with the mean age of first seizures occurring between 6 weeks – 4.5 years (133–136). The triad of epilepsy (usually gelastic (laughing) or dacrystic (crying) seizures), central precocious puberty, and developmental delay is classic with the seizure semiology eventually evolving into multiple, more severe seizure types (130). Rarely, they are associated with Pallister-Hall syndrome, an autosomal dominant disorder characterized by polydactyly and other midline defects (imperforate anus, bifid epiglottis, panhypopituitarism and dysmorphic facies) (132,137), or with SOX2 mutations (138).
The intrinsic epileptogenicity of these lesions (139,140), the trend towards evolving seizure semiology, the worsening of behavioral and psychiatric co-morbidities, and the general failure of anti-epileptic drug therapy has led clinicians to explore the options of surgical or stereotactic radiosurgical resection despite their deep-seated location, with variably reported success in the remission of seizure activity and behavioral disturbances, but more modest improvements in cognitive function (130,131,141–143). Li et al.'s (144) case series reported successful remission of central precocious puberty (CPP) and little, if any, late-onset endocrinopathy; but a larger cohort study by Freeman et al. (145) suggested that clinically silent endocrine dysfunction (particularly GH and TSH deficiency) is common both at diagnosis and postoperatively. Transient posterior pituitary dysfunction leading to DI and the syndrome of inappropriate antidiuretic hormone secretion (SIADH) has also been described (145,146). One adult cohort study corroborates these findings, showing that >1/3 of these patients had endocrine dysfunction and approximately 2/3 experienced excessive weight gain postoperatively (147). More recently laser induced thermal therapy (LiTT) of these lesions has shown promising results with regards to seizure control, with little late onset additional endocrinopathies (148,149).
Langerhans Cell Histiocytosis (LCH)

Figure 6.
T1-weighted MRI image demonstrating the appearances of a contrast-enhancing suprasellar LCH lesion. There is a small anterior pituitary and absent posterior pituitary bright spot in keeping with the known panhypopituitarism (including central DI) present at diagnosis.
LCH (previously “histiocytosis X”) is one of the three major histiocyte disorders, and involves clonal proliferation of bone marrow-derived dendritic antigen-presenting (“Langerhans”) cells which accumulate in various organs (150). It is a rare disease with an incidence of 2.6-8.9 cases/million/year, the majority presenting in infancy (median age at diagnosis 2-3.8 years, incidence at age <1 year 9.0-15.3 cases/million/year vs. age >5 years 0.7-4.5 cases/million/year) with no sex predilection (151–154). The variability in organ involvement causes a spectrum of clinical features ranging from a single self-healing cutaneous lesion to fatal multiorgan disease, particularly if the liver, spleen, lungs, and hemopoietic system (the “risk” organs) are involved (150). Multisystem involvement is present in 27-56% of cases, of which 28-80% have “risk” organ involvement (151–153,155,156). LCH can thus be considered a primary hematological disorder which, in a proportion of cases, infiltrates the CNS, although its etiology, whether neoplastic or reactive, remain poorly understood (155). More than half of biopsied lesions contain BRAF mutations (157).
In the CNS, the hypothalamo-pituitary region is involved in up to 25% of cases, which almost invariably leads to DI (previously known as Hand-Schuller-Christian disease if associated with orbital and bony lesions) (151,152,154,158,159). Commonly associated radiological findings include thickening of the pituitary stalk with progression to space-occupying tumors and an absence of the posterior pituitary bright spot (159). Indeed, LCH is the commonest underlying diagnosis in patients with central DI and an intracranial mass, occurring in 70% of this cohort (160). The presence of multisystem involvement, particularly if involving “risk” organs, craniofacial bones, gastrointestinal tract, skin, or genitalia) is a particular risk factor for DI (159,161).
Treatment is dependent on the number of organs involved and may range from biopsy/curettage, intralesional steroids, indomethacin, and radiotherapy/UV phototherapy for single bone and cutaneous lesions to systemic chemotherapy with steroids and vinblastine for multisystem disease (155,162,163). Refractory cases have been treated with cytarabine, cladribine, clofarabine, hemopoietic stem cell transplantation, or BRAF inhibitors (164–168). Notably, no treatment protocol has been shown to reverse existing or prevent future DI or other endocrinopathies (159), though current therapeutic recommendations are aimed at preventing disease progression and limiting endocrinopathy with prolonged, low-dose systemic chemotherapy (155,169–171). Overall, 5-year survival remains relatively high at 71-95%, but 3-25% of patients experience at least one endocrinopathy (particularly GH deficiency), with no current chemotherapeutic regimens showing superior overall- or endocrine event-free survival (151,156,158,161).
Pituitary Stalk Thickening

Figure 7.
T1-weighted MRI image illustrating the appearances of a contrast-enhancing thickened pituitary stalk lesion (arrow) and an absent posterior pituitary bright spot in a patient presenting with central DI. The differential diagnosis included germinoma and LCH. However, approximately one year after diagnosis, the pituitary stalk lesion resolved completely, although the patient has been left with GH deficiency and central DI.
A thickened pituitary stalk (TPS) may be discovered either as part of the evaluation of a patient presenting with central DI, visual impairment, or other endocrine dysfunction or incidentally on neuroimaging performed for other purposes. It is discussed here as it is an important differential for germ cell tumors and Langerhans cell histiocytosis (LCH), resulting frequently in diagnostic and management dilemmas, due to a number of reasons:
- 1.
There is no clear consensus as to what constitutes abnormality for children; previous adult studies have shown that the 95th centile for the transverse dimensions of the infundibulum at the optic chiasm and pituitary insertion are 4.21-4.35 mm and 2.69-2.89 mm respectively (upper limit 4.21-4.58 mm and 2.93-3.04 mm) (172,173). Raybaud and Barkovich suggest using a pediatric threshold thickness of 3.8 mm at the optic chiasm and 2.7 mm at the pituitary insertion for investigating further pathology, particularly if there are interruptions in the normal smooth tapering of the infundibulum from median eminence to pituitary insertion (174).
- 2.
The radiological appearances of a TPS, LCH and germinomas cannot be easily differentiated and there is substantial overlap (Table 2). The normal infundibulum lacks a blood-brain barrier and therefore always enhances with contrast, obscuring neoplastic processes. TPS is the commonest initial radiological finding in both LCH and germinomas, and concurrent absence of the posterior pituitary bright spot is inconsistent (123,175,176). Similarly, the two commonest causes of TPS in the pediatric age group are LCH and germinomas, accounting for 7-75% and 9-71% of TPS cases respectively (176–179). Other common causes of TPS in adults such as lymphocytic hypophysitis and neurosarcoidosis are rare in children (176).
- 3.
Biopsies of the TPS to obtain a definitive histological diagnosis can be inconclusive and lead to further substantial endocrine morbidity, including panhypopituitarism with DI, and are thus generally avoided (178).
- 4.
The interval from the time of initial symptoms to diagnostic MRI can be prolonged, particularly for germinomas (up to 21 years), occasionally with initially normal neuroimaging (123,124,180,181). An initially normal MRI does not therefore preclude an occult germinoma or other pathological process in the presence of idiopathic central DI, leading some authors to recommend serial 3-6 monthly scans and follow-up, although the duration of serial scanning is unclear (174). Additionally, there have been cases of occult germinomas masquerading as radiologically or even histologically diagnosed lymphocytic hypophysitis in children (182,183).
In an attempt to define which patients with isolated TPS are at risk of neoplasia and therefore require more intensive follow-up or biopsy, Robison et al. suggest risk factors such as the presence of DI (strongest risk factor), the coexistence of DI with anterior pituitary dysfunction or a progressive increase in infundibular size of >15% from baseline (178). Apart from size, no other particular MRI appearances have been found to be specific for pediatric-related tumor processes (184). Various proposed diagnostic pathways have been proposed for the management of TPS and idiopathic DI (178,184,185) but most recently a national consensus-based guideline has been developed in the UK by the CCLG, RCPCH and BSPED to help achieve a more consistent approach to this finding (186).
Miscellaneous Non-Neoplastic Hypothalamo-Pituitary Masses
Other hypothalamo-pituitary malformations can mimic neoplastic processes in the suprasellar region, and should therefore be considered in the differential diagnosis particularly before commencing oncological therapies:
- Pituitary hyperplasia – Hypothalamic releasing hormones are trophic on the pituitary gland, hence hypersecretion of these hormones (e.g., GHRH from a pancreatic tumor in children with MEN1 syndrome) can cause anterior pituitary enlargement and mimic a true mass. The commonest physiological cause of pituitary hyperplasia is puberty, where the maximal height of the gland can be 10 mm in girls and 7 mm in boys (187,188). Pituitary hyperplasia can also occur pathologically, for instance in chronic primary hypothyroidism leading to thyrotroph hyperplasia due to a lack of negative feedback (24,187). It is also important to note that pituitary enlargement can be associated with certain congenital forms of hypopituitarism (PROP1, LHX3, SOX3 mutations (189,190).
- Rathke’s cleft cysts (RCCs) – RCCs are congenital cystic epithelial remnants of Rathke’s pouch which fail to involute during pituitary development, hence arising in the pars intermedia but often extending superiorly (24). Although often incidental and asymptomatic (occurring in 11% of autopsy cases (191)), cystic growth can lead to visual deficits and endocrinopathies, requiring surgical marsupialization (resection exacerbates endocrine dysfunction) (192). Unlike craniopharyngiomas (the other common cystic suprasellar lesion), RCCs do not calcify.
- Arachnoid cysts – These are congenital collections of cerebrospinal fluid (CSF) arising from the splitting and/ or duplication of the arachnoid membranes. 16% are suprasellar and these cysts can present with symptoms of raised intracranial pressure, visual deterioration, endocrinopathies, or developmental delay (193–197). Treatment is by endoscopic fenestration (196,198,199).
- Rare entities – In contrast to adults where autoimmune lymphocytic hypophysitis is the commonest cause of isolated thickened pituitary stalk (TPS), this is exceptionally rare in children, but should be considered in the differential together with other granulomatous diseases (neurosarcoidosis, tuberculosis (24,200).
Table 2.
The Differential Diagnosis of Pediatric Suprasellar Masses by Radiological Features
| Tumor | Primary location | T1 intensity§ | T2 intensity§ | Contrast enhancement | Other features |
|---|---|---|---|---|---|
| Craniopharyngioma | Supra>intrasellar | Variable, heterogenous | High | Yes (cystic rims) | Cysts, heterogenous, calcification |
| LGG | Suprasellar, optic pathways | Low | High | Yes | Generally homogenous |
| Pituitary adenoma | Intrasellar (intrapituitary) | Low | Low | No | Sella turcica expansion |
| Germinoma* | Suprasellar, pituitary stalk | Isointense – low | Isointense – low | Yes | Loss of posterior pituitary bright spot, coexistent pineal tumor |
| Hamartoma | Suprasellar (tuber cinereum) | Isointense | Isointense – high | No | - |
| LCH* | Suprasellar, pituitary stalk | Isointense | Isointense | Yes | Loss of posterior pituitary bright spot, coexistent osseous lesions |
| Lymphocytic hypophysitis* | Suprasellar, pituitary stalk, intrasellar | Isointense | Isointense | Yes | Loss of posterior pituitary bright spot |
| Pituitary hyperplasia | Intrasellar | Isointense | Isointense | Yes | Homogenous |
| RCC | Intrasellar | Isointense – high | Isointense – low | No | Round & smooth walled |
| Granuloma (sarcoidosis, TB) | Suprasellar, pituitary stalk | Isointense – low | Low – isointense | Yes | Coexistent parenchymal and leptomeningeal lesions |
| Arachnoid cyst | Suprasellar | Very low (isointense with CSF) | High (isointense with CSF) | No | - |
NEUROENDOCRINE DYSFUNCTION AT DIAGNOSIS OF HYPOTHALAMO-PITUITARY TUMORS
Neurological Syndromes
RAISED INTRACRANIAL PRESSURE (RICP)
The proximity of hypothalamo-pituitary tumors to the floor of the third ventricle and optic chiasm accounts for the high frequency of RICP and visual symptoms at presentation. RICP symptoms (headache, vomiting, and/or papilloedema) are the commonest presenting feature of any pediatric brain tumor (30-60%) (202,203), but occur with even greater frequency in suprasellar lesions such as craniopharyngiomas (78%) and LGGs (86%) (37,66). Children may therefore present to acute neurosurgical units as a neurosurgical emergency or subacutely with a chronic course that may initially be misdiagnosed as tension/ migrainous headaches or infective gastroenteritis with unrecognized concurrent visual disturbances. Current UK recommendations are to scan all children with vomiting persisting <2 weeks, and/ or headaches occurring in children <4 years, on waking or during sleep, in association with confusion and/ or disorientation, or persisting >4 weeks (9). Persistent vomiting in the absence of other features suggestive of gastroenteritis (diarrhea, pyrexia) should also prompt consideration of an intracranial lesion. It is important to note that due to the delayed fusion of cranial sutures, children <4 years of age with hydrocephalus more often (41%) present with a rapidly increasing head circumference than classical RICP symptoms (203).
VISUAL DETERIORATION
Visual field loss and/or worsening visual acuity are the second commonest presenting feature, particularly in LGGs, where up to 100% of cases may have visual impairment due to direct involvement of the optic pathway (75). Other suprasellar tumors affect visual function by mass effect on the optic chiasm, occurring in up to 50-70% of craniopharyngiomas and 15% of pituitary adenomas (38,44,102). Contrastingly, visual symptoms are rare (~5-7%) in children with other CNS tumors (203). Other common ophthalmological symptoms that warrant urgent neuroimaging include new onset nystagmus, incomitant (paralytic) squints, optic atrophy, and proptosis, particularly given the difficulties in assessing visual function in young children and the danger of passing off a squint as being “normal” in childhood without detailed examination (9,203,204). Parinaud’s syndrome, a combination of upward gaze palsy, convergence-retraction nystagmus, and pupillary dilatation with light-near dissociation is a rare particular presentation of bifocal suprasellar/pineal germinomas due to pressure of the pineal tumor on the tectal plate (124,205). Although the aim of oncological therapy in many of these low-grade tumors is to preserve vision, this has not been generally successful, most likely due to nerve fiber dropout and optic atrophy (84), or the fact that anatomical tumor characteristics correlate poorly with the degree of visual loss at diagnosis (206).
SEIZURES
Seizures are an uncommon presenting clinical feature of pediatric hypothalamo-pituitary tumors, occurring in <10% of craniopharyngiomas, LGGs, and germinomas (35,39,124,207,208), and are more often the result of reversible metabolic causes such as hypoglycemia (from cortisol and/or GH insufficiency), hypernatremia (from DI), or hyponatremia (from SIADH). Gelastic or dacrystic (laughing or crying, from the Greek gelos and dakryon respectively) seizures are notoriously difficult to diagnose but are characteristic of hypothalamic hamartomas (80-90%) due to the intrinsic epileptogenicity of these lesions that are essentially disorders of neuronal migration (134,139,147).
OTHER NEUROLOGICAL AND COGNITIVE SYMPTOMS
Hemiparesis and ataxia are less common but significant presenting features of intracranial tumors, as are cognitive impairment, delayed development, behavioral changes, and psychiatric symptoms, all of which mandate detailed neuro-ophthalmological examination in such cases, particularly in the presence of the neurocutaneous stigmata of tumor-predisposing syndromes such as neurofibromatosis and tuberous sclerosis.
Endocrine Dysfunction
Although neuro-ophthalmological symptoms are the commonest presenting feature of hypothalamo-pituitary lesions, they are often preceded by symptoms associated with undiagnosed endocrinopathies in as many as two-thirds of patients (209). Endocrine dysfunction may be due to hormone excess (e.g., secreting pituitary adenomas, central precocious puberty) or hormone deficiency from pituitary invasion or compression by tumor mass, disrupting the various hypothalamo-pituitary endocrine pathways. The incidence of dysfunction in each of the hypothalamo-pituitary axes is partly dependent on the lesion (Table 3) though the reasons for the specificity of these presentations are largely unknown.
GH deficiency (GHD) and gonadotrophin dysfunction (either central precocious puberty (CPP) or gonadotrophin deficiency (GnD, i.e., pubertal delay/arrest)) are often the initial and commonest endocrinopathies at presentation of both craniopharyngiomas (GHD – up to 100%; GnD – up to 85%, CPP – up to 3%) and LGGs (CPP – up to 56%; GHD – up to 27%; GnD – up to 12%) (37,41,42,66,210). CPP is particularly prevalent in LGGs as it can occur in the context of NF-1 even in the absence of a hypothalamo-pituitary lesion (211). It is also one of key components of the hypothalamic hamartoma clinical triad, present in up to 45% of patients at diagnosis (131,145). In both these cases it is presumed to result from premature activation of hypothalamic GnRH, unlike its occurrence in up to 35% of germinomas, where gonadotrophin-independent CPP can occur due to secretion of β-hCG which shares a common alpha subunit with LH and FSH and thus stimulates the same receptors (124,126).
Other anterior pituitary deficits evolve only with extensive disease, and are usually only seen at presentation with craniopharyngiomas, although more subtle deficits may have previously been under-recognized with other tumors. ACTH deficiency (secondary hypoadrenalism) is particularly important to diagnose and treat pre-operatively, and is present at diagnosis in up to 71% of craniopharyngiomas, 19% of germinomas, 10% of hamartomas and 3% of LGGs (41,124,145,212). Similarly, TSH/TRH deficiency (secondary/central hypothyroidism) is present in up to 32% of craniopharyngiomas, 19% of germinomas and 10% of LGGs and hamartomas(45,124,145,213). Mild to moderate hyperprolactinemia (<2000 mU/l) is common in all non-prolactinoma hypothalamo-pituitary lesions, needs to be distinguished from true prolactinomas (>5000 mU/l), and does not usually lead to clinically significant galactorrhea.
Posterior pituitary dysfunction, particularly central (“cranial”) DI, is the hallmark endocrinopathy of germinomas and Langerhans cell histiocytosis (LCH), being present in up to 90% and 40% of patients respectively at diagnosis (123,158). However, DI can also occur as a presenting clinical feature for other suprasellar lesions which may be missed if symptoms of polyuria and polydipsia are not elucidated.
Table 1.
Common Endocrinopathies at Presentation of Various Hypothalamo-Pituitary Lesions
| Tumor | Commonest endocrinopathy at presentation |
|---|---|
| Craniopharyngioma | GH deficiency, pubertal delay/arrest |
| Optic pathway LGG | Central precocious puberty |
| Pituitary adenoma | Hyperprolactinemia (prolactinomas) |
| Suprasellar germinoma | Central diabetes insipidus, gonadotrophin-independent central precocious puberty (hCG-secreting) |
| Hypothalamic hamartoma | Central precocious puberty |
| Langerhans cell histiocytosis | Central diabetes insipidus |
GH, growth hormone; LGG, low-grade glioma; hCG, human chorionic gonadotrophin.
Endocrine dysfunction is under-recognized at presentation, as demonstrated by the discrepancies between spontaneous reports of growth retardation, weight loss/gain, polyuria and polydipsia compared to their true incidence based on direct enquiry or assessment (44). Longitudinal retrospective studies have shown that growth failure and weight gain can occur up to 3 years before the diagnosis of a craniopharyngioma, especially in the presence of hypothalamic infiltration (214). Since the diagnosis of GH deficiency requires dynamic endocrine testing, and idiopathic CPP can be a normal variant in young girls, a significant underlying lesion may be missed without mandatory neuroimaging, despite studies showing that 14-45% of female patients with CPP have a hypothalamo-pituitary mass (215–217). DI may remain occult in the ACTH-deficient patient, or unrecognized until the patient is water-deprived or rendered effectively adipsic by general anesthesia, coma or further hypothalamic damage sustained during surgery, with potentially fatal consequences. Lethargy, recurrent infections, somnolence, and cold intolerance may be subtle symptoms of ACTH and/or TSH deficiencies, whilst hypothalamic dysfunction (discussed below) manifesting as hyperphagia, escalating obesity, sleep-wake cycle disturbance, and temperature dysregulation may be mistaken for psychosocial dysfunction.
PRE-OPERATIVE ENDOCRINE ASSESSMENT AND MANAGEMENT OF HYPOTHALAMO-PITUITARY TUMORS
Due to their relative rarity and a general lack of data on optimum treatment strategies, all pediatric hypothalamo-pituitary tumors should be discussed in a multidisciplinary forum which comprises, at minimum, a neuro-oncologist, neuroradiologist, pediatric endocrinologist, and pituitary surgeon. Careful endocrine assessment with appropriate neuroimaging is vital before definitive therapy (Table 4). Early morning cortisol/ACTH measurements should ideally be performed before any dexamethasone is given for cerebral oedema, alongside paired urine and plasma osmolarities & electrolytes as these will influence perioperative fluid management. Plasma tumor markers (prolactin, β-hCG, α-fetoprotein) should be obtained prior to any surgical intervention regardless of radiological appearances, as both prolactinomas and germinomas can be treated medically without requiring a biopsy. In some cases, cerebrospinal fluid β-hCG and α-fetoprotein may be required to aid diagnosis. Early access to a pediatric endocrinologist enhances diagnostic decision-making and ensures appropriate peri-operative fluid management particularly in the presence of life-threatening salt/water and hypocortisolemic crises. If dexamethasone has not been commenced for peritumoral edema and where a patient’s hypothalamo-pituitary-adrenal status is unknown, parenteral hydrocortisone (2 mg/kg) should be given at anesthetic induction and 6-8 hourly thereafter for 48-72 hours (or via a continuous hydrocortisone infusion), weaning to maintenance doses over 5-10 days according to clinical status until this axis can be formally assessed with a synacthen test. Clinicians should be aware of cortisol’s permissive effects on the renal tubule for free water clearance; thus, its replacement will unmask occult DI. In this situation, precise fluid balance measurements and the judicious use of desmopressin by an experienced endocrinologist are required. GH, thyroxine and estradiol/ testosterone supplementation may also be necessary. It is important to note that thyroid hormone replacement should not be commenced until a patient is cortisol replete for at least 48 hours to avoid precipitating an adrenal crisis.
Table 4.
Recommended Minimum Pre-Treatment Endocrine Assessment for Hypothalamo-Pituitary Tumors
| Clinical assessment |
| Height Weight Sitting height BMI Tanner pubertal stage Bone age |
| Endocrine biochemistry |
| IGF-1/IGF-BP3 LH, FSH, estradiol/testosterone TSH, free T4 ± free T3 Early morning (8-9 am) cortisol & ACTH Early morning paired urine & plasma osmolarities & electrolytes |
| Tumor markers |
| PRL AFP β-hCG |
BMI, body mass index; IGF-1, insulin-like growth factor 1; IGF-BP3, insulin-like growth factor binding protein 3; LH, luteinizing hormone; FSH, follicle-stimulating hormone; TSH, thyroid stimulating hormone; T4, thyroxine; T3, triiodothyronine; ACTH, adrenocorticotrophic hormone; PRL, prolactin; AFP, alpha-fetoprotein; β-hCG, beta-human chorionic gonadotrophin.
Rare Emaciation/Failure To Thrive Syndromes
DIENCEPHALIC SYNDROME (DS)
DS is a rare syndrome of severe emaciation first described over 60 years ago typically seen in infants <2 years of age in the presence of a hypothalamic tumor (218). The original description incorporated four “major” criteria – profound emaciation (often leading to a multitude of misdirected investigations for failure to thrive), preserved (or accelerated) linear growth, hyperactivity, and euphoria – and three “minor” features: pallor without anemia, hypoglycemia, and hypotension. There is marked loss of subcutaneous fat despite increased caloric intake. Other associated features result from either tumor location (nystagmus, papilloedema, optic atrophy, vomiting, ataxia) or increased sympathetic tone (sweatiness, tremor). Classically, DS occurs in <10% of hypothalamic LGGs (11,209), but has also been described in suprasellar high grade gliomas (77,219), germinomas (220,221), teratomas (222), ependymomas (223), craniopharyngiomas (224), epidermoid cysts (225), and rarely with non-suprasellar lesions such as brainstem gliomas (226). Since Russell’s original description, however, the definition for DS has now too loosely broadened to include all cancer-related cachexia (227), with <4% of patients with DS having onset of symptoms at >2 years of age (220,228), and some publications reporting adult-onset DS where growth velocity is irrelevant (224,229). It is therefore becoming increasingly difficult to determine whether the patients described in these cases truly have DS or not. Its pathophysiology remains poorly understood, although the most consistent biochemical finding is of high random plasma GH concentrations that is neither suppressed by an oral glucose tolerance test, nor further stimulated by insulin-induced hypoglycemia, with low or normal IGF-1 concentrations, indicative of a GH-resistant state (77,230,231). Studies showing increased resting energy expenditure (232,233) support the theory of a dysregulated metabolism rather than abnormal caloric intake. At the time of writing, the next LGG trial is being designed to incorporate an international study of this rare entity, which is an independent risk factor for death, progression (11) and severe endocrine morbidity (25).
ANOREXIA AND EATING DISORDERS
Anorexia nervosa is an over-represented symptom in multiple published case reports of patients with hypothalamic lesions (particularly slow-growing germ cell tumors), with an average delay in diagnosis of nearly 3 years (234), though symptoms tend to resolve with appropriate therapy. Given the ventromedial and lateral hypothalamic location of the hunger and satiety centers, it is reasonable to postulate the effect of a suprasellar lesion on appetite. However, current understanding of the orexigenic and anorexigenic neuroendocrine regulators of tumor-related anorexia is still incomplete, and reports of non-suprasellar CNS tumors presenting with anorexia (227,235,236) suggest dysregulation beyond the hypothalamus, whilst the effect of inflammatory cytokines present in disseminated disease (tumor necrosis factor-α (TNF- α), interleukin-1 (IL-1), interleukin-6 (IL-6), interferon-γ (IFN- γ)), may also play a role (227). An intracranial lesion needs to be differentiated from true anorexia nervosa, which should fulfil DSM-V or ICD-10 criteria (237,238)), in all patients presenting with anorexia and weight loss. A full auxological, pubertal and endocrine biochemical assessment should be performed to exclude neuroendocrine disease, particularly in boys where the lower prevalence of anorexia nervosa requires mandatory pituitary neuroimaging. Anorexia nervosa presenting with amenorrhea may be due to a suprasellar tumor causing hypogonadotrophic hypogonadism (239), and initially normal imaging may not exclude an eventual diagnosis of a tumor, particularly for germinomas (235). Severe weight loss at diagnosis may be a predictor for future hypothalamic obesity (240).
NEUROENDOCRINE DYSFUNCTION AFTER DIAGNOSIS AND/OR TREATMENT
The Evolution Of Endocrinopathy And Its Association With Treatment
Whilst the initial endocrinopathies present at diagnosis are fairly typical for particular tumor subtypes, the pattern of post-treatment endocrine dysfunction in survivors of these lesions is interestingly very similar in frequency and timing. It has long been recognized that there is an evolution in the incidence of dysfunction in each of the hypothalamo-pituitary axes over time, closely mimicking that seen in congenital neurodevelopmental disorders such as septo-optic dysplasia (241). Although the various axes are differentially sensitive to irradiation, with the GH axis being the most susceptible (even at doses as low as 20 Gy), and the ACTH axis being the most robust (38,242,243), the similar evolutionary pattern of endocrine dysfunction seen in patients with a wide range of hypothalamo-pituitary lesions even in the absence of therapeutic irradiation suggests that the pattern of deficits is related most strongly to the position of the tumor (and thus recurrent disease) rather than treatment. GH deficiency is thus commonest, followed by gonadotrophin dysfunction (either central precocious puberty or hypogonadotrophic hypogonadism), ACTH and TSH deficiency, and least commonly posterior pituitary dysfunction, usually presenting as central DI (which is never seen after similar pituitary irradiation doses administered to non-suprasellar tumors) (25,37,45,145,158,244–247). Hence, lifelong endocrine follow-up of these survivors with regular clinical and biochemical assessments is vital as all patients with such tumors remain at high-risk for the development of these deficits. National guidelines on the neuroendocrine long-term follow-up of tumors such as craniopharyngiomas have been developed in the UK (49).
GH Deficiency
GH deficiency affects virtually all survivors of pediatric hypothalamo-pituitary lesions at some stage. If not already present at diagnosis, it is virtually guaranteed to occur after pituitary-directed therapy such as radiotherapy or surgery (45,248). Diagnosis of GH deficiency requires dynamic endocrine testing with the gold standard being the insulin tolerance test, although this is contraindicated in patients with a history of seizures. It is worth noting that the GHRH stimulation test should not be used in this context as it will not detect GH deficiency of hypothalamic origin (249). Serum IGF-1 and its binding protein IGF-BP3 are less accurate markers of GH deficiency, although they may be useful in severe growth failure in the context of a hypothalamo-pituitary tumor where GH testing is considered too hazardous (250,251). They should not be used in the context of suspected GH deficiency in the context of previous irradiation (252–254). Occasionally, GH deficiency may initially present with abnormal spontaneous secretion but normal peak responses to stimulation tests (termed “neurosecretory dysfunction”) (255), although testing for this with overnight GH profiling is not currently recommended by the GH Research Society (256).
Paradoxical normal growth may continue despite GH deficiency either due to precocious or accelerated puberty, or the syndrome of “growth without growth hormone”, where secondary hyperinsulinemia occurs due to the rapid weight gain observed post-treatment (257). Growth failure may also be masked by concurrent central precocious puberty. Both situations deserve prompt investigation and GH substitution which, in replacement doses, does not increase tumor recurrence (25,258–260), but promotes anabolism and lean body mass. This should therefore not be delayed beyond 12 months after definitive therapy (although this cut-off is arbitrary) (261), particularly in patients who have irreversible loss of height from spinal irradiation (e.g., for germinomas) (262).
Gonadotrophin Dysfunction
Gonadotrophin dysfunction may manifest in three ways. Firstly, central precocious puberty (CPP) (defined as a testicular volume of ≥4 ml in a boy <9 years or breast budding in a girl <8 years) which, if not already present at diagnosis (e.g., hamartomas, LGGs, germinomas) is increased particularly by radiotherapy (243). There is no evidence that surgical resection of hypothalamic hamartomas, the commonest lesion associated with CPP, improves these symptoms, despite ameliorating the seizures (145). As mentioned above, coexistence of an early puberty with GH deficiency may mask the latter as height velocity may initially appear to be maintained or even accelerated, but not when corrected for bone age. Any child in puberty should therefore concurrently have an urgent assessment of GH secretion and consideration of replacement to restore height in combination with GnRH analogues to delay skeletal maturation if it is felt psychosocially appropriate. It is worth noting that prior CPP does not preclude later pubertal delay or arrest and may in fact be a risk factor (25). Therefore, careful monitoring is required even after the cessation of GnRH analogues.
Pubertal delay or arrest may either be due to hypogonadotrophic hypogonadism from tumor- or treatment-related injury to the hypothalamus (causing GnRH and/or LH/FSH deficiency) or to primary gonadal failure from systemic chemotherapy (hypergonadotrophic hypogonadism). Patients may fail to enter puberty altogether by the expected age (14 years in boys, 13 years in girls), enter puberty normally and subsequently fail to progress, or demonstrate secondary amenorrhea (girls) or sexual dysfunction (boys). In this situation concurrent GH deficiency can be corrected simultaneously or 6 months prior to commencing sex steroid replacement to initiate an appropriately-timed pubertal growth spurt. There is no advantage to adult height in delaying sex steroid replacement any further, particularly in the light of the benefits on bone mineral accretion (263).
Most chemotherapeutic drugs used in CNS tumor regimens (e.g., carboplatin, vincristine, etoposide) are not considered gonadotoxic, but other high-risk agents such as cyclophosphamide, temozolomide, and cisplatin are occasionally used, with their effects being modulated by age at exposure and gender (264). Since it is possible to protect future fertility in boys even as young as 12 years with some masculinization (Tanner stage 3+ and/or testicular volume of 8+ mls) by sperm cryopreservation, this should be considered before definitive therapy, even in those not receiving chemotherapy (265). By contrast, girls who have achieved regular spontaneous menses should be warned of the reduced window of reproductive capacity and a premature menopause due to a reduced ovarian follicular reserve (266). Notably, patients with hypothalamo-pituitary tumors who have received chemotherapy can potentially have concurrent hypogonadotrophic hypogonadism and primary gonadal failure, compounding the future risk of subfertility.
ACTH Deficiency/Central Adrenal Insufficiency
The hypothalamo-pituitary-adrenal (HPA) axis is fortunately relatively robust to irradiation and chemotherapeutic damage. However, in the context of a hypothalamo-pituitary tumor, the most important diagnostic challenge is to accurately determine adrenal reserve and differentiate reversible dexamethasone-induced ACTH suppression (after treatment for cerebral edema) from true, permanent ACTH deficiency. Given the lifelong implications of the latter, it is our opinion that the diagnosis should be carefully made ideally with the gold standard insulin tolerance test (ITT) and repeatedly reviewed with time. This may additionally necessitate regular plasma morning cortisol and ACTH measurements and 24-hour cortisol day curves. Although the standard synacthen test (SST) is often used to test adrenal integrity in adults, this supraphysiological stimulus does not test the entire pathway and the integrity of the hypothalamus or pituitary. There is evidence to suggest that in CNS tumor survivors the SST may be less sensitive than the ITT or low dose synacthen stimulation in detecting more subtle degrees of deficiency (267–269). In patients who have received peri-operative dexamethasone for peritumoral edema, formal testing of the HPA axis may be best left until 2-3 months after substitution with maintenance hydrocortisone as doses can be more safely omitted whilst testing is performed. Testing should be performed in a tertiary pediatric endocrinology unit used to managing patients with multiple endocrinopathies, with routine glucose rescue at 25-30 minutes and hydrocortisone at the end of low-dose (0.1 units/kg) insulin-induced hypoglycemia or glucagon stimulation. Treatment of adrenal insufficiency with glucocorticoids may unmask occult DI, and the coexistence of ACTH deficiency, DI, and adipsia due to hypothalamic damage can be fatal and should be avoided where possible.
TRH/TSH Deficiency/Central Hypothyroidism
The thyroid, like the hypothalamo-pituitary-gonadal axis, can be rendered underactive by either central TRH/TSH deficiency (inappropriately normal/low TSH for a low free T4 or T3) due to the tumor itself or surgery, or primary hypothyroidism (high TSH with a normal (compensated/subclinical) or low (frank) free T4) from spinal irradiation and/or chemotherapy, with the potential for the two states coexisting in some patients. There is little evidence for the role of irradiation in the former. In the adult cohort studied by Littley et al., no patients treated with low-dose radiotherapy alone experienced TSH deficiency (242). Similarly, Gan et al. found that the only independent risk factor for TSH deficiency in LGGs was hypothalamic involvement of the tumor (25). TRH stimulation tests may not differentiate hypothalamic (tertiary) from pituitary (secondary) damage, and serial thyroid function tests with two consecutive low free T4 concentrations in association with a low or inappropriately normal TSH concentration confirm the diagnosis without the need for further testing (270–272).
Primary hypothyroidism can present many years after the initial irradiation or chemotherapeutic insult. Annual thyroid function tests in at-risk children are important for early detection of subclinical hypothyroidism and institution of early treatment, particularly in light of the known effects on the developing brain. Given the known risk of radiation-associated second primary thyroid cancers, the carcinogenicity of nuclear fallouts, and the long-term cardiovascular mortality risk of subclinical hypothyroidism, few clinicians would leave a persistently raised TSH in such a patient cohort untreated (273).
Hyperprolactinemia
The importance of serum prolactin (PRL) measurements in the diagnosis of prolactinomas has already been discussed. Similarly, a rise in PRL levels can occur post-treatment in two situations. In the presence of a prolactinoma, this can indicate tumor “escape” from dopamine agonist (cabergoline/bromocriptine) control requiring further therapy. The more common situation arises where hyperprolactinemia is due to stalk compression by a progressive sellar or suprasellar tumor or hypothalamic damage. In this situation PRL concentrations are usually <2000 mU/l (274) and patients are unlikely to be symptomatic, with galactorrhea being unusual (25). Chronic severe primary hypothyroidism will also lead to hyperprolactinemia due to the stimulatory effects of a raised TRH on the lactotroph.
Posterior Pituitary Dysfunction (PPD)
Posterior pituitary dysfunction can present itself in three ways – DI, SIADH, or cerebral salt-wasting syndrome (CSW), the latter attributed to hypersecretion of cerebral atrial natriuretic (ANP) and brain natriuretic peptides (BNP) in response to plasma volume expansion by ADH. The latter two syndromes are rare outside the context of an acute cerebral insult and are usually transient, whilst DI may be a presenting feature and/or a permanent post-operative deficit. The absence of a posterior pituitary bright spot on MRI is a relatively sensitive marker of a lack of neurohypophyseal integrity (275–277). DI does not develop after cranial irradiation in the absence of a hypothalamo-pituitary tumor or surgery to the area (25,99). Whilst PPD is the least common form of endocrinopathy, the rapid shifts from hyper- to hyponatremia in the acute setting can prove life-threatening, as evidenced by a recent retrospective cohort study of optic pathway LGGs with high survival showing showed that nearly 50% of the deaths that occurred were associated with uncontrolled PPD (25). This risk is further increased by coexistent ACTH deficiency, hypothalamic adipsia, and treatment with anti-epileptic medications, which have SIADH-like effects.
After hypothalamo-pituitary surgery, PPD presents as a well-described triphasic response in ADH secretion: firstly, immediate but transient DI up to day 2; secondly, SIADH from day 1-14; and finally, a second phase of DI, which is usually permanent if it persists beyond 21 days, the preceding SIADH is prolonged or severe, or if extensive surgery has been performed (278,279). This triphasic response is thought to result from necrosis of hypothalamic ADH-secreting magnocellular neurons and is seen more often in children than adults (23% vs. 14% in one craniopharyngioma study) (280). The three phases may also occur independently, and cerebral salt-wasting syndrome may coexist and complicate diagnosis and management. Dramatic changes in sodium concentrations can therefore occur with the inherent risk of seizures, cerebral edema and death; such patients require high intensity care with precise fluid management supervised by an experienced pediatric endocrinologist. The measurement of plasma and urinary arginine-vasopressin (AVP) may help differentiate between these different disorders, but these assays are not widely available and careful sample processing is required prior to analysis (281). More recently, measurement of plasma copeptin, the more stable by-product of cleavage of AVP from its carrier protein neurophysin II, is becoming more widely available and has been shown to be a more easily measurable, sensitive, and specific surrogate marker of AVP secretion (282–284).
Detailed management of these disorders is beyond the scope of this chapter, but can be summarized in the algorithm seen in Figure 8.

The Hypothalamic Syndrome
The hypothalamic “syndrome” is loosely defined and usually refers to a constellation of features attributed to hypothalamic dysfunction. Central to it is hypothalamic obesity, a morbid, inexorably escalating obesity (BMI usually >+3 SDS) first described over a century ago (285). It occurs in up to 77% of craniopharyngiomas, 53% of optic pathway LGGs, 40% of pituitary adenomas, 40% of germinomas, and 23% of hamartomas (64,145,286–288), with some symptoms occurring at diagnosis prior to any treatment. Despite this, its pathophysiology is still poorly understood, although it is becoming increasingly evident that both hyperphagia and a dysregulation of anorexigenic and orexigenic hormones contribute (289). Young age at diagnosis, hypothalamic injury by tumor, high dose irradiation or surgery (including biopsies), and multiple endocrinopathies are all risk factors (278,289). Unlike common obesity, the weight gain is largely resistant to caloric restriction, lifestyle interventions, medical and surgical therapies (290–295). Several authors have recently trialed GLP-1 agonists in hypothalamic obesity with some success (296–298), but a randomized control trial is needed to confirm these findings in the longer-term, particularly given the newly published data demonstrating long-term success with common obesity (299). More recently, the combination of tesofensine (a monoamine reuptake inhibitor) and metoprolol has shown promising results in a phase 2 trial (300).
Other hypothalamic symptoms include sleep-wake cycle disturbances, adipsia, temperature dysregulation, cognitive (particularly memory loss), and behavioral (particularly autistic) disorders. Children with disturbed sleep and/or behavioral difficulties should be referred to a specialist sleep laboratory and behavioral neuropsychopharmacology unit. These disorders are even more difficult to treat than replacement of the endocrine deficits. Where endocrine deficits, particularly ACTH deficiency and DI coexist, hypothalamic adipsia is potentially fatal particularly during intercurrent illness and surgery, requiring careful day-to-day fluid management with obligate daily fluid intake and desmopressin dose adjustments. The difficulties in managing patients with panhypopituitarism with concurrent hypothalamic dysfunction should not be underestimated, therefore avoiding these complications must be an important aim of initial therapy.
CONCLUSIONS
Pediatric hypothalamo-pituitary tumors are uncommon, and may present with occult or unusual clinical features posing diagnostic dilemmas that incur treatment delays or necessitate prolonged MRI surveillance. Notwithstanding their generally high survival rates, tumor- or treatment-related neuroendocrine morbidity is very significant and not always simply reversible by hormone replacement therapy. Consequently, treatment decision-making should aim to preserve not only visual, but also hypothalamo-pituitary function. Pediatric endocrinologists and pituitary surgeons should be part of the decision-making multidisciplinary team, with radiological, visual, and biochemical assessments together aiding management planning. A detailed baseline endocrine assessment is paramount to both diagnosis and treatment and will ultimately improve long-term outcome monitoring, the clarification of tumor- and treatment-related consequences and management of lifelong morbidity. Given the potentially significant reduction in health-related quality of survival, lifelong, age-appropriate follow-up and management within a dedicated multidisciplinary neuroendocrine unit familiar with the complexity of patients’ needs is recommended. To achieve this, rehabilitation, reproductive, neuropsychological, and vocational services need developing further in parallel with appropriate transition processes to adult services if we are to better manage and improve outcomes for this high-risk group of young patients. Evidence- and consensus-based guidelines are increasingly being developed to define a standard of best practice for the management of these rare tumors.
REFERENCES
- 1.
- Baade PD, Youlden DR, Valery PC, Hassall T, Ward L, Green AC, et al. Trends in incidence of childhood cancer in Australia, 1983-2006. Br J Cancer [Internet]. 2010/01/07. 2010;102(3):620–6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20051948. [PMC free article: PMC2822940] [PubMed: 20051948] - 2.
- Childhood Cancer Research Group . The National Registry of Childhood Tumours. Oxford: Childhood Cancer Research Group; 2012.
- 3.
- Stiller C. Childhood cancer in Britain: incidence, survival, mortality. Oxford: Oxford University Press; 2007.
- 4.
- Department of Health ., Macmillan Cancer Support ., NHS Improvement . Living with and beyond cancer: taking action to improve outcomes. London: National Cancer Survivorship Initiative (NCSI), Department of Health; 2013.
- 5.
- Ward EM, Thun MJ, Hannan LM, Jemal A. Interpreting cancer trends. Ann N Y Acad Sci. 2006 Nov 23;2006(1076):29–53. http://www
.ncbi.nlm.nih .gov/pubmed/17119192 [Internet] Available from. [PubMed: 17119192] - 6.
- Adamson P, Law G, Roman E. Assessment of trends in childhood cancer incidence. Lancet [Internet]. 2005/03/01. 2005;365(9461):753. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15733714. [PubMed: 15733714] - 7.
- Steliarova-Foucher E, Stiller C, Kaatsch P, Berrino F, Coebergh JW. Trends in childhood cancer incidence in Europe, 1970-99. Lancet [Internet]. 2005/06/21. 2005;365(9477):2088. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15964441. [PubMed: 15964441] - 8.
- Hjalmars U, Kulldorff M, Wahlqvist Y, Lannering B. Increased incidence rates but no space-time clustering of childhood astrocytoma in Sweden, 1973-1992: a population-based study of pediatric brain tumors. Cancer [Internet]. 1999/05/01. 1999;85(9):2077–90. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/10223251. [PubMed: 10223251] - 9.
- Royal College of Paediatrics & Child Health ., Samantha Dickson Brain Tumour Trust ., Children’s Brain Tumour Research Centre ., The Health Foundation . The diagnosis of brain tumours in children: an evidence-based guideline to assist healthcare professionals in the assessment of children presenting with symptoms and signs that may be due to a brain tumour. 3rd ed. Nottingham: Children’s Brain Tumour Research Centre; 2011.
- 10.
- Gatta G, Capocaccia R, Stiller C, Kaatsch P, Berrino F, Terenziani M. Childhood cancer survival trends in Europe: a EUROCARE Working Group study. J Clin Oncol [Internet]. 2005/06/01. 2005;23(16):3742–51. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15923571. [PubMed: 15923571] - 11.
- Gnekow AK, Falkenstein F, von Hornstein S, Zwiener I, Berkefeld S, Bison B, et al. Long-term follow-up of the multicenter, multidisciplinary treatment study HIT-LGG-1996 for low-grade glioma in children and adolescents of the German Speaking Society of Pediatric Oncology and Hematology. Neuro Oncol [Internet]. 2012/09/04. 2012;14(10):1265–84. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22942186. [PMC free article: PMC3452343] [PubMed: 22942186] - 12.
- Stokland T, Liu JF, Ironside JW, Ellison DW, Taylor R, Robinson KJ, et al. A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro Oncol [Internet]. 2010/09/24. 2010;12(12):1257–68. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20861086. [PMC free article: PMC3018938] [PubMed: 20861086] - 13.
- Skinner R, Wallace WH, Levitt G. Long-term follow-up of children treated for cancer: why is it necessary, by whom, where and how? Arch Dis Child [Internet]. 2007/03/06. 2007;92(3):257–60. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17337686. [PMC free article: PMC2083428] [PubMed: 17337686] - 14.
- Skinner R, Wallace WHB, Levitt GA. Therapy based long-term follow-up. 2nd ed. UK Children’s Cancer Study Group (UK CCSG) Late Effects Group; 2005.
- 15.
- Wallace WH, Thompson L, Anderson RA. Long term follow-up of survivors of childhood cancer: summary of updated SIGN guidance. BMJ [Internet]. 2013/03/29. 2013;346:f1190. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23535255. [PubMed: 23535255] - 16.
- Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, et al. Chronic health conditions in adult survivors of childhood cancer. N Engl J Med [Internet]. 2006/10/13. 2006;355(15):1572–82. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17035650. [PubMed: 17035650] - 17.
- Brignardello E, Felicetti F, Castiglione A, Chiabotto P, Corrias A, Fagioli F, et al. Endocrine health conditions in adult survivors of childhood cancer: the need for specialized adult-focused follow-up clinics. Eur J Endocrinol [Internet]. 2012/12/22. 2013;168(3):465–72. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23258270. [PubMed: 23258270] - 18.
- Regal M, Paramo C, Sierra SM. Garcia-Mayor R v. Prevalence and incidence of hypopituitarism in an adult Caucasian population in northwestern Spain. Clin Endocrinol (Oxf). 2001;55(6):735–40. http://www
.ncbi.nlm.nih .gov/pubmed/11895214 [Internet] Available from. [PubMed: 11895214] - 19.
- Tanriverdi F, Dokmetas HS, Kebapci N, Kilicli F, Atmaca H, Yarman S, et al. Etiology of hypopituitarism in tertiary care institutions in Turkish population: analysis of 773 patients from Pituitary Study Group database. Endocrine. 2014;47(1):198–205. http://www
.ncbi.nlm.nih .gov/pubmed/24366641 [Internet] Available from. [PubMed: 24366641] - 20.
- Andoniadou CL, Gaston-Massuet C, Reddy R, Schneider RP, Blasco MA, le Tissier P, et al. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol [Internet]. 2012/02/22. 2012;124(2):259–71. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22349813. [PMC free article: PMC3400760] [PubMed: 22349813] - 21.
- Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, et al. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A [Internet]. 2011/06/04. 2011;108(28):11482–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21636786. [PMC free article: PMC3136310] [PubMed: 21636786] - 22.
- Muller HL, Emser A, Faldum A, Bruhnken G, Etavard-Gorris N, Gebhardt U, et al. Longitudinal study on growth and body mass index before and after diagnosis of childhood craniopharyngioma. J Clin Endocrinol Metab [Internet]. 2004/07/09. 2004;89(7):3298–305. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15240606. [PubMed: 15240606] - 23.
- Arora RS, Alston RD, Eden TO, Estlin EJ, Moran A, Birch JM. Age-incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro Oncol [Internet]. 2008/11/27. 2009;11(4):403–13. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/19033157. [PMC free article: PMC2743220] [PubMed: 19033157] - 24.
- Schroeder JW, Vezina LG. Pediatric sellar and suprasellar lesions. Pediatr Radiol [Internet]. 2011/01/27. 2011;41(3):287–98. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21267556. [PubMed: 21267556] - 25.
- Gan HW, Phipps K, Aquilina K, Gaze MN, Hayward R, Spoudeas HA. Neuroendocrine Morbidity After Pediatric Optic Gliomas: A Longitudinal Analysis of 166 Children Over 30 Years. J Clin Endocrinol Metab. 2015;100(10):3787–99. http://www
.ncbi.nlm.nih .gov/pubmed/26218754 [Internet] Available from. [PubMed: 26218754] - 26.
- Warmuth-Metz M, Gnekow AK, Muller H, Solymosi L. Differential diagnosis of suprasellar tumors in children. Klin Padiatr [Internet]. 2004/11/27. 2004;216(6):323–30. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15565547. [PubMed: 15565547] - 27.
- Kaatsch P, Rickert CH, Kuhl J, Schuz J, Michaelis J. Population-based epidemiologic data on brain tumors in German children. Cancer. 2001;92(12):3155–64. http://www
.ncbi.nlm.nih .gov/pubmed/11753995 [Internet] Available from. [PubMed: 11753995] - 28.
- May JA, Krieger MD, Bowen I, Geffner ME. Craniopharyngioma in childhood. Adv Pediatr. 2006;53:183–209. http://www
.ncbi.nlm.nih .gov/pubmed/17089867 [Internet] Available from. [PubMed: 17089867] - 29.
- Bunin GR, Surawicz TS, Witman PA, Preston-Martin S, Davis F, Bruner JM. The descriptive epidemiology of craniopharyngioma. J Neurosurg [Internet]. 1998/10/07. 1998;89(4):547–51. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/9761047. [PubMed: 9761047] - 30.
- Stiller CA, Nectoux J. International incidence of childhood brain and spinal tumours. Int J Epidemiol [Internet]. 1994/06/01. 1994;23(3):458–64. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7960369. [PubMed: 7960369] - 31.
- Nielsen EH, Feldt-Rasmussen U, Poulsgaard L, Kristensen LO, Astrup J, Jorgensen JO, et al. Incidence of craniopharyngioma in Denmark (n = 189) and estimated world incidence of craniopharyngioma in children and adults. J Neurooncol [Internet]. 2011/02/22. 2011;104(3):755–63. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21336771. [PubMed: 21336771] - 32.
- Zhang YQ, Wang CC, Ma ZY. Pediatric craniopharyngiomas: clinicomorphological study of 189 cases. Pediatr Neurosurg [Internet]. 2002/03/15. 2002;36(2):80–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/11893889. [PubMed: 11893889] - 33.
- Boch AL, van Effenterre R, Kujas M. Craniopharyngiomas in two consanguineous siblings: case report. Neurosurgery. 1997;41(5):1185–7. http://www
.ncbi.nlm.nih .gov/pubmed/9361074 [Internet] Available from. [PubMed: 9361074] - 34.
- Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46(2):161–5. [PMC free article: PMC3982316] [PubMed: 24413733]
- 35.
- Caldarelli M, Massimi L, Tamburrini G, Cappa M, di Rocco C. Long-term results of the surgical treatment of craniopharyngioma: the experience at the Policlinico Gemelli, Catholic University, Rome. Childs Nerv Syst [Internet]. 2005/07/05. 2005;21(8–9):747–57. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15995885. [PubMed: 15995885] - 36.
- Hoffman HJ, de Silva M, Humphreys RP, Drake JM, Smith ML, Blaser SI. Aggressive surgical management of craniopharyngiomas in children. J Neurosurg [Internet]. 1992/01/01. 1992;76(1):47–52. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/1727168. [PubMed: 1727168] - 37.
- Karavitaki N, Brufani C, Warner JT, Adams CB, Richards P, Ansorge O, et al. Craniopharyngiomas in children and adults: systematic analysis of 121 cases with long-term follow-up. Clin Endocrinol (Oxf) [Internet]. 2005/04/06. 2005;62(4):397–409. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15807869. [PubMed: 15807869] - 38.
- Merchant TE, Kiehna EN, Sanford RA, Mulhern RK, Thompson SJ, Wilson MW, et al. Craniopharyngioma: the St. Jude Children’s Research Hospital experience 1984-2001. Int J Radiat Oncol Biol Phys [Internet]. 2002/06/14. 2002;53(3):533–42. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12062594. [PubMed: 12062594] - 39.
- Puget S, Garnett M, Wray A, Grill J, Habrand JL, Bodaert N, et al. Pediatric craniopharyngiomas: classification and treatment according to the degree of hypothalamic involvement. J Neurosurg [Internet]. 2007/01/20. 2007;106(1 Suppl):3–12. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17233305. [PubMed: 17233305] - 40.
- van Effenterre R, Boch AL. Craniopharyngioma in adults and children: a study of 122 surgical cases. J Neurosurg [Internet]. 2002/07/24. 2002;97(1):3–11. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12134929. [PubMed: 12134929] - 41.
- de Vries L, Lazar L, Phillip M. Craniopharyngioma: presentation and endocrine sequelae in 36 children. J Pediatr Endocrinol Metab [Internet]. 2003/07/26. 2003;16(5):703–10. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12880119. [PubMed: 12880119] - 42.
- Hetelekidis S, Barnes PD, Tao ML, Fischer EG, Schneider L, Scott RM, et al. 20-year experience in childhood craniopharyngioma. Int J Radiat Oncol Biol Phys [Internet]. 1993/09/30. 1993;27(2):189–95. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8407391. [PubMed: 8407391] - 43.
- Lin LL, el Naqa I, Leonard JR, Park TS, Hollander AS, Michalski JM, et al. Long-term outcome in children treated for craniopharyngioma with and without radiotherapy. J Neurosurg Pediatr [Internet]. 2008/03/21. 2008;1(2):126–30. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/18352781. [PubMed: 18352781] - 44.
- Muller HL. Childhood craniopharyngioma--current concepts in diagnosis, therapy and follow-up. Nat Rev Endocrinol [Internet]. 2010/09/30. 2010;6(11):609–18. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20877295. [PubMed: 20877295] - 45.
- DeVile CJ, Grant DB, Hayward RD, Stanhope R. Growth and endocrine sequelae of craniopharyngioma. Arch Dis Child [Internet]. 1996/08/01. 1996;75(2):108–14. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8869189. [PMC free article: PMC1511642] [PubMed: 8869189] - 46.
- Sorva R, Heiskanen O, Perheentupa J. Craniopharyngioma surgery in children: endocrine and visual outcome. Childs Nerv Syst [Internet]. 1988/04/01. 1988;4(2):97–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/3401877. [PubMed: 3401877] - 47.
- de Vries L, Weintrob N, Phillip M. Craniopharyngioma presenting as precocious puberty and accelerated growth. Clin Pediatr (Phila). 2003;42(2):181–4. http://www
.ncbi.nlm.nih .gov/pubmed/12659393 [Internet] Available from. [PubMed: 12659393] - 48.
- Molla E, Marti-Bonmati L, Revert A, Arana E, Menor F, Dosda R, et al. Craniopharyngiomas: identification of different semiological patterns with MRI. Eur Radiol [Internet]. 2002/07/12. 2002;12(7):1829–36. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12111075. [PubMed: 12111075] - 49.
- Children’s Cancer & Leukaemia Group (CCLG) . Craniopharyngioma: guideline for the management of children and young people (CYP) aged <19 years. Leicester, UK: CCLG; 2021.
- 50.
- Flitsch J, Muller HL, Burkhardt T. Surgical strategies in childhood craniopharyngioma. Front Endocrinol (Lausanne) [Internet]. 2011/01/01. 2011;2:96. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22645514. [PMC free article: PMC3355821] [PubMed: 22645514] - 51.
- de Vile CJ, Grant DB, Kendall BE, Neville BG, Stanhope R, Watkins KE, et al. Management of childhood craniopharyngioma: can the morbidity of radical surgery be predicted? J Neurosurg [Internet]. 1996/07/01. 1996;85(1):73–81. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8683285. [PubMed: 8683285] - 52.
- Muller HL, Albanese A, Calaminus G, Hargrave D, Garre ML, Gebhardt U, et al. Consensus and perspectives on treatment strategies in childhood craniopharyngioma: results of a meeting of the Craniopharyngioma Study Group (SIOP), Genova, 2004. J Pediatr Endocrinol Metab. 2006;19 Suppl 1:453–4. http://www
.ncbi.nlm.nih .gov/pubmed/16700324 [Internet] Available from. [PubMed: 16700324] - 53.
- Spoudeas HA, Harrison B, Spoudeas HA, Harrison B. Paediatric Endocrine Tumours: A Multidisciplinary Consensus Statement of Best Practice from a Working Group Convened Under the Auspices of the BSPED and UKCCSG (rare tumour working groups). 1st ed. Crawley: Novo Nordisk Ltd.; 2005.
- 54.
- Clark AJ, Cage TA, Aranda D, Parsa AT, Sun PP, Auguste KI, et al. A systematic review of the results of surgery and radiotherapy on tumor control for pediatric craniopharyngioma. Childs Nerv Syst. 2013;29(2):231–8. http://www
.ncbi.nlm.nih .gov/pubmed/23089933 [Internet] Available from. [PubMed: 23089933] - 55.
- Iannalfi A, Fragkandrea I, Brock J, Saran F. Radiotherapy in craniopharyngiomas. Clin Oncol (R Coll Radiol). 2013;25(11):654–67. https://www
.ncbi.nlm .nih.gov/pubmed/23910225 [Internet] Available from. [PubMed: 23910225] - 56.
- Stripp DC, Maity A, Janss AJ, Belasco JB, Tochner ZA, Goldwein JW, et al. Surgery with or without radiation therapy in the management of craniopharyngiomas in children and young adults. Int J Radiat Oncol Biol Phys [Internet]. 2004/02/18. 2004;58(3):714–20. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/14967425. [PubMed: 14967425] - 57.
- Bishop AJ, Greenfield B, Mahajan A, Paulino AC, Okcu MF, Allen PK, et al. Proton beam therapy versus conformal photon radiation therapy for childhood craniopharyngioma: multi-institutional analysis of outcomes, cyst dynamics, and toxicity. Int J Radiat Oncol Biol Phys. 2014;90(2):354–61. http://www
.ncbi.nlm.nih .gov/pubmed/25052561 [Internet] Available from. [PMC free article: PMC4194252] [PubMed: 25052561] - 58.
- Leroy R, Benahmed N, Hulstaert F, van Damme N, de Ruysscher D. Proton Therapy in Children: A Systematic Review of Clinical Effectiveness in 15 Pediatric Cancers. Int J Radiat Oncol Biol Phys. 2016;95(1):267–78. https://www
.ncbi.nlm .nih.gov/pubmed/27084646 [Internet] Available from. [PubMed: 27084646] - 59.
- Bremer AM, Nguyen TQ, Balsys R. Therapeutic benefits of combination chemotherapy with vincristine, BCNU, and procarbazine on recurrent cystic craniopharyngioma. A case report. J Neurooncol [Internet]. 1984/01/01. 1984;2(1):47–51. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/6470759. [PubMed: 6470759] - 60.
- Lippens RJ, Rotteveel JJ, Otten BJ, Merx H. Chemotherapy with Adriamycin (doxorubicin) and CCNU (lomustine) in four children with recurrent craniopharyngioma. Eur J Paediatr Neurol [Internet]. 2000/03/22. 1998;2(5):263–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/10726829. [PubMed: 10726829] - 61.
- Bartels U, Laperriere N, Bouffet E, Drake J. Intracystic therapies for cystic craniopharyngioma in childhood. Front Endocrinol (Lausanne) [Internet]. 2012/06/02. 2012;3:39. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22654864. [PMC free article: PMC3356106] [PubMed: 22654864] - 62.
- Cavalheiro S, di Rocco C, Valenzuela S, Dastoli PA, Tamburrini G, Massimi L, et al. Craniopharyngiomas: intratumoral chemotherapy with interferon-alpha: a multicenter preliminary study with 60 cases. Neurosurg Focus [Internet]. 2010/04/07. 2010;28(4):E12. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20367356. [PubMed: 20367356] - 63.
- Crom DB, Smith D, Xiong Z, Onar A, Hudson MM, Merchant TE, et al. Health status in long-term survivors of pediatric craniopharyngiomas. J Neurosci Nurs [Internet]. 2011/01/07. 2010;42(6):323–8; quiz 329–30. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21207770. [PMC free article: PMC4895693] [PubMed: 21207770] - 64.
- Armstrong GT, Conklin HM, Huang S, Srivastava D, Sanford R, Ellison DW, et al. Survival and long-term health and cognitive outcomes after low-grade glioma. Neuro Oncol [Internet]. 2010/12/24. 2011;13(2):223–34. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21177781. [PMC free article: PMC3064628] [PubMed: 21177781] - 65.
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol [Internet]. 2007/07/10. 2007;114(2):97–109. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17618441. [PMC free article: PMC1929165] [PubMed: 17618441] - 66.
- Bataini JP, Delanian S, Ponvert D. Chiasmal gliomas: results of irradiation management in 57 patients and review of literature. Int J Radiat Oncol Biol Phys [Internet]. 1991/08/01. 1991;21(3):615–23. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/1907959. [PubMed: 1907959] - 67.
- Gnekow AK, Kortmann RD, Pietsch T, Emser A. Low grade chiasmatic-hypothalamic glioma-carboplatin and vincristin chemotherapy effectively defers radiotherapy within a comprehensive treatment strategy -- report from the multicenter treatment study for children and adolescents with a low grade glioma -- HIT-LGG 1996 -- of the Society of Pediatric Oncology and Hematology (GPOH). Klin Padiatr [Internet]. 2004/11/27. 2004;216(6):331–42. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15565548. [PubMed: 15565548] - 68.
- Janss AJ, Grundy R, Cnaan A, Savino PJ, Packer RJ, Zackai EH, et al. Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with a 6-year follow-up. Cancer [Internet]. 1995/02/15. 1995;75(4):1051–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7842408. [PubMed: 7842408] - 69.
- Medlock MD, Madsen JR, Barnes PD, Anthony DS, Cohen LE, Scott RM. Optic chiasm astrocytomas of childhood. 1. Long-term follow-up. Pediatr Neurosurg [Internet]. 1998/04/21. 1997;27(3):121–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/9548522. [PubMed: 9548522] - 70.
- Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res [Internet]. 2005/01/25. 2005;65(1):236–45. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15665300. [PubMed: 15665300] - 71.
- Lawson AR, Tatevossian RG, Phipps KP, Picker SR, Michalski A, Sheer D, et al. RAF gene fusions are specific to pilocytic astrocytoma in a broad paediatric brain tumour cohort. Acta Neuropathol [Internet]. 2010/05/11. 2010;120(2):271–3. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20454969. [PubMed: 20454969] - 72.
- Hargrave DR, Bouffet E, Tabori U, Broniscer A, Cohen KJ, Hansford JR, et al. Efficacy and Safety of Dabrafenib in Pediatric Patients with BRAF V600 Mutation-Positive Relapsed or Refractory Low-Grade Glioma: Results from a Phase I/IIa Study. Clin Cancer Res. 2019;25(24):7303–11. https://www
.ncbi.nlm .nih.gov/pubmed/31811016 [Internet] Available from. [PubMed: 31811016] - 73.
- Campagna M, Opocher E, Viscardi E, Calderone M, Severino SM, Cermakova I, et al. Optic pathway glioma: long-term visual outcome in children without neurofibromatosis type-1. Pediatr Blood Cancer [Internet]. 2010/10/28. 2010;55(6):1083–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20979170. [PubMed: 20979170] - 74.
- Jaing TH, Lin KL, Tsay PK, Hsueh C, Hung PC, Wu CT, et al. Treatment of optic pathway hypothalamic gliomas in childhood: experience with 18 consecutive cases. J Pediatr Hematol Oncol [Internet]. 2008/04/01. 2008;30(3):222–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/18376285. [PubMed: 18376285] - 75.
- Tao ML, Barnes PD, Billett AL, Leong T, Shrieve DC, Scott RM, et al. Childhood optic chiasm gliomas: radiographic response following radiotherapy and long-term clinical outcome. Int J Radiat Oncol Biol Phys [Internet]. 1997/10/23. 1997;39(3):579–87. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/9336136. [PubMed: 9336136] - 76.
- Brauner R, Trivin C, Zerah M, Souberbielle JC, Doz F, Kalifa C, et al. Diencephalic syndrome due to hypothalamic tumor: a model of the relationship between weight and puberty onset. J Clin Endocrinol Metab [Internet]. 2006/04/20. 2006;91(7):2467–73. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16621905. [PubMed: 16621905] - 77.
- Fleischman A, Brue C, Poussaint TY, Kieran M, Pomeroy SL, Goumnerova L, et al. Diencephalic syndrome: a cause of failure to thrive and a model of partial growth hormone resistance. Pediatrics [Internet]. 2005/06/03. 2005;115(6):e742-8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15930202. [PubMed: 15930202] - 78.
- Suarez JC, Viano JC, Zunino S, Herrera EJ, Gomez J, Tramunt B, et al. Management of child optic pathway gliomas: new therapeutical option. Childs Nerv Syst [Internet]. 2006/01/04. 2006;22(7):679–84. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16389565. [PubMed: 16389565] - 79.
- Picariello S, Cerbone M, D’Arco F, Gan HW, O’Hare P, Aquilina K, et al. A 40-Year Cohort Study of Evolving Hypothalamic Dysfunction in Infants and Young Children (<3 years) with Optic Pathway Gliomas. Cancers (Basel). 2022 Jan 31;14(3) [PMC free article: PMC8833541] [PubMed: 35159015]
- 80.
- Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol [Internet]. 2004/07/03. 2004;5(7):399–408. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15231246. [PubMed: 15231246] - 81.
- Friedman DL, Whitton J, Leisenring W, Mertens AC, Hammond S, Stovall M, et al. Subsequent neoplasms in 5-year survivors of childhood cancer: the Childhood Cancer Survivor Study. J Natl Cancer Inst [Internet]. 2010/07/17. 2010;102(14):1083–95. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20634481. [PMC free article: PMC2907408] [PubMed: 20634481] - 82.
- Taylor AJ, Little MP, Winter DL, Sugden E, Ellison DW, Stiller CA, et al. Population-based risks of CNS tumors in survivors of childhood cancer: the British Childhood Cancer Survivor Study. J Clin Oncol [Internet]. 2010/11/17. 2010;28(36):5287–93. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21079138. [PMC free article: PMC4809645] [PubMed: 21079138] - 83.
- Ullrich NJ, Robertson R, Kinnamon DD, Scott RM, Kieran MW, Turner CD, et al. Moyamoya following cranial irradiation for primary brain tumors in children. Neurology [Internet]. 2007/03/21. 2007;68(12):932–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17372129. [PubMed: 17372129] - 84.
- Dalla Via P, Opocher E, Pinello ML, Calderone M, Viscardi E, Clementi M, et al. Visual outcome of a cohort of children with neurofibromatosis type 1 and optic pathway glioma followed by a pediatric neuro-oncology program. Neuro Oncol [Internet]. 2007/08/21. 2007;9(4):430–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17704361. [PMC free article: PMC1994100] [PubMed: 17704361] - 85.
- Wen PY, Stein A, van den Bent M, de Greve J, Wick A, de Vos FYFL, et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutant low-grade and high-grade glioma (ROAR): a multicentre, open-label, single-arm, phase 2, basket trial. Lancet Oncol. 2022;23(1):53–64. [PubMed: 34838156]
- 86.
- Perreault S, Larouche V, Tabori U, Hawkin C, Lippe S, Ellezam B, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway: TRAM-01. BMC Cancer. 2019;19(1):1250. https://www
.ncbi.nlm .nih.gov/pubmed/31881853 [Internet] Available from. [PMC free article: PMC6935133] [PubMed: 31881853] - 87.
- Selt F, van Tilburg CM, Bison B, Sievers P, Harting I, Ecker J, et al. Response to trametinib treatment in progressive pediatric low-grade glioma patients. J Neurooncol. 2020 Sep;149(3):499–510. [PMC free article: PMC7609413] [PubMed: 33026636]
- 88.
- Gan HW. Management of Craniopharyngiomas in the Era of Molecular Oncological Therapies: Not a Panacea. J Endocr Soc. 2021 Jul 1;5(7):bvab094. [PMC free article: PMC8212679] [PubMed: 34159290]
- 89.
- Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev [Internet]. 2006/05/18. 2006;27(5):485–534. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16705142. [PubMed: 16705142] - 90.
- Fideleff HL, Boquete HR, Suarez MG, Azaretzky M. Prolactinoma in children and adolescents. Horm Res [Internet]. 2009/09/30. 2009;72(4):197–205. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/19786791. [PubMed: 19786791] - 91.
- Harrington MH, Casella SJ. Pituitary tumors in childhood. Curr Opin Endocrinol Diabetes Obes [Internet]. 2011/12/14. 2012;19(1):63–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22157404. [PubMed: 22157404] - 92.
- Colao A, Loche S. Prolactinomas in children and adolescents. Endocr Dev. 2009 Dec 04;2010(17):146–59. http://www
.ncbi.nlm.nih .gov/pubmed/19955764 [Internet] Available from. [PubMed: 19955764] - 93.
- Diamond Jr. FB. Pituitary adenomas in childhood: development and diagnosis. Fetal Pediatr Pathol [Internet]. 2007/08/19. 2006;25(6):339–56. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17696045. [PubMed: 17696045] - 94.
- Beckers A, Rostomyan L, Daly AF. Overview of genetic testing in patients with pituitary adenomas. Ann Endocrinol (Paris) [Internet]. 2012/04/17. 2012;73(2):62–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22503805. [PubMed: 22503805] - 95.
- Gan HW, Bulwer C, Jeelani O, Levine MA, Korbonits M, Spoudeas HA. Treatment-resistant pediatric giant prolactinoma and multiple endocrine neoplasia type 1. Int J Pediatr Endocrinol. 2015;2015(1):15. https://www
.ncbi.nlm .nih.gov/pubmed/26180530 [Internet] Available from. [PMC free article: PMC4503293] [PubMed: 26180530] - 96.
- Korbonits M, Storr H. Kumar A v. Familial pituitary adenomas - who should be tested for AIP mutations? Clin Endocrinol (Oxf). 2012;77(3):351–6. http://www
.ncbi.nlm.nih .gov/pubmed/22612670 [Internet] Available from. [PubMed: 22612670] - 97.
- Alband N, Korbonits M. Familial pituitary tumors. Handb Clin Neurol. 2014;124:339–60. http://www
.ncbi.nlm.nih .gov/pubmed/25248598 [Internet] Available from. [PubMed: 25248598] - 98.
- Melmed S, Casanueva FF, Hoffman AR, Kleinberg DL, Montori VM, Schlechte JA, et al. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab [Internet]. 2011/02/08. 2011;96(2):273–88. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21296991. [PubMed: 21296991] - 99.
- Moraes AB, Silva CM, Vieira Neto L, Gadelha MR. Giant prolactinomas: the therapeutic approach. Clin Endocrinol (Oxf) [Internet]. 2013/05/15. 2013;79(4):447–56. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23662975. [PubMed: 23662975] - 100.
- Schade R, Andersohn F, Suissa S, Haverkamp W, Garbe E. Dopamine agonists and the risk of cardiac-valve regurgitation. N Engl J Med. 2007 Jan 4;356(1):29–38. [PubMed: 17202453]
- 101.
- Bulwer C, Gan HW, Stern E, Powell M, Jeelani O, Korbonits M, et al. Managing rare, resistant, macro- and giant prolactinomas causing raised intracranial pressure in children: lessons learnt at a single centre. Horm Res Paediatr. 2013;80 Suppl 1:165.
- 102.
- Steele CA, MacFarlane IA, Blair J, Cuthbertson DJ, Didi M, Mallucci C, et al. Pituitary adenomas in childhood, adolescence and young adulthood: presentation, management, endocrine and metabolic outcomes. Eur J Endocrinol [Internet]. 2010/08/06. 2010;163(4):515–22. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20685833. [PubMed: 20685833] - 103.
- Mindermann T, Wilson CB. Pediatric pituitary adenomas. Neurosurgery. 1995;36(2):259–68. http://www
.ncbi.nlm.nih .gov/pubmed/7731505 [Internet] Available from. [PubMed: 7731505] - 104.
- Savage MO, Storr HL, Chan LF, Grossman AB. Diagnosis and treatment of pediatric Cushing’s disease. Pituitary. 2007;10(4):365–71. http://www
.ncbi.nlm.nih .gov/pubmed/17570065 [Internet] Available from. [PubMed: 17570065] - 105.
- Joshi SM, Hewitt RJ, Storr HL, Rezajooi K, Ellamushi H, Grossman AB, et al. Cushing’s disease in children and adolescents: 20 years of experience in a single neurosurgical center. Neurosurgery. 2005;57(2):281–5. http://www
.ncbi.nlm.nih .gov/pubmed/16094156 [Internet] Available from. [PubMed: 16094156] - 106.
- Guemes M, Murray PG, Brain CE, Spoudeas HA, Peters CJ, Hindmarsh PC, et al. Management of Cushing syndrome in children and adolescents: experience of a single tertiary centre. Eur J Pediatr. 2016;175(7):967–76. http://www
.ncbi.nlm.nih .gov/pubmed/27169546 [Internet] Available from. [PubMed: 27169546] - 107.
- Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics. 2007;120(3):e575–86. http://www
.ncbi.nlm.nih .gov/pubmed/17698579 [Internet] Available from. [PubMed: 17698579] - 108.
- Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526–40. https://www
.ncbi.nlm .nih.gov/pubmed/18334580 [Internet] Available from. [PMC free article: PMC2386281] [PubMed: 18334580] - 109.
- Pecori Giraldi F, Pivonello R, Ambrogio AG, de Martino MC, de Martin M, Scacchi M, et al. The dexamethasone-suppressed corticotropin-releasing hormone stimulation test and the desmopressin test to distinguish Cushing’s syndrome from pseudo-Cushing’s states. Clin Endocrinol (Oxf). 2007;66(2):251–7. https://www
.ncbi.nlm .nih.gov/pubmed/17223996 [Internet] Available from. [PubMed: 17223996] - 110.
- Wood PJ, Barth JH, Freedman DB, Perry L, Sheridan B. Evidence for the low dose dexamethasone suppression test to screen for Cushing’s syndrome--recommendations for a protocol for biochemistry laboratories. Ann Clin Biochem. 1997;34(Pt 3):222–9. https://www
.ncbi.nlm .nih.gov/pubmed/9158818 [Internet] Available from. [PubMed: 9158818] - 111.
- Magiakou MA, Mastorakos G, Oldfield EH, Gomez MT, Doppman JL, Cutler GB Jr, et al. Cushing’s syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med. 1994;331(10):629–36. https://www
.ncbi.nlm .nih.gov/pubmed/8052272 [Internet] Available from. [PubMed: 8052272] - 112.
- Hopwood NJ, Kenny FM. Incidence of Nelson’s syndrome after adrenalectomy for Cushing’s disease in children: results of a nationwide survey. Am J Dis Child. 1977;131(12):1353–6. http://www
.ncbi.nlm.nih .gov/pubmed/930887 [Internet] Available from. [PubMed: 930887] - 113.
- Atkinson AB, Kennedy A, Wiggam MI, McCance DR, Sheridan B. Long-term remission rates after pituitary surgery for Cushing’s disease: the need for long-term surveillance. Clin Endocrinol (Oxf). 2005;63(5):549–59. http://www
.ncbi.nlm.nih .gov/pubmed/16268808 [Internet] Available from. [PubMed: 16268808] - 114.
- Devoe DJ, Miller WL, Conte FA, Kaplan SL, Grumbach MM, Rosenthal SM, et al. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab. 1997;82(10):3196–202. http://www
.ncbi.nlm.nih .gov/pubmed/9329338 [Internet] Available from. [PubMed: 9329338] - 115.
- Storr HL, Afshar F, Matson M, Sabin I, Davies KM, Evanson J, et al. Factors influencing cure by transsphenoidal selective adenomectomy in paediatric Cushing’s disease. Eur J Endocrinol. 2005;152(6):825–33. http://www
.ncbi.nlm.nih .gov/pubmed/15941921 [Internet] Available from. [PubMed: 15941921] - 116.
- Kane LA, Leinung MC, Scheithauer BW, Bergstralh EJ, Laws Jr. ER, Groover R v, et al. Pituitary adenomas in childhood and adolescence. J Clin Endocrinol Metab [Internet]. 1994;79(4):1135–40. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7525627. [PubMed: 7525627] - 117.
- Eugster EA, Pescovitz OH. Gigantism. J Clin Endocrinol Metab. 1999;84(12):4379–84. http://www
.ncbi.nlm.nih .gov/pubmed/10599691 [Internet] Available from. [PubMed: 10599691] - 118.
- van der Lely AJ, Biller BM, Brue T, Buchfelder M, Ghigo E, Gomez R, et al. Long-term safety of pegvisomant in patients with acromegaly: comprehensive review of 1288 subjects in ACROSTUDY. J Clin Endocrinol Metab. 2012;97(5):1589–97. http://www
.ncbi.nlm.nih .gov/pubmed/22362824 [Internet] Available from. [PubMed: 22362824] - 119.
- Eugster E. Gigantism. In: de Groot LJ, Beck-Peccoz P, Chrousos G, Dungan K, Grossman A, Hershman JM, et al., editors. Endotext [Internet]. South Dartmouth (MA); 2000. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/25905378. - 120.
- Surawicz TS, McCarthy BJ, Kupelian V, Jukich PJ, Bruner JM, Davis FG. Descriptive epidemiology of primary brain and CNS tumors: results from the Central Brain Tumor Registry of the United States, 1990-1994. Neuro Oncol [Internet]. 2001/09/14. 1999;1(1):14–25. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/11554386. [PMC free article: PMC1919458] [PubMed: 11554386] - 121.
- Murray MJ, Horan G, Lowis S, Nicholson JC. Highlights from the Third International Central Nervous System Germ Cell Tumour symposium: laying the foundations for future consensus. Ecancermedicalscience [Internet]. 2013/07/19. 2013;7:333. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23861728. [PMC free article: PMC3709531] [PubMed: 23861728] - 122.
- Maity A, Shu HK, Janss A, Belasco JB, Rorke L, Phillips PC, et al. Craniospinal radiation in the treatment of biopsy-proven intracranial germinomas: twenty-five years’ experience in a single center. Int J Radiat Oncol Biol Phys [Internet]. 2004/03/06. 2004;58(4):1165–70. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15001260. [PubMed: 15001260] - 123.
- Phi JH, Kim SK, Lee YA, Shin CH, Cheon JE, Kim IO, et al. Latency of intracranial germ cell tumors and diagnosis delay. Childs Nerv Syst [Internet]. 2013/07/03. 2013;29(10):1871–81. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23811803. [PubMed: 23811803] - 124.
- Sethi R v, Marino R, Niemierko A, Tarbell NJ, Yock TI, Macdonald SM. Delayed diagnosis in children with intracranial germ cell tumors. J Pediatr [Internet]. 2013/07/31. 2013;163(5):1448–53. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23896184. [PubMed: 23896184] - 125.
- Cancer Research UK . CancerStats: Childhood Cancer - Great Britain & UK. London: Cancer Research UK; 2010.
- 126.
- Wang Y, Zou L, Gao B. Intracranial germinoma: clinical and MRI findings in 56 patients. Childs Nerv Syst [Internet]. 2010/07/29. 2010;26(12):1773–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20665036. [PubMed: 20665036] - 127.
- da Silva NS, Cappellano AM, Diez B, Cavalheiro S, Gardner S, Wisoff J, et al. Primary chemotherapy for intracranial germ cell tumors: results of the third international CNS germ cell tumor study. Pediatr Blood Cancer [Internet]. 2010/01/12. 2010;54(3):377–83. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20063410. [PubMed: 20063410] - 128.
- Calaminus G, Kortmann R, Worch J, Nicholson JC, Alapetite C, Garre ML, et al. SIOP CNS GCT 96: final report of outcome of a prospective, multinational nonrandomized trial for children and adults with intracranial germinoma, comparing craniospinal irradiation alone with chemotherapy followed by focal primary site irradiation for patients with localized disease. Neuro Oncol [Internet]. 2013/03/06. 2013;15(6):788–96. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23460321. [PMC free article: PMC3661100] [PubMed: 23460321] - 129.
- O’Neil S, Ji L, Buranahirun C, Azoff J, Dhall G, Khatua S, et al. Neurocognitive outcomes in pediatric and adolescent patients with central nervous system germinoma treated with a strategy of chemotherapy followed by reduced-dose and volume irradiation. Pediatr Blood Cancer [Internet]. 2011/04/16. 2011;57(4):669–73. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21495164. [PubMed: 21495164] - 130.
- Maixner W. Hypothalamic hamartomas--clinical, neuropathological and surgical aspects. Childs Nerv Syst [Internet]. 2006/06/10. 2006;22(8):867–73. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16763856. [PubMed: 16763856] - 131.
- Brandberg G, Raininko R, Eeg-Olofsson O. Hypothalamic hamartoma with gelastic seizures in Swedish children and adolescents. Eur J Paediatr Neurol [Internet]. 2004/03/17. 2004;8(1):35–44. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15023373. [PubMed: 15023373] - 132.
- Ng YT, Kerrigan JF, Prenger EC, White WL, Rekate HL. Successful resection of a hypothalamic hamartoma and a Rathke cleft cyst. Case report. J Neurosurg [Internet]. 2005/10/07. 2005;102(1 Suppl):78–80. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16206738. [PubMed: 16206738] - 133.
- Weissenberger AA, Dell ML, Liow K, Theodore W, Frattali CM, Hernandez D, et al. Aggression and psychiatric comorbidity in children with hypothalamic hamartomas and their unaffected siblings. J Am Acad Child Adolesc Psychiatry [Internet]. 2001/06/08. 2001;40(6):696–703. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/11392348. [PubMed: 11392348] - 134.
- Castano De La Mota C, Martin Del Valle F, Perez Villena A, Calleja Gero ML, Losada Del Pozo R, Ruiz-Falco Rojas ML. [Hypothalamic hamartoma in paediatric patients: clinical characteristics, outcomes and review of the literature]. Neurologia [Internet]. 2012/02/22. 2012;27(5):268–76. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22341983. [PubMed: 22341983] - 135.
- Papayannis CE, Consalvo D, Seifer G, Kauffman MA, Silva W, Kochen S. Clinical spectrum and difficulties in management of hypothalamic hamartoma in a developing country. Acta Neurol Scand [Internet]. 2008/05/09. 2008;118(5):313–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/18462479. [PubMed: 18462479] - 136.
- Tassinari C, Riguzzi P, Rizzi R. Gelastic seizures. In: Tuxhom I, Holthausen H, Boenigk K, editors. Paediatric Epilepsy Syndromes and Their Surgical Management. London: John Libbey; 1997. p. 429–46.
- 137.
- Graham JM Jr, Saunders R, Fratkin J, Spiegel P, Harris M, Klein RZ. A cluster of Pallister-Hall syndrome cases, (congenital hypothalamic hamartoblastoma syndrome). Am J Med Genet Suppl. 1986 Jan 01;1986(2):53–63. http://www
.ncbi.nlm.nih .gov/pubmed/3146300 [Internet] Available from. [PubMed: 3146300] - 138.
- Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, et al. Mutations within Sox2/SOX2 are associated with abnormalities in the hypothalamo-pituitary-gonadal axis in mice and humans. J Clin Invest. 2006;116(9):2442–55. https://www
.ncbi.nlm .nih.gov/pubmed/16932809 [Internet] Available from. [PMC free article: PMC1551933] [PubMed: 16932809] - 139.
- Wu J, Xu L, Kim DY, Rho JM, St John PA, Lue LF, et al. Electrophysiological properties of human hypothalamic hamartomas. Ann Neurol [Internet]. 2005/09/01. 2005;58(3):371–82. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16130091. [PubMed: 16130091] - 140.
- Munari C, Kahane P, Francione S, Hoffmann D, Tassi L, Cusmai R, et al. Role of the hypothalamic hamartoma in the genesis of gelastic fits (a video-stereo-EEG study). Electroencephalogr Clin Neurophysiol [Internet]. 1995/09/01. 1995;95(3):154–60. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7555906. [PubMed: 7555906] - 141.
- Wethe J v, Prigatano GP, Gray J, Chapple K, Rekate HL, Kerrigan JF. Cognitive functioning before and after surgical resection for hypothalamic hamartoma and epilepsy. Neurology [Internet]. 2013/08/16. 2013;81(12):1044–50. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23946307. [PubMed: 23946307] - 142.
- Mittal S, Mittal M, Montes JL, Farmer JP, Andermann F. Hypothalamic hamartomas. Part 2. Surgical considerations and outcome. Neurosurg Focus [Internet]. 2013/06/04. 2013;34(6):E7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23724841. [PubMed: 23724841] - 143.
- Kerrigan JF, Ng YT, Chung S, Rekate HL. The hypothalamic hamartoma: a model of subcortical epileptogenesis and encephalopathy. Semin Pediatr Neurol [Internet]. 2005/08/24. 2005;12(2):119–31. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16114178. [PubMed: 16114178] - 144.
- Li CD, Luo SQ, Gong J, Ma ZY, Jia G, Zhang YQ, et al. Surgical treatment of hypothalamic hamartoma causing central precocious puberty: long-term follow-up. J Neurosurg Pediatr [Internet]. 2013/06/12. 2013;12(2):151–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23746126. [PubMed: 23746126] - 145.
- Freeman JL, Zacharin M, Rosenfeld J v, Harvey AS. The endocrinology of hypothalamic hamartoma surgery for intractable epilepsy. Epileptic Disord [Internet]. 2004/02/21. 2003;5(4):239–47. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/14975793. [PubMed: 14975793] - 146.
- Abla AA, Wait SD, Forbes JA, Pati S, Johnsonbaugh RE, Kerrigan JF, et al. Syndrome of alternating hypernatremia and hyponatremia after hypothalamic hamartoma surgery. Neurosurg Focus [Internet]. 2011/02/03. 2011;30(2):E6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21284452. [PubMed: 21284452] - 147.
- Drees C, Chapman K, Prenger E, Baxter L, Maganti R, Rekate H, et al. Seizure outcome and complications following hypothalamic hamartoma treatment in adults: endoscopic, open, and Gamma Knife procedures. J Neurosurg [Internet]. 2012/06/12. 2012;117(2):255–61. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22680243. [PubMed: 22680243] - 148.
- Burrows AM, Marsh WR, Worrell G, Woodrum DA, Pollock BE, Gorny KR, et al. Magnetic resonance imaging-guided laser interstitial thermal therapy for previously treated hypothalamic hamartomas. Neurosurg Focus. 2016;41(4):E8. [PubMed: 27690651]
- 149.
- Du VX. Gandhi S v, Rekate HL, Mehta AD. Laser interstitial thermal therapy: a first line treatment for seizures due to hypothalamic hamartoma? Epilepsia. 2017;58 Suppl 2:77–84. [PubMed: 28591480]
- 150.
- Henter JI, Tondini C, Pritchard J. Histiocyte disorders. Crit Rev Oncol Hematol [Internet]. 2004/05/26. 2004;50(2):157–74. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15157664. [PubMed: 15157664] - 151.
- Alston RD, Tatevossian RG, McNally RJ, Kelsey A, Birch JM, Eden TO. Incidence and survival of childhood Langerhans cell histiocytosis in Northwest England from 1954 to 1998. Pediatr Blood Cancer [Internet]. 2006/05/03. 2007;48(5):555–60. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16652350. [PubMed: 16652350] - 152.
- Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer [Internet]. 2008/02/09. 2008;51(1):71–5. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/18260117. [PubMed: 18260117] - 153.
- Salotti JA, Nanduri V, Pearce MS, Parker L, Lynn R, Windebank KP. Incidence and clinical features of Langerhans cell histiocytosis in the UK and Ireland. Arch Dis Child [Internet]. 2008/12/09. 2009;94(5):376–80. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/19060008. [PubMed: 19060008] - 154.
- Stalemark H, Laurencikas E, Karis J, Gavhed D, Fadeel B, Henter JI. Incidence of Langerhans cell histiocytosis in children: a population-based study. Pediatr Blood Cancer [Internet]. 2008/02/13. 2008;51(1):76–81. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/18266220. [PubMed: 18266220] - 155.
- Abla O, Egeler RM, Weitzman S. Langerhans cell histiocytosis: Current concepts and treatments. Cancer Treat Rev [Internet]. 2010/03/02. 2010;36(4):354–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20188480. [PubMed: 20188480] - 156.
- Kim BE, Koh KN, Suh JK, Im HJ, Song JS, Lee JW, et al. Clinical Features and Treatment Outcomes of Langerhans Cell Histiocytosis: A Nationwide Survey From Korea Histiocytosis Working Party. J Pediatr Hematol Oncol. 2013 Nov 28;2013 http://www
.ncbi.nlm.nih .gov/pubmed/24276037 [Internet] Available from. [PubMed: 24276037] - 157.
- Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–23. https://www
.ncbi.nlm .nih.gov/pubmed/20519626 [Internet] Available from. [PMC free article: PMC3173987] [PubMed: 20519626] - 158.
- Donadieu J, Rolon MA, Thomas C, Brugieres L, Plantaz D, Emile JF, et al. Endocrine involvement in pediatric-onset Langerhans’ cell histiocytosis: a population-based study. J Pediatr [Internet]. 2004/03/06. 2004;144(3):344–50. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15001940. [PubMed: 15001940] - 159.
- Grois N, Potschger U, Prosch H, Minkov M, Arico M, Braier J, et al. Risk factors for diabetes insipidus in langerhans cell histiocytosis. Pediatr Blood Cancer [Internet]. 2005/07/28. 2006;46(2):228–33. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16047354. [PubMed: 16047354] - 160.
- Varan A, Atas E, Aydin B, Yalcin B, Akyuz C, Kutluk T, et al. Evaluation of patients with intracranial tumors and central diabetes insipidus. Pediatr Hematol Oncol [Internet]. 2013/08/31. 2013;30(7):668–73. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23988090. [PubMed: 23988090] - 161.
- Haupt R, Nanduri V, Calevo MG, Bernstrand C, Braier JL, Broadbent V, et al. Permanent consequences in Langerhans cell histiocytosis patients: a pilot study from the Histiocyte Society-Late Effects Study Group. Pediatr Blood Cancer [Internet]. 2004/03/30. 2004;42(5):438–44. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15049016. [PubMed: 15049016] - 162.
- Arico M. Langerhans cell histiocytosis in children: from the bench to bedside for an updated therapy. Br J Haematol [Internet]. 2016; Available from: http://www
.ncbi.nlm.nih .gov/pubmed/26913480. [PubMed: 26913480] - 163.
- Braier J, Rosso D, Pollono D, Rey G, Lagomarsino E, Latella A, et al. Symptomatic bone langerhans cell histiocytosis treated at diagnosis or after reactivation with indomethacin alone. J Pediatr Hematol Oncol. 2014;36(5):e280–4. http://www
.ncbi.nlm.nih .gov/pubmed/24977402 [Internet] Available from. [PubMed: 24977402] - 164.
- Bernard F, Thomas C, Bertrand Y, Munzer M, Landman Parker J, Ouache M, et al. Multi-centre pilot study of 2-chlorodeoxyadenosine and cytosine arabinoside combined chemotherapy in refractory Langerhans cell histiocytosis with haematological dysfunction. Eur J Cancer. 2005;41(17):2682–9. http://www
.ncbi.nlm.nih .gov/pubmed/16291085 [Internet] Available from. [PubMed: 16291085] - 165.
- Heritier S, Jehanne M, Leverger G, Emile JF, Alvarez JC, Haroche J, et al. Vemurafenib Use in an Infant for High-Risk Langerhans Cell Histiocytosis. JAMA Oncol. 2015;1(6):836–8. http://www
.ncbi.nlm.nih .gov/pubmed/26180941 [Internet] Available from. [PubMed: 26180941] - 166.
- Simko SJ, McClain KL, Allen CE. Up-front therapy for LCH: is it time to test an alternative to vinblastine/prednisone? Br J Haematol. 2015;169(2):299–301. http://www
.ncbi.nlm.nih .gov/pubmed/25400231 [Internet] Available from. [PMC free article: PMC4791956] [PubMed: 25400231] - 167.
- Simko SJ, Tran HD, Jones J, Bilgi M, Beaupin LK, Coulter D, et al. Clofarabine salvage therapy in refractory multifocal histiocytic disorders, including Langerhans cell histiocytosis, juvenile xanthogranuloma and Rosai-Dorfman disease. Pediatr Blood Cancer. 2014;61(3):479–87. http://www
.ncbi.nlm.nih .gov/pubmed/24106153 [Internet] Available from. [PMC free article: PMC4474604] [PubMed: 24106153] - 168.
- Veys PA, Nanduri V, Baker KS, He W, Bandini G, Biondi A, et al. Haematopoietic stem cell transplantation for refractory Langerhans cell histiocytosis: outcome by intensity of conditioning. Br J Haematol. 2015;169(5):711–8. http://www
.ncbi.nlm.nih .gov/pubmed/25817915 [Internet] Available from. [PMC free article: PMC4433436] [PubMed: 25817915] - 169.
- Grois N, Fahrner B, Arceci RJ, Henter JI, McClain K, Lassmann H, et al. Central nervous system disease in Langerhans cell histiocytosis. J Pediatr [Internet]. 2010/05/04. 2010;156(6):873–81, 881 e1. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20434166. [PubMed: 20434166] - 170.
- Abla O, Weitzman S, Minkov M, McClain KL, Visser J, Filipovich A, et al. Diabetes insipidus in Langerhans cell histiocytosis: When is treatment indicated? Pediatr Blood Cancer [Internet]. 2009/01/15. 2009;52(5):555–6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/19142995. [PubMed: 19142995] - 171.
- Gadner H, Minkov M, Grois N, Potschger U, Thiem E, Arico M, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. Blood. 2013;121(25):5006–14. http://www
.ncbi.nlm.nih .gov/pubmed/23589673 [Internet] Available from. [PubMed: 23589673] - 172.
- Satogami N, Miki Y, Koyama T, Kataoka M, Togashi K. Normal pituitary stalk: high-resolution MR imaging at 3T. AJNR Am J Neuroradiol [Internet]. 2009/10/03. 2010;31(2):355–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/19797792. [PMC free article: PMC7964128] [PubMed: 19797792] - 173.
- Simmons GE, Suchnicki JE, Rak KM, Damiano TR. MR imaging of the pituitary stalk: size, shape, and enhancement pattern. AJR Am J Roentgenol [Internet]. 1992/08/01. 1992;159(2):375–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/1632360. [PubMed: 1632360] - 174.
- Raybaud C, Barkovich AJ. Intracranial, orbital and neck masses of childhood. In: Barkovich AJ, Raybaud C, editors. Pediatric Neuroimaging. Philadelphia: Wolters Kluwer Health/ Lippincott Wiliams & Wilkins; 2012. p. 714–5.
- 175.
- Varan A, Cila A, Akyuz C, Kale G, Kutluk T, Buyukpamukcu M. Radiological evaluation of patients with pituitary langerhans cell histiocytosis at diagnosis and at follow-up. Pediatr Hematol Oncol [Internet]. 2008/08/30. 2008;25(6):567–74. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/18728976. [PubMed: 18728976] - 176.
- Hamilton BE, Salzman KL, Osborn AG. Anatomic and pathologic spectrum of pituitary infundibulum lesions. AJR Am J Roentgenol [Internet]. 2007/02/22. 2007;188(3):W223-32. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17312027. [PubMed: 17312027] - 177.
- Jinguji S, Nishiyama K, Yoshimura J, Yoneoka Y, Harada A, Sano M, et al. Endoscopic biopsies of lesions associated with a thickened pituitary stalk. Acta Neurochir (Wien) [Internet]. 2012/10/31. 2013;155(1):119–24; discussion 124. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23108562. [PubMed: 23108562] - 178.
- Robison NJ, Prabhu SP, Sun P, Chi SN, Kieran MW, Manley PE, et al. Predictors of neoplastic disease in children with isolated pituitary stalk thickening. Pediatr Blood Cancer [Internet]. 2013/05/15. 2013;60(10):1630–5. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23670879. [PubMed: 23670879] - 179.
- Beni-Adani L, Sainte-Rose C, Zerah M, Brunelle F, Constantini S, Renier D, et al. Surgical implications of the thickened pituitary stalk accompanied by central diabetes insipidus. J Neurosurg [Internet]. 2005/12/24. 2005;103(2 Suppl):142–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16370280. [PubMed: 16370280] - 180.
- Biller BM, Colao A, Petersenn S, Bonert VS, Boscaro M. Prolactinomas, Cushing’s disease and acromegaly: debating the role of medical therapy for secretory pituitary adenomas. BMC Endocr Disord [Internet]. 2010/05/19. 2010;10:10. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20478050. [PMC free article: PMC2887860] [PubMed: 20478050] - 181.
- Mootha SL, Barkovich AJ, Grumbach MM, Edwards MS, Gitelman SE, Kaplan SL, et al. Idiopathic hypothalamic diabetes insipidus, pituitary stalk thickening, and the occult intracranial germinoma in children and adolescents. J Clin Endocrinol Metab [Internet]. 1997/05/01. 1997;82(5):1362–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/9141516. [PubMed: 9141516] - 182.
- Mikami-Terao Y, Akiyama M, Yanagisawa T, Takahashi-Fujigasaki J, Yokoi K, Fukuoka K, et al. Lymphocytic hypophysitis with central diabetes insipidus and subsequent hypopituitarism masking a suprasellar germinoma in a 13-year-old girl. Childs Nerv Syst. 2006;22(10):1338–43. http://www
.ncbi.nlm.nih .gov/pubmed/16565852 [Internet] Available from. [PubMed: 16565852] - 183.
- Nishiuchi T, Imachi H, Murao K, Fujiwara M, Sato M, Nishiuchi Y, et al. Suprasellar germinoma masquerading as lymphocytic hypophysitis associated with central diabetes insipidus, delayed sexual development, and subsequent hypopituitarism. Am J Med Sci [Internet]. 2010/01/07. 2010;339(2):195–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20051818. [PubMed: 20051818] - 184.
- Turcu AF, Erickson BJ, Lin E, Guadalix S, Schwartz K, Scheithauer BW, et al. Pituitary stalk lesions: the Mayo Clinic experience. J Clin Endocrinol Metab [Internet]. 2013/03/28. 2013;98(5):1812–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23533231. [PubMed: 23533231] - 185.
- di Iorgi N, Napoli F, Allegri AE, Olivieri I, Bertelli E, Gallizia A, et al. Diabetes insipidus--diagnosis and management. Horm Res Paediatr [Internet]. 2012/03/22. 2012;77(2):69–84. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22433947. [PubMed: 22433947] - 186.
- Cerbone M, Visser J, Bulwer C, Ederies A, Vallabhaneni K, Ball S, et al. Management of children and young people with idiopathic pituitary stalk thickening, central diabetes insipidus, or both: a national clinical practice consensus guideline. Lancet Child Adolesc Health. 2021;5(9):662–76. [PubMed: 34214482]
- 187.
- Aquilina K, Boop FA. Nonneoplastic enlargement of the pituitary gland in children. J Neurosurg Pediatr [Internet]. 2011/05/03. 2011;7(5):510–5. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21529191. [PubMed: 21529191] - 188.
- Elster AD, Chen MY, Williams 3rd DW, Key LL. Pituitary gland: MR imaging of physiologic hypertrophy in adolescence. Radiology [Internet]. 1990/03/01. 1990;174(3 Pt 1):681–5. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/2305049. [PubMed: 2305049] - 189.
- Alatzoglou KS, Kelberman D, Cowell CT, Palmer R, Arnhold IJ, Melo ME, et al. Increased transactivation associated with SOX3 polyalanine tract deletion in a patient with hypopituitarism. J Clin Endocrinol Metab. 2011;96(4):E685–90. http://www
.ncbi.nlm.nih .gov/pubmed/21289259 [Internet] Available from. [PubMed: 21289259] - 190.
- Gan HW, Alatzoglou KS, Dattani MT. Disorders of Hypothalamo-pituitary Axis. In: Dattani MT, Brook CGD, editors. Brook’s Clinical Pediatric Endocrinology. 7th ed. Oxford, UK: John Wiley & Sons Ltd; 2020. p. 133–98.
- 191.
- Teramoto A, Hirakawa K, Sanno N, Osamura Y. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology [Internet]. 1994/10/01. 1994;193(1):161–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8090885. [PubMed: 8090885] - 192.
- Han SJ, Rolston JD, Jahangiri A, Aghi MK. Rathke’s cleft cysts: review of natural history and surgical outcomes. J Neurooncol. 2013 Oct 23;2013 http://www
.ncbi.nlm.nih .gov/pubmed/24146189 [Internet] Available from. [PubMed: 24146189] - 193.
- Dubuisson AS, Stevenaert A, Martin DH, Flandroy PP. Intrasellar arachnoid cysts. Neurosurgery. 2007;61(3):505–13. http://www
.ncbi.nlm.nih .gov/pubmed/17881962 [Internet] Available from. [PubMed: 17881962] - 194.
- Ali ZS, Lang SS, Bakar D, Storm PB, Stein SC. Pediatric intracranial arachnoid cysts: comparative effectiveness of surgical treatment options. Childs Nerv Syst. 2014;30(3):461–9. http://www
.ncbi.nlm.nih .gov/pubmed/24162618 [Internet] Available from. [PubMed: 24162618] - 195.
- Ozek MM, Urgun K. Neuroendoscopic management of suprasellar arachnoid cysts. World Neurosurg [Internet]. 2013;79(2 Suppl):S19 e13-8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22381821. [PubMed: 22381821] - 196.
- El-Ghandour NM. Endoscopic treatment of suprasellar arachnoid cysts in children. J Neurosurg Pediatr. 2011;8(1):6–14. http://www
.ncbi.nlm.nih .gov/pubmed/21721882 [Internet] Available from. [PubMed: 21721882] - 197.
- Adan L, Bussieres L, Dinand V, Zerah M, Pierre-Kahn A, Brauner R. Growth, puberty and hypothalamic-pituitary function in children with suprasellar arachnoid cyst. Eur J Pediatr. 2000;159(5):348–55. http://www
.ncbi.nlm.nih .gov/pubmed/10834520 [Internet] Available from. [PubMed: 10834520] - 198.
- McCrea HJ, George E, Settler A, Schwartz TH, Greenfield JP. Pediatric Suprasellar Tumors. J Child Neurol [Internet]. 2015; Available from: http://www
.ncbi.nlm.nih .gov/pubmed/26676303. [PubMed: 26676303] - 199.
- Ogiwara H, Morota N, Joko M, Hirota K. Endoscopic fenestrations for suprasellar arachnoid cysts. J Neurosurg Pediatr. 2011;8(5):484–8. http://www
.ncbi.nlm.nih .gov/pubmed/22044374 [Internet] Available from. [PubMed: 22044374] - 200.
- Howlett TA, Levy MJ, Robertson IJ. How reliably can autoimmune hypophysitis be diagnosed without pituitary biopsy. Clin Endocrinol (Oxf) [Internet]. 2009/12/31. 2010;73(1):18–21. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20039888. [PubMed: 20039888] - 201.
- Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. AJR Am J Roentgenol [Internet]. 2004/01/23. 2004;182(2):289–95. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/14736648. [PubMed: 14736648] - 202.
- Wilne S, Collier J, Kennedy C, Jenkins A, Grout J, Mackie S, et al. Progression from first symptom to diagnosis in childhood brain tumours. Eur J Pediatr [Internet]. 2011/05/20. 2012;171(1):87–93. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21594769. [PubMed: 21594769] - 203.
- Wilne S, Collier J, Kennedy C, Koller K, Grundy R, Walker D. Presentation of childhood CNS tumours: a systematic review and meta-analysis. Lancet Oncol [Internet]. 2007/07/24. 2007;8(8):685–95. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17644483. [PubMed: 17644483] - 204.
- Royal College of Ophthalmologists . Guidelines for the management of strabismus in childhood. London: Royal College of Ophthalmologists; 2012.
- 205.
- Hawley DP, Walker DA. A symptomatic journey to the centre of the brain. Arch Dis Child Educ Pract Ed [Internet]. 2010/03/31. 2010;95(2):59–64. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20351153. [PubMed: 20351153] - 206.
- Aquilina K, Daniels DJ, Spoudeas H, Phipps K, Gan HW, Boop FA. Optic pathway glioma in children: does visual deficit correlate with radiology in focal exophytic lesions? Childs Nerv Syst. 2015;31(11):2041–9. http://www
.ncbi.nlm.nih .gov/pubmed/26277358 [Internet] Available from. [PubMed: 26277358] - 207.
- Chateil JF, Soussotte C, Pedespan JM, Brun M, le Manh C, Diard F. MRI and clinical differences between optic pathway tumours in children with and without neurofibromatosis. Br J Radiol [Internet]. 2001/03/03. 2001;74(877):24–31. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/11227773. [PubMed: 11227773] - 208.
- Grill J, Laithier V, Rodriguez D, Raquin MA, Pierre-Kahn A, Kalifa C. When do children with optic pathway tumours need treatment? An oncological perspective in 106 patients treated in a single centre. Eur J Pediatr [Internet]. 2000/10/03. 2000;159(9):692–6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/11014471. [PubMed: 11014471] - 209.
- Taylor M, Couto-Silva AC, Adan L, Trivin C, Sainte-Rose C, Zerah M, et al. Hypothalamic-pituitary lesions in pediatric patients: endocrine symptoms often precede neuro-ophthalmic presenting symptoms. J Pediatr [Internet]. 2012/06/26. 2012;161(5):855–63. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22727865. [PubMed: 22727865] - 210.
- Rodriguez LA, Edwards MS, Levin VA. Management of hypothalamic gliomas in children: an analysis of 33 cases. Neurosurgery [Internet]. 1990/02/01. 1990;26(2):242–6; discussion 246-7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/2308672. [PubMed: 2308672] - 211.
- Virdis R, Sigorini M, Laiolo A, Lorenzetti E, Street ME, Villani AR, et al. Neurofibromatosis type 1 and precocious puberty. J Pediatr Endocrinol Metab. 2000 Sep 02;2000(13) Suppl 1:841–4. http://www
.ncbi.nlm.nih .gov/pubmed/10969931 [Internet] Available from. [PubMed: 10969931] - 212.
- Ahn Y, Cho BK, Kim SK, Chung YN, Lee CS, Kim IH, et al. Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst [Internet]. 2006/04/22. 2006;22(9):1136–42. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16628460. [PubMed: 16628460] - 213.
- Cappelli C, Grill J, Raquin M, Pierre-Kahn A, Lellouch-Tubiana A, Terrier-Lacombe MJ, et al. Long-term follow up of 69 patients treated for optic pathway tumours before the chemotherapy era. Arch Dis Child [Internet]. 1999/01/06. 1998;79(4):334–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/9875044. [PMC free article: PMC1717725] [PubMed: 9875044] - 214.
- Muller HL, Kaatsch P, Warmuth-Metz M, Flentje M, Sorensen N. Kraniopharyngeom im Kindes-und Jugendalter: Diagnostische und therapeutische Strategien (Childhood craniopharyngioma - diagnostic and therapeutic strategies). Monatsschrift Kindheilkunde. 2003;151:1056–63.
- 215.
- Cisternino M, Arrigo T, Pasquino AM, Tinelli C, Antoniazzi F, Beduschi L, et al. Etiology and age incidence of precocious puberty in girls: a multicentric study. J Pediatr Endocrinol Metab. 2000 Sep 02;2000(13) Suppl 1:695–701. http://www
.ncbi.nlm.nih .gov/pubmed/10969911 [Internet] Available from. [PubMed: 10969911] - 216.
- Faizah M, Zuhanis A, Rahmah R, Raja A, Wu L, Dayang A, et al. Precocious puberty in children: A review of imaging findings. Biomed Imaging Interv J [Internet]. 2012/09/13. 2012;8(1):e6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22970062. [PMC free article: PMC3432225] [PubMed: 22970062] - 217.
- Mogensen SS, Aksglaede L, Mouritsen A, Sorensen K, Main KM, Gideon P, et al. Diagnostic work-up of 449 consecutive girls who were referred to be evaluated for precocious puberty. J Clin Endocrinol Metab [Internet]. 2011/02/25. 2011;96(5):1393–401. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21346077. [PubMed: 21346077] - 218.
- Russell A. A diencephalic syndrome of emaciation in infancy and childhood. Arch Dis Child. 1951;26(127):270–5.
- 219.
- Waga S, Shimizu T, Sakakura M. Diencephalic syndrome of emaciation (Russell’s syndrome). Surg Neurol [Internet]. 1982/02/01. 1982;17(2):141–6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/6803375. [PubMed: 6803375] - 220.
- Burr IM, Slonim AE, Danish RK, Gadoth N, Butler IJ. Diencephalic syndrome revisited. J Pediatr [Internet]. 1976/03/01. 1976;88(3):439–44. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/1245953. [PubMed: 1245953] - 221.
- Mohan SM, Dharmalingam M, Prasanna Kumar KM, Verma RG, Balaji Pai S, Krishna KN, et al. Suprasellar germ cell tumor presenting as diencephalic syndrome and precocious puberty. J Pediatr Endocrinol Metab [Internet]. 2003/04/23. 2003;16(3):443–6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12705371. [PubMed: 12705371] - 222.
- Chipkevitch E, Fernandes AC. Hypothalamic tumor associated with atypical forms of anorexia nervosa and diencephalic syndrome. Arq Neuropsiquiatr [Internet]. 1993/06/01. 1993;51(2):270–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8274094. [PubMed: 8274094] - 223.
- Addy DP, Hudson FP. Diencephalic syndrome of infantile emaciation. Analysis of literature and report of further 3 cases. Arch Dis Child [Internet]. 1972/06/01. 1972;47(253):338–43. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/5034666. [PMC free article: PMC1648126] [PubMed: 5034666] - 224.
- Sharma RR, Chandy MJ, Lad SD. Diencephalic syndrome of emaciation in an adult associated with a suprasellar craniopharyngioma--a case report. Br J Neurosurg [Internet]. 1990/01/01. 1990;4(1):77–80. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/2334532. [PubMed: 2334532] - 225.
- Eliash A, Roitman A, Karp M, Reichental E, Manor RS, Shalit M, et al. Diencephalic syndrome due to a suprasellar epidermoid cyst. Case report. Childs Brain [Internet]. 1983/01/01. 1983;10(6):414–8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/6661939. [PubMed: 6661939] - 226.
- Maroon JC, Albright L. “Failure to thrive” due to pontine glioma. Arch Neurol [Internet]. 1977/05/01. 1977;34(5):295–7. Available from: http://www
.ncbi.nlm.nih.gov/pubmed/67836. [PubMed: 67836] - 227.
- Ramos EJ, Suzuki S, Marks D, Inui A, Asakawa A, Meguid MM. Cancer anorexia-cachexia syndrome: cytokines and neuropeptides. Curr Opin Clin Nutr Metab Care [Internet]. 2004/06/12. 2004;7(4):427–34. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15192446. [PubMed: 15192446] - 228.
- DeSousa AL, Kalsbeck JE, Mealey Jr. J, Fitzgerald J. Diencephalic syndrome and its relation to opticochiasmatic glioma: review of twelve cases. Neurosurgery [Internet]. 1979/03/01. 1979;4(3):207–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/460550. [PubMed: 460550] - 229.
- Miyoshi Y, Yunoki M, Yano A, Nishimoto K. Diencephalic syndrome of emaciation in an adult associated with a third ventricle intrinsic craniopharyngioma: case report. Neurosurgery [Internet]. 2002/12/21. 2003;52(1):224–7; discussion 227. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12493122. [PubMed: 12493122] - 230.
- Hager A, Thorell JI. Studies on growth hormone secretion in a patient with the diencephalic syndrome of emaciation. Acta Paediatr Scand. 1973;62(3):231–40. https://www
.ncbi.nlm .nih.gov/pubmed/4703018 [Internet] Available from. [PubMed: 4703018] - 231.
- Pimstone BL, Sobel J, Meyer E, Eale D. Secretion of growth hormone in the diencephalic syndrome of childhood. J Pediatr [Internet]. 1970/06/01. 1970;76(6):886–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/5444580. [PubMed: 5444580] - 232.
- Kilday JP, Bartels U, Huang A, Barron M, Shago M, Mistry M, et al. Favorable survival and metabolic outcome for children with diencephalic syndrome using a radiation-sparing approach. J Neurooncol [Internet]. 2013/11/13. 2014;116(1):195–204. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/24218181. [PubMed: 24218181] - 233.
- Vlachopapadopoulou E, Tracey KJ, Capella M, Gilker C, Matthews DE. Increased energy expenditure in a patient with diencephalic syndrome. J Pediatr [Internet]. 1993/06/01. 1993;122(6):922–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8501572. [PubMed: 8501572] - 234.
- Chipkevitch E. Brain tumors and anorexia nervosa syndrome. Brain Dev [Internet]. 1994/05/01. 1994;16(3):175–9, discussion 180-2. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7943600. [PubMed: 7943600] - 235.
- de Vile CJ, Sufraz R, Lask BD, Stanhope R. Occult intracranial tumours masquerading as early onset anorexia nervosa. BMJ [Internet]. 1995/11/18. 1995;311(7016):1359–60. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7496292. [PMC free article: PMC2551253] [PubMed: 7496292] - 236.
- Houy E, Debono B, Dechelotte P, Thibaut F. Anorexia nervosa associated with right frontal brain lesion. Int J Eat Disord [Internet]. 2007/08/09. 2007;40(8):758–61. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/17683096. [PubMed: 17683096] - 237.
- American Psychiatric Association . Diagnostic and Statistic Manual of Mental Disorders (DSM-5). 5th ed. Arlington, VA, USA: American Psychiatric Publishing; 2013.
- 238.
- World Health Organisation . The ICD-10 Classification of Mental and Behavioural Disorders: Clinical descriptions and diagnostic guidelines. 10th ed. Geneva, Switzerland: World Health Organisation; 1992.
- 239.
- Diamanti A, Ubertini GM, Basso MS, Caramadre AM, Alterio A, Panetta F, et al. Amenorrhea and weight loss: not only anorexia nervosa. Eur J Obstet Gynecol Reprod Biol [Internet]. 2011/12/27. 2012;161(1):111–2. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22197307. [PubMed: 22197307] - 240.
- de Vile CJ, Grant DB, Hayward RD, Kendall BE, Neville BG, Stanhope R. Obesity in childhood craniopharyngioma: relation to post-operative hypothalamic damage shown by magnetic resonance imaging. J Clin Endocrinol Metab [Internet]. 1996/07/01. 1996;81(7):2734–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/8675604. [PubMed: 8675604] - 241.
- Webb EA, Dattani MT. Septo-optic dysplasia. Eur J Hum Genet. 2010;18(4):393–7. http://www
.ncbi.nlm.nih .gov/pubmed/19623216 [Internet] Available from. [PMC free article: PMC2987262] [PubMed: 19623216] - 242.
- Littley MD, Shalet SM, Beardwell CG, Robinson EL, Sutton ML. Radiation-induced hypopituitarism is dose-dependent. Clin Endocrinol (Oxf) [Internet]. 1989/09/01. 1989;31(3):363–73. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/2559824. [PubMed: 2559824] - 243.
- Adan L, Trivin C, Sainte-Rose C, Zucker JM, Hartmann O, Brauner R. GH deficiency caused by cranial irradiation during childhood: factors and markers in young adults. J Clin Endocrinol Metab [Internet]. 2001/11/10. 2001;86(11):5245–51. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/11701685. [PubMed: 11701685] - 244.
- Talbot L, Spoudeas H. Late effects in relation to childhood cancer. In: Estlin EJ, Gilbertson RJ, Wynn RF, editors. Pediatric Hematology and Oncology: Scientific Principles & Clinical Practice. Oxford: Wiley-Blackwell; 2010. p. 367–91.
- 245.
- Collet-Solberg PF, Sernyak H, Satin-Smith M, Katz LL, Sutton L, Molloy P, et al. Endocrine outcome in long-term survivors of low-grade hypothalamic/chiasmatic glioma. Clin Endocrinol (Oxf) [Internet]. 1997/07/01. 1997;47(1):79–85. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/9302376. [PubMed: 9302376] - 246.
- Grabenbauer GG, Schuchardt U, Buchfelder M, Rodel CM, Gusek G, Marx M, et al. Radiation therapy of optico-hypothalamic gliomas (OHG)--radiographic response, vision and late toxicity. Radiother Oncol [Internet]. 2000/03/30. 2000;54(3):239–45. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/10738082. [PubMed: 10738082] - 247.
- Nanduri VR, Bareille P, Pritchard J, Stanhope R. Growth and endocrine disorders in multisystem Langerhans’ cell histiocytosis. Clin Endocrinol (Oxf). 2000;53(4):509–15. http://www
.ncbi.nlm.nih .gov/pubmed/11012577 [Internet] Available from. [PubMed: 11012577] - 248.
- Huguenin M, Trivin C, Zerah M, Doz F, Brugieres L, Brauner R. Adult height after cranial irradiation for optic pathway tumors: relationship with neurofibromatosis. J Pediatr [Internet]. 2003/07/03. 2003;142(6):699–703. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12838200. [PubMed: 12838200] - 249.
- Sklar CA, Antal Z, Chemaitilly W, Cohen LE, Follin C, Meacham LR, et al. Hypothalamic-Pituitary and Growth Disorders in Survivors of Childhood Cancer: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103(8):2761–84. https://www
.ncbi.nlm .nih.gov/pubmed/29982476 [Internet] Available from. [PubMed: 29982476] - 250.
- Hindmarsh PC, Swift PG. An assessment of growth hormone provocation tests. Arch Dis Child [Internet]. 1995/04/01. 1995;72(4):362–7; discussion 367-8. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/7539245. [PMC free article: PMC1511229] [PubMed: 7539245] - 251.
- Shah A, Stanhope R, Matthew D. Hazards of pharmacological tests of growth hormone secretion in childhood. BMJ [Internet]. 1992/01/18. 1992;304(6820):173–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/1737165. [PMC free article: PMC1881200] [PubMed: 1737165] - 252.
- Sfeir JG, Kittah NEN, Tamhane SU, Jasim S, Chemaitilly W, Cohen LE, et al. Diagnosis of GH deficiency as a late effect of radiotherapy in survivors of childhood cancers. J Clin Endocrinol Metab. 2018;103(8):2785–93. [PubMed: 29982753]
- 253.
- Cattoni A, Clarke E, Albanese A. The predictive value of insulin-like growth factor 1 in irradiation-dependent growth hormone deficiency in childhood cancer survivors. Horm Res Paediatr. 2018;90(5):314–25. [PubMed: 30645996]
- 254.
- Sklar C, Sarafoglou K, Whittam E. Efficacy of insulin-like growth factor binding protein 3 in predicting the growth hormone response to provocative testing in children treated with cranial irradiation. Acta Endocrinol (Copenh). 1993;129(6):511–5. [PubMed: 7509100]
- 255.
- Murray PG, Dattani MT, Clayton PE. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Arch Dis Child. 2016;101(1):96–100. https://www
.ncbi.nlm .nih.gov/pubmed/26153506 [Internet] Available from. [PubMed: 26153506] - 256.
- Growth Hormone Research S. Consensus guidelines for the diagnosis and treatment of growth hormone (GH) deficiency in childhood and adolescence: summary statement of the GH Research Society. GH Research Society. J Clin Endocrinol Metab. 2000;85(11):3990–3. https://www
.ncbi.nlm .nih.gov/pubmed/11095419 [Internet] Available from. [PubMed: 11095419] - 257.
- Phillip M, Moran O, Lazar L. Growth without growth hormone. J Pediatr Endocrinol Metab. 2003 Jan 04;2002(15) Suppl 5:1267–72. http://www
.ncbi.nlm.nih .gov/pubmed/12510977 [Internet] Available from. [PubMed: 12510977] - 258.
- Moshang T Jr, Rundle AC, Graves DA, Nickas J, Johanson A, Meadows A. Brain tumor recurrence in children treated with growth hormone: the National Cooperative Growth Study experience. J Pediatr. 1996;128(5 Pt 2):S4–7. http://www
.ncbi.nlm.nih .gov/pubmed/8627468 [Internet] Available from. [PubMed: 8627468] - 259.
- Muller HL, Gebhardt U, Schroder S, Pohl F, Kortmann RD, Faldum A, et al. Analyses of treatment variables for patients with childhood craniopharyngioma--results of the multicenter prospective trial KRANIOPHARYNGEOM 2000 after three years of follow-up. Horm Res Paediatr [Internet]. 2010/03/04. 2010;73(3):175–80. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20197669. [PubMed: 20197669] - 260.
- Karavitaki N, Warner JT, Marland A, Shine B, Ryan F, Arnold J, et al. GH replacement does not increase the risk of recurrence in patients with craniopharyngioma. Clin Endocrinol (Oxf). 2006;64(5):556–60. https://www
.ncbi.nlm .nih.gov/pubmed/16649976 [Internet] Available from. [PubMed: 16649976] - 261.
- Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, et al. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin-Like Growth Factor-I Deficiency. Horm Res Paediatr. 2016;86(6):361–97. https://www
.ncbi.nlm .nih.gov/pubmed/27884013 [Internet] Available from. [PubMed: 27884013] - 262.
- Lerner SE, Huang GJ, McMahon D, Sklar CA, Oberfield SE. Growth hormone therapy in children after cranial/craniospinal radiation therapy: sexually dimorphic outcomes. J Clin Endocrinol Metab [Internet]. 2004/12/08. 2004;89(12):6100–4. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15579765. [PubMed: 15579765] - 263.
- Carel JC. Management of short stature with GnRH agonist and co-treatment with growth hormone: a controversial issue. Mol Cell Endocrinol [Internet]. 2006/06/22. 2006;254–255:226–33. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16787697. [PubMed: 16787697] - 264.
- Brougham MF, Wallace WH. Subfertility in children and young people treated for solid and haematological malignancies. Br J Haematol [Internet]. 2005/10/04. 2005;131(2):143–55. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16197443. [PubMed: 16197443] - 265.
- Gan HW, Spoudeas HA. Preserving reproductive capacity in young boys with cancer. Trends Urol Men’s Health. 2013/5/23. 2013;4(3):8–12.
- 266.
- Wallace WH, Kelsey TW. Ovarian reserve and reproductive age may be determined from measurement of ovarian volume by transvaginal sonography. Hum Reprod [Internet]. 2004/06/19. 2004;19(7):1612–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/15205396. [PubMed: 15205396] - 267.
- Crowley S, Hindmarsh PC, Holownia P, Honour JW, Brook CG. The use of low doses of ACTH in the investigation of adrenal function in man. J Endocrinol [Internet]. 1991/09/01. 1991;130(3):475–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/1940720. [PubMed: 1940720] - 268.
- Patterson BC, Truxillo L, Wasilewski-Masker K, Mertens AC, Meacham LR. Adrenal function testing in pediatric cancer survivors. Pediatr Blood Cancer [Internet]. 2009/07/29. 2009;53(7):1302–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/19637328. [PubMed: 19637328] - 269.
- Schmiegelow M, Feldt-Rasmussen U, Rasmussen AK, Lange M, Poulsen HS, Muller J. Assessment of the hypothalamo-pituitary-adrenal axis in patients treated with radiotherapy and chemotherapy for childhood brain tumor. J Clin Endocrinol Metab [Internet]. 2003/07/05. 2003;88(7):3149–54. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12843158. [PubMed: 12843158] - 270.
- Persani L, Brabant G, Dattani M, Bonomi M, Feldt-Rasmussen U, Fliers E, et al. 2018 European Thyroid Association (ETA) Guidelines on the Diagnosis and Management of Central Hypothyroidism. Eur Thyroid J. 2018;7(5):225–37. https://www
.ncbi.nlm .nih.gov/pubmed/30374425 [Internet] Available from. [PMC free article: PMC6198777] [PubMed: 30374425] - 271.
- Mehta A, Hindmarsh PC, Stanhope RG, Brain CE, Preece MA, Dattani MT. Is the thyrotropin-releasing hormone test necessary in the diagnosis of central hypothyroidism in children. J Clin Endocrinol Metab [Internet]. 2003/12/13. 2003;88(12):5696–703. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/14671155. [PubMed: 14671155] - 272.
- Crofton PM, Tepper LA, Kelnar CJ. An evaluation of the thyrotrophin-releasing hormone stimulation test in paediatric clinical practice. Horm Res. 2008;69(1):53–9. https://www
.ncbi.nlm .nih.gov/pubmed/18059084 [Internet] Available from. [PubMed: 18059084] - 273.
- Rodondi N, den Elzen WP, Bauer DC, Cappola AR, Razvi S, Walsh JP, et al. Subclinical hypothyroidism and the risk of coronary heart disease and mortality. JAMA [Internet]. 2010/09/23. 2010;304(12):1365–74. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20858880. [PMC free article: PMC3923470] [PubMed: 20858880] - 274.
- Karavitaki N, Thanabalasingham G, Shore HC, Trifanescu R, Ansorge O, Meston N, et al. Do the limits of serum prolactin in disconnection hyperprolactinaemia need re-definition? A study of 226 patients with histologically verified non-functioning pituitary macroadenoma. Clin Endocrinol (Oxf) [Internet]. 2006/09/21. 2006;65(4):524–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/16984247. [PubMed: 16984247] - 275.
- Edate S, Albanese A. Management of electrolyte and fluid disorders after brain surgery for pituitary/suprasellar tumours. Horm Res Paediatr. 2015;83(5):293–301. https://www
.ncbi.nlm .nih.gov/pubmed/25677941 [Internet] Available from. [PubMed: 25677941] - 276.
- Liu SY, Tung YC, Lee CT, Liu HM, Peng SF, Wu MZ, et al. Clinical characteristics of central diabetes insipidus in Taiwanese children. J Formos Med Assoc. 2013;112(10):616–20. https://www
.ncbi.nlm .nih.gov/pubmed/23916565 [Internet] Available from. [PubMed: 23916565] - 277.
- Maghnie M, Villa A, Arico M, Larizza D, Pezzotta S, Beluffi G, et al. Correlation between magnetic resonance imaging of posterior pituitary and neurohypophyseal function in children with diabetes insipidus. J Clin Endocrinol Metab. 1992;74(4):795–800. https://www
.ncbi.nlm .nih.gov/pubmed/1548343 [Internet] Available from. [PubMed: 1548343] - 278.
- Ghirardello S, Hopper N, Albanese A, Maghnie M. Diabetes insipidus in craniopharyngioma: postoperative management of water and electrolyte disorders. J Pediatr Endocrinol Metab. 2006 May 17;2006(19) Suppl 1:413–21. http://www
.ncbi.nlm.nih .gov/pubmed/16700319 [Internet] Available from. [PubMed: 16700319] - 279.
- Finken MJ, Zwaveling-Soonawala N, Walenkamp MJ, Vulsma T, van Trotsenburg AS, Rotteveel J. Frequent occurrence of the triphasic response (diabetes insipidus/hyponatremia/diabetes insipidus) after surgery for craniopharyngioma in childhood. Horm Res Paediatr. 2011;76(1):22–6. http://www
.ncbi.nlm.nih .gov/pubmed/21701131 [Internet] Available from. [PubMed: 21701131] - 280.
- Pratheesh R, Swallow DM, Rajaratnam S, Jacob KS, Chacko G, Joseph M, et al. Incidence, predictors and early post-operative course of diabetes insipidus in paediatric craniopharygioma: a comparison with adults. Childs Nerv Syst [Internet]. 2013/02/07. 2013;29(6):941–9. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23386174. [PubMed: 23386174] - 281.
- Shimura N. Urinary arginine vasopressin (AVP) measurement in children: water deprivation test incorporating urinary AVP. Acta Paediatr Jpn. 1993;35(4):320–4. https://www
.ncbi.nlm .nih.gov/pubmed/8379325 [Internet] Available from. [PubMed: 8379325] - 282.
- de Fost M, Oussaada SM, Endert E, Linthorst GE, Serlie MJ, Soeters MR, et al. The water deprivation test and a potential role for the arginine vasopressin precursor copeptin to differentiate diabetes insipidus from primary polydipsia. Endocr Connect. 2015;4(2):86–91. http://www
.ncbi.nlm.nih .gov/pubmed/25712898 [Internet] Available from. [PMC free article: PMC4401105] [PubMed: 25712898] - 283.
- Fenske W, Quinkler M, Lorenz D, Zopf K, Haagen U, Papassotiriou J, et al. Copeptin in the differential diagnosis of the polydipsia-polyuria syndrome--revisiting the direct and indirect water deprivation tests. J Clin Endocrinol Metab. 2011;96(5):1506–15. http://www
.ncbi.nlm.nih .gov/pubmed/21367924 [Internet] Available from. [PubMed: 21367924] - 284.
- Timper K, Fenske W, Kuhn F, Frech N, Arici B, Rutishauser J, et al. Diagnostic Accuracy of Copeptin in the Differential Diagnosis of the Polyuria-polydipsia Syndrome: A Prospective Multicenter Study. J Clin Endocrinol Metab. 2015;100(6):2268–74. http://www
.ncbi.nlm.nih .gov/pubmed/25768671 [Internet] Available from. [PubMed: 25768671] - 285.
- Babinski MJ. Tumeur du corps pituitaire san acromegalie et avec arret de developpement des organs genitaux. Rev Neurol (Paris). 1900;8:531–3.
- 286.
- Steele CA, Cuthbertson DJ, MacFarlane IA, Javadpour M, Das KS, Gilkes C, et al. Hypothalamic obesity: prevalence, associations and longitudinal trends in weight in a specialist adult neuroendocrine clinic. Eur J Endocrinol [Internet]. 2013/01/08. 2013;168(4):501–7. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23293322. [PubMed: 23293322] - 287.
- Lustig RH. Hypothalamic obesity after craniopharyngiomas: mechanisms, diagnosis and treatment. Frontiers in Endocrinology. 2011;2:60. [PMC free article: PMC3356006] [PubMed: 22654817]
- 288.
- Pratheesh R, Rajaratnam S, Prabhu K, Mani SE, Chacko G, Chacko AG. The current role of transcranial surgery in the management of pituitary adenomas. Pituitary [Internet]. 2012/10/19. 2013;16(4):419–34. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/23076713. [PubMed: 23076713] - 289.
- Lustig RH, Post SR, Srivannaboon K, Rose SR, Danish RK, Burghen GA, et al. Risk factors for the development of obesity in children surviving brain tumors. J Clin Endocrinol Metab [Internet]. 2003/02/08. 2003;88(2):611–6. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12574189. [PubMed: 12574189] - 290.
- Hamilton JK, Conwell LS, Syme C, Ahmet A, Jeffery A, Daneman D. Hypothalamic Obesity following Craniopharyngioma Surgery: Results of a Pilot Trial of Combined Diazoxide and Metformin Therapy. Int J Pediatr Endocrinol [Internet]. 2011/05/24. 2011;2011:417949. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/21603206. [PMC free article: PMC3198743] [PubMed: 21603206] - 291.
- Harz KJ, Muller HL, Waldeck E, Pudel V, Roth C. Obesity in patients with craniopharyngioma: assessment of food intake and movement counts indicating physical activity. J Clin Endocrinol Metab [Internet]. 2003/11/07. 2003;88(11):5227–31. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/14602754. [PubMed: 14602754] - 292.
- Lustig RH, Hinds PS, Ringwald-Smith K, Christensen RK, Kaste SC, Schreiber RE, et al. Octreotide therapy of pediatric hypothalamic obesity: a double-blind, placebo-controlled trial. J Clin Endocrinol Metab [Internet]. 2003/06/06. 2003;88(6):2586–92. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12788859. [PubMed: 12788859] - 293.
- Mason PW, Krawiecki N, Meacham LR. The use of dextroamphetamine to treat obesity and hyperphagia in children treated for craniopharyngioma. Arch Pediatr Adolesc Med [Internet]. 2002/08/29. 2002;156(9):887–92. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/12197795. [PubMed: 12197795] - 294.
- Muller HL, Gebhardt U, Maroske J, Hanisch E. Long-term follow-up of morbidly obese patients with childhood craniopharyngioma after laparoscopic adjustable gastric banding (LAGB). Klin Padiatr [Internet]. 2011/11/05. 2011;223(6):372–3. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/22052635. [PubMed: 22052635] - 295.
- Rakhshani N, Jeffery AS, Schulte F, Barrera M, Atenafu EG, Hamilton JK. Evaluation of a comprehensive care clinic model for children with brain tumor and risk for hypothalamic obesity. Obesity (Silver Spring) [Internet]. 2010/01/09. 2010;18(9):1768–74. Available from: http://www
.ncbi.nlm.nih .gov/pubmed/20057368. [PubMed: 20057368] - 296.
- Zoicas F, Droste M, Mayr B, Buchfelder M, Schofl C. GLP-1 analogues as a new treatment option for hypothalamic obesity in adults: report of nine cases. Eur J Endocrinol. 2013;168(5):699–706. https://www
.ncbi.nlm .nih.gov/pubmed/23392214 [Internet] Available from. [PubMed: 23392214] - 297.
- Ando T, Haraguchi A, Matsunaga T, Natsuda S, Yamasaki H, Usa T, et al. Liraglutide as a potentially useful agent for regulating appetite in diabetic patients with hypothalamic hyperphagia and obesity. Intern Med. 2014;53(16):1791–5. http://www
.ncbi.nlm.nih .gov/pubmed/25130112 [Internet] Available from. [PubMed: 25130112] - 298.
- Lomenick JP, Buchowski MS, Shoemaker AH. A 52-week pilot study of the effects of exenatide on body weight in patients with hypothalamic obesity. Obesity (Silver Spring). 2016;24(6):1222–5. https://www
.ncbi.nlm .nih.gov/pubmed/27133664 [Internet] Available from. [PMC free article: PMC4882247] [PubMed: 27133664] - 299.
- Wilding JPH, Batterham RL, Calanna S, Davies M, van Gaal LF, Lingvay I, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. New England Journal of Medicine. 2021 Mar 18;384(11):989–1002. [PubMed: 33567185]
- 300.
- Huynh K, Klose M, Krogsgaard K, Drejer J, Byberg S, Madsbad S, et al. Randomized controlled trial of Tesomet for weight loss in hypothalamic obesity. European Journal of Endocrinology. 2022 Jun 1;186(6):687–700. [PMC free article: PMC9175551] [PubMed: 35294397]
- Review Pituitary Tumors in Childhood.[Endotext. 2000]Review Pituitary Tumors in Childhood.Colao A, Pirchio R. Endotext. 2000
- Clinical, radiological and pathological features of patients with Rathke's cleft cysts: tumors that may recur.[J Clin Endocrinol Metab. 1997]Clinical, radiological and pathological features of patients with Rathke's cleft cysts: tumors that may recur.Mukherjee JJ, Islam N, Kaltsas G, Lowe DG, Charlesworth M, Afshar F, Trainer PJ, Monson JP, Besser GM, Grossman AB. J Clin Endocrinol Metab. 1997 Jul; 82(7):2357-62.
- Risk factor for pituitary dysfunction in children and adolescents with Rathke's cleft cysts.[Korean J Pediatr. 2010]Risk factor for pituitary dysfunction in children and adolescents with Rathke's cleft cysts.Lim HH, Yang SW. Korean J Pediatr. 2010 Jul; 53(7):759-65. Epub 2010 Jul 31.
- Review [Central diabetes insipidus after resection of sellar-suprasellar tumors: prevalence and predictors of manifestation].[Zh Vopr Neirokhir Im N N Burde...]Review [Central diabetes insipidus after resection of sellar-suprasellar tumors: prevalence and predictors of manifestation].Badmaeva IN, Astafieva LI, Kalinin PL, Kadashev BA, Kutin MA. Zh Vopr Neirokhir Im N N Burdenko. 2021; 85(6):111-118.
- Review Surgical Treatment of Pituitary Adenomas.[Endotext. 2000]Review Surgical Treatment of Pituitary Adenomas.Jane JA Jr, Catalino MP, Laws ER Jr. Endotext. 2000
- Pituitary and Hypothalamic Tumor Syndromes in Childhood - EndotextPituitary and Hypothalamic Tumor Syndromes in Childhood - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...