

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Feingold KR, Anawalt B, Blackman MR, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-.

ABSTRACT
The two major goals of the treatment of hypertriglyceridemia are the prevention of cardiovascular disease and pancreatitis. Here we discuss the drugs used for the treatment of hypertriglyceridemia: (niacin, fibrates, omega-3-fatty acids, volanesorsen (available in Europe) and lipoprotein lipase gene therapy (alipogene tiparvovec- no longer available). Niacin decreases total cholesterol, TGs (20-50% decrease), LDL-C, and Lp(a). Additionally, niacin decreases small dense LDL resulting in a shift to large, buoyant LDL particles. Moreover, niacin increases HDL-C. Skin flushing, insulin resistance, and other side effects have limited the use of niacin. The enthusiasm for niacin has greatly decreased with the failure of AIM-HIGH and HPS-2 Thrive to decrease cardiovascular events when niacin was added to statin therapy. The omega-3-fatty acids eicosapentaenoic acid (C20:5n-3) (EPA) and docosahexaenoic acid (C22:6n-3) (DHA) lower TGs by 10-50% but do not affect total cholesterol, HDL-C, or Lp(a). LDL-C may increase with EPA + DHA when the TG levels are markedly elevated (>500mg/dL). EPA alone does not increase LDL-C. Omega-3-fatty acids have few side effects, drug interactions, or contraindications. Numerous studies of low dose omega-3-fatty acids on cardiovascular outcomes have failed to demonstrate a benefit. However, in the JELIS trial and REDUCE-IT trial high doses of EPA alone reduced cardiovascular events while in the STRENGTH trial high dose EPA+DHA did not reduce cardiovascular events. Fibrates reduce TG levels by 25-50% and increase HDL-C by 5-20%. The effect on LDL-C is variable. If the TG levels are very high (>500mg/dL), fibrate therapy may result in an increase in LDL-C, whereas if TGs are not markedly elevated fibrates decrease LDL-C by 10-30%. Fibrates also reduce apolipoprotein B, LDL particle number, and non-HDL-C and there may be a shift from small dense LDL towards large LDL particles. Fibrates do not have any major effects on Lp(a). Monotherapy with fibrates appears to reduce cardiovascular events in patients with high TG and low HDL-C levels. Whether the addition of fibrates to statin therapy will reduce cardiovascular disease is uncertain. In patients with diabetes fibrates appear to slow the progression of microvascular disease. Volanesorsen is an antisense oligonucleotide that inhibits the production of apolipoprotein C-III. In patients with the familial chylomicronemia syndrome (FCS) volanesorsen decreases TG by 77% (mean decrease of 1712 mg/dL) with 77% of the patients having TG levels less than 750 mg/dL. In addition, volanesorsen treatment resulted in decreases in non–HDL-C by 46%, and VLDL-C by 58% and increases in HDL-C by 46%, LDL-C by 136%, (LDL-C increased from 28 to 61 mg/dL), and total apolipoprotein B by 20%. Studies have suggested that volanesorsen may reduce episodes of pancreatitis. Patients with FCS have also reported that volanesorsen improved symptoms and reduced interference of FCS with work/school responsibilities. Of concern has been decreases in platelet levels with 47% of patients treated with volanesorsen developing platelet counts below100 x 109/L. Thus, a number of drugs are available for the treatment of hypertriglyceridemia and may be employed when lifestyle changes are not sufficient. For complete coverage of all related areas of Endocrinology, please visit our on-line FREE web-text, WWW.ENDOTEXT.ORG.
INTRODUCTION
The two primary goals of the treatment of hypertriglyceridemia are the prevention of cardiovascular disease and the prevention of pancreatitis. The evaluation and guidelines for the management of hypertriglyceridemia are discussed in detail in the Endotext chapter “Risk of Fasting and Non-Fasting Hypertriglyceridemia in Coronary Vascular Disease and Pancreatitis” (1) and the approach to evaluating a patient with hypertriglyceridemia is discussed in the Endotext chapter “Approach to the Patient with Dyslipidemia” (2). The treatment of hypertriglyceridemia by diet and weight loss are discussed in detail in the Endotext chapter “The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels” and “Obesity and Dyslipidemia” (3,4). Lifestyle changes are recommended as the first line for therapy of hypertriglyceridemia, but drug therapy is often required. In this chapter we will discuss the drugs used for the treatment of elevated plasma TG levels. Statins, ezetimibe, PCSK9 inhibitors, bempedoic acid, lomitapide, mipomersen, and evinacumab, which are primarily used to lower LDL-C, are discussed in the chapter “Cholesterol Lowering Drugs” (5).
NIACIN
Introduction
Niacin was the first drug approved to treat dyslipidemia. In 1955, Altschul et al showed that pharmacologic doses of niacin decreased plasma cholesterol levels (6). Several forms of niacin are available for clinical use. Immediate release niacin has a short duration of action and is typically given two or three times per day with meals, whereas sustained release niacin and extended-release niacin are once a day drugs usually given at bedtime. The extended release form of niacin exhibits release rates that are intermediate between immediate release niacin and sustained release niacin (7). While the effects of the various forms of niacin on plasma lipid levels are similar, the side effect profiles are different. Because of an increased risk of serious liver toxicity with sustained release niacin this preparation is no longer widely used to treat dyslipidemia. Over-the- counter “No flush” niacin is also available but is generally ineffective as a lipid-modifying agent because most of these preparations do not contain active nicotinic acid.
Effect of Niacin on Lipid and Lipoprotein Levels
Table 1.
Effect of Niacin on Lipid and Lipoproteins
| Decreases Total Cholesterol |
| Decreases LDL-C |
| Decreases TGs |
| Decreases Non-HDL-C |
| Decreases Lp(a) |
| Increases HDL-C |
| Decreases Apolipoprotein B |
| Shifts Small Dense LDL to Large Buoyant LDL |
Niacin decreases all the pro-atherogenic lipid and lipoprotein particles including total cholesterol, TG, LDL-C, and Lp(a) levels (Table 1) (8,9). Additionally, niacin has been shown to decrease small dense LDL resulting in a shift to large, buoyant LDL particles (10). Moreover, niacin increases HDL-C levels (8,9).
In a meta-analysis of 30 trials with 4,749 subjects treatment with immediate release, sustained release, or extended release niacin decreased total cholesterol by 10%, decreased TGs by 20%, decreased LDL-C by 14%, and increased HDL-C by 16% (11). All three niacin preparations were effective in decreasing total cholesterol, TG, and LDL-C levels and increasing HDL-C levels (11). At a dose of 1.5 grams per day, immediate release niacin and extended release niacin produced similar decreases in total cholesterol, TGs, and LDL-C and a similar increase in HDL-C (12). A meta-analysis of 14 studies with 9,013 subjects reported a 23% decrease in Lp(a) with extended release niacin treatment (13).
A small meta-analysis of 5 trials in 432 subjects compared the response to extended release niacin in men and women (14). The effect of niacin on LDL-C was greater in women than men at all niacin doses (1,000mg 6.8% decrease in women vs 0.2% in men, p = 0.006; 1,500mg 11.3% decrease vs 5.6% decrease, p = 0.013; 2,000 mg 14.8% decrease vs 6.9% decrease, p = 0.010; 3,000mg 28.7% decrease vs 17.7% decrease, p = 0.006). The effect of niacin on plasma TG levels also tended to be greater in women but the difference only reached statistical significance at the 1,500mg dose (28.6% vs 20.4%, p = 0.040). The mechanism for the more robust decrease in LDL-C and TGs in women is unknown but might be due to a smaller body mass in women leading to increased circulating niacin levels and hence a greater response. However, the effect of niacin on HDL-C and Lp(a) levels were similar in males and females. Not unexpectedly the effect of niacin is dose dependent with higher doses having a greater effect on plasma lipid and lipoprotein levels (Table 2) (14).
Table 2.
Effect of Niacin Dose on Lipid and Lipoprotein Response in Women (percent change)
| Niacin Dose | LDL-C | TG | HDL-C | Lp(a) |
|---|---|---|---|---|
| 500mg | -5.2 | -9.5 | 7.7 | -2.6 |
| 1000mg | -6.8 | -14.5 | 17.6 | -11.5 |
| 1500mg | -11.3 | -28.6 | 21.1 | -4.0 |
| 2000mg | -14.8 | -37.3 | 25.2 | -24.7 |
| 2500mg | -28.7 | -45.6 | 34.5 | -28.6 |
| 3000mg | -28.7 | -51.0 | 28.7 | -29.9 |
Numerous studies have examined the effect of the addition of niacin to statin therapy. Combination therapy typically results in further reductions in atherogenic lipoprotein particles and an increase in HDL-C levels. An example of such a study is shown in Table 3 (15).
Table 3.
Effect of the Addition of Niacin to Statin Therapy on Lipid and Lipoprotein Levels
| Change in Lipids with Addition of Extended-Release Niacin 2000mg/day to Simvastatin 20mg/day | |
|---|---|
| LDL-C | 7.1% Decrease |
| HDL-C | 18.2% Increase |
| TG | 22.7% Decrease |
| Non-HDL-C | 15.1% Decrease |
| Lp(a) | 17.4% Decrease |
While a literature search did not find any studies comparing the combination of ezetimibe + niacin vs. monotherapy there is a large trial that has examined the effect of adding 2 grams niacin to ezetimibe/simvastatin 10/20 (16). In this study the addition of niacin improved the lipid profile with a marked decrease in TGs and an increase in HDL-C levels (table 4).
Table 4.
Effect of the Addition of Niacin to Ezetimibe/Statin Therapy on Lipid and Lipoprotein Levels
| Change in Lipids with Addition of Niacin 2000mg/day to Ezetimibe/Simvastatin 10/20mg/day | |
|---|---|
| LDL-C | 4.8% Decrease |
| HDL-C | 21.5% Increase |
| TG | 17.6% Decrease |
| Non-HDL-C | 7.3% Decrease |
In patients with marked hypertriglyceridemia combining niacin with other drugs that also lower plasma TGs can be considered. Sixty patients with the metabolic syndrome were randomized to 16 weeks of treatment with placebo, omega-3-fatty acids (Lovaza 4 g/day), extended release niacin (2 g/day), or both drugs in combination (17). In the niacin group TGs were decreased by 30%, in the omega-3-fatty acids group by 22%, and in the combination group by 42% compared to the placebo group. Of note the beneficial effects of niacin on decreasing LDL and non-HDL-C levels were blunted by omega-3-fatty acids, which are known to raise LDL-C levels in patients with marked hypertriglyceridemia (see below). These results show that the combination of niacin and fish oil will lower TG levels more than either drug individually but at the expense of diminishing the effect of niacin on LDL and non-HDL-C levels.
Surprisingly there are few large randomized trials examining the effect of combination therapy with niacin + fibrate vs. monotherapy. One very small trial reported that while both niacin monotherapy and bezafibrate monotherapy were effective in lowering serum TGs there was no statistically significant added benefit of combination therapy in reducing serum TG levels (18). However, a larger trial in HIV+ patients reported that the combination of niacin and fenofibrate was better at lowering TGs and non-HDL-C and increasing HDL-C levels than monotherapy with either niacin or fenofibrate (19). It would be informative if additional trials of combination therapy were carried out in patients with marked hypertriglyceridemia that can often be difficult to control with lifestyle changes and monotherapy.
Mechanisms Accounting for the Niacin Induced Lipid Effects
TRIGLYCERIDES
Early studies demonstrated that niacin inhibited the release of free fatty acids from cultured adipocytes and decreased circulating free fatty acid levels (20-22). The ability of niacin to inhibit adipose tissue lipolysis is mediated by the activation of GPR109A (hydroxycarboxylic acid 2 receptor), a G protein-coupled receptor that is highly expressed in adipose tissue (22-24). It was initially thought that the decrease in plasma TGs induced by niacin therapy was due to niacin inhibiting lipolysis in adipose tissue resulting in a decrease in the transport of fatty acids to the liver leading to the decreased availability of fatty acids for hepatic TG synthesis. However, studies have shown that while niacin acutely decreases plasma free fatty acid levels this inhibition is not sustained (25). Additionally, studies in mice lacking GPR109A have shown that niacin does not inhibit lipolysis but still decreases plasma TG and LDL-C levels (26). Moreover, studies in humans using GPR109A agonists lowered plasma free fatty acid levels but did not cause the expected effects on plasma TGs and LDL-C (26). Thus, the effects of niacin on adipose tissue lipolysis are no longer thought to mediate the niacin induced decrease in plasma TG levels.
Niacin has been shown to inhibit diglycerol acyltransferase 2 (DGAT2) activity in the liver (22,27). DGAT2 is the key enzyme that catalyzes the final step in TG synthesis. Inhibition of DGAT2 will reduce hepatic TG synthesis and the availability of TG for VLDL assembly and secretion (22). A decrease in TG will result in an increase in apolipoprotein B degradation in the liver. Kinetic studies in humans have shown that treatment with niacin decreases VLDL TG production (28,29).
In addition, in animal models, niacin reduces the hepatic expression of apolipoprotein C-III, which could result in the accelerated clearance of TG rich lipoproteins (30). Whether this plays a significant role in mediating the decrease in plasma TG levels induced by niacin therapy remains to be determined.
LOW DENSITY LIPOPROTEIN
The decrease in plasma LDL-C with niacin therapy is thought to be secondary to a reduction in VLDL and LDL formation and secretion by the liver (22).
HIGH DENSITY LIPOPROTEIN
There are multiple potential mechanisms by which niacin may increase HDL-C levels. Some of these changes may be anti-atherogenic while others may be pro-atherogenic. One hypothesis for the increase in HDL induced by niacin therapy is a decrease in the surface expression of hepatocyte beta chain ATP synthase, a receptor that has been proposed to be involved in the uptake of HDL particles by the liver (31). Studies have further shown that niacin inhibits HDL protein degradation by cultured hepatocytes but does not inhibit the selective uptake of cholesterol esters carried in HDL (22,32).
Some kinetic studies have shown that niacin decreases HDL and apolipoprotein A1 fractional catabolic rate (33,34). In contrast, other kinetic studies have shown that niacin increase apolipoprotein AI production (35).
In addition, in monocytes, niacin also increased the expression of ABCA1 and CD36 resulting in an increase in cholesterol efflux to HDL, which would increase HDL-C levels and likely have anti-atherogenic effects (36). Similarly, in vitro studies suggest that niacin may increase the transport of cholesterol and phospholipids via ABCA1 from the liver to lipid poor apolipoprotein A1 particles thereby decreasing the clearance of apolipoprotein A1, which might not be anti-atherogenic (22,37).
Finally, decreasing plasma TG levels may result in a reduction in CETP mediated exchange of TGs on VLDL for cholesterol on HDL leading to an increase in HDL-C levels. Additionally, studies have shown that niacin decreases the expression of CETP (38).
Pharmacokinetics
Oral niacin is well absorbed with immediate release niacin resulting in a rapid increase in plasma levels while extended release and sustained release niacin result in a delayed peak in plasma levels. Niacin undergoes metabolism in the liver by two primary pathways; conjugation or amidation (7,42). The conjugative pathway is low affinity and high capacity that metabolizes niacin to nicotinuric acid while the amidation pathway is high affinity and low capacity that converts niacin into several oxidative-reductive intermediates, which can induce hepatic toxicity (7,42) (Figure 1). The clinical importance is that immediate release niacin results in high levels of niacin and therefore is primarily metabolized by the conjugative pathway (low affinity, high capacity), which does not result in toxic intermediates that can cause liver damage. In contrast, sustained release niacin results in lower levels of niacin for a longer period and therefore metabolism via the amidation pathway (high affinity, low capacity) is dominant leading to an increase in the formation of toxic intermediates that can induce hepatic injury (7,42). Extended-release niacin would be metabolized midway between immediate release and sustained release
niacin (42).
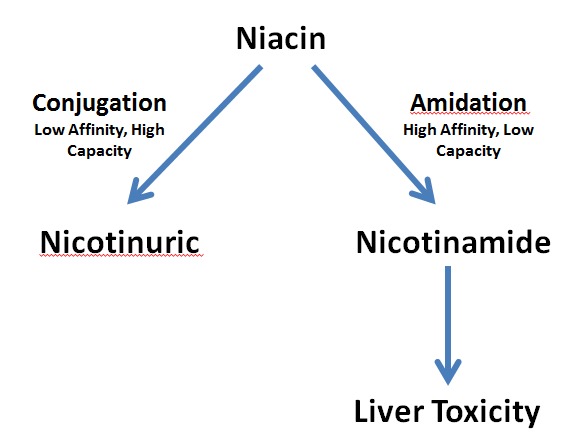
Figure 1.
Pathways of Niacin Metabolism.
Effect of Niacin on Cardiovascular Outcomes
MONOTHERAPY
The Coronary Drug Project, conducted between 1966 and 1975, was the first large randomized, double-blind clinical trial to show that lowering lipids reduced cardiovascular disease (43). This trial determined the effect of clofibrate (1.8g/day), dextrothyroxine (6mg/day), two doses of oral estrogen (2.5 or 5mg per day), or immediate release niacin (3 grams/day) vs. placebo in 8,341 men 30 to 64 years of age with an electrocardiogram documented myocardial infarction. The mean baseline total cholesterol level was 251mg/dL and the TG level was 183mg/dL. The two estrogen regimens and dextrothyroxine treatment groups were discontinued early because of increased adverse effects. Clofibrate treatment did not demonstrate clinical benefit. In the niacin treated patients there was an average 10% decrease in serum cholesterol and 26% decrease in serum TGs despite modest compliance with the study medication. Moreover, niacin treatment (n=1,119) decreased recurrent myocardial infarctions by 26%, stroke by 24%, and revascularization by 67% compared to placebo (n=2,789) but did not decrease total mortality, which was the primary endpoint. Long term follow-up (6.2 years during the study and 8.8 years post study after niacin was discontinued in most participants) demonstrated an 11% decrease in mortality in the niacin group vs. the placebo group (52.0 versus 58.2%; p = 0.0004) (44). The majority of this difference in mortality was accounted for by a decrease in coronary heart disease mortality (36.5% vs. 41.3%; p=0.005). Further analysis revealed that niacin reduced the risk of 6-year recurrent myocardial infarction and coronary heart disease death and 15-year total mortality similarly in patients at all levels of baseline fasting plasma glucose, including those with glucose levels ≥126mg/dL (i.e. patients with diabetes) (45). Additionally, the beneficial effect of niacin on cardiovascular events and total mortality was not diminished, even among those with one hour plasma glucose levels > 220mg/dL (45). Moreover, the beneficial effects of niacin on recurrent myocardial infarction and total mortality were similar in patients with or without the metabolic syndrome at baseline (46). These results demonstrate that immediate release niacin monotherapy decreases recurrent atherosclerotic cardiovascular events in a broad spectrum of patients with pre-existing cardiovascular disease (secondary prevention).
COMBINATION WITH FIBRATES
In the Stockholm Ischemic Heart Disease Secondary Prevention Study survivors of a myocardial infarction below 70 years of age were randomized to a control group (n = 276) (no placebo) and a group treated with clofibrate (2 grams) and immediate release nicotinic acid (up to 3 grams) (n = 279) (47). Serum cholesterol and TG was lowered by 13% and 19%, respectively, in the treatment group compared to the control group. Recurrent myocardial infarction was reduced by 50% within one year (48). Total mortality was decreased by 26% in the group treated with clofibrate + niacin (p< 0.05) while ischemic heart disease mortality was decreased by 36% (p< 0.01). Notably, the benefit of clofibrate + niacin was only observed in patients with a baseline TG level > 143mg/dL. In the age of statins, the clinical implications of this early study are unclear.
COMBINATION WITH STATINS
The AIM-HIGH trial was designed to determine if the addition of Niaspan, an extended-release form of niacin, to aggressive statin therapy would result in a further reduction in cardiovascular events in patients with pre-existing cardiovascular disease (49). In this trial 3,314 patients were randomized to extended-release Niaspan (1500-2000mg/day) vs. placebo that contained 100-150mg of immediate release niacin. On trial, LDL-C levels were in the 60-70mg/dL range in both groups. As expected, HDL-C levels were increased in the Niaspan treated group (approximately 44mg/dL vs. 38mg/dL), while TGs were decreased (approximately 121mg/dL vs. 155mg/dL). However, there were no differences in the primary endpoint between the control and Niaspan treated groups (Primary endpoint consisted of the first event of death from coronary heart disease, nonfatal myocardial infarction, ischemic stroke, hospitalization for an acute coronary syndrome, or symptom-driven coronary or cerebral revascularization). There were also no differences in secondary endpoints except for a possible increase in strokes in the Niaspan treated group. The addition of Niaspan to statin therapy did not result in a significant increase in either muscle or liver toxicity. Thus, this study does not provide support for the addition of niacin to statins. However, most of the patients included in this study did not have a lipid profile that one would typically consider treating with niacin therapy. In the subset of patients with TG > 198mg/dL and HDL-C < 33mg/dL Niaspan treatment showed a trend towards benefit (hazard ratio 0.74; p=0.073), suggesting that if the appropriate patient population was studied the results may have been different (50).
HPS 2 Thrive also studied the effect of niacin added to statin therapy (51). This trial utilized extended-release niacin (2000mg/day) combined with laropiprant, a prostaglandin D2 receptor antagonist, which reduces the flushing side effect of niacin treatment. HPS 2 Thrive was a very large trial with over 25,000 patients randomized to either niacin therapy or placebo. As in the AIM HIGH study, the baseline LDL-C levels were low at 63mg/dL, the HDL-C levels were 44mg/dL, and the TGs were 125mg/dL at baseline. As expected, niacin therapy resulted in a modest reduction in LDL-C (10mg/dL), a modest increase in HDL-C (6mg/dL), and a marked reduction in TGs (33mg/dL) compared to placebo. However, despite these lipid changes there were no significant differences in major cardiovascular events between the niacin and control group (risk ratio 0.96 CI 0.90- 1.03). It is unknown whether laropiprant, the prostaglandin D2 receptor antagonist, might have effects that worsen atherosclerosis and increase event rates. Mice deficient in the prostaglandin D2 receptor have been noted to have an increase in atherogenesis in response to angiotensin II (52). Similar to the AIM-HIGH study, the group of patients included in the HPS 2 Thrive trial may not have been the ideal patient population to study for the beneficial effects of niacin treatment added to statin therapy. Ideally, patients with high TGs and high non-HDL-C levels coupled with low HDL-C levels should be studied.
Thus, these two studies have failed to demonstrate that adding niacin to statin therapy results in a decrease in cardiovascular events. It should be recognized that both the AIM-HIGH study and the HPS-2 Thrive study had limitations. First, the patient populations that were included in these studies were not ideal as the TG and non-HDL-C levels were not elevated in a range that one would usually consider adding niacin therapy. Second, in both trials a significant percentage of patients stopped niacin therapy (AIM-HIGH 25.4% discontinued niacin; HPS-2 Thrive 25.4% discontinued niacin). Third, the duration of these studies was relatively short and it is possible that the beneficial effects of niacin take longer to occur (AIM-HIGH 3 years; HPS-2 Thrive 3.9 years). Fourth, in the HPS-2 Thrive it is possible, as noted earlier, that laropiprant had adverse effects that increased the risk of cardiovascular events. Fifth, in the AIM-HIGH study the placebo contained a low dose of niacin, which may have resulted in beneficial effects. Finally, both of these trials used extended-release niacin, whereas the Coronary Drug Project and the Stockholm Ischemic Heart Disease Secondary Prevention Study used immediate release niacin. It is possible that these different formulations of niacin have different effects on cardiovascular events. Additional studies are required to definitively determine the effect of niacin added to a statin therapy on cardiovascular events.
Effect of Niacin on Atherosclerosis
Many of the initial niacin therapy imaging studies combined niacin with other drugs and compared these combinations vs. placebo. These studies showed that niacin in combination with other drugs reduced the progression and/or increased the regression of atherosclerosis. However, because of the use of other drugs it is impossible to determine if niacin therapy per se was beneficial (Table 5).
Table 5.
Niacin Angiography Imaging Studies
| Combination Studies | Drugs |
|---|---|
| Cholesterol Lowering Atherosclerosis Study (CLAS) (53) | Niacin + colestipol vs. placebo |
| Familial Atherosclerosis Treatment Study (FATS) (54) | Niacin + colestipol or lovastatin + colestipol vs. placebo |
| UCSF-SCORE (55) | Niacin + colestipol +/- lovastatin vs. placebo +/- low dose colestipol |
| HDL Atherosclerosis Study (HATS) (56) | Niacin + simvastatin vs. placebo |
| Armed Forces Regression Study (57) | Niacin + gemfibrozil + cholestyramine vs. placebo |
| Harvard Atherosclerosis Reversibility Project (HARP) (58) | Niacin + pravastatin + cholestyramine + gemfibrozil as needed vs. placebo |
However, there are studies that compared niacin to placebo or other drugs added to standard statin therapy that do provide useful insights (Table 6).
Table 6.
Effect of Niacin Added to Statin Therapy on Atherosclerosis
The ARBITER 2 Trial was a double-blind randomized study of extended-release niacin (1000mg) vs. placebo added to background statin therapy in 167 patients with coronary heart disease and low HDL-C levels (<45mg/dL) (60). At the initiation of the study mean LDL-C levels were < 100mg/dL. The primary end point was the change in common carotid intima-media thickness (CIMT). As expected, plasma TGs decreased and HDL-C levels increased with niacin therapy. LDL-C levels were unchanged. After 12 months, mean CIMT increased significantly in the placebo group (P<0.001) and was unchanged in the niacin group (P=0.23). The overall difference in CIMT progression between the niacin and placebo groups was almost statistically significant (P=0.08). Cardiovascular events occurred in 3 patients treated with niacin (3.8%) and 7 patients treated with placebo (9.6%; P=0.20). ARBITER 3 was a 12-month extension and in the 57 patients that continued on niacin therapy there was an additional regression of CIMT (p = 0.001 vs. placebo) (59).
In ARBITER 6, patients with coronary heart disease or a coronary heart disease risk equivalent on long-term statin therapy with LDL-C level < 100mg/dL and an HDL-C level < 50mg/dL for men or 55mg/dL for women were randomly assigned to receive either extended-release niacin (target dose, 2000mg per day) or ezetimibe (10mg per day) (61). The primary end point was the change from baseline in the mean CIMT. LDL-C levels decreased in the ezetimibe group by −18mg/dL (~ 20%) and by −10.0mgdl (~ 12%) in the niacin group (P=0.01) while HDL-C levels were slightly decreased in the ezetimibe group −2.8mg/dL and increased by 7.5mg/dL (~18%) in the niacin group (P<0.001). TG levels were not markedly altered in the ezetimibe group but decreased by ~ 15-20% in the niacin group. Notably niacin therapy resulted in a significant reduction of both mean (P = 0.001) and maximal CIMT (P < 0.001) while ezetimibe therapy significantly increased CIMT (P < 0.001). The incidence of major cardiovascular events was lower in the niacin group than in the ezetimibe group (1% vs. 5%, P = 0.04).
In a trial by Thoenes and colleagues fifty patients with the metabolic syndrome not on statin therapy were randomized to either extended-release niacin (1000mg/day) or placebo (62). Treatment with niacin decreased LDL-C by 17% and TGs by 23% and increased HDL-C levels by 24% without significant changes in the placebo group. After 52 weeks of treatment, there was an increase in CIMT of +0.009 +/- 0.003 mm in the placebo group and a decrease in CIMT of -0.005 +/- 0.002 mm in the niacin group (p = 0.021 between groups).
Finally, Lee and colleagues performed a double-blind, randomized study of 2 g daily modified-release niacin or placebo added to statin therapy in 71 patients with low HDL-C (<40mg/dL) and either: 1) type 2 diabetes with coronary heart disease; or 2) carotid/peripheral atherosclerosis (63). The primary end point was the change in carotid artery wall area, quantified by magnetic resonance imaging, after 1 year. Treatment with niacin increased HDL-C by 23% and decreased LDL-C by 19% and TGs by 11%. At 12 months, niacin significantly reduced carotid wall area compared with placebo (Mean change in carotid wall area was -1.1 +/- 2.6 mm2 for niacin vs +1.2 +/- 3.0 mm2 for placebo).
While these imaging studies provide data suggesting that niacin therapy when added to statin therapy may reduce atherosclerotic cardiovascular disease, one must recognize that the studies described above were relatively small studies and that decreases or the lack of progression in CIMT or carotid wall area are surrogate markers, which may not necessarily indicate that cardiovascular events will be decreased.
Side Effects
Treatment with niacin frequently results in side effects and these side effects are a major limitation of niacin therapy.
SKIN FLUSHING
This is a very common side effect and is characterized by redness and warmth due to vasodilation of the blood vessels in the skin (8,64). It is often most apparent in the head and neck region. Itching can occur and a tingling and burning sensation may also be noted. Niacin induced flushing is usually not accompanied by diaphoresis. The cutaneous flushing usually lasts for approximately one hour and in some patients is extremely annoying. In a review of 30 studies, it was noted that flushing occurred in 85% of participants treated with immediate release niacin, 66% of participants treated with extended release niacin, and 26% of participants treated with slow release niacin (11). The occurrence of flushing is related to a rapid increase in plasma nicotinic acid levels, which differs depending upon the niacin preparation. Flushing was the primary reason that subjects discontinued niacin therapy during studies and with either immediate release or extended release niacin approximately 20% of study participants discontinue niacin, which is twice the rate of discontinuation observed in the placebo groups (11). Continuous administration of niacin for approximately one- week results in tachyphylaxis and the flushing decreases. Unfortunately, if a patient skips taking niacin for a few days this tachyphylaxis is lost and the flushing returns.
The mechanism for the niacin induced skin flushing has been partially elucidated (8,64). Niacin activates GPR109A in dermal Langerhan cells (macrophages in the skin), which leads to the increased production of prostaglandin D2. Additionally, niacin activates GPR 109A in keratinocytes, which leads to the production of prostaglandin E2. The prostaglandins then interact with prostaglandin receptors on blood vessels resulting in vasodilation and the flushing phenomena. Aspirin and nonsteroidal anti-inflammatory drugs (NSAIDS) taken prior to niacin administration can decrease flushing by inhibiting the synthesis of prostaglandins (8,65). Laropiprant decreases flushing by blocking the D prostanoid receptor (8). Since flushing is related to rapid increases in plasma nicotinamide levels taking immediate release niacin with food slows absorption and thereby reduces flushing. Extended-release niacin is typically taken at bedtime so that the flushing will occur when the patient is asleep. Conditions that predispose to cutaneous vasodilatation such as alcohol intake, hot liquids, spicy foods, or hot showers should be avoided. One should increase the dose of niacin slowly to reduce the severity of flushing reactions and allow tolerance to develop.
HEPATIC TOXICITY
Sustained release niacin has a much greater propensity to induce hepatic toxicity than other niacin preparations and therefore is no longer widely used (7,42,66). The explanation for this difference is due to the increased metabolism of sustained release niacin by the amidation pathway described in the pharmacokinetics section, which results in toxic compounds that injure the liver (7,42). Patients who have developed signs of liver toxicity on sustained release niacin can often be treated with immediate release niacin without developing liver problems (67). Extended-release niacin can induce liver dysfunction but the rate is much lower than sustained release niacin. Because of the potential for liver disease, serum transaminase levels (SGOT and SGPT) should be monitored before treatment begins, every 6 to 12 weeks for the first year, and periodically thereafter (e.g., at approximately 6-month intervals).
It should be noted that there is some evidence that niacin may be beneficial for non-alcoholic fatty liver disease (NAFLD) but further studies are required (68).
MUSCLE SYMPTOMS
Myalgias and myopathy have not been a significant adverse effect with niacin monotherapy (11). In combination with statins, an increased risk of muscle symptoms has been observed in some studies. In the HPS-2 Thrive study the combination of simvastatin and extended-release niacin increased the risk of myopathy four-fold (1.2% of patients on combined therapy) (51). Of note, this increase occurred predominantly in Chinese participants. In contrast, in the AIM-HIGH trial muscle related symptoms were not increased with the simvastatin + niacin combination (49,69).
HYPERGLYCEMIA
It has been recognized for many years that niacin induces insulin resistance (70). The mechanisms by which niacin induces insulin resistance are unknown but possible mechanisms include a rebound increase in free fatty acids with niacin therapy or the accumulation of diacylglycerol (29,71). A recent analysis of the AIM-HIGH trial demonstrated that in subjects with normal glucose metabolism, subjects with impaired fasting glucose, and subjects with diabetes, treatment with extended release niacin resulted in only small increases in fasting glucose levels but increased serum insulin levels due to an increase in insulin resistance (72). Additionally, there was an increased risk of progressing from normal to impaired fasting glucose in subjects treated with niacin in the AIM-HIGH trial (niacin 58.6% vs placebo 41.5%; P < .001) (72).
A meta-analysis examined the effect of niacin therapy on the development of new onset diabetes (73). In 11 trials with 26,340 non-diabetic participants, niacin therapy was associated with a 34% increased risk of developing diabetes (RR of 1.34; 95% CIs 1.21 to 1.49). This increased risk results in one additional case of diabetes per 43 initially non-diabetic individuals who are treated with niacin for 5 years (0.47% ten-year risk or 4.7 per 1000 patient years). Results were similar in patients who were receiving niacin therapy in combination with statin therapy.
Studies have shown that niacin is usually well tolerated in diabetic subjects who are in good glycemic control (74,75). In patients with poor glycemic control, niacin is more likely to adversely impact glucose levels. A meta-analysis of 7 studies with 838 patients with diabetes found that niacin therapy did not result in a significant increase in fasting glucose levels in short term studies but in long term studies there was a very small increase in fasting glucose levels (1.5mg/dL) that was not clinically significant (76). An important caveat is that in most of these trials adjustments in diabetes therapy was permitted, which could blunt worsening of glycemic control. In contrast to these findings, the HPS-2 Thrive Trial reported that in the 8,299 participants who had diabetes at the time of randomization, treatment with niacin–laropiprant was associated with a 55% increase in serious disturbances in diabetes control, most of which led to hospitalization (11.1% vs. 7.5%, P<0.001) (51). The extent to which the latter was due to laropiprant is unknown. Thus, care must be used in treating patients with diabetes with niacin. In patients in whom adjustments in diabetic therapy can easily be carried out the risk of adverse effects will likely be limited whereas in patients in whom adjustments in diabetic therapy will be difficult the risks of niacin therapy are likely to be increased. Careful patient selection and education are important steps to reduce the risks of niacin therapy in patients with diabetes.
Thus, while niacin therapy may adversely affect glucose homeostasis one needs to balance these adverse effects with the potential benefits of niacin therapy. One should note that in the Coronary Drug Project participants with abnormal glucose metabolism also demonstrated a decrease in cardiovascular events with niacin therapy (45).
URIC ACID
Niacin may increase uric acid levels by inhibiting the secretion of uric acid (8,77). In susceptible patients niacin therapy can precipitate gouty attacks (8).
GASTROINTESTINAL SYMPTOMS
Niacin therapy can induce heartburn, indigestion, nausea, diarrhea, and abdominal discomfort (8). High dose niacin is more likely to cause these gastrointestinal disturbances. The mechanism for these symptoms is not clear.
MISCELLANEOUS
Recent trials have reported an increased incidence of infections with niacin therapy (51,69). A trial of niacin in combination with laropiprant found increased bleeding (51). The increased bleeding could be due to the approximately 10% decrease in platelet levels that can occur with niacin (see Niaspan Package Insert). However, a very large observational study that compared rates of major gastrointestinal bleeding and intracranial hemorrhage in patients treated with niacin (>200,000 subjects) to propensity matched subjects on fenofibrate did not observe an increase in bleeding (78). Niacin has been reported to induce cystoid macular edema, which resolves when the drug is stopped (79).
Contraindications
There are a number of contraindications to niacin therapy (Table 7).
Table 7.
Contraindications for Niacin Therapy
| Active gastritis or peptic ulcer disease |
| Impaired liver function (elevated transaminases 2-3X the upper limit or cholestasis) |
| Uncontrolled gout |
| Pregnancy |
| Lactation |
| Poorly controlled diabetes |
| Active bleeding |
Summary
The enthusiasm for the use of niacin has greatly decreased with the failure of AIM-HIGH and HPS-2 Thrive to show a decrease in cardiovascular events when niacin was added to statin therapy. In the absence of definitive data showing benefits from niacin therapy when added to a statin it is hard to justify the use of this drug given the frequent side effects. The availability of ezetimibe, bempedoic acid, and PCSK9 inhibitors has greatly reduced the need to use niacin to lower LDL-C levels. Additionally, in patients with markedly elevated TG levels (>500mg/dL), niacin can be employed in combination with other drugs to reduce the risk of pancreatitis but fibrates and omega-3-fatty acids are the initial choices.
OMEGA-3-FATTY ACIDS (FISH OIL)
Introduction
The lipid lowering effects of fish oil are mediated by two omega-3-fatty acids; eicosapentaenoic acid (C20:5n-3) (EPA) and docosahexaenoic acid (C22:6n-3) (DHA). There are four prescription products approved by the FDA which contain various amounts of EPA and DHA (Table 8). Lovaza and Omacor contain a mixture of EPA and DHA fatty acid esters (ethyl esters), Vascepa contains only EPA fatty acid esters (ethyl esters), and Epanova contains a mixture of EPA and DHA free fatty acids (Epanova is currently not available in the US).
Table 8.
Prescription Omega-3-Fatty Acid Products (data from package inserts)
| Generic Name | Omega-3-ethyl esters | Icosapent ethyl | Omega-3-carboxylic acid |
|---|---|---|---|
| Brand Name | Lovaza or Omacor | Vascepa | Epanova |
| EPA/capsule | 0.465g | 1.0g | See below |
| DHA/capsule | 0.375g | --- | See below |
| Daily Dose | 4 capsules/day | 4 capsules/day | 2-4 capsules/day |
1-gram capsules of Epanova contain at least 850mg of fish oil derived fatty acids including multiple omega-3-fatty acids with EPA and DHA being the most abundant.
Fish oil is also sold as a food supplement. It should be recognized that dietary fish oil supplements are not approved by the FDA and quality control will not meet the same rigorous standards as prescription or over the counter drugs. The amount of EPA and DHA can vary greatly in these supplements and one needs to read the labels carefully, as products can contain less than 100mg of EPA/DHA per 1 gram capsule (80). It is helpful to have the patient bring their fish oil supplements to the clinic for verification of the actual amount of EPA and DHA in the product. Moreover, the amount of EPA and DHA indicated on the label may not be accurate (81). One needs to take a sufficient number of capsules to provide 2-4 grams of EPA/DHA per day to effectively lower plasma TG levels. Depending upon the fish oil supplement, the patient may be required to take a large number of capsules to obtain 2-4 grams of EPA/DHA per day. Furthermore, these supplements may contain other compounds in addition to omega-3-fatty acids, such as cholesterol, oxidized lipids, and saturated fatty acids. The major advantage of fish oil supplements is that they are much less expensive than prescription omega-3-fatty acid drugs. If one elects to use fish oil supplements, one should have the patient use a single brand to try to ensure as much consistency as possible.
Some omega-3 supplements contain alpha linolenic acid (C18:3n-3) (ALA), a plant omega-3-fatty acid rather than EPA/DHA. ALA can be converted to EPA and DHA but the conversion is limited and hence it is ineffective in lowering plasma TG levels or altering other lipid or lipoprotein levels (82).
Effect of Omega-3-Fatty Acids on Lipid and Lipoprotein Levels
Table 9.
Effect of Fish Oil Supplements on Lipids and Lipoproteins
| Decreases TGs |
| No Change in Total Cholesterol |
| No Change in LDL-C; if TGs are very high may increase LDL-C |
| No Change in HDL-C |
| No Change in Lp(a); modest decrease in some studies |
| Shift from Small Dense LDL to Large Buoyant LDL |
Several meta-analyses have examined the effect of fish oil supplements on lipid and lipoprotein levels. A meta-analysis by Eslick and colleagues of 47 studies with 16,511 participants found that fish oil supplements significantly decreased plasma TG levels by approximately 14% without resulting in clinically significant changes in total, LDL-C, or HDL-C levels (83). These authors also reported that the reduction in plasma TG levels was directly related to baseline plasma TG levels (i.e., the higher the baseline TG level the greater the reduction in TGs with fish oil). Additionally, the higher the dose of EPA/DHA, the greater the reduction in plasma TGs, with clinically significant reductions occurring with approximately 3.25 grams per day. A meta-analysis by Balk and colleagues of 21 studies also found minimal effects of fish oil supplements on total, LDL-C, and HDL-C levels (< 5% change) with significant decreases in plasma TG levels (most of the studies in this meta-analysis had at least a 15% decrease) (84). Similar to the meta-analysis by Eslick et al, the higher the baseline TG levels the greater the reduction in TG levels.
Several meta-analyses have focused on specific patient populations. In a meta-analysis of patients with diabetes, twenty three trials with1075 participants were analyzed and similar to patients without diabetes the major effect of fish oil supplements was a reduction in plasma TG levels with no change in total cholesterol or HDL-C (85). A small increase in LDL-C was observed (4.3mg/dL). Of note, fish oil supplementation did not alter fasting glucose or glycated hemoglobin levels indicating that fish oil supplementation does not adversely affect glycemic control. In a meta-analysis that included patients with type 2 diabetes or impaired glucose metabolism a decrease in TGs was observed without significant changes in total cholesterol, LDL-C, or HDL-C levels (86). Again, no adverse effects on glycemic control were observed.
In patients with end stage renal disease several meta-analyses have consistently shown a decrease in plasma TGs with fish oil administration but the effect on total, LDL-C, and HDL-C has been variable (87-89). This variability was likely due to the small changes that were observed. In patients with nephrotic syndrome a study has shown a reduction in plasma TGs and an increase in LDL-C levels without a change in total cholesterol or HDL-C levels (90). In patients with non-alcoholic fatty liver disease, omega-3-fatty acids have also been shown to decrease plasma TG levels (91). Finally, In HIV infected subjects, fish oil supplementation was also effective in lowering plasma TG levels (92,93).
Thus, fish oil supplementation in a variety of different patient populations lowers plasma TG levels. In patients with elevated TG levels treated with 3-4 grams of EPA/DHA one can expect an approximate 25% decrease. Total plasma cholesterol levels are usually not altered by fish oil supplementation. The exceptions are patients with high chylomicron and/or VLDL levels where a substantial portion of the plasma cholesterol is carried on these TG rich lipoproteins. Fish oil supplementation will decrease the levels of these TG rich lipoproteins and thereby result in a decrease in total plasma cholesterol levels. LDL-C levels are not markedly affected by fish oil supplementation except in patients with very high TG levels (>500mg/dL) where increases in LDL-C levels have been observed (94-96). If there are sufficient reductions in plasma TG levels a shift from small dense LDL to large buoyant LDL may be observed (97,98). The effect of fish oil supplements on HDL-C levels is minimal except if the patient has very high TG levels where significant elevations (>10%) have been reported (94-96). Finally, some but not all studies have shown that the administration of fish oil modestly lowers Lp(a) levels (99-103)
During the development of pharmacological omega-3-fatty acid drugs for approval by the FDA, extensive clinical trials were carried out and will be reviewed below (Tables 10 and 11). It should be noted that these studies are not directly comparable as they studied different patient populations at different times.
EPA + DHA FATTY ACID ESTERS (LOVAZA)
In patients with marked elevations in plasma TG levels (500-2000mg/dL) a 6 week trial of EPA + DHA esters resulted in a 31% decrease in plasma TGs, a 21% increase in LDL-C levels, and a 12% increase in HDL-C levels compared to the placebo group (96). In a 16 week trial TG concentrations were decreased by 45% and LDL-C and HDL-C were increased by 31% and 13%, respectively (94). Studies have also been carried out in patients with moderate hypertriglyceridemia (200-500mg/dL) who were on statin therapy (104). EPA + DHA esters resulted in a 23% decrease in plasma TGs and a 7% decrease in non-HDL-C levels, and a 4.6% increase in HDL-C levels (104).
EPA FATTY ACID ESTER ALONE (VASCEPA)
In patients with marked elevations in plasma TGs (500-2000mg/dL), 4 grams of EPA ester alone significantly decreased TG levels by 33.1% and non-HDL-C levels by 17.7% (105). In contrast to EPA and DHA fatty acid esters, LDL-C and HDL-C levels were not significantly altered by EPA fatty acid esters alone (105). Studies have also been carried out in patients with moderate hypertriglyceridemia (200-500mg/dL) who were on statin therapy. EPA esters resulted in a 21.5% decrease in plasma TGs, 13.6% decrease in non-HDL-C, 6.2% decrease in LDL-C, and a 4.5% decrease in HDL-C levels (106)
EPA + DHA FATTY ACIDS (EPANOVA)
In patients with marked elevations in plasma TGs (500-2000mg/dL), 4 grams of EPA + DHA fatty acids decreased plasma TGs by 31% and non-HDL-C by 9.6% and increased LDL-C by 19% and HDL-C by 5.8% (107). Studies have also been carried out in patients with moderate hypertriglyceridemia (200-500mg/dL) who were on statin therapy. EPA + DHA fatty acids resulted in a 20.6% decrease in plasma TGs, 6.9% decrease in non-HDL-C with no significant changes in LDL-C or HDL-C levels (95).
These studies demonstrate that in patients on statin therapy with moderate elevations in plasma TG levels the effects of these three pharmaceutical products on lipids and lipoprotein levels are similar (table 11). However, in patients with marked elevations in plasma TG levels EPA ethyl esters alone do not increase LDL-C levels whereas products containing EPA and DHA result in a substantial increase in LDL-C levels (table 10). It should also be noted that the ability of omega-3-fatty acids to reduce plasma TGs and increase HDL-C levels is enhanced if baseline TG levels are markedly elevated.
Table 10.
Effect of Omega-3-Fatty Acids on Lipids and Lipoprotein in Patients with Marked Hypertriglyceridemia (500-2000mg/dL)
| TGs | Non-HDL-C | LDL-C | HDL-C | |
|---|---|---|---|---|
| EPA+DHA ethyl esters- 6 weeks | 31% decrease | ND | 21% increase | 12% increase |
| EPA+DHA ethyl esters 12 weeks | 45% decrease | ND | 31% increase | 13% increase |
| EPA ethyl esters | 33% decrease | 18% decrease | NS | NS |
| EPA+DHA fatty acids | 31% decrease | 9.6% decrease | 19% increase | 5.8% increase |
ND- not determined; NS- no significant change.
Table 11.
Effect of Omega-3-Fatty Acids on Lipids and Lipoprotein in Patients with Moderate Hypertriglyceridemia (200-500mg/dL) on Statin Therapy
| TGs | Non-HDL-C | LDL-C | HDL-C | |
|---|---|---|---|---|
| EPA+DHA ethyl esters | 23% decrease | 7% decrease | __ | 4.6% increase |
| EPA ethyl esters | 22% decrease | 14% decrease | 6.2% increase | 4.5% decrease |
| EPA+DHA fatty acids | 21% decrease | 6.9% decrease | NS | NS |
NS- no significant change.
HEAD-TO-HEAD COMPARISONS
A meta-analysis of six studies has compared the effect of EPA alone vs. DHA alone on plasma lipids and lipoproteins (108). Administration of DHA increased LDL-C by 4.6mg/dL compared to EPA (95% CI 2.2- 7.1). In contrast, DHA reduced plasma TG levels to a greater extent than EPA (6.1mg/dL; 95% CI 2.5- 9.8). Finally, DHA increased HDL-C levels more than EPA (3.7mg/dL; 95% CI: 2.4- 5.1). Whether these very modest differences are clinically significant is unknown.
Tatsuno et al compared the effect of DHA + EPA ethyl esters vs. EPA ethyl esters alone on lipid and lipoprotein levels in patients with mean baseline plasma TG of 250-270mg/dL and mean LDL-C levels of 125-135mg/dL (109,110). These authors found that at equivalent doses there were no differences in effect on plasma TG, LDL-C, or HDL-C levels between DHA + EPA ethyl ester or EPA ethyl ester treatment.
These head-to-head studies indicate that in subjects with moderate hypertriglyceridemia the effects of EPA and DHA on lipid and lipoprotein levels are similar. Perhaps if the baseline TGs were markedly elevated differences in response might have been observed.
IN COMBINATION WITH FENOFIBRATE
In patients with marked hypertriglyceridemia a single drug is often not sufficient to lower TGs into the desired range. In patients with TG levels > 500mg/dL the combination of fenofibrate 130mg per day and 4 grams of Lovaza reduced TG levels to a greater extent than monotherapy with fenofibrate alone (7% decrease; P = 0.059) (111). Not unexpectedly, LDL-C levels were increased to a greater extent with combination therapy (9% increase; p= 0.03). When subjects who had received 8 weeks of fenofibrate monotherapy were treated with Lovaza during the 8-week, open-label extension study, TG levels were reduced by an additional 17.5% (P = 0.003). These results indicate that the addition of omega-3-fatty acids to fenofibrate will further decrease TG levels.
IN COMBINATION WITH NIACIN
Sixty patients with the metabolic syndrome were randomized to 16 weeks of treatment with placebo, Lovaza (4 g/day), extended release niacin (2 g/day), or both drugs in combination (17). In the niacin group TGs were decreased by 30%, in the omega-3-fatty acids group by 22%, and in the combination group by 42% compared to the placebo group. Of note, the beneficial effects of niacin on decreasing LDL and non-HDL-C were blunted by omega-3-fatty acids. These results show that the combination of niacin and fish oil will lower TG levels more than either drug individually but at the expense of diminishing the effect of niacin on LDL and non-HDL-C levels.
Mechanism Accounting for the Omega-3-Fatty Acid Induced Lipid Effects
As noted above, the major effect of fish oil is to lower plasma TG levels. The predominant cause of the reduction in plasma TG levels is a decrease in the hepatic production and secretion of TG rich lipoproteins (112-115). In cultured hepatocytes, omega-3-fatty acids inhibit the assembly and secretion of VLDL and apolipoprotein B 100 (113,115-117). The incorporation of TGs into VLDL is a key regulatory step in determining the rate of formation and secretion of VLDL and there are a number of mechanisms by which omega-3 fatty acids reduce the level of hepatic TGs available for VLDL formation (112,113,115). Studies in animal models have demonstrated that omega-3-fatty acids inhibit fatty acid synthesis and stimulate fatty acid oxidation in the liver, which would reduce the availability of fatty acids for TG synthesis (112-115). The increase in fatty acid oxidation is due to omega-3-fatty acids activating PPAR alpha, which stimulates fatty acid oxidation in the liver and other tissues (112,114,115,118). The decrease in fatty acid synthesis is due to omega-3-fatty acids inhibiting the expression of SREBP-1c, a key transcription factor that regulates fatty acid synthesis (114,115,118). In addition, omega-3-fatty acids decrease TG synthesis, which may be due to the decreased availability of fatty acids and an inhibition of the activity of DGAT, a key enzyme required for TG synthesis (112,114,115). Finally, omega-3-fatty acids also decrease the flux of free fatty acids from adipose tissue to the liver, which will lead to a decreased quantity of fatty acids available for TG synthesis in the liver (112). This decrease in flux of free fatty acids is due to omega-3-fatty acids reducing hormone sensitive lipase mediated intracellular lipolysis in adipose tissue (112). It is likely that these and perhaps other factors lead to the decreased availability of TGs resulting in a reduction in VLDL formation and secretion. In addition, the peroxidation of omega-3-fatty acids may stimulate the degradation of apolipoprotein B-100, which would provide another pathway that could contribute to a decrease in VLDL formation and secretion (115).
While not the primary mechanism for the decrease in plasma TGs, studies have shown that omega-3-fatty acids may increase the clearance of TG rich lipoproteins (112,119). Post heparin lipoprotein lipase activity is not increased by omega-3-fatty acid administration but the lipolytic activity of non-stimulated plasma is enhanced (112,119). Additionally, apolipoprotein C-III levels are decreased with omega-3-fatty acid administration which could also contribute to an increase in the clearance of TG rich lipoproteins (120-123).
The increase in LDL-C levels that occurs in patients with marked hypertriglyceridemia treated with omega-3-fatty acids is thought to be due to the enhanced conversion of VLDL to LDL (114). The increase in HDL-C observed in studies in patients with very high TG levels may be due to the increased clearance of TG rich lipoproteins.
Pharmacokinetics and Drug Interactions
Omega-3 ethyl esters and fatty acids are absorbed by the GI tract similar to other dietary lipids. It is worth noting that omega-3-free fatty acids (Epanova) are directly absorbed by the small intestine and are not dependent on pancreatic lipases for absorption. Thus, absorption of omega-3-fatty acids is not decreased in patients with pancreatic insufficiency and therefore may be preferred in patients with pancreatic disease. Additionally, the bioavailability of omega-3-fatty acids with a low fat diet was greater than omega-3-ethyl esters while there was little difference between these different formulations with a high fat diet (124,125).
Drug interactions have not been seen with omega-3-fatty acids (Package Inserts for Lovaza, Vascepa, and Epanova).
Effect of Low Dose Omega-3-Fatty Acids on Clinical Outcomes
Initial studies of the effect of low dose fish oil administration on cardiovascular outcomes were favorable, demonstrating a reduction in events including all-cause mortality. However, more recent studies have failed to confirm these favorable results. In these more recent studies the use of other drugs, such as statins, that reduce cardiovascular disease were more intensively utilized. The outcomes studies that will be described below were carried out with doses of EPA and DHA that are lower than the doses used to lower plasma TGs. We will limit our discussion to the administration of fish oil as a drug and not discuss diet studies, such as DART, which had patients increase fatty fish intake (126,127).
1) GISSI-Prevenzione trial was a randomized trial of 850-882mg of EPA and DHA ethyl esters per day in 11,323 participants with a recent myocardial infarction (< 3 months) for 3.5 years (128). The primary endpoint was death, non-fatal myocardial infarction, and stroke. No change in total cholesterol, LDL-C, or HDL-C was observed but plasma TG levels were decreased by 5%. Patients treated with EPA/DHA had a significant decreased risk of major cardiovascular events (RR 0.90), cardiac death (RR 0.78), and sudden death (RR 0.74). The decrease in sudden death occurred very quickly and was noted as early as 4 months after initiation of therapy. Interestingly, non-fatal cardiovascular events were not affected by EPA/DHA treatment (RR 0.98). The decrease in total mortality was driven by a reduction in sudden death suggesting an anti-arrhythmic effect of EPA/DHA.
2) GISSI-Heart Failure (GISSI-HF) trial was a randomized, double-blind, placebo-controlled trial in patients with chronic heart failure who were randomly assigned to 850-882mg of EPA and DHA ethyl esters per day (n=3,494) or placebo (n=3,481) (129). Patients were followed for a median of 3.9 years. Primary endpoints were time to death, and time to death or admission to the hospital for cardiovascular reasons. Omega-3-fatty acid treatment at these low doses resulted in a slight decrease in plasma TG levels with no change in total, LDL-C or HDL-C levels. In the omega-3-fatty acid group 27% patients died from any cause vs. 29% in the placebo group (HR 0.91; p=0.041). In the omega-3-fatty acid group 57% of patients died or were admitted to hospital for cardiovascular reasons vs. 59% in the placebo group (HR 0.92; p=0.009). No significant differences were observed in fatal or non-fatal myocardial infarctions or strokes. In this trial, similar to the GISSI-Prevenzione trial, the benefit was primarily due to a reduction in arrhythmic events and little benefit on atherothrombotic events was noted.
3) OMEGA was a randomized, placebo-controlled, double-blind, trial in 3,851 survivors of an acute myocardial infarction (130). Patients were randomized 3 to 14 days after an acute myocardial infarction to omega-3-acid ethyl esters, 1 gram/day (460mg EPA and 380mg DHA) or placebo capsules containing 1 gram of olive oil and followed for one year. The primary endpoint was rate of sudden death and secondary end points were total mortality and nonfatal clinical events. No significant differences were seen in the primary or secondary endpoints.
4) Alpha Omega was a double-blind, placebo-controlled trial in 4,837 patients between 60 and 80 years of age (78% men) who had had a myocardial infarction (131). Patients were randomized to receive for 40 months one of four trial margarines: a margarine supplemented with a combination of EPA and DHA (with a targeted additional daily intake of 400mg of EPA-DHA; actual intake 226mg EPA and 150mg DHA), a margarine supplemented with alpha-linolenic acid (ALA) (with a targeted additional daily intake of 2g of ALA), a margarine supplemented with EPA-DHA and ALA, or a placebo margarine. The primary end point was the rate of major cardiovascular events, which comprised fatal and nonfatal cardiovascular events and cardiac interventions. Neither low dose EPA-DHA, ALA, nor the combination of EPA/DHA and ALA significantly reduced the rate of major cardiovascular events or cardiac interventions.
5) SU.FOL.OM3 Study was a double blind, randomized, placebo-controlled trial in 2,501 patients with a history of a myocardial infarction, unstable angina, or ischemic stroke in the past 12 months (132). Patients were randomized to a daily dietary supplement containing 5-methyltetrahydrofolate (560μg), vitamin B-6 (3mg), and vitamin B-12 (20μg) or placebo; and a dietary supplement containing omega 3 fatty acids (600mg of EPA and DHA) or placebo. Median duration of treatment was 4.7 years. The primary outcome was a composite of non-fatal myocardial infarction, stroke, or death from cardiovascular disease. Treatment with B vitamins or omega 3 fatty acids had no significant effect on major vascular events.
6) Origin was a double-blind study in 12,536 patients at high risk for cardiovascular disease who had impaired fasting glucose, impaired glucose tolerance, or diabetes (133). Patients were randomized to receive a 1-gram capsule containing at least 900mg of ethyl esters of omega-3 fatty acids (EPA 465mg and DHA 375mg) or placebo for approximately 6 years. The primary outcome was death from cardiovascular causes. TG levels were reduced by 14.5mg/dL in the group receiving omega-3-fatty acids compared to the placebo group (P<0.001), without a significant effect on other lipids. The incidence of the primary outcome was not significantly decreased among patients receiving omega-3-fatty acids as compared with those receiving placebo. The use of omega-3-fatty acids also had no significant effect on the rates of major vascular events, death from any cause, or death from arrhythmia.
7) Risk and Prevention Study was a double-blind, placebo-controlled trial in 12,513 men and women with multiple cardiovascular risk factors or atherosclerotic vascular disease but not myocardial infarctions (134). Patients were randomly assigned to 1-gram daily omega-3 fatty acids (EPA and DHA content not <85%,) or placebo (olive oil) for 5 years. The initially specified primary end point was the rate of death, nonfatal myocardial infarction, and nonfatal stroke. At 1 year, after the event rate was found to be lower than anticipated, the primary end point was revised as time to death from cardiovascular causes or admission to the hospital for cardiovascular causes. Plasma TG levels decreased slightly more in the omega−3-fatty acid group than in those who received placebo (−28.2±1.3mg/dL vs. −20.1±1.3mg/dL; P<0.001). Total, LDL, and HDL-C levels were similar in the omega-3-fatty acid and placebo groups. No significant differences were observed between the omega-3-fatty acid group and placebo group for the primary endpoint or any of the secondary endpoints.
8) A Study of Cardiovascular Events in Diabetes (ASCEND) was a randomized, placebo controlled, double blind trial of 1-gram omega-3-fattys acids (400mg EPA and 300mg DHA ethyl esters) vs. olive oil placebo in 15,480 patients with diabetes without a history of cardiovascular disease (primary prevention trial) (135). The primary end point was serious vascular events (non-fatal myocardial infarction, non-fatal stroke, transient ischemic attack, or vascular death). Total cholesterol, HDL-C, and non-HDL-C levels were not significantly altered by omega-3-fatty acid treatment (changes in TG levels were not reported). After a mean follow-up of 7.4 years the composite outcome of a serious vascular event or revascularization occurred in 882 patients (11.4%) on omega-3-fatty acids and 887 patients (11.5%) on placebo (rate ratio, 1.00; 95% CI, 0.91 to 1.09). Serious adverse events were similar in placebo and omega-3-fatty acid treated groups.
9) The Vitamin D and Omega-3 Trial (Vital) was a randomized, double blind, placebo-controlled trial of 1-gram omega-3 fatty acids (465mg EPA and 375mg DHA ethyl esters) vs. placebo in 25,875 men (>50 years of age) and women (>55 years of age) that were not selected on the basis of an elevated risk (primary prevention) (136). Changes in lipid levels were not reported. The primary end point was major cardiovascular events, a composite of myocardial infarction, stroke, or death from cardiovascular causes. After a median follow-up of 5.3 years, major cardiovascular event occurred in 386 participants in the omega-3 fatty acid group and in 419 in the placebo group (hazard ratio, 0.92; 95% confidence interval (CI), 0.80 to 1.06; P=0.24). Serious adverse events were similar in placebo and omega-3-fatty acid treated groups.
10) Summary: The above results indicate that low dose fish oil (doses that do not greatly affect lipid levels) do not consistently reduce the risk of cardiovascular disease.
Effect of High Dose Omega-3-Fatty Acids on Clinical Outcomes
1) Japan EPA Lipid Intervention Study (JELIS) was an open label study without a placebo in patients with total cholesterol levels > 254mg/dL with (n= 3,664) or without cardiovascular disease (n=14,981) who were randomly assigned to be treated with 1800 mg of EPA (Vascepa) + statin (n=9,326) or statin alone (n= 9,319) with a 5-year follow-up (130). The primary endpoint was any major coronary event, including sudden cardiac death, fatal and non-fatal myocardial infarction, and other non-fatal events including unstable angina pectoris, angioplasty, stenting, or coronary artery bypass grafting. Total, LDL-C, and HDL-C levels were similar in the two groups but plasma TGs were modestly decreased in the EPA treated group (5% decrease in EPA group compared to controls; p = 0.0001). In the EPA group the primary endpoint occurred in 2.8% of the patients vs. 3.5% of the patients in the statin alone group (19% decrease; p = 0.011). Unstable angina and non-fatal coronary events were also significantly reduced in the EPA group but in this study sudden cardiac death and coronary death did not differ between groups. Unstable angina was the main component contributing to the primary endpoint and this is a more subjective endpoint than other endpoints such as a myocardial infarction, stroke, or cardiovascular death. In patients with high TG levels (>150 mg/dL) and low HDL-C levels (<40 mg/dL EPA treatment decreased the risk of CAD by 53% (HR: 0.47; P=0.043) (137). A subjective endpoint has the potential to be an unreliable endpoint in an open label study and is a limitation of the JELIS Study.
2) The Reduction of Cardiovascular Events with EPA – Intervention Trial (REDUCE-IT) was a randomized, double blind trial of 2 grams twice per day of EPA ethyl ester (icosapent ethyl) (Vascepa) vs. mineral oil placebo in 8,179 patients with hypertriglyceridemia (135mg/dL to 499mg/dL) and established cardiovascular disease or high cardiovascular disease risk (diabetes plus one risk factor) who were on stable statin therapy (138). The primary end point was a composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina. The key secondary end point was a composite of cardiovascular death, nonfatal myocardial infarction, or nonfatal stroke. At baseline, the median LDL-C level was 75.0 mg/dL, HDL-C level was 40.0 mg/dL, and TG level was 216.0 mg/dL. The median change in TG level from baseline to 1 year was a decrease of 18.3% (−39.0 mg/dL) in the EPA group and an increase of 2.2% (4.5 mg/dL) in the placebo group. After a median of 4.9 years the primary end-point occurred in 17.2% of the patients in the EPA group vs. 22.0% of the patients in the placebo group (hazard ratio, 0.75; P<0.001), indicating a 25% decrease in events. The number needed to treat to avoid one primary end-point event was 21. The reduction in cardiovascular events was noted after approximately 2 years of EPA treatment. Additionally, the rate of cardiovascular death was decreased by 20% in the EPA group (4.3% vs. 5.2%; hazard ratio, 0.80; P=0.03). The cardiovascular benefits of EPA were similar across baseline levels of TGs (<150, ≥150 to <200, and ≥200 mg per deciliter). Moreover, the cardiovascular benefits of EPA appeared to occur irrespective of the attained TG level at 1 year (≥150 or <150 mg/dL), suggesting that the cardiovascular risk reduction was not associated with attainment of a normal TG level. An increase in hospitalization for atrial fibrillation or flutter (3.1% vs. 2.1%, P=0.004) occurred in the EPA group. In addition, serious bleeding events occurred in 2.7% of the patients in the EPA group and in 2.1% in the placebo group (P=0.06). There were no fatal bleeding events in either group and the rates of hemorrhagic stroke, serious central nervous system bleeding, and serious gastrointestinal bleeding were not significantly higher in the EPA group than in the placebo group.
It should be noted that in this trial mineral oil was used as the placebo. In the placebo group the LDL-C, non-HDL-C, and CRP levels were increased compared to the EPA group during the trial (LDL-C 96mg/dL vs 85mg/dL; non-HDL-C 130mg/dL vs. 113mg/dL; hsCRP 2.8mg/L vs. 1.8mg/L). The impact of these adverse changes on clinical outcomes is uncertain and whether they contributed to the apparent beneficial effects observed in the individuals treated with EPA is unknown.
3) The STRENGTH Trial was a double-blind, randomized, trial comparing 4 grams per day of a carboxylic acid formulation of omega-3 fatty acids (EPA and DHA; Epanova) (n = 6,539)) vs. corn oil placebo (n = 6539) in statin-treated participants with high cardiovascular risk, hypertriglyceridemia, and low levels of HDL-C (139). Approximately 55% of patients had established cardiovascular disease and approximately 70% had diabetes. Median LDL-C level was 75.0 mg/dL, median TG level was 240 mg/dL and median HDL-C level was 36 mg/dL. There were minimal differences in the change in LDL-C and HDL-C levels between the treated and placebo groups after treatment for 12 months but as expected there was a greater reduction in TG levels in the group treated with omega-3-fatty acids (−19.0% vs −0.9%). The primary endpoint was a composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization which occurred in 12.0% of individuals treated with omega-3 CA vs. 12.2% treated with corn oil (hazard ratio, 0.99; P = .84). There were no significant differences between the treatment groups with regard to the risk of the individual components of the primary end point over the 3-4 years of the study. Similar to the REDUCE-IT trial atrial fibrillation was increased with EPA + DHA treatment (HR 1.69 CI 1.29- 2.21). Thus, in contrast the JELIS and REDUCE-IT trials the STRENGTH trial did not demonstrate a benefit of treatment with a mixture of omega-3-fatty acids (EPA + DHA).
4) The OMEMI trial was a randomized trial of 1.8 grams per day of omega-3-fatty acids (930 mg EPA and 660 mg DHA) (n= 505) vs. corn oil placebo (509) in patients aged 70 to 82 years with a recent myocardial infarction (2-8 weeks) (140). Baseline LDL-C was approximately 76mg/dL, HDL-C was 49mg/dL, and TGs 110mg/dL. The primary endpoint was a composite of nonfatal myocardial infarction, unscheduled revascularization, stroke, all-cause death, and heart failure hospitalization after 2 years of follow-up. The primary endpoint occurred in 21.4% of patients on omega-3-fatty acids vs. 20.0% on placebo (hazard ratio, 1.08; P=0.60). TGs levels decreased 8.1% in the omega-3-fatty acid group and increased 5.1% in the placebo group (between group difference 13.2%; P<0.001) while changes in LDL-C were minimal in both groups. Thus, similar to the STRENGTH trial no benefits on cardiovascular disease were observed with EPA + DHA treatment.
Summary of Omega-3-Fatty Acid Clinical Outcome Trials
1) Low dose omega-3-fatty acids are not effective at decreasing cardiovascular outcomes.
2) High dose EPA (JELIS and REDUCE-IT) reduced cardiovascular outcomes while high dose EPA+DHA (STENGTH and OMEMI) did not decrease cardiovascular outcomes.
3) The decrease in TG levels is not a major contributor to the beneficial effect of high dose EPA as the combination of high EPA+DHA lowers TG levels to the same degree as EPA alone without benefit. Additionally, the JELIS trial only lowered TG levels by 5% but nevertheless reduced cardiovascular events. It is likely that the beneficial effects of EPA seen in the JELIS and REDUCE-IT trials are multifactorial with TG lowering making only a small contribution to the decrease in cardiovascular disease. Other actions of EPA, such as decreasing platelet function, anti-inflammation, decreasing lipid oxidation, stabilizing membranes, etc. could account for or contribute to the reduction in cardiovascular events (141). A large meta-analysis, excluding the REDUCE-IT trial, demonstrated that a 40mg/dL decrease in triglyceride levels resulted in a relative risk reduction of only 0.96 (4% decrease) indicating that one needs to markedly lower triglyceride levels to reduce cardiovascular events (142).
4) Whether EPA has special properties that resulted in the reduction in cardiovascular events in the REDUCE-IT trial or there were flaws in the trial design (the use of mineral oil as the placebo) is uncertain and debated. It should be noted that in the REDUCE-IT trial LDL-C and non-HDL-C levels were increased by approximately 10% in the mineral oil placebo group (138). Additionally, Apo B levels were increased by 7% (6mg/dL) by mineral oil (138). Finally, an increase in hsCRP (20-30%) and other biomarkers of atherosclerosis (oxidized LDL-C, IL-6, IL-1 beta, and lipoprotein-associated phospholipase A2) were noted in the mineral oil group (138,143). In the STRENGTH trial there were no differences in LDL-C, Non-HDL-C, HDL-C, Apo B, or hsCRP levels between the treated vs. placebo groups (139). Whether EPA has special properties compared to DHA leading to a reduction in cardiovascular events or the mineral oil placebo resulted in adverse changes increasing ASCVD in the placebo resulting in an artifactual decrease in the EPA group is debated (144,145). Ideally, another large randomized cardiovascular trial with EPA ethyl ester (icosapent ethyl) (Vascepa) using a placebo other than mineral oil would help resolve this controversy. In the meantime, clinicians will need to use their clinical judgement on whether to treat patients with modest elevations in TG levels with EPA (icosapent ethyl; Vascepa) balancing the potential benefits of treatment vs. the potential side effects.
Side Effects
Gastrointestinal side effects such as diarrhea, nausea, dyspepsia, abdominal discomfort, and eructation have been observed with fish oil therapy (Package Inserts for Lovaza, Vascepa, and Epanova).
At very high doses, omega-3-fatty acids can inhibit platelets and prolong bleeding time. However, at the recommended doses this has not been a major clinical problem but nevertheless when patients are on anti-platelet drugs one should be alert for the possibility of bleeding problems (Package Inserts for Lovaza, Vascepa, and Epanova). Increased bleeding was noted in the REDUCE-IT trial in the patients treated with icosapent ethyl 4 grams/day (EPA) (see above discussion of this trial). A recent review found no evidence for discontinuing the use of omega-3 fatty acid treatment before invasive procedures or when given in combination with other agents that affect bleeding (146).
As noted above an increase in atrial fibrillation was observed in the REDUCE-IT trial in the patients treated with icosapent ethyl 4 grams/day (EPA) and in the STRENGTH trial in the patients treated with EPA + DHA.
Contraindications
There are no contraindications to the use of omega-3-fatty acids. Lovaza, Omacor, and Vascepa are pregnancy category C drugs and they should only be used if the benefits to the mother outweigh the potential risks to the fetus.
Conclusions
Omega-3-fatty acids are effective drugs in reducing TG levels with few significant side effects, drug interactions, or contraindications. High dose EPA (4 grams/day) reduced cardiovascular disease events in the REDUCE-IT trial and a moderate dose of EPA (1.8 grams/day) reduced cardiovascular events in the JELIS trial but trials of EPA and DHA have not produced cardiovascular benefits. The basis for these differences is debated and discussed in the “Summary of Omega-3-Fatty Acid Clinical Outcome Trials” section above. Finally, omega-3-fatty acids are effective in lowering TGs in patients with marked hypertriglyceridemia and while not proven will likely reduce the risk of development of pancreatitis.
FIBRATES
Introduction
The fibrate drug class includes clofibrate, gemfibrozil, fenofibrate, bezafibrate, and ciprofibrate. Clofibrate was developed in the 1960s and was the first member of this class. Clofibrate is no longer available because of an increased risk of adverse effects. Gemfibrozil and fenofibrate are available in the United States while gemfibrozil, fenofibrate, bezafibrate, and ciprofibrate are available in Europe. All of the fibrates work via activation of the nuclear hormone receptor PPAR alpha.
Effect of Fibrates on Lipid and Lipoprotein Levels
Table 12.
Effect of Fibrates on Lipids and Lipoproteins
| Decreases TG |
| Increases HDL-C |
| Decreases LDL-C; if TGs Very High can Increase LDL-C |
| Decreases Non-HDL-C |
| Decreases Apolipoprotein B |
| Decreases LDL Particle Number |
| Shift Small Dense LDL to Large Buoyant LDL |
| No Effect on Lp(a) |
Fibrates reduce fasting TG levels by 25-50% (147-149). The magnitude of the reduction in TGs is dependent on the baseline TG levels. Patients with marked elevations in TGs have a greater reduction in TG levels (147,149,150). In addition, fibrates increase HDL-C levels by 5-20% (148,149). The increase in HDL-C levels is more robust if the TG levels are elevated and/or if the HDL-C levels are low (150). The effect on LDL-C is more variable (149). If the TG levels are very high (>400-500mg/dL), fibrate therapy may result in an increase in LDL-C levels whereas if TGs are not elevated fibrates decrease LDL-C by 10-30% (147). Given the decrease in plasma TGs and LDL-C levels, fibrates also reduce apolipoprotein B, LDL particle number, and non-HDL-C levels (149). Depending upon the TG level there may be a shift from small dense LDL towards large LDL particles (149). Fibrates do not have any major or consistent effects on Lp(a) levels (151). Table 13 below shows the effect of fenofibrate on lipid and lipoprotein levels in patients with different lipid profiles and illustrates some of the principles outlined above.
Table 13.
Effect of Fenofibrate on Lipid and Lipoprotein Levels
| TGs | LDL-C | HDL-C | |
|---|---|---|---|
| Elevated TG Levels | |||
| Baseline Levels | ~404mg/dL | ~125mg/dL | ~35mg/dL |
| Change with Fenofibrate | 45% Decrease | 2.5% Increase | 16% Increase |
| Elevated LDL-C and TG Levels | |||
| Baseline Levels | 232mg/dL | 220mg/dL | 46.7mg/dL |
| Change with Fenofibrate | 37% Decrease | 13% Decrease | 12% Increase |
| Elevated LDL-C and Normal TG Levels | |||
| Baseline Levels | 102mg/dL | 228mg/dL | 58.1mg/dL |
| Change with Fenofibrate | 35% Decrease | 29% Decrease | 7% Increase |
The values are adjusted for changes in the placebo group. Data modified from Tricor Package Insert.
In large, randomized, fibrate outcome trials similar changes in lipid and lipoprotein levels were noted (Table 14). These trials are discussed in detail in the effect of fibrates on cardiovascular outcomes section presented below.
Table 14.
Effect of Fibrates on Lipid and Lipoprotein Levels in Large Outcome Studies*
| TGs | LDL-C | HDL-C | |
|---|---|---|---|
| Helsinki Heart Study- Gemfibrozil (152) | 35% Decrease | 11% Decrease | 10% Increase |
| VA-HIT Study Gemfibrozil (153) | 31% Decrease | No Change | 6% Increase |
| BIP Study Bezafibrate (154) | 21% Decrease | 7% Decrease | 18% Increase |
| Leader Study Bezafibrate (155) | 23% Decrease | 8% Decrease | 8% Increase |
| Field Study Fenofibrate (156) | 29% Decrease | 12% Decrease | 5% Increase |
- *
The values are adjusted for changes in the placebo group.
The different fibrates in general cause similar changes in lipid and lipoprotein levels. There are only a few comparative trials of fibrates comparing their effects on lipid and lipoprotein levels and these trials have been very small. Comparisons of ciprofibrate and gemfibrozil have not shown any major differences between these two fibrates (157,158). In contrast, two very small trials have compared gemfibrozil vs. fenofibrate and reported that fenofibrate was more efficacious in lowering LDL levels than gemfibrozil (159,160).
In very rare instances fibrates can cause a paradoxical marked decrease in HDL-C levels (161-164). This rare paradoxical decrease in HDL-C typically occurs when fibrates are used in combination with a thiazolidinedione (rosiglitazone and pioglitazone) but can occur when fibrates are used alone or with ezetimibe (161-165). The decrease in HDL-C can be extreme with decreases of 50% to 88% reported and recovery to normal can take weeks after the fibrate is discontinued (162). The mechanism for this paradoxical effect is unknown.
Effect of Fibrates in Combination with Other Lipid Lowering Drugs on Lipid and Lipoprotein Levels
STATINS
Statins are the primary drugs used to treat most patients with dyslipidemia. Statins are very effective in lowering LDL-C levels but have only modest effects on TG and HDL-C levels. Therefore, it is appealing to add a fibrate to patients who on statin therapy have LDL-C levels at goal but still have elevated non-HDL-C and TG levels and decreased HDL-C levels. Therefore, there have been numerous studies examining the effect of the combination of statins and fibrates on lipid and lipoprotein levels. An example is the Safari Trial which compared the effect of simvastatin only (n=207) vs. simvastatin + fenofibrate (n=411) in patients with combined hyperlipidemia (166). The results of this trial are shown in table 15. As anticipated, adding a fibrate results in a further lowering of LDL-C, non-HDL-C, and TG levels with a further increase in HDL-C.
Table 15.
Effect of Simvastatin Alone vs. Simvastatin + Fenofibrate on Lipid and Lipoprotein Levels
| LDL | TG | Non-HDLC | HDL | |
|---|---|---|---|---|
| Simvastatin | -26% | -20% | -26% | +10% |
| Simvastatin + Fenofibrate | -31% | -43% | -35% | +19% |
A meta-analysis of 9 studies with over 1,200 participants compared the effect of statin alone vs. statin + fibrate on lipid and lipoprotein levels (167). The combination of statins and fibrates provided significantly greater reductions in total cholesterol, LDL-C, and TGs, and a significantly greater increase in HDL-C than treatment with statins alone. A larger meta-analysis of 13 randomized controlled trials, involving 7,712 patients, similarly demonstrated significant decreases in LDL-C (8.8mg/dL), TGs (58mg/dL), and total cholesterol (11.2mg/dL), and increases in HDL-C (4.65mg/dL) in patients receiving the combination of statins + fibrates compared with statin therapy alone (168). The combination of statins + fibrates also result in a shift of LDL particles from small dense particles to large buoyant particles whereas no change in LDL particle size was observed with statin monotherapy (169).
A recent meta-analysis of 6 studies with over 400 participants compared the effect of adding a statin to fibrate therapy (fibrate alone vs. fibrate + statin) and showed similar changes (170). The fibrate-statin combination produced significantly greater reductions in the levels of total cholesterol, LDL-C, and TGs compared to fibrate alone. In contrast there was no significant difference in HDL-C levels in the fibrate vs. fibrate + statins group.
EZETIMIBE
In patients unable to tolerate statin therapy one needs to use other drugs to treat dyslipidemia. In a study comparing the effect of ezetimibe 10mg alone, fenofibrate 145mg alone, or ezetimibe + fenofibrate the combination had a better effect on the lipid profile resulting in a greater decrease in LDL-C levels and increase in HDL-C levels than either drug alone (Table 16) (171).
Table 16.
Effect of the Combination of Ezetimibe and Fenofibrate on Lipid and Lipoprotein Levels
| LDL-C | HDL-C | TG | |
|---|---|---|---|
| Ezetimibe | 23% Decrease | 2.2% Increase | 10% Decrease |
| Fenofibrate | 22% Decrease | 7.5% Increase | 38% Decrease |
| Ezetimibe + Fenofibrate | 34% Decrease | 11.5% Increase | 38% Decrease |
Similar results were observed in another randomized trial of ezetimibe 10mg and fenofibrate 160mg (172). Moreover, both fibrate therapy and the combination of ezetimibe and fenofibrate results in a shift of LDL particles from small dense LDL particles to large buoyant particles (172).
EZETIMIBE + STATIN
A large randomized trial has compared the effect of ezetimibe /simvastatin 10mg/20mg, fenofibrate 160mg, or ezetimibe/simvastatin + fenofibrate on lipid and lipoprotein levels. As one would expect triple drug therapy had a better effect on the lipid profile (Table 17) (173). While ezetimibe/simvastatin was very effective in lowering LDL-C levels and fenofibrate was very effective in lowering TGs and raising HDL-C levels the combination resulted in more favorable changes in TGs. In a similar study the addition of fenofibrate 135mg to atorvastatin 40 mg + ezetimibe 10 mg resulted in a greater reduction in TGs (-57% vs. -40%; p<0.001) and a greater increase in HDL (13% vs. 4.2%; p<0.001) than placebo (174). Fibrate therapy and ezetimibe/simvastatin + fenofibrate also resulted in a shift of LDL particles from small dense LDL particles to large buoyant particles (173).
Table 17.
Effect of the Combination of Ezetimibe/Simvastatin and Fenofibrate on Lipid and Lipoprotein Levels
| LDL-C | HDL-C | TG | |
|---|---|---|---|
| Placebo | -3.5% | +1.1 | -3.1% |
| Ezetimibe/Simvastatin | -47% | +9.3% | -29% |
| Fenofibrate | -16% | +18.2 | -41 |
| Eze/Simva + Fenofibrate | -46% | +18.7 | -50% |
BILE ACID SEQUESTRANT
Studies have also examined the effect of fibrates in combination with bile acid sequestrants. Participants receiving fenofibrate 160 mg/day were randomized to receive either colesevelam HCl 3.75 g/day or placebo (175). No significant differences in TG or HDL-C levels were observed between the two groups. However, LDL-C levels were decreased in the fenofibrate + colesevelam group compared to the fenofibrate + placebo group (12.4% greater decrease: p<0.001). A study of the combination of fenofibrate and colestipol also demonstrated a more marked decrease in LDL-C with that combination compared to either drug alone (colestipol -18%; fenofibrate -17%, colestipol + fenofibrate 37%) (176). The combination of both drugs did not blunt the effects of fenofibrate on VLDL and HDL. Other studies of the combination of a fibrate with a bile acid sequestrant have also demonstrated an enhanced effect in lowering LDL-C levels (177-179).
NIACIN
Surprisingly there are few large randomized trials examining the effect of combination therapy with niacin + fibrate vs. monotherapy. One very small trial reported that while both niacin monotherapy and bezafibrate monotherapy were effective in lowering serum TGs there was no added benefit of combination therapy in reducing serum TG level although a large variance may have reduced the ability to detect statistically significant results (16). A larger trial in HIV+ patients reported that the combination of niacin and fenofibrate was better at lowering TGs and non-HDL-C and increasing HDL-C levels than monotherapy with either niacin or fenofibrate (17). It would be informative if additional trials of fibrate + niacin combination therapy were carried out in patients with marked hypertriglyceridemia that can often be difficult to control with lifestyle changes and monotherapy.
FISH OIL
In patients with TG levels > 500mg/dL the combination of fenofibrate 130mg per day and 4 grams of Lovaza (DHA and EPA) reduced TG levels to a greater extent than monotherapy with fenofibrate alone (7% decrease; P = 0.059) (103). Not unexpectedly, LDL levels were increased to a greater extent with combination therapy (9% increase; p= 0.03). When subjects who had received 8 weeks of fenofibrate monotherapy were treated with Lovaza (DHA and EPA) during the 8-week, open-label extension study TG levels were reduced by an additional 17.5% (P = 0.003). These results indicate that the addition of omega-3-fatty acids to fenofibrate will further decrease TG levels.
Mechanisms Accounting for the Fibrate Induced Lipid Effects
Fibrates are ligands that bind and activate PPAR alpha, a member of the family of nuclear hormone receptors that are activated by lipids (180,181). PPAR alpha is highly expressed in the liver and other tissues important in fatty acid metabolism. PPAR alpha forms a heterodimer with RXR and together the PPAR alpha:RXR complex when activated binds to the PPAR response elements in a large number of genes and regulates the expression of these genes (180,181). The natural ligands of PPAR alpha are fatty acid derivatives formed during lipolysis, lipogenesis, or fatty acid catabolism (180,181).
TRIGLYCERIDES
Fibrates lower plasma TG levels by decreasing VLDL production and by increasing the clearance of TG rich lipoproteins (182,183). The decrease in VLDL production is primarily due to PPAR alpha activation of the beta oxidation of fatty acids, which reduces the substrate available for the synthesis of TGs and the formation of VLDL (180,183). Additionally, a decrease in hepatic fatty acid synthesis may also contribute to the decrease in fatty acids (180,183). The increased clearance of TG rich lipoproteins is due to PPAR alpha stimulating the transcription of lipoprotein lipase, the key enzyme that catabolizes the TGs carried by VLDL and chylomicrons (180,183). In addition, activation of PPAR alpha also inhibits the transcription of APO C-III, which inhibits lipoprotein lipase activity (180,183). A decrease in Apo C-III enhances the clearance of TG rich lipoproteins by increasing lipoprotein lipase activity. Notably, a decrease in Apo C-III also decreases TG levels in patients deficient in lipoprotein lipase indicating that there are multiple mechanisms for its effects on TG metabolism (184). Recent studies suggest that Apo C-III inhibits the uptake of TG rich lipoproteins into the liver by the LDL receptors/ LDLR-related protein 1 axis (185). PPAR alpha activation also increases the transcription of Apo A-V, which would also facilitate the activity of lipoprotein lipase (180).
HIGH DENSITY LIPOPROTEINS
The increase in HDL induced by fibrates is due to PPAR alpha activation stimulating Apo A-I and A-II transcription (180,183). This leads to the increased production of HDL (182). In addition, a decrease in TG rich lipoproteins may result in a reduction in CETP mediated transfer of cholesterol from HDL to VLDL and of TG from VLDL to HDL (183). This would lead to less TG enrichment of HDL and a decrease in the opportunity of hepatic lipase to remove TG leading to small HDL particles that may be rapidly catabolized.
LOW DENSITY LIPOPROTEINS
As noted above the effect of fibrates on LDL-C levels is variable with increases in LDL seen in patients with high TG levels (>400mg/dL) and decreases in LDL-C levels in patients with lower TG levels. In patients with modest elevations in plasma TG levels the clearance of LDL is enhanced (182). The mechanism for this enhanced clearance could be due to a decrease in Apo C-III, as increased levels of this protein inhibits LDL receptor activity (185,186). Additionally, the shift from small dense LDL to large buoyant LDL would enhance the uptake of LDL by the LDL receptor (187). In patients with TG levels > 400mg/dL fibrate therapy decreases LDL clearance (182). Prior to treatment, patients with marked hypertriglyceridemia have hypercatabolism of LDL, which is likely due to increased uptake by the reticuloendothelial system (182). This increased clearance is LDL receptor independent. Treatment with fibrates lowers the plasma TGs leading to normalization of reticuloendothelial cell function and a decrease in LDL clearance resulting in an increase in LDL-C levels with fibrate therapy (182). In addition, the metabolism of VLDL to LDL may be enhanced by fibrates when the TG levels are markedly elevated.
Effect of Monotherapy with Fibrates on Cardiovascular Outcomes
There have been a number of studies that have examined the effect of monotherapy with a variety of different fibrates on cardiovascular disease. We will describe the major studies below.
1) Coronary Drug Project (CDP): CDP conducted between 1966 and 1975, was a randomized, double-blind clinical trial that determined the effect of clofibrate (1.8g/day), dextrothyroxine (6mg/day), two doses of oral estrogen (2.5 or 5mg per day), or immediate release niacin (3 grams/day) vs. placebo in 8,341 men aged 30 to 64 years of age with an electrocardiogram documented myocardial infarction on cardiovascular events and mortality (43). The mean baseline total cholesterol level was 251mg/dL and TG level was 183mg/dL. The two estrogen regimens and dextrothyroxine treatment groups were discontinued early because of increased adverse effects. Clofibrate treatment (n= 1,051) compared to placebo (n= 2,680) also did not demonstrate clinical benefit. The five-year mortality in subjects treated with clofibrate was 20.0% as compared with 20.9% in subjects on placebo therapy (P = 0.55). The results with niacin are discussed above in the section on niacin and cardiovascular outcomes.
2) WHO: WHO was a double-blind trial in middle-aged men, age 30-59 years of age, without evidence of heart or other major disease, who were treated with 1.6 grams/day clofibrate (n=5,000) or placebo (n=5,000) for an average of 5.3 years (188). Average serum cholesterol levels were approximately 248mg/dL and a mean reduction of approximately 9 per cent occurred in the clofibrate group. The incidence of ischemic heart disease was decreased by 20% in the clofibrate group compared to the control group (P <0.05). This decrease was confined to non-fatal myocardial infarcts which were reduced by 25% while the incidence of fatal heart attacks and angina was similar in the clofibrate and placebo groups. Importantly, the numbers of deaths and crude mortality rates from all causes were increased in the clofibrate-treated group compared to the control group (P < 0.05). The excess deaths were partially accounted for by increased deaths due to liver, biliary tract, and intestinal disease. There was also an increase in cholecystectomies in subjects treated with clofibrate. Because of increased toxicity clofibrate is no longer available.
3) Helsinki Heart Study (HHS): HSS was a randomized double-blind trial in middle aged men age 40-55 years of age without cardiovascular who had non-HDL-C levels greater than or equal to 200mg/dL (152). Subjects were randomized to receive 600mg gemfibrozil twice a day (n=2,051) or placebo (n=2,030) for five years. At initiation of the study total cholesterol was 289mg/dL, HDL-C 47mg/dL, non-HDL-C 242mg/dL, and TGs 176mg/dL. Gemfibrozil caused an increase in HDL-C (approximately 10%) and reductions in total (~10%), LDL-C (~11%), non-HDL-C (~14%), and TG levels (~35%). There were minimal changes in serum lipid levels in the placebo group. Fatal and non-fatal myocardial infarctions and cardiac death were the principal end points and the cumulative rate of these cardiac end points were reduced 34% in the gemfibrozil group (27.3 per 1,000 in the gemfibrozil group vs. 41.4 per 1,000 in the placebo group; P< 0.02). The decrease in cardiovascular disease in the gemfibrozil group became evident in the second year and continued throughout the remainder of the study. There was no difference in mortality between the gemfibrozil and placebo groups. The benefit of gemfibrozil therapy was greatest in participants with elevated TGs and decreased HDL-C levels (189,190). Risk reduction with gemfibrozil was 78% (P = .002) among those with BMI > 26 kg/m2 and dyslipidemia (TGs > ~200mg/dL and HDL-C < 42mg/dL) suggesting that certain types of patients are likely to derive greater benefit from fibrate treatment (191).
4) Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT): VA-HIT was a double-blind trial in men with coronary heart disease who had an HDL-C level <40mg/dL and LDL-C level <140mg/dL (153). Subjects were randomized to gemfibrozil 1200mg per day (n=1,264) or placebo (n=1,267) for 5.1 years. Mean lipid levels at study initiation were HDL-C 32mg/dL, LDL-C 111mg/dL, total cholesterol 175mg/dL, and TGs 160mg/dL. At one year, the mean HDL-C level was 6 percent higher, the mean TG level was 31 percent lower, and the mean total cholesterol level was 4 percent lower in the gemfibrozil group than in the placebo group. LDL-C levels did not differ significantly between the groups. The primary study outcome was nonfatal myocardial infarction or death from coronary causes. The primary outcome occurred in 21.7% of patients in the placebo group and 17.3% of patients in the gemfibrozil group (22 percent decrease; P=0.006). A 24% reduction in the combined outcome of death from coronary heart disease, nonfatal myocardial infarction, and stroke was observed in the gemfibrozil group (P< 0.001). There were no significant differences in the rates of coronary revascularization, hospitalization for unstable angina, death from any cause, and cancer. Similar to HHS the beneficial effect of gemfibrozil did not become apparent until approximately two years after treatment. A low HDL-C (<33.5mg/dL) and high TGs (>180mg/dL) at baseline predicted a beneficial response to gemfibrozil therapy (192).
5) Bezafibrate Infarction Prevention Study (BIP): BIP was a double-blind study in male and female patients aged 45-74 with a previous myocardial infarction or stable angina (154). Patients were randomized to receive either 400 mg of bezafibrate per day (n=1,548) or a placebo (n=1,542) and were followed for 6.2 years. At the initiation of the study total cholesterol was 212mg/dL, LDL-C was 148mg/dL, HDL-C was 34.6mg/dL, and TGs were145mg/dL. Bezafibrate increased HDL-C by 18% and reduced TGs by 21%. There was a small 7% decrease in LDL-C. The primary end point was fatal or nonfatal myocardial infarction or sudden death. The primary end point occurred in 13. 6% of the bezafibrate group vs. 15.0% of the placebo (9.4% reduction; P=0.26). Total and non-cardiac mortality rates were similar. In a post hoc analysis in the subgroup with high baseline TGs (> or =200 mg/dL), the reduction in the primary end point in the bezafibrate group was 39.5% (P=0.02). Additionally, bezafibrate reduced cardiovascular events in patients with the metabolic syndrome (193). These results again suggest that patients with high TGs are likely to derive benefit from fibrate therapy.
6) Leader Trial: The Leader trial was a double blind placebo controlled randomized trial in men age 35 to 92 with lower extremity arterial disease (194,195). Subjects were randomized to bezafibrate 400mg per day (n=783) or placebo (n=785). At baseline total cholesterol levels were 218mg/dL, LDL-C levels 132mg/dL, HDL-C levels 44mg/dL, and TGs 187mg/dL. Bezafibrate therapy reduced total cholesterol levels by 7.6%, LDL-C by 8.1%, and TGs by 23% and increased HDL-C levels by 8%. The primary endpoint of coronary heart disease and strokes was not reduced by bezafibrate treatment. Neither major coronary events nor strokes were significantly reduced.
.
7) Fenofibrate Intervention and Event Lowering in Diabetes Trial (FIELD): In the FIELD Trial patients with Type 2 diabetes between the ages of 50 and 75 with or without pre-existing cardiovascular disease not taking statin therapy were randomized to fenofibrate 200 mg daily (n=4,895) or placebo (n=4,900) and followed for approximately 5 years (156). At initiation of the study total cholesterol was 196mg/dL, LDL-C was 120mg/dL, HDL-C was 43mg/dL, and TGs were 152mg/dL. Fenofibrate therapy resulted in an 11% decrease in total cholesterol, a 12% decrease in LDL-C, a 29% decrease in TGs, and a 5% increase in HDL-C levels. The primary outcome was coronary events (coronary heart disease death and non-fatal MI), which were reduced by 11% in the fenofibrate group but this difference did not reach statistical significance (p= 0.16). However, there was a 24% decrease in non-fatal MI in the fenofibrate treated group (p=0.01) and a non-significant increase in coronary heart disease mortality. Total cardiovascular disease events (coronary events plus stroke and coronary or carotid revascularization) were reduced 11% (p=0.035). These beneficial effects of fenofibrate therapy on cardiovascular disease were observed in patients without a previous history of cardiovascular disease. In patients with a previous history of cardiovascular disease no benefits were observed. Additionally, the beneficial effect of fenofibrate therapy was seen only in those subjects less than 65 years of age. The beneficial effects of fenofibrate in this study may have been blunted by the increased use of statins in the placebo group, which reduced the differences in lipid levels between the placebo and fenofibrate groups. If one adjusted for the addition of lipid-lowering therapy, fenofibrate reduced the risk of coronary heart disease events by 19% (p=0.01) and of total cardiovascular disease events by 15% (p=0.004). Additionally, many patients in the Field trial did not have elevations in TGs and decreased HDL-C levels. In a post hoc analysis, patients with high TGs 200mg/dL) and low HDL levels (<40mg for men and <50mg/dL for women) derived greater benefit from fenofibrate therapy (196).
8) Summary: While the above monotherapy fibrate studies suggest that fibrates reduce cardiovascular event, particularly in patients with high TG and low HDL levels, the results are not as robust or consistent as the beneficial effects of statins on cardiovascular outcomes (5).
Effect of Combination Therapy of Fibrates and Statins on Cardiovascular Outcomes
Given the marked benefits of statin therapy it is essential to know if adding fibrates to statin therapy further reduces cardiovascular events. Two large trials described below have addressed this key question.
1) ACCORD LIPID Trial: The ACCORD-LIPID Trial was designed to determine if the addition of fenofibrate to aggressive statin therapy would result in a further reduction in cardiovascular disease in patients with Type 2 diabetes (197). In this trial, 5,518 patients on statin therapy were randomized to placebo or fenofibrate therapy. The patients had diabetes for approximately 10 years and either had pre-existing cardiovascular disease or were at high risk for developing cardiovascular disease. During the trial, LDL-C levels were approximately 80mg/dL. There was only a small difference in HDL-C with the fenofibrate groups having a mean HDL-C of 41.2mg/dL while the control group had an HDL-C of 40.5mg/dL. Differences in TG levels were somewhat more impressive with the fenofibrate group having a mean TG level of 122mg/dL while the control group had a TG level of 144mg/dL. First occurrence of nonfatal myocardial infarction, nonfatal stroke, or death from cardiovascular causes was the primary outcome and there was no statistical difference between the fenofibrate treated group and the placebo group. Additionally, there were also no statistically significant differences between the groups with regards to any of the secondary outcome measures of cardiovascular disease. Of note, the addition of fenofibrate to statin therapy did not result in an increase in either muscle or liver side effects. On further analysis there was a suggestion of benefit with fenofibrate therapy in the patients in whom the baseline TG levels were elevated (>204mg/dL) and HDL-C levels decreased (<34mg/dL). While this was a negative study, it must be recognized that most of the patients included in this study did not have the lipid profile that would typically lead to treatment with fibrates.
2) PROMINENT Trial: The PROMINENT trial studied the effect of pemafibrate, a new selective PPAR-alpha activator, in reducing cardiovascular outcomes in 10,497 patients (66.9% with previous ASCVD) with diabetes (198). This was a double-blind, randomized, controlled trial, in patients with Type 2 diabetes, with mild-to-moderate hypertriglyceridemia (TG level, 200 to 499 mg/dL), LDL-C < 100mg/dL, and HDL-C levels < 40 mg/dL who received either pemafibrate (0.2-mg tablets twice daily) or placebo in addition to statin therapy (96% on statins). The primary end point was a composite of nonfatal MI, ischemic stroke, coronary revascularization, or death from cardiovascular causes. Baseline fasting TG was 271 mg/dL, HDL-C 33 mg/dL, and LDL-C 78 mg/dL. Compared with placebo, pemafibrate decreased TG by 26.2%, while HDL-C increased 5.1% and LDL-C increased 12.3%. Notably non-HDL-C levels were unchanged and Apo B levels increased 4.8%. The primary endpoint was similar in the pemafibrate and placebo group (HR 1.03; 95% CI 0.91 to 1.15). The increase in LDL-C and Apo B levels may have accounted for the failure to reduce cardiovascular events.
3) Summary: The results of the ACCORD and PROMINENT trials were disappointing and have greatly reduced the enthusiasm for adding fibrates to statin therapy to cardiovascular events.
Effect of Fibrates on Non-Cardiovascular Outcomes
DIABETIC RETINOPATHY
Small studies in the 1960’s presented suggestive evidence that treatment with clofibrate improved diabetic retinopathy (199,200). Randomized trials have confirmed these observations.
The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study was a randomized trial in patients with Type 2 diabetes mellitus. Patients were randomly assigned to receive either fenofibrate 200 mg/day (n=4,895) or placebo (n=4,900). Laser treatment for retinopathy was significantly lower in the fenofibrate group than in the placebo group (3.4% patients on fenofibrate vs 4.9% on placebo; p=0.0002) (201). Fenofibrate therapy reduced the need for laser therapy to a similar extent for maculopathy (31% decrease) and for proliferative retinopathy (30% decrease). In the ophthalmology sub-study (n=1,012), the primary endpoint of 2-step progression of retinopathy grade did not differ significantly between the fenofibrate and control groups (9.6% patients on fenofibrate vs 12.3% on placebo; p=0.19). In patients without pre-existing retinopathy there was no difference in progression (11.4% vs 11.7%; p=0.87). However, in patients with pre-existing retinopathy, significantly fewer patients on fenofibrate had a 2-step progression than did those on placebo (3.1% patients vs 14.6%; p=0.004). A composite endpoint of 2-step progression of retinopathy grade, macular edema, or laser treatments was significantly reduced in the fenofibrate group (HR 0.66, 95% CI 0.47-0.94; p=0.022).
In the ACCORD Lipids Study a subgroup of participants were evaluated for the progression of diabetic retinopathy by 3 or more steps on the Early Treatment Diabetic Retinopathy Study Severity Scale or the development of diabetic retinopathy necessitating laser photocoagulation or vitrectomy over a four-year period (202). At 4 years, the rates of progression of diabetic retinopathy were 6.5% with fenofibrate therapy (n=806) vs. 10.2% with placebo (n=787) (adjusted odds ratio, 0.60; 95% CI, 0.42 to 0.87; P = 0.006). Of note, this reduction in the progression of diabetic retinopathy was of a similar magnitude as intensive glycemic treatment vs. standard therapy.
A double-blind, randomized, placebo-controlled study in 296 patients with type 2 diabetes mellitus evaluated the effect of placebo or etofibrate on diabetic retinopathy (203). After 12 months an improvement in ocular pathology was more frequent in the etofibrate group vs the placebo group ((46% versus 32%; p< 0.001).
The MacuFen study was a small double-blind, randomized, placebo-controlled study in 110 subjects with diabetic macular edema who did not require immediate photocoagulation or intraocular treatment (204). Patients were randomized to fenofibric acid or placebo for 1 year. Patients treated with fenofibric acid had a modest improvement in total macular volume that was not statistically significant compared to the placebo group.
Taken together these results indicate that fibrates have beneficial effects on the progression of diabetic retinopathy (205). The mechanisms by which fibrates decrease diabetic retinopathy are unknown, and whether decreases in serum TG levels plays an important role is uncertain. Fibrates activate PPAR alpha, which is expressed in the retina (206). Diabetic PPARα KO mice developed more severe DR while overexpression of PPARα in the retina of diabetic rats significantly alleviated diabetes-induced retinal vascular leakage and retinal inflammation, suggesting that fibrates could have direct effects on the retina to reduce diabetic retinopathy (206).
DIABETIC KIDNEY DISEASE
The Diabetes Atherosclerosis Intervention Study (DAIS) evaluated the effect of fenofibrate therapy (n= 155) vs. placebo (n=159) on changes in urinary albumin excretion in patients with Type 2 diabetes (207). Fenofibrate significantly reduced the worsening of albumin excretion (fenofibrate 8% vs. placebo 18%; P < 0.05). This effect was primarily due to reduced progression from normal albumin excretion to microalbuminuria (fenofibrate 3% vs. 18% placebo; P < 0.001).
In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) trial, Type 2 diabetic patients aged 50 to 75 years were randomly assigned to fenofibrate (n = 4,895) or placebo (n = 4,900) for 5 years (208). Fenofibrate reduced urine albumin/creatinine ratio by 24% vs 11% in placebo group (p < 0.001), with 14% less progression and 18% more albuminuria regression (p < 0.001) in the fenofibrate group than in participants on placebo. As expected, fenofibrate therapy acutely increased plasma creatinine levels and decreased eGFR (209). However, over the long-term, the increase in plasma creatinine was lower in the fenofibrate group compared to the placebo group (14% decrease; p=0.01). Similarly, there was a slower annual decrease in eGFR in the fenofibrate group (1.19 vs 2.03 ml/min/1.73 m2 annually, p < 0.001). End-stage renal disease, dialysis, renal transplant, and renal death were similar in the fenofibrate and placebo groups, likely due to the small number of events.
In the ACCORD-LIPID trial, 5,518 patients on statin therapy were randomized to placebo or fenofibrate therapy (197). The patients had diabetes for approximately 10 years and either had pre-existing cardiovascular disease or were at high risk for developing cardiovascular disease. The post-randomization incidence of microalbuminuria was 38.2% in the fenofibrate group and 41.6% in the placebo group (p=0.01) and post-randomization incidence of macroalbumuria was 10.5% in the fibrate group and 12.3% in the placebo group (p=0.04) indicating a modest reduction in the development of proteinuria in patients treated with fenofibrate (197). There was no significant difference in the incidence of end-stage renal disease or need for dialysis between the fenofibrate group and the placebo group, likely due to the small number of events.
A small randomized study in patients with Type 2 diabetes and hypertriglyceridemia compared the effect of fenofibrate (200mg/day) (n=28) vs. no treatment (n=28) on urinary albumin excretion (210). After 180 days urinary albumin/creatine ratio was decreased in the fenofibrate group vs. controls (control -8.15 vs fenofibrate -44.05 mg/g; P<0.05).
These studies suggest that fibrates may have a beneficial effect on diabetic kidney disease (211). One should recognize that reducing proteinuria is a surrogate marker and may not indicate a reduction in the development of end stage renal disease. The mechanisms accounting for the decrease in proteinuria are unknown.
AMPUTATIONS
In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study, patients aged 50-75 years with Type 2 diabetes were randomly assigned to receive fenofibrate 200 mg per day (n=4,895) or matching placebo (n=4,900) for 5 years' duration (212). The risk of first amputation was decreased by 36% (p=0.02) and minor amputation events without known large-vessel disease by 47% (p=0.027) in the fenofibrate treated group (212). The reduction in amputations was independent of glucose control or dyslipidemia. No difference between the risks of major amputations was seen in the placebo and fenofibrate groups. The basis for this reduction in amputations is unknown.
GOUT
In the Field trial treatment, fenofibrate reduced uric acid levels by 20% and reduced episodes of gout by approximately 50% compared to placebo (HR 0·48, 95% CI 0·37-0·60; p<0·0001) (213). Interestingly, a meta-analysis of fibrate trials found that fenofibrate but not bezafibrate reduced serum uric acid levels suggesting that the reduction in uric acid levels is not a class effect (214).
SUMMARY
The above studies provide substantial evidence that fibrates have a favorable effect on diabetic microvascular disease (155). While fibrates are not approved specifically for the prevention or treatment of diabetic microvascular disease one should consider these potential beneficial effects when deciding on treatment choices. For example, in a patient with diabetes and microvascular disease and hypertriglyceridemia needing therapy one might elect to use fibrates to lower plasma TGs given their potential beneficial effects on slowing the progression of microvascular disease.
Side Effects
RENAL
Fibrate therapy leads to an increase in serum creatinine and cystatin C levels (215-217). For example, in the Field Trial serum creatinine levels increased from 0.88mg/dL to 0.99mg/dL, a 12% increase (156). This increase in creatinine has been seen with all fibrates but appears to be less profound with gemfibrozil (215). The increase in cystatin C occurs with fenofibrate but not with other fibrates (216). It must be recognized that this increase in creatinine is reversible on stopping fibrate therapy and does not reflect kidney damage (215). In fact, careful measurements of renal function have not demonstrated a decrease in glomerular filtration rate despite the increase in serum creatinine (209,218,219). As discussed above, studies of renal function in patients with diabetes actually suggests that treatment with fibrates may be protective. The precise mechanism by which fibrates increase serum creatinine levels is unknown.
In patients with chronic renal disease fibrates should be used with caution and at lower doses (215). Fibrates are all excreted by the kidneys and thus the excretion of fibrates is decreased in patients with renal dysfunction (215). Therefore, one needs to adjust the fibrate dose depending upon renal function. The National Kidney Foundation recommends the dose adjustments shown in Table 18 (220).
Table 18.
Fibrate Dose Adjustments in Renal Disease
| No Kidney Disease | GFR 30-60 | GFR < 30 | Kidney Transplant | |
|---|---|---|---|---|
| Bezafibrate | 400-600mg | 200mg | Avoid | Avoid |
| Ciprofibrate | 1000-2000mg | ? | Avoid | Avoid |
| Fenofibrate | 150-200mg | 40-60mg | Avoid | Avoid |
| Gemfibrozil | 1200mg | 1200mg | 600mg | 600mg |
GALLBLADDER DISEASE
It is clear that clofibrate increases the risk of gallbladder disease. In both the WHO trial and the Coronary Drug Project, cholecystectomies occurred two to three times more often in the patients treated with clofibrate compared to placebo (43,188,221). Whether gemfibrozil, fenofibrate, or other fibrates increases the risk of gallbladder disease is uncertain. In the large randomized outcome studies presented earlier (Effect of fibrates on cardiovascular outcomes section) a statistically significant increase in either gallbladder disease or cholecystectomies were not observed. However, in a sub-study of 450 Helsinki Heart Study participants a trend toward a greater prevalence of gallstones during the study in the gemfibrozil group was observed (7.5% versus 4.9% for the placebo group, a 55% excess for the gemfibrozil group) (Lopid Package Insert). A trend toward a greater incidence of gallbladder surgery was also observed in the gemfibrozil group (17 versus 11 subjects, a 54% excess) (Lopid Package Insert). In a single epidemiological trial fibrate treatment independently correlated with the presence of gallstones with a relative risk of 1.7 (p=0.04) (222).
All fibrates alter the composition of bile resulting in an increase in the concentration of cholesterol, which will predispose to the formation of cholesterol gallstones (215). In a comparison of clofibrate and gemfibrozil it was observed that clofibrate resulted in changes in bile composition that would be more lithogenic than gemfibrozil (223).
The effect of combining fibrates with statins on the risk of gallbladder disease is unknown. An increased risk of gallbladder disease or cholecystectomies was not reported in the ACCORD-LIPID trial where fenofibrate was added to statin therapy or the PROMINENT trial where pemafibrate was added to statin therapy (197,198).
While it is clear that clofibrate increases the risk of gallbladder disease the effect of other fibrates either as monotherapy or in combination with other drugs is less well defined.
PANCREATITIS
In a meta-analysis of 7 fibrate trials involving 40,162 participants conducted over 5.3 years, 144 participants developed pancreatitis (84 assigned to fibrate therapy, 60 assigned to placebo) (RR, 1.39 (95% CI, 1.00-1.95; P = .053) (224). These observations raise the possibility that fibrates may increase the risk of pancreatitis.
CANCER
A large meta-analysis of 17 randomized controlled trials, involving 44,929 participants, with an average follow-up of 5.2 years has examined if fibrates lead to an increased risk of cancer. No increase in either cancer incidence (RR = 1.02, 95% CI 0.92-1.12) or cancer death (RR = 1.06, 95% CI: 0.92-1.22) was noted with fibrate treatment (225).
LIVER DISEASE
Fenofibrate has rarely been associated with idiosyncratic hepatotoxicity manifesting as hepatocellular to cholestatic disorders (226). The hepatitis may be acute self-limited or persistent chronic hepatitis. Liver abnormalities are very rare and in large trials such as the FIELD trial described above liver function test abnormalities were similar in the fenofibrate and placebo groups (156).
GLYCEMIC PARAMETERS
A meta-analysis of 22 randomized placebo-controlled trials involving a total of 11,402 subjects demonstrated that fibrate therapy significantly decreased fasting plasma glucose, insulin levels, and insulin resistance measured by HOMA-IR, but did not effect HbA1c levels (227).
MUSCLE DISORDERS
Fibrate monotherapy has been reported to cause myopathy (215). In a large epidemiological study the incidence of hospitalization for rhabdomyolysis per 10,000 person-years for monotherapy with a fibrate was 2.82 (95% CI, 0.58-8.24) while in patients not exposed to lipid lowering drugs the incidence was 0 (95% CI, 0-0.48) (228). The risk of rhabdomyolysis was greater with gemfibrozil therapy than with fenofibrate. Interestingly the incidence of rhabdomyolysis was greater for patients treated with fibrate monotherapy than for patients treated with statin monotherapy (incidence for atorvastatin, pravastatin, or simvastatin was only 0.44 per 10,000 person-years). In an epidemiological study focusing on myopathy similar results were observed (229). The relative risks of myopathy in current users of fibrates and statins compared with nonusers were 42.4 (95% CI = 11.6-170.5) and 7.6 (95% CI = 1.4-41.3), respectively. It should be recognized though that in large randomized clinical trials the risk of muscle symptoms was low in patients treated with fibrates and not dissimilar to that seen in the patients treated with placebo (215). For example, in the Helsinki Heart Study over 2,000 patients were treated and in the VA-HIT over 1,000 patients were treated with gemfibrozil for five years and no cases of myopathy were reported in either trial (152,153). In the Bezafibrate Infarction Prevention Study, seven patients in the placebo group and five patients in the bezafibrate group reported muscle pain, while CPK levels greater than 2x the upper range of normal was seen in four patients in the bezafibrate group and one patient in the placebo group (154). Finally, in the Field Trial, patients with diabetes were treated with fenofibrate (n=4,895) or placebo (n=4,900) (156). Myositis was observed in one patient treated with placebo and two patients treated with fenofibrate while rhabdomyolysis was observed in one patient treated with placebo and three patients treated with fenofibrate. Elevations in CPK levels values > 10x the upper range of normal were seen in three patients on placebo and 4 patients treated with fenofibrate. Thus, while fibrates can lead to significant muscle dysfunction this is a rare event and appears to occur only slightly more often in patients treated with a fibrate than in patients treated with a placebo. The risk of serious muscle disease appears to be increased in patients with renal failure, hypothyroidism, and in the elderly (215). The mechanism by which fibrates predispose to muscle disorders is unknown.
The effect of fibrates in combination with statins on muscle disorders will be discussed in detail in the section on drug interactions below.
Drug Interactions
STATINS
The combination a fibrate and a statin may increase the risk of developing muscle symptoms (215). The degree of risk is dependent on both the specific statin and the specific fibrate that is being used in combination (215). For example, the average incidence per 10,000 person-years for hospitalization for rhabdomyolysis with monotherapy with atorvastatin, pravastatin, or simvastatin was 0.44 (95% CI, 0.20-0.84); with fibrate alone was 2.82 (95% CI, 0.58-8.24); and with combined therapy of atorvastatin, pravastatin, or simvastatin with a fibrate was 5.98 (95% CI, 0.72-216.0) (228). Of note, the average incidence per 10,000 person-years for hospitalization for rhabdomyolysis with the combination of cerivastatin with a fibrate was 1035 (95% CI, 389-2117), clearly demonstrating an increased risk of the cerivastatin-fibrate combination compared to other statin-fibrate combinations (228). A study by Alsheikh-Ali and colleagues looking at cases of rhabdomyolysis reported to the FDA relative to the total number of prescriptions reached the conclusion that the combination of cerivastatin with a fibrate markedly increased the risk of this complication (230). Additionally, it was noted that the risk of rhabdomyolysis was greater with gemfibrozil compared to fenofibrate and that the combination of cerivastatin and gemfibrozil was particularly toxic (230). Other studies have also noted a marked risk with the combination of cerivastatin and gemfibrozil (231). Cerivastatin is no longer available.
Studies comparing the risk of rhabdomyolysis with gemfibrozil-statin combination therapy compared to fenofibrate-statin combination therapy have shown an increased risk with gemfibrozil (215). For example, the number of cases of rhabdomyolysis reported with fenofibrate and statins other than cerivastatin was 0.58 per million prescriptions whereas with gemfibrozil and statins other than cerivastatin was 8.6 per million prescriptions (232). Reviews of the FDA’s adverse events reporting system database have estimated that the risk of myopathy for the combination of gemfibrozil with a statin was much greater than the risk with the combination of fenofibrate with a statin (230,232). Additionally, studies that employed the combination of gemfibrozil and statins have reported a significant occurrence of muscle related symptoms whereas studies of fenofibrate in combination with statins have not shown an increase in muscle related symptoms (215). For example, the rate of myopathy in over 4,000 patients taking lovastatin was only 0.4% but in patients on the combination of lovastatin and gemfibrozil the frequency increased to 5% (233). In contrast, in the ACCORD-LIPID Trial over 5,000 patients on statin therapy were randomized to fenofibrate or placebo for 4.7 years and no increase in the incidence of muscle related symptoms was observed with fenofibrate therapy (197). Similarly, in the Field Trial approximately 1,000 patients were taking fenofibrate and a statin and with 5 years of follow-up no cases of rhabdomyolysis were reported (156). Finally, a meta-analysis by Geng and colleagues identified 13 randomized trials with 7,712 patients receiving combination fenofibrate-statin therapy compared with statin therapy alone (168). The incidence of elevated creatine kinase levels, muscle-associated adverse events, or withdrawals attributed to muscle dysfunction did not differ significantly between the fenofibrate + statin patients vs. the statin alone patients (168). The American College of Cardiology and American Heart Association Guidelines recommend against using the combination of a statin and gemfibrozil but recognize that the use of a statin and fenofibrate is appropriate under certain circumstances (234).
The increased risk of combining gemfibrozil with statins is due to alterations in statin metabolism leading to increases in the serum levels of statins and hence an increased risk of myopathy (215,235). In contrast, fenofibrate does not alter statin metabolism and therefore can be safely combined with statins (Table 19) (235).
Table 19.
| Statin | Gemfibrozil | Fenofibrate |
|---|---|---|
| Atorvastatin | Increase in C-Max by 1.5-Fold | No Change |
| Simvastatin | Increase in C-Max by 2-Fold | No Change |
| Pravastatin | Increase in C-Max by 2-Fold | No Change |
| Rosuvastatin | Increase in C-Max by 2-Fold | No Change |
| Lovastatin | Increase in C-Max by 2.8-Fold | No Change |
| Pitavastatin | Increase in C-Max by 41% | Unknown |
| Fluvastatin | No Change | No Change |
The explanation for the difference between gemfibrozil and fenofibrate is that gemfibrozil uses the same family of glucuronidation enzymes as the statins thereby inhibiting statin metabolism (215,237). In contrast, fenofibrate uses a different family of glucuronidation enzymes and does not inhibit statin metabolism (215).
COUMADIN ANTI-COAGULANTS
Gemfibrozil and fenofibrate can potentiate the effect of coumadin anti-coagulants leading to a prolongation of prothrombin time and an increased risk of bleeding. When starting a fibrate in patients on coumadin therapy the dose of coumadin should be decreased and prothrombin times should be closely monitored (Lopid and Tricor Package Inserts).
REPAGLINIDE
Gemfibrozil in combination with rapaglinide increases blood levels of rapaglinide and therefore this combination should not be used because of the increased risk of hypoglycemia (Lopid Package Insert).
Contraindications
Fibrates are contraindicated in patients with severe hepatic dysfunction. Additionally, patients with pre-existing gallstones should not be treated with fibrates. Fenofibrate and gemfibrozil are pregnancy category C drugs and should only be used if the potential benefit justifies the potential risk to the fetus. The combination of gemfibrozil and a statin should be avoided.
Conclusions
Fibrates are effective drugs in reducing TG levels and modestly increase HDL-C levels. Additionally, they also reduce LDL-C and non-HDL-C levels. Fibrates have a number of side effects and one should avoid using gemfibrozil in combination with statins. In contrast, fenofibrate can be used in combination with statins. Studies have not consistently demonstrated that fibrate monotherapy therapy reduces cardiovascular events and the combination of fibrates and statins in two studies has not been shown to be beneficial. Therefore, enthusiasm to use fibrates to reduce cardiovascular events has markedly diminished. In patients with diabetes fibrates appear to slow the progression of microvascular disease. Finally, fibrates are effective in lowering TGs in patients with marked hypertriglyceridemia and while not proven will likely reduce the risk of the development of pancreatitis.
VOLANESORSEN
Introduction
Volanesorsen (Waylivra) is an antisense oligonucleotide inhibitor of apolipoprotein C-III (apo C-III) mRNA that is approved in Europe for the treatment of familial chylomicronemia syndrome (FCS). This drug has not been approved by the FDA for use in the United States. FCS is a rare metabolic disorder involving the impaired function of lipoprotein lipase (LPL) due to mutations in LPL, Apo C-II, Apo A-V, lipase maturation factor 1, and glycosylphosphatidylinositol-anchored high-density lipoprotein-binding protein 1 (GPIHBP1) (238,239). For a detailed discussion of the diagnosis and treatment of FCS see the following references (238-240).
Effect of Volanesorsen on Lipid and Lipoprotein Levels
FAMILIAL CHYLOMICRONEMIA SYNDROME (FCS)
A double-blind, randomized 52-week trial (APPROACH study) evaluated the ability of volanesorsen (300 mg subcutaneously once weekly) vs. placebo to decrease TG levels in 66 patients with FCS (baseline TGs 2,209mg/dL) (241). The primary end point was the percentage change in fasting TG levels at 3 months. As expected, there was a marked reduction in Apo C-III levels (84% decrease) in the volanesorsen group and a small increase (6%) in the placebo group. Most importantly patients treated with volanesorsen had a 77% decrease at 3 months in TG levels (mean decrease of 1,712 mg/dL) whereas patients receiving placebo had an 18% increase in TG levels. The decrease in TGs in patients treated with volanesorsen persisted for 24 months (242). Significantly, 77% of the patients in the volanesorsen group vs. only 10% of patients in the placebo group had TG levels of less than 750 mg/dL, a level that would greatly reduce the risk of pancreatitis. In addition, patients who received volanesorsen had decreases in levels of chylomicron TG by 83%, apolipoprotein B-48 by 76%, non–HDL-C by 46%, and VLDL-C by 58% and increases in levels of HDL-C by 46%, apolipoprotein A1 by 14%, LDL-C by 136% (note LDL-C increased from 28 to 61 mg/dL), and total apolipoprotein B by 20%.
While the APPROACH study was not powered to examine the effect of volanesorsen on pancreatitis, during the study three patients in the placebo group had four episodes of acute pancreatitis, whereas one patient in the volanesorsen group had one episode. In patients with a history of recurrent pancreatitis events (≥ 2 events in the 5 years prior to study, n = 11), a reduction in pancreatitis attacks was seen in patients treated with volanesorsen compared with placebo (none of the 7 patients in the volanesorsen group and 3 of the 4 patients in the placebo group experienced a pancreatitis attack over the 52-week study period).
In a retrospective global web-based survey open to all patients with the FCS who received volanesorsen for ≥3 months, 22 patients responded and reported reductions in steatorrhea, pancreatic pain, and constant worry about an attack of pain/ acute pancreatitis (243). The patients also reported that volanesorsen improved overall management of symptoms and reduced interference of FCS with work/school responsibilities. Decreases in the negative impact of FCS on personal, social, and professional life were also reported.
HYPERTRIGLYCERIDEMIA
A randomized, double-blind, placebo-controlled, study evaluated volanesorsen in patients with hypertriglyceridemia (244). Patients who were not receiving TG-lowering therapy (n=57) were eligible if they had fasting TG level between 350 mg/dL and 2000 mg/dL and were assigned to volanesorsen 100, 200, or 300 mg or placebo. Patients who were receiving a fibrate (n=28) were eligible if they had a fasting TG level between 225 mg/dL and 2000 mg/dL and were randomly assigned to volanesorsen 200 or 300 mg or placebo. The study drug was administered as a single subcutaneous injection once a week for 13 weeks. Baseline TG levels were 581±291 mg/dL in patients not on fibrates and 376±188 mg/dL in patients on fibrates. In patients not on fibrates volanesorsen 300 mg decreased Apo C-III levels by 79.6% vs. an increase of 4.2% in the placebo group (P<0.001) and decreased TG levels by 70.9% compared with an increase of 20.1% in the placebo group (P<0.001). Additionally, HDL-C levels increased by 45.7% from baseline in the 300 mg group, as compared with an increase of 0.7% in the placebo group (P<0.001). LDL-C levels increased from 79.5±29.9 mg/dL to 127.8±44.9 mg/dL with 300 mg of volanesorsen and was associated with an increase in LDL particle size. However, non-HDL-C and total apo B levels remained relatively unchanged and similar to those in the placebo group. Similar changes in Apo C-III, TGs, HDL-C, non-HDL-C, VLDL-C, and total apo B levels were observed in the patients on fibrates treated with volanesorsen. Of note, LDL-C levels did not increase in the patients on fibrates treated with volanesorsen perhaps due to the lower baseline TG levels.
The COMPASS study randomized 113 patients with fasting TGs ≥500 mg/dL (mean TG 1,261mg/dL) to receive either volanesorsen 300 mg or placebo subcutaneously once weekly for 26 weeks (245). Most of these patients had the multifactorial chylomicronemia syndrome but a small number had FCS. A 71% reduction in TGs from baseline after 3 months was observed in patients treated with volanesorsen vs. a 0.9% reduction in placebo-treated patients (P<0.0001). LDL-C levels increased 96% (64 to 111mg/dL), HDL-C increased 61% (25 to 39mg/dL) and non-HDL-C decreased 27% (232 to 158mg/dL) Notably pancreatitis episodes were reduced with 5 events in 3 patients occurring in the placebo group vs. none with volanesorsen treatment (P=0.036).
DIABETES
A randomized, double-blind, placebo-controlled trial of volanesorsen 300 mg weekly or placebo was performed in 15 adult patients with type 2 diabetes (HbA1c >7.5%) and hypertriglyceridemia (TG >200 and <500 mg/dL) (246). Treatment with volanesorsen significantly reduced plasma apo C-III (-88%, P = 0.02) and TG (-69%, P = 0.02) levels and raised HDL-C (42%, P = 0.03) without altering LDL-C levels compared with placebo. These changes were accompanied by a 57% improvement in whole-body insulin sensitivity (P < 0.001) and decreases in HbA1c (-0.44%, P = 0.025) 3 months postdosing. The improvement in insulin sensitivity was strongly related to the decrease in plasma apo C-III and TGs.
FAMILIAL PARTIAL LIPODYSTROPY (FPL)
Patients with FPL were randomized to volanesorsen 300mg weekly (n=21) or placebo (n=19) (247). Median TG level was 781mg/dL in the placebo group and 749mg/dL in the volanesorsen group. Volanesorsen treatment at 3 months resulted in an 88% decrease in TG levels while in the placebo group TG levels decreased by 22% (net difference of −67%; P=0.0009). Non-HDL-HDL-C levels decreased while LDL-C and HDL-C levels increased.
Mechanisms Accounting for the Volanesorsen Induced Lipid Effects
Volanesorsen binds to apo C-III mRNA leading to increased degradation and thereby inhibits the hepatic synthesis of apo C-III protein resulting in a reduction in plasma apo C-III levels (248,249). Apo C-III has a number of important effects on the metabolism of TG rich lipoproteins (250). Apo C-III is an inhibitor of LPL and therefore decreasing apo C-III levels will enhance LPL activity. In patients with FCS this will not be important because patients with this disorder have defects in components of the LPL complex that result in the inability to increase LPL activity. However, in patients with increased TG levels not due FCS this would accelerate the clearance of TG rich lipoproteins. Studies have also shown that apo C-III stimulates the production and secretion of VLDL by the liver. This effect is also not likely to be of primary importance in patients with FCS as the very high TG levels are primarily due to chylomicrons and not VLDL. However, in other situations increased hepatic secretion of VLDL may be an important contributor to the hypertriglyceridemia. Whether apo C-III regulates chylomicron secretion by the intestine is unknown. Finally, Apo C-III inhibits the binding of TG rich lipoproteins to hepatic LDL receptors and LDL receptor–related protein 1 decreasing the clearance of TG rich lipoprotein particles. A decrease in apo C-III will accelerate the clearance of TG rich lipoproteins, which likely accounts for the ability of volanesorsen to decrease TG levels in patients with FCS.
Drug Administration and Pharmacokinetics
The recommended starting dose is 285 mg injected subcutaneously once weekly for 3 months after which the dose should be reduced to 285 mg every 2 weeks. If serum TGs decrease by less than 25% or are not below 2000 mg/dL (22.6 mmol/L) after 3 months on volanesorsen 285 mg weekly, treatment should be discontinued (package insert; https://www.ema.europa.eu/en/documents/product-information/waylivra-epar-product-information_en.pdf).
After 6 months of treatment one can consider increasing the dose frequency back to 285 mg weekly if the serum TG response has been inadequate and the platelet counts are in the normal range. Patients should return to 285 mg every 2 weeks if the higher 285 mg once weekly dose does not provide a significant additional TG reduction after 9 months (package insert).
Effect on Clinical Outcomes
As described above in the description of the effect of volanesorsen on lipid/lipoprotein levels in patients with FCS and marked hypertriglyceridemia there is suggestive evidence that lowering the very high TG levels with volanesorsen treatment will reduce the risk of pancreatitis and improve the quality of life.
Volanesorsen treatment reduced hepatic fat assessed by MRI in patients with FCS, severe hypertriglyceridemia, and familial partial lipodystrophy (251). The greater the hepatic fat the greater the decrease induced by volanesorsen.
The effect of volanesorsen on cardiovascular disease has not been determined. However, epidemiologic studies have demonstrated that increased Apo C-III levels are associated with an increased risk of cardiovascular events (252-254) and coronary artery calcification (255). Moreover, carriers of rare heterozygous loss-of-function mutations in Apo C-III have reduced TG levels and reduced cardiovascular disease risk (256-258). One can speculate that lowering Apo C-III and TG levels with volanesorsen will have beneficial effects on the development of cardiovascular disease.
Side Effects
Treatment with volanesorsen is very commonly associated with reductions in platelet count in patients with the FCS and may result in thrombocytopenia (package insert; https://www.ema.europa.eu/en/documents/product-information/waylivra-epar-product-information_en.pdf). Platelet counts below 140 x 109/L were observed in 75% of patients treated with volanesorsen vs. 24% of placebo patients. Reductions to below 100 x 109/L were observed in 47% of patients treated with volanesorsen compared with none of the patients in the placebo group. Bleeding secondary to low platelets may occur. Careful monitoring for thrombocytopenia is important during treatment and recommendations for adjustments to monitoring frequency and dosing are shown in table 20 (package insert). Platelet counts recover following drug discontinuation and administration of glucocorticoids where medically indicated.
Table 20.
Volanesorsen Monitoring and Treatment Recommendations
| Platelet Count (x109/L) | Dose | Monitoring Frequency |
|---|---|---|
| Normal (≥140) | Starting dose: Weekly After 3 months: Every 2 weeks | Every 2 weeks |
| 100-139 | Every 2 weeks | Weekly |
| 75-99 | Pause treatment for ≥4 weeks and resume treatment after platelet levels ≥ 100 x 109/L | Weekly |
| 50-74 | Pause treatment for ≥4 weeks and resume treatment after platelet levels ≥ 100 x 109/L | Every 2-3 days |
| Less than 50 | Discontinue treatment Glucocorticoids recommended | Daily |
Renal toxicity has been observed after administration of volanesorsen. Monitoring for evidence of nephrotoxicity by routine urine dipstick is recommended on a quarterly basis. In the case of a positive assessment, one should measure serum creatinine and collect a 24-hour urine collection to quantify the proteinuria and assess creatinine clearance. Treatment should be discontinued if proteinuria ≥ 500 mg/24 hour is present, or an increase in serum creatinine ≥ 0.3 mg/dL that is >ULN occurs, or the creatinine clearance estimated by the CKD-EPI equation is ≤ 30 mL/min/1.73m2 (package insert).
Elevations of liver enzymes have been observed after administration of volanesorsen. Serum liver enzymes and bilirubin should be monitored every 3 months. Treatment should be discontinued if there is a single increase in ALT or AST > 8 x ULN, or an increase > 5 x ULN, which persists for ≥ 2 weeks, or lesser increases in ALT or AST that are associated with total bilirubin > 2 x ULN or INR > 1.5 (package insert).
As expected, injection site reactions are frequently observed and were reported in 82% of patients (erythema, pain, pruritus, or local swelling) (package insert).
Contraindications
Treatment should not be initiated in patients with thrombocytopenia (platelet count <140 x 109/L). Safety and efficacy have not been established in patients with severe renal disease or patients with hepatic impairment (package insert). There are no data on the use of volanesorsen in pregnant women and it is preferable to avoid the use of volanesorsen during pregnancy (package insert).
Drug Interactions
Discontinuation of antiplatelet drugs/NSAIDs/anticoagulants should be considered for
platelet levels < 75 x 109/L. Treatment with these products must be discontinued at platelet levels < 50 x 109/L. No other drug interactions have been described (package insert)
Conclusions
Volanesorsen is a useful drug in patients with the FCS, particularly in patients who have repeated episodes of acute pancreatitis. Whether volanesorsen will be useful for the treatment of less severe hypertriglyceridemia remains to be determined, particularly given its potential side effects. Drugs similar to volanesorsen (Olezarsen) that do not adversely affect platelets are underdevelopment (259).
ALIPOGENE TIPARVOVEC (GLYBERA)
Introduction
Alipogene tiparvovec is a gene therapy that was approved in Europe for adult patients with Familial Lipoprotein Lipase deficiency and a history of multiple or severe episodes of pancreatitis who have failed dietary therapy (260). The diagnosis of Familial Lipoprotein Lipase with loss of function mutations must be confirmed by genetic testing but patients need to have detectable levels of lipoprotein lipase protein (to avoid immunological reactions) (260). Alipogene tiparvovec is an adeno-associated virus gene therapy that results in the expression of the naturally occurring S447X variant of the human lipoprotein lipase gene that has increased lipoprotein lipase activity compared to “normal” lipoprotein lipase (260). Approximately 20% of Caucasians express this gene variant and these individuals have lower plasma TG levels and an increase in HDL-C levels (261,262). Because of the lack of long-term efficacy alipogene tiparvovec is no longer clinically available.
Effect of Alipogene Tiparvovec on Lipid and Lipoprotein Levels
In patients with plasma TG levels > 880mg/d, treatment with alipogene tiparvovec resulted in an approximately 40% decrease in fasting plasma TGs with half of the patients having > 40% decrease in fasting plasma TG levels at 3-12 weeks post treatment (263). By week 16-26, fasting TG levels returned to baseline values but chylomicron levels were reduced (263). While fasting TG levels returned to baseline, postprandial TG levels were reduced by approximately 60% suggesting that there are long term effects that are not reflected by fasting TG levels (264). In fact, in some patients treated with alipogene tiparvovec, lipoprotein lipase expression was demonstrated in muscle biopsies at 26 weeks (263).
Mechanisms Accounting for the Alipogene Tiparvovec Induced Lipid Effects
Gene therapy with alipogene tiparvovec results in the expression of lipoprotein lipase in muscle, which accelerates the clearance of chylomicrons (260,263). Studies have demonstrated a reduced peak level and a reduced area under the curve for postprandial chylomicrons (264).
Drug Administration and Pharmacokinetics
Alipogene tiparvovec is administered by multiple intramuscularly injections in the legs given at a single visit (260). The number of injections is > 40 and therefore the injections are given under spinal anesthesia (263). From 3 days before administration until 12 weeks after administration patients may be treated with cyclosporine (3mg/kg/day) and mycophenolate (2g/day) and on the day of administration methylprednisolone 1mg/kg) may be administered IV (260,263).
Effect on Clinical Outcomes
In patients with Familial Lipoprotein Lipase Deficiency the outcome of interest is pancreatitis. In a retrospective study of 19 patients treated with alipogene tiparvovec an approximate 50% decrease in pancreatitis was observed (265). In addition, patients treated with alipogene tiparvovec have reported benefits including discontinuing lipoprotein apheresis, increased energy, and the ability to liberalize their diet, which is difficult to comply with due to the marked limitation in dietary fat (263,266).
Conclusions
Alipogene tiparvovec may be a useful treatment for the rare patient with Familial Lipoprotein Lipase deficiency but the lack of long-term efficacy and the difficulty of giving the required injections led to this drug being removed from the market. Because of the rarity of this disorder the information on patients treated with this drug is limited and randomized trials are impossible.
EVINACUMAB (EVKEEZA)
Introduction
Evinacumab is a human monoclonal antibody against angiopoietin-like protein 3 (ANGPTL3). It is approved for the treatment of Homozygous Familial Hypercholesterolemia. Evinacumab decreases LDL-C levels by mechanisms independent of LDL receptor activity. The recommended dose of evinacumab is 15 mg/kg administered by intravenous infusion over 60 minutes every 4 weeks. While it is not approved for TG lowering it is effective in lowering TG levels.
Effect on Evinacumab on TG Levels
For information on the effect of evinacumab on LDL-C levels see the Endotext chapter on “Cholesterol Lowering Drugs (5). Because of the difficulty in treating severe hypertriglyceridemia, I have focused on evinacumab in this group of patients. Phase 1 studies have shown that various doses of evinacumab lower TG levels in individuals with TG levels between 150-450mg/dL with maximal effects of approximately 80% reductions (267). As one would expect LDL-C and HDL-C levels also decreased in these individuals with modest hypertriglyceridemia.
A phase 2 study evaluated evinacumab in three groups of patients with severe hypertriglyceridemia; FCS patients with bi-allelic loss-of-function mutations in the lipoprotein lipase (LPL) pathway (n = 17), multifactorial chylomicronemia syndrome (MFCS) with heterozygous loss-of-function LPL pathway mutations (n = 15), and MFCS without LPL pathway mutations (n = 19) (268). Patients were randomized to evinacumab 15 mg/kg IV or placebo every 4 weeks over 12-weeks. The effect on TG and non-HDL-C levels are shown in table 21. Despite the very small number of patients the results suggest that evinacumab can lower TG levels in patients with MFCS but not in patients with FCS. This result Is not surprising based on the proposed mechanism of action of inhibiting ANGPTL3 (see below).
Table 21.
Change in Lipid/Lipoprotein Parameters
| FCS | MFCS/heterozygous LPL pathway mutations | MFCS/ without LPL pathway mutations | ||||
|---|---|---|---|---|---|---|
| Placebo (n=5) | Evinacumab (n=12) | Placebo (n=8) | Evinacumab ((n=9) | Placebo (n=5) | Evinacumab (n=14) | |
| Fasting TG | ||||||
| Baseline | 3,918mg/dL | 3,140mg/dL | 1,351mg/dL | 1,238mg/dL | 1,030mg/dL | 1,917mg/dL |
| % change | −22.9 | −27.7 | 9.4 | −64.8* | 80.9 | −81.7** |
| Non-HDL-C | ||||||
| Baseline | 356mg/dL | 345mg/dL | 202mg/dL | 220mg/dL | 209mg/dL | 296mg/dL |
| % change | −15.2 | −34.2^ | 8.0 | −31.0^^ | 48.4 | −38.5^^^ |
- *
p= 0.0076, **p= 0.0418, ^p= 0.0074, ^^p= 0.0677, ^^^p= 0.1016.
FCS= familial chylomicronemia syndrome, MFCS= multifactorial chylomicronemia syndrome.
Mechanism Accounting for the Evinacumab Induced Decrease in TG
ANGPTL3 inhibits lipoprotein lipase (LPL) activity thereby slowing the clearance of VLDL and chylomicrons resulting in an increase in plasma triglyceride levels (269,270). Mice deficient in ANGPTL3 have lower plasma triglyceride levels while mice overexpressing ANGPTL3 have elevated plasma triglyceride levels (270). Evinacumab by inhibiting the ability of ANGPTL3 to decrease LPL activity results in an increases in LPL activity, which accelerates the clearance of TG rich lipoproteins decreasing plasma triglyceride levels (270). In patients with FCS who lack a functioning lipoprotein lipase clearance system evinacumab will not accelerate the clearance of TG rich lipoproteins. For information on the mechanism by which evinacumab lowers LDL-C and HDL-C see the Endotext chapter on “Cholesterol Lowering Drugs” (5).
Pharmacokinetics and Drug Interactions
There are no significant drug interactions.
Effect of Evinacumab on Clinical Outcomes
There are no cardiovascular outcome studies.
Homozygosity for loss-of-function mutations in ANGPTL3 is associated with significantly lower plasma levels of LDL-C, HDL-C, and triglycerides (familial combined hypolipidemia) (270,271). Heterozygous carriers of loss-of-function mutations in ANGPTL3, which occur at a frequency of about 1:300, have significantly lower total cholesterol, LDL-C, and triglyceride levels than noncarriers (270). Moreover, patients carrying loss-of-function variants in ANGPTL3 have a significantly lower risk of coronary artery disease (272,273). Additionally, in an animal model of atherosclerosis treatment with evinacumab decreased atherosclerotic lesion area and necrotic content (272). Taken together these observations suggest that inhibiting ANGPTL3 with evinacumab will reduce cardiovascular disease.
Side Effects
Serious hypersensitivity reactions have occurred with evinacumab. In clinical trials, 1 (1%) of evinacumab treated patients experienced anaphylaxis vs. 0% of patients who received placebo (package insert).
Contraindications
Based on animal studies, evinacumab may cause fetal harm when administered to pregnant patients (package insert). Patients should be advised of the potential risks to the fetus of pregnancy. Patients who may become pregnant should be advised to use effective contraception during treatment with evinacumab and for at least 5 months following the last dose.
Summary
Evinacumab lowers triglyceride levels in patients with severe hypertriglyceridemia due to multifactorial chylomicronemia syndrome and could be useful in selected patients with hypertriglyceridemia. Note it is not approved to treat severe hypertriglyceridemia and administration intravenously every 4 weeks will limit its use to special circumstances.
CLINICAL USE OF TRIGLYCERIDE LOWERING DRUGS
Marked Hypertriglyceridemia (>500mg/dL); Prevention of Pancreatitis
In patients with marked elevations in TG levels (>500-1000mg/dL) the major concern is an increased risk of pancreatitis (274,275). Because of this increased risk it is imperative to lower TG levels. The initial steps are to 1) treat any disease states that could be leading to an elevation in plasma TG levels, 2) if possible, discontinue any drugs that could be leading to an elevation in plasma TGs, and 3) initiate lifestyle changes (Table 22) (2,276).
Table 22.
Causes of Secondary Hypertriglyceridemia
| Lifestyle | Diseases | Medications |
|---|---|---|
| Excess calories | Poorly controlled diabetes | Corticosteroids |
| Excess dietary fat intake | Hypothyroidism | Oral estrogen |
| Excess simple sugars | Renal disease | Retinoic acid derivatives |
| Overweight/Obesity | HIV infection | Beta adrenergic blockers |
| Alcohol intake | Cushing’s syndrome | Thiazide diuretics |
| Pregnancy | Acromegaly | Protease inhibitors |
| Growth hormone deficiency | Bile acid sequestrants | |
| Lipodystrophy | Anti-psychotic drugs | |
| Paraproteinemia | Cyclosporine/tacrolimus | |
| Nephrotic Syndrome | L-asparaginase | |
| Inflammatory Disorders | Interferon alpha 2b | |
| Cyclophosphamide |
These initial steps are often sufficient to result in marked reductions in plasma TG levels eliminating the need for TG lowering medications. For example, in patients with diabetes in very poor glycemic control, treatment that results in good glycemic control can markedly lower TG levels (277). Similarly, the restoration of euthyroidism in a hypothyroid patient can also markedly lower lipid levels (278). If these initial steps do not result in a lowering of TGs into an acceptable range, then the use of drugs to lower plasma TG levels is indicated. There have been no randomized controlled trials demonstrating that treatment diminishes pancreatitis but most experienced clinicians believe that lowering TG levels to below 500-1000mg/dL reduces the risk of developing pancreatitis (274,275). The addition of either fibrates or fish oil to lifestyle changes are commonly used to lower markedly elevated TG levels. In some patients, combination therapy is required to lower plasma TGs to an acceptable range. In patients with Familial Chylomicronemia syndrome volanesorsen is a promising therapeutic tool.
Moderate Hypertriglyceridemia (150-500mg/dL); Prevention of Cardiovascular Disease
In the era of statin therapy, it is uncertain whether lowering TG levels in patients on statin therapy will further reduce cardiovascular events. As discussed in detail in the sections on individual drugs, the studies carried out so far have not shown that adding niacin or fibrates to statin therapy is beneficial with regards to cardiovascular disease. As also discussed, some of the available studies have major limitations because many of the patients in these outcome studies did not have substantial elevations in TGs. Nevertheless, at this time there is little enthusiasm for adding either fibrates or niacin to statins to lower the risk of cardiovascular event.
Notably, the REDUCE-IT trial, which tested the effect of high dose EPA (4 grams per day) in patients with elevated TG levels on statin therapy demonstrated a 25% reduction in cardiovascular events. However, the decrease in cardiovascular events was considerably greater than one would expect based on the reduction in TG levels suggesting that the decrease in cardiovascular events was not solely due to lowering TG levels and that other effects of EPA likely played a role. Additionally, as discussed in detail in the section discussing cardiovascular trials in the omega-3-fatty acid section there are concerns that the use of mineral oil as the placebo in the REDUCE-IT trial may have caused harmful effects leading to increased events. Thus, the role of EPA in reducing cardiovascular events is debated with some experts feeling that it is beneficial while others feeling that the evidence for benefit is very weak. Clearly additional studies are required to resolve this controversy. In the meantime, clinicians will need to use their clinical judgement on whether to treat patients with modest elevations in TG levels with EPA (icosapent ethyl; Vascepa) balancing the potential benefits of treatment vs. the potential side effects.
Some guidelines use non-HDL-C as a therapeutic goal and thus the use of omega-3-fatty acids and fibrates will often be required to lower TG levels to achieve these non-HDL-C goals. In contrast, other guidelines focus on LDL-C levels and the use of statins and thus de-emphasize the use of omega-3-fatty acids and fibrates. Given the absence of definitive data one needs to use clinical judgement. Consideration should also be given to the use of fenofibrate in hypertriglyceridemic patients with diabetes at high risk for microvascular disease given the studies that have shown that fibrates reduce the microvascular complications of diabetes. Because of the side effects of niacin, the use of niacin to lower TG levels has markedly diminished. In the past we used to use niacin to lower both LDL-C levels and TGs but with the availability of ezetimibe, bempedoic acid, and PCSK9 inhibitors the need to use niacin to lower LDL-C levels has markedly decreased.
ACKNOWLEDGEMENTS
This work was supported by grants from the Northern California Institute for Research and Education.
REFERENCES
- 1.
- Geier RR, Tannock LR. Risk of Fasting and Non-Fasting Hypertriglyceridemia in Coronary Vascular Disease and Pancreatitis. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2022.
- 2.
- Feingold KR. Approach to the Patient with Dyslipidemia. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA) 2023.
- 3.
- Feingold KR. Obesity and Dyslipidemia. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- 4.
- Feingold KR. The Effect of Diet on Cardiovascular Disease and Lipid and Lipoprotein Levels. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2021.
- 5.
- Feingold KR. Cholesterol Lowering Drugs. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hershman JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A, eds. Endotext. South Dartmouth (MA) 2021.
- 6.
- Altschul R, Hoffer A, Stephen JD. Influence of nicotinic acid on serum cholesterol in man. Arch Biochem Biophys 1955; 54:558-559 [PubMed: 14350806]
- 7.
- Cooper DL, Murrell DE, Roane DS, Harirforoosh S. Effects of formulation design on niacin therapeutics: mechanism of action, metabolism, and drug delivery. Int J Pharm 2015; 490:55-64 [PubMed: 25987211]
- 8.
- Song WL, FitzGerald GA. Niacin, an old drug with a new twist. J Lipid Res 2013; 54:2586-2594 [PMC free article: PMC3770072] [PubMed: 23948546]
- 9.
- Julius U. Niacin as antidyslipidemic drug. Can J Physiol Pharmacol 2015; 93:1043-1054 [PubMed: 26370906]
- 10.
- Morgan JM, Capuzzi DM, Baksh RI, Intenzo C, Carey CM, Reese D, Walker K. Effects of extended-release niacin on lipoprotein subclass distribution. Am J Cardiol 2003; 91:1432-1436 [PubMed: 12804729]
- 11.
- Birjmohun RS, Hutten BA, Kastelein JJ, Stroes ES. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: a meta-analysis of randomized controlled trials. J Am Coll Cardiol 2005; 45:185-197 [PubMed: 15653014]
- 12.
- Knopp RH, Alagona P, Davidson M, Goldberg AC, Kafonek SD, Kashyap M, Sprecher D, Superko HR, Jenkins S, Marcovina S. Equivalent efficacy of a time-release form of niacin (Niaspan) given once-a-night versus plain niacin in the management of hyperlipidemia. Metabolism 1998; 47:1097-1104 [PubMed: 9751239]
- 13.
- Sahebkar A, Reiner Z, Simental-Mendia LE, Ferretti G, Cicero AF. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016; 65:1664-1678 [PubMed: 27733255]
- 14.
- Goldberg AC. A meta-analysis of randomized controlled studies on the effects of extended-release niacin in women. Am J Cardiol 2004; 94:121-124 [PubMed: 15219522]
- 15.
- Ballantyne CM, Davidson MH, McKenney J, Keller LH, Bajorunas DR, Karas RH. Comparison of the safety and efficacy of a combination tablet of niacin extended release and simvastatin vs simvastatin monotherapy in patients with increased non-HDL cholesterol (from the SEACOAST I study). Am J Cardiol 2008; 101:1428-1436 [PubMed: 18471454]
- 16.
- Fazio S, Guyton JR, Polis AB, Adewale AJ, Tomassini JE, Ryan NW, Tershakovec AM. Long-term safety and efficacy of triple combination ezetimibe/simvastatin plus extended-release niacin in patients with hyperlipidemia. Am J Cardiol 2010; 105:487-494 [PubMed: 20152243]
- 17.
- Shearer GC, Pottala JV, Hansen SN, Brandenburg V, Harris WS. Effects of prescription niacin and omega-3 fatty acids on lipids and vascular function in metabolic syndrome: a randomized controlled trial. J Lipid Res 2012; 53:2429-2435 [PMC free article: PMC3466011] [PubMed: 22892157]
- 18.
- Pradhan B, Neopane A, Karki S, Karki DB. Effectiveness of nicotinic acid and bezafibrate alone and in combination for reducing serum triglyceride level. Kathmandu Univ Med J (KUMJ) 2005; 3:411-414 [PubMed: 16449845]
- 19.
- Balasubramanyam A, Coraza I, Smith EO, Scott LW, Patel P, Iyer D, Taylor AA, Giordano TP, Sekhar RV, Clark P, Cuevas-Sanchez E, Kamble S, Ballantyne CM, Pownall HJ. Combination of niacin and fenofibrate with lifestyle changes improves dyslipidemia and hypoadiponectinemia in HIV patients on antiretroviral therapy: results of "heart positive," a randomized, controlled trial. J Clin Endocrinol Metab 2011; 96:2236-2247 [PMC free article: PMC3135191] [PubMed: 21565796]
- 20.
- Carlson LA. Studies on the effect of nicotinic acid on catecholamine stimulated lipolysis in adipose tissue in vitro. Acta Med Scand 1963; 173:719-722 [PubMed: 14018705]
- 21.
- Carlson LA, Oro L. The effect of nicotinic acid on the plasma free fatty acid; demonstration of a metabolic type of sympathicolysis. Acta Med Scand 1962; 172:641-645 [PubMed: 14018702]
- 22.
- Kamanna VS, Ganji SH, Kashyap ML. Recent advances in niacin and lipid metabolism. Curr Opin Lipidol 2013; 24:239-245 [PubMed: 23619367]
- 23.
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, Offermanns S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med 2003; 9:352-355 [PubMed: 12563315]
- 24.
- Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, Ignar DM, Murdock PR, Steplewski K, Green A, Brown AJ, Dowell SJ, Szekeres PG, Hassall DG, Marshall FH, Wilson S, Pike NB. Molecular identification of high and low affinity receptors for nicotinic acid. J Biol Chem 2003; 278:9869-9874 [PubMed: 12522134]
- 25.
- Wang W, Basinger A, Neese RA, Christiansen M, Hellerstein MK. Effects of nicotinic acid on fatty acid kinetics, fuel selection, and pathways of glucose production in women. Am J Physiol Endocrinol Metab 2000; 279:E50-59 [PubMed: 10893322]
- 26.
- Lauring B, Taggart AK, Tata JR, Dunbar R, Caro L, Cheng K, Chin J, Colletti SL, Cote J, Khalilieh S, Liu J, Luo WL, Maclean AA, Peterson LB, Polis AB, Sirah W, Wu TJ, Liu X, Jin L, Wu K, Boatman PD, Semple G, Behan DP, Connolly DT, Lai E, Wagner JA, Wright SD, Cuffie C, Mitchel YB, Rader DJ, Paolini JF, Waters MG, Plump A. Niacin lipid efficacy is independent of both the niacin receptor GPR109A and free fatty acid suppression. Sci Transl Med 2012; 4:148ra115 [PubMed: 22914621]
- 27.
- Ganji SH, Tavintharan S, Zhu D, Xing Y, Kamanna VS, Kashyap ML. Niacin noncompetitively inhibits DGAT2 but not DGAT1 activity in HepG2 cells. J Lipid Res 2004; 45:1835-1845 [PubMed: 15258194]
- 28.
- Grundy SM, Mok HY, Zech L, Berman M. Influence of nicotinic acid on metabolism of cholesterol and triglycerides in man. J Lipid Res 1981; 22:24-36 [PubMed: 7217784]
- 29.
- Blond E, Rieusset J, Alligier M, Lambert-Porcheron S, Bendridi N, Gabert L, Chetiveaux M, Debard C, Chauvin MA, Normand S, Roth H, de Gouville AC, Krempf M, Vidal H, Goudable J, Laville M, Niacin" Study G. Nicotinic acid effects on insulin sensitivity and hepatic lipid metabolism: an in vivo to in vitro study. Horm Metab Res 2014; 46:390-396 [PubMed: 24806747]
- 30.
- Hernandez C, Molusky M, Li Y, Li S, Lin JD. Regulation of hepatic ApoC3 expression by PGC-1beta mediates hypolipidemic effect of nicotinic acid. Cell Metab 2010; 12:411-419 [PMC free article: PMC2950832] [PubMed: 20889132]
- 31.
- Zhang LH, Kamanna VS, Zhang MC, Kashyap ML. Niacin inhibits surface expression of ATP synthase beta chain in HepG2 cells: implications for raising HDL. J Lipid Res 2008; 49:1195-1201 [PubMed: 18316796]
- 32.
- Jin FY, Kamanna VS, Kashyap ML. Niacin decreases removal of high-density lipoprotein apolipoprotein A-I but not cholesterol ester by Hep G2 cells. Implication for reverse cholesterol transport. Arterioscler Thromb Vasc Biol 1997; 17:2020-2028 [PubMed: 9351367]
- 33.
- Blum CB, Levy RI, Eisenberg S, Hall M, 3rd, Goebel RH, Berman M. High density lipoprotein metabolism in man. J Clin Invest 1977; 60:795-807 [PMC free article: PMC372427] [PubMed: 197124]
- 34.
- Shepherd J, Packard CJ, Patsch JR, Gotto AM, Jr., Taunton OD. Effects of nicotinic acid therapy on plasma high density lipoprotein subfraction distribution and composition and on apolipoprotein A metabolism. J Clin Invest 1979; 63:858-867 [PMC free article: PMC372026] [PubMed: 221531]
- 35.
- Lamon-Fava S, Diffenderfer MR, Barrett PH, Buchsbaum A, Nyaku M, Horvath KV, Asztalos BF, Otokozawa S, Ai M, Matthan NR, Lichtenstein AH, Dolnikowski GG, Schaefer EJ. Extended-release niacin alters the metabolism of plasma apolipoprotein (Apo) A-I and ApoB-containing lipoproteins. Arterioscler Thromb Vasc Biol 2008; 28:1672-1678 [PMC free article: PMC2761712] [PubMed: 18566298]
- 36.
- Rubic T, Trottmann M, Lorenz RL. Stimulation of CD36 and the key effector of reverse cholesterol transport ATP-binding cassette A1 in monocytoid cells by niacin. Biochem Pharmacol 2004; 67:411-419 [PubMed: 15037193]
- 37.
- Zhang LH, Kamanna VS, Ganji SH, Xiong XM, Kashyap ML. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J Lipid Res 2012; 53:941-950 [PMC free article: PMC3329393] [PubMed: 22389325]
- 38.
- van der Hoorn JW, de Haan W, Berbee JF, Havekes LM, Jukema JW, Rensen PC, Princen HM. Niacin increases HDL by reducing hepatic expression and plasma levels of cholesteryl ester transfer protein in APOE*3Leiden.CETP mice. Arterioscler Thromb Vasc Biol 2008; 28:2016-2022 [PubMed: 18669886]
- 39.
- Seed M, O'Connor B, Perombelon N, O'Donnell M, Reaveley D, Knight BL. The effect of nicotinic acid and acipimox on lipoprotein(a) concentration and turnover. Atherosclerosis 1993; 101:61-68 [PubMed: 8216503]
- 40.
- Croyal M, Ouguerram K, Passard M, Ferchaud-Roucher V, Chetiveaux M, Billon-Crossouard S, de Gouville AC, Lambert G, Krempf M, Nobecourt E. Effects of Extended-Release Nicotinic Acid on Apolipoprotein (a) Kinetics in Hypertriglyceridemic Patients. Arterioscler Thromb Vasc Biol 2015; 35:2042-2047 [PubMed: 26160958]
- 41.
- Chennamsetty I, Kostner KM, Claudel T, Vinod M, Frank S, Weiss TS, Trauner M, Kostner GM. Nicotinic acid inhibits hepatic APOA gene expression: studies in humans and in transgenic mice. J Lipid Res 2012; 53:2405-2412 [PMC free article: PMC3466008] [PubMed: 22930813]
- 42.
- Pieper JA. Overview of niacin formulations: differences in pharmacokinetics, efficacy, and safety. Am J Health Syst Pharm 2003; 60:S9-14; quiz S25 [PubMed: 12901025]
- 43.
- Clofibrate and niacin in coronary heart disease. JAMA 1975; 231:360-381 [PubMed: 1088963]
- 44.
- Canner PL, Berge KG, Wenger NK, Stamler J, Friedman L, Prineas RJ, Friedewald W. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol 1986; 8:1245-1255 [PubMed: 3782631]
- 45.
- Canner PL, Furberg CD, Terrin ML, McGovern ME. Benefits of niacin by glycemic status in patients with healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2005; 95:254-257 [PubMed: 15642562]
- 46.
- Canner PL, Furberg CD, McGovern ME. Benefits of niacin in patients with versus without the metabolic syndrome and healed myocardial infarction (from the Coronary Drug Project). Am J Cardiol 2006; 97:477-479 [PubMed: 16461040]
- 47.
- Carlson LA, Rosenhamer G. Reduction of mortality in the Stockholm Ischaemic Heart Disease Secondary Prevention Study by combined treatment with clofibrate and nicotinic acid. Acta Med Scand 1988; 223:405-418 [PubMed: 3287837]
- 48.
- Carlson LA, Danielson M, Ekberg I, Klintemar B, Rosenhamer G. Reduction of myocardial reinfarction by the combined treatment with clofibrate and nicotinic acid. Atherosclerosis 1977; 28:81-86 [PubMed: 911371]
- 49.
- AIM-HIGH Investigators, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med 2011; 365:2255-2267 [PubMed: 22085343]
- 50.
- Guyton JR, Slee AE, Anderson T, Fleg JL, Goldberg RB, Kashyap ML, Marcovina SM, Nash SD, O'Brien KD, Weintraub WS, Xu P, Zhao XQ, Boden WE. Relationship of lipoproteins to cardiovascular events: the AIM-HIGH Trial (Atherothrombosis Intervention in Metabolic Syndrome With Low HDL/High Triglycerides and Impact on Global Health Outcomes). J Am Coll Cardiol 2013; 62:1580-1584 [PMC free article: PMC3862446] [PubMed: 23916935]
- 51.
- Hps Thrive Collaborative Group, Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med 2014; 371:203-212 [PubMed: 25014686]
- 52.
- Song WL, Stubbe J, Ricciotti E, Alamuddin N, Ibrahim S, Crichton I, Prempeh M, Lawson JA, Wilensky RL, Rasmussen LM, Pure E, FitzGerald GA. Niacin and biosynthesis of PGD(2)by platelet COX-1 in mice and humans. J Clin Invest 2012; 122:1459-1468 [PMC free article: PMC3314457] [PubMed: 22406532]
- 53.
- Cashin-Hemphill L, Mack WJ, Pogoda JM, Sanmarco ME, Azen SP, Blankenhorn DH. Beneficial effects of colestipol-niacin on coronary atherosclerosis. A 4-year follow-up. JAMA 1990; 264:3013-3017 [PubMed: 2243429]
- 54.
- Brown G, Albers JJ, Fisher LD, Schaefer SM, Lin JT, Kaplan C, Zhao XQ, Bisson BD, Fitzpatrick VF, Dodge HT. Regression of coronary artery disease as a result of intensive lipid-lowering therapy in men with high levels of apolipoprotein B. N Engl J Med 1990; 323:1289-1298 [PubMed: 2215615]
- 55.
- Kane JP, Malloy MJ, Ports TA, Phillips NR, Diehl JC, Havel RJ. Regression of coronary atherosclerosis during treatment of familial hypercholesterolemia with combined drug regimens. JAMA 1990; 264:3007-3012 [PubMed: 2243428]
- 56.
- Brown BG, Zhao XQ, Chait A, Fisher LD, Cheung MC, Morse JS, Dowdy AA, Marino EK, Bolson EL, Alaupovic P, Frohlich J, Albers JJ. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med 2001; 345:1583-1592 [PubMed: 11757504]
- 57.
- Whitney EJ, Krasuski RA, Personius BE, Michalek JE, Maranian AM, Kolasa MW, Monick E, Brown BG, Gotto AM, Jr. A randomized trial of a strategy for increasing high-density lipoprotein cholesterol levels: effects on progression of coronary heart disease and clinical events. Ann Intern Med 2005; 142:95-104 [PubMed: 15657157]
- 58.
- Sacks FM, Pasternak RC, Gibson CM, Rosner B, Stone PH. Effect on coronary atherosclerosis of decrease in plasma cholesterol concentrations in normocholesterolaemic patients. Harvard Atherosclerosis Reversibility Project (HARP) Group. Lancet 1994; 344:1182-1186 [PubMed: 7934538]
- 59.
- Taylor AJ, Lee HJ, Sullenberger LE. The effect of 24 months of combination statin and extended-release niacin on carotid intima-media thickness: ARBITER 3. Curr Med Res Opin 2006; 22:2243-2250 [PubMed: 17076985]
- 60.
- Taylor AJ, Sullenberger LE, Lee HJ, Lee JK, Grace KA. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation 2004; 110:3512-3517 [PubMed: 15537681]
- 61.
- Taylor AJ, Villines TC, Stanek EJ, Devine PJ, Griffen L, Miller M, Weissman NJ, Turco M. Extended-release niacin or ezetimibe and carotid intima-media thickness. N Engl J Med 2009; 361:2113-2122 [PubMed: 19915217]
- 62.
- Thoenes M, Oguchi A, Nagamia S, Vaccari CS, Hammoud R, Umpierrez GE, Khan BV. The effects of extended-release niacin on carotid intimal media thickness, endothelial function and inflammatory markers in patients with the metabolic syndrome. Int J Clin Pract 2007; 61:1942-1948 [PubMed: 17935553]
- 63.
- Lee JM, Robson MD, Yu LM, Shirodaria CC, Cunnington C, Kylintireas I, Digby JE, Bannister T, Handa A, Wiesmann F, Durrington PN, Channon KM, Neubauer S, Choudhury RP. Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function: a randomized, placebo-controlled, magnetic resonance imaging study. J Am Coll Cardiol 2009; 54:1787-1794 [PubMed: 19874992]
- 64.
- Dunbar RL, Gelfand JM. Seeing red: flushing out instigators of niacin-associated skin toxicity. J Clin Invest 2010; 120:2651-2655 [PMC free article: PMC2912206] [PubMed: 20664168]
- 65.
- Dunn RT, Ford MA, Rindone JP, Kwiecinski FA. Low-Dose Aspirin and Ibuprofen Reduce the Cutaneous Reactions Following Niacin Administration. Am J Ther 1995; 2:478-480 [PubMed: 11850694]
- 66.
- McKenney JM, Proctor JD, Harris S, Chinchili VM. A comparison of the efficacy and toxic effects of sustained- vs immediate-release niacin in hypercholesterolemic patients. JAMA 1994; 271:672-677 [PubMed: 8309029]
- 67.
- Henkin Y, Johnson KC, Segrest JP. Rechallenge with crystalline niacin after drug-induced hepatitis from sustained-release niacin. JAMA 1990; 264:241-243 [PubMed: 2355446]
- 68.
- Kashyap ML, Ganji S, Nakra NK, Kamanna VS. Niacin for treatment of nonalcoholic fatty liver disease (NAFLD): novel use for an old drug? J Clin Lipidol 2019; 13:873-879 [PubMed: 31706905]
- 69.
- Anderson TJ, Boden WE, Desvigne-Nickens P, Fleg JL, Kashyap ML, McBride R, Probstfield JL, AIM_HIGH Investigators. Safety profile of extended-release niacin in the AIM-HIGH trial. N Engl J Med 2014; 371:288-290 [PMC free article: PMC4156937] [PubMed: 25014706]
- 70.
- Miettinen TA, Taskinen MR, Pelkonen R, Nikkila EA. Glucose tolerance and plasma insulin in man during acute and chronic administration of nicotinic acid. Acta Med Scand 1969; 186:247-253 [PubMed: 5378103]
- 71.
- Poynten AM, Gan SK, Kriketos AD, O'Sullivan A, Kelly JJ, Ellis BA, Chisholm DJ, Campbell LV. Nicotinic acid-induced insulin resistance is related to increased circulating fatty acids and fat oxidation but not muscle lipid content. Metabolism 2003; 52:699-704 [PubMed: 12800094]
- 72.
- Goldberg RB, Bittner VA, Dunbar RL, Fleg JL, Grunberger G, Guyton JR, Leiter LA, McBride R, Robinson JG, Simmons DL, Wysham C, Xu P, Boden WE. Effects of Extended-Release Niacin Added to Simvastatin/Ezetimibe on Glucose and Insulin Values in AIM-HIGH. Am J Med 2016; 129:753 e713-722 [PMC free article: PMC4914402] [PubMed: 27036394]
- 73.
- Goldie C, Taylor AJ, Nguyen P, McCoy C, Zhao XQ, Preiss D. Niacin therapy and the risk of new-onset diabetes: a meta-analysis of randomised controlled trials. Heart 2016; 102:198-203 [PMC free article: PMC4752613] [PubMed: 26370223]
- 74.
- Elam MB, Hunninghake DB, Davis KB, Garg R, Johnson C, Egan D, Kostis JB, Sheps DS, Brinton EA. Effect of niacin on lipid and lipoprotein levels and glycemic control in patients with diabetes and peripheral arterial disease: the ADMIT study: A randomized trial. Arterial Disease Multiple Intervention Trial. JAMA 2000; 284:1263-1270 [PubMed: 10979113]
- 75.
- Garg A, Grundy SM. Nicotinic acid as therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. JAMA 1990; 264:723-726 [PubMed: 2374275]
- 76.
- Ding Y, Li Y, Wen A. Effect of niacin on lipids and glucose in patients with type 2 diabetes: A meta-analysis of randomized, controlled clinical trials. Clin Nutr 2015; 34:838-844 [PubMed: 25306426]
- 77.
- Gershon SL, Fox IH. Pharmacologic effects of nicotinic acid on human purine metabolism. J Lab Clin Med 1974; 84:179-186 [PubMed: 4367231]
- 78.
- Gagne JJ, Houstoun M, Reichman ME, Hampp C, Marshall JH, Toh S. Safety assessment of niacin in the US Food and Drug Administration's mini-sentinel system. Pharmacoepidemiol Drug Saf 2018; 27:30-37 [PubMed: 29108128]
- 79.
- Domanico D, Verboschi F, Altimari S, Zompatori L, Vingolo EM. Ocular Effects of Niacin: A Review of the Literature. Med Hypothesis Discov Innov Ophthalmol 2015; 4:64-71 [PMC free article: PMC4458328] [PubMed: 26060832]
- 80.
- Zargar A, Ito MK. Long chain omega-3 dietary supplements: a review of the National Library of Medicine Herbal Supplement Database. Metab Syndr Relat Disord 2011; 9:255-271 [PubMed: 21787228]
- 81.
- Kleiner AC, Cladis DP, Santerre CR. A comparison of actual versus stated label amounts of EPA and DHA in commercial omega-3 dietary supplements in the United States. J Sci Food Agric 2015; 95:1260-1267 [PubMed: 25044306]
- 82.
- Wendland E, Farmer A, Glasziou P, Neil A. Effect of alpha linolenic acid on cardiovascular risk markers: a systematic review. Heart 2006; 92:166-169 [PMC free article: PMC1860766] [PubMed: 15890766]
- 83.
- Eslick GD, Howe PR, Smith C, Priest R, Bensoussan A. Benefits of fish oil supplementation in hyperlipidemia: a systematic review and meta-analysis. Int J Cardiol 2009; 136:4-16 [PubMed: 18774613]
- 84.
- Balk EM, Lichtenstein AH, Chung M, Kupelnick B, Chew P, Lau J. Effects of omega-3 fatty acids on serum markers of cardiovascular disease risk: a systematic review. Atherosclerosis 2006; 189:19-30 [PubMed: 16530201]
- 85.
- Hartweg J, Perera R, Montori V, Dinneen S, Neil HA, Farmer A. Omega-3 polyunsaturated fatty acids (PUFA) for type 2 diabetes mellitus. Cochrane Database Syst Rev 2008:CD003205 [PMC free article: PMC9006221] [PubMed: 18254017]
- 86.
- Zheng T, Zhao J, Wang Y, Liu W, Wang Z, Shang Y, Zhang W, Zhang Y, Zhong M. The limited effect of omega-3 polyunsaturated fatty acids on cardiovascular risk in patients with impaired glucose metabolism: a meta-analysis. Clin Biochem 2014; 47:369-377 [PubMed: 24342751]
- 87.
- Chi H, Lin X, Huang H, Zheng X, Li T, Zou Y. Omega-3 fatty acid supplementation on lipid profiles in dialysis patients: meta-analysis. Arch Med Res 2014; 45:469-477 [PubMed: 25010561]
- 88.
- Pei J, Zhao Y, Huang L, Zhang X, Wu Y. The effect of n-3 polyunsaturated fatty acids on plasma lipids and lipoproteins in patients with chronic renal failure--a meta-analysis of randomized controlled trials. J Ren Nutr 2012; 22:525-532 [PubMed: 22698988]
- 89.
- Zhu W, Dong C, Du H, Zhang H, Chen J, Hu X, Hu F. Effects of fish oil on serum lipid profile in dialysis patients: a systematic review and meta-analysis of randomized controlled trials. Lipids Health Dis 2014; 13:127 [PMC free article: PMC4266905] [PubMed: 25106703]
- 90.
- Hall AV, Parbtani A, Clark WF, Spanner E, Huff MW, Philbrick DJ, Holub BJ. Omega-3 fatty acid supplementation in primary nephrotic syndrome: effects on plasma lipids and coagulopathy. J Am Soc Nephrol 1992; 3:1321-1329 [PubMed: 1477328]
- 91.
- Spadaro L, Magliocco O, Spampinato D, Piro S, Oliveri C, Alagona C, Papa G, Rabuazzo AM, Purrello F. Effects of n-3 polyunsaturated fatty acids in subjects with nonalcoholic fatty liver disease. Dig Liver Dis 2008; 40:194-199 [PubMed: 18054848]
- 92.
- Oliveira JM, Rondo PH. Omega-3 fatty acids and hypertriglyceridemia in HIV-infected subjects on antiretroviral therapy: systematic review and meta-analysis. HIV Clin Trials 2011; 12:268-274 [PubMed: 22180524]
- 93.
- De Truchis P, Kirstetter M, Perier A, Meunier C, Zucman D, Force G, Doll J, Katlama C, Rozenbaum W, Masson H, Gardette J, Melchior JC. Reduction in triglyceride level with N-3 polyunsaturated fatty acids in HIV-infected patients taking potent antiretroviral therapy: a randomized prospective study. J Acquir Immune Defic Syndr 2007; 44:278-285 [PubMed: 17179770]
- 94.
- Harris WS, Ginsberg HN, Arunakul N, Shachter NS, Windsor SL, Adams M, Berglund L, Osmundsen K. Safety and efficacy of Omacor in severe hypertriglyceridemia. J Cardiovasc Risk 1997; 4:385-391 [PubMed: 9865671]
- 95.
- Maki KC, Orloff DG, Nicholls SJ, Dunbar RL, Roth EM, Curcio D, Johnson J, Kling D, Davidson MH. A highly bioavailable omega-3 free fatty acid formulation improves the cardiovascular risk profile in high-risk, statin-treated patients with residual hypertriglyceridemia (the ESPRIT trial). Clin Ther 2013; 35:1400-1411 e1401-1403 [PubMed: 23998969]
- 96.
- Pownall HJ, Brauchi D, Kilinc C, Osmundsen K, Pao Q, Payton-Ross C, Gotto AM, Jr., Ballantyne CM. Correlation of serum triglyceride and its reduction by omega-3 fatty acids with lipid transfer activity and the neutral lipid compositions of high-density and low-density lipoproteins. Atherosclerosis 1999; 143:285-297 [PubMed: 10217357]
- 97.
- Calabresi L, Donati D, Pazzucconi F, Sirtori CR, Franceschini G. Omacor in familial combined hyperlipidemia: effects on lipids and low density lipoprotein subclasses. Atherosclerosis 2000; 148:387-396 [PubMed: 10657575]
- 98.
- Minihane AM, Khan S, Leigh-Firbank EC, Talmud P, Wright JW, Murphy MC, Griffin BA, Williams CM. ApoE polymorphism and fish oil supplementation in subjects with an atherogenic lipoprotein phenotype. Arterioscler Thromb Vasc Biol 2000; 20:1990-1997 [PubMed: 10938022]
- 99.
- Haglund O, Mehta JL, Saldeen T. Effects of fish oil on some parameters of fibrinolysis and lipoprotein(a) in healthy subjects. Am J Cardiol 1994; 74:189-192 [PubMed: 8023790]
- 100.
- Beil FU, Terres W, Orgass M, Greten H. Dietary fish oil lowers lipoprotein(a) in primary hypertriglyceridemia. Atherosclerosis 1991; 90:95-97 [PubMed: 1839209]
- 101.
- Herrmann W, Biermann J, Kostner GM. Comparison of effects of N-3 to N-6 fatty acids on serum level of lipoprotein(a) in patients with coronary artery disease. Am J Cardiol 1995; 76:459-462 [PubMed: 7653444]
- 102.
- Shinozaki K, Kambayashi J, Kawasaki T, Uemura Y, Sakon M, Shiba E, Shibuya T, Nakamura T, Mori T. The long-term effect of eicosapentaenoic acid on serum levels of lipoprotein (a) and lipids in patients with vascular disease. J Atheroscler Thromb 1996; 2:107-109 [PubMed: 9225217]
- 103.
- Eritsland J, Arnesen H, Berg K, Seljeflot I, Abdelnoor M. Serum Lp(a) lipoprotein levels in patients with coronary artery disease and the influence of long-term n-3 fatty acid supplementation. Scand J Clin Lab Invest 1995; 55:295-300 [PubMed: 7569731]
- 104.
- Davidson MH, Stein EA, Bays HE, Maki KC, Doyle RT, Shalwitz RA, Ballantyne CM, Ginsberg HN. Efficacy and tolerability of adding prescription omega-3 fatty acids 4 g/d to simvastatin 40 mg/d in hypertriglyceridemic patients: an 8-week, randomized, double-blind, placebo-controlled study. Clin Ther 2007; 29:1354-1367 [PubMed: 17825687]
- 105.
- Bays HE, Ballantyne CM, Kastelein JJ, Isaacsohn JL, Braeckman RA, Soni PN. Eicosapentaenoic acid ethyl ester (AMR101) therapy in patients with very high triglyceride levels (from the Multi-center, plAcebo-controlled, Randomized, double-blINd, 12-week study with an open-label Extension [MARINE] trial). Am J Cardiol 2011; 108:682-690 [PubMed: 21683321]
- 106.
- Ballantyne CM, Bays HE, Kastelein JJ, Stein E, Isaacsohn JL, Braeckman RA, Soni PN. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study). Am J Cardiol 2012; 110:984-992 [PubMed: 22819432]
- 107.
- Kastelein JJ, Maki KC, Susekov A, Ezhov M, Nordestgaard BG, Machielse BN, Kling D, Davidson MH. Omega-3 free fatty acids for the treatment of severe hypertriglyceridemia: the EpanoVa fOr Lowering Very high triglyceridEs (EVOLVE) trial. J Clin Lipidol 2014; 8:94-106 [PubMed: 24528690]
- 108.
- Wei MY, Jacobson TA. Effects of eicosapentaenoic acid versus docosahexaenoic acid on serum lipids: a systematic review and meta-analysis. Curr Atheroscler Rep 2011; 13:474-483 [PubMed: 21975919]
- 109.
- Tatsuno I, Saito Y, Kudou K, Ootake J. Efficacy and safety of TAK-085 compared with eicosapentaenoic acid in Japanese subjects with hypertriglyceridemia undergoing lifestyle modification: the omega-3 fatty acids randomized double-blind (ORD) study. J Clin Lipidol 2013; 7:199-207 [PubMed: 23725919]
- 110.
- Tatsuno I, Saito Y, Kudou K, Ootake J. Long-term safety and efficacy of TAK-085 in Japanese subjects with hypertriglyceridemia undergoing lifestyle modification: the omega-3 fatty acids randomized long-term (ORL) study. J Clin Lipidol 2013; 7:615-625 [PubMed: 24314359]
- 111.
- Roth EM, Bays HE, Forker AD, Maki KC, Carter R, Doyle RT, Stein EA. Prescription omega-3 fatty acid as an adjunct to fenofibrate therapy in hypertriglyceridemic subjects. J Cardiovasc Pharmacol 2009; 54:196-203 [PubMed: 19597368]
- 112.
- Shearer GC, Savinova OV, Harris WS. Fish oil -- how does it reduce plasma triglycerides? Biochim Biophys Acta 2012; 1821:843-851 [PMC free article: PMC3563284] [PubMed: 22041134]
- 113.
- Harris WS, Bulchandani D. Why do omega-3 fatty acids lower serum triglycerides? Curr Opin Lipidol 2006; 17:387-393 [PubMed: 16832161]
- 114.
- Ooi EM, Watts GF, Ng TW, Barrett PH. Effect of dietary Fatty acids on human lipoprotein metabolism: a comprehensive update. Nutrients 2015; 7:4416-4425 [PMC free article: PMC4488792] [PubMed: 26043038]
- 115.
- Davidson MH. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am J Cardiol 2006; 98:27i-33i [PubMed: 16919514]
- 116.
- Wang H, Chen X, Fisher EA. N-3 fatty acids stimulate intracellular degradation of apoprotein B in rat hepatocytes. J Clin Invest 1993; 91:1380-1389 [PMC free article: PMC288110] [PubMed: 8473489]
- 117.
- Lang CA, Davis RA. Fish oil fatty acids impair VLDL assembly and/or secretion by cultured rat hepatocytes. J Lipid Res 1990; 31:2079-2086 [PubMed: 2150855]
- 118.
- Jump DB, Tripathy S, Depner CM. Fatty acid-regulated transcription factors in the liver. Annu Rev Nutr 2013; 33:249-269 [PMC free article: PMC3940310] [PubMed: 23528177]
- 119.
- Park Y, Harris WS. Omega-3 fatty acid supplementation accelerates chylomicron triglyceride clearance. J Lipid Res 2003; 44:455-463 [PubMed: 12562865]
- 120.
- Davidson MH, Maki KC, Bays H, Carter R, Ballantyne CM. Effects of prescription omega-3-acid ethyl esters on lipoprotein particle concentrations, apolipoproteins AI and CIII, and lipoprotein-associated phospholipase A(2) mass in statin-treated subjects with hypertriglyceridemia. J Clin Lipidol 2009; 3:332-340 [PubMed: 21291831]
- 121.
- Phillipson BE, Rothrock DW, Connor WE, Harris WS, Illingworth DR. Reduction of plasma lipids, lipoproteins, and apoproteins by dietary fish oils in patients with hypertriglyceridemia. N Engl J Med 1985; 312:1210-1216 [PubMed: 3990714]
- 122.
- Skulas-Ray AC, Alaupovic P, Kris-Etherton PM, West SG. Dose-response effects of marine omega-3 fatty acids on apolipoproteins, apolipoprotein-defined lipoprotein subclasses, and Lp-PLA2 in individuals with moderate hypertriglyceridemia. J Clin Lipidol 2015; 9:360-367 [PMC free article: PMC4677829] [PubMed: 26073395]
- 123.
- Sahebkar A, Simental-Mendia LE, Mikhailidis DP, Pirro M, Banach M, Sirtori CR, Reiner Z. Effect of omega-3 supplements on plasma apolipoprotein C-III concentrations: a systematic review and meta-analysis of randomized controlled trials. Ann Med 2018:1-11 [PubMed: 30102092]
- 124.
- Davidson MH, Johnson J, Rooney MW, Kyle ML, Kling DF. A novel omega-3 free fatty acid formulation has dramatically improved bioavailability during a low-fat diet compared with omega-3-acid ethyl esters: the ECLIPSE (Epanova((R)) compared to Lovaza((R)) in a pharmacokinetic single-dose evaluation) study. J Clin Lipidol 2012; 6:573-584 [PubMed: 23312053]
- 125.
- Offman E, Marenco T, Ferber S, Johnson J, Kling D, Curcio D, Davidson M. Steady-state bioavailability of prescription omega-3 on a low-fat diet is significantly improved with a free fatty acid formulation compared with an ethyl ester formulation: the ECLIPSE II study. Vasc Health Risk Manag 2013; 9:563-573 [PMC free article: PMC3794864] [PubMed: 24124374]
- 126.
- Burr ML, Ashfield-Watt PA, Dunstan FD, Fehily AM, Breay P, Ashton T, Zotos PC, Haboubi NA, Elwood PC. Lack of benefit of dietary advice to men with angina: results of a controlled trial. Eur J Clin Nutr 2003; 57:193-200 [PubMed: 12571649]
- 127.
- Burr ML, Fehily AM, Gilbert JF, Rogers S, Holliday RM, Sweetnam PM, Elwood PC, Deadman NM. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: diet and reinfarction trial (DART). Lancet 1989; 2:757-761 [PubMed: 2571009]
- 128.
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell'Infarto miocardico. Lancet 1999; 354:447-455 [PubMed: 10465168]
- 129.
- Tavazzi L, Maggioni AP, Marchioli R, Barlera S, Franzosi MG, Latini R, Lucci D, Nicolosi GL, Porcu M, Tognoni G, Gissi HFI. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet 2008; 372:1223-1230 [PubMed: 18757090]
- 130.
- Rauch B, Schiele R, Schneider S, Diller F, Victor N, Gohlke H, Gottwik M, Steinbeck G, Del Castillo U, Sack R, Worth H, Katus H, Spitzer W, Sabin G, Senges J, Omega Study Group. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation 2010; 122:2152-2159 [PubMed: 21060071]
- 131.
- Kromhout D, Giltay EJ, Geleijnse JM, Alpha Omega Trial Group. n-3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med 2010; 363:2015-2026 [PubMed: 20929341]
- 132.
- Galan P, Kesse-Guyot E, Czernichow S, Briancon S, Blacher J, Hercberg S. Effects of B vitamins and omega 3 fatty acids on cardiovascular diseases: a randomised placebo controlled trial. BMJ 2010; 341:c6273 [PMC free article: PMC2993045] [PubMed: 21115589]
- 133.
- Investigators OT, Bosch J, Gerstein HC, Dagenais GR, Diaz R, Dyal L, Jung H, Maggiono AP, Probstfield J, Ramachandran A, Riddle MC, Ryden LE, Yusuf S. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med 2012; 367:309-318 [PubMed: 22686415]
- 134.
- Risk, Prevention Study Collaborative Group. n-3 fatty acids in patients with multiple cardiovascular risk factors. N Engl J Med 2013; 368:1800-1808 [PubMed: 23656645]
- 135.
- Ascend Study Collaborative Group, Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G, Barton J, Murphy K, Aung T, Haynes R, Cox J, Murawska A, Young A, Lay M, Chen F, Sammons E, Waters E, Adler A, Bodansky J, Farmer A, McPherson R, Neil A, Simpson D, Peto R, Baigent C, Collins R, Parish S, Armitage J. Effects of n-3 Fatty Acid Supplements in Diabetes Mellitus. N Engl J Med 2018; 379:1540-1550 [PubMed: 30146932]
- 136.
- Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Albert CM, Gordon D, Copeland T, D'Agostino D, Friedenberg G, Ridge C, Bubes V, Giovannucci EL, Willett WC, Buring JE, Group VR. Marine n-3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N Engl J Med 2019; 380: 23-32 [PMC free article: PMC6392053] [PubMed: 30415637]
- 137.
- Saito Y, Yokoyama M, Origasa H, Matsuzaki M, Matsuzawa Y, Ishikawa Y, Oikawa S, Sasaki J, Hishida H, Itakura H, Kita T, Kitabatake A, Nakaya N, Sakata T, Shimada K, Shirato K, Jelis Investigators Japan. Effects of EPA on coronary artery disease in hypercholesterolemic patients with multiple risk factors: sub-analysis of primary prevention cases from the Japan EPA Lipid Intervention Study (JELIS). Atherosclerosis 2008; 200:135-140 [PubMed: 18667204]
- 138.
- Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, Doyle RT, Jr., Juliano RA, Jiao L, Granowitz C, Tardif JC, Ballantyne CM, REDUCE-IT Investigators. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N Engl J Med 2019; 380:11-22 [PubMed: 30415628]
- 139.
- Nicholls SJ, Lincoff AM, Garcia M, Bash D, Ballantyne CM, Barter PJ, Davidson MH, Kastelein JJP, Koenig W, McGuire DK, Mozaffarian D, Ridker PM, Ray KK, Katona BG, Himmelmann A, Loss LE, Rensfeldt M, Lundstrom T, Agrawal R, Menon V, Wolski K, Nissen SE. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA 2020; 324:2268-2280 [PMC free article: PMC7667577] [PubMed: 33190147]
- 140.
- Kalstad AA, Myhre PL, Laake K, Tveit SH, Schmidt EB, Smith P, Nilsen DWT, Tveit A, Fagerland MW, Solheim S, Seljeflot I, Arnesen H, Omemi Investigators. Effects of n-3 Fatty Acid Supplements in Elderly Patients After Myocardial Infarction: A Randomized, Controlled Trial. Circulation 2021; 143:528-539 [PubMed: 33191772]
- 141.
- Mason RP, Libby P, Bhatt DL. Emerging Mechanisms of Cardiovascular Protection for the Omega-3 Fatty Acid Eicosapentaenoic Acid. Arterioscler Thromb Vasc Biol 2020:ATVBAHA119313286 [PMC free article: PMC7176343] [PubMed: 32212849]
- 142.
- Marston NA, Giugliano RP, Im K, Silverman MG, O'Donoghue ML, Wiviott SD, Ference BA, Sabatine MS. Association Between Triglyceride Lowering and Reduction of Cardiovascular Risk Across Multiple Lipid-Lowering Therapeutic Classes: A Systematic Review and Meta-Regression Analysis of Randomized Controlled Trials. Circulation 2019; 140:1308-1317 [PMC free article: PMC6791781] [PubMed: 31530008]
- 143.
- Ridker PM, Rifai N, MacFadyen J, Glynn RJ, Jiao L, Steg PG, Miller M, Brinton EA, Jacobson TA, Tardif JC, Ballantyne CM, Mason RP, Bhatt DL. Effects of Randomized Treatment With Icosapent Ethyl and a Mineral Oil Comparator on Interleukin-1beta, Interleukin-6, C-Reactive Protein, Oxidized Low-Density Lipoprotein Cholesterol, Homocysteine, Lipoprotein(a), and Lipoprotein-Associated Phospholipase A2: A REDUCE-IT Biomarker Substudy. Circulation 2022; 146:372-379 [PubMed: 35762321]
- 144.
- Goff ZD, Nissen SE. N-3 polyunsaturated fatty acids for cardiovascular risk. Curr Opin Cardiol 2022; 37:356-363 [PubMed: 35275889]
- 145.
- Mason RP, Sherratt SCR, Eckel RH. Omega-3-fatty acids: Do they prevent cardiovascular disease? Best Pract Res Clin Endocrinol Metab 2023; 37:101681 [PubMed: 35739003]
- 146.
- Wachira JK, Larson MK, Harris WS. n-3 Fatty acids affect haemostasis but do not increase the risk of bleeding: clinical observations and mechanistic insights. Br J Nutr 2014; 111:1652-1662 [PubMed: 24472372]
- 147.
- Zimetbaum P, Frishman WH, Kahn S. Effects of gemfibrozil and other fibric acid derivatives on blood lipids and lipoproteins. J Clin Pharmacol 1991; 31:25-37 [PubMed: 2045526]
- 148.
- Loomba RS, Arora R. Prevention of cardiovascular disease utilizing fibrates--a pooled meta-analysis. Am J Ther 2010; 17:e182-188 [PubMed: 20535009]
- 149.
- Rosenson RS. Fenofibrate: treatment of hyperlipidemia and beyond. Expert Rev Cardiovasc Ther 2008; 6:1319-1330 [PubMed: 19018684]
- 150.
- Miller M, Bachorik PS, McCrindle BW, Kwiterovich PO, Jr. Effect of gemfibrozil in men with primary isolated low high-density lipoprotein cholesterol: a randomized, double-blind, placebo-controlled, crossover study. Am J Med 1993; 94:7-12 [PubMed: 8420303]
- 151.
- Berthold HK, Gouni-Berthold I. Hyperlipoproteinemia(a): clinical significance and treatment options. Atheroscler Suppl 2013; 14:1-5 [PubMed: 23357133]
- 152.
- Frick MH, Elo O, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P, Manninen V, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. N Engl J Med 1987; 317:1237-1245 [PubMed: 3313041]
- 153.
- Rubins HB, Robins SJ, Collins D, Fye CL, Anderson JW, Elam MB, Faas FH, Linares E, Schaefer EJ, Schectman G, Wilt TJ, Wittes J. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. N Engl J Med 1999; 341:410-418 [PubMed: 10438259]
- 154.
- Bezafibrate Infarction Prevention s. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation 2000; 102:21-27 [PubMed: 10880410]
- 155.
- Czupryniak L, Joshi SR, Gogtay JA, Lopez M. Effect of micronized fenofibrate on microvascular complications of type 2 diabetes: a systematic review. Expert Opin Pharmacother 2016; 17:1463-1473 [PubMed: 27267786]
- 156.
- Keech A, Simes RJ, Barter P, Best J, Scott R, Taskinen MR, Forder P, Pillai A, Davis T, Glasziou P, Drury P, Kesaniemi YA, Sullivan D, Hunt D, Colman P, d'Emden M, Whiting M, Ehnholm C, Laakso M, investigators Fs. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet 2005; 366:1849-1861 [PubMed: 16310551]
- 157.
- Aguilar-Salinas CA, Fanghanel-Salmon G, Meza E, Montes J, Gulias-Herrero A, Sanchez L, Monterrubio-Flores EA, Gonzalez-Valdez H, Gomez Perez FJ. Ciprofibrate versus gemfibrozil in the treatment of mixed hyperlipidemias: an open-label, multicenter study. Metabolism 2001; 50:729-733 [PubMed: 11398153]
- 158.
- Knipscheer HC, de Valois JC, van den Ende B, Wouter ten Cate J, Kastelein JJ. Ciprofibrate versus gemfibrozil in the treatment of primary hyperlipidaemia. Atherosclerosis 1996; 124 Suppl:S75-81 [PubMed: 8831919]
- 159.
- Insua A, Massari F, Rodriguez Moncalvo JJ, Ruben Zanchetta J, Insua AM. Fenofibrate of gemfibrozil for treatment of types IIa and IIb primary hyperlipoproteinemia: a randomized, double-blind, crossover study. Endocr Pract 2002; 8:96-101 [PubMed: 11942772]
- 160.
- Jen SL, Chen JW, Lee WL, Wang SP. Efficacy and safety of fenofibrate or gemfibrozil on serum lipid profiles in Chinese patients with type IIb hyperlipidemia: a single-blind, randomized, and cross-over study. Zhonghua Yi Xue Za Zhi (Taipei) 1997; 59:217-224 [PubMed: 9216117]
- 161.
- Ebcioglu Z, Morgan J, Carey C, Capuzzi D. Paradoxical lowering of high-density lipoprotein cholesterol level in 2 patients receiving fenofibrate and a thiazolidinedione. Ann Intern Med 2003; 139:W80 [PubMed: 14597479]
- 162.
- Shetty C, Balasubramani M, Capps N, Milles J, Ramachandran S. Paradoxical HDL-C reduction during rosiglitazone and fibrate treatment. Diabet Med 2007; 24:94-97 [PubMed: 17227331]
- 163.
- Collinson PO, Hjelm CJ, Canepo-Anson R. Paradoxical high-density lipoprotein reduction induced by fenofibrate and ciprofibrate. Ann Clin Biochem 1996; 33 (Pt 2):159-161 [PubMed: 8729728]
- 164.
- Schofield JD, Liu Y, France MW, Sandle L, Soran H. A review of paradoxical HDL-C responses to fenofibrate, illustrated by a case report. J Clin Lipidol 2014; 8:455-459 [PubMed: 25110229]
- 165.
- Nobecourt E, Cariou B, Lambert G, Krempf M. Severe decrease in high-density lipoprotein cholesterol with the combination of fibrates and ezetimibe: A case series. J Clin Lipidol 2017; 11:289-293 [PubMed: 28391898]
- 166.
- Grundy SM, Vega GL, Yuan Z, Battisti WP, Brady WE, Palmisano J. Effectiveness and tolerability of simvastatin plus fenofibrate for combined hyperlipidemia (the SAFARI trial). Am J Cardiol 2005; 95:462-468 [PubMed: 15695129]
- 167.
- Choi HD, Shin WG. Safety and efficacy of statin treatment alone and in combination with fibrates in patients with dyslipidemia: a meta-analysis. Curr Med Res Opin 2014; 30:1-10 [PubMed: 24063624]
- 168.
- Geng Q, Ren J, Chen H, Lee C, Liang W. Adverse events following statin-fenofibrate therapy versus statin alone: a meta-analysis of randomized controlled trials. Clin Exp Pharmacol Physiol 2013; 40:219-226 [PubMed: 23324122]
- 169.
- Kontopoulos AG, Athyros VG, Papageorgiou AA, Hatzikonstandinou HA, Mayroudi MC, Boudoulas H. Effects of simvastatin and ciprofibrate alone and in combination on lipid profile, plasma fibrinogen and low density lipoprotein particle structure and distribution in patients with familial combined hyperlipidaemia and coronary artery disease. Coron Artery Dis 1996; 7:843-850 [PubMed: 8993943]
- 170.
- Choi HD, Shin WG, Lee JY, Kang BC. Safety and efficacy of fibrate-statin combination therapy compared to fibrate monotherapy in patients with dyslipidemia: a meta-analysis. Vascul Pharmacol 2015; 65-66:23-30 [PubMed: 25451563]
- 171.
- Ansquer JC, Bekaert I, Guy M, Hanefeld M, Simon A. Efficacy and safety of coadministration of fenofibrate and ezetimibe compared with each as monotherapy in patients with type IIb dyslipidemia and features of the metabolic syndrome: a prospective, randomized, double-blind, three-parallel arm, multicenter, comparative study. Am J Cardiovasc Drugs 2009; 9:91-101 [PubMed: 19331437]
- 172.
- Farnier M, Freeman MW, Macdonell G, Perevozskaya I, Davies MJ, Mitchel YB, Gumbiner B, Ezetimibe Study Group. Efficacy and safety of the coadministration of ezetimibe with fenofibrate in patients with mixed hyperlipidaemia. Eur Heart J 2005; 26:897-905 [PubMed: 15781429]
- 173.
- Farnier M, Roth E, Gil-Extremera B, Mendez GF, Macdonell G, Hamlin C, Perevozskaya I, Davies MJ, Kush D, Mitchel YB, Ezetimibe/Simvastatin + Fenofibrate Study G. Efficacy and safety of the coadministration of ezetimibe/simvastatin with fenofibrate in patients with mixed hyperlipidemia. Am Heart J 2007; 153:335 e331-338 [PubMed: 17239698]
- 174.
- Jones PH, Goldberg AC, Knapp HR, Kelly MT, Setze CM, Stolzenbach JC, Sleep DJ. Efficacy and safety of fenofibric acid in combination with atorvastatin and ezetimibe in patients with mixed dyslipidemia. Am Heart J 2010; 160:759-766 [PubMed: 20934572]
- 175.
- McKenney J, Jones M, Abby S. Safety and efficacy of colesevelam hydrochloride in combination with fenofibrate for the treatment of mixed hyperlipidemia. Curr Med Res Opin 2005; 21:1403-1412 [PubMed: 16197659]
- 176.
- Weisweiler P. Low-dose colestipol plus fenofibrate: effects on plasma lipoproteins, lecithin:cholesterol acyltransferase, and postheparin lipases in familial hypercholesterolemia. Metabolism 1989; 38:271-274 [PubMed: 2918846]
- 177.
- Curtis LD, Dickson AC, Ling KL, Betteridge J. Combination treatment with cholestyramine and bezafibrate for heterozygous familial hypercholesterolaemia. BMJ 1988; 297:173-175 [PMC free article: PMC1834216] [PubMed: 3044508]
- 178.
- Series JJ, Caslake MJ, Kilday C, Cruickshank A, Demant T, Lorimer AR, Packard CJ, Shepherd J. Effect of combined therapy with bezafibrate and cholestyramine on low-density lipoprotein metabolism in type IIa hypercholesterolemia. Metabolism 1989; 38:153-158 [PubMed: 2643751]
- 179.
- Stein EA, Heimann KW. Colestipol, clofibrate, cholestyramine and combination therapy in the treatment of familial hyperbetalipoproteinaemia. S Afr Med J 1975; 49:1252-1256 [PubMed: 168650]
- 180.
- Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARalpha action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015; 62:720-733 [PubMed: 25450203]
- 181.
- Staels B, Maes M, Zambon A. Fibrates and future PPARalpha agonists in the treatment of cardiovascular disease. Nat Clin Pract Cardiovasc Med 2008; 5:542-553 [PubMed: 18628776]
- 182.
- Shah A, Rader DJ, Millar JS. The effect of PPAR-alpha agonism on apolipoprotein metabolism in humans. Atherosclerosis 2010; 210:35-40 [PubMed: 20005515]
- 183.
- Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation 1998; 98:2088-2093 [PubMed: 9808609]
- 184.
- Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM, Witztum JL. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 2014; 371:2200-2206 [PubMed: 25470695]
- 185.
- Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL, Esko JD. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through LDL family receptors. J Clin Invest 2016; 126:2855-2866 [PMC free article: PMC4966320] [PubMed: 27400128]
- 186.
- Clavey V, Lestavel-Delattre S, Copin C, Bard JM, Fruchart JC. Modulation of lipoprotein B binding to the LDL receptor by exogenous lipids and apolipoproteins CI, CII, CIII, and E. Arterioscler Thromb Vasc Biol 1995; 15:963-971 [PubMed: 7600129]
- 187.
- Nigon F, Lesnik P, Rouis M, Chapman MJ. Discrete subspecies of human low density lipoproteins are heterogeneous in their interaction with the cellular LDL receptor. J Lipid Res 1991; 32:1741-1753 [PubMed: 1770294]
- 188.
- A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. Report from the Committee of Principal Investigators. Br Heart J 1978; 40:1069-1118 [PMC free article: PMC483536] [PubMed: 361054]
- 189.
- Manninen V, Elo MO, Frick MH, Haapa K, Heinonen OP, Heinsalmi P, Helo P, Huttunen JK, Kaitaniemi P, Koskinen P, et al. Lipid alterations and decline in the incidence of coronary heart disease in the Helsinki Heart Study. JAMA 1988; 260:641-651 [PubMed: 3164788]
- 190.
- Manninen V, Tenkanen L, Koskinen P, Huttunen JK, Manttari M, Heinonen OP, Frick MH. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation 1992; 85:37-45 [PubMed: 1728471]
- 191.
- Tenkanen L, Manttari M, Manninen V. Some coronary risk factors related to the insulin resistance syndrome and treatment with gemfibrozil. Experience from the Helsinki Heart Study. Circulation 1995; 92:1779-1785 [PubMed: 7671361]
- 192.
- Robins SJ, Collins D, Wittes JT, Papademetriou V, Deedwania PC, Schaefer EJ, McNamara JR, Kashyap ML, Hershman JM, Wexler LF, Rubins HB, Trial V-HSGVAH-DLI. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA 2001; 285:1585-1591 [PubMed: 11268266]
- 193.
- Tenenbaum A, Motro M, Fisman EZ, Tanne D, Boyko V, Behar S. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med 2005; 165:1154-1160 [PubMed: 15911729]
- 194.
- Meade T, Zuhrie R, Cook C, Cooper J. Bezafibrate in men with lower extremity arterial disease: randomised controlled trial. BMJ 2002; 325:1139 [PMC free article: PMC133451] [PubMed: 12433762]
- 195.
- Meade TW, For the British Medical Research Council General Practice Research Framework, participating vascular clinics. Design and intermediate results of the Lower Extremity Arterial Disease Event Reduction (LEADER)* trial of bezafibrate in men with lower extremity arterial disease [ISRCTN4119421]. Curr Control Trials Cardiovasc Med 2001; 2:195-204 [PMC free article: PMC57751] [PubMed: 11806795]
- 196.
- Scott R, O'Brien R, Fulcher G, Pardy C, D'Emden M, Tse D, Taskinen MR, Ehnholm C, Keech A, Fenofibrate Intervention and Event Lowering in Diabetes Study. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009; 32:493-498 [PMC free article: PMC2646035] [PubMed: 18984774]
- 197.
- ACCORD Study Group, Ginsberg HN, Elam MB, Lovato LC, Crouse JR, 3rd, Leiter LA, Linz P, Friedewald WT, Buse JB, Gerstein HC, Probstfield J, Grimm RH, Ismail-Beigi F, Bigger JT, Goff DC, Jr., Cushman WC, Simons-Morton DG, Byington RP. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010; 362:1563-1574 [PMC free article: PMC2879499] [PubMed: 20228404]
- 198.
- Das Pradhan A, Glynn RJ, Fruchart JC, MacFadyen JG, Zaharris ES, Everett BM, Campbell SE, Oshima R, Amarenco P, Blom DJ, Brinton EA, Eckel RH, Elam MB, Felicio JS, Ginsberg HN, Goudev A, Ishibashi S, Joseph J, Kodama T, Koenig W, Leiter LA, Lorenzatti AJ, Mankovsky B, Marx N, Nordestgaard BG, Pall D, Ray KK, Santos RD, Soran H, Susekov A, Tendera M, Yokote K, Paynter NP, Buring JE, Libby P, Ridker PM. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N Engl J Med 2022; 387:1923-1934 [PubMed: 36342113]
- 199.
- Harrold BP, Marmion VJ, Gough KR. A double-blind controlled trial of clofibrate in the treatment of diabetic retinopathy. Diabetes 1969; 18:285-291 [PubMed: 4894161]
- 200.
- Duncan LJ, Cullen JF, Ireland JT, Nolan J, Clarke BF, Oliver MF. A three-year trial of atromid therapy in exudative diabetic retinopathy. Diabetes 1968; 17:458-467 [PubMed: 4875170]
- 201.
- Keech AC, Mitchell P, Summanen PA, O'Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d'Emden MC, Crimet DC, O'Connell RL, Colman PG. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet 2007; 370:1687-1697 [PubMed: 17988728]
- 202.
- ACCORD Study Group, ACCORD Eye Study Group, Chew EY, Ambrosius WT, Davis MD, Danis RP, Gangaputra S, Greven CM, Hubbard L, Esser BA, Lovato JF, Perdue LH, Goff DC, Jr., Cushman WC, Ginsberg HN, Elam MB, Genuth S, Gerstein HC, Schubart U, Fine LJ. Effects of medical therapies on retinopathy progression in type 2 diabetes. N Engl J Med 2010; 363:233-244 [PMC free article: PMC4026164] [PubMed: 20587587]
- 203.
- Emmerich KH, Poritis N, Stelmane I, Klindzane M, Erbler H, Goldsteine J, Gortelmeyer R. [Efficacy and safety of etofibrate in patients with non-proliferative diabetic retinopathy]. Klin Monbl Augenheilkd 2009; 226:561-567 [PubMed: 19644802]
- 204.
- Massin P, Peto T, Ansquer JC, Aubonnet P, Macu FENSIFT. Effects of fenofibric acid on diabetic macular edema: the MacuFen study. Ophthalmic Epidemiol 2014; 21:307-317 [PubMed: 25133794]
- 205.
- Knickelbein JE, Abbott AB, Chew EY. Fenofibrate and Diabetic Retinopathy. Curr Diab Rep 2016; 16:90 [PMC free article: PMC8323449] [PubMed: 27525681]
- 206.
- Hu Y, Chen Y, Ding L, He X, Takahashi Y, Gao Y, Shen W, Cheng R, Chen Q, Qi X, Boulton ME, Ma JX. Pathogenic role of diabetes-induced PPAR-alpha down-regulation in microvascular dysfunction. Proc Natl Acad Sci U S A 2013; 110:15401-15406 [PMC free article: PMC3780907] [PubMed: 24003152]
- 207.
- Ansquer JC, Foucher C, Rattier S, Taskinen MR, Steiner G, DAIS Investigators. Fenofibrate reduces progression to microalbuminuria over 3 years in a placebo-controlled study in type 2 diabetes: results from the Diabetes Atherosclerosis Intervention Study (DAIS). Am J Kidney Dis 2005; 45:485-493 [PubMed: 15754270]
- 208.
- Davis TM, Ting R, Best JD, Donoghoe MW, Drury PL, Sullivan DR, Jenkins AJ, O'Connell RL, Whiting MJ, Glasziou PP, Simes RJ, Kesaniemi YA, Gebski VJ, Scott RS, Keech ACi. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia 2011; 54:280-290 [PubMed: 21052978]
- 209.
- Ansquer JC, Dalton RN, Causse E, Crimet D, Le Malicot K, Foucher C. Effect of fenofibrate on kidney function: a 6-week randomized crossover trial in healthy people. Am J Kidney Dis 2008; 51:904-913 [PubMed: 18501783]
- 210.
- Sun X, Liu J, Wang G. Fenofibrate decreased microalbuminuria in the type 2 diabetes patients with hypertriglyceridemia. Lipids Health Dis 2020; 19:103 [PMC free article: PMC7245839] [PubMed: 32446306]
- 211.
- Kouroumichakis I, Papanas N, Zarogoulidis P, Liakopoulos V, Maltezos E, Mikhailidis DP. Fibrates: therapeutic potential for diabetic nephropathy? Eur J Intern Med 2012; 23:309-316 [PubMed: 22560376]
- 212.
- Rajamani K, Colman PG, Li LP, Best JD, Voysey M, D'Emden MC, Laakso M, Baker JR, Keech AC. Effect of fenofibrate on amputation events in people with type 2 diabetes mellitus (FIELD study): a prespecified analysis of a randomised controlled trial. Lancet 2009; 373:1780-1788 [PMC free article: PMC2687887] [PubMed: 19465233]
- 213.
- Waldman B, Ansquer JC, Sullivan DR, Jenkins AJ, McGill N, Buizen L, Davis TME, Best JD, Li L, Feher MD, Foucher C, Kesaniemi YA, Flack J, d'Emden MC, Scott RS, Hedley J, Gebski V, Keech AC. Effect of fenofibrate on uric acid and gout in type 2 diabetes: a post-hoc analysis of the randomised, controlled FIELD study. Lancet Diabetes Endocrinol 2018; 6:310-318 [PubMed: 29496472]
- 214.
- Derosa G, Maffioli P, Sahebkar A. Plasma uric acid concentrations are reduced by fenofibrate: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol Res 2015; 102:63-70 [PubMed: 26384444]
- 215.
- Davidson MH, Armani A, McKenney JM, Jacobson TA. Safety considerations with fibrate therapy. Am J Cardiol 2007; 99:3C-18C [PubMed: 17368275]
- 216.
- Sahebkar A, Simental-Mendia LE, Pirro M, Montecucco F, Carbone F, Banach M, Barreto GE, Butler AE. Impact of fibrates on circulating cystatin C levels: a systematic review and meta-analysis of clinical trials. Ann Med 2018:1-9 [PubMed: 29957074]
- 217.
- Hadjivasilis A, Kouis P, Kousios A, Panayiotou A. The Effect of Fibrates on Kidney Function and Chronic Kidney Disease Progression: A Systematic Review and Meta-Analysis of Randomised Studies. J Clin Med 2022; 11 [PMC free article: PMC8836930] [PubMed: 35160220]
- 218.
- Hottelart C, El Esper N, Rose F, Achard JM, Fournier A. Fenofibrate increases creatininemia by increasing metabolic production of creatinine. Nephron 2002; 92:536-541 [PubMed: 12372935]
- 219.
- Hottelart C, el Esper N, Achard JM, Pruna A, Fournier A. [Fenofibrate increases blood creatinine, but does not change the glomerular filtration rate in patients with mild renal insufficiency]. Nephrologie 1999; 20:41-44 [PubMed: 10081035]
- 220.
- National Kidney Foundation. KDOQI Clinical Practice Guideline for Diabetes and CKD: 2012 Update. Am J Kidney Dis 2012; 60:850-886 [PubMed: 23067652]
- 221.
- Gallbladder disease as a side effect of drugs influencing lipid metabolism. Experience in the Coronary Drug Project. N Engl J Med 1977; 296:1185-1190 [PubMed: 323705]
- 222.
- Caroli-Bosc FX, Le Gall P, Pugliese P, Delabre B, Caroli-Bosc C, Demarquay JF, Delmont JP, Rampal P, Montet JC. Role of fibrates and HMG-CoA reductase inhibitors in gallstone formation: epidemiological study in an unselected population. Dig Dis Sci 2001; 46:540-544 [PubMed: 11318529]
- 223.
- Hall MJ, Nelson LM, Russell RI, Howard AN. Gemfibrozil--the effect of biliary cholesterol saturation of a new lipid-lowering agent and its comparison with clofibrate. Atherosclerosis 1981; 39:511-516 [PubMed: 6942844]
- 224.
- Preiss D, Tikkanen MJ, Welsh P, Ford I, Lovato LC, Elam MB, LaRosa JC, DeMicco DA, Colhoun HM, Goldenberg I, Murphy MJ, MacDonald TM, Pedersen TR, Keech AC, Ridker PM, Kjekshus J, Sattar N, McMurray JJ. Lipid-modifying therapies and risk of pancreatitis: a meta-analysis. JAMA 2012; 308:804-811 [PubMed: 22910758]
- 225.
- Bonovas S, Nikolopoulos GK, Bagos PG. Use of fibrates and cancer risk: a systematic review and meta-analysis of 17 long-term randomized placebo-controlled trials. PLoS One 2012; 7:e45259 [PMC free article: PMC3446944] [PubMed: 23028888]
- 226.
- Ahmad J, Odin JA, Hayashi PH, Chalasani N, Fontana RJ, Barnhart H, Cirulli ET, Kleiner DE, Hoofnagle JH. Identification and Characterization of Fenofibrate-Induced Liver Injury. Dig Dis Sci 2017; 62:3596-3604 [PMC free article: PMC5694705] [PubMed: 29119413]
- 227.
- Simental-Mendia LE, Simental-Mendia M, Sanchez-Garcia A, Banach M, Atkin SL, Gotto AM, Jr., Sahebkar A. Effect of fibrates on glycemic parameters: A systematic review and meta-analysis of randomized placebo-controlled trials. Pharmacol Res 2018; 132:232-241 [PubMed: 29292213]
- 228.
- Graham DJ, Staffa JA, Shatin D, Andrade SE, Schech SD, La Grenade L, Gurwitz JH, Chan KA, Goodman MJ, Platt R. Incidence of hospitalized rhabdomyolysis in patients treated with lipid-lowering drugs. JAMA 2004; 292:2585-2590 [PubMed: 15572716]
- 229.
- Gaist D, Rodriguez LA, Huerta C, Hallas J, Sindrup SH. Lipid-lowering drugs and risk of myopathy: a population-based follow-up study. Epidemiology 2001; 12:565-569 [PubMed: 11505177]
- 230.
- Alsheikh-Ali AA, Kuvin JT, Karas RH. Risk of adverse events with fibrates. Am J Cardiol 2004; 94:935-938 [PubMed: 15464682]
- 231.
- Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med 2002; 346:539-540 [PubMed: 11844864]
- 232.
- Jones PH, Davidson MH. Reporting rate of rhabdomyolysis with fenofibrate + statin versus gemfibrozil + any statin. Am J Cardiol 2005; 95:120-122 [PubMed: 15619408]
- 233.
- Tobert JA. Efficacy and long-term adverse effect pattern of lovastatin. Am J Cardiol 1988; 62:28J-34J [PubMed: 3055921]
- 234.
- Stone NJ, Robinson JG, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC, Jr., Watson K, Wilson PW, Eddleman KM, Jarrett NM, LaBresh K, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC, Jr., Tomaselli GF, American College of Cardiology/American Heart Association Task Force on Practice Group. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014; 129:S1-45 [PubMed: 24222016]
- 235.
- Kellick KA, Bottorff M, Toth PP, The National Lipid Association's Safety Task Force. A clinician's guide to statin drug-drug interactions. J Clin Lipidol 2014; 8:S30-46 [PubMed: 24793440]
- 236.
- Whitfield LR, Porcari AR, Alvey C, Abel R, Bullen W, Hartman D. Effect of gemfibrozil and fenofibrate on the pharmacokinetics of atorvastatin. J Clin Pharmacol 2011; 51:378-388 [PubMed: 20413454]
- 237.
- Prueksaritanont T, Tang C, Qiu Y, Mu L, Subramanian R, Lin JH. Effects of fibrates on metabolism of statins in human hepatocytes. Drug Metab Dispos 2002; 30:1280-1287 [PubMed: 12386136]
- 238.
- Chait A, Subramanian S. Hypertriglyceridemia: Pathophysiology, Role of Genetics, Consequences, and Treatment. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA) 2019.
- 239.
- Shah AS, Wilson DP. Genetic Disorders Causing Hypertriglyceridemia in Children and Adolescents. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, eds. Endotext. South Dartmouth (MA) 2020.
- 240.
- Chait A, Eckel RH. The Chylomicronemia Syndrome Is Most Often Multifactorial: A Narrative Review of Causes and Treatment. Ann Intern Med 2019; 170:626-634 [PubMed: 31035285]
- 241.
- Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, Yang Q, Hughes SG, Geary RS, Arca M, Stroes ESG, Bergeron J, Soran H, Civeira F, Hemphill L, Tsimikas S, Blom DJ, O'Dea L, Bruckert E. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N Engl J Med 2019; 381:531-542 [PubMed: 31390500]
- 242.
- Witztum JL, Gaudet D, Arca M, Jones A, Soran H, Gouni-Berthold I, Stroes ESG, Alexander VJ, Jones R, Watts L, Xia S, Tsimikas S. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome: Long-term efficacy and safety data from patients in an open-label extension trial. J Clin Lipidol 2023; 17:342-355 [PubMed: 37100699]
- 243.
- Arca M, Hsieh A, Soran H, Rosenblit P, O'Dea L, Stevenson M. The effect of volanesorsen treatment on the burden associated with familial chylomicronemia syndrome: the results of the ReFOCUS study. Expert Rev Cardiovasc Ther 2018; 16:537-546 [PubMed: 29889589]
- 244.
- Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, Geary RS, Hughes SG, Viney NJ, Graham MJ, Crooke RM, Witztum JL, Brunzell JD, Kastelein JJ. Antisense Inhibition of Apolipoprotein C-III in Patients with Hypertriglyceridemia. N Engl J Med 2015; 373:438-447 [PubMed: 26222559]
- 245.
- Gouni-Berthold I, Alexander VJ, Yang Q, Hurh E, Steinhagen-Thiessen E, Moriarty PM, Hughes SG, Gaudet D, Hegele RA, O'Dea LSL, Stroes ESG, Tsimikas S, Witztum JL. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol 2021; 9:264-275 [PubMed: 33798466]
- 246.
- Digenio A, Dunbar RL, Alexander VJ, Hompesch M, Morrow L, Lee RG, Graham MJ, Hughes SG, Yu R, Singleton W, Baker BF, Bhanot S, Crooke RM. Antisense-Mediated Lowering of Plasma Apolipoprotein C-III by Volanesorsen Improves Dyslipidemia and Insulin Sensitivity in Type 2 Diabetes. Diabetes Care 2016; 39:1408-1415 [PubMed: 27271183]
- 247.
- Oral EA, Garg A, Tami J, Huang EA, O'Dea LSL, Schmidt H, Tiulpakov A, Mertens A, Alexander VJ, Watts L, Hurh E, Witztum JL, Geary RS, Tsimikas S. Assessment of efficacy and safety of volanesorsen for treatment of metabolic complications in patients with familial partial lipodystrophy: Results of the BROADEN study: Volanesorsen in FPLD; The BROADEN Study. J Clin Lipidol 2022; 16:833-849 [PubMed: 36402670]
- 248.
- Graham MJ, Lee RG, Bell TA, 3rd, Fu W, Mullick AE, Alexander VJ, Singleton W, Viney N, Geary R, Su J, Baker BF, Burkey J, Crooke ST, Crooke RM. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res 2013; 112:1479-1490 [PubMed: 23542898]
- 249.
- Paik J, Duggan S. Volanesorsen: First Global Approval. Drugs 2019; 79:1349-1354 [PubMed: 31301033]
- 250.
- Hegele RA, Tsimikas S. Lipid-Lowering Agents. Circ Res 2019; 124:386-404 [PubMed: 30702996]
- 251.
- Prohaska TA, Alexander VJ, Karwatowska-Prokopczuk E, Tami J, Xia S, Witztum JL, Tsimikas S. APOC3 inhibition with volanesorsen reduces hepatic steatosis in patients with severe hypertriglyceridemia. J Clin Lipidol 2023; 17:406-411 [PubMed: 37164837]
- 252.
- Lee SJ, Campos H, Moye LA, Sacks FM. LDL containing apolipoprotein CIII is an independent risk factor for coronary events in diabetic patients. Arterioscler Thromb Vasc Biol 2003; 23:853-858 [PubMed: 12637336]
- 253.
- Luc G, Fievet C, Arveiler D, Evans AE, Bard JM, Cambien F, Fruchart JC, Ducimetiere P. Apolipoproteins C-III and E in apoB- and non-apoB-containing lipoproteins in two populations at contrasting risk for myocardial infarction: the ECTIM study. Etude Cas Temoins sur 'Infarctus du Myocarde. J Lipid Res 1996; 37:508-517 [PubMed: 8728314]
- 254.
- Sacks FM, Alaupovic P, Moye LA, Cole TG, Sussex B, Stampfer MJ, Pfeffer MA, Braunwald E. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation 2000; 102:1886-1892 [PubMed: 11034934]
- 255.
- Qamar A, Khetarpal SA, Khera AV, Qasim A, Rader DJ, Reilly MP. Plasma apolipoprotein C-III levels, triglycerides, and coronary artery calcification in type 2 diabetics. Arterioscler Thromb Vasc Biol 2015; 35:1880-1888 [PMC free article: PMC4556282] [PubMed: 26069232]
- 256.
- Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, Post W, McLenithan JC, Bielak LF, Peyser PA, Mitchell BD, Miller M, O'Connell JR, Shuldiner AR. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008; 322:1702-1705 [PMC free article: PMC2673993] [PubMed: 19074352]
- 257.
- Tg, Hdl Working Group of the Exome Sequencing Project NHL, Blood Institute, Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, Masca N, Stirrups K, Kanoni S, Do R, Jun G, Hu Y, Kang HM, Xue C, Goel A, Farrall M, Duga S, Merlini PA, Asselta R, Girelli D, Olivieri O, Martinelli N, Yin W, Reilly D, Speliotes E, Fox CS, Hveem K, Holmen OL, Nikpay M, Farlow DN, Assimes TL, Franceschini N, Robinson J, North KE, Martin LW, DePristo M, Gupta N, Escher SA, Jansson JH, Van Zuydam N, Palmer CN, Wareham N, Koch W, Meitinger T, Peters A, Lieb W, Erbel R, Konig IR, Kruppa J, Degenhardt F, Gottesman O, Bottinger EP, O'Donnell CJ, Psaty BM, Ballantyne CM, Abecasis G, Ordovas JM, Melander O, Watkins H, Orho-Melander M, Ardissino D, Loos RJ, McPherson R, Willer CJ, Erdmann J, Hall AS, Samani NJ, Deloukas P, Schunkert H, Wilson JG, Kooperberg C, Rich SS, Tracy RP, Lin DY, Altshuler D, Gabriel S, Nickerson DA, Jarvik GP, Cupples LA, Reiner AP, Boerwinkle E, Kathiresan S. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med 2014; 371:22-31 [PMC free article: PMC4180269] [PubMed: 24941081]
- 258.
- Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014; 371:32-41 [PubMed: 24941082]
- 259.
- Gouni-Berthold I, Schwarz J, Berthold HK. Updates in Drug Treatment of Severe Hypertriglyceridemia. Curr Atheroscler Rep 2023; 25:701-709 [PMC free article: PMC10564803] [PubMed: 37642858]
- 260.
- Scott LJ. Alipogene tiparvovec: a review of its use in adults with familial lipoprotein lipase deficiency. Drugs 2015; 75:175-182 [PubMed: 25559420]
- 261.
- Fisher RM, Humphries SE, Talmud PJ. Common variation in the lipoprotein lipase gene: effects on plasma lipids and risk of atherosclerosis. Atherosclerosis 1997; 135:145-159 [PubMed: 9430364]
- 262.
- Rip J, Nierman MC, Ross CJ, Jukema JW, Hayden MR, Kastelein JJ, Stroes ES, Kuivenhoven JA. Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler Thromb Vasc Biol 2006; 26:1236-1245 [PubMed: 16574898]
- 263.
- Gaudet D, Methot J, Dery S, Brisson D, Essiembre C, Tremblay G, Tremblay K, de Wal J, Twisk J, van den Bulk N, Sier-Ferreira V, van Deventer S. Efficacy and long-term safety of alipogene tiparvovec (AAV1-LPLS447X) gene therapy for lipoprotein lipase deficiency: an open-label trial. Gene Ther 2013; 20:361-369 [PMC free article: PMC4956470] [PubMed: 22717743]
- 264.
- Carpentier AC, Frisch F, Labbe SM, Gagnon R, de Wal J, Greentree S, Petry H, Twisk J, Brisson D, Gaudet D. Effect of alipogene tiparvovec (AAV1-LPL(S447X)) on postprandial chylomicron metabolism in lipoprotein lipase-deficient patients. J Clin Endocrinol Metab 2012; 97:1635-1644 [PubMed: 22438229]
- 265.
- Gaudet D, Stroes ES, Methot J, Brisson D, Tremblay K, Bernelot Moens SJ, Iotti G, Rastelletti I, Ardigo D, Corzo D, Meyer C, Andersen M, Ruszniewski P, Deakin M, Bruno MJ. Long-Term Retrospective Analysis of Gene Therapy with Alipogene Tiparvovec and Its Effect on Lipoprotein Lipase Deficiency-Induced Pancreatitis. Hum Gene Ther 2016; 27:916-925 [PubMed: 27412455]
- 266.
- Kassner U, Hollstein T, Grenkowitz T, Wuhle-Demuth M, Salewsky B, Demuth I, Dippel M, Steinhagen-Thiessen E. Gene Therapy in Lipoprotein Lipase Deficiency: Case Report on the First Patient Treated with Alipogene Tiparvovec Under Daily Practice Conditions. Hum Gene Ther 2018; 29:520-527 [PubMed: 29641318]
- 267.
- Ahmad Z, Banerjee P, Hamon S, Chan KC, Bouzelmat A, Sasiela WJ, Pordy R, Mellis S, Dansky H, Gipe DA, Dunbar RL. Inhibition of Angiopoietin-Like Protein 3 With a Monoclonal Antibody Reduces Triglycerides in Hypertriglyceridemia. Circulation 2019; 140:470-486 [PMC free article: PMC6686956] [PubMed: 31242752]
- 268.
- Rosenson RS, Gaudet D, Ballantyne CM, Baum SJ, Bergeron J, Kershaw EE, Moriarty PM, Rubba P, Whitcomb DC, Banerjee P, Gewitz A, Gonzaga-Jauregui C, McGinniss J, Ponda MP, Pordy R, Zhao J, Rader DJ. Evinacumab in severe hypertriglyceridemia with or without lipoprotein lipase pathway mutations: a phase 2 randomized trial. Nat Med 2023; 29:729-737 [PMC free article: PMC10033404] [PubMed: 36879129]
- 269.
- Mattijssen F, Kersten S. Regulation of triglyceride metabolism by Angiopoietin-like proteins. Biochim Biophys Acta 2012; 1821:782-789 [PubMed: 22063269]
- 270.
- Kersten S. New insights into angiopoietin-like proteins in lipid metabolism and cardiovascular disease risk. Curr Opin Lipidol 2019; 30:205-211 [PubMed: 30893111]
- 271.
- Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, Fennell T, Banks E, Ambrogio L, Cibulskis K, Kernytsky A, Gonzalez E, Rudzicz N, Engert JC, DePristo MA, Daly MJ, Cohen JC, Hobbs HH, Altshuler D, Schonfeld G, Gabriel SB, Yue P, Kathiresan S. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med 2010; 363:2220-2227 [PMC free article: PMC3008575] [PubMed: 20942659]
- 272.
- Dewey FE, Gusarova V, Dunbar RL, O'Dushlaine C, Schurmann C, Gottesman O, McCarthy S, Van Hout CV, Bruse S, Dansky HM, Leader JB, Murray MF, Ritchie MD, Kirchner HL, Habegger L, Lopez A, Penn J, Zhao A, Shao W, Stahl N, Murphy AJ, Hamon S, Bouzelmat A, Zhang R, Shumel B, Pordy R, Gipe D, Herman GA, Sheu WHH, Lee IT, Liang KW, Guo X, Rotter JI, Chen YI, Kraus WE, Shah SH, Damrauer S, Small A, Rader DJ, Wulff AB, Nordestgaard BG, Tybjaerg-Hansen A, van den Hoek AM, Princen HMG, Ledbetter DH, Carey DJ, Overton JD, Reid JG, Sasiela WJ, Banerjee P, Shuldiner AR, Borecki IB, Teslovich TM, Yancopoulos GD, Mellis SJ, Gromada J, Baras A. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med 2017; 377:211-221 [PMC free article: PMC5800308] [PubMed: 28538136]
- 273.
- Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, Natarajan P, Klarin D, Emdin CA, Zekavat SM, Nomura A, Erdmann J, Schunkert H, Samani NJ, Kraus WE, Shah SH, Yu B, Boerwinkle E, Rader DJ, Gupta N, Frossard PM, Rasheed A, Danesh J, Lander ES, Gabriel S, Saleheen D, Musunuru K, Kathiresan S. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol 2017; 69:2054-2063 [PMC free article: PMC5404817] [PubMed: 28385496]
- 274.
- Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, Stalenhoef AF, Endocrine Society. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2012; 97:2969-2989 [PMC free article: PMC3431581] [PubMed: 22962670]
- 275.
- Jacobson TA, Ito MK, Maki KC, Orringer CE, Bays HE, Jones PH, McKenney JM, Grundy SM, Gill EA, Wild RA, Wilson DP, Brown WV. National lipid association recommendations for patient-centered management of dyslipidemia: part 1--full report. J Clin Lipidol 2015; 9:129-169 [PubMed: 25911072]
- 276.
- Chait A, Feingold KR. Approach to patients with hypertriglyceridemia. Best Pract Res Clin Endocrinol Metab 2023; 37:101659 [PubMed: 35459627]
- 277.
- Feingold KR. Dyslipidemia in Patients with Diabetes. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- 278.
- Feingold KR. The Effect of Endocrine Disorders on Lipids and Lipoproteins. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, de Herder WW, Dhatariya K, Dungan K, Hofland J, Kalra S, Kaltsas G, Kapoor N, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, Levy M, McGee EA, McLachlan R, New M, Purnell J, Sahay R, Shah AS, Singer F, Sperling MA, Stratakis CA, Trence DL, Wilson DP, eds. Endotext. South Dartmouth (MA) 2023.
- Review TRIGLYCERIDES, ATHEROSCLEROSIS, AND CARDIOVASCULAR OUTCOME STUDIES: FOCUS ON OMEGA-3 FATTY ACIDS.[Endocr Pract. 2017]Review TRIGLYCERIDES, ATHEROSCLEROSIS, AND CARDIOVASCULAR OUTCOME STUDIES: FOCUS ON OMEGA-3 FATTY ACIDS.Handelsman Y, Shapiro MD. Endocr Pract. 2017 Jan; 23(1):100-112. Epub 2016 Nov 7.
- Review Targeting low HDL-cholesterol to decrease residual cardiovascular risk in the managed care setting.[J Manag Care Pharm. 2008]Review Targeting low HDL-cholesterol to decrease residual cardiovascular risk in the managed care setting.Cziraky MJ, Watson KE, Talbert RL. J Manag Care Pharm. 2008 Oct; 14(8 Suppl):S3-28; quiz S30-1.
- Review Long-chain omega-3 fatty acids, fibrates and niacin as therapeutic options in the treatment of hypertriglyceridemia: a review of the literature.[Atherosclerosis. 2015]Review Long-chain omega-3 fatty acids, fibrates and niacin as therapeutic options in the treatment of hypertriglyceridemia: a review of the literature.Ito MK. Atherosclerosis. 2015 Oct; 242(2):647-56. Epub 2015 Jun 11.
- Review Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and their mechanisms of action on apolipoprotein B-containing lipoproteins in humans: a review.[Lipids Health Dis. 2017]Review Omega-3 fatty acids eicosapentaenoic acid and docosahexaenoic acid and their mechanisms of action on apolipoprotein B-containing lipoproteins in humans: a review.Oscarsson J, Hurt-Camejo E. Lipids Health Dis. 2017 Aug 10; 16(1):149. Epub 2017 Aug 10.
- Review Eicosapentaenoic Acid Versus Docosahexaenoic Acid as Options for Vascular Risk Prevention: A Fish Story.[Am J Ther. 2016]Review Eicosapentaenoic Acid Versus Docosahexaenoic Acid as Options for Vascular Risk Prevention: A Fish Story.Singh S, Arora RR, Singh M, Khosla S. Am J Ther. 2016 May-Jun; 23(3):e905-10.
- Triglyceride Lowering Drugs - EndotextTriglyceride Lowering Drugs - Endotext
Your browsing activity is empty.
Activity recording is turned off.
See more...