

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
Madame Curie Bioscience Database [Internet]. Austin (TX): Landes Bioscience; 2000-2013.

The mitogen- and stress-activated kinases-1 and -2 (MSK1 and MSK2) are ubiquitous serine/ threonine kinases that mediate intracellular signal transduction by mitogen-activated protein kinases (MAPKs) of the p38 and ERK1/2 families. The structure of MSK is complex, featuring a substrate-phosphorylating N-terminal kinase domain, a regulatory C-terminal kinase domain as well as many regulatory phosphorylation sites and motifs. Here we review all aspects of the structure-function and mechanism of activation of MSK. First, we summarize the numerous and diverse environmental stimuli, ranging from physiological signaling molecules to cellular stresses that activate ERK1/2, p38α or both to effect activation of MSK in cells. Next is described the series of sequential phosphorylation events that first activates the C-terminal kinase domain and finally the N-terminal kinase domain of MSK in an activation mechanism that can be divided into four stages. We proceed by detailing the activation mechanism from a 3-dimensional perspective, focusing on the roles of each of the phosphorylation events. Throughout, parallels are drawn to similarities with activation mechanisms of the kinase super family in general and with kinases of the AGC and CaMK groups in particular, to which the N- and C-terminal kinase domains, respectively, belong. Finally, topics like MSK interactions with MAPK docking sites, MSK substrate specificity, monitoring of MSK activity in cells and mutant MSK as research tools are discussed.
Introduction
The mitogen and stress-activated kinases-1 and -2 (MSK1 and MSK2; gene symbols RPS6KA5 and RPS6KA4, respectively) where discovered in 1998. MSK1 was identified by homology cloning1;2, whereas MSK2 (originally termed RSK-B) was discovered in a yeast two-hybrid screen.3 These seminal studies immediately established MSKs as protein kinases that mediate signal transduction by mitogen-activated protein kinase (MAPK) cascades that rank among the most important signaling pathways in eukaryotic cells. Accordingly, MSK is ubiquitously expressed, albeit often at rather low levels, and highly conserved in vertebrates. MSK orthologs have also been identified in A. gambiae (mosquito), D. melanogaster (fruit fly) and C. elegans (nematode), but seem absent from yeast and plants. Til date, all published reports have studied MSK from human, mouse or fly. The fly ortholog Jil-1, however, seems to have diverged significantly and may not be considered a true ortholog of vertebrate MSK with respect to regulation and function. Therefore, this review will focus on the mammalian MSKs that are also the topic of the other chapters in this MSK edition. It should be borne in mind that the large majority of published MSK studies have investigated MSK1. Thus, the informations conveyed in this review are biased toward this homolog. However, many reports studying activation of MSK1 have exploited phosphospecific antibodies whose homolog specificity may not have been rigorously tested. Given the high degree of sequence conservation around the phosphorylation sites of the two MSK homologs, it is uncertain whether such studies in reality were monitoring MSK1, MSK2 or both.
MSK Is Activated by ERK1/2 and p38α MAPKs in Response to Numerous Environmental Stimuli
MAPKs are intracellular transducers of signals by a vast array of environmental stimuli in eukaryotic cells. They function as effector kinases of so-called MAPK cascades that are modules composed of three kinases: the environmental stimulus impinges on a cognate receptor to activate a MAP kinase kinase kinase (MAPKKK), leading to activation of a MAP kinase kinase (MAPKK) and finally the MAPK that mounts a proper cellular response via phosphorylation of protein substrates. Mammalian MAPKs can be grouped into seven families: ERK1/2, p38α/β/γ/δ, JNK1/2/3, ERK5, ERK3/4, ERK8 (the rat homolog is termed ERK7) and NLK.
MSK1 and MSK2 are activated by members of two MAPK families, namely the ERK1/2 (extracellular signal regulated kinase) and the p38 family (Fig. 1). These are major and ubiquitous MAPK families that regulate a plethora of physiological processes by controlling the function of hundreds of cellular proteins via phosphorylation, either directly or via other kinases such as MSK.
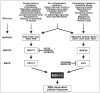
The ERK1/2 cascade responds to growth factors, neurotrophins, signal peptides, neurotransmitters and numerous other stimuli that are capable of activating the main entry point of this cascade, which is the small GTPase RAS. RAS activates RAF family kinases (MAPKKKs) that activate MEK1 and MEK2 (MAPKKs) that finally activate ERK1 and ERK2. The RAS-ERK pathway is constitutively activated in most human cancers due to gain-of-function mutations in, for example, RAS, RAF or growth factor receptors, meaning that MSK may likewise be constitutively activated in most cancer cells. The ERK1/2 cascade can also be activated by more specialized entry points such as Tpl2 that is the major MAPKKK in Toll-like receptor signaling in response to pathogen-derived molecules (pathogen-associated molecular patterns; PAMPs). Other specialized entry points are Mos, a MAPKKK that functions in oocytes mainly, and PAK that can serve as a MAPKKK downstream of the small GTPases Rac and cdc42.
The role of ERK1/2 in activation of MSK in vivo has been demonstrated by using pharmacological inhibitors of MEK1/2 such as PD98059, U0126 and PD184352. Thus, the first published MSK study established that inhibition of MEK blocks activation of MSK1 in response to stimuli that activate ERK1/2, but not p38 in the cell type studied (HEK 293), such as phorbol 12-myristate 13-acetate (PMA) or epidermal growth factor.1 Other examples of stimuli that activate MSK predominantly via the ERK1/2 pathway in the particular cell types analyzed include brain-derived neurotrophic factor and neurotrophin-3 in mouse cortical neurons,4 lysophosphatidic acid or serum in mouse embryonic stem cells,5 erythropoietin in the human ASE2 erythroid cell line6 and progesterone in the human T47D breast cancer cell line.7 One question remains unanswered regarding activation of MSK by the ERK1/2 pathway: MEK inhibitors block activation of ERK1 as well as ERK2 and it has not been resolved whether one or both homologs are involved in activation of MSK. Since either ERK can coimmunoprecipitate MSK, both may have a role. However, since ERK2 is the higher expressed homolog in most cell types, this ERK may be the principle activator of MSK downstream of MEK.
The p38 MAPK cascade is activated by a broad spectrum of cellular stress stimuli, including UV-C or ionizing irradiation, hyperosmotic, heat or oxidative stress, hypoxia, PAMPs, poisons like heavy metals and arsenite and certain antibiotics such as anisomycin. However, the cascade is also activated by a variety of physiological signaling molecules, including pro-inflammatory cytokines and retinoic acid. As might be expected, such diverse stimuli employ several distinct MAPKKKs to activate the pathway. These various MAPKKKs converge on MKK3 and MKK6 (MAPKKs) that finally activates the p38 family members p38α, p38β, p38γ and p38δ.
The role of p38 in activation of MSK in vivo has been demonstrated by using knockdown, knockout and pharmacological inhibitors of p38, such as SB203580 and SB202190. The seminal studies on MSK demonstrated that inhibition of p38 blocks activation of MSK in response to stimuli that activate the p38, but not the ERK1/2 pathway in the cells studied (HEK 293), such as arsenite, UV irradiation, H2O2 or transfection with a constitutively active MKK6 mutant.1,3 SB203580 and SB22190 inhibit p38α and p38β, but not p38γ or p38δ 8, indicating that the latter p38 homologs are dispensable for activation of MSK. In vitro, MSK1 could be activated equally well by p38α and p38β, suggesting that both of these homologous mediate activation of MSK.1 However, analysis of mouse embryonic fibroblasts (MEFs) from knockout mice revealed that p38β knockout did not affect activation of MSK1 by anisomycin,9 whereas p38α knockout essentially abolished activation of MSK1 by this stimulus.10 Furthermore, arsenite-induced activation of MSK1 was completely eliminated in p38α knockout MEFs.11 Finally, in MCF7 mammary carcinoma cells, siRNA knockdown of p38α essentially abolished activation of MSK by retinoic acid treatment.12 These observations suggest that p38α is the most important p38 homolog for activation of MSK, possibly due to the fact that p38α is highly abundant, whereas p38β seems to be expressed at low levels in most cell types.13 Other examples of stimuli that activate MSK predominantly via the p38 pathway in the cells studied include interleukin-1β in primary human keratinocytes14 and the human HaCaT keratinocyte cell line,15 C. difficile toxin-A in the human NCM460 colon epithelial cell line,16 manganese in the human BL41 lymphoma cell line17 and the S. griseolus antibiotic anisomycin in HEK 293 cells.18
A number of stimuli are capable of activating both the ERK1/2 and the p38 pathway, sometimes in a cell type-specific manner, and employ both pathways to activate MSK. The first MSK1 study demonstrated that for tumor necrosis factor-α and nerve growth factor, administered to the human HeLa cervical carcinoma and rat PC12 pheochromocytoma cell line, respectively, inhibition of either ERK1/2 or p38 only partially suppressed activation of MSK, whereas complete suppression required inhibition of both MAPKs.1 Similar observations were made with PAMPs for several Toll-like receptors (lipopolysaccharide, lipoteichoic acid, Pam3CSK4, CpG DNA) in primary mouse macrophages,19 with interleukin-1β and tumor necrosis factor-α in the human NCI-H292 lung carcinoma cell line20 and with the H. pylori protein HP0175 in the human THP-1 monocytic leukemia cell line.21 Deviations from above scheme have been reported. Thus in the mouse C2 myoblast cell line, activation of MSK by H2O2 was reduced by ∼50% by either PD98059 or SB203580 but unexpectedly, no further suppression was observed upon administration of both inhibitors.22 Furthermore, in primary rat cardiomyocytes, activation of MSK by α1-adrenergic agonists or endothelin-1 was almost completely suppressed by pre-incubation with either PD98059 or SB203580 alone.23 The latter observation could be explained by the possibility that ERK and p38 are each essential for activation of MSK in this particular context, e.g., via phosphorylation of distinct regulatory sites in MSK, but evidence for such a mechanism has so far not been reported.
In conclusion, the above cited and numerous other studies seem to show that whenever ERK1/2 and/or p38α become activated in cells, MSK typically does likewise.
While above studies were performed on cultured cells, a few investigations have also demonstrated activation of MSK in whole animals in response to various stimuli. Thus, MSK1/2 were found to become activated in human exercised muscle.24 Exposure of mice to light was shown to activate MSK1 in the suprachiasmatic nucleus, the major circadian pacemaker, which mainly occurred via the ERK1/2 pathway, but with some contribution from p38.25 Administration of cocaine to mice resulted in activation of MSK1 in the striatum via the ERK1/2 pathway.26 In mice, levels of active MSK1 increased in neurons in several brain regions during ischemic/hypoxic preconditioning.27 Finally, in mouse hippocampal CA1 pyramidal neurons, MSK1 became activated during training for contextual memory, likely via the ERK1/2 pathway.28
Do MAPKs other than ERK1/2 and p38α play a role in activation of MSK? The scattered evidence pertaining to the issue generally argues against this possibility: JNK is activated by many stimuli that also activate p38, including arsenite, anisomycin, UV/radioactive irradiation and pro-inflammatory cytokines. However, activation of MSK by such stimuli is typically completely prevented by SB203580 that does not inhibit JNKs.8 Furthermore, active JNK1 or JNK2 did not phosphorylate MSK1 in vitro under conditions where active ERK2 and p38α did2,3. Finally, JNK1 failed to be co-immunoprecipitated by MSK2 under conditions where ERK1/2 and p38α were co-precipitated.3 ERK5 is activated by many stimuli that also activate ERK1/2 and, MEK5 (its MAPKK) is inhibited by PD98059, U0126 and PD184352 at high concentrations.29 However, a low concentration (2 μM) of PD184352 that does not inhibit activation of MEK5 and ERK5,29 fully blocked activation of ERK1/2 and MSK,30 suggesting that the ERK5 pathway does not play a role in activation of MSK. On the other hand, it cannot be ruled out that p38β, due to its effective activation of MSK in vitro, may serve as an MSK activator in, so far unidentified cell types where it may be the major p38 homolog.
Functional Domains, Motifs and Phosphorylation Sites of MSK
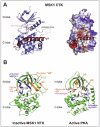
MSK1 and MSK2 are complex enzymes that contain several different domains, motifs and phosphorylation sites (Figs. 2, 4). MSKs are unusual among protein kinases by having two functional kinase domains in the same polypeptide, a feature shared only with the closely related RSK family of kinases.31 Both domains posses serine/threonine kinase activity and belong to the super-family of classical eukaryotic protein kinases that comprises almost 500 members in human, all possessing a similar 3-dimensional structure.32 The N-terminal kinase (NTK) domain of MSK belongs to the AGC kinase group, named after the members protein kinase A (PKA), protein kinase G (PKG) and protein kinase C (PKC). The C-terminal kinase (CTK) domain of MSK belongs to the CaMK group, named after its calcium-calmodulin-dependent protein kinase family members.
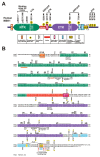
The NTK-domain of MSK is thought to be responsible for the phosphorylation of all known MSK substrates, whereas the only established function of the CTK-domain is activation of the NTK-domain (Fig. 3). Thus, the CTK and NTK-domains form components of a kinase cascade that fused during evolution. Summarizing the structural features that control kinase activity and cellular functions, MSK begins with a short tail that, at least in MSK1, contains an element (β1L0) that represses the activity of the NTK-domain. Within the NTK-domain is an activating phosphorylation site located in a so-called activation loop. The NTK-domain is followed by a linker sequence that functions to stimulate the NTK-domain activity when phosphorylated at several sites, one of which is located in a hydrophobic motif (HM) that is characterized by three aromatic residues surrounding the phosphorylated serine (underlined): F-x-x-Y-S-F. The CTK-domain also contains an activation loop with a regulatory phosphorylation site and is followed by a relatively long tail that contains several functional motifs and phosphorylation sites: immediately following the CTK-domain, the tail contains two a helices, αK and αL, that are thought to function as repressors of CTK-domain activity, and of these, αK possesses a CTK-domain-activating phosphorylation site. Next is found a functional nuclear localization signal (NLS).33 MSKs appear to typically exhibit strict nuclear localization regardless of activation status,1,3,33 consistent with the fact that all of the well-validated MSK substrates function within the nucleus. However, glucocorticoids have been shown to cause nuclear extrusion of MSK1, via direct or indirect interaction between MSK1 and the glucocorticoid receptor, which may serve as a mechanism whereby glucocorticoids can antagonize MSK signaling.34 Overlapping with the NLS is found a so-called D-motif MAPK docking site capable of binding ERK and p38. Finally, MSK ends with a C-terminal sequence containing 3 phosphorylation sites of unknown function.
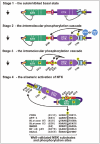
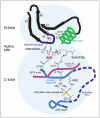
Figure 3.
Cartoon model of the MSK activation mechanism via multi-site phosphorylation. The transition from completely inactive to fully active MSK can be grouped into 4 stages, as indicated. See, Figure. 2A for key to the various functional elements.
Except for the extreme N- and C-terminal tails, MSK1 and MSK2 show a high degree of amino acid identity/similarity and most of the features described above are present in both homologs (Fig. 2B, 4). However, the β1L0 element is not easily discernible in MSK2 (Fig. 2B) and the phosphorylation sites S-381, S-750, S-752 and S-758 have only been mapped/investigated in MSK1, although they are likely to also exist in MSK2 (unless otherwise stated, amino acid numbering refers to human MSK1 in this review). The few known dissimilarities between the two MSK homologs are discussed in a separate section below.
The Cartoon Model of the MSK Activation Mechanism via Multi-Site Phosphorylation
MSK is activated by a series of sequential phosphorylation events that serve to ultimately activate the NTK-domain. Of note, in vitro incubation of MSK1 or MSK2 with active ERK1/2 or p38 can initiate and complete this series of phosphorylation events in the absence of any other cellular components.1,30,35,36 This suggests that in vivo, the sole requirement for activation of MSK is activation of a MAPK that can interact with and phosphorylate MSK at the proper sites.
The activation mechanism of MSK can be grouped into four distinct stages (Fig. 3). In stage 1, MSK is tightly locked in an inactive state due to the autoinhibitory elements, β1L0 and αK/L and the lack of activating phosphorylations. In stage 2, active ERK1/2 or p38α, bound to the MAPK docking site in MSK, phosphorylates S-360 in the linker region, T-581 in the activation loop of the CTK-domain and T-700 in the autoinhibitory αK helix.30,35,36 Phosphorylation at T-581 and T-700 activates the CTK-domain.30,35,36 In stage 3, the activated CTK-domain phosphorylates S-212 in the activation loop of the NTK-domain, S-376 in HM of the linker as well as S-381.30 Hereafter, the CTK-domain has likely full-filled its role in activation of MSK. In stage 4, phospho-S-212, -S-360, -S-376 and -S-381 finally collaborate to allosterically activate the NTK-domain: the S-376 phosphoryl group of HM interacts with a phosphate-binding site within the NTK-domain which enables the three aromatic residues of HM to bind and stabilize a nearby hydrophobic pocket (sometimes called the PDK1-interacting fragment (PIF) pocket in the AGC kinase literature).37 The phosphoryl group of S-360 interacts with another nearby phosphate-binding site within the NTK-domain.38 This increases the binding interaction between the linker and the NTK-domain which serves to stabilize the phosphorylated HM in its binding pocket. The phosphoryl group of S-381 most likely stimulates the NTK-domain activity by a mechanism similar to that of S-360, but so far direct evidence of this is lacking. Once HM occupies its binding pocket, it stabilizes residues in the active site of the NTK-domain in a position optimal for catalysis. Similarly, the phosphoryl group of S-212 in the activation loop of the NTK-domain serves to stabilize residues in the active site in an optimal position, and combined, the phosphorylation events at S-212 and S-376 thereby activate the NTK-domain in an allosteric and synergistic manner.37 The NTK-domain can now autophosphorylate MSK at S-750, S-752 and S-75830 and phosphorylate the downstream substrates of MSK.
The various MSK phosphorylation sites are of differing quantitative importance with respect to the events leading to activation of the NTK-domain. For both MSK homologous, the two activation loop sites (S-212, T-581) and the HM site (S-376) are essential, as evidenced by > 95% reduced NTK-domain kinase activity upon their mutation to Ala.30,36 In MSK2, the S-343 site (= S-360 in MSK1) is also essential for kinase activity.36 By contrast, in MSK1, the S-360 as well as the S-381 sites are modulatory, since their mutation to Ala reduced activation of the NTK-domain by ∼60%30,38 and ∼40%,30 respectively.
Relationship to Other AGC Kinase and CaMK Activation Mechanisms
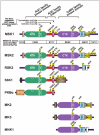
Although MSK possesses relatively low overall amino acid identity to the most closely related AGC and CaMK group members, it shares several key regulatory elements and phosphorylation sites with these kinases.
Regarding the NTK-domain, three of the major phosphorylation sites of MSK are also present in > 20 other AGC kinases and the sites may therefore be called the three conserved AGC kinase phosphorylation sites (Fig. 4). The first one is the activation loop site (S-212 in MSK1). The second and third ones, known as the "turn motif site" (S-360 in MSK1) and the "HM site" (S-376 in MSK1), respectively, are located in the linker and HM, structures that are also conserved in other AGC kinases. In several AGC kinases, as for example PKB, the linker is known as the "tail", since it is not followed by any functional domains (see Fig. 4). Finally, the binding sites for phospho-S-360, phospho-S-376 and HM within the kinase domain are conserved. Thus, as might be expected, all these AGC kinases share a common structural mechanism of activation of their catalytic domain based on the three phosphorylation sites. What distinguishes these AGC kinases with respect to regulation of their activity is the fact that the upstream kinases responsible for phosphorylating the three conserved sites often differ (as indicated in Fig. 4). In addition, the various AGC kinases contain distinct signaling modules flanking the kinase domain which render the kinases responsive to distinct upstream pathways including the kinases that phosphorylate the three conserved sites.
The CTK-domain belongs functionally and phylogenetically to a sub-family of the CaMK group called the MAPKAPKs (MAPK-activated protein kinases). As indicated by the name, these kinases serve as effectors of ERK1/2 and/or p38 and include MSK1/2 CTK, RSK1-4 CTK, MK2/3 and MNK1/2 (reviewed in ref. 39). Common to these kinases is the presence of a C-terminal D-motif MAPK docking site that likely dictates which MAPK can activate the individual members as well as a regulatory MAPK phosphorylation site in the activation loop (Fig. 4). In addition, these MAPKAPKs also possess C-terminal autoinhibitory α-helices, often regulated by a phosphorylation site located within or nearby.
The More Detailed and Structural View of the Activation Mechanism of MSK
Crystal structures of full-length MSK in inactive, active or intermediate states have not been solved. Nevertheless, a credible and fairly detailed model how the sequential phosphorylation events act at the 3-dimensional structural level to stimulate MSK catalytic activity can be proposed. This model is based on the crystal structures of the isolated the NTK (inactive) and CTK (activation status unclear) domains of MSK1 as well as molecular modeling of the NTK-domain combined with biochemical analyses. Furthermore, the model can draw on knowledge gained from related AGC kinases and CaMKs due to the intimate structural relationships alluded to in the section above.
We will first outline some general principles regarding activation of the classical eukaryotic protein kinases that often were appreciated for the first time through studies on PKA, which remains one of the structurally best-characterized kinases (for review of below general principles, please see refs. 40-44) As mentioned above, these enzymes exhibit a very similar 3-dimensional structure in their active state. The reason for a shared active conformation is logical: all the kinases catalyze the same enzymatic reaction, namely the transfer of the γ-phosphoryl group of ATP onto hydroxyl groups of protein substrates. Consequently, kinases contain many conserved residues involved in catalysis or binding of ATP and Mg2+ co-factor ions. These residues, scattered throughout the active site, must hold the same 3-dimensional position in the active enzyme, meaning that the active site and the overall fold must be very similar in the various active kinase conformations. By contrast, no such constraints exist regarding the inactive conformation that, accordingly, is often found to differ substantially among kinases. In essence, kinase activation therefore typically entails re-structuring of the catalytic domain from a disorganized conformation that is kinase-specific to a catalytically competent conformation that is shared by kinases. This restructuring can be induced by, for example, phosphorylation or regulated interactions of the kinase domain with flanking elements. Both such mechanisms operate in MSK.
The classical eukaryotic protein kinases consist of two lobes: a small N-terminal lobe (N-lobe) predominantly composed of β-strands, and a large C-terminal lobe (C-lobe), mainly composed of α-helices (for examples, see the kinase structures in Figs. 6 and 7). The β-strands and α-helices are designated relative to those identified in PKA, the first crystallized kinase, and are highly, but not fully conserved, making their designation somewhat inconsistent across the various kinases. The secondary structures present in MSK1 are indicated in Figure. 2B. Between the two lobes is situated the deep, ATP-binding active site.

The N-lobe contains the αC-helix that encompasses a conserved Glu that in the active kinase conformation must ion pair with a conserved Lys in the β3-strand, enabling the Lys residue to position the α,β-phosphates, and thereby also the γ-phosphate of ATP for phosphoryl transfer. Figure 5 shows a cartoon illustrating these interactions in the active kinase fold with amino acid numbering referring to the MSK1 NTK-domain (the residues are also indicated in Fig. 2B). The αC-helix frequently functions as an important regulatory element in activation of kinases, including the NTK-domain of MSK. Thus, in many inactive kinase conformations, the αC-helix is disordered or malpositioned and activation involves reordering/positioning of the helix to allow the interactions described above. Furthermore, the N-lobe contains the so-called glycine-rich loop that is also involved in binding and aligning the phosphates of ATP for catalysis.
The C-lobe contains the so-called activation segment (bordered by conserved DFG and APE motifs) that is a key element in allosteric activation of numerous kinases, including MSK the NTK and CTK-domains. The 3-dimensional structure of the segment often differs greatly between inactive and active kinase conformations. Typically, phosphorylation of the so-called activation loop within the activation segment accounts for reordering to the active conformation. Specifically, the phosphate interacts with and stabilizes conserved positively charged residues located within the activation segment and within the so-called catalytic loop and sometimes the phosphate also binds a basic residue within the αC-helix of the N-lobe. As a consequence, several key residues neighboring the positively charged residues become positioned optimally for catalysis. Thus, the binding of the activation loop phosphate to an Arg residue in the catalytic loop serves to stabilize a neighboring and invariant Asp that functions as a catalytic base in the phosphorylation reaction (Fig. 5). Furthermore, the binding of the activation loop phosphate to a Lys residue in the β9-strand of the activation segment serves to stabilize the adjacent DFG motif, allowing its Asp residue (via a Mg2+ ion) to position the γ-phosphate of ATP for transfer onto the substrate. In AGC kinases, this activation loop phosphate/Lys interaction may additionally stabilize proper αC-helix positioning, since the DFG motif and the β9-strand bind the αC-helix in the active PKA and PKB conformations. Finally, in AGC kinases, the activation loop phosphate may also help positioning the αC-helix by interacting with a His residue in this helix, again suggested by the active structures of PKA and PKB.
All the functional elements and nearly all of the residues described above are conserved in the NTK and CTK-domains of MSK (Fig. 2B), where they can be assumed to perform the listed functions. We will next discuss the 4-stage activation mechanism of MSK in the context of inducing the prototypical active kinase conformation.
Stage 1—the Autoinhibited Basal State
In unstimulated cells, MSK is kept at low basal activity, as evidenced by the fact that ERK/ p38-activating signals are typically found to increase MSK kinase activities by 10- to 50-fold.1,30,36-38 The low basal activity is due to the absence of phosphorylation, since the aforementioned increases in kinase activities are almost invariably associated with large increases in the phosphorylation level at the activating sites.30,35-38 In some cases, these large relative increases may reflect switch-like modulation of the majority of MSK molecules in the cell from a fully-off to a fully-on state. Thus, prolonged SDS-PAGE that allows detection of phosphorylation-induced electrophoretic mobility shifts has shown strong stimuli to completely shift MSK to a hyperphosphorylated species.18 In unstimulated cells, phosphatases therefore seem capable of fully suppressing MSK activation by the basal activities of ERK/p38.
The αK/L-helices in the C-terminal tail of MSK are most likely involved in suppressing the basal activity of CTK and thereby keeping MSK activity low in unstimulated cells. Analogous helices had previously been identified in CaMKI, MK2 and RSK2 CTK-domains and shown to function as autoinhibitory elements, apparently by interfering with ATP and/or protein substrate binding.45-47 For MSK, the first evidence of C-terminal autoinhibitory elements was provided with MSK2, where a mutant truncated immediately prior to the predicted αK/L-helices could be activated by co-expressed MKK6 and p38, whereas as a mutant truncated immediately after αK/L could not.33 These authors noted an AFN motif in αL that apparently corresponds to a similar motif in the analogous helix of CaMKI. In the inactive CaMKI conformation, the Phe of this motif is inserted into the ATP-binding site, greatly attributing to its obstruction.45 Interestingly, Ala mutation of the Phe residue in full-length MSK2 (F-709) resulted in greatly elevated basal and moderately enhanced stimulated kinase activity. The actual existence of αK/L-helices in MSK was later demonstrated by the crystal structure of MSK1 CTK which also revealed that these helices occupy deep grooves in the CTK-domain (Fig. 6A), holding exactly the same position as the analogous helices in MK2 and RSK CTK-domain.48 By contrast, the position of αL differs greatly between MSK1 the CTK-domain and inactive CaMKI. Whether this reflects the circumstance that the MSK1 CTK-domain crystallized may adopt an (at least partly) active conformation and that αL and F-709 would hold a CaMKI-like position in inactive MSK, remains to be investigated. Curiously, the study reporting the CTK-domain crystal structure and demonstrating the existence of the αK/L-helices in MSK did not directly test for an inhibitory role of αK/L by performing e.g., truncation analysis of αK/L or point mutation of F-722 (= MSK2 F-709), as performed by Tomás-Zuber et al., (2001)33 for MSK2. Interestingly, however, the study showed that an isolated CTK-domain truncated shortly after αK/L displayed much higher in vitro kinase activity than an isolated CTK-domain encompassing the entire C-terminal tail. This suggests that the C-terminal tail may contain an CTK-domain inhibitory element(s) in addition to αK/L. In conclusion, whereas biochemical data and analogies with related kinases strongly support autoinhibition of MSK by the C-terminal tail, further definition of the inhibitory elements and mechanisms are desirable.
The β1L0 element identified in the N-terminal tail of MSK1 also seems to serve an important function in keeping MSK activity low in unstimulated cells. This was first suggested by the crystal structure of the MSK1 NTK-domain that revealed an autoinhibited conformation of this domain in which β1L0 played a central structural role.49 Specifically, β1L0 engaged with residues derived from the αB-helix (converting it into a β-strand) that in turn engaged with the β9-strand in the activation segment to create a three-stranded β-sheet on the surface of the N-lobe (Fig. 6B; in Fig. 2B, the sequences that form the inhibitory β-sheet are indicated by orange bars). The β1L0 element thereby completely disrupts the structure and position of the activation segment and the αC-helix, creating a conformation of the NTK domain that is completely incompatible with catalytic activity The gross-disruptive actions of β1L0 is evident when comparing the position and conformation of the activation segment and αC-helix in the inactive MSK1 NTK-domain vs. active PKA, assuming that the NTK-domain folds like PKA, when active (Fig. 6B). An analogous autoinhibited conformation based on an inhibitory β-sheet has not been observed in any other inactive AGC kinase crystal, possibly because the β1L0 element may be unique to MSK. Strong functional evidence of an autoinhibitory role of the N-terminal tail has been obtained with MSK1. Thus, an MSK1 mutant lacking the first 41 residues, including β1L0, exhibited vastly higher kinase activity upon incubation with active p38 in vitro as compared with wild-type MSK1.50 Since, the β1L0 element is hard to discern in the MSK2 primary structure, similar functional evidence of its possible existence in this homolog is warranted.
Stage 2—the Intermolecular Phosphorylation Cascade
The association of active ERK1/2 or p38α with the MAPK docking motif of MSK initiates the phosphorylation cascade leading to activation of MSK. The MAPK in question phosphorylates three sites in MSK, namely S-360 in the linker, T-581 in the CTK-domain activation loop and T-700 in αK.30,35,36 Phosphorylation of T-581 and T-700 serves to activate the CTK-domain and enable the transition to stage 3, whereas phospho-S-360 awaits stage 4 to exert its function. The mechanistic roles of the T-581 and T-700 sites cannot be inferred from the crystal structure of the MSK1 CTK-domain.48 First, although T-700 was visible in the crystal, it was not phosphorylated. Second, although T-581 phosphorylation was detectable by immunoblotting in the preparation used for crystallization, 200 ng and 1000 ng of the CTK-domain protein were used for the immunoblot analysis. These are very large sample amounts considering the high sensitivity of immunoblotting analysis, suggesting a low stoichiometric phosphorylation of T-581 in the CTK preparation and that the crystal therefore was composed primarily (perhaps even exclusively) of non-T-581-phosphorylated CTK-domain. In support of this idea, neither phosphorylated T-581 was visible in the crystal, nor was the activation loop that could be anticipated to be stabilized and visible, had T-581 been stoichiometrically phosphorylated. Thus, it remains unclear whether the CTK-domain crystal structure represents a T-581-phosphorylated, partially active conformation or a completely non-phosphorylated and thus assumably inactive conformation. In any case, the role of the T-581 and T-700 phosphorylation sites must be inferred from biochemical studies on MSK and knowledge about analogous sites in other kinases.
In this regard, T-581 phosphorylation most likely stimulates the CTK-domain activity by organizing active site residues as described above and illustrated in Figure 5. Thus, the positively charged residues that bind the activation loop phosphate group for stabilization of the catalytic Asp (MSK1 D-544) and the DFG motif, respectively, are conserved (MSK1 R-543 and R-570). In any case, the CTK-domain activation loop phosphorylation site is absolutely essential as its mutation to Ala abolishes activation of either MSK homolog.30,36
The mechanism whereby T-700 phosphorylation stimulates the CTK-domain is more elusive, since structural information on an analogous phosphorylation site in related kinases is not available. Nevertheless, biochemical evidence point to a clear role of this site in regulation of the CTK-domain activity. Thus in MSK2, phosphomimicking Glu mutation of T-687 ( = T-700 in MSK1) greatly enhanced basal kinase activity.33 Similarly, Asp mutation of T-700 significantly increased basal as well as stimulated MSK1 activity, in particular at early time-points of stimulation.35 Apart from providing further evidence that the αK/L helices function to suppress basal MSK activity as described under stage 1, these findings also suggest that phosphorylation of αK serves to relieve this inhibition as part of the activation mechanism.
Perhaps surprisingly, Ala mutation of MSK1 T-700 produced the same effects as Asp mutation.35 This might be explained by the possibility that the T-700 side chain plays a direct role in inhibition of the CTK-domain by αK/L which can be relieved by phosphorylation of this residue, as occurring during activation of MSK, or by substitution with side chains that mimick phosphorylation (Asp) or lack inhibitory capacity (Ala). More unexpectedly, Ala or Asp mutation of T-700 greatly decreased the stimulated phosphorylation levels at T-581. Nevertheless, at the same time, T-700 mutation clearly increased phosphorylation levels at S-212 and S-376, indicating that net CTK-domain activity was not decreased, but rather increased by mutation of T-700. The authors proposed that T-700 phosphorylation (or mutation) does not prevent CTK-domain phosphorylation at T-581 but increases the turn-over of this phosphorylation site. Thus the net outcome of T-700 phosphorylation (or mutation) is increased CTK-domain activity, phosphorylation of S-212 and S-376, and ultimately increased basal and stimulated MSK activity. Whatever the mechanism, the pronounced interplay between T-700 phosphorylation/mutation and T-581 phosphorylation bears evidence of an intimate structural and functional interaction of these sites, reinforcing the idea of an important role of αK/L phosphorylation in activation of MSK. Finally, two additional phosphorylation sites, S-657 and S-695, have been identified in the C-terminal part of the CTK-domain, both of which affected MSK activity in a manner rather similar to T-700 when mutated, but the responsible kinase(s) remains unknown.35
The αC-helix is ordered and properly positioned in the CTK-domain crystal structure despite the absence of an organized and phosphorylated activation segment. This suggests that there is no major structural collaboration between the αC-helix and activation loop phosphorylation with respect to inducing the active conformation of the CTK-domain.
In conclusion, phosphorylation of the CTK-domain at T-581 and αK at T-700 (and possibly the additional nearby S-657 and S-695 sites in the CTK-domain) activates this domain leading to stage 3 of the activation mechanism. However at the present, the roles of these phosphorylation sites are mechanistically the least understood events in the MSK activation mechanism.
Stage 3—the Intramolecular Phosphorylation Cascade
Once activated, the CTK-domain catalyzes the autophosphorylation of MSK at three sites, namely S-212 in the NTK-domain activation loop, S-376 in the HM and, S-381 immediately after HM.30 From what we currently know, the CTK-domain has now full-filled its role in activation of MSK. It may be noted that MSK is unusual among its closest AGC kinase relatives, such as RSK, S6K, PKB, SGK and PKC, in that MSK autophosphorylates the NTK activation loop,30,51 whereas the analogous site of the other kinases is typically phosphorylated by PDK1.
Stage 4—the Allosteric Activation of NTK
This stage is characterized by a dramatic transition of NTK from an autoinhibited, inactive conformation to a fully active conformation that marks the completion of the multi-step activation mechanism of MSK. This final step is driven by S-212 phosphorylation of the NTK-domain activation loop in addition to the phosphorylation of the linker on S-360, S-376 and S-381 that converts it to an allosteric activator of the NTK-domain. These two phosphorylated structures are thought to cooperate to synergistically activate the NTK-domain according to the model described below. The model is a variation of the common core model of AGC kinase activation based on the three conserved phosphorylation sites in the activation loop and the linker (Fig. 4) that act via phosphate-binding sites within the kinase domain to stabilize the active conformation, as illustrated in Figure. 7. The elucidation of this common model began with the crystallization of PKA52 and matured over the subsequent 15 y through studies on several AGC kinases, including MSK.
With respect to the linker, the key activating principle is HM. This motif exerts its function by inserting its aromatic residues into a hydrophobic pocket in the N-lobe of the NTK-domain that is formed by residues from the αB and αC-helices as well as the β5-strand. This pocket is a key feature of most AGC kinases that, in fact, evolved a αB-helix, absent from other kinases, to make the pocket large enough to accommodate the phosphorylated HM. The major consequence of HM binding to the pocket is stabilization of the αC-helix in the position held in the active kinase conformation, where the conserved αC-helix Glu (MSK1 E-101) can ion pair with the conserved Lys residue in β3-strand (MSK1 K-81) allowing the Lys to properly position the phosphates of ATP, as outlined above. However as described under stage 1, the β1L0 element has disorganized and malpositioned the αB and αC-helices in the inactive MSK1 NTK-domain, resulting in complete disruption of the hydrophobic pocket. Thus apparently, the job of HM is to reorganize the hydrophobic pocket. However, the non-phosphorylated HM lacks this capability, as evidenced by its inability to activate the NTK-domain even in the presence of S-212 phosphorylation.37 Apparently, the non-phosphorylated HM lacks sufficient binding energy to dissolve the autoinhibitory β-sheet created by β1L0 and reorganize the hydrophobic pocket. This is where the linker phosphorylation sites enter the scene: upon phosphorylation, their phosphate-groups engage with phosphate-binding sites in the close vicinity of the HM-binding pocket (Fig. 7). This most likely provides critical binding energy that enables the aromatic residues of HM to dissolve the inhibitory β-sheet and reorganize the hydrophobic pocket, including the crucial αC-helix.
With respect to the activation loop, S-212 phosphorylation can be expected to stimulate NTK-domain activity by interacting with the prototypical conserved basic residues to organize important active site residues as illustrated in Figurs. 5 and 7. Thus, the positively charged residues in the catalytic loop and the β9-strand of the activation segment that bind the activation loop phosphate for stabilization of the catalytic Asp (MSK1 D-177) and the DFG motif are conserved in MSK NTK (MSK1 R-176 and K-200, respectively).
How may the activation loop and linker phosphorylation sites co-operate to synergistically activate the NTK-domain? Several mechanisms can be envisioned. Most intriguingly, phosphorylation of the activation loop may assist the phosphorylated HM in disassembling the inhibitory β-sheet. The reason is that β9-strand, including K-200, is part of the inhibitory β-sheet. It therefore seems plausible that the interaction of phospho-S-212 with K-200 will act to promote dissolution of the inhibitory β-sheet by favoring the relocation of β9-strand to its position in the active kinase conformation (compare the position of the β9-strand in the inactive MSK1 NTK-domain vs. active PKA in Fig. 6B). In this scenario, the phosphorylated HM and phospho-S-212 thus act on the αB-helix (a β-strand in the inhibitory β-sheet) and the β9-strand, respectively, to dismantle the inhibitory β-sheet. This would provide an elegant mechanism whereby the activation loop and HM, when phosphorylated, can co-operate to induce the active conformation of the NTK-domain of MSK. Furthermore, once phospho-S-212 has rearranged the catalytic loop, the Phe of the DFG motif may interact with, and stabilize αC-helix, since such an interaction has been observed in the active structures of PKA and PKB.52,53 Finally, phospho-S-212 may interact with, and stabilize the αC-helix via binding to H97 (MSK1) within the helix (Fig. 7), since an analogous interaction exists in the active structures of PKA and PKB.52,53
We will now summarize the various experimental evidence that support above model. The essential roles of the phosphorylation sites in HM and the NTK-domain activation loop were revealed by the complete loss of MSK NTK-domain activity upon their mutation to Ala.30,36 The molecular mechanism of action of the phosphorylated HM (pHM) as an allosteric activator of the MSK NTK-domain was demonstrated by Frodin et al.37 First, molecular modeling suggested the existence of a hydrophobic binding pocket for HM in the active conformation of the NTK-domain, formed by the αB and αC-helices and the β5-strand. In addition, the modeling suggested the existence of an adjacent binding site for the phosphate group of pHM, formed by two basic residues, K-43 and R-102. The predicted phosphate-binding site appeared functional, since an R-102A mutation decreased MSK activity to the same extent as mutation of the HM phosphorylation site. Interestingly, R-102 is located in the αC-helix immediately adjacent to E-101, thus enabling pHM to tightly control the position of this important residue. The role of pHM and its interplay with activation loop phosphorylation in activation of the NTK-domain was demonstrated directly using an in vitro reconstitution assay: thus, the kinase activity of the isolated NTK-domain protein truncated before HM was analyzed unphosphorylated or phosphorylated at S-212 and in the absence or presence of short synthetic HM or pHM peptides. The results based on this defined system demonstrated that the phosphorylated HM cannot activate the NTK-domain by itself, whereas S-212 phosphorylation in the activation loop induces modest activation. Combined, however, activation loop and HM phosphorylation synergistically activate NTK to high levels. Finally, non-phosphorylated HM has no activating effect under any conditions and the effect of pHM was completely dependent on the predicted binding site for the phosphate group of pHM. In the same study, a virtually identical interplay between activation loop and HM phosphorylation was demonstrated in the related AGC kinases RSK, PKB, S6K, SGK and PKC. In concurrent studies, crystallization and biochemical analysis of PKB and PDK1 (also an AGC kinase) confirmed this mode of action of pHM in activation of AGC kinases.53-55These latter studies also identified an additional residue (Gln) that binds the phosphate group of HM and this residue is conserved in MSK (MSK1 Q-122).
In a subsequent study, the mechanism of action of the S-360 phosphorylation site in the linker was characterized.38 In the AGC kinase literature, this site is termed the "turn motif" site. The designation derives from the fact that this conserved AGC kinase linker site was thought to be equivalent to phospho-S-338 of PKA that binds residues within the PKA tail, thereby promoting a turn of this structure.52 However, as demonstrated by the study by Hauge et al.,38 the PKA S-338 phosphorylation site is distinct from the generally conserved AGC kinase linker phosphorylation site. Thus, in contrast to the PKA turn motif site, the phosphate group of the generally conserved linker site was shown to interact with a phosphate-binding site formed by a cluster of basic residues located above the glycine-rich loop in the N-lobe, rather close to the hydrophobic pocket, as demonstrated with MSK, PKB, S6K, RSK, PRK and PKC. In MSK1, this phosphate-binding site is composed of K-62, K-84 and K-126. The binding interaction was found to be important, since mutation of either S-360 or its phosphate-binding residues (K-62, K-126) decreased activation of MSK1 to the same extent (∼60%). Notably, the binding interaction does not seem to stimulate kinase activity directly, as revealed through analysis of the analogous phosphorylation site in the AGC kinase S6K. Rather, the interaction serves to stabilize pHM in the hydrophobic pocket and in this way stimulate kinase activity. This idea was supported by results with MSK1 showing that mutation of S-36030,38 or the corresponding phosphate-binding site38 greatly reduced phoshorylation levels in HM. Presumably, these mutations destabilized the interaction of pHM with its binding site, thereby rendering phospho-S-376 more exposed to phosphatase action. In conclusion, the various lines of evidence support the model that phosphorylation of S-360 functions to increase the affinity of the linker for the NTK-domain and thereby assist pHM in dissolving the inhibitory β-sheet and re-organizing the hydrophobic pocket.
What is the mechanism of action of the final linker phosphorylation site, S-381, the mutation of which reduced MSK1 activation by ∼40%30? Most likely the site has a function analogous to that of S-360, i.e., stabilizing pHM in its binding pocket by interacting with a phosphate-binding site adjacent to the pHM-binding pocket. This idea is supported by the immediate proximity of S-381 to HM and the finding that S-381 to Ala mutation reduced HM phosphorylation levels in MSK1,30 exactly as was observed upon mutation of S360 or the binding site for phospho-S-360.30,38 For phospho-S-381, however, the predicted binding site remains to be identified.
The Inter-Dependency of MSK Phosphorylation Sites
As evident from above, the phosphorylation sites of MSK are inter-dependent in two ways. The first way is direct and is the result of the sequential phosphorylation cascade that activates MSK: thus, since the S-212, S-376 and S-381 phosphorylation events are catalyzed by the CTK-domain, naturally they depend on the phosphorylation events at T-581 and T-700 which activate the CTK-domain. The second way is indirect, yet still very important, and derived from the fact that the various conformational states of MSK likely render the various phosphorylation sites accessible to phosphatases to different extents. For the NTK-domain and the linker, the fully phosphorylated and active conformation is predicted to be the most phosphatase-resistant state. In this conformation, the phosphates are each thought to make intricate interactions with several phosphate-binding residues, occupying rather buried positions, which protect them from phosphatase action (see Fig. 7). Quite direct evidence that the phosphate-binding sites protect against dephosphorylation has been obtained for pHM of MSK1. Thus, R-102 to Ala mutation of the binding site for phospho-S-376 resulted in a dramatic loss of phosphorylation of this residue.37 By extrapolation, other phosphorylation events that promote the interaction of pHM with its binding site should protect phospho-S-376 from dephosphorylation. Evidence for this also exists, since mutation of either S-360, the phospho-S-360-binding site or S-381 (the latter albeit only being assumed to stabilize pHM at the present) all reduced the phosphorylation levels at S-376 in MSK1, as also mentioned above.30,38 This inter-dependency logically should go both ways. Thus, binding of pHM to the hydrophobic pocket is expected to stabilize phospho-S-360 and phospho-S-381 in their known and predicted binding sites, respectively. Supporting this idea, mutation of S-376 to Ala substantially reduced the phosphorylation levels at S-360 and S-381.30 Finally, since the phosphorylation sites in the linker and NTK-domain activation loop communicate structurally regarding stabilization of the active conformation of the NTK-domain, the sites in these elements would also be expected to exhibit inter-dependency. Again supportive evidence exists, as Ala mutation of S-376 reduced S-212 phosphorylation levels and vice versa.30
Similar observations that mutation of one of the three conserved AGC kinase phosphorylation site affects the phosphorylation levels at the other sites have been reported countless times in the literature on diverse AGC kinases. Earlier, these observations seemed confusing or hinting at complex, unknown regulatory mechanisms. With our recent knowledge on AGC kinase structure and function, most of these observations can most likely be explained by the simple and unified model that the sites cooperate to promote a common, active and phosphatase-resistant conformation, and that perturbation of one site therefore will influence the phosphorylation status at the other sites.
Nevertheless, studies on MSK have also revealed inter-dependency of phosphorylation sites that currently cannot be explained. One example described above, is the loss of T-581 phosphorylation upon mutation of T-700.35 Another example is the loss of T-581 phosphorylation observed after mutation of S-212, S-360 or S-376.30
The MAPK Docking Motif of MSK Helps Dictating by Which MAPKs It Can Be Activated
So-called consensus phosphorylation motifs provide one major determinant of substrate specificity of protein kinases. These motifs are composed of specific amino acid patterns in the primary structure immediately surrounding the phosphorylated residue in the substrate and they bind specific residues in the kinase near or within the active site, thereby promoting a selective substrate recognition and phosphorylation reaction. However, all members of the ERK1/2, p38 and JNK MAPK families phosphorylate Ser/Thr residues in the same minimalistic consensus motif: Ψ-x-x-S/T-P, in which Ψ is a Pro or aliphatic residue, x is a variable amino acid and, the phosphorylated Ser/Thr is underlined. The MAPK phosphorylation sites in MSK conform to a P-x-x-S/T-P motif (Fig. 2A). Clearly, this consensus motif cannot account for specificity in MAPK signaling to MSKs or other substrates.
As one important means to increase specificity, MAPKs have evolved docking sites that bind specific, linear docking motifs present in many, though not all of their substrates (reviewed in: 56;57). Two major classes of MAPK docking motifs have been identified. One is the so-called DEF motif that conforms to variations over an F/Y-x-F/Y-P pattern. This motif seems absent from MSK. However, MSK contains the other motif, called the D-motif that conforms to variations over an ΦH(R/K)1-3-(x)1-6-ΦA-x-ΦB pattern, in which Φ is a hydrophobic residue, usually Leu, Iso or Val. The basic residues of the D-motif bind an area rich in negatively charged residues in the MAPK distant to the active site. This area can be sub-divided in the common docking (CD) site and the Glu/Asp (ED) site.58,59 The hydrophobic residues of the ΦA-x-ΦB sequence bind a hydrophobic groove adjacent to the CD/ED sites.60 A number of crystallographical studies on D-motif/MAPK interactions have confirmed the overall generality of this binding mode of D-motifs to MAPKs.60-64 Importantly however, these studies also revealed that sequence variations in D-motifs and MAPK docking sites create large heterogeneity regarding the exact interactions that take place between particular MAPKs and D-motifs. These variations appear to provide key specificity determinants that help dictating which MAPKs can interact with which proteins and phosphorylate them in the minimal MAPK consensus motif. This was actually predicted by early biochemical studies, demonstrating that swopping of specific residues in the D-motif or its docking site could redirect the substrate specificity of a particular MAPK toward that of another MAPK.59
The D-motif variants of MSK1 and MSK2 are predicted by alignment to be LΦH-x-K-R-R-K-MΦA-x-KΦB and LΦH-x-K-R-R-K-QΦA-x-LΦB, respectively (Fig. 2A). The presence of K and Q in the ΦA-x-ΦB sequence is unusual for D-motifs. Whether this means that MSK possesses an atypical or no functional ΦA-x-ΦB sequence is not known. In any case, the D-motif MAPK docking site is functional. Thus, in MSK1, phosphorylation of the MAPK sites and activation was substantially reduced by double-mutation of the underlined basic residues: LΦH-x-K-R-R-K-MΦA-x-KΦB and partially reduced by single-mutation of LΦH.30 Furthermore, either of the mutations abolished the ability of exogenous MSK1 to coimmunoprecipitate endogenous ERK1/2 or p38α. In MSK2, deletion of the D-motif abolished the ability of MSK2 to coimmunoprecipitate or to be activated by p38α 33. With respect to the docking site for the D-motif within ERK1/2 and p38, the CD and the ED sites have both been shown to contribute to the affinity and specificity of MAPK interactions with MSK. Thus, Asn mutation of the acidic residues of the CD site disrupted the ability of p38α to be coprecipitated by MSK1 or MSK2 and ERK2 to be co-precipitated by MSK1.59 Furthermore, ERK1/2 has threonines at the ED site and only two acidic residues in the CD site, contrasting to p38α that has three. It was noted early that ERK binds weaker to MSK2 than does p38α 3 and the reduced negative charge in the docking site of ERK contributes to this fact.59 Thus, introduction of the lacking acidic residues in ERK2 enables it to be co-precipitated by MSK2 as efficiently as p38α. Conversely, mutating the three acidic residues in p38α to the neutral ones present in ERK2 decreased the ability of p38α to associate with MSK2. Interestingly, the same mutations produced overlapping as well as distinct effects on the interaction between MSK1 and ERK2 or p38α. Thus, the MAPK docking interactions of MSK1 and MSK2 are not identical.
In conclusion, D-motifs increase specificity by increasing the interaction between appropriate MAPKs and substrates. This increases the efficiency of substrate phosphorylation by MAPKs via raising the local concentration of substrate around the kinase. This feature likely underlies the observation that mutation of the non-essential LΦH mildly reduced phosphorylation and activation of MSK1 by a strong stimulus (PMA) but strongly suppressed these events in response to a weaker stimulus (UV-C).30
What is the strength of the MAPK/MSK interaction? Several direct and indirect studies suggest that the interaction between MAPKs and the D-motif of many substrates typically is of low affinity, having a Kd in the 10-100 μM range, which precludes the formation of stable complexes.65-70 Interestingly, however, binding experiments with isolated MSK1 CTK and ERK1 suggested a Kd around 70 nM.48 To our knowledge, this affinity is exceeded only by that of the interaction between MK2 and p38α (Kd in the lower nanomolar range) that do form stable complexes in unstimulated cells. It would be interesting to investigate whether MSK exists in semi-stable complexes with ERK1/2 and/or p38α, which would distinguish MSKs from the large majority of other D-motif containing MAPK substrates and point to some functional importance of such complexes.
Inactivation of MSK
Only a few reports have compared the kinetics of dephosphorylation/inactivation of MSK and its upstream MAP kinases. Studies in some settings observed close coupling between inactivation of MSK and MAPK,1,12 whereas in another setting, MSK inactivation showed a significant delay as compared with MAPK.36 Thus, while MSK inactivation is controlled by phosphatases in cells, these may act in a context- and/or MSK homolog-specific manner. When co-expressed, exogenous MSK1 or MSK2 can co-immunoprecipitate exogenous protein phosphatase-2Cδ 71. However, RNAi or other functional evidence demonstrating a requirement of this phosphatase in dephosphorylation of MSK is lacking. Thus, the phosphatase(s) that terminates MSK signaling remains to be identified.
Substrate Specificity of MSK
The consensus phosphorylation motif of MSK is poorly defined. This is due to the lack of a co-crystal structure of MSK NTK and a substrate as well as the paucity of known MSK substrate phosphorylation sites. Nevertheless, inspection of the sequences surrounding the few well-validated MSK phosphorylation sites is informative (CREB S-133, ATF-1 S-63, histone H3 S-10 and S-28, HMGN1 S-6, NF-κB p65 S-276 and RARa1 S-369—see Fig. 3). Surprisingly, the consensus phosphorylation motif of MSK does not seem to conform to the one preferred by the AGG kinases most closely related to the MSK NTK-domain, namely RSK NTK-domain, S6K, PKB or SGK. For these kinases, the large majority of phosphorylation sites are thought to be situated in an R-x-R-x-x-S/T-Φ motif, in which x can be a variable amino acid, Φ a hydrophobic amino acid and the phosphorylated Ser/Thr is underlined.72 Testing of a limited set of peptides as MSK1 substrates suggested that Arg in the -5 position relative to the phosphorylated Ser is not required and that Lys at the -3 position greatly increases Km compared with Arg.1 Based on these experiments and the validated MSK substrates, a very tentative MSK consensus motif could be R-R-x-S or R-K-S. Notably, a Pro is tolerated at the -1 position, which is rarely observed for the other aforementioned AGC kinases.72 This could indicate that MSK may have a substrate specificity quite distinct from that of RSK, S6K, PKB and S6K that, in contrast, are known to share several substrates. Rather, the consensus motif of MSK resembles that of PKA.73 In agreement with these considerations, neither RSK, S6K, PKB nor S6K are thought to phosphorylate any of the MSK substrates shown in Figure 3 in vivo while several (CREB, ATF1, RARα1 and NF-κB) can also be phosphorylated by PKA.
Analysis of MEFs from mice with individual or double-knockout of MSK1 and MSK2 revealed that both kinases contributed to essentially the same extent to the phosphorylation of CREB and ATF1 in response to anisomycin, UV irradiation, PMA or epidermal growth factor.74 By contrast, in identical analyses of MEFs, MSK2 appeared to be the major MSK homolog for phosphorylation of histone H3 and HMGN1 in response to anisomycin or PMA.75 Analysis of cultured cortical neurons from individual or double MSK knockout mice revealed that MSK1 almost exclusively mediate CREB phosphorylation in response to brain-derived neurotrophic factor.4 Given the similar activity of both MSKs against CREB in MEFs, the latter finding may be due to the fact that MSK1 is the predominant MSK in cortical neurons, as evidenced by ∼16-fold higher mRNA levels for MSK1 as compared with MSK2. Altogether, the data from the knockout cells suggest that MSK1 and MSK2 can utilize a similar or very overlapping consensus motif, as evidenced with CREB and ATF1, and that the relative expression levels of the individual MSKs may be an important determinant regarding which MSK homolog that phosphorylates a given substrate in a given cell type. However, the MEF experiments also reveal that in cells expressing shared MSK substrates like CREB and ATF1, other substrates like histone H3 and HMGN1 are preferentially phosphorylated by one homolog, i.e., MSK2. It remains to be investigated whether this difference is due to the fact that histone H3 and HMGN1 possess motifs that are poorly recognized by MSK1 or whether e.g., MSK2 is more efficiently targeted to sites of histone H3 and HMGN1 phosphorylation in chromatin.
Structure, Function and Activation of MSK1 vs. MSK2: What Is the Difference?
The reported similarities greatly out-number the dissimilarities regarding structure-function and regulation of the two MSK homologs. So far, only a few distinctions have been observed, the most striking of which may be the selectivity of MSK2 over MSK1 for phosphorylation of histone H3 and HMGN1, as described above. Additionally, in HeLa cells exposed to tumor necrosis factor-α, activation of MSK1 was transient (gone by 30 min) and followed the kinetics of p38 and ERK1/2 activation closely.1 By contrast, in the same context, high MSK2 activity persisted for at least three hours after p38 and ERK1/2 activity had returned to base-line and apparently was sustained by low basal p38 activity.36 Although the underlying mechanism is unknown, it would profoundly distinguish MSK1 and MSK2 signaling functions in this context. As another example of dissimilarity, MSK2 may be under control of the protein kinase CK2, since pharmacological inhibition or knockdown of this kinase greatly suppressed UV-induced activation of MSK2 in MDA-MB-231 and HEK293 cells, whereas MSK1 remained unaffected.76 CK2 was proposed to regulate MSK2 via phosphorylation of S-324, a site absent from MSK1 (Fig. 2B). CK2 is considered constitutively active suggesting that S-324 typically would be constitutively phosphorylated in cells and that activation of ERK1/2 or p38α in reality is the sole determinant for stimulus induced-activation of MSK2. A requirement for CK2 phosphorylation of MSK2 is however at odds with the finding that in vitro incubation with active p38 is sufficient to activate MSK2.36 The possibility therefore remains that CK2 may regulate UV-induced activation of MSK2 via a mechanism other than phosphorylation of S-324, for example by acting upstream of MSK2 in the UV signaling pathway. Finally, the extreme N- and C-terminal ends of MSK1 and MSK2 differ substantially compared with the rest of the molecules and would represent key candidates to underlie potential MSK homolog-specific functions.
Monitoring MSK Activity in Cells
Since the activity of MSK NTK is governed by the relative phosphorylation levels at several phosphorylation sites, the most reliable way of determining MSK activity in cells, in principle, is to immunoprecipitate the kinase from cell extracts and measure its activity in vitro in immune-complex kinase assays using [γ-32P]ATP and a suitable protein/peptide substrate as phospho-acceptor. Most frequently, the peptide Crosstide encompassing the phosphorylation site S-9 of GSK3 (GRPRTSSFAEG) has been used as phosphoryl-acceptor. Even though GSK3 is not a physiological substrate of MSK, this assay appears to faithfully reflect the level of NTK-domain activity.1,30 Alternatively, CREBtide encompassing the phosphorylation site S-133 of CREB (EILSRRPSYRK; the phosphorylated S is underlined) can be used.1,36 Phosphospecific antibodies to key activating phosphorylation sites of MSK are also an option, since the available evidence suggests that they are coordinately phosphorylated, partly due to their interdependency as discussed above. For instance, notably the phosphorylation levels of S-212, S-360, S-376, S-381 and T-581 were all found to be induced or inhibited to a remarkably similar relative extent in response to various stimuli and inhibitors and, correlated closely with in vitro kinase activity measurements using Crosstide as a substrate.30 Nevertheless, it should be noted that the phosphorylation kinetics of these sites have not been studied in detail as to what extent they are coordinated over time. With our current knowledge, phosphospecific antibodies to the S-212 and S-376 phosphorylation sites would be reasonable tools for assessing MSK activity by immunoblotting. When monitoring endogenous MSKs by immunoblotting, it may be necessary to first immunoprecipitate the kinases, since MSKs are not abundant proteins and rather similar phosphorylation sites are present in RSK, which runs at roughly the same size as MSK.
Use of MSK Mutants as Experimental Tools: Structure-Function Considerations
Mutants of protein kinases are extensively used experimental tools for elucidating the cellular functions of their wild-type counterparts. However, our deepening knowledge on the structure-function relationships of protein kinases is raising serious concerns regarding this approach, and this is also the case with MSK.
Kinases rendered catalytically inactive, typically by point mutating critical active site residues, have been used to test for a requirement of a specific kinase in a given processes. MSK1 and MSK2 have been rendered completely inactive by Ala mutation of the Asp in the DFG motif (MSK1 D-195, D-565) and the α,β-phosphate-coordinating Lys in β3-strand (MSK1 K-65, K-440), respectively, in either kinase domain.1,3 Inactive mutant is then overexpressed in cells, where it is supposed to inhibit the endogenous kinase in a dominantly interfering manner. Obviously, this strategy is artifact-prone in the case of MSK, since when overexpressed, its D-motif would be expected to massively sequester ERK1/2 and p38 via their D-motif binding sites and thereby globally interfere with the interaction of these MAPKs with other D-motif-containing proteins in the nucleus. This suggests that in dominant negative MSK mutants, the MAPK docking motif should be mutated (and an additional NLS be introduced, for example, N-terminally, to compensate for the cognate NLS that overlaps the MAPK docking motif ). Whether such MSK mutants work as dominant negatives remain to be shown.
Overexpression of wild-type or constitutively active mutants of kinases has been used to support an involvement of a specific kinase in a given process, if overexpression further enhances the process or rescues its modulation after kinase knockdown. A constitutively active MSK1 mutant can be generated be replacing the HM of MSK with that of another AGC kinase, namely PRK2 that has a phospho-mimicking Asp in place of a phosphorylation site in its HM.77 Although CTK is not modified in this mutant, the very high, constitutive activity of NTK likely derives from two factors: first, the HM of PRK2 appears to possess particularly high ability to interact with the conserved AGC kinase HM-binding pocket for reasons that are not clear.37,55 Second, when introduced into MSK, the PRK2 HM likely recruits the kinase PDK1 enabling it to phosphorylate the NTK activation loop, since HM has this function in PRK2, RSK, S6K and certain other AGC kinases, thereby by-passing a need for CTK for phosphorylation of S-212. As for the dominant negative MSK approach, overexpression of wild-type or constitutively active MSK can cause artifacts by general interference with MAPK signaling by the D-motif in MSK. Furthermore, protein kinases typically loose substrate specificity when overexpressed. Consequently, very modest overexpression of wild-type or constitutively active MSK constructs is advisable, when these are used as experimental tools.
Conclusion
At this point, MSKs have been firmly established as ubiquitous mediators of ERK1/2 and p38α signal transduction. Furthermore, a substantial amount of biochemical and structural data obtained with MSK and related kinases have enabled construction of a credible model for the activation mechanism of MSK. Nevertheless, several important issues remain to be addressed in future studies on the structure-function and regulation of MSK, of which some are outstanding: crystal structures of full-length MSK1 and MSK2 in the various phosphorylation states will be essential to validate and extend the model for the activation mechanism of MSK. The phosphatase(s) that inactivate MSK need be identified. Finally, since the large majority of MSK reports have focused on MSK1, further studies on the structure-function and regulation of MSK2 are particularly warranted.
Acknowledgments
We thank Dr. Camilla Hauge from our lab for helpful comments on the manuscript. M.F. acknowledges support from the Danish Cancer Society (R2-A132-02-S2) and the Danish National Research Foundation.
References
- 1.
- Deak M, Clifton AD, Lucocq LM, Alessi DR. Mitogen- and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J. 1998;17:4426–41. http://dx.doi.org/10.1093/emboj/17.15.4426 . [PMC free article: PMC1170775] [PubMed: 9687510]
- 2.
- New L, Zhao M, Li Y, Bassett WW, Feng Y, Ludwig S, et al. Cloning and characterization of RLPK, a novel RSK-related protein kinase. J Biol Chem. 1999;274:1026–32. http://dx.doi.org/10.1074/jbc.274.2.1026 . [PubMed: 9873047]
- 3.
- Pierrat B, Correia JS, Mary JL, Tomas-Zuber M, Lesslauer W. RSK-B, a novel ribosomal S6 kinase family member, is a CREB kinase under dominant control of p38alpha mitogen-activated protein kinase (p38alphaMAPK). J Biol Chem. 1998;273:29661–71. http://dx.doi.org/10.1074/ jbc.273.45.29661 . [PubMed: 9792677]
- 4.
- Arthur JS, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, et al. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24:4324–32. http://dx.doi.org/10.1523/ JNEUROSCI.5227-03.2004 . [PMC free article: PMC6729446] [PubMed: 15128846]
- 5.
- Schuck S, Soloaga A, Schratt G, Arthur JS, Nordheim A. The kinase MSK1 is required for induction of c-fos by lysophosphatidic acid in mouse embryonic stem cells. BMC Mol Biol. 2003;4:6. http://dx.doi.org/10.1186/1471-2199-4-6 . [PMC free article: PMC161794] [PubMed: 12769834]
- 6.
- Wierenga AT, Vogelzang I, Eggen BJ, Vellenga E. Erythropoietin-induced serine 727 phosphorylation of STAT3 in erythroid cells is mediated by a MEK-, ERK-, and MSK1-dependent pathway. Exp Hematol. 2003;31:398–405. http://dx.doi.org/10.1016/S0301-472X(03)00045-6 . [PubMed: 12763138]
- 7.
- Vicent GP, Ballare C, Nacht AS, Clausell J, Subtil-Rodrguez A, Quiles I, et al. Induction of progesterone target genes requires activation of Erk and Msk kinases and phosphorylation of histone H3. Mol Cell. 2006;24:367–81. http://dx.doi.org/10.1016/j.molcel.2006.10.011 . [PubMed: 17081988]
- 8.
- Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. http://dx.doi. org/10.1042/BJ20070797 . [PMC free article: PMC2267365] [PubMed: 17850214]
- 9.
- Beardmore VA, Hinton HJ, Eftychi C, Apostolaki M, Armaka M, Darragh J, et al. Generation and characterization of p38beta (MAPK11) gene-targeted mice. Mol Cell Biol. 2005;25:10454–64. http://dx.doi.org/10.1128/MCB.25.23.10454-10464.2005 . [PMC free article: PMC1291241] [PubMed: 16287858]
- 10.
- Darragh J, Soloaga A, Beardmore VA, Wingate AD, Wiggin GR, Peggie M, et al. MSKs are required for the transcription of the nuclear orphan receptors Nur77, Nurr1 and Nor1 downstream of MAPK signalling. Biochem J. 2005;390:749–59. http://dx.doi.org/10.1042/BJ20050196 . [PMC free article: PMC1199668] [PubMed: 15910281]
- 11.
- Kannan-Thulasiraman P, Katsoulidis E, Tallman MS, Arthur JS, Platanias LC. Activation of the mitogenand stress-activated kinase 1 by arsenic trioxide. J Biol Chem. 2006;281:22446–52. http://dx.doi.org/10.1074/jbc.M603111200 . [PubMed: 16762916]
- 12.
- Bruck N, Vitoux D, Ferry C, Duong V, Bauer A, de Thé H, et al. A coordinated phosphorylation cascade initiated by p38MAPK/MSK1 directs RARalpha to target promoters. EMBO J. 2009;28:34–47. http://dx.doi.org/10.1038/emboj.2008.256 . [PMC free article: PMC2633082] [PubMed: 19078967]
- 13.
- Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–49. http://dx.doi.org/10.1038/nrc2694 . [PubMed: 19629069]
- 14.
- Funding AT, Johansen C, Kragballe K, Otkjaer K, Jensen UB, Madsen MW, et al. Mitogen- and stress-activated protein kinase 1 is activated in lesional psoriatic epidermis and regulates the expression of pro-inflammatory cytokines. J Invest Dermatol. 2006;126:1784–91. http://dx.doi. org/10.1038/sj.jid.5700252 . [PubMed: 16543895]
- 15.
- Schiller M, Bohm M, Dennler S, Ehrchen JM, Mauviel A. Mitogen- and stress-activated protein kinase 1 is critical for interleukin-1-induced, CREB-mediated, c-fos gene expression in keratinocytes. Oncogene. 2006;25:4449–57. http://dx.doi.org/10.1038/sj.onc.1209479 . [PubMed: 16532028]
- 16.
- Kim H, Rhee SH, Kokkotou E, Na X, Savidge T, Moyer MP, et al. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J Biol Chem. 2005;280:21237–45. http://dx.doi.org/10.1074/jbc.M413842200 . [PubMed: 15767259]
- 17.
- El Mchichi MB, Hadji A, Vazquez A, Leca G. p38 MAPK and MSK1 mediate caspase-8 activation in manganese-induced mitochondria-dependent cell death. Cell Death Differ. 2007;14:1826–36. http://dx.doi.org/10.1038/sj.cdd.4402187 . [PubMed: 17585337]
- 18.
- Dümmler BA, Hauge C, Silber J, Yntema HG, Kruse LS, Kofoed B, et al. Functional characterization of human RSK4, a new 90-kDa ribosomal S6 kinase, reveals constitutive activation in most cell types. J Biol Chem. 2005;280:13304–14. http://dx.doi.org/10.1074/jbc.M408194200 . [PubMed: 15632195]
- 19.
- Ananieva O, Darragh J, Johansen C, Carr JM, McIlrath J, Park JM, et al. The kinases MSK1 and MSK2 act as negative regulators of Toll-like receptor signaling. Nat Immunol. 2008;9:1028–36. http://dx.doi.org/10.1038/ni.1644 . [PubMed: 18690222]
- 20.
- Song KS, Lee WJ, Chung KC, Koo JS, Yang EJ, Choi JY, et al. Interleukin-1 beta and tumor necrosis factor-alpha induce MUC5AC overexpression through a mechanism involving ERK/p38 mitogen-activated protein kinases-MSK1-CREB activation in human airway epithelial cells. J Biol Chem. 2003;278:23243–50. http://dx.doi.org/10.1074/jbc.M300096200 . [PubMed: 12690113]
- 21.
- Pathak SK, Basu S, Bhattacharyya A, Pathak S, Banerjee A, Basu J, et al. TLR4-dependent NF-kappaB activation and mitogen- and stress-activated protein kinase 1-triggered phosphorylation events are central to Helicobacter pylori peptidyl prolyl cis-, trans-isomerase (HP0175)-mediated induction of IL-6 release from macrophages. J Immunol. 2006;177:7950–8. [PubMed: 17114467]
- 22.
- Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal. 2006;18:2238–51. http://dx.doi.org/10.1016/j.cellsig.2006.05.004 . [PubMed: 16806820]
- 23.
- Markou T, Lazou A. Phosphorylation and activation of mitogen- and stress-activated protein kinase-1 in adult rat cardiac myocytes by G-protein-coupled receptor agonists requires both extracellular-signal-regulated kinase and p38 mitogen-activated protein kinase. Biochem J. 2002;365:757–63. [PMC free article: PMC1222733] [PubMed: 11994045]
- 24.
- Krook A, Widegren U, Jiang XJ, Henriksson J, Wallberg-Henriksson H, Alessi D, et al. Effects of exercise on mitogen- and stress-activated kinase signal transduction in human skeletal muscle. Am. J. Physiol Regul Integr Comp Physiol. 2000;279:R1716–21. [PubMed: 11049854]
- 25.
- Butcher GQ, Lee B, Cheng HY, Obrietan K. Light stimulates MSK1 activation in the suprachiasmatic nucleus via a PACAP-ERK/MAP kinase-dependent mechanism. J Neurosci. 2005;25:5305–13. http://dx.doi.org/10.1523/JNEUROSCI.4361-04.2005 . [PMC free article: PMC6724997] [PubMed: 15930378]
- 26.
- Brami-Cherrier K, Valjent E, Herve D, Darragh J, Corvol JC, Pages C, et al. Parsing molecular and behavioral effects of cocaine in mitogen- and stress-activated protein kinase-1-deficient mice. J Neurosci. 2005;25:11444–54. http://dx.doi.org/10.1523/JNEUROSCI.1711-05.2005 . [PMC free article: PMC6725898] [PubMed: 16339038]
- 27.
- Huang P, Qi Z, Bu X, Zhang N, Han S, Fang L, et al. Neuron-specific phosphorylation of mitogenand stress-activated protein kinase-1 involved in cerebral hypoxic preconditioning of mice. J Neurosci Res. 2007;85:1279–87. http://dx.doi.org/10.1002/jnr.21242 . [PubMed: 17330274]
- 28.
- Sindreu CB, Scheiner ZS, Storm DR. Ca2+ -stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron. 2007;53:79–89. http://dx.doi. org/10.1016/j.neuron.2006.11.024 . [PMC free article: PMC1858648] [PubMed: 17196532]
- 29.
- Mody N, Leitch J, Armstrong C, Dixon J, Cohen P. Effects of MAP kinase cascade inhibitors on the MKK5/ERK5 pathway. FEBS Lett. 2001;502:21–4. http://dx.doi.org/10.1016/ S0014-5793(01)02651-5 . [PubMed: 11478941]
- 30.
- McCoy CE, Campbell DG, Deak M, Bloomberg GB, Arthur JS. MSK1 activity is controlled by multiple phosphorylation sites. Biochem J. 2005;387:507–17. http://dx.doi.org/10.1042/ BJ20041501 . [PMC free article: PMC1134980] [PubMed: 15568999]
- 31.
- Hauge C, Frodin M. RSK and MSK in MAP kinase signalling. J Cell Sci. 2006;119:3021–3. http://dx.doi.org/10.1242/jcs.02950 . [PubMed: 16868029]
- 32.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. http://dx.doi.org/10.1126/science.1075762 . [PubMed: 12471243]
- 33.
- Tomás-Zuber M, Mary JL, Lamour F, Bur D, Lesslauer W. C-terminal elements control location, activation threshold, and p38 docking of ribosomal S6 kinase B (RSKB). J Biol Chem. 2001;276:5892–9. http://dx.doi.org/10.1074/jbc.M005822200 . [PubMed: 11035004]
- 34.
- Beck IM, Vanden Berghe W, Vermeulen L, Bougarne N, Vander Cruyssen B, Haegeman G, et al. Altered subcellular distribution of MSK1 induced by glucocorticoids contributes to NF-kappaB inhibition. EMBO J. 2008;27:1682–93. http://dx.doi.org/10.1038/emboj.2008.95 . [PMC free article: PMC2435130] [PubMed: 18511904]
- 35.
- McCoy CE, MacDonald A, Morrice NA, Campbell DG, Deak M, Toth R, et al. Identification of novel phosphorylation sites in MSK1 by precursor ion scanning MS. Biochem J. 2007;402:491–501. http://dx.doi.org/10.1042/BJ20061183 . [PMC free article: PMC1863562] [PubMed: 17117922]
- 36.
- Tomás-Zuber M, Mary JL, Lesslauer W. Control sites of ribosomal S6 kinase B and persistent activation through tumor necrosis factor. J Biol Chem. 2000;275:23549–58. http://dx.doi. org/10.1074/jbc.M002586200 . [PubMed: 10806207]
- 37.
- Fördin M, Antal TL, Dummler BA, et al. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 2002;21:5396–407. http://dx.doi.org/10.1093/emboj/cdf551 . [PMC free article: PMC129083] [PubMed: 12374740]
- 38.
- Hauge C, Antal TL, Hirschberg D, et al. Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 2007;26:2251–61. http://dx.doi. org/10.1038/sj.emboj.7601682 . [PMC free article: PMC1864980] [PubMed: 17446865]
- 39.
- Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. http://dx.doi.org/10.1128/ MMBR.00031-10 . [PMC free article: PMC3063353] [PubMed: 21372320]
- 40.
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–82. http://dx.doi.org/10.1016/S0092-8674(02)00741-9 . [PubMed: 12015977]
- 41.
- Johnson DA, Akamine P, Radzio-Andzelm E, Madhusudan M, Taylor SS. Dynamics of cAMP-dependent protein kinase. Chem Rev. 2001;101:2243–70. http://dx.doi.org/10.1021/cr000226k . [PubMed: 11749372]
- 42.
- Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–58. http://dx.doi.org/10.1016/S0092-8674(00)81092-2 . [PubMed: 8612268]
- 43.
- Kornev AP, Taylor SS. Defining the conserved internal architecture of a protein kinase . Biochim Biophys Acta. 2010;1804:440–444. [PMC free article: PMC3435107] [PubMed: 19879387]
- 44.
- Shi Z, Resing KA, Ahn NG. Networks for the allosteric control of protein kinases. Curr Opin Struct Biol. 2006;16:686–92. http://dx.doi.org/10.1016/j.sbi.2006.10.011 . [PubMed: 17085044]
- 45.
- Goldberg J, Nairn AC, Kuriyan J. Structural basis for the autoinhibition of calcium/calmodulin-dependent protein kinase I. Cell. 1996;84:875–87. http://dx.doi.org/10.1016/S0092-8674(00)81066-1 . [PubMed: 8601311]
- 46.
- Malakhova M, Tereshko V, Lee SY, et al. Structural basis for activation of the autoinhibitory C-terminal kinase domain of p90 RSK2. Nat Struct Mol Biol. 2008;15:112–3. http://dx.doi.org/10.1038/nsmb1347 . [PMC free article: PMC2864125] [PubMed: 18084304]
- 47.
- Meng W, Swenson LL, Fitzgibbon MJ, et al. Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export. J Biol Chem. 2002;277:37401–5. http://dx.doi.org/10.1074/jbc.C200418200 . [PubMed: 12171911]
- 48.
- Malakhova M, D'Angelo I, Kim HG, Kurinov I, Bode AM, Dong Z. The crystal structure of the active form of the C-terminal kinase domain of mitogen- and stress-activated protein kinase 1. J Mol Biol. 2010;399:41–52. http://dx.doi.org/10.1016/j.jmb.2010.03.064 . [PMC free article: PMC2897027] [PubMed: 20382163]
- 49.
- Smith KJ, Carter PS, Bridges A, Horrocks P, Lewis C, Pettman G, et al. The structure of MSK1 reveals a novel autoinhibitory conformation for a dual kinase protein. Structure. 2004;12:1067–77. http://dx.doi.org/10.1016/j.str.2004.02.040 . [PubMed: 15274926]
- 50.
- Shimada M, Nakadai T, Fukuda A, Hisatake K. cAMP-response element-binding protein (CREB) controls MSK1-mediated phosphorylation of histone H3 at the c-fos promoter in vitro. J Biol Chem. 2010;285:9390–401. http://dx.doi.org/10.1074/jbc.M109.057745 . [PMC free article: PMC2843188] [PubMed: 20089855]
- 51.
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, et al. The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol. 2000;10:439–48. http://dx.doi.org/10.1016/S0960-9822(00)00441-3 . [PubMed: 10801415]
- 52.
- Knighton DR, Zheng JH, Ten Eyck LF, Ashford VA, Xuong NH, Taylor SS, et al. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–14. http://dx.doi.org/10.1126/science.1862342 . [PubMed: 1862342]
- 53.
- Yang J, Cron P, Good VM, Thompson V, Hemmings BA, Barford D. Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol. 2002;9:940–4. http://dx.doi.org/10.1038/nsb870 . [PubMed: 12434148]
- 54.
- Biondi RM, Komander D, Thomas CC, Lizcano JM, Deak M, Alessi DR, et al. High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J. 2002;21:4219–28. http://dx.doi.org/10.1093/emboj/cdf437 . [PMC free article: PMC126174] [PubMed: 12169624]
- 55.
- Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, et al. Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell. 2002;9:1227–40. http://dx.doi.org/10.1016/S1097-2765(02)00550-6 . [PubMed: 12086620]
- 56.
- Akella R, Moon TM, Goldsmith EJ. Unique MAP Kinase binding sites. Biochem Biophys Acta. 2008;1784:48–55. [PMC free article: PMC2266891] [PubMed: 18068683]
- 57.
- Ubersax JA, Ferrell JEJr. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–41. http://dx.doi.org/10.1038/nrm2203 . [PubMed: 17585314]
- 58.
- Tanoue T, Adachi M, Moriguchi T, Nishida E. A conserved docking motif in MAP kinases common to substrates, activators and regulators. Nat Cell Biol. 2000;2:110–6. http://dx.doi.org/10.1038/35000065 . [PubMed: 10655591]
- 59.
- Tanoue T, Maeda R, Adachi M, Nishida E. Identification of a docking groove on ERK and p38 MAP kinases that regulates the specificity of docking interactions. EMBO J. 2001;20:466–79. http://dx.doi.org/10.1093/emboj/20.3.466 . [PMC free article: PMC133461] [PubMed: 11157753]
- 60.
- Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell. 2002;9:1241–9. http://dx.doi.org/10.1016/S1097-2765(02)00525-7 . [PubMed: 12086621]
- 61.
- Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, et al. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–95. http://dx.doi.org/10.1038/sj.emboj.7600212 . [PMC free article: PMC419904] [PubMed: 15141161]
- 62.
- Liu S, Sun JP, Zhou B, Zhang ZY. Structural basis of docking interactions between ERK2 and MAP kinase phosphatase 3. Proc Natl Acad Sci USA. 2006;103:5326–31. http://dx.doi.org/10.1073/pnas.0510506103 . [PMC free article: PMC1459354] [PubMed: 16567630]
- 63.
- Reményi A, Good MC, Bhattacharyya RP, Lim WA. The role of docking interactions in mediating signaling input, output, and discrimination in the yeast MAPK network. Mol Cell. 2005;20:951–62. http://dx.doi.org/10.1016/j.molcel.2005.10.030 . [PubMed: 16364919]
- 64.
- Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006;14:1011–9. http://dx.doi.org/10.1016/j. str.2006.04.006 . [PubMed: 16765894]
- 65.
- Bardwell AJ, Flatauer LJ, Matsukuma K, Thorner J, Bardwell L. A conserved docking site in MEKs mediates high-affinity binding to MAP kinases and cooperates with a scaffold protein to enhance signal transmission. J Biol Chem. 2001;276:10374–86. http://dx.doi.org/10.1074/ jbc.M010271200 . [PMC free article: PMC3021106] [PubMed: 11134045]
- 66.
- Bardwell AJ, Abdollahi M, Bardwell L. Docking sites on mitogen-activated protein kinase (MAPK) kinases, MAPK phosphatases and the Elk-1 transcription factor compete for MAPK binding and are crucial for enzymic activity. Biochem J. 2003;370:1077–85. http://dx.doi.org/10.1042/ BJ20021806 . [PMC free article: PMC1223246] [PubMed: 12529172]
- 67.
- Bardwell L, Shah K. Analysis of mitogen-activated protein kinase activation and interactions with regulators and substrates. Methods. 2006;40:213–23. http://dx.doi.org/10.1016/j. ymeth.2006.06.008 . [PMC free article: PMC3017500] [PubMed: 16884917]
- 68.
- Gaestel M, Kracht M. Peptides as signaling inhibitors for mammalian MAP kinase cascades. Curr Pharm Des. 2009;15:2471–80. http://dx.doi.org/10.2174/138161209788682299 . [PubMed: 19601844]
- 69.
- Ho DT, Bardwell AJ, Abdollahi M, Bardwell L. A docking site in MKK4 mediates high affinity binding to JNK MAPKs and competes with similar docking sites in JNK substrates. J Biol Chem. 2003;278:32662–72. http://dx.doi.org/10.1074/jbc.M304229200 . [PMC free article: PMC3017503] [PubMed: 12788955]
- 70.
- Ho DT, Bardwell AJ, Grewal S, Iverson C, Bardwell L. Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J Biol Chem. 2006;281:13169–79. http://dx.doi.org/10.1074/jbc.M601010200 . [PMC free article: PMC3017509] [PubMed: 16533805]
- 71.
- Doehn U, Gammeltoft S, Shen SH, Jensen CJ. p90 ribosomal S6 kinase 2 is associated with and dephosphorylated by protein phosphatase 2Cdelta. Biochem J. 2004;382:425–31. http://dx.doi.org/10.1042/BJ20040948 . [PMC free article: PMC1133798] [PubMed: 15206906]
- 72.
- Moritz A, Li Y, Guo A, Villén J, Wang Y, MacNeill J, et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 2010;3:ra64. http://dx.doi.org/10.1126/scisignal.2000998 . [PMC free article: PMC3137639] [PubMed: 20736484]
- 73.
- Shabb JB. Physiological substrates of cAMP-dependent protein kinase. Chem Rev. 2001;101:2381–411. http://dx.doi.org/10.1021/cr000236l . [PubMed: 11749379]
- 74.
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–81. http://dx.doi.org/10.1128/MCB.22.8.2871-2881.2002 . [PMC free article: PMC133730] [PubMed: 11909979]
- 75.
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, et al. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–97. http://dx.doi.org/10.1093/emboj/cdg273 . [PMC free article: PMC156769] [PubMed: 12773393]
- 76.
- Jacks KA, Koch CA. Differential regulation of mitogen- and stress-activated protein kinase-1 and -2 (MSK1 and MSK2) by CK2 following UV radiation. J Biol Chem. 2010;285:1661–70. http://dx.doi.org/10.1074/jbc.M109.083808 . [PMC free article: PMC2804324] [PubMed: 19933278]
- 77.
- Doehn U, Hauge C, Frank SR, Jensen CJ, Duda K, Nielsen JV, et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol Cell. 2009;35:511–22. http://dx.doi.org/10.1016/j.molcel.2009.08.002 . [PMC free article: PMC3784321] [PubMed: 19716794]
- Introduction
- MSK Is Activated by ERK1/2 and p38α MAPKs in Response to Numerous Environmental Stimuli
- Functional Domains, Motifs and Phosphorylation Sites of MSK
- The Cartoon Model of the MSK Activation Mechanism via Multi-Site Phosphorylation
- Relationship to Other AGC Kinase and CaMK Activation Mechanisms
- The More Detailed and Structural View of the Activation Mechanism of MSK
- Stage 1—the Autoinhibited Basal State
- Stage 2—the Intermolecular Phosphorylation Cascade
- Stage 3—the Intramolecular Phosphorylation Cascade
- Stage 4—the Allosteric Activation of NTK
- The Inter-Dependency of MSK Phosphorylation Sites
- The MAPK Docking Motif of MSK Helps Dictating by Which MAPKs It Can Be Activated
- Inactivation of MSK
- Substrate Specificity of MSK
- Structure, Function and Activation of MSK1 vs. MSK2: What Is the Difference?
- Monitoring MSK Activity in Cells
- Use of MSK Mutants as Experimental Tools: Structure-Function Considerations
- Conclusion
- Acknowledgments
- References
- Stimuli That Activate MSK in Cells and the Molecular Mechanism of Activation - M...Stimuli That Activate MSK in Cells and the Molecular Mechanism of Activation - Madame Curie Bioscience Database
Your browsing activity is empty.
Activity recording is turned off.
See more...