ABSTRACT
Primary hyperparathyroidism (PHPT) is characterized by hypercalcemia and elevated or inappropriately normal parathyroid hormone (PTH) levels. PHPT results from excessive secretion of PTH from one or more of the parathyroid glands. The clinical presentation of PHPT has evolved since the 1970’s with the advent of the routine measurement of serum calcium at that time. Classical PHPT, with its associated severe hypercalcemia, osteitis fibrosa cystica, nephrolithiasis, and neuropsychological symptoms, once common is now infrequent. Today most patients are asymptomatic and have mild hypercalcemia, but may have evidence of subclinical skeletal and renal sequelae such as osteoporosis and hypercalciuria as well as vertebral fractures and nephrolithiasis both of which may be asymptomatic. Parathyroidectomy is the only curative treatment for PHPT and is recommended in patients with symptoms and those with asymptomatic disease who have evidence of end-organ sequelae. Parathyroidectomy results in an increase in BMD and a reduction in nephrolithiasis. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
Primary hyperparathyroidism (PHPT) is characterized by hypercalcemia and elevated or inappropriately normal parathyroid hormone (PTH) levels. The disorder today bears few similarities to the severe condition described by Fuller Albright and others as a “disease of stones, bones, and groans” in the 1930s (1-3). The skeletal hallmark of PHPT was osteitis fibrosa cystica, radiographically characterized by brown tumors of the long bones, subperiosteal bone resorption, distal tapering of the clavicles and phalanges, and “salt-and-pepper” erosions of the skull (4). Nephrocalcinosis and nephrolithiasis were present in the majority of patients, and neuromuscular dysfunction with muscle weakness was also common. With the advent of the automated serum chemistry autoanalyzer in the 1970s, the diagnosis of PHPT was increasingly recognized, leading to a four- to five-fold increase in incidence (5-7). Classic symptomatology, concomitantly, became much less frequent. In the United States and elsewhere in the developed world, symptomatic PHPT is now the exception and more than three fourths of patients having no symptoms attributable to their disease, making PHPT a disease that has “evolved” from its classic presentation (Table 1) (8). Symptomatic nephrolithiasis is still observed, although much less frequently than in the past. Now, radiologically evident bone disease is rare, but subclinical skeletal involvement can be readily detected by bone densitometry (9). This chapter describes the modern presentation, diagnosis and management of PHPT.
Table 1.
Changing Clinical Profile of Primary Hyperparathyroidism
RISK FACTORS, PATHOLOGY, AND ANATOMICAL LOCATION
PHPT results from excessive secretion of PTH from one or more of the parathyroid glands. The underlying cause of sporadic PHPT is unknown in most cases. While external neck radiation and lithium therapy are risk factors for the development of sporadic PHPT, most patients do not report these exposures (10-12). Chronically low calcium intake and higher body weight have also been recently described to be risk factors (13,14). The genetic pathogenesis of sporadic PHPT is unclear in most patients but genes regulating the cell cycle are thought to be important given the clonal nature of sporadic parathyroid adenomas.
By far the most common pathological finding in patients with PHPT is a solitary parathyroid adenoma, occurring in 80% of patients (15). In 2-4% of patients, PHPT is due to multiple adenomas (16). In approximately 15% of patients, all four parathyroid glands are involved (15,17). Parathyroid carcinoma accounts for <1% of all cases of PHPT(18). The etiology of four-gland parathyroid hyperplasia is multifactorial. There are no clinical features that definitively differentiate single versus multiglandular disease, but risk factors include inherited genetic syndromes such as multiple endocrine neoplasia (MEN) type 1 or type 2a and lithium exposure (17).
Parathyroid adenomas can be found in many unexpected anatomic locations. Parathyroid tissue embryonal migration patterns account for a plethora of possible sites of ectopic parathyroid adenomas. The most common atypical locations are within the thyroid gland, the superior mediastinum, and within the thymus (19). Occasionally, adenomas are identified in the retroesophageal space, the pharynx, the lateral neck, and even in the alimentary submucosa of the esophagus (20-22). On histologic examination, most parathyroid adenomas are encapsulated and are composed of parathyroid chief cells. Adenomas containing mainly oxyphilic or oncocytic cells are rare, but can give rise to clinical PHPT (23). Very rarely, PHPT may be due to parathyromatosis. This refers to an uncommon condition in which benign hyperfunctioning parathyroid tissue is scattered throughout the neck and/or in the superior mediastinum (see Unusual Presentations) (24).
EPIDEMIOLOGY
The incidence of PHPT has changed dramatically over the last half century (5,6,25,26). Before the advent of the multichannel autoanalyzer in the early 1970s, Heath et al reported an incidence of 7.8 cases per 100,000 persons in Rochester, Minnesota (5). With the introduction of routine calcium measurements in the mid-1970s, this rate rose dramatically to 51.1 cases per 100,000 in the same community. After prevalent cases were diagnosed, the incidence declined to approximately 27 per 100,000 persons per year in the United States until 1998, at which time another sharp increase was noted (25,27,28). This second peak has been attributed to the introduction of osteoporosis screening guidelines and targeted testing in those with osteoporosis (28). Recent works shows the incidence of PHPT increases with age and is higher in women and African-Americans than in men and other racial groups, respectively (29).
Greater appreciation of the catabolic potential of PTH in postmenopausal women with osteoporosis has led to measurement of PTH even in subjects who do not have hypercalcemia. This trend has led to the emergence of a new entity, normocalcemic PHPT or NPHPT(30). This condition is characterized by normal serum calcium, elevated PTH, and exclusion of known causes of secondary hyperparathyroidism. The incidence of NPHPT is unknown, but recent studies suggest a prevalence ranging from 0.2-3.1% (31,32).
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
The diagnosis of PHPT is made when hypercalcemia and elevated PTH levels are present.
PTH levels that are inappropriately normal are also consistent with the diagnosis. The other major cause of hypercalcemia, malignancy, is readily discriminated from PHPT by a suppressed PTH level. Further, both the clinical presentation and biochemical profile of PHPT and hypercalcemia of malignancy help distinguish them. Patients with hypercalcemia of malignancy typically have severe and symptomatic hypercalcemia and advanced cancers that are clinically obvious. On the other hand, in PHPT most patients are asymptomatic and the serum calcium level is typically mildly elevated (within 1 mg/dl of the upper limit of normal). Extremely rarely, a patient with malignancy will be shown to have elevated PTH levels resulting from ectopic secretion of native PTH from the tumor itself (33). Much more commonly, the malignancy is associated with the secretion of parathyroid hormone–related protein (PTHrP), a molecule that does not cross-react in intact PTH assay (discussed below). Finally, it is possible that a malignancy is present in association with PHPT. When the PTH level is elevated in someone with a malignancy, this is more likely to be the case than a true ectopic PTH syndrome.
While ninety percent of patients with hypercalcemia have either PHPT or malignancy, the differential diagnosis of hypercalcemia includes a number of other etiologies such as vitamin D intoxication, granulomatous disease, and others (33). With the exception of lithium and thiazide use and familial hypocalciuric hypercalcemia (FHH), virtually all other causes of hypercalcemia are associated with suppressed levels of PTH. If lithium and/or thiazides can be safely withdrawn, serum calcium and PTH levels that continue to be elevated 3-6 months later, confirm the diagnosis of PHPT. FHH, on the other hand, is differentiated from PHPT by family history, typically (but not always) low urinary calcium excretion, and mutations in the calcium sensing receptor (CASR) or more recently associated GNA11 and AP2S1 genes (34-36). In addition, virtual complete genetic penetrance leads to its clinical appearance typically before the age of 30. It is extremely unusual for FHH to present without an antecedent history after the age of 50.
To distinguish PTH-mediated from non-PTH mediated causes of hypercalcemia, PTH should be measured with an intact immunoradiometric (IRMA) or immunochemiluminometric (ICMA) assay, which readily discriminates between PHPT and hypercalcemia of malignancy. In PHPT, PTH concentrations are usually frankly elevated, but most often within 2 times the upper limit of normal. A minority may have PTH levels in the normal range, typically in the upper range of normal. In PHPT, such values, although within the normal range, are clearly abnormal in a hypercalcemic setting. Several factors affect the PTH level in those with and without PHPT, including age, vitamin D levels, and renal function. Because PTH levels normally rise with age, the broad normal range (typically 10-65 pg/mL) reflects values for the entire population. In the younger individual (< 45 years), one expects a narrower and lower normal range (10-45 pg/mL). Occasionally, the PTH level as measured will be as low as 20-30 pg/mL. Such unusual examples require a more careful consideration of other causes of hypercalcemia, but such individuals will usually be shown to have PHPT because hypercalcemia that is not PTH-mediated suppresses the PTH concentration to levels that are either undetectable or at the lower limits of the reference range. Souberbielle et al (37) have illustrated that the normal range is dependent on whether or not the reference population is or is not vitamin D deficient. When vitamin D–deficient individuals were excluded, the upper limit of the PTH reference interval decreased. Patients with PHPT and vitamin D deficiency have a “heightened” PTH levels compared to those who are vitamin D sufficient (38).
On the other hand, renal dysfunction tends to elevate PTH levels via a number of mechanisms, including reduced clearance and degradation of PTH. Indeed, patients with PHPT and severe renal dysfunction (glomerular filtration rate < 30ml/min), may also have higher PTH levels compared to those with better renal function (39). In addition, the “intact” IRMA for PTH overestimates the concentration of biologically active PTH, particularly in renal failure. In 1998, Lepage et al (40) demonstrated a large non-(1–84) PTH fragment that comigrated with a large aminoterminally truncated fragment (PTH[7–84]) and showed substantial cross-reactivity in commercially available IRMAs. This large, inactive moiety constituted as much as 50% of immunoreactivity by IRMA for PTH in individuals with chronic renal failure (41). Recognition of this molecule led to the development of a new IRMA using affinity-purified polyclonal antibodies to PTH (39–84) and to the extreme N-terminal amino acid regions, PTH (1–4) (42,43). This “whole PTH” or third generation assay detects only the full-length PTH molecule, PTH (1–84). This assay has clear utility in uremic patients, but in PHPT, both assays are equally useful (40,44-46). Using the third-generation assay for PTH (1-84), a second molecular form of PTH(1-84) that is immunologically intact at both extremes has been identified. This molecule reacts only poorly in second-generation PTH assays. It represents less than 10% of the immunoreactivity in normal individuals and up to 15% in renal failure patients. In a limited number of patients with a severe form of PHPT or with parathyroid cancer, it may be over-expressed (47).
PHPT can be discriminated from secondary and tertiary hyperparathyroidism by its different biochemical profile. Secondary hyperparathyroidism is associated with an appropriate elevation in PTH in response to a hypocalcemic provocation and either a frankly low or normal serum calcium level. Secondary hyperparathyroidism is often due to vitamin D deficiency. Other causes include malabsorption, kidney disease, or hypercalciuria. Infrequently, patients with secondary hyperparathyroidism may become hypercalcemic, and will ultimately be found to have PHPT, when the underlying condition (for example, vitamin D deficiency) is corrected (48). In these cases, the hypercalcemia of PHPT was ‘masked’ by the co-existing condition. On the other hand, tertiary hyperparathyroidism describes a condition in which prolonged, severe secondary hyperparathyroidism (as in end-stage renal disease) evolves into a hypercalcemic state due to the development of autonomous functioning of one or more of the hyperplastic parathyroid glands. This can be observed in patients on dialysis or after renal transplant. Tertiary hyperparathyroidism is usually obvious from the history.
Normocalcemic primary hyperparathyroidism (NPHPT) describes a condition characterized by normal serum albumin-corrected calcium levels and ionized calcium values with an elevated PTH level. This condition can only be diagnosed when all known causes of secondary hyperparathyroidism have been excluded. Patients with NPHPT typically are diagnosed when PTH is measured in the course of an evaluation for low bone mass. NPHPT may represent the earliest manifestations of PHPT, a “forme fruste” of the disease. Several reports have appeared describing these individuals, with some patients progressing to overt hypercalcemia while under observation (30,32,49,50).
Although the term “normocalcemic PHPT” has been in use for decades, there has been considerable controversy concerning the accuracy of this designation. In many cases, the increases in PTH levels were attributable to the limitations of available assay technology. The older midmolecule radioimmunoassay for PTH, previously in common use, measured hormone fragments in addition to the intact molecule. Spuriously elevated PTH levels, particularly in those with renal insufficiency in whom clearance of hormone fragments is impaired, were seen. Alternative explanations for hyperparathyroidism with NPHPT have been discovered, including medications, hypercalciuria, renal insufficiency, and certain forms of liver and gastrointestinal disease. In recent years, it has become clear that many patients designated as having NPHPT were vitamin D deficient. Vitamin D deficiency with coexisting PHPT can give the semblance of normal calcium levels when in fact they would have been hypercalcemic if the vitamin D levels were normal. Since a possible view of NPHPT is a condition fostered by an element of vitamin D resistance, it is important to ensure vitamin D sufficiency. While the Institute of Medicine states that normal levels of vitamin D, as measured by 25-hydroxyvitamin D, are 20 ng/ml, it did not address conditions of abnormal mineral metabolism, such as PHPT. In particular, in NPHPT, we and others recommend that levels of 25-hydorxyvitamin D be raised, if necessary, to > 30 ng/mL for at least 3 months in order to rule out an element of vitamin D insufficiency in this population. Biochemical profiles for the various causes of hypercalcemia and hyperparathyroidism are shown in Table 2.
Table 2.
Biochemical Profiles for Various Causes of Hypercalcemia and Hyperparathyroidism
OTHER BIOCHEMICAL FEATURES
In PHPT, serum phosphorus tends to be in the lower range of normal, but frank hypophosphatemia is present in less than one fourth of patients. Hypophosphatemia, when present, is due to the phosphaturic actions of PTH. Average total urinary calcium excretion is at the upper end of the normal range, with about 40% of all patients experiencing hypercalciuria. Serum 25-hydroxyvitamin D levels tend to be in the lower end of the normal range. Although mean values of 1,25-dihydroxyvitamin D are in the high-normal range, approximately one third of patients have frankly elevated levels of 1,25-dihydroxyvitamin D (51). This pattern is a result of the actions of PTH to increase expression of the 1-alpha hydroxylase that converts 25-hydroxyvitamin D to 1,25-dihydroxyvitamin D. A typical biochemical profile is shown in Table 3.
Table 3.
Biochemical Profile in Primary Hyperparathyroidism (n = 137)
CLINICAL PRESENTATION
PHPT typically occurs in individuals in their middle years, with a peak incidence between ages 50 and 60 years. However, the condition can occur at any age. Women are affected more frequently than men, in a ratio of approximately 3-4:1. Several different presentations of PHPT are possible and were originally described successively in time (2,30,52). The classical symptomatic presentation was described first; later, asymptomatic PHPT emerged due to biochemical screening, and most recently the normocalcemic variant was discovered as described above. However, these three forms of PHPT contemporaneously exist today. Which presentation predominates depends upon population- and geographic-specific screening practices. It is also postulated that vitamin D deficiency may affect clinical presentation. Vitamin D deficiency heightens PTH elevations and this can worsen the hyperparathyroid process (53). In regions of the world and populations where biochemical screening is not routine and incidentally where vitamin D deficiency is endemic, symptomatic PHPT is the most common form and PHPT will appear to be uncommon because it is only discovered when symptomatic (54-64). In areas and populations where screening is routine, asymptomatic PHPT will predominate and the incidence of PHPT is higher (52,65).
This chapter focuses on asymptomatic PHPT, as it is the predominant form in the United States and in most of the developed world. At the time of diagnosis, most patients with PHPT do not exhibit classic symptoms or signs associated with disease. Clinically overt kidney stones and fractures are rare (66). Constitutional complaints such as weakness, easy fatigability, depression, and intellectual weariness are seen with some regularity (see later discussion) (67). The physical examination is generally unremarkable. Band keratopathy, a hallmark of classic PHPT, occurs because of deposition of calcium phosphate crystals in the cornea, but is virtually never seen grossly. Even by slit-lamp examination, this finding is rare. The neck shows no masses. The neuromuscular system is normal. The sections below provide a detailed description of the multi-systemic manifestations of PHPT.
Diseases associated epidemiologically with PHPT have included hypertension (68-70), peptic ulcer disease, gout, or pseudogout (71,72). More recently celiac disease has been associated with PHPT (73). Some concomitant disorders such as hypertension are commonly seen, but it is not established that any of these associated disorders are etiologically linked to the disease.
The Skeleton
The classic radiologic bone disease of PHPT, osteitis fibrosa cystica, is rarely seen today in the United States. Most series place the incidence of osteitis fibrosa cystica at less than 2% of patients with PHPT. The absence of classic radiographic features (salt-and-pepper skull, tapering of the distal third of the clavicle, subperiosteal bone resorption of the phalanges, brown tumors) does not mean that the skeleton is not affected. With more sensitive techniques, it has become clear that skeletal involvement in the hyperparathyroid process is actually quite common. This section reviews the profile of the skeleton in PHPT as it is reflected in assays for bone markers, bone densitometry, bone histomorphometry, and new skeletal imaging techniques.
BONE TURNOVER MARKERS
PTH stimulates both bone resorption and bone formation. Markers of bone turnover, which reflect those dynamics, provide clues to the extent of skeletal involvement in PHPT (74).
Bone Formation Markers
Osteoblast products, including bone-specific alkaline phosphatase activity, osteocalcin, and serum amino-terminal propeptide of type I collagen (P1NP), reflect bone formation (74). In PHPT, alkaline phosphatase levels, the most widely clinically available marker, can be mildly elevated, but in many patients, total alkaline phosphatase values are within normal limits (75,76). In a small study from our group (77), bone-specific alkaline phosphatase activity correlated with PTH levels and BMD at the lumbar spine and femoral neck. Osteocalcin is also generally increased in patients with PHPT (77-79). Sclerostin is an important regulator of bone formation. Patients with PHPT have low sclerostin levels, suggesting PTH down regulates sclerostin (80). As expected the bone formation marker, serum amino-terminal propeptide of type I collagen (P1NP), is negatively associated with sclerostin in PHPT (81). In a small series of 27 patients followed for up to a year post-PTX, circulating sclerostin increases shortly after post-surgery but return to the age reference range within 10 days (82).
Bone Resorption Markers
Markers of bone resorption include the osteoclast product, tartrate-resistant acid phosphatase (TRAP), and collagen breakdown products such as hydroxyproline, hydroxypyridinium cross-links of collagen, and N- and C-telopeptides of type 1 collagen (NTX and CTX) (74). Urinary hydroxyproline, once the only available marker of bone resorption, no longer offers sufficient sensitivity or specificity to make it useful. Although urinary hydroxyproline was frankly elevated in patients with osteitis fibrosa cystica, in mild asymptomatic PHPT it is generally normal. Hydroxypyridinium cross-links of collagen, pyridinoline (PYD), and deoxypyridinoline (DPD), on the other hand, are often elevated in PHPT. They return to normal after parathyroidectomy (83). DPD and PYD both correlate positively with PTH concentrations. Studies of NTX, CTX and TRAP are limited, although levels of the latter have been shown to be elevated (48). Thus, sensitive assays of bone formation and bone resorption are both elevated in mild PHPT.
Longitudinal Bone Turnover Marker Studies
Studies of bone turnover markers in the longitudinal follow-up of patients with PHPT indicate a reduction in these markers following parathyroidectomy. Information from our group (83,84), Guo et al (85), and Tanaka et al (86) all report declining levels of bone markers following surgery. The kinetics of change in bone resorption versus bone formation following parathyroidectomy provide insight into skeletal recovery. We have found that markers of bone resorption decline rapidly following successful parathyroidectomy, whereas indices of bone formation follow a more gradual decrease (83). Urinary PYD and DPD decreased significantly as early as 2 weeks following parathyroid surgery, preceding reductions in alkaline phosphatase. Similar data were reported from Tanaka et al (86), who demonstrated a difference in time course between changes in NTX (reflecting bone resorption) and osteocalcin (reflecting bone formation) following parathyroidectomy, and Minisola et al (87), who reported a drop in bone resorptive markers and no significant change in alkaline phosphatase or osteocalcin. The persistence of elevated bone formation markers coupled with rapid declines in bone resorption markers indicate a shift in the coupling between bone formation and bone resorption toward an accrual of bone mineral postoperatively. More recent data indicate that levels of preoperative markers of bone turnover (formation and resorption) are positively associated with the extent of bone accrual after parathyroidectomy, though some of the patients included in this study had more severe PHPT than is typically seen in the United States today (88).
BONE DENSITOMETRY
The advent of bone mineral densitometry as a major diagnostic tool for osteoporosis occurred at a time when the clinical profile of PHPT was changing from a symptomatic to an asymptomatic disease. This fortuitous timing allowed questions about skeletal involvement in PHPT to be addressed when specific gross radiologic features of PHPT had all but disappeared. Observations of skeletal health in PHPT made by bone densitometry have established the importance of this technology in the evaluation of all patients with PHPT. The Consensus Development Conference on Asymptomatic Primary Hyperparathyroidism in 1990 implicitly acknowledged this point when bone mineral densitometry was included as a separate criterion for clinical decision making (89). Since that time, bone densitometry has become an indispensable component of both evaluating the patient and establishing clinical guidelines for management and monitoring.
The known physiologic proclivity of PTH to be catabolic at sites of cortical bone make a cortical site essential to any complete densitometric study of PHPT. By convention, the distal third of the radius is the site used. The early densitometric studies in PHPT also showed another physiologic property of PTH, namely, to preserve bone at cancellous sites. The lumbar spine is an important site to measure not only because it is predominantly cancellous bone, but also because postmenopausal women are at risk for cancellous bone loss. In PHPT, bone density at the distal third of the radius is diminished (90,91) while at the lumbar spine it is only minimally reduced (Figure 1). The hip region, containing relatively equal amounts of cortical and cancellous elements, shows bone density intermediate between the cortical and cancellous sites. The results support not only the notion that PTH is catabolic for cortical bone but also the view that PTH is generally protective against bone loss in cancellous bone (92-94). In postmenopausal women, the same pattern was observed (91). Postmenopausal women with PHPT, therefore, show a reversal of the pattern typically associated with postmenopausal bone loss. Rather than preferential loss of cancellous bone at the lumbar spine, the cortical site of the distal radius is more often affected in postmenopausal women with PHPT.

Figure 1.
The pattern of bone loss in primary hyperparathyroidism. A typical pattern of bone loss is seen in asymptomatic patients with primary hyperparathyroidism. The lumbar spine is relatively well preserved while the distal radius (1/3 site) is preferentially affected. (Reprinted with permission from Silverberg SJ, Shane E, DeLaCruz L, et al. Skeletal disease in primary hyperparathyroidism. J Bone Mineral Res 1989;4:283-291).
The bone density profile in which there is relative preservation of skeletal mass at the vertebrae and reduction at the more cortical distal radius is not always seen in PHPT. Although this pattern is evident in the vast majority of patients, small groups of patients show evidence of vertebral osteopenia at the time of presentation. In our natural-history study, approximately 15% of patients had a lumbar spine Z score of less than –1.5 at the time of diagnosis (95). Only half of these patients were postmenopausal women, so not all vertebral bone loss could be attributed entirely to estrogen deficiency. These patients are of interest with regard to changes in bone density following parathyroidectomy and are discussed in further detail later. The extent of vertebral bone involvement will vary as a function of disease severity. In the typical mild form of the disease, the pattern described earlier is seen. When PHPT is more advanced, there will be more generalized involvement, with involvement of the lumbar bone. When PHPT is severe or more symptomatic, all bones can be extensively involved. Vitamin D deficiency in mild asymptomatic PHPT seems to have minimal effect on BMD with only slightly reduced BMD at the 1/3 radius in those with low vitamin D (96,97).
BONE HISTOMORPHOMETRY
Analyses of percutaneous bone biopsies from patients with PHPT have provided direct information that could only be indirectly surmised by bone densitometry and by bone markers. Both static and dynamic parameters present a picture of cortical thinning, maintenance of cancellous bone volume, and a very dynamic process associated with high turnover and accelerated bone remodeling. Cortical thinning, inferred by bone mineral densitometry, is clearly documented in a quantitative manner by iliac crest bone biopsy (98,99). Van Doorn et al (100) demonstrated a positive correlation between PTH levels and cortical porosity. These findings are consistent with the known effect of PTH to be catabolic at endocortical surfaces of bone. Osteoclasts are thought to erode more deeply along the corticomedullary junction under the influence of PTH.
Histomorphometric studies have also contributed information about cancellous bone in PHPT (100). Again, as suggested by bone densitometry, cancellous bone volume is well preserved in PHPT. This is seen as well among postmenopausal women with PHPT. Several studies have shown that cancellous bone is actually increased in PHPT as compared to normal subjects (101,102). When cancellous bone volume is compared among age- and sex-matched subjects with PHPT or postmenopausal osteoporosis, a dramatic difference is evident. Whereas postmenopausal women with osteoporosis have reduced cancellous bone volume, women with PHPT have higher cancellous bone volume (101). The region(s) of bone loss in PHPT is (are) directed toward the cortical bone compartment, with good maintenance of cancellous bone volume unless the PHPT is unusually active.
In PHPT, age-related bone loss appears to be mitigated. In a study of 27 patients with PHPT (10 men and 17 women), static parameters of bone turnover (osteoid surface, osteoid volume, and eroded surface) were increased, as expected, in patients relative to control subjects (103). However, in control subjects, trabecular number varied inversely with age, whereas trabecular separation increased with advancing age. These observations are expected concomitants of aging. In marked contrast, in the patients with PHPT, no such age dependency was seen. There was no relationship between trabecular number or separation and age in PHPT, suggesting that the actual plates and their connections were being maintained over time more effectively than one would have expected by aging per se. Thus, PHPT seems to retard the normal age-related processes associated with trabecular loss.
In PHPT, indices of trabecular connectivity are greater than expected, whereas indices of disconnectivity are decreased. When three matched groups of postmenopausal women were assessed (a normal group, a group with postmenopausal osteoporosis, and a group with PHPT), women with PHPT were shown to have trabeculae with less evidence of disconnectivity compared with normal, despite increased levels of bone turnover (102,103). Thus, cancellous bone is preserved in PHPT through the maintenance of well-connected trabecular plates. To determine the mechanism of cancellous bone preservation in PHPT, static and dynamic histomorphometric indices were compared between normal and hyperparathyroid postmenopausal women. In normal postmenopausal women, there is an imbalance in bone formation and resorption, which favors excess bone resorption. In postmenopausal women with PHPT, on the other hand, the adjusted apposition rate is increased. Bone formation, thus favored, may explain the efficacy of PTH at cancellous sites in patients with osteoporosis (92,104-106). Assessment of bone remodeling variables in patients with PHPT shows increases in the active bone-formation period (101) (Table 4). The increased bone formation rate and total formation period may explain the preservation of cancellous bone seen in this disease.
Table 4.
Wall Width and Remodeling Variables in PHPT and Control Groups (Mean ± SEM)
More recently, further analysis of trabecular microarchitecture has taken advantage of newer technologies that have largely been confirmatory. In a three-dimensional analysis of transiliac bone biopsies using microCT technology, a highly significant correlation was observed with the conventional histomorphometry described earlier (107). In comparison to age-matched control subjects without PHPT, postmenopausal women with PHPT had higher bone volume (BV/TV), higher bone surface area (BS/TV), higher connectivity density (Conn.D), and lower trabecular separation (Tb.Sp.). There were also less marked age-related declines in BV/TV and Conn.D as compared to controls, with no decline in BS/TV. Using the technique of backscattered electron imaging (qBEI) to evaluate trabecular BMD distribution (BMDD) in iliac crest bone biopsies, Roschger et al (108) showed reduced average mineralization density and an increase in the heterogeneity of the degree of mineralization, consistent with reduced mean age of bone tissue. Studies of collagen maturity using Fourier Transform Infrared Spectroscopy provide further support for these observations (109). Bone strength, therefore, in PHPT has to take into account a number of factors related to skeletal properties of bone besides BMD (110).
NEW IMAGING TECHNIQUES
Newer non-invasive skeletal imaging technologies offer new insight into the skeletal manifestations of PHPT beyond observations made by DXA and radiography. The trabecular bone score or TBS provides an indirect assessment of trabecular microstructure from the DXA image. In those without PHPT, it has been shown to predicts fracture independently of BMD (111). In PHPT, TBS reveals trabecular microstructural deterioration at the spine, despite preserved BMD by DXA at this site (112). High resolution peripheral quantitative CT (HRpQCT) is a technology that noninvasively and directly measures skeletal microstructure at the distal radius and tibia. Utilizing this technology, studies in patients with PHPT indicate not only cortical thinning, but additionally trabecular deficits at the radius and tibia (Figure 2) (113-115). At the radius, trabeculae were fewer, thinner, more widely and heterogeneously spaced. At the tibia, trabeculae were more heterogeneously spaced (116). These deficits led to reduced stiffness when images were analyzed using microfinite element analysis, a technique that integrates structural and denisitometric information from the HRpQCT image into an estimated of mechanical competence. These recent findings, pointing to abnormalities in the trabecular compartment of bone, help to account for the increased risk of vertebral fractures (see below) observed in PHPT that had remained unexplained prior to the advent of such technologies (117-120). The difference in microskeletal abnormalities between the iliac crest bone biopsy data and HRpQCT may well reflect site-specific sampling differences.

Figure 2.
High-resolution peripheral quantitative CT images of the radius in a patient with primary hyperparathyroidism (PHPT; left) and a normal control (right). Trabecular deterioration is evident in PHPT. Image from Stein EM, Silva BC, Boutroy S, et al. Primary hyperparathyroidism is associated with abnormal cortical and trabecular microstructure and reduced bone stiffness in postmenopausal women. J Bone Miner Res 2012
FRACTURES
Fractures were a common clinical event in classic PHPT. In modern PHPT, one would anticipate, based on the BMD patterns observed with DXA, an increased risk of peripheral fractures, but a reduction in vertebral fractures. While not all studies are consistent and much of the data is retrospective and/or cross-sectional, the majority of studies suggest an increased risk for vertebral fractures in patients with PHPT (117-123). Moreover, recent data indicates that many vertebral fractures are in fact clinically silent (118,124). The paradox of increased vertebral fracture risk despite preserved lumbar spine BMD in PHPT had remained unclear until the advent of TBS and HRpQCT, which clearly document trabecular deficits in addition to previously recognized cortical skeletal deterioration. Using the Danish National Patient registry and a nested case-control design, Ejlsmark-Svensson et al. recently showed that vertebral fractures in patients with PHPT occur at a higher BMD than in patients without PHPT, again pointing to the importance of other elements of bone quality in PHPT (125). In one recent study, among asymptomatic PHPT patients, only those who met surgical guidelines showed a higher incidence of vertebral fractures compared with controls (118).
The risk for hip fracture is not clearly increased in PHPT. In a study that focused on hip fracture, a population-based prospective analysis (mean of 17 years’ duration; 23,341 person years) showed women with PHPT in Sweden not to be at increased risk (126). The Mayo Clinic experience with PHPT and risk of fracture reviewed 407 cases of PHPT recognized during the 28-year period between 1965 and 1992 (117). Fracture risk was assessed by comparing fractures at a number of sites with numbers of fractures expected based on gender and age from the general population. The clinical presentation of these patients with PHPT was typical of the mild form of the disease, with the serum calcium being only modestly elevated at 10.9 ± 0.6 mg/dL. The data from this retrospective epidemiologic study indicate that overall fracture risk was significantly increased at many sites such as the vertebral spine, the distal forearm, the ribs, and the pelvis. There was no increase in hip fractures. One might expect to see an increased incidence of distal forearm fractures as seen in the May study, because the hyperparathyroid process tends to lead to a reduction of cortical bone (distal forearm) in preference to cancellous bone (vertebral spine). Unfortunately, there were no densitometric data provided in this study, so one could not relate bone density to fracture incidence.
The impact of PHPT on fracture incidence appears complex and may be site-specific. This relationship is likely influenced by site-specific changes in areal bone density, bone size, and microstructure. Excess PTH would induce cortical thinning due to endosteal bone resorption but would also increase periosteal apposition, thus increasing bone diameter. Decreased areal bone density would increase fracture risk, while increased bone diameter and preserved microstructure at certain sites, might protect against fractures. Prospective studies are needed to elucidate the site-specific risk of fracture in PHPT.
Nephrolithiasis and Renal Function
In the past, classic clinical descriptions of PHPT emphasized kidney stones as a principal complication of the disease (127). The cause of nephrolithiasis in PHPT is probably multifactorial. An increase in the amount of calcium filtered at the glomerulus resulting from hypercalcemia may lead to hypercalciuria despite the physiologic actions of PTH to facilitate calcium reabsorption. A component of absorptive hypercalciuria exists in this disorder. The enhanced intestinal calcium absorption is likely a result of increased production of 1,25-dihydroxyvitamin d, a consequence of PTH’s action to increase the synthesis of this active metabolite (128) (129). Indeed, urinary calcium excretion is correlated with 1,25-dihydroxyvitamin d levels (129,130). The skeleton provides yet another possible source for the increased levels of calcium in the glomerular filtrate. Hyperparathyroid bone resorption might contribute to hypercalciuria, and subsequently to nephrolithiasis, even though there is no convincing evidence to support this hypothesis (131). Finally, alteration in local urinary factors, such as a reduction in inhibitor activity or an increase in stone-promoting factors, may predispose some patients with PHPT to nephrolithiasis (131,132). It remains unclear whether the urine of patients with hyperparathyroid stone disease is different in this regard from that of other stone formers.
Studies in the 1970s and 1980s documented a higher incidence of renal stone disease than do reports of more recent experience. Although the incidence of symptomatic nephrolithiasis today is much less common than it was in classical PHPT, kidney stones remain the most common manifestation of symptomatic PHPT (see Table 1). Estimates suggest symptomatic kidney stones in 15% to 20% of all patients (133). Screening for asymptomatic nephrolithiasis, indicates that the prevalence is actually much higher and this is now recommended in the most recent set of guidelines on the management of asymptomatic primary hyperparathyroidism (134-138).
The etiology of why stones develop in some but not others with PHPT has been postulated since the 1930s, but is still not well understood. In the 1930s, it was generally accepted that bone and stone disease did not coexist in the same patient with classic PHPT (2,139). Albright and Reifenstein (139) theorized that a low dietary calcium intake led to bone disease, whereas adequate or high dietary calcium levels caused stone disease. Dent et al (140), who provided convincing evidence against this construct, proposed the existence of two forms of circulating PTH, one causing renal stones and the other causing bone disease. A host of mechanisms, including differences in dietary calcium, calcium absorption, forms of circulating PTH, and levels of 1,25-dihydroxyvitamin d, were proposed to account for the clinical distinction between bone and stone disease in PHPT (131,140). Today, there is no clear evidence for two distinct subtypes of PHPT or that the process affecting the skeleton and kidneys occur in different subsets of patients (127). Cortical bone demineralization is as common and as extensive in those with and without nephrolithiasis (127,131).
Although more recent work has suggested that 1,25-dihydroxyvitamin d plays an etiologic role in the development of nephrolithiasis in PHPT, not all studies are consistent with this premise (127,131,132,138,141). Other investigations have shown risk factors for nephrolithiasis include younger age and male sex, whereas degree of hypercalcemia and hypercalciuria, PTH levels and other urinary factors have less consistently been associated (38,135,136,138,141-143). Hypomagnesuria has recently been associated with silent kidney stones in PHPT (144).
Other renal manifestations of PHPT include hypercalciuria, which is seen in approximately 40% of patients, and nephrocalcinosis. The frequency of nephrocalcinosis is unknown, but it appears to be relatively uncommon today (135). Though there were clear reports of renal impairment in early descriptions of PHPT, the prevalence of renal dysfunction (estimated glomerular filtration rate (eGFR) <60 ml/min) today in patients with mild PHPT is low with recent studies suggesting rates of 15–17% (39,145,146). Neither the severity of PHPT nor having a history of nephrolithiasis were risk factors for reduced eGFR in a 2014 study in those with mild PHPT; instead, traditional risk factors, such as age, hypertension, use of antihypertensive medication, and fasting glucose levels were associated with poorer kidney function (145). Longitudinal data is reassuring in this regard, as renal function remains stable in PHPT over long periods of follow-up (52,147).
Other Organ Involvement
CARDIOVASCULAR SYSTEM
Interest in the effect of PHPT on cardiovascular function is rooted in pathophysiologic observations of the hypercalcemic state. Hypercalcemia has been associated with increases in blood pressure, left ventricular hypertrophy, heart muscle hypercontractility, and arrhythmias (148,149). Furthermore, evidence of calcium deposition has been documented in the form of calcifications in the myocardium, heart valves, and coronary arteries. The association of overt cardiovascular symptomatology with modern-day PHPT is unclear because of inconsistencies between studies. An explanation for the inconsistent results reported in the literature on the cardiovascular manifestations of PHPT relates to the fact that the clinical profile of the disease has changed. As a result, the cohorts that have been studied have varied greatly in the severity of their underlying disease. This is particularly true in terms of the serum calcium and parathyroid hormone concentrations, with data from cohorts with marked hypercalcemia and hyperparathyroidism showing the most cardiovascular involvement. Because it is thought that both calcium and PTH can independently affect the cardiovascular system, such variability among cohorts can give rise to inconsistent results. Recent studies have focused on not only cardiovascular mortality, but also hypertension, coronary artery disease (CAD), valve calcification, left ventricular hypertrophy (LVH), carotid disease, and vascular stiffness.
Cardiovascular Mortality
There is little doubt that in very active PHPT, cardiovascular mortality is increased (150-153). Of some interest are the postoperative observations in which the higher cardiovascular mortality rate persists for years after cure (154). These observations differ markedly from those in which asymptomatic PHPT has been studied. Although limited, the studies have not shown any increase in mortality (155,156). The Mayo Clinic studies help to bring these observations together. In the mildly hypercalcemic individuals, overall and cardiovascular mortality was reduced, but in those whose serum calcium was in the highest quartile, cardiovascular mortality was increased (156). The idea that the more common asymptomatic form of PHPT is not associated with increased mortality is supported by data from Nilsson et al (157) and by other studies (158,159) in which more recently enrolled subjects had better survival than those who entered earlier and presumably had more active disease.
Hypertension
Hypertension, a common feature of PHPT when it is part of a MEN syndrome with pheochromocytoma or hyperaldosteronism, has also been reported as more prevalent in sporadic, asymptomatic PHPT than in appropriately matched control groups. The mechanism of this association is unknown, and the condition does not clearly remit following cure of the hyperparathyroid state (68,70,160-163).
Coronary Artery Disease (CAD) and Valve Calcification
Both calcium and PTH have independently been shown to be associated with coronary heart disease (164,165). Aside from autopsy studies such as those of Roberts and Waller (166), in which coronary atherosclerosis was seen in patients with marked hypercalcemia (16.8 to 27.4 mg/dL), the more recent literature has been controversial. When CAD is present in PHPT, it is most likely due to traditional risk factors rather than the disease itself (159,167,168). Some studies have actually shown that in mild PHPT, there is better exercise tolerance as determined by the electrocardiogram (169). Valve calcification, which is present in severe PHPT, has been shown to be more extensive (greater valve area) when present in those with mild PHPT versus controls (149,170,171), and is associated with increased PTH levels but it is not reversible with parathyroidectomy (171).
Left Ventricular Hypertrophy
Left ventricular hypertrophy (LVH) is a strong predictor of cardiovascular disease and mortality (172,173). LVH has been associated with PHPT in many, but not all, studies (174). A 2015 meta-analysis indicated that parathyroidectomy is associated with a decline in left ventricular mass and that higher levels of PTH pre-operatively predict a greater cardiovascular benefit. However, dissociating disease severity from study design (RCTs included individuals with lower levels of calcium and PTH than those included in observational studies) was not possible (175).
Vascular Function
Conflicting data exist regarding whether intima media thickness is increased in PHPT (176-180). Multiple studies have reported increased vascular stiffness, sometimes associated with PTH levels, in mild PHPT, but its reversibility with parathyroidectomy is inconsistent (180-183). Given conflicting data, most experts do not consider cardiovascular disease to be an indication for parathyroidectomy (137).
NEUROLOGICAL, PSYCHOLOGICAL, AND COGNITIVE FEATURES
Descriptions of classical PHPT do indeed indicate neuropsychological features (2,184). The extent to which these features remain a part of the modern picture of PHPT as well as the exact mechanisms underlying them is unclear. Perhaps the most common complaints have been those of weakness and easy fatigability (67). Classic PHPT was formerly associated with a distinct neuromuscular syndrome characterized by type II muscle cell atrophy (185). Originally described by Vicale in 1949 (186), the syndrome consisted of easy fatigability, symmetric proximal muscle weakness, and muscle atrophy. Both the clinical and electromyographic features of this disorder were reversible after parathyroid surgery (187,188). In the milder, less symptomatic form of the disease that is common today, this disorder is rarely seen (189). In a group of 42 patients with mild disease, none had complaints consistent with the classic neuromuscular dysfunction described previously. Although more than half of all patients expressed nonspecific complaints of paresthesias and muscle cramps, electromyographic studies did not confirm the picture of past observations.
The “psychic groans” described by early observers of patients with classic PHPT remain a source of controversy today. Patients with PHPT often report some degree of behavioral and/or psychiatric symptomatology. A retrospective inquiry of patients with more severe disease showed a 23% incidence of psychiatric symptomatology (n = 441) (190). A number of studies suggest, however, that even ‘mild PHPT’ (serum calcium <12 mg/dl) is associated with non-specific symptoms such as depression, anxiety, fatigue, decreased quality of life (QOL), sleep disturbance, and cognitive dysfunction. Many, but not all, observational studies have indicated these features improve after parathyroidectomy (191). Three RCTs have investigated the reversibility of reduced QOL and psychiatric symptoms (192-194). Despite being of similar design and using similar assessment tools, all three randomized controlled trials came to different conclusions; one randomized controlled trial suggested parathyroidectomy prevents worsening of QOL and improves psychiatric symptoms (193); another randomized controlled trial indicated no benefit; and the third randomized controlled trial demonstrated improvement in QOL (192,194). One randomized controlled trial investigated changes in cognition after parathyroidectomy, but its small size precluded definitive conclusions being drawn (195).
While less well studied, some, but not all, studies have demonstrated reduced memory or impairment in other cognitive domains (195-201). It is unclear if cognition improves after parathyroidectomy because results of studies in which longitudinal control groups are compared to those undergoing parathyroidectomy are inconsistent (197,199,202-204). Recent work has turned to the potential mechanisms that contribute to cognitive dysfunction in PHPT. Our latest studies have addressed this issue. We hypothesized that cerebrovascular dysfunction (i.e., vascular stiffness) might underlie cognitive changes in patients with PHPT. While PTH correlated with cerebrovascular function as measured by transcranial Doppler, there was no consistent association between cerebrovascular function and cognitive performance (205). In a separate study, we utilized functional magnetic resonance imaging to assess if cerebral activation was altered by PHPT. Functional magnetic resonance imaging, or fMRI, is a non-invasive tool that maps brain function based on changes in blood flow (206). We found that PHPT was associated with differences in task-related neural activation patterns but no difference in cognitive performance. This may indicate compensation to maintain the same cognitive function, but there was no clear improvement in neural activation after parathyroidectomy (206). At present, most experts do not recognize cognitive or psychiatric symptoms as a sole indication for parathyroidectomy. Reasons for this include the failure to clearly demonstrate reversibility in randomized controlled trials, the inability to predict which patients might improve and a lack of a clear mechanism (137).
GASTROINTESTINAL MANIFESTATIONS
Primary hyperparathyroidism has long been considered associated with an increased incidence of peptic ulcer disease. Most recent studies suggest that the incidence of peptic ulcer disease in PHPT is approximately 10%, a figure similar to its percentage in the general population. An increased incidence of peptic ulcer disease is seen in patients with PHPT resulting from MEN1, in which approximately 40% of patients have clinically apparent gastrinomas (Zollinger-Ellison syndrome). In those patients, PHPT is associated with increased clinical severity of gastrinoma, and treatment of the associated PHPT has been reported to benefit patients with Zollinger-Ellison syndrome (207,208). Despite this, current recommendations (Consensus Conference Guidelines for Therapy of MEN1) state that the coexistence of Zollinger-Ellison syndrome does not represent sufficient indication for parathyroidectomy, because medical therapy is so successful (208).
Although hypercalcemia can underlie pancreatitis, most large series have not reported an increased incidence of pancreatitis in patients with PHPT with serum calcium levels less than 12 mg/dL. The Mayo Clinic experience from 1950 to 1975 showed that only 1.5% of those with PHPT exhibited coexisting pancreatitis, and alternative explanations for pancreatitis were found for several patients. Regarding pancreatitis in pregnancy in patients with PHPT, these conditions may coexist, but there is no evidence for a causal relationship between the disorders (209).
OTHER SYSTEMIC INVOLVEMENT
Many organ systems were affected by the hyperparathyroid state in the past. Anemia, band keratopathy, and loose teeth are no longer part of the clinical syndrome of PHPT. Gout and pseudogout are seen infrequently, and their etiologic relationship to PHPT is not clear.
Unusual Presentations
NEONATAL PRIMARY HYPERPARATHYROIDISM
Neonatal PHPT is a rare form of the disorder caused by homozygous inactivation of the calcium-sensing receptor (210). When present in a heterozygous form, it is a benign hypercalcemic state known as familial hypercalciuric hypercalcemia (FHH). However, in the homozygous neonatal form, hypercalcemia is severe and the outcome is fatal unless recognized early. The treatment of choice is early subtotal parathyroidectomy to remove the majority of hyperplastic parathyroid tissue.
PRIMARY HYPERPARATHYROISM IN PREGNANCY
Primary hyperparathyroidism in pregnancy is primarily of concern for its potential effect on the fetus and neonate (211,212). Potential complications of PHPT in pregnancy include spontaneous abortion, low birth weight, supravalvular aortic stenosis, and neonatal tetany. The last condition is a result of fetal parathyroid gland suppression by high levels of maternal calcium, which readily crosses the placenta during pregnancy. These infants, accustomed to hypercalcemia in utero, have functional hypoparathyroidism after birth, and can develop hypocalcemia and tetany in the first few days of life. Today, with most patients (pregnant or not) presenting with a mild form of PHPT, an individualized approach to the management of the pregnant patient with PHPT is advised. A recent retrospective study suggested that PHPT did not increase the risk of abortion, birth weight, length, or Apgar score (213). Thus, many of those with mild disease can be followed safely, with successful neonatal outcomes without surgery. However, parathyroidectomy during the second trimester remains the traditional recommendation for this condition.
ACUTE PRIMARY HYPERPARATHYROIDISM
Acute PHPT is known variously as parathyroid crisis, parathyroid poisoning, parathyroid intoxication, and parathyroid storm. Acute PHPT describes an episode of life-threatening hypercalcemia of sudden onset in a patient with PHPT (214,215). Clinical manifestations of acute PHPT are mainly those associated with severe hypercalcemia. Nephrocalcinosis or nephrolithiasis is frequently seen. Radiologic evidence of subperiosteal bone resorption is also commonly present. Laboratory evaluation is remarkable not only for very high serum calcium levels but also for extremely high levels in PTH to approximately 20 times normal (215). In this way, acute PHPT resembles, in biochemical terms, parathyroid carcinoma. A history of persistent mild hypercalcemia has been reported in 25% of patients. However, given the rarity of this condition, the risk of developing acute PHPT in a patient with mild asymptomatic PHPT is very low. Intercurrent medical illness with immobilization may precipitate acute PHPT. Early diagnosis, with aggressive medical management followed by surgical cure, is essential for a successful outcome. The initial impression of patients who present in this manner, without an antecedent history of hypercalcemia, is often mistaken for malignancy. However, the parathyroid hormone level usually quickly clarifies the diagnosis to PHPT in most situations.
PARATHYROID CANCER
An indolent yet potentially fatal disease, parathyroid carcinoma accounts for less than 0.5% of cases of PHPT. In contrast to patients with PHPT due to benign parathyroid pathology, patients with parathyroid carcinoma typically have marked elevations in serum calcium and PHT. The cause of the disease is unknown, and no clear risk factors have been identified except for hereditary syndromes. There is no evidence to support the malignant degeneration of previously benign parathyroid adenomas (216). Parathyroid carcinoma has been reported particularly in hyperparathyroidism-jaw tumor (HPT-JT) syndrome (217-221), a rare autosomal disorder in which as many as 15% of patients will have malignant parathyroid disease. Because cystic changes are common, this disorder has also been referred to as cystic parathyroid adenomatosis (222). In HPT-JT, ossifying fibromas of the maxilla and mandible are seen in 30% of cases. Less commonly, kidney lesions, including cysts, polycystic disease, hamartomas, or Wilms’ tumors, can be present (223). Parathyroid carcinoma has also been reported in familial isolated hyperparathyroidism (218,224). Parathyroid carcinoma, as defined pathologically, has been reported in MEN1 syndrome and with somatic MEN1 mutations (225-227). However, recurrent parathyroid disease in MEN1 may mimic but might not actually be a result of malignancy. Only one case of parathyroid carcinoma has been reported in the MEN2A syndrome (228).
Loss of the retinoblastoma tumor suppressor gene was formerly considered a marker for parathyroid cancer (229), but more recent studies do not unequivocally support this impression (230). Work by Shattuck et al (231,232) has provided new insights into the molecular pathogenesis of parathyroid cancer. Parathyroid carcinomas from 10 of 15 patients with sporadic parathyroid cancer carried a mutation in the HRPT2 gene. The HRPT2 gene encodes for the parafibromin protein that was shown to be mutated in a substantial number of patients with parathyroid cancer. Marcocci et al (216) have reviewed this topic, pointing out a potential role for parafibromin in parathyroid cancer. In three of 15 patients with parathyroid cancer, Shattuck et al (231) showed that the mutation was in the germline. The presence of the mutation in the germline suggests that this disease might be related in some way to the HPT-JT syndrome, in which this gene has been implicated (231). In addition, there is an increased risk of parathyroid cancer in the HPT-JT syndrome. In fact, certain clinical features in patients with a germline mutation and in their relatives are indicative of the HPT-JT syndrome or phenotypic variants (220,223,224).
Manifestations of hypercalcemia are the primary effects of parathyroid cancer. The disease tends not to have a bulk tumor effect, spreading slowly in the neck. Metastatic disease is a late finding, with lung (40%), liver (10%), and lymph node (30%) involvement seen most commonly. The clinical profile of parathyroid cancer differs from that of benign PHPT in several important ways (216). First, no female predominance is seen among patients with carcinoma. Second, elevations in serum calcium and PTH are far greater. Consequently, the hyperparathyroid disease tends to be much more severe, with the classic targets of PTH excess involved in most cases. Nephrolithiasis or nephrocalcinosis is seen in up to 60% of patients; overt radiologic evidence of skeletal involvement is seen in 35% to 90% of patients. A palpable neck mass, distinctly unusual in benign PHPT, has been reported in 30% to 76% of patients with parathyroid cancer (233). Grossly, malignant glands are large, often exceeding 12 g. They tend to be adherent to adjacent structures. Microscopically, thick, fibrous bands divide the trabecular arrangement of the tumor cells. Capsular and blood vessel invasion is common by these cells, which often contain mitotic figures (233). Treatment is reviewed below.
Parathyromatosis
Originally reported in 1975, fewer than one-hundred cases of parathyromatosis have been described in the literature (234,235). The condition is characterized histologically by small collections or nodules of parathyroid cells embedded within surrounding soft tissue outside the parathyroid gland capsule margins (24,236). Parathyromatosis may rarely be embryologic in origin or, more often, is secondary to tissue seeding during parathyroid surgery or fine needle aspiration (24,234,237). The majority of cases have been described in those who have undergone parathyroid surgery for secondary hyperparathyroidism associated with end-stage renal disease (24,235). While clinically and biochemically similar to primary hyperparathyroidism, parathyromatosis is associated with recurrent or persistent disease (24). The diagnosis is typically made at the time of surgery, although pre-operative imaging has been reported to be diagnostically helpful (238,239). Management is challenging and complete cure is uncommon. Treatment involves complete surgical excision of all parathyromatosis nodules and/or parathyroid tissue (24,240). Intra-operative parathyroid hormone level monitoring and pathologic review of frozen sections at the time of surgery may be helpful to increase surgical success. Successful accounts of medical therapy with calcimimetics and bisphosphonates have been reported (24,235).
EVALUATION
The diagnosis of PHPT is confirmed by demonstrating an elevated or inappropriately normal PTH level in the face of hypercalcemia. Further biochemical assessment should include serum phosphorus, alkaline phosphatase activity, vitamin D metabolites, albumin, and creatinine. A morning 2-hour or 24-hour urine collection should be obtained for calcium and creatinine. A urinary bone resorption marker such as serum CTX or urinary N-telopeptide can be helpful. Bone densitometry is performed in all patients. It is important to obtain densitometry at three sites: the lumbar spine, the hip, and the distal third of the radius. Because of the differing amounts of cortical and cancellous bone at the three sites and the different effects of PTH on cortical and cancellous bone, measurement at all three sites gives the most accurate clinical assessment of skeletal involvement in PHPT. Bone biopsy is not routinely obtained in the evaluation of PHPT, but is essential in research. In the most recent guidelines, spinal imaging is recommended to assess for clinically silent vertebral fractures (137). This can be vertebral X-rays, vertebral fracture assessment or TBS score, the latter two obtained by the DXA image. While symptomatic kidney stones are present in 15% to 20% of patients by history, the finding that many more have clinically silent nephrolithiasis has led to the recommendation to obtain renal ultrasound, CT, or abdominal x-ray to assess for either nephrolithiasis or nephrocalcinosis.
NATURAL HISTORY
Since the early 1990s, new knowledge of the natural history of PHPT with or without surgery has been very helpful in guiding decisions regarding surgery in patients with asymptomatic PHPT. The authors and their colleagues have conducted the longest prospective observational trial (52,241). This project began in 1984 in an effort to define the natural history of asymptomatic PHPT. The study included detailed analyses of pathophysiologic, densitometric, histomorphometric, and other skeletal features of PHPT (52,241). Much of the information gleaned from that study has been summarized already in this chapter. The 15-year follow-up to this study constitutes the longest natural-history study of this disorder (241).
Recommendations for surgery or observation were made based on the 1990 set of National Institutes of Health guidelines, but both groups included patients who did or did not meet surgical guidelines. This is because some patients opted for surgery even if they did not meet the guidelines, whereas others opted for a conservative approach even if they did meet guidelines for surgery. As will be described in the following sections, this imperfect design was followed by three studies that were truly randomized but were of much shorter duration. The results with regard to natural history from all studies are remarkably concordant.
Natural History with Surgery
Successful parathyroidectomy results in permanent normalization of the serum calcium and PTH levels. Postoperatively, there is a marked improvement in BMD at all sites (lumbar spine, femoral neck, and distal one-third radius) amounting to gains greater than 10% (52) (Figure 3), The improvement is most rapid at the lumbar spine, followed by gains at the hip regions and the distal 1/3 radius during the 15-year follow-up (241). The improvements were seen in those who met and did not meet surgical criteria at study entry, confirming the salutary effect of parathyroidectomy in this regard on all patients.
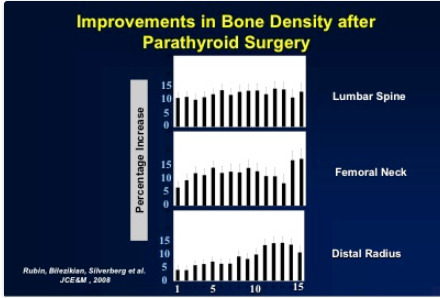
Figure 3.
Improvement in bone density after parathyroid surgery. Data shown are the cumulative percentage changes from baseline over 15 years of follow-up in patients who underwent parathyroidectomy.
Natural History Without Surgery
In subjects who did not undergo parathyroid surgery, serum calcium remained stable for about 12 years, with a tendency for the serum calcium level to rise in years 13 to 15 (241). Other biochemical indices such as PTH, vitamin D metabolites, and urinary calcium did not change for the entire 15 years of follow-up in the group as a whole. Bone density at all three sites remained stable for the first 8 to 10 years. However, after this period of stability, declining cortical BMD was seen at the hip and more dramatically at the distal one-third radius site. Although the numbers became limiting after 10 years of follow-up, it is noteworthy that a small majority of the subjects lost more than 10% of their BMD during the 15 years of observation. Even though this decline was observed in the majority of subjects, only 37% of subjects met one or more guidelines for surgery after the 15 years of follow-up.
Randomized Studies of the Natural History of Asymptomatic Primary Hyperparathyroidism
The long natural history study of asymptomatic PHPT has added much to our knowledge about this disease throughout time. Subsequent randomized trials confirm the observational data, but are limited by their shorter duration. In 2004, Rao et al (242) reported on 53 subjects, assigned either to parathyroid surgery (n = 25) or to no surgery (n = 28). The follow-up lasted for at least 2 years. There was a significant effect of parathyroidectomy on BMD at the hip and femoral neck but not the spine or forearm. Bollerslev et al (192) reported in 2007 the interim results of their randomized trial of parathyroidectomy versus no surgery. This study from three Scandinavian countries was larger, with 191 patients who were randomized to medical observation or to surgery. After surgery, biochemical indices normalized and BMD increased. In the group that did not undergo parathyroid surgery, BMD did not change. Also, in 2007, Ambrogini et al (194) reported the results of their randomized controlled trial of parathyroidectomy versus observation. Surgery was associated with a significant increase in BMD of the lumbar spine and hip after 1 year.
Whether fracture risk decreases after parathyroidectomy is not clear. The study by Bollerslev reported on vertebral fracture risk reduction at 5 years after initial treatment allocation. That study indicated that successful parathyroidectomy versus observation was associated with a reduction in vertebral fracture risk that was of borderline statistical significance (243).
GUIDELINES FOR PARATHYROIDECTOMY
Parathyroidectomy remains the only currently available option to cure PHPT. As the disease profile has changed, questions have arisen concerning the advisability of surgery in asymptomatic patients. If asymptomatic patients have a benign natural history, the surgical alternative is not an attractive one. On the other hand, asymptomatic patients may display levels of hypercalcemia or hypercalciuria that cause concern for the future. Similarly, bone-mass measurements can be frankly low. In an effort to address such issues, there have been four consensus development conferences (in 1991, 2002, 2008, and 2013) on the management of asymptomatic PHPT (89,137,244-247). The most recent guidelines that emerged from the 2013 conference are helpful to the clinician faced with the asymptomatic hyperparathyroid patient: All symptomatic patients are advised to undergo parathyroidectomy. Surgery is advised in asymptomatic patients with (1) serum calcium greater than 1 mg/dL higher than the upper limit of normal; (2) renal guidelines: reduction in creatinine clearance to less than 60 mL/min; urinary calcium excretion >400 mg/24 h with increased stone risk; or presence of nephrolithiasis or nephrocalcinosis on renal imaging; (3) skeletal guidelines: reduced bone density T-score < –2.5 at any site; or vertebral compression fracture on an imaging study; and (4) age younger than 50 years. The most recent guidelines are shown in Table 5. A noteworthy change in the guidelines reflects the fact that asymptomatic kidney stones and vertebral compression fractures are now considered as indications for parathyroidectomy.
Table 5.
Comparison of New and Old Guidelines for Surgery in Asymptomatic Primary Hyperparathyroidism
A number of points were discussed that did not lead to specific recommendations, including the issues of the neurocognitive and cardiovascular aspects of PHPT. The workshop panel also acknowledged a potential role of vitamin D deficiency in fueling processes associated with abnormal parathyroid glandular activity. Finally, the panel also reaffirmed the entity of normocalcemic PHPT, but noted that there are insufficient data to provide evidence-based guidelines for management.
SURGERY
A large percentage of those patients who meet the surgical guidelines listed in Table 5 are asymptomatic. Some asymptomatic patients who meet surgical guidelines elect not to have surgery for varying reasons including personal choice, intercurrent medical conditions, and previous unsuccessful parathyroid surgery. Conversely, there are patients who meet none of the NIH guidelines for parathyroidectomy but opt for surgery nevertheless. Physician and patient input remain important factors in the decision regarding parathyroid surgery.
Preoperative Localization of Hyperfunctioning Parathyroid Tissue
A number of imaging tests have been developed and have been applied singly or in combination to address the challenge of preoperative localization. The rationale for locating abnormal parathyroid tissue before surgery is that the glands can be notoriously unpredictable in their location. Although most parathyroid adenomas are identified in regions proximate to their embryologically intended position (the four poles of the thyroid gland), many are not. In such situations, previous surgical experience and skill are needed to locate the ectopic parathyroid gland. In such hands, 95% of abnormal parathyroid glands will be discovered and removed at the time of initial parathyroid surgery. However, in the patient with previous neck surgery, even expert parathyroid surgeons do not generally achieve such high success rates. Preoperative localization of the abnormal parathyroid tissue can be extremely helpful under these circumstances. Preoperative imaging is also necessary for any patient who will undergo parathyroidectomy using a minimally invasive approach. Imaging studies should not be used for the diagnosis of PHPT because the sensitivity and specificity of various imaging modalities varies with some having false-positive rates as high as 25% (248).
NONINVASIVE IMAGING
Noninvasive parathyroid imaging studies include technetium (Tc)-99m sestamibi scintigraphy, ultrasound, computed tomography (CT) scanning, magnetic resonance imaging (MRI), and positron emission tomography (PET) scanning. Tc-99m sestamibi is generally regarded to be the most sensitive and specific imaging modality, especially when it is combined with single-photon emission CT (SPECT). For the single parathyroid adenoma, sensitivity has ranged from 80% to 100%, with a 5% to 10% false-positive rate. On the other hand, sestamibi scintigraphy and the other localization tests have a relatively poor record in the context of multiglandular disease (249). The success of ultrasonography is highly operator dependent (250). In centers where there is great expertise, this noninvasive approach is most attractive. Abnormalities identified by ultrasound as possible parathyroid tissue may prove to be a thyroid nodule or lymph node, which underscores the importance of the skill and experience of the ultrasonographer. Rapid spiral thin-slice CT scanning of the neck and mediastinum with evaluation of axial, coronal, and sagittal views can add much to the search for elusive parathyroid tissue, albeit with attendant higher radiation exposure (251). Four-dimensional (4D) CT has emerged as a promising method and consists of multiphase CT acquired at non-contrast, contrast-enhanced, arterial and delayed phases. In a recent study, 4D CT was superior compared with sestamibi SPECT/CT (252). MRI can also identify abnormal parathyroid tissue, but it is time consuming and expensive. It is also less sensitive than the other noninvasive modalities. It can nonetheless be useful when the search with these other noninvasive approaches has been unsuccessful. PET with or without simultaneous CT scan (PET/CT) can be used, but like MRI, it is expensive and does not have the kind of experiential basis that make it attractive. There are also specificity issues because FDG, the scanning agent, accumulates in the thyroid, making differentiation between parathyroid adenoma and thyroid nodules difficult. Recently, 18F-fluorocholine (FCH) positron emission tomography (PET) has been employed for the detection of parathyroid adenomas.
INVASIVE IMAGING
Parathyroid Fine-Needle Aspiration
Fine-needle aspiration (FNA) of a parathyroid gland, identified by any of the aforementioned modalities, can be performed and the aspirate analyzed for PTH. This technique is not recommended for routine de novo cases.(253) A theoretical concern with this approach is the possibility that parathyroid cells could be deposited outside the parathyroid gland in the course of the aspiration. Autoseeding of parathyroid tissue would be an unwanted consequence of this procedure if it were to occur.
Arteriography and Selective Venous Sampling for Parathyroid Hormone
In situations where the gland has not been identified by any of the techniques described, the combination of arteriography and selective venous sampling can provide both anatomic and functional localization of abnormal parathyroid tissue. This approach, however, is costly and requires an experienced interventional radiologist. It is also performed in only a few centers in the United States. This approach is reserved for those individuals who have undergone previous unsuccessful parathyroid surgery in whom all other localization techniques have failed (254).
Surgical Approach
In the hands of an expert parathyroid surgeon, parathyroidectomy is a successful with infrequent complications. A standard surgical approach is the four-gland parathyroid exploration under general or local anesthesia, with or without preoperative localization. This approach has been reported to lead to surgical cure in more than 95% of cases (255). Several alternative approaches have emerged that focus on the single gland and not the total four-gland neck exploration that was routinely used in the past. Unilateral approaches are appealing for a disease in which most often only a single gland is involved. These procedures include a unilateral operation in which the gland on the same side that harbors the adenoma is ascertained to be normal. Because multiple parathyroid adenomas are unusual, a normal parathyroid gland is considered by some to be sufficient evidence for single-gland disease. Another limited surgical approach that has emerged in many centers as the approach of choice is the minimally invasive parathyroidectomy (MIP) (256,257). Preoperative parathyroid imaging is necessary, and the procedure is directed only to the site where the abnormal parathyroid gland has been visualized (258). Preoperative blood is obtained for comparison of the PTH concentration with an intraoperative sample(s) obtained after removal of the “abnormal” parathyroid gland. The availability of a rapid PTH assay in or near the operating room is necessary for this procedure. If the ten-minute post-excision PTH level falls by more than 50% compared to baseline, and into the normal range, the gland that has been removed is considered to be the sole source of overactive parathyroid tissue, and the operation is terminated. If the PTH level does not fall by more than 50%, into the normal range, the operation is extended to a more traditional one in a search for other overactive parathyroid tissue. There is a risk (albeit small) that the minimally invasive procedure may miss other overactive gland(s) that are suppressed in the presence of a dominant gland. The MIP procedure seems to be as successful, in the range of 95% to 98%, as more standard approaches (259,260). According to a recent meta-analysis that included more than 12,000 patients, MIP was associated with similar rates of success, disease recurrence, persistence, overall failure, and reoperation (261). The operative time was significantly shorter, with a lower overall complication rate for MIP compared to bilateral neck exploration. In Europe, MIP is being performed with an endoscopic camera, but this does not offer any advantage other than a smaller incision (262,263). Yet another variation on this theme is the use of preoperative sestamibi scanning with an intraoperative gamma probe to help locate enlarged parathyroid glands.
Immediate Postoperative Course
After surgery, biochemical indices return rapidly to normal (52,264). Serum calcium and PTH levels normalize, and urinary calcium excretion falls by as much as 50%. Serum calcium levels no longer fall rapidly into the hypocalcemic range, a situation characteristic of an earlier time when PHPT was a symptomatic disease with overt skeletal involvement. The acute reversal of PHPT was associated with a robust deposition of calcium into the skeleton at a pace that could not be compensated for by supplemental calcium. Thus, postoperative hypocalcemia was routine and was sometimes a serious short-term complication (“hungry bone syndrome”). Occasionally, postoperative hypocalcemia still occurs, especially if preoperative bone turnover markers are markedly elevated or there is concomitant vitamin D deficiency. More typically, however, the early postoperative course is not complicated by symptomatic hypocalcemia.
After successful parathyroid surgery, biochemical indices of the disease return to normal and BMD improves, as mentioned. The capacity of the skeleton to restore itself is seen dramatically in young patients with severe PHPT. Kulak et al (265) reported two patients with osteitis fibrosa cystica who experienced increases in bone density that ranged from 260% to 430% in a period of 3 to 4 years following surgery. Tritos and Hartzband (266) and DiGregorio (267) have made similar observations.
MEDICAL MANAGEMENT
Patients who do not meet any surgical guidelines are often followed without intervention. The most recent guidelines for management of asymptomatic PHPT restated the position that it is reasonable to pursue a nonsurgical course of management for those who do not meet criteria for surgery, at least for a period of years. In those patients who are not going to have parathyroid surgery, the Workshop (137) suggested a set of monitoring steps that are summarized in Table 6. This includes annual measurements of the serum calcium concentration, a calculated creatinine clearance, and regular monitoring of BMD.
Table 6.
Comparison of New and Old Management Guidelines for Patients with Asymptomatic Primary Hyperparathyroidism Who Do Not Undergo Parathyroid Surgery
Ideal medical therapy of PHPT would provide the equivalent to a medical parathyroidectomy. Such an agent would normalize serum calcium and PTH levels as well as urinary calcium excretion, increase BMD and lower fracture risk, and reduce the risk of kidney stones. Unfortunately, no currently available single drug meets all these criteria. The following medications can achieve some of these goals and might be considered in patients not having surgery in whom it is desirable to lower serum or urinary calcium levels or increase BMD.
General Measures
Patients should be instructed to remain well hydrated and to avoid, if possible, medications that can increase serum calcium (e.g. thiazide diuretics). Prolonged immobilization, which can raise the serum calcium concentration further and induce hypercalciuria, should also be avoided.
DIET AND SUPPLEMENTS
Dietary management of PHPT has long been an area of controversy. Many patients are advised to limit their dietary calcium intake because of the hypercalcemia. However, it is well known that low dietary calcium can lead to increased PTH levels in normal individuals (268-270). In patients with PHPT, even though the abnormal PTH tissue is not as sensitive to slight perturbations in the circulating calcium concentration, it is still possible that PTH levels will rise when dietary calcium is tightly restricted. Conversely, diets enriched in calcium could suppress PTH levels in PHPT, as shown by Insogna et al (271). Dietary calcium could also be variably influenced by ambient levels of 1,25-dihydroxyvitamin d. In patients with normal levels of 1,25-dihydroxyvitamin d, Locker et al (272) noted no difference in urinary calcium excretion between those on high (1000 mg/day) and low (500 mg/day) calcium intake diets. On the other hand, in those with elevated levels of 1,25-dihydroxyvitamin d, high calcium diets were associated with worsening hypercalciuria. This observation suggests that dietary calcium intake in patients can be liberalized to 1000 mg/day if 1,25-dihydroxyvitamin d levels are not increased but should be more tightly controlled if 1,25-dihydroxyvitamin d levels are elevated. Although calcium supplements are not specifically recommended in those with PHPT and osteoporosis, small doses do not seem to exacerbate hypercalcemia or hypercalciuria if the diet is deficient (273). Most experts recommend that patients with PHPT who are going to be followed without surgery have an intake of calcium that is consistent with nutritional guidelines for a normal population.
Recent guidelines recommend maintaining 25-hydroxyvitamin D to levels of 21–30 ng/ml with conservative doses of vitamin D (600–1000 IU daily) based on data showing that vitamin D repletion lowers PTH levels (274). Higher levels of vitamin D might be beneficial. A 2014 RCT of cholecalciferol (2,800 IU daily versus placebo) indicated that treatment increased 25-hydroxyvitamin d levels from 20 ng/ml to 37.8 ng/ml, lowered levels of PTH, and increased lumbar spine BMD without having a deleterious effect on serum or urinary calcium levels (275).
PHARMACEUTICALS
Phosphate
Oral phosphate can lower the serum calcium by up to 1 mg/dL (276,277). A complex interplay of mechanisms leads to this moderating effect of oral phosphate. First, calcium absorption falls in the presence of intestinal phosphorus. Second, concomitant increases in serum phosphorus will tend to reduce circulating 1,25-dihydroxyvitamin d levels. Third, phosphate can be an antiresorptive agent. Finally, increased serum phosphorus reciprocally lowers serum calcium. Problems with oral phosphate include limited gastrointestinal tolerance, possible further increase in PTH levels, and the possibility of soft-tissue calcifications after long-term use. It is essentially not used any longer in the management of PHPT.
Estrogens and Selective Estrogen-Receptor Modulators
The earliest studies on the use of estrogen replacement therapy in PHPT date back to the early 1970s. A 0.5 to 1.0 mg/dL reduction in total serum calcium levels in postmenopausal women with PHPT who received estrogen was seen along with a lowering of urinary calcium (278,279). Most studies indicated no change in PTH (279-281). A randomized controlled trial of conjugated estrogen (0.625 mg daily plus medroxyprogesterone 5 mg daily) versus placebo indicated that hormone-replacement therapy effectively increases BMD at all skeletal sites in patients with PHPT, with the greatest increases at the lumbar spine (281). This randomized controlled trials, however, did not confirm the calcium-lowering effect of earlier uncontrolled studies (274). In view of concerns expressed about chronic estrogen use in the Women’s Health Study, estrogen use is not often recommended for medical management of hyperparathyroidism.
Raloxifene, a selective estrogen-receptor modulator, has been studied in PHPT, but the data are sparse. In a short-term (8-week) trial of 18 postmenopausal women, raloxifene (60 mg/day) was associated with a statistically significant although small (0.5 mg/dL) reduction in the serum calcium concentration and in markers of bone turnover (282). No long-term data or data on bone density are available.
Bisphosphonates and Denosumab
Bisphosphonates are conceptually attractive in PHPT because they are antiresorptive agents with an overall effect of reducing bone turnover. Although they do not affect PTH secretion directly, bisphosphonates could reduce serum and urinary calcium levels. Early studies with the first-generation bisphosphonates were disappointing. Etidronate has no effect (283). Clodronate use was associated in several studies with a reduction in serum and urinary calcium (284), but the effect was transient.
Alendronate has been studied most extensively in PHPT. Studies by Rossini et al (285) and Hassani et al (286) were followed by those of Chow et al (287), Parker et al (288), and Kahn et al (289). These studies were all characterized by a randomized, controlled design. Typically, BMD of the lumbar spine and hip regions increases along with reductions in bone turnover markers (Figure 4). Except for the study of Chow et al (287), serum calcium was unchanged. These results suggest that bisphosphonates may be useful in patients with low bone density in whom parathyroid surgery is not to be performed. One small study suggests that denosumab increases BMD in women with PHPT to a greater extent than patients without PHPT but with osteoporosis (290).
Figure 4.
The effect of alendronate on bone mineral density in primary hyperparathyroidism. With alendronate, bone mineral density increases significantly after 1 year, while the placebo group shows no change until it is crossed over to alendronate in year 2. (Modified from reference Khan AA, Bilezikian JP, Kung AWC, et al: Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab 2004;89:3319-3325).
Inhibition of Parathyroid Hormone
The most specific pharmacologic approach to PHPT is to inhibit the synthesis and secretion of PTH from the parathyroid glands, such as those that act on the parathyroid cell calcium-sensing receptor. This G protein–coupled receptor recognizes calcium as its cognate ligand (291-293). When activated by increased extracellular calcium, the calcium-sensing receptor signals the cell via a G protein–transducing pathway to raise the intracellular calcium concentration, which inhibits PTH secretion. Molecules that mimic the effect of extracellular calcium by altering the affinity of calcium for the receptor could activate this receptor and inhibit parathyroid cell function. The phenylalkylamine (R)-N(3-methoxy-a-phenylethyl)-3-(2-chlorophenyl)-1-propylamine (R-568) is one such calcimimetic compound. R-568 was found to increase cytoplasmic calcium and to reduce PTH secretion in vitro, as well as in normal postmenopausal women (294,295). This drug was also shown to inhibit PTH secretion in postmenopausal women with PHPT (296). A second-generation ligand, cinacalcet, has been the subject of more extensive investigations in PHPT and is now approved for the treatment of severe hypercalcemia in PHPT when surgery cannot be pursued. Studies conducted by the authors and their colleagues (297-299) indicate that this drug can reduce the serum calcium concentration to normal in PHPT, but despite normalization of the serum calcium concentration, PTH levels do not return to normal; they do fall by 35% to 50% after administration of the drug. Urinary calcium excretion does not change; serum phosphorus levels increase but are maintained in the lower range of normal; and 1,25-dihydroxyvitamin D levels do not change. The average BMD does not change, even after 5 years of administration of cinacalcet (300). Marcocci et al (299) have shown that cinacalcet is effective in subjects with intractable PHPT. Silverberg et al (301) have shown that cinacalcet reduces calcium levels effectively in inoperable parathyroid carcinoma.
Hydrochlorothiazide
Though avoiding agents that exacerbate hypercalcemia is generally recommended in patients with PHPT, a recent study has recently reexamined the role of thiazides in patients with PHPT. This retrospective analysis of 72 patients suggested that thiazides might not increase serum levels of calcium in PHPT as they can do in normal individuals. Hydrochlorothiazide (12.5–50 mg daily for 3.1 years on average) was associated with a decrease in urinary calcium excretion but no change in serum levels of calcium (302). Smaller and cross-sectional studies have suggested similar results, although it is unclear if hydrochlorothiazide reduces the risk of nephrolithiasis (303,304). Given the heterogeneity of doses used, and the absence of larger, (preferably) randomized trial data, recommending thiazide use routinely in PHPT is premature. However, thiazides could be considered in those who refuse surgery or are poor surgical candidates but at high risk of nephrolithiasis in whom the benefit is thought to outweigh the risk as long as serum levels of calcium are monitored regularly.
TREATMENT OF PARATHYROID CANCER
Surgery is the only effective therapy currently available for parathyroid cancer. The greatest chance for cure occurs with the first operation. After the disease recurs, cure is unlikely, although the disease may smolder for many years thereafter. The tumor is not radiosensitive, although there are isolated reports of tumor regression with localized radiation therapy. Traditional chemotherapeutic agents have not been useful. When metastasis occurs, isolated removal is an option, especially if only one or two nodules are found in the lung. Such isolated metastasectomies are never curative but they can lead to prolonged remissions, sometimes lasting for several years. Similarly, local debulking of tumor tissue in the neck can be palliative, although malignant tissue is invariably left behind.
Chemotherapy has had a very limited role in this disease. Bradwell and Harvey (305) have attempted an immunotherapeutic approach by injecting a patient who had severe hypercalcemia resulting from parathyroid cancer with antibodies raised to their own circulating PTH. Coincident with a rise in antibody titer to PTH, previously refractory hypercalcemia fell impressively. A more recent report (306) provided evidence of the antitumor effect in a single case of PTH immunization in metastatic parathyroid cancer.
Attention has been focused instead on control of hypercalcemia. Intravenous bisphosphonates have been used to treat severe hypercalcemia. Although efficacious in the short term, they do not provide an approach that allows long-term outpatient normalization of serum calcium levels. Denosumab has more recently been reported to treat resistant hypercalcemia in patients with parathyroid carcinoma (307,308).
The calcimimetic agents offer a newer approach. Our group (309) reported on a single patient treated with the calcimimetic, R-568; despite widely metastatic disease, the patient showed serum calcium levels that were maintained within a range that allowed him to return to normal functioning for nearly 2 years. A wider experience by Silverberg et al (301) showed that cinacalcet is useful in the management of parathyroid cancer. The U.S. Food and Drug Administration approved this calcimimetic for the treatment of hypercalcemia in patients with parathyroid cancer. Use of this agent in parathyroid cancer led to improvement in serum calcium levels and a decrease in symptoms of nausea, vomiting, and mental lethargy, which are common concomitants of marked hypercalcemia. There are no data on the effect of cinacalcet on tumor growth in parathyroid cancer. Similarly, there are no data on the use of a combination of cinacalcet and bisphosphonates in parathyroid cancer, the former used to decrease PTH secretion from the cancer, and the latter used to decrease release of calcium from the skeleton. Cinacalcet offers an option for control of intractable hypercalcemia when surgical removal of cancerous tissue is no longer an option.
SUMMARY
This chapter has presented a comprehensive summary of the modern-day presentation of PHPT. Typically, an asymptomatic disorder in countries that are economically more developed, the disorder’s presentation has raised issues regarding the extent to which such patients may nevertheless show subclinical target organ involvement, who should be recommended for parathyroid surgery, who can be safely followed without surgical intervention, as well as questions regarding the role of medical therapy. Questions about the natural history and pathophysiology of the disorder continue to be of great interest. Inasmuch as this disorder continues to evolve, it is clear that additional careful studies are required continually to gain new insights into this disease.
ACKNOWLEDGEMENTS
This work was supported in part by National Institutes of Health grants NIDDK 32333, DK084986, RR 00645, and R21DK104105. With permission, this chapter is adapted from:
Walker MD, Bilezikian JP. Primary hyperparathyroidism. IN: Endocrinology, 8th edition (Jameson JL, Robertson P, eds) Saunders, Elsevier (in press), 2021.
REFERENCES
- 1.
- Albright F, Bauer W, Claflin D, Cockrill JR. STUDIES IN PARATHYROID PHYSIOLOGY: III. The Effect of Phosphate Ingestion in Clinical Hyperparathyroidism. J Clin Invest. 1932;11:411–435. [PMC free article: PMC435825] [PubMed: 16694050]
- 2.
- Albright F, Aub J, Bauer W. Hyperparathyroidism: common and polymorphic condition as illustrated by seventeen proven cases in one clinic. JAMA. 1934;102:1276.
- 3.
- Bauer W, Federman DD. Hyperparathyroidism epitomized: the case of Captain Charles E. Martell. Metabolism. 1962;11:21–29. [PubMed: 13865885]
- 4.
- Silverberg S, Bilezikian JP. Clinical presentation of primary hyperparathyroidism in the United States. New York: Academic Press.
- 5.
- Heath H 3rd, Hodgson SF, Kennedy MA. Primary hyperparathyroidism. Incidence, morbidity, and potential economic impact in a community. N Engl J Med. 1980;302:189–193. [PubMed: 7350459]
- 6.
- Mundy GR, Cove DH, Fisken R. Primary hyperparathyroidism: changes in the pattern of clinical presentation. Lancet. 1980;1:1317–1320. [PubMed: 6104128]
- 7.
- Scholz DA, Purnell DC. Asymptomatic primary hyperparathyroidism. 10-year prospective study. Mayo Clin Proc. 1981;56:473–478. [PubMed: 7266058]
- 8.
- Silverberg SJ, Bilezikian JP. The diagnosis and management of asymptomatic primary hyperparathyroidism. Nat Clin Pract Endocrinol Metab. 2006;2:494–503. [PubMed: 16957763]
- 9.
- Misiorowski W, Czajka-Oraniec I, Kochman M, Zgliczynski W, Bilezikian JP. Osteitis fibrosa cystica-a forgotten radiological feature of primary hyperparathyroidism. Endocrine. 2017;58:380–385. [PMC free article: PMC5671544] [PubMed: 28900835]
- 10.
- Bendz H, Sjodin I, Toss G, Berglund K. Hyperparathyroidism and long-term lithium therapy--a cross-sectional study and the effect of lithium withdrawal. J Intern Med. 1996;240:357–365. [PubMed: 9010382]
- 11.
- Rao SD, Frame B, Miller MJ, Kleerekoper M, Block MA, Parfitt AM. Hyperparathyroidism following head and neck irradiation. Arch Intern Med. 1980;140:205–207. [PubMed: 7352815]
- 12.
- Nordenstrom J, Strigard K, Perbeck L, Willems J, Bagedahl-Strindlund M, Linder J. Hyperparathyroidism associated with treatment of manic-depressive disorders by lithium. Eur J Surg. 1992;158:207–211. [PubMed: 1352133]
- 13.
- Vaidya A, Curhan GC, Paik JM, Wang M, Taylor EN. Body Size and the Risk of Primary Hyperparathyroidism in Women: A Cohort Study. J Bone Miner Res. 2017;32:1900–1906. [PMC free article: PMC5555811] [PubMed: 28488734]
- 14.
- Paik JM, Curhan GC, Taylor EN. Calcium intake and risk of primary hyperparathyroidism in women: prospective cohort study. BMJ. 2012;345:e6390. [PMC free article: PMC3475985] [PubMed: 23080543]
- 15.
- Golden SH, Robinson KA, Saldanha I, Anton B, Ladenson PW. Clinical review: Prevalence and incidence of endocrine and metabolic disorders in the United States: a comprehensive review. J Clin Endocrinol Metab. 2009;94:1853–1878. [PMC free article: PMC5393375] [PubMed: 19494161]
- 16.
- Attie JN, Bock G, Auguste LJ. Multiple parathyroid adenomas: report of thirty-three cases. Surgery. 1990;108:1014–1019. [PubMed: 2247825]
- 17.
- Barczynski M, Branstrom R, Dionigi G, Mihai R. Sporadic multiple parathyroid gland disease--a consensus report of the European Society of Endocrine Surgeons (ESES). Langenbecks Arch Surg. 2015;400:887–905. [PMC free article: PMC4747992] [PubMed: 26542689]
- 18.
- Bilezikian JP, Bandeira L, Khan A, Cusano NE. Hyperparathyroidism. Lancet. 2018;391:168–178. [PubMed: 28923463]
- 19.
- Nudelman IL, Deutsch AA, Reiss R. Primary hyperparathyroidism due to mediastinal parathyroid adenoma. Int Surg. 1987;72:104–108. [PubMed: 3610535]
- 20.
- Sloane JA, Moody HC. Parathyroid adenoma in submucosa of esophagus. Arch Pathol Lab Med. 1978;102:242–243. [PubMed: 580560]
- 21.
- Joseph MP, Nadol JB, Pilch BZ, Goodman ML. Ectopic parathyroid tissue in the hypopharyngeal mucosa (pyriform sinus). Head Neck Surg. 1982;5:70–74. [PubMed: 7174345]
- 22.
- Gilmour JR. Some developmental abnormalities of the thymus and parathyroids. J Pathol Bacteriol. 1941;52:213–218.
- 23.
- Fleischer J, Becker C, Hamele-Bena D, Breen TL, Silverberg SJ. Oxyphil parathyroid adenoma: a malignant presentation of a benign disease. J Clin Endocrinol Metab. 2004;89:5948–5951. [PubMed: 15579742]
- 24.
- Jain M, Krasne DL, Singer FR, Giuliano AE. Recurrent primary hyperparathyroidism due to Type 1 parathyromatosis. Endocrine. 2017;55:643–650. [PubMed: 27743301]
- 25.
- Wermers RA, Khosla S, Atkinson EJ, Hodgson SF, O'Fallon WM, Melton LJ 3rd. The rise and fall of primary hyperparathyroidism: a population-based study in Rochester, Minnesota, 1965-1992. Ann Intern Med. 1997;126:433–440. [PubMed: 9072928]
- 26.
- Melton LJ 3rd. The epidemiology of primary hyperparathyroidism in North America. J Bone Miner Res. 2002;17 Suppl 2:N12–17. [PubMed: 12412772]
- 27.
- Wermers RA, Khosla S, Atkinson EJ, Achenbach SJ, Oberg AL, Grant CS, Melton LJ 3rd. Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993-2001: an update on the changing epidemiology of the disease. J Bone Miner Res. 2006;21:171–177. [PubMed: 16355286]
- 28.
- Griebeler ML, Kearns AE, Ryu E, Hathcock MA, Melton LJ 3rd, Wermers RA. Secular trends in the incidence of primary hyperparathyroidism over five decades (1965-2010). Bone. 2015;73:1–7. [PMC free article: PMC4445941] [PubMed: 25497786]
- 29.
- Yeh MW, Ituarte PH, Zhou HC, Nishimoto S, Liu IL, Harari A, Haigh PI, Adams AL. Incidence and prevalence of primary hyperparathyroidism in a racially mixed population. J Clin Endocrinol Metab. 2013;98:1122–1129. [PMC free article: PMC3590475] [PubMed: 23418315]
- 30.
- Lowe H, McMahon DJ, Rubin MR, Bilezikian JP, Silverberg SJ. Normocalcemic primary hyperparathyroidism: further characterization of a new clinical phenotype. J Clin Endocrinol Metab. 2007;92:3001–3005. [PubMed: 17536001]
- 31.
- Cusano NE, Maalouf NM, Wang PY, Zhang C, Cremers SC, Haney EM, Bauer DC, Orwoll ES, Bilezikian JP. Normocalcemic hyperparathyroidism and hypoparathyroidism in two community-based nonreferral populations. J Clin Endocrinol Metab. 2013;98:2734–2741. [PMC free article: PMC3701271] [PubMed: 23690312]
- 32.
- Schini M, Jacques RM, Oakes E, Peel NFA, Walsh JS, Eastell R. Normocalcemic Hyperparathyroidism: Study of its Prevalence and Natural History. J Clin Endocrinol Metab. 2020:105. [PMC free article: PMC7069345] [PubMed: 32072184]
- 33.
- Jacobs TP, Bilezikian JP. Clinical review: Rare causes of hypercalcemia. J Clin Endocrinol Metab. 2005;90:6316–6322. [PubMed: 16131579]
- 34.
- Eastell R, Arnold A, Brandi ML, Brown EM, D'Amour P, Hanley DA, Rao DS, Rubin MR, Goltzman D, Silverberg SJ, Marx SJ, Peacock M, Mosekilde L, Bouillon R, Lewiecki EM. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the third international workshop. J Clin Endocrinol Metab. 2009;94:340–350. [PubMed: 19193909]
- 35.
- Nesbit MA, Hannan FM, Howles SA, Babinsky VN, Head RA, Cranston T, Rust N, Hobbs MR, Heath H 3rd, Thakker RV. Mutations affecting G-protein subunit alpha11 in hypercalcemia and hypocalcemia. N Engl J Med. 2013;368:2476–2486. [PMC free article: PMC3773604] [PubMed: 23802516]
- 36.
- Nesbit MA, Hannan FM, Howles SA, Reed AA, Cranston T, Thakker CE, Gregory L, Rimmer AJ, Rust N, Graham U, Morrison PJ, Hunter SJ, Whyte MP, McVean G, Buck D, Thakker RV. Mutations in AP2S1 cause familial hypocalciuric hypercalcemia type 3. Nat Genet. 2013;45:93–97. [PMC free article: PMC3605788] [PubMed: 23222959]
- 37.
- Souberbielle JC, Cormier C, Kindermans C, Gao P, Cantor T, Forette F, Baulieu EE. Vitamin D status and redefining serum parathyroid hormone reference range in the elderly. J Clin Endocrinol Metab. 2001;86:3086–3090. [PubMed: 11443171]
- 38.
- Walker MD. Cong E, Lee JA, Kepley A, Zhang C, McMahon DJ, Silverberg SJ. Vitamin D in Primary Hyperparathyroidism: Effects on Clinical, Biochemical, and Densitometric Presentation. J Clin Endocrinol Metab. 2015;100:3443–3451. [PMC free article: PMC4570160] [PubMed: 26079779]
- 39.
- Tassone F, Gianotti L, Emmolo I, Ghio M, Borretta G. Glomerular filtration rate and parathyroid hormone secretion in primary hyperparathyroidism. J Clin Endocrinol Metab. 2009;94:4458–4461. [PubMed: 19808852]
- 40.
- Lepage R, Roy L, Brossard JH, Rousseau L, Dorais C, Lazure C, D'Amour P. A non-(1-84) circulating parathyroid hormone (PTH) fragment interferes significantly with intact PTH commercial assay measurements in uremic samples. Clin Chem. 1998;44:805–809. [PubMed: 9554492]
- 41.
- Quarles LD, Lobaugh B, Murphy G. Intact parathyroid hormone overestimates the presence and severity of parathyroid-mediated osseous abnormalities in uremia. J Clin Endocrinol Metab. 1992;75:145–150. [PubMed: 1619003]
- 42.
- John MR, Goodman WG, Gao P, Cantor TL, Salusky IB, Juppner H. A novel immunoradiometric assay detects full-length human PTH but not amino-terminally truncated fragments: implications for PTH measurements in renal failure. J Clin Endocrinol Metab. 1999;84:4287–4290. [PubMed: 10566687]
- 43.
- Gao P, Scheibel S, D'Amour P, John MR, Rao SD, Schmidt-Gayk H, Cantor TL. Development of a novel immunoradiometric assay exclusively for biologically active whole parathyroid hormone 1-84: implications for improvement of accurate assessment of parathyroid function. J Bone Miner Res. 2001;16:605–614. [PubMed: 11315988]
- 44.
- Slatopolsky E, Finch J, Clay P, Martin D, Sicard G, Singer G, Gao P, Cantor T, Dusso A. A novel mechanism for skeletal resistance in uremia. Kidney Int. 2000;58:753–761. [PubMed: 10916099]
- 45.
- Silverberg SJ, Gao P, Brown I, LoGerfo P, Cantor TL, Bilezikian JP. Clinical utility of an immunoradiometric assay for parathyroid hormone (1-84) in primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:4725–4730. [PubMed: 14557447]
- 46.
- D'Amour P, Brossard JH, Rousseau L, Roy L, Gao P, Cantor T. Amino-terminal form of parathyroid hormone (PTH) with immunologic similarities to hPTH(1-84) is overproduced in primary and secondary hyperparathyroidism. Clin Chem. 2003;49:2037–2044. [PubMed: 14633875]
- 47.
- Rubin MR, Silverberg SJ, D'Amour P, Brossard JH, Rousseau L, Sliney J Jr, Cantor T, Bilezikian JP. An N-terminal molecular form of parathyroid hormone (PTH) distinct from hPTH(1 84) is overproduced in parathyroid carcinoma. Clin Chem. 2007;53:1470–1476. [PubMed: 17599957]
- 48.
- Silverberg SJ, Shane E, Dempster DW, Bilezikian JP. The effects of vitamin D insufficiency in patients with primary hyperparathyroidism. Am J Med. 1999;107:561–567. [PubMed: 10625024]
- 49.
- Hagag P, Revet-Zak I, Hod N, Horne T, Rapoport MJ, Weiss M. Diagnosis of normocalcemic hyperparathyroidism by oral calcium loading test. J Endocrinol Invest. 2003;26:327–332. [PubMed: 12841540]
- 50.
- Silverberg SJ, Bilezikian JP. "Incipient" primary hyperparathyroidism: a "forme fruste" of an old disease. J Clin Endocrinol Metab. 2003;88:5348–5352. [PubMed: 14602772]
- 51.
- Vieth R, Bayley TA, Walfish PG, Rosen IB, Pollard A. Relevance of vitamin D metabolite concentrations in supporting the diagnosis of primary hyperparathyroidism. Surgery. 1991;110:1043–1046. [PubMed: 1745973]
- 52.
- Silverberg SJ, Shane E, Jacobs TP, Siris E, Bilezikian JP. A 10-year prospective study of primary hyperparathyroidism with or without parathyroid surgery. N Engl J Med. 1999;341:1249–1255. [PubMed: 10528034]
- 53.
- Lumb GA, Stanbury SW. Parathyroid function in human vitamin D deficiency and vitamin D deficiency in primary hyperparathyroidism. Am J Med. 1974;56:833–839. [PubMed: 4831321]
- 54.
- Liu JM, Cusano NE, Silva BC, Zhao L, He XY, Tao B, Sun LH, Zhao HY, Fan WW, Romano ME, Ning G, Bilezikian JP. Primary Hyperparathyroidism: A Tale of Two Cities Revisited - New York and Shanghai. Bone research. 2013;1:162–169. [PMC free article: PMC4472111] [PubMed: 26273500]
- 55.
- Malabu UH, Founda MA. Primary hyperparathyroidism in Saudi Arabia: a review of 46 cases. The Medical journal of Malaysia. 2007;62:394–397. [PubMed: 18705474]
- 56.
- Paruk IM, Esterhuizen TM, Maharaj S, Pirie FJ, Motala AA. Characteristics, management and outcome of primary hyperparathyroidism in South Africa: a single-centre experience. Postgraduate medical journal. 2013;89:626–631. [PubMed: 23824345]
- 57.
- Shah VN, Bhadada S, Bhansali A, Behera A, Mittal BR. Changes in clinical & biochemical presentations of primary hyperparathyroidism in India over a period of 20 years. Indian J Med Res. 2014;139:694–699. [PMC free article: PMC4140033] [PubMed: 25027078]
- 58.
- Zhao L, Liu JM, He XY, Zhao HY, Sun LH, Tao B, Zhang MJ, Chen X, Wang WQ, Ning G. The changing clinical patterns of primary hyperparathyroidism in Chinese patients: data from 2000 to 2010 in a single clinical center. J Clin Endocrinol Metab. 2013;98:721–728. [PubMed: 23365127]
- 59.
- Castellano E, Attanasio R, Boriano A, Pellegrino M, Garino F, Gianotti L, Borretta G. Sex Difference in the Clinical Presentation of Primary Hyperparathyroidism: Influence of Menopausal Status. J Clin Endocrinol Metab. 2017;102:4148–4152. [PubMed: 28938410]
- 60.
- Pradeep PV, Mishra A, Agarwal G, Agarwal A, Verma AK, Mishra SK. Long-term outcome after parathyroidectomy in patients with advanced primary hyperparathyroidism and associated vitamin D deficiency. World J Surg. 2008;32:829–835. [PubMed: 18204946]
- 61.
- Bandeira F, Cassibba S. Hyperparathyroidism and Bone Health. Current rheumatology reports. 2015;17:48. [PubMed: 26105042]
- 62.
- Harinarayan CV, Gupta N, Kochupillai N. Vitamin D status in primary hyperparathyroidism in India. Clin Endocrinol (Oxf). 1995;43:351–358. [PubMed: 7586606]
- 63.
- Meng X, Xing X, Liu S. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 1994;16:13–19. [The diagnosis of primary hyperparathyroidism--analysis of 134 cases] [PubMed: 7954961]
- 64.
- Luong KV, Nguyen LT. Coexisting hyperthyroidism and primary hyperparathyroidism with vitamin D-deficient osteomalacia in a Vietnamese immigrant. Endocr Pract. 1996;2:250–254. [PubMed: 15251523]
- 65.
- Usta A, Alhan E, Cinel A, Turkyilmaz S, Erem C. A 20-year study on 190 patients with primary hyperparathyroidism in a developing country: Turkey experience. Int Surg. 2015;100:648–655. [PMC free article: PMC4400933] [PubMed: 25875546]
- 66.
- Silverberg SJ, Bilezikian JP. Primary Hyperparathyroidism. Primer on Metabolic Bone Diseases and Disorders of Mineral Metbolism. Washington D. C.: American Society for Bone and Mineral Research:302-306.
- 67.
- Silverberg SJ. Non-classical target organs in primary hyperparathyroidism. J Bone Miner Res. 2002;17 Suppl 2:N117–125. [PubMed: 12412788]
- 68.
- Ringe JD. Reversible hypertension in primary hyperparathyroidism--pre- and posteroperative blood pressure in 75 cases. Klin Wochenschr. 1984;62:465–469. [PubMed: 6748560]
- 69.
- Broulik PD, Horky K, Pacovsky V. Blood pressure in patients with primary hyperparathyroidism before and after parathyroidectomy. Exp Clin Endocrinol. 1985;86:346–352. [PubMed: 4092745]
- 70.
- Rapado A. Arterial hypertension and primary hyperparathyroidism. Incidence and follow-up after parathyroidectomy. Am J Nephrol. 1986;6 Suppl 1:49–50. [PubMed: 3826152]
- 71.
- Bilezikian JP, Connor TB, Aptekar R, Freijanes J, Aurbach GD, Pachas WN, Wells SA, Decker JL. Pseudogout after parathyroidectomy. Lancet. 1973;1:445–446. [PubMed: 4120363]
- 72.
- Geelhoed GW, Kelly TR. Pseudogout as a clue and complication in primary hyperparathyroidism. Surgery. 1989;106:1036–1041, discussion 1041-1032. [PubMed: 2588110]
- 73.
- Ludvigsson JF, Kampe O, Lebwohl B, Green PH, Silverberg SJ, Ekbom A. Primary hyperparathyroidism and celiac disease: a population-based cohort study. J Clin Endocrinol Metab. 2012;97:897–904. [PMC free article: PMC3319223] [PubMed: 22238405]
- 74.
- Silverberg SJ, Bilezikian JP. Primary Hyperparathyroidism. In: Seibel MJ, Robins SP, Bilezikian JP, eds. Dynamics of bone and cartilage metabolism. San Diego: Elsevier:767–778.
- 75.
- Silverberg SJ, Gartenberg F, Jacobs TP, Shane E, Siris E, Staron RB, Bilezikian JP. Longitudinal measurements of bone density and biochemical indices in untreated primary hyperparathyroidism. J Clin Endocrinol Metab. 1995;80:723–728. [PubMed: 7883823]
- 76.
- Seibel MJ. Molecular markers of bone metabolism in primary hyperparathyroidism. In: Bilezikian JP, ed. The parathyroids: basic and clinical concepts. New York: Academic Press:399-410.
- 77.
- Price PA, Parthemore JG, Deftos LJ. New biochemical marker for bone metabolism. Measurement by radioimmunoassay of bone GLA protein in the plasma of normal subjects and patients with bone disease. J Clin Invest. 1980;66:878–883. [PMC free article: PMC371521] [PubMed: 6968755]
- 78.
- Deftos LJ, Parthemore JG, Price PA. Changes in plasma bone GLA protein during treatment of bone disease. Calcif Tissue Int. 1982;34:121–124. [PubMed: 6805917]
- 79.
- Ebeling PR, Peterson JM, Riggs BL. Utility of type I procollagen propeptide assays for assessing abnormalities in metabolic bone diseases. J Bone Miner Res. 1992;7:1243–1250. [PubMed: 1466250]
- 80.
- van Lierop AH, Witteveen JE, Hamdy NA, Papapoulos SE. Patients with primary hyperparathyroidism have lower circulating sclerostin levels than euparathyroid controls. Eur J Endocrinol. 2010;163:833–837. [PubMed: 20817762]
- 81.
- Costa AG, Cremers S, Rubin MR, McMahon DJ, Sliney J Jr, Lazaretti-Castro M, Silverberg SJ, Bilezikian JP. Circulating sclerostin in disorders of parathyroid gland function. J Clin Endocrinol Metab. 2011;96:3804–3810. [PMC free article: PMC3232608] [PubMed: 21937621]
- 82.
- Ardawi MS, Al-Sibiany AM, Bakhsh TM, Rouzi AA, Qari MH. Decreased serum sclerostin levels in patients with primary hyperparathyroidism: a cross-sectional and a longitudinal study. Osteoporos Int. 2011 [PubMed: 22041864]
- 83.
- Seibel MJ, Gartenberg F, Silverberg SJ, Ratcliffe A, Robins SP, Bilezikian JP. Urinary hydroxypyridinium cross-links of collagen in primary hyperparathyroidism. J Clin Endocrinol Metab. 1992;74:481–486. [PubMed: 1740480]
- 84.
- Seibel MJ, Woitge HW, Pecherstorfer M, Karmatschek M, Horn E, Ludwig H, Armbruster FP, Ziegler R. Serum immunoreactive bone sialoprotein as a new marker of bone turnover in metabolic and malignant bone disease. J Clin Endocrinol Metab. 1996;81:3289–3294. [PubMed: 8784085]
- 85.
- Guo CY, Thomas WE, al-Dehaimi AW, Assiri AM, Eastell R. Longitudinal changes in bone mineral density and bone turnover in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:3487–3491. [PubMed: 8855790]
- 86.
- Tanaka Y, Funahashi H, Imai T, Tominaga Y, Takagi H. Parathyroid function and bone metabolic markers in primary and secondary hyperparathyroidism. Semin Surg Oncol. 1997;13:125–133. [PubMed: 9088068]
- 87.
- Minisola S, Romagnoli E, Scarnecchia L, Rosso R, Pacitti MT, Scarda A, Mazzuoli G. Serum carboxy-terminal propeptide of human type I procollagen in patients with primary hyperparathyroidism: studies in basal conditions and after parathyroid surgery. Eur J Endocrinol. 1994;130:587–591. [PubMed: 8205259]
- 88.
- Ohe MN, Bonansea TCP, Santos RO, Neves MCD, Santos LM, Rosano M, Kunii IS, Castro ML, Vieira JGH. Prediction of bone mass changes after successful parathyroidectomy using biochemical markers of bone metabolism in primary hyperparathyroidism: is it clinically useful? Arch Endocrinol Metab. 2019;63:394–401. [PMC free article: PMC10528649] [PubMed: 31365627]
- 89.
- NIH conference. Diagnosis and management of asymptomatic primary hyperparathyroidism: consensus development conference statement. Ann Intern Med. 1991;114:593–597. [PubMed: 1900404]
- 90.
- Silverberg SJ, Shane E, de la Cruz L, Dempster DW, Feldman F, Seldin D, Jacobs TP, Siris ES, Cafferty M, Parisien MV, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res. 1989;4:283–291. [PubMed: 2763869]
- 91.
- Bilezikian JP, Silverberg SJ, Shane E, Parisien M, Dempster DW. Characterization and evaluation of asymptomatic primary hyperparathyroidism. J Bone Miner Res. 1991;6 Suppl 2:S85–89. [PubMed: 1662460]
- 92.
- Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R. Anabolic actions of parathyroid hormone on bone. Endocr Rev. 1993;14:690–709. [PubMed: 8119233]
- 93.
- Bilezikian JP, Rubin MR, Finkelstein JS. Parathyroid hormone as an anabolic therapy for women and men. J Endocrinol Invest. 2005;28:41–49. [PubMed: 16329201]
- 94.
- Canalis E, Giustina A, Bilezikian JP. Mechanisms of anabolic therapies for osteoporosis. N Engl J Med. 2007;357:905–916. [PubMed: 17761594]
- 95.
- Silverberg SJ, Locker FG, Bilezikian JP. Vertebral osteopenia: a new indication for surgery in primary hyperparathyroidism. J Clin Endocrinol Metab. 1996;81:4007–4012. [PubMed: 8923852]
- 96.
- Walker MD, Saeed I, Lee JA, Zhang C, Hans D, Lang T, Silverberg SJ. Effect of concomitant vitamin D deficiency or insufficiency on lumbar spine volumetric bone mineral density and trabecular bone score in primary hyperparathyroidism. Osteoporos Int. 2016;27:3063–3071. [PMC free article: PMC5555733] [PubMed: 27198233]
- 97.
- Walker MD, Nishiyama KK, Zhou B, Cong E, Wang J, Lee JA, Kepley A, Zhang C, Guo XE, Silverberg SJ. Effect of Low Vitamin D on Volumetric Bone Mineral Density, Bone Microarchitecture, and Stiffness in Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2016;101:905–913. [PMC free article: PMC4803169] [PubMed: 26745256]
- 98.
- Parisien M, Silverberg SJ, Shane E, de la Cruz L, Lindsay R, Bilezikian JP, Dempster DW. The histomorphometry of bone in primary hyperparathyroidism: preservation of cancellous bone structure. J Clin Endocrinol Metab. 1990;70:930–938. [PubMed: 2318948]
- 99.
- Parfitt AM. Accelerated cortical bone loss: primary and secondary hyperparathyroidism. In: Uhthoff H, Stahl E, eds. Current Concepts in Bone Fragility. New York: Mary Ann Liebert:7-14.
- 100.
- van Doorn L, Lips P, Netelenbos JC, Hackeng WH. Bone histomorphometry and serum concentrations of intact parathyroid hormone (PTH(1-84)) in patients with primary hyperparathyroidism. Bone Miner. 1993;23:233–242. [PubMed: 8148668]
- 101.
- Dempster DW, Parisien M, Silverberg SJ, Liang XG, Schnitzer M, Shen V, Shane E, Kimmel DB, Recker R, Lindsay R, Bilezikian JP. On the mechanism of cancellous bone preservation in postmenopausal women with mild primary hyperparathyroidism. J Clin Endocrinol Metab. 1999;84:1562–1566. [PubMed: 10323380]
- 102.
- Parisien M, Cosman F, Mellish RW, Schnitzer M, Nieves J, Silverberg SJ, Shane E, Kimmel D, Recker RR, Bilezikian JP, et al. Bone structure in postmenopausal hyperparathyroid, osteoporotic, and normal women. J Bone Miner Res. 1995;10:1393–1399. [PubMed: 7502712]
- 103.
- Parisien M, Mellish RW, Silverberg SJ, Shane E, Lindsay R, Bilezikian JP, Dempster DW. Maintenance of cancellous bone connectivity in primary hyperparathyroidism: trabecular strut analysis. J Bone Miner Res. 1992;7:913–919. [PubMed: 1442205]
- 104.
- Lindsay R, Nieves J, Formica C, Henneman E, Woelfert L, Shen V, Dempster D, Cosman F. Randomised controlled study of effect of parathyroid hormone on vertebral-bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;350:550–555. [PubMed: 9284777]
- 105.
- Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, Shane E, Plavetic K, Muller R, Bilezikian J, Lindsay R. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res. 2001;16:1846–1853. [PubMed: 11585349]
- 106.
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, Wang O, Mitlak BH. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–1441. [PubMed: 11346808]
- 107.
- Dempster DW, Muller R, Zhou H, Kohler T, Shane E, Parisien M, Silverberg SJ, Bilezikian JP. Preserved three-dimensional cancellous bone structure in mild primary hyperparathyroidism. Bone. 2007;41:19–24. [PMC free article: PMC2721277] [PubMed: 17490921]
- 108.
- Roschger P, Dempster DW, Zhou H, Paschalis EP, Silverberg SJ, Shane E, Bilezikian JP, Klaushofer K. New observations on bone quality in mild primary hyperparathyroidism as determined by quantitative backscattered electron imaging. J Bone Miner Res. 2007;22:717–723. [PubMed: 17266396]
- 109.
- Zoehrer R, Dempster DW, Bilezikian JP, Zhou H, Silverberg SJ, Shane E, Roschger P, Paschalis EP, Klaushofer K. Bone quality determined by Fourier transform infrared imaging analysis in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2008;93:3484–3489. [PMC free article: PMC2567864] [PubMed: 18593769]
- 110.
- Bilezikian JP. Bone strength in primary hyperparathyroidism. Osteoporos Int. 2003;14 Suppl 5:S113–115. [PubMed: 14504715]
- 111.
- Hans D, Goertzen AL, Krieg MA, Leslie WD. Bone microarchitecture assessed by TBS predicts osteoporotic fractures independent of bone density: the Manitoba study. J Bone Miner Res. 2011;26:2762–2769. [PubMed: 21887701]
- 112.
- Silva BC, Boutroy S, Zhang C, McMahon DJ, Zhou B, Wang J, Udesky J, Cremers S, Sarquis M, Guo XD, Hans D, Bilezikian JP. Trabecular bone score (TBS)--a novel method to evaluate bone microarchitectural texture in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2013;98:1963–1970. [PMC free article: PMC3644593] [PubMed: 23526463]
- 113.
- Stein EM, Silva BC, Boutroy S, Zhou B, Wang J, Udesky J, Zhang C, McMahon DJ, Romano M, Dworakowski E, Costa AG, Cusano N, Irani D, Cremers S, Shane E, Guo XE, Bilezikian JP. Primary hyperparathyroidism is associated with abnormal cortical and trabecular microstructure and reduced bone stiffness in postmenopausal women. J Bone Miner Res. 2013;28:1029–1040. [PMC free article: PMC3631282] [PubMed: 23225022]
- 114.
- Hansen S, Hauge EM, Rasmussen L, Jensen JE, Brixen K. Parathyroidectomy improves bone geometry and microarchitecture in female patients with primary hyperparathyroidism. A 1-year prospective controlled study using high resolution peripheral quantitative computed tomography. J Bone Miner Res. 2012 [PubMed: 22228118]
- 115.
- Vu TD, Wang XF, Wang Q, Cusano NE, Irani D, Silva BC, Ghasem-Zadeh A, Udesky J, Romano ME, Zebaze R, Jerums G, Boutroy S, Bilezikian JP, Seeman E. New insights into the effects of primary hyperparathyroidism on the cortical and trabecular compartments of bone. Bone. 2013;55:57–63. [PMC free article: PMC4308951] [PubMed: 23541782]
- 116.
- Stein EM, Silva BC, Boutroy S, Zhou B, Wang J, Udesky J, Zhang C, McMahon DJ, Romano M, Dworakowski E, Costa AG, Cusano N, Irani D, Cremers S, Shane E, Guo XE, Bilezikian JP. Primary hyperparathyroidism is associated with abnormal cortical and trabecular microstructure and reduced bone stiffness in postmenopausal women. J Bone Miner Res. 2012 [PMC free article: PMC3631282] [PubMed: 23225022]
- 117.
- Khosla S, Melton LJ 3rd, Wermers RA, Crowson CS, O'Fallon W, Riggs B. Primary hyperparathyroidism and the risk of fracture: a population-based study. J Bone Miner Res. 1999;14:1700–1707. [PubMed: 10491217]
- 118.
- Vignali E, Viccica G, Diacinti D, Cetani F, Cianferotti L, Ambrogini E, Banti C, Del Fiacco R, Bilezikian JP, Pinchera A, Marcocci C. Morphometric vertebral fractures in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2009;94:2306–2312. [PMC free article: PMC3214277] [PubMed: 19401378]
- 119.
- Mosekilde L. Primary hyperparathyroidism and the skeleton. Clin Endocrinol (Oxf). 2008;69:1–19. [PubMed: 18167138]
- 120.
- De Geronimo S, Romagnoli E, Diacinti D, D'Erasmo E, Minisola S. The risk of fractures in postmenopausal women with primary hyperparathyroidism. Eur J Endocrinol. 2006;155:415–420. [PubMed: 16914595]
- 121.
- Dauphine RT, Riggs BL, Scholz DA. Back pain and vertebral crush fractures: an unemphasized mode of presentation for primary hyperparathyroidism. Ann Intern Med. 1975;83:365–367. [PubMed: 126654]
- 122.
- Kochersberger G, Buckley NJ, Leight GS, Martinez S, Studenski S, Vogler J, Lyles KW. What is the clinical significance of bone loss in primary hyperparathyroidism? Arch Intern Med. 1987;147:1951–1953. [PubMed: 3675096]
- 123.
- Wilson RJ, Rao S, Ellis B, Kleerekoper M, Parfitt AM. Mild asymptomatic primary hyperparathyroidism is not a risk factor for vertebral fractures. Ann Intern Med. 1988;109:959–962. [PubMed: 3195878]
- 124.
- Liu M, Williams J, Kuo J, Lee JA, Silverberg SJ, Walker MD. Risk factors for vertebral fracture in primary hyperparathyroidism. Endocrine. 2019;66:682–690. [PubMed: 31583576]
- 125.
- Ejlsmark-Svensson H, Bislev LS, Lajlev S, Harslof T, Rolighed L, Sikjaer T, Rejnmark L. Prevalence and Risk of Vertebral Fractures in Primary Hyperparathyroidism: A Nested Case-Control Study. J Bone Miner Res. 2018;33:1657–1664. [PubMed: 29734476]
- 126.
- Larsson K, Ljunghall S, Krusemo UB, Naessen T, Lindh E, Persson I. The risk of hip fractures in patients with primary hyperparathyroidism: a population-based cohort study with a follow-up of 19 years. J Intern Med. 1993;234:585–593. [PubMed: 8258750]
- 127.
- Silverberg SJ, Shane E, Jacobs TP, Siris ES, Gartenberg F, Seldin D, Clemens TL, Bilezikian JP. Nephrolithiasis and bone involvement in primary hyperparathyroidism. Am J Med. 1990;89:327–334. [PubMed: 2393037]
- 128.
- Pak CY, Oata M, Lawrence EC, Snyder W. The hypercalciurias. Causes, parathyroid functions, and diagnostic criteria. J Clin Invest. 1974;54:387–400. [PMC free article: PMC301566] [PubMed: 4367891]
- 129.
- Kaplan RA, Haussler MR, Deftos LJ, Bone H, Pak CY. The role of 1 alpha, 25-dihydroxyvitamin D in the mediation of intestinal hyperabsorption of calcium in primary hyperparathyroidism and absorptive hypercalciuria. J Clin Invest. 1977;59:756–760. [PMC free article: PMC372282] [PubMed: 192763]
- 130.
- Broadus AE, Horst RL, Lang R, Littledike ET, Rasmussen H. The importance of circulating 1,25-dihydroxyvitamin D in the pathogenesis of hypercalciuria and renal-stone formation in primary hyperparathyroidism. N Engl J Med. 1980;302:421–426. [PubMed: 7351950]
- 131.
- Pak CY, Nicar MJ, Peterson R, Zerwekh JE, Snyder W. A lack of unique pathophysiologic background for nephrolithiasis of primary hyperparathyroidism. J Clin Endocrinol Metab. 1981;53:536–542. [PubMed: 7263839]
- 132.
- Pak CY. Effect of parathyroidectomy on crystallization of calcium salts in urine of patients with primary hyperparathyroidism. Invest Urol. 1979;17:146–148. [PubMed: 468514]
- 133.
- Klugman VA, Favus M, Pak CY. Nephrolithiasis in primary hyperparathyroidism. In: Bilezikian JP, ed. The parathyroids: basic and clinical concepts. New York: Academic Press:437-450.
- 134.
- Cipriani C, Biamonte F, Costa AG, Zhang C, Biondi P, Diacinti D, Pepe J, Piemonte S, Scillitani A, Minisola S, Bilezikian JP. Prevalence of kidney stones and vertebral fractures in primary hyperparathyroidism using imaging technology. J Clin Endocrinol Metab. 2015;100:1309–1315. [PMC free article: PMC4399306] [PubMed: 25646791]
- 135.
- Rejnmark L, Vestergaard P, Mosekilde L. Nephrolithiasis and renal calcifications in primary hyperparathyroidism. J Clin Endocrinol Metab. 2011;96:2377–2385. [PubMed: 21646371]
- 136.
- Cassibba S, Pellegrino M, Gianotti L, Baffoni C, Baralis E, Attanasio R, Guarnieri A, Borretta G, Tassone F. Silent renal stones in primary hyperparathyroidism: prevalence and clinical features. Endocr Pract. 2014;20:1137–1142. [PubMed: 24936557]
- 137.
- Bilezikian JP, Brandi ML, Eastell R, Silverberg SJ, Udelsman R, Marcocci C, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3561–3569. [PMC free article: PMC5393490] [PubMed: 25162665]
- 138.
- Tay YD, Liu M, Bandeira L, Bucovsky M, Lee JA, Silverberg SJ, Walker MD. Occult urolithiasis in asymptomatic primary hyperparathyroidism. Endocr Res. 2018;43:106–115. [PMC free article: PMC6042842] [PubMed: 29400579]
- 139.
- Albright F, Reifenstein EC. The parathyroid glands and metabolic bone disease. Baltimore: Williams and Wilkins.
- 140.
- Dent CE, Hartland BV, Hicks J, Sykes ED. Calcium intake in patients with primary hyperparathyroidism. Lancet. 1961;2:336–338. [PubMed: 13721925]
- 141.
- Berger AD, Wu W, Eisner BH, Cooperberg MR, Duh QY, Stoller ML. Patients with primary hyperparathyroidism--why do some form stones? J Urol. 2009;181:2141–2145. [PubMed: 19296981]
- 142.
- Odvina CV, Sakhaee K, Heller HJ, Peterson RD, Poindexter JR, Padalino PK, Pak CY. Biochemical characterization of primary hyperparathyroidism with and without kidney stones. Urological research. 2007;35:123–128. [PubMed: 17476495]
- 143.
- Saponaro F, Cetani F, Mazoni L, Apicella M, Di Giulio M, Carlucci F, Scalese M, Pardi E, Borsari S, Bilezikian JP, Marcocci C. Hypercalciuria: its value as a predictive risk factor for nephrolithiasis in asymptomatic primary hyperparathyroidism? J Endocrinol Invest. 2020;43:677–682. [PubMed: 31873910]
- 144.
- Saponaro F, Marcocci C, Apicella M, Mazoni L, Borsari S, Pardi E, Di Giulio M, Carlucci F, Scalese M, Bilezikian JP, Cetani F. Hypomagnesuria is Associated With Nephrolithiasis in Patients With Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2020:105. [PubMed: 32369583]
- 145.
- Walker MD, Nickolas T, Kepley A, Lee JA, Zhang C, McMahon DJ, Silverberg SJ. Predictors of renal function in primary hyperparathyroidism. J Clin Endocrinol Metab. 2014;99:1885–1892. [PMC free article: PMC4010697] [PubMed: 24527717]
- 146.
- Walker MD, Dempster DW, McMahon DJ, Udesky J, Shane E, Bilezikian JP, Silverberg SJ. Effect of renal function on skeletal health in primary hyperparathyroidism. J Clin Endocrinol Metab. 2012;97:1501–1507. [PMC free article: PMC3339888] [PubMed: 22399521]
- 147.
- Rao DS, Wilson RJ, Kleerekoper M, Parfitt AM. Lack of biochemical progression or continuation of accelerated bone loss in mild asymptomatic primary hyperparathyroidism: evidence for biphasic disease course. J Clin Endocrinol Metab. 1988;67:1294–1298. [PubMed: 3192682]
- 148.
- Symons C, Fortune F, Greenbaum RA, Dandona P. Cardiac hypertrophy, hypertrophic cardiomyopathy, and hyperparathyroidism--an association. Br Heart J. 1985;54:539–542. [PMC free article: PMC481943] [PubMed: 2932135]
- 149.
- Stefenelli T, Mayr H, Bergler-Klein J, Globits S, Woloszczuk W, Niederle B. Primary hyperparathyroidism: incidence of cardiac abnormalities and partial reversibility after successful parathyroidectomy. Am J Med. 1993;95:197–202. [PubMed: 8356983]
- 150.
- Palmer M, Adami HO, Bergstrom R, Akerstrom G, Ljunghall S. Mortality after surgery for primary hyperparathyroidism: a follow-up of 441 patients operated on from 1956 to 1979. Surgery. 1987;102:1–7. [PubMed: 3589970]
- 151.
- Ronni-Sivula H. Causes of death in patients previously operated on for primary hyperparathyroidism. Ann Chir Gynaecol. 1985;74:13–18. [PubMed: 4015016]
- 152.
- Hedback G, Tisell LE, Bengtsson BA, Hedman I, Oden A. Premature death in patients operated on for primary hyperparathyroidism. World J Surg. 1990;14:829–835. [PubMed: 2256355]
- 153.
- Ljunghall S, Jakobsson S, Joborn C, Palmer M, Rastad J, Akerstrom G. Longitudinal studies of mild primary hyperparathyroidism. J Bone Miner Res. 1991;6 Suppl 2:S111–116. [PubMed: 1763661]
- 154.
- Hedback G, Oden A, Tisell LE. The influence of surgery on the risk of death in patients with primary hyperparathyroidism. World J Surg. 1991;15:399–405. [PubMed: 1853620]
- 155.
- Soreide JA, van Heerden JA, Grant CS, Yau Lo C, Schleck C, Ilstrup DM. Survival after surgical treatment for primary hyperparathyroidism. Surgery. 1997;122:1117–1123. [PubMed: 9426427]
- 156.
- Wermers RA, Khosla S, Atkinson EJ, Grant CS, Hodgson SF, O'Fallon WM, Melton LJ 3rd. Survival after the diagnosis of hyperparathyroidism: a population-based study. Am J Med. 1998;104:115–122. [PubMed: 9528728]
- 157.
- Nilsson IL, Yin L, Lundgren E, Rastad J, Ekbom A. Clinical presentation of primary hyperparathyroidism in Europe--nationwide cohort analysis on mortality from nonmalignant causes. J Bone Miner Res. 2002;17 Suppl 2:N68–74. [PubMed: 12412780]
- 158.
- Hedback G, Oden A. Increased risk of death from primary hyperparathyroidism--an update. Eur J Clin Invest. 1998;28:271–276. [PubMed: 9615902]
- 159.
- Vestergaard P, Mollerup CL, Frokjaer VG, Christiansen P, Blichert-Toft M, Mosekilde L. Cardiovascular events before and after surgery for primary hyperparathyroidism. World J Surg. 2003;27:216–222. [PubMed: 12616440]
- 160.
- Bradley EL 3rd, Wells JO. Primary hyperparathyroidism and hypertension. Am Surg. 1983;49:569–570. [PubMed: 6638692]
- 161.
- Dominiczak AF, Lyall F, Morton JJ, Dargie HJ, Boyle IT, Tune TT, Murray G, Semple PF. Blood pressure, left ventricular mass and intracellular calcium in primary hyperparathyroidism. Clin Sci (Lond). 1990;78:127–132. [PubMed: 2155737]
- 162.
- Nainby-Luxmoore JC, Langford HG, Nelson NC, Watson RL, Barnes TY. A case-comparison study of hypertension and hyperparathyroidism. J Clin Endocrinol Metab. 1982;55:303–306. [PubMed: 7085856]
- 163.
- Lind L, Jacobsson S, Palmer M, Lithell H, Wengle B, Ljunghall S. Cardiovascular risk factors in primary hyperparathyroidism: a 15-year follow-up of operated and unoperated cases. J Intern Med. 1991;230:29–35. [PubMed: 2066709]
- 164.
- Lind L, Skarfors E, Berglund L, Lithell H, Ljunghall S. Serum calcium: a new, independent, prospective risk factor for myocardial infarction in middle-aged men followed for 18 years. J Clin Epidemiol. 1997;50:967–973. [PubMed: 9291883]
- 165.
- Kamycheva E, Sundsfjord J, Jorde R. Serum parathyroid hormone level is associated with body mass index. The 5th Tromso study. Eur J Endocrinol. 2004;151:167–172. [PubMed: 15296470]
- 166.
- Roberts WC, Waller BF. Effect of chronic hypercalcemia on the heart. An analysis of 18 necropsy patients. Am J Med. 1981;71:371–384. [PubMed: 7282728]
- 167.
- Streeten EA, Munir K, Hines S, Mohamed A, Mangano C, Ryan KA, Post W. Coronary artery calcification in patients with primary hyperparathyroidism in comparison with control subjects from the multi-ethnic study of atherosclerosis. Endocr Pract. 2008;14:155–161. [PubMed: 18308652]
- 168.
- Kepez A, Harmanci A, Hazirolan T, Isildak M, Kocabas U, Ates A, Ciftci O, Tokgozoglu L, Gurlek A. Evaluation of subclinical coronary atherosclerosis in mild asymptomatic primary hyperparathyroidism patients. Int J Cardiovasc Imaging. 2009;25:187–193. [PubMed: 18784985]
- 169.
- Nilsson IL, Aberg J, Rastad J, Lind L. Maintained normalization of cardiovascular dysfunction 5 years after parathyroidectomy in primary hyperparathyroidism. Surgery. 2005;137:632–638. [PubMed: 15933631]
- 170.
- Iwata S, Walker MD, Di Tullio MR, Hyodo E, Jin Z, Liu R, Sacco RL, Homma S, Silverberg SJ. Aortic valve calcification in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2012;97:132–137. [PMC free article: PMC3251929] [PubMed: 22031523]
- 171.
- Walker MD, Rundek T, Homma S, DiTullio M, Iwata S, Lee JA, Choi J, Liu R, Zhang C, McMahon DJ, Sacco RL, Silverberg SJ. Effect of parathyroidectomy on subclinical cardiovascular disease in mild primary hyperparathyroidism. Eur J Endocrinol. 2012;167:277–285. [PMC free article: PMC3668344] [PubMed: 22660025]
- 172.
- Nuzzo V, Tauchmanova L, Fonderico F, Trotta R, Fittipaldi MR, Fontana D, Rossi R, Lombardi G, Trimarco B, Lupoli G. Increased intima-media thickness of the carotid artery wall, normal blood pressure profile and normal left ventricular mass in subjects with primary hyperparathyroidism. Eur J Endocrinol. 2002;147:453–459. [PubMed: 12370105]
- 173.
- Nilsson IL, Aberg J, Rastad J, Lind L. Left ventricular systolic and diastolic function and exercise testing in primary hyperparathyroidism-effects of parathyroidectomy. Surgery. 2000;128:895–902. [PubMed: 11114621]
- 174.
- Walker MD, Silverberg SJ. Cardiovascular aspects of primary hyperparathyroidism. J Endocrinol Invest. 2008;31:925–931. [PMC free article: PMC6056175] [PubMed: 19092300]
- 175.
- McMahon DJ, Carrelli A, Palmeri N, Zhang C, DiTullio M, Silverberg SJ, Walker MD. Effect of Parathyroidectomy Upon Left Ventricular Mass in Primary Hyperparathyroidism: A Meta-Analysis. J Clin Endocrinol Metab. 2015;100:4399–4407. [PMC free article: PMC4667168] [PubMed: 26445115]
- 176.
- Nilsson IL, Aberg J, Rastad J, Lind L. Endothelial vasodilatory dysfunction in primary hyperparathyroidism is reversed after parathyroidectomy. Surgery. 1999;126:1049–1055. [PubMed: 10598187]
- 177.
- Kosch M, Hausberg M, Vormbrock K, Kisters K, Rahn KH, Barenbrock M. Studies on flow-mediated vasodilation and intima-media thickness of the brachial artery in patients with primary hyperparathyroidism. Am J Hypertens. 2000;13:759–764. [PubMed: 10933566]
- 178.
- Lumachi F, Ermani M, Frego M, Pilon F, Filosa T, Di Cristofaro L, De Lotto F, Fallo F. Intima-media thickness measurement of the carotid artery in patients with primary hyperparathyroidism. A prospective case-control study and long-term follow-up. In Vivo. 2006;20:887–890. [PubMed: 17203784]
- 179.
- Fallo F, Camporese G, Capitelli E, Andreozzi GM, Mantero F, Lumachi F. Ultrasound evaluation of carotid artery in primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:2096–2099. [PubMed: 12727960]
- 180.
- Walker MD, Fleischer J, Rundek T, McMahon DJ, Homma S, Sacco R, Silverberg SJ. Carotid vascular abnormalities in primary hyperparathyroidism. J Clin Endocrinol Metab. 2009;94:3849–3856. [PMC free article: PMC2758727] [PubMed: 19755478]
- 181.
- Rosa J, Raska I Jr, Wichterle D, Petrak O, Strauch B, Somloova Z, Zelinka T, Holaj R, Widimsky J Jr. Pulse wave velocity in primary hyperparathyroidism and effect of surgical therapy. Hypertens Res. 2011;34:296–300. [PubMed: 21107330]
- 182.
- Schillaci G, Pucci G, Pirro M, Monacelli M, Scarponi AM, Manfredelli MR, Rondelli F, Avenia N, Mannarino E. Large-artery stiffness: a reversible marker of cardiovascular risk in primary hyperparathyroidism. Atherosclerosis. 2011;218:96–101. [PubMed: 21645899]
- 183.
- Rubin MR, Maurer MS, McMahon DJ, Bilezikian JP, Silverberg SJ. Arterial stiffness in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2005;90:3326–3330. [PubMed: 15769995]
- 184.
- Cope O. The study of hyperparathyroidism at the Massachusetts General Hospital. N Engl J Med. 1966;274:1174–1182. [PubMed: 5327350]
- 185.
- Patten BM, Bilezikian JP, Mallette LE, Prince A, Engel WK, Aurbach GD. Neuromuscular disease in primary hyperparathyroidism. Ann Intern Med. 1974;80:182–193. [PubMed: 4811792]
- 186.
- Vicale CT. Diagnostic features of muscular syndrome resulting from hyperparathyroidism, osteomalacia owing to renal tubular acidosis and perhaps related disorders of calcium metabolism. Trans Am Neurol Assoc. 1949;74:143–147.
- 187.
- Frame B, Heinze EG Jr, Block MA, Manson GA. Myopathy in primary hyperparathyroidism. Observations in three patients. Ann Intern Med. 1968;68:1022–1027. [PubMed: 5646807]
- 188.
- Rollinson RD, Gilligan BS. Primary hyperparathyroidism presenting as a proximal myopathy. Aust N Z J Med. 1977;7:420–421. [PubMed: 270996]
- 189.
- Turken SA, Cafferty M, Silverberg SJ, De La Cruz L, Cimino C, Lange DJ, Lovelace RE, Bilezikian JP. Neuromuscular involvement in mild, asymptomatic primary hyperparathyroidism. Am J Med. 1989;87:553–557. [PubMed: 2816971]
- 190.
- Joborn C, Hetta J, Johansson H, Rastad J, Agren H, Akerstrom G, Ljunghall S. Psychiatric morbidity in primary hyperparathyroidism. World J Surg. 1988;12:476–481. [PubMed: 2458656]
- 191.
- Walker MD, Silverberg SJ. Parathyroidectomy in asymptomatic primary hyperparathyroidism: improves "bones" but not "psychic moans". J Clin Endocrinol Metab. 2007;92:1613–1615. [PubMed: 17483374]
- 192.
- Bollerslev J, Jansson S, Mollerup CL, Nordenstrom J, Lundgren E, Torring O, Varhaug JE, Baranowski M, Aanderud S, Franco C, Freyschuss B, Isaksen GA, Ueland T, Rosen T. Medical observation, compared with parathyroidectomy, for asymptomatic primary hyperparathyroidism: a prospective, randomized trial. J Clin Endocrinol Metab. 2007;92:1687–1692. [PubMed: 17284629]
- 193.
- Talpos GB, Bone HG 3rd, Kleerekoper M, Phillips ER, Alam M, Honasoge M, Divine GW, Rao DS. Randomized trial of parathyroidectomy in mild asymptomatic primary hyperparathyroidism: patient description and effects on the SF-36 health survey. Surgery. 2000;128:1013–1020. [PubMed: 11114637]
- 194.
- Ambrogini E, Cetani F, Cianferotti L, Vignali E, Banti C, Viccica G, Oppo A, Miccoli P, Berti P, Bilezikian JP, Pinchera A, Marcocci C. Surgery or surveillance for mild asymptomatic primary hyperparathyroidism: a prospective, randomized clinical trial. J Clin Endocrinol Metab. 2007;92:3114–3121. [PubMed: 17535997]
- 195.
- Perrier ND, Balachandran D, Wefel JS, Jimenez C, Busaidy N, Morris GS, Dong W, Jackson E, Weaver S, Gantela S, Evans DB, Grubbs EG, Lee JE. Prospective, randomized, controlled trial of parathyroidectomy versus observation in patients with "asymptomatic" primary hyperparathyroidism. Surgery. 2009;146:1116–1122. [PubMed: 19879613]
- 196.
- Walker MD, McMahon DJ, Inabnet WB, Lazar RM, Brown I, Vardy S, Cosman F, Silverberg SJ. Neuropsychological features in primary hyperparathyroidism: a prospective study. J Clin Endocrinol Metab. 2009;94:1951–1958. [PMC free article: PMC2690425] [PubMed: 19336505]
- 197.
- Roman SA, Sosa JA, Mayes L, Desmond E, Boudourakis L, Lin R, Snyder PJ, Holt E, Udelsman R. Parathyroidectomy improves neurocognitive deficits in patients with primary hyperparathyroidism. Surgery. 2005;138:1121–1128. [PubMed: 16360399]
- 198.
- Benge JF, Perrier ND, Massman PJ, Meyers CA, Kayl AE, Wefel JS. Cognitive and affective sequelae of primary hyperparathyroidism and early response to parathyroidectomy. J Int Neuropsychol Soc. 2009;15:1002–1011. [PubMed: 19807940]
- 199.
- Chiang CY, Andrewes DG, Anderson D, Devere M, Schweitzer I, Zajac JD. A controlled, prospective study of neuropsychological outcomes post parathyroidectomy in primary hyperparathyroid patients. Clin Endocrinol (Oxf). 2005;62:99–104. [PubMed: 15638877]
- 200.
- Numann PJ, Torppa AJ, Blumetti AE. Neuropsychologic deficits associated with primary hyperparathyroidism. Surgery. 1984;96:1119–1123. [PubMed: 6505965]
- 201.
- Babinska D, Barczynski M, Stefaniak T, Oseka T, Babinska A, Babinski D, Sworczak K, Lachinski AJ, Nowak W, Sledzinski Z. Evaluation of selected cognitive functions before and after surgery for primary hyperparathyroidism. Langenbecks Arch Surg. 2012;397:825–831. [PubMed: 22160326]
- 202.
- Lourida I, Thompson-Coon J, Dickens CM, Soni M, Kuzma E, Kos K, Llewellyn DJ. Parathyroid hormone, cognitive function and dementia: a systematic review. PLoS One. 2015;10:e0127574. [PMC free article: PMC4444118] [PubMed: 26010883]
- 203.
- Dotzenrath CM, Kaetsch AK, Pfingsten H, Cupisti K, Weyerbrock N, Vossough A, Verde PE, Ohmann C. Neuropsychiatric and cognitive changes after surgery for primary hyperparathyroidism. World J Surg. 2006;30:680–685. [PubMed: 16680584]
- 204.
- Goyal A, Chumber S, Tandon N, Lal R, Srivastava A, Gupta S. Neuropsychiatric manifestations in patients of primary hyperparathyroidism and outcome following surgery. Indian J Med Sci. 2001;55:677–686. [PubMed: 12024994]
- 205.
- Liu M, Sum M, Cong E, Colon I, Bucovsky M, Williams J, Kepley A, Kuo J, Lee JA, Lazar RM, Marshall R, Silverberg S, Walker MD. Cognition and cerebrovascular function in primary hyperparathyroidism before and after parathyroidectomy. J Endocrinol Invest. 2020;43:369–379. [PMC free article: PMC7275118] [PubMed: 31621051]
- 206.
- Gazes Y, Liu M, Sum M, Cong E, Kuo J, Lee JA, Silverberg S, Stern Y, Walker M. Functional magnetic resonance imaging in primary hyperparathyroidism. Eur J Endocrinol. 2020;183:21–30. [PMC free article: PMC7472931] [PubMed: 32348956]
- 207.
- Marx S. Multiple endocrine neoplasia type 1. In: Bilezikian JP, ed. The parathyroids. New York: Academic Press:535-584.
- 208.
- Brandi ML, Gagel RF, Angeli A, Bilezikian JP, Beck-Peccoz P, Bordi C, Conte-Devolx B, Falchetti A, Gheri RG, Libroia A, Lips CJ, Lombardi G, Mannelli M, Pacini F, Ponder BA, Raue F, Skogseid B, Tamburrano G, Thakker RV, Thompson NW, Tomassetti P, Tonelli F, Wells SA Jr, Marx SJ. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab. 2001;86:5658–5671. [PubMed: 11739416]
- 209.
- Khoo TK, Vege SS, Abu-Lebdeh HS, Ryu E, Nadeem S, Wermers RA. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab. 2009;94:2115–2118. [PMC free article: PMC2730346] [PubMed: 19318456]
- 210.
- Marx SJ, Fraser D, Rapoport A. Familial hypocalciuric hypercalcemia. Mild expression of the gene in heterozygotes and severe expression in homozygotes. Am J Med. 1985;78:15–22. [PubMed: 3966479]
- 211.
- Kristoffersson A, Dahlgren S, Lithner F, Jarhult J. Primary hyperparathyroidism in pregnancy. Surgery. 1985;97:326–330. [PubMed: 3975852]
- 212.
- Lowe DK, Orwoll ES, McClung MR, Cawthon ML, Peterson CG. Hyperparathyroidism and pregnancy. Am J Surg. 1983;145:611–619. [PubMed: 6342432]
- 213.
- Abood A, Vestergaard P. Pregnancy outcomes in women with primary hyperparathyroidism. Eur J Endocrinol. 2014;171:69–76. [PubMed: 24743398]
- 214.
- Fitzpatrick LA, Bilezikian JP. Acute primary hyperparathyroidism. Am J Med. 1987;82:275–282. [PubMed: 3812520]
- 215.
- Bayat-Mokhtari F, Palmieri GM, Moinuddin M, Pourmand R. Parathyroid storm. Arch Intern Med. 1980;140:1092–1095. [PubMed: 7396615]
- 216.
- Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, Bilezikian JP. Parathyroid carcinoma. J Bone Miner Res. 2008;23:1869–1880. [PMC free article: PMC3276344] [PubMed: 19016595]
- 217.
- Streeten EA, Weinstein LS, Norton JA, Mulvihill JJ, White BJ, Friedman E, Jaffe G, Brandi ML, Stewart K, Zimering MB, et al. Studies in a kindred with parathyroid carcinoma. J Clin Endocrinol Metab. 1992;75:362–366. [PubMed: 1639936]
- 218.
- Wassif WS, Moniz CF, Friedman E, Wong S, Weber G, Nordenskjold M, Peters TJ, Larsson C. Familial isolated hyperparathyroidism: a distinct genetic entity with an increased risk of parathyroid cancer. J Clin Endocrinol Metab. 1993;77:1485–1489. [PubMed: 7903311]
- 219.
- Yoshimoto K, Endo H, Tsuyuguchi M, Tanaka C, Kimura T, Iwahana H, Kato G, Sano T, Itakura M. Familial isolated primary hyperparathyroidism with parathyroid carcinomas: clinical and molecular features. Clin Endocrinol (Oxf). 1998;48:67–72. [PubMed: 9509070]
- 220.
- Marx SJ. F. SW, K. AS. Hyperparathyroidism in hereditary syndromes: special expressions and special managements. Journal of Bone and Mineral Research. 2002;17(N37-N):43. [PubMed: 12412776]
- 221.
- Chen JD, Morrison C, Zhang C, Kahnoski K, Carpten JD, Teh BT. Hyperparathyroidism-jaw tumour syndrome. J Intern Med. 2003;253:634–642. [PubMed: 12755959]
- 222.
- Mallette LE, Malini S, Rappaport MP, Kirkland JL. Familial cystic parathyroid adenomatosis. Ann Intern Med. 1987;107:54–60. [PubMed: 3592449]
- 223.
- Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, Bailey-Wilson J, Simonds WF, Gillanders EM, Kennedy AM, Chen JD, Agarwal SK, Sood R, Jones MP, Moses TY, Haven C, Petillo D, Leotlela PD, Harding B, Cameron D, Pannett AA, Hoog A, Heath H 3rd, James-Newton LA, Robinson B, Zarbo RJ, Cavaco BM, Wassif W, Perrier ND, Rosen IB, Kristoffersson U, Turnpenny PD, Farnebo LO, Besser GM, Jackson CE, Morreau H, Trent JM, Thakker RV, Marx SJ, Teh BT, Larsson C, Hobbs MR. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism-jaw tumor syndrome. Nat Genet. 2002;32:676–680. [PubMed: 12434154]
- 224.
- Simonds WF, James-Newton LA, Agarwal SK, Yang B, Skarulis MC, Hendy GN, Marx SJ. Familial isolated hyperparathyroidism: clinical and genetic characteristics of 36 kindreds. Medicine (Baltimore). 2002;81:1–26. [PubMed: 11807402]
- 225.
- Dionisi S, Minisola S, Pepe J, De Geronimo S, Paglia F, Memeo L, Fitzpatrick LA. Concurrent parathyroid adenomas and carcinoma in the setting of multiple endocrine neoplasia type 1: presentation as hypercalcemic crisis. Mayo Clin Proc. 2002;77:866–869. [PubMed: 12173721]
- 226.
- Agha A, Carpenter R, Bhattacharya S, Edmonson SJ, Carlsen E, Monson JP. Parathyroid carcinoma in multiple endocrine neoplasia type 1 (MEN1) syndrome: two case reports of an unrecognised entity. J Endocrinol Invest. 2007;30:145–149. [PubMed: 17392605]
- 227.
- Haven CJ, van Puijenbroek M, Tan MH, Teh BT, Fleuren GJ, van Wezel T, Morreau H. Identification of MEN1 and HRPT2 somatic mutations in paraffin-embedded (sporadic) parathyroid carcinomas. Clin Endocrinol (Oxf). 2007;67:370–376. [PubMed: 17555500]
- 228.
- Jenkins PJ, Satta MA, Simmgen M, Drake WM, Williamson C, Lowe DG, Britton K, Chew SL, Thakker RV, Besser GM. Metastatic parathyroid carcinoma in the MEN2A syndrome. Clin Endocrinol (Oxf). 1997;47:747–751. [PubMed: 9497883]
- 229.
- Cryns VL, Thor A, Xu HJ, Hu SX, Wierman ME, Vickery AL Jr, Benedict WF, Arnold A. Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med. 1994;330:757–761. [PubMed: 7906387]
- 230.
- Dotzenrath C, Teh BT, Farnebo F, Cupisti K, Svensson A, Toell A, Goretzki P, Larsson C. Allelic loss of the retinoblastoma tumor suppressor gene: a marker for aggressive parathyroid tumors? J Clin Endocrinol Metab. 1996;81:3194–3196. [PubMed: 8784068]
- 231.
- Shattuck TM, Valimaki S, Obara T, Gaz RD, Clark OH, Shoback D, Wierman ME, Tojo K, Robbins CM, Carpten JD, Farnebo LO, Larsson C, Arnold A. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med. 2003;349:1722–1729. [PubMed: 14585940]
- 232.
- Weinstein LS, Simonds WF. HRPT2, a marker of parathyroid cancer. N Engl J Med. 2003;349:1691–1692. [PubMed: 14585935]
- 233.
- LiVolsi V. Morphology of the parathyroid glands. In: Becker KL, ed. Principles and Practice of Endocrinology and Metabolism. Philadelphia: Lipincott Williams and Wilkins.
- 234.
- Palmer JA, Brown WA, Kerr WH, Rosen IB, Watters NA. The surgical aspects of hyperparathyroidism. Arch Surg. 1975;110:1004–1007. [PubMed: 1156145]
- 235.
- Hage MP, Salti I, El-Hajj Fuleihan G. Parathyromatosis: a rare yet problematic etiology of recurrent and persistent hyperparathyroidism. Metabolism. 2012;61:762–775. [PubMed: 22221828]
- 236.
- Baloch ZW. LiVolsi VA. Pathology of the parathyroid glands in hyperparathyroidism. Semin Diagn Pathol. 2013;30:165–177. [PubMed: 24144286]
- 237.
- Reddick RL, Costa JC, Marx SJ. Parathyroid hyperplasia and parathyromatosis. Lancet. 1977;1:549. [PubMed: 65648]
- 238.
- Pinnamaneni N, Shankar PR, Muthukrishnan A. (99m)Tc MIBI SPECT findings in parathyromatosis--a rare entity causing recurrent hyperparathyroidism. Clin Nucl Med. 2013;38:e443–445. [PubMed: 23603581]
- 239.
- Tublin ME, Yim JH, Carty SE. Recurrent hyperparathyroidism secondary to parathyromatosis: clinical and imaging findings. J Ultrasound Med. 2007;26:847–851. [PubMed: 17526617]
- 240.
- Kollmorgen CF, Aust MR, Ferreiro JA, McCarthy JT, van Heerden JA. Parathyromatosis: a rare yet important cause of persistent or recurrent hyperparathyroidism. Surgery. 1994;116:111–115. [PubMed: 8023257]
- 241.
- Rubin MR, Bilezikian JP, McMahon DJ, Jacobs T, Shane E, Siris E, Udesky J, Silverberg SJ. The natural history of primary hyperparathyroidism with or without parathyroid surgery after 15 years. J Clin Endocrinol Metab. 2008;93:3462–3470. [PMC free article: PMC2567863] [PubMed: 18544625]
- 242.
- Rao DS, Phillips ER, Divine GW, Talpos GB. Randomized controlled clinical trial of surgery versus no surgery in patients with mild asymptomatic primary hyperparathyroidism. J Clin Endocrinol Metab. 2004;89:5415–5422. [PubMed: 15531491]
- 243.
- Lundstam K, Heck A, Mollerup C, Godang K, Baranowski M, Pernow Y, Varhaug JE, Hessman O, Rosen T, Nordenstrom J, Jansson S, Hellstrom M, Bollerslev J, Group SS. Effects of parathyroidectomy versus observation on the development of vertebral fractures in mild primary hyperparathyroidism. J Clin Endocrinol Metab. 2015;100:1359–1367. [PubMed: 25636048]
- 244.
- Bilezikian JP, Potts JT Jr, Fuleihan Gel H, Kleerekoper M, Neer R, Peacock M, Rastad J, Silverberg SJ, Udelsman R, Wells SA. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century. J Clin Endocrinol Metab. 2002;87:5353–5361. [PubMed: 12466320]
- 245.
- Khan AA, Bilezikian JP, Potts JT Jr. The diagnosis and management of asymptomatic primary hyperparathyroidism revisited. J Clin Endocrinol Metab. 2009;94:333–334. [PubMed: 19193907]
- 246.
- Bilezikian JP, Khan AA, Potts JT Jr. Guidelines for the management of asymptomatic primary hyperparathyroidism: summary statement from the third international workshop. J Clin Endocrinol Metab. 2009;94:335–339. [PMC free article: PMC3214274] [PubMed: 19193908]
- 247.
- Silverberg SJ, Clarke BL, Peacock M, Bandeira F, Boutroy S, Cusano NE, Dempster D, Lewiecki EM, Liu JM, Minisola S, Rejnmark L, Silva BC, Walker MD, Bilezikian JP. Current issues in the presentation of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J Clin Endocrinol Metab. 2014;99:3580–3594. [PMC free article: PMC5393491] [PubMed: 25162667]
- 248.
- Cheung K, Wang TS, Farrokhyar F, Roman SA, Sosa JA. A meta-analysis of preoperative localization techniques for patients with primary hyperparathyroidism. Ann Surg Oncol. 2012;19:577–583. [PubMed: 21710322]
- 249.
- Civelek AC, Ozalp E, Donovan P, Udelsman R. Prospective evaluation of delayed technetium-99m sestamibi SPECT scintigraphy for preoperative localization of primary hyperparathyroidism. Surgery. 2002;131:149–157. [PubMed: 11854692]
- 250.
- Van Husen R, Kim LT. Accuracy of surgeon-performed ultrasound in parathyroid localization. World J Surg. 2004;28:1122–1126. [PubMed: 15490067]
- 251.
- Mortenson MM, Evans DB, Lee JE, Hunter GJ, Shellingerhout D, Vu T, Edeiken BS, Feng L, Perrier ND. Parathyroid exploration in the reoperative neck: improved preoperative localization with 4D-computed tomography. J Am Coll Surg. 2008;206:888–895. [PubMed: 18471717]
- 252.
- Yeh R, Tay YD, Tabacco G, Dercle L, Kuo JH, Bandeira L, McManus C, Leung DK, Lee JA, Bilezikian JP. Diagnostic Performance of 4D CT and Sestamibi SPECT/CT in Localizing Parathyroid Adenomas in Primary Hyperparathyroidism. Radiology. 2019;291:469–476. [PMC free article: PMC7440755] [PubMed: 30835187]
- 253.
- Maser C, Donovan P, Santos F, Donabedian R, Rinder C, Scoutt L, Udelsman R. Sonographically guided fine needle aspiration with rapid parathyroid hormone assay. Ann Surg Oncol. 2006;13:1690–1695. [PubMed: 17009140]
- 254.
- Udelsman R, Donovan PI. Remedial parathyroid surgery: changing trends in 130 consecutive cases. Ann Surg. 2006;244:471–479. [PMC free article: PMC1856539] [PubMed: 16926573]
- 255.
- Clark OH. How should patients with primary hyperparathyroidism be treated? J Clin Endocrinol Metab. 2003;88:3011–3014. [PubMed: 12843135]
- 256.
- Udelsman R. Six hundred fifty-six consecutive explorations for primary hyperparathyroidism. Ann Surg. 2002;235:665–670. [PMC free article: PMC1422492] [PubMed: 11981212]
- 257.
- Irvin GL 3rd, Solorzano CC, Carneiro DM. Quick intraoperative parathyroid hormone assay: surgical adjunct to allow limited parathyroidectomy, improve success rate, and predict outcome. World J Surg. 2004;28:1287–1292. [PubMed: 15517474]
- 258.
- Udelsman R, Donovan PI, Sokoll LJ. One hundred consecutive minimally invasive parathyroid explorations. Ann Surg. 2000;232:331–339. [PMC free article: PMC1421147] [PubMed: 10973383]
- 259.
- Westerdahl J, Bergenfelz A. Unilateral versus bilateral neck exploration for primary hyperparathyroidism: five-year follow-up of a randomized controlled trial. Ann Surg. 2007;246:976–980. [PubMed: 18043099]
- 260.
- Russell CF, Dolan SJ, Laird JD. Randomized clinical trial comparing scan-directed unilateral versus bilateral cervical exploration for primary hyperparathyroidism due to solitary adenoma. Br J Surg. 2006;93:418–421. [PubMed: 16392100]
- 261.
- Jinih M, O'Connell E, O'Leary DP, Liew A, Redmond HP. Focused Versus Bilateral Parathyroid Exploration for Primary Hyperparathyroidism: A Systematic Review and Meta-analysis. Ann Surg Oncol. 2017;24:1924–1934. [PubMed: 27896505]
- 262.
- Miccoli P, Berti P, Materazzi G, Ambrosini CE, Fregoli L, Donatini G. Endoscopic bilateral neck exploration versus quick intraoperative parathormone assay (qPTHa) during endoscopic parathyroidectomy: A prospective randomized trial. Surg Endosc. 2008;22:398–400. [PubMed: 17522920]
- 263.
- Henry JF, Sebag F, Tamagnini P, Forman C, Silaghi H. Endoscopic parathyroid surgery: results of 365 consecutive procedures. World J Surg. 2004;28:1219–1223. [PubMed: 15517493]
- 264.
- Silverberg SJ, Gartenberg F, Jacobs TP, Shane E, Siris E, Staron RB, McMahon DJ, Bilezikian JP. Increased bone mineral density after parathyroidectomy in primary hyperparathyroidism. J Clin Endocrinol Metab. 1995;80:729–734. [PubMed: 7883824]
- 265.
- Kulak CA, Bandeira C, Voss D, Sobieszczyk SM, Silverberg SJ, Bandeira F, Bilezikian JP. Marked improvement in bone mass after parathyroidectomy in osteitis fibrosa cystica. J Clin Endocrinol Metab. 1998;83:732–735. [PubMed: 9506717]
- 266.
- Tritos NA, Hartzband P. Rapid improvement of osteoporosis following parathyroidectomy in a premenopausal woman with acute primary hyperparathyroidism. Arch Intern Med. 1999;159:1495–1498. [PubMed: 10399902]
- 267.
- DiGregorio S. Hiperparatiroidismo primario: dramatico incremento de la masa ostea post paratiroidectomia. Diagn Osteol. 1999;1:11–15.
- 268.
- Dawson-Hughes B, Stern DT, Shipp CC, Rasmussen HM. Effect of lowering dietary calcium intake on fractional whole body calcium retention. J Clin Endocrinol Metab. 1988;67:62–68. [PubMed: 3132484]
- 269.
- Barger-Lux MJ, Heaney RP. Effects of calcium restriction on metabolic characteristics of premenopausal women. J Clin Endocrinol Metab. 1993;76:103–107. [PubMed: 8421072]
- 270.
- Calvo MS, Kumar R, Heath H. Persistently elevated parathyroid hormone secretion and action in young women after four weeks of ingesting high phosphorus, low calcium diets. J Clin Endocrinol Metab. 1990;70:1334–1340. [PubMed: 2335575]
- 271.
- Insogna KL, Mitnick ME, Stewart AF, Burtis WJ, Mallette LE, Broadus AE. Sensitivity of the parathyroid hormone-1,25-dihydroxyvitamin D axis to variations in calcium intake in patients with primary hyperparathyroidism. N Engl J Med. 1985;313:1126–1130. [PubMed: 2995810]
- 272.
- Locker FG, Silverberg SJ, Bilezikian JP. Optimal dietary calcium intake in primary hyperparathyroidism. Am J Med. 1997;102:543–550. [PubMed: 9217669]
- 273.
- Jorde R, Szumlas K, Haug E, Sundsfjord J. The effects of calcium supplementation to patients with primary hyperparathyroidism and a low calcium intake. Eur J Nutr. 2002;41:258–263. [PubMed: 12474069]
- 274.
- Marcocci C, Bollerslev J, Khan AA, Shoback DM. Medical management of primary hyperparathyroidism: proceedings of the fourth International Workshop on the Management of Asymptomatic Primary Hyperparathyroidism. J Clin Endocrinol Metab. 2014;99:3607–3618. [PubMed: 25162668]
- 275.
- Rolighed L, Rejnmark L, Sikjaer T, Heickendorff L, Vestergaard P, Mosekilde L, Christiansen P. Vitamin D treatment in primary hyperparathyroidism: a randomized placebo controlled trial. J Clin Endocrinol Metab. 2014;99:1072–1080. [PubMed: 24423366]
- 276.
- Purnell DC, Scholz DA, Smith LH, Sizemore GW, Black MB, Goldsmith RS, Arnaud CD. Treatment of primary hyperparathyroidism. Am J Med. 1974;56:800–809. [PubMed: 4831317]
- 277.
- Broadus AE, Magee JS, Mallette LE, Horst RL, Lang R, Jensen PS, Gertner JM, Baron R. A detailed evaluation of oral phosphate therapy in selected patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 1983;56:953–961. [PubMed: 6300178]
- 278.
- Gallagher JC, Nordin BE. Treatment with with oestrogens of primary hyperparathyroidism in post-menopausal women. Lancet. 1972;1:503–507. [PubMed: 4110019]
- 279.
- Marcus R, Madvig P, Crim M, Pont A, Kosek J. Conjugated estrogens in the treatment of postmenopausal women with hyperparathyroidism. Ann Intern Med. 1984;100:633–640. [PubMed: 6324624]
- 280.
- Selby PL, Peacock M. Ethinyl estradiol and norethindrone in the treatment of primary hyperparathyroidism in postmenopausal women. N Engl J Med. 1986;314:1481–1485. [PubMed: 3754618]
- 281.
- Grey AB, Stapleton JP, Evans MC, Tatnell MA, Reid IR. Effect of hormone replacement therapy on bone mineral density in postmenopausal women with mild primary hyperparathyroidism. A randomized, controlled trial. Ann Intern Med. 1996;125:360–368. [PubMed: 8702086]
- 282.
- Rubin MR, Lee KH, McMahon DJ, Silverberg SJ. Raloxifene lowers serum calcium and markers of bone turnover in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:1174–1178. [PubMed: 12629102]
- 283.
- Kaplan RA, Geho WB, Poindexter C, Haussler M, Dietz GW, Pak CY. Metabolic effects of diphosphonate in primary hyperparathyroidism. J Clin Pharmacol. 1977;17:410–419. [PubMed: 195983]
- 284.
- Shane E, Baquiran DC, Bilezikian JP. Effects of dichloromethylene diphosphonate on serum and urinary calcium in primary hyperparathyroidism. Ann Intern Med. 1981;95:23–27. [PubMed: 6264825]
- 285.
- Rossini M, Gatti D, Isaia G, Sartori L, Braga V, Adami S. Effects of oral alendronate in elderly patients with osteoporosis and mild primary hyperparathyroidism. J Bone Miner Res. 2001;16:113–119. [PubMed: 11149474]
- 286.
- Hassani S, Braunstein GD, Seibel MJ, Brickman AS, Geola F, Pekary AE, Hershman JM. Alendronate therapy of primary hyperparathyroidism. Endocrinologist. 2001;11:459–464.
- 287.
- Chow CC, Chan WB, Li JK, Chan NN, Chan MH, Ko GT, Lo KW, Cockram CS. Oral alendronate increases bone mineral density in postmenopausal women with primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:581–587. [PubMed: 12574184]
- 288.
- Parker CR, Blackwell PJ, Fairbairn KJ, Hosking DJ. Alendronate in the treatment of primary hyperparathyroid-related osteoporosis: a 2-year study. J Clin Endocrinol Metab. 2002;87:4482–4489. [PubMed: 12364423]
- 289.
- Khan AA, Bilezikian JP, Kung AW, Ahmed MM, Dubois SJ, Ho AY, Schussheim D, Rubin MR, Shaikh AM, Silverberg SJ, Standish TI, Syed Z, Syed ZA. Alendronate in primary hyperparathyroidism: a double-blind, randomized, placebo-controlled trial. J Clin Endocrinol Metab. 2004;89:3319–3325. [PubMed: 15240609]
- 290.
- Eller-Vainicher C, Palmieri S, Cairoli E, Goggi G, Scillitani A, Arosio M, Falchetti A, Chiodini I. Protective Effect of Denosumab on Bone in Older Women with Primary Hyperparathyroidism. J Am Geriatr Soc. 2018;66:518–524. [PubMed: 29364518]
- 291.
- Nemeth EF, Scarpa A. Rapid mobilization of cellular Ca2+ in bovine parathyroid cells evoked by extracellular divalent cations. Evidence for a cell surface calcium receptor. J Biol Chem. 1987;262:5188–5196. [PubMed: 3558389]
- 292.
- Brown EM, Gamba G, Riccardi D, Lombardi M, Butters R, Kifor O, Sun A, Hediger MA, Lytton J, Hebert SC. Cloning and characterization of an extracellular Ca(2+)-sensing receptor from bovine parathyroid. Nature. 1993;366:575–580. [PubMed: 8255296]
- 293.
- Brown EM, Pollak M, Seidman CE, Seidman JG, Chou YH, Riccardi D, Hebert SC. Calcium-ion-sensing cell-surface receptors. N Engl J Med. 1995;333:234–240. [PubMed: 7791841]
- 294.
- Fox J, Hadfield S, Petty BA, Nemeth EF. A first generation calcimimetic compound (NPS R-568) that acts on the parathyroid cell calcium receptor: a novel therapeutic approach for hyperparathyroidism. J Bone Miner Res. 1993:8.
- 295.
- Heath H, Sanguinetti EL, Oglseby S, Marriott TB. Inhibition of human parathyroid hormone secretion in vivo by NPS R-568, a calcimimetic drug that targets the parathyroid cell surface calcium receptor. Bone. 1995;16:85S.
- 296.
- Silverberg SJ, Bone HG 3rd, Marriott TB, Locker FG, Thys-Jacobs S, Dziem G, Kaatz S, Sanguinetti EL, Bilezikian JP. Short-term inhibition of parathyroid hormone secretion by a calcium-receptor agonist in patients with primary hyperparathyroidism. N Engl J Med. 1997;337:1506–1510. [PubMed: 9366582]
- 297.
- Shoback DM, Bilezikian JP, Turner SA, McCary LC, Guo MD, Peacock M. The calcimimetic cinacalcet normalizes serum calcium in subjects with primary hyperparathyroidism. J Clin Endocrinol Metab. 2003;88:5644–5649. [PubMed: 14671147]
- 298.
- Peacock M, Bilezikian JP, Klassen PS, Guo MD, Turner SA, Shoback D. Cinacalcet hydrochloride maintains long-term normocalcemia in patients with primary hyperparathyroidism. J Clin Endocrinol Metab. 2005;90:135–141. [PubMed: 15522938]
- 299.
- Marcocci C, Chanson P, Shoback D, Bilezikian J, Fernandez-Cruz L, Orgiazzi J, Henzen C, Cheng S, Sterling LR, Lu J, Peacock M. Cinacalcet reduces serum calcium concentrations in patients with intractable primary hyperparathyroidism. J Clin Endocrinol Metab. 2009;94:2766–2772. [PMC free article: PMC3214593] [PubMed: 19470620]
- 300.
- Peacock M, Bolognese MA, Borofsky M, Scumpia S, Sterling LR, Cheng S, Shoback D. Cinacalcet treatment of primary hyperparathyroidism: biochemical and bone densitometric outcomes in a five-year study. J Clin Endocrinol Metab. 2009;94:4860–4867. [PubMed: 19837909]
- 301.
- Silverberg SJ, Rubin MR, Faiman C, Peacock M, Shoback DM, Smallridge RC, Schwanauer LE, Olson KA, Klassen P, Bilezikian JP. Cinacalcet hydrochloride reduces the serum calcium concentration in inoperable parathyroid carcinoma. J Clin Endocrinol Metab. 2007;92:3803–3808. [PubMed: 17666472]
- 302.
- Tsvetov G, Hirsch D, Shimon I, Benbassat C, Masri-Iraqi H, Gorshtein A, Herzberg D, Shochat T, Shraga-Slutzky I, Diker-Cohen T. Thiazide Treatment in Primary Hyperparathyroidism - A New Indication for an Old Medication? J Clin Endocrinol Metab. 2016:jc20162481. [PubMed: 28388724]
- 303.
- Riss P, Kammer M, Selberherr A, Bichler C, Kaderli R, Scheuba C, Niederle B. The influence of thiazide intake on calcium and parathyroid hormone levels in patients with primary hyperparathyroidism. Clin Endocrinol (Oxf). 2016;85:196–201. [PubMed: 26921840]
- 304.
- Farquhar CW, Spathis GS, Barron JL, Levin GE. Failure of thiazide diuretics to increase plasma calcium in mild primary hyperparathyroidism. Postgraduate medical journal. 1990;66:714–716. [PMC free article: PMC2426866] [PubMed: 2235800]
- 305.
- Bradwell AR, Harvey TC. Control of hypercalcaemia of parathyroid carcinoma by immunisation. Lancet. 1999;353:370–373. [PubMed: 9950443]
- 306.
- Betea D, Bradwell AR, Harvey TC, Mead GP, Schmidt-Gayk H, Ghaye B, Daly AF, Beckers A. Hormonal and biochemical normalization and tumor shrinkage induced by anti-parathyroid hormone immunotherapy in a patient with metastatic parathyroid carcinoma. J Clin Endocrinol Metab. 2004;89:3413–3420. [PubMed: 15240624]
- 307.
- Vellanki P, Lange K, Elaraj D, Kopp PA, El Muayed M. Denosumab for management of parathyroid carcinoma-mediated hypercalcemia. J Clin Endocrinol Metab. 2014;99:387–390. [PMC free article: PMC3913820] [PubMed: 24178790]
- 308.
- Nadarasa K, Theodoraki A, Kurzawinski TR, Carpenter R, Bull J, Chung TT, Drake WM. Denosumab for management of refractory hypercalcaemia in recurrent parathyroid carcinoma. Eur J Endocrinol. 2014;171:L7–8. [PubMed: 24939719]
- 309.
- Collins MT, Skarulis MC, Bilezikian JP, Silverberg SJ, Spiegel AM, Marx SJ. Treatment of hypercalcemia secondary to parathyroid carcinoma with a novel calcimimetic agent. J Clin Endocrinol Metab. 1998;83:1083–1088. [PubMed: 9543122]
Publication Details
Author Information and Affiliations
Vagelos College of Physicians and Surgeons, Columbia University, New York, NY 10032
Publication History
Last Update: April 19, 2021.
Copyright
This electronic version has been made freely available under a Creative Commons (CC-BY-NC-ND) license. A copy of the license can be viewed at http://creativecommons.org/licenses/by-nc-nd/2.0/.
Publisher
MDText.com, Inc., South Dartmouth (MA)
NLM Citation
Walker MD, Bilezikian JP. Primary Hyperparathyroidism. [Updated 2021 Apr 19]. In: Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.