Summary
Clinical characteristics.
Autoimmune lymphoproliferative syndrome (ALPS), caused by defective lymphocyte homeostasis, is characterized by the following:
- Non-malignant lymphoproliferation (lymphadenopathy, hepatosplenomegaly with or without hypersplenism) that often improves with age
- Autoimmune disease, mostly directed toward blood cells
- Lifelong increased risk for both Hodgkin and non-Hodgkin lymphoma
In ALPS-FAS (the most common and best-characterized type of ALPS, associated with heterozygous germline pathogenic variants in FAS), non-malignant lymphoproliferation typically manifests in the first years of life, inexplicably waxes and wanes, and then often decreases without treatment in the second decade of life; in many affected individuals, however, neither splenomegaly nor the overall expansion of lymphocyte subsets in peripheral blood decreases. Although autoimmunity is often not present at the time of diagnosis or at the time of the most extensive lymphoproliferation, autoantibodies can be detected before autoimmune disease manifests clinically.
In ALPS-FAS caused by homozygous or compound heterozygous (biallelic) pathogenic variants in FAS, severe lymphoproliferation occurs before, at, or shortly after birth, and usually results in death at an early age.
ALPS-sFAS, resulting from somatic FAS pathogenic variants in selected cell populations, notably the alpha/beta double-negative T cells (α/β-DNT cells), appears to be similar to ALPS-FAS resulting from heterozygous germline pathogenic variants in FAS, although lower incidence of splenectomy and lower lymphocyte counts have been reported in ALPS-sFAS and no cases of lymphoma have yet been published.
Diagnosis/testing.
The diagnosis of ALPS is based on the following:
- Clinical findings
- Laboratory abnormalities:
- Abnormal biomarker testing (soluble interleukin-10 [IL-10], Fas ligand [FasL], IL-18, and vitamin B12)
- Defective in vitro tumor necrosis factor receptor superfamily member 6 (Fas)-mediated apoptosis
- T cells that express the alpha/beta T-cell receptor but lack both CD4 and CD8 (so-called "α/β-DNT cells")
- Identification of pathogenic variants in genes relevant for the Fas pathway of apoptosis. These genes include FAS (either germline or somatic pathogenic variants), CASP10, and FASGL.Up to 20% of those with clinical ALPS have not had a genetic etiology identified.
Management.
Treatment of manifestations: Current management is focused on monitoring for and treatment of lymphoproliferation, hypersplensim, and lymphomas and management of cytopenias and other autoimmune diseases. Corticosteroids and immunosuppressive therapy do not decrease lymphadenopathy long term and are generally reserved for severe complications of lymphoproliferation (e.g., airway obstruction, significant hypersplenism associated with splenomegaly) and/or autoimmune manifestations. Experience with sirolimus suggests that it is the preferred agent in treating lymphoproliferation in a more sustained manner, including maintenance of remission following a period of discontinued use of sirolimus; however, sirolimus is not without side effects. Lymphoma is treated with conventional protocols. Autoimmune cytopenias and other autoimmune diseases are typically treated by immune suppression with corticosteroids as well as corticosteroid-sparing agents if prolonged treatment of autoimmune cytopenias is required and/or in cases of refractory cytopenias.
Splenectomy is reserved as an option of last resort in the treatment of life-threatening refractory cytopenias and/or severe hypersplenia because of the high risk of recurrence of cytopenias and sepsis post-splenectomy in persons with ALPS.
Prevention of primary manifestations: Bone marrow (hematopoietic stem cell) transplantation (BMT/HSCT), the only curative treatment for ALPS, has to date mostly been performed on those with severe clinical phenotypes such as ALPS-FAS caused by biallelic pathogenic variants, those with severe and/or refractory autoimmune cytopenias, those with lymphoma, and those who have developed complications from (often long-term) immunosuppressive therapy.
Prevention of secondary complications: Vaccinations pre-splenectomy (with consideration of post-splenectomy boost vaccinations) and penicillin prophylaxis are strongly recommended for individuals who undergo splenectomy.
Surveillance: Clinical assessment and imaging and laboratory studies for manifestations of lymphoproliferation and autoimmunity; specialized imaging studies to detect malignant transformation.
Agents/circumstances to avoid: Splenectomy is discouraged as it typically does not lead to permanent remission of autoimmunity and is associated with increased risk of infection. Aspirin and other nonsteroidal anti-inflammatory drugs should be used with caution in individuals with immune thrombocytopenia as they can interfere with platelet function.
Evaluation of relatives at risk: If the pathogenic variant(s) have been identified in a family member with ALPS, it is appropriate to perform molecular genetic testing on at-risk relatives to allow for early diagnosis and treatment.
Pregnancy management: Assessment of the risks and benefits of treating a woman who has ALPS with corticosteroids, mycophenylate mofitil, or sirolimus during pregnancy must take into consideration the potential teratogenic risks to the fetus.
Genetic counseling.
Inheritance of ALPS-CASP10, most cases of ALPS-FAS, and some cases of ALPS-FASLG is autosomal dominant. Each child of an individual with autosomal dominant ALPS has a 50% chance of inheriting the pathogenic variant. Inheritance of most cases of ALPS-FASLG and severe ALPS associated with biallelic FAS pathogenic variants is autosomal recessive. The parents of an individual with autosomal recessive ALPS are likely to be heterozygotes, in which case each has one FAS pathogenic variant; these parents may have ALPS-related findings or may be clinically asymptomatic.
Prenatal testing for a pregnancy at increased risk is possible if the pathogenic variant(s) have been identified in an affected family member.
ALPS-FAS can also be the result of somatic mosaicism. Somatic pathogenic variants have not been reported in ALPS-FASLG or ALPS-CASP10 to date.
Diagnosis
Suggestive Findings
The diagnosis of autoimmune lymphoproliferative syndrome (ALPS) is based on a constellation of clinical findings, laboratory abnormalities, and identification of pathogenic variants in genes relevant for the tumor necrosis factor receptor superfamily member 6 (Fas) pathway of apoptosis.
ALPS should be suspected in individuals with combinations of the following [Bleesing 2003, Rieux-Laucat et al 2003]:
- Chronic non-malignant lymphoproliferation
- Chronic and/or recurrent lymphadenopathy
- Splenomegaly with/without hypersplenism
- Hepatomegaly
- Lymphocytic interstitial pneumonia (less common)
- Autoimmune disease
- Cytopenia, particularly combinations of autoimmune hemolytic anemia (AIHA), immune thrombocytopenia (ITP), and autoimmune neutropeniaNote: The combination of AIHA and ITP is often referred to as Evans syndrome.
- Other, including autoimmune hepatitis, autoimmune glomerulonephritis, autoimmune thyroiditis and (less commonly) uveitis and Guillain-Barré syndrome
- Lymphoma, both Hodgkin lymphoma and non-Hodgkin lymphoma
- Skin rashes, often but not exclusively of an urticarial nature
- Family history of ALPS or ALPS-like features
Establishing the Diagnosis
The diagnosis of ALPS is established in a proband who meets the clinical diagnostic criteria, which may include identification of a heterozygous pathogenic variant or biallelic pathogenic variants in one of the genes listed in Table 1.
A revised set of diagnostic criteria have been proposed [Oliveira et al 2010]:
- A definitive diagnosis of ALPS is based on the presence of both required criteria and one primary accessory criterion (see following).
- A probable diagnosis is based on the presence of both required criteria plus one secondary accessory criterion.
Required criteria
- Chronic (>6 months) non-malignant, noninfectious lymphadenopathy and/or splenomegaly
- Elevated α/β-DNT cells with normal or elevated lymphocyte counts
Primary accessory criteria
- Defective lymphocyte apoptosis (repeated at least once)
- Germline or somatic pathogenic variants in CASP10, FAS, or FASLG
Secondary accessory criteria
- Elevated levels of one of the following:
- Plasma soluble FASL
- Plasma interleukin-10
- Serum vitamin B12
- Plasma interleukin-18
- Typical immunohistologic findings as determined by an experienced hematopathologist
- Autoimmune cytopenias with elevated (polyclonal) immunoglobulin G levels
- Positive family history
Laboratory Findings
Although no specific laboratory abnormality alone is diagnostic of ALPS, the detection of the following facilitates the diagnosis [Bleesing 2003, Magerus-Chatinet et al 2009, Caminha et al 2010, Oliveira et al 2010, Rensing-Ehl et al 2013]:
- Defective Fas-mediated apoptosis in vitro
- T cells that express the alpha/beta T-cell receptor but lack both CD4 and CD8 (so-called alpha/beta double-negative T cells [α/β-DNT cells] in peripheral blood or tissue specimens). Detected by flow cytometric immunophenotyping, these terminally differentiated in vivo-activated T cells are rare in healthy individuals and other immune-mediated (lymphoproliferative) disorders; typically they constitute less than 2% of the lymphocyte pool.
- Increased levels of the ALPS-specific biomarkers: soluble IL-10, IL-18, FasL, and vitamin B12 in plasma/serum [Bowen et al 2012]
Secondary laboratory findings in ALPS [Lim et al 1998, Carter et al 2000, Bleesing et al 2001,Bleesing 2003, Bleesing 2005, Maric et al 2005, Magerus-Chatinet et al 2009, Caminha et al 2010, Oliveira et al 2010, Bowen et al 2012, Neven et al 2014]:
- Hematology
- Lymphocytosis, lymphopenia (primary or secondary in response to treatment)
- Coombs-positive hemolytic anemia
- Dyserythropoiesis
- Reticulocytosis
- Thrombocytopenia
- Neutropenia
- Eosinophilia
- Immunology
- Expansion of other lymphocyte subsets
- Gamma/delta-DNT cells
- CD8+/CD57+ T cells
- HLA-DR+ T cells
- CD5+ B cells
- Decreased numbers of CD4+/CD25+ T cells
- Decreased numbers of CD27+ B cells
- Elevated concentration of IL-10 and IL-18 in serum/plasma
- Elevated concentrations of IgG, IgA, and IgE; normal or decreased concentrations of IgM
- Autoantibodies (most often positive direct or indirect antiglobulin test, antiplatelet antibody, antineutrophil antibody, antiphospholipid antibody, antinuclear antibody, rheumatoid factor)
- Lymph node pathology (paracortical expansion with immunoblasts/plasma cells and DNT cells in interfollicular areas, florid follicular hyperplasia, progressive transformation of germinal centers [PTGC])
- Other
- Increased soluble CD25 (sIL-2R alpha), CD27, CD30, and Fas ligand (FasL)
- Monoclonal gammopathy
- Decreased antibody responses to polysaccharide antigens [Neven et al 2014]
- Chemistry
- Liver function abnormalities (in case of autoimmune hepatitis)
- Proteinuria (in case of glomerulonephritis)
- Elevated serum concentration of vitamin B12
Normal findings in (typical) ALPS
- Neutrophil function
- Complement factors concentrations and function
- In vitro proliferative responses of T cells (e.g., in response to common mitogens and antigens)
- NK-cell and cytotoxic T-lymphocyte (CTL) function; possibly decreased CTL activity in ALPS on the basis of defective FasL (i.e., ALPS-FASLG).
- Antibody responses to protein antigens (e.g., diphtheria, tetanus)
Note: (1) The abnormal and normal laboratory findings listed have been most reliably established for individuals with ALPS caused by either germline or somatic pathogenic variants in FAS. (2) Cell surface expression of Fas (CD95) can be normal, increased, or decreased and is in general not helpful in the diagnosis of ALPS. (3) When interpreting laboratory data of individuals with (suspected) ALPS, the influence of concurrent immunosuppressive agents at the time of testing needs to be considered.
Molecular Genetic Testing
Molecular genetic testing approaches can include serial single-gene testing, use of a multigene panel, and more comprehensive genomic testing.
Serial single-gene testing. Sequence analysis of the gene of interest is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
- For this disorder, it is recommended that FAS be tested first. See Figure 1 for algorithm.
- In the absence of a germline FAS pathogenic variant, FAS sequencing in sorted α/β-DNT cells to detect somatic pathogenic variants should be performed. If present, the diagnosis of ALPS-sFAS is established.
- If neither a germline nor a somatic FAS pathogenic variant is identified, CASP10 and FASLG should be tested next. The detection of germline pathogenic variants in either CASP10 or FASLG establishes the diagnosis of ALPS-CASP10 or ALPS-FASLG, respectively.
- A Fas-mediated apoptosis assay should be performed if germline pathogenic variants in CASP10 or FASLG are not identified (repeat if necessary, noting the influence of concomitant immunosuppressive therapy). If abnormal, the diagnosis of ALPS-U is established.
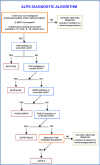
Figure 1.
One proposed algorithm for the diagnostic evaluation of an individual suspected of having ALPS
Notes: (1) Absence of a positive family history is suggestive of ALPS-sFAS. (2) Loss of heterozygosity of FAS pathogenic variants has been observed in blood cells. (3) FAS somatic pathogenic variants in selected cell populations, including α/β-DNT cells, produce a phenotype similar to that caused by FAS germline pathogenic variants. (4) The presence of elevated biomarkers has not been reliably established in CASP10 or FASLG-related ALPS. (5) Thus far, somatic pathogenic variants in FAS only have been reported to cause ALPS; however, it is theoretically possible that somatic pathogenic variants in CASP10 and FASLG may also be causative.
A multigene panel that includes CASP10, FAS, FASLG, and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in ALPS
Clinical Characteristics
Clinical Description
Autoimmune lymphoproliferative syndrome (ALPS) can be considered a prototypic disorder of defective lymphocyte homeostasis [Sneller et al 1992, Fisher et al 1995, Rieux-Laucat et al 1995].
The manifestations are lymphadenopathy, hepatosplenomegaly with or without hypersplenism, and autoimmune disease, mostly directed toward blood cells. In addition, the risk of lymphoma is increased.
Summary of Clinical Manifestations of ALPS
Lymphoproliferation of non-malignant lymphoid cells
- Lymphadenopathy
- Splenomegaly (+/- hypersplenism)
- Hepatomegaly
Autoimmunity
- Autoimmune hemolytic anemia
- Autoimmune thrombocytopenia
- Autoimmune neutropenia
- Glomerulonephritis
- Autoimmune hepatitis
- Guillain Barré syndrome
- Uveitis, iridocyclitis
- Other autoimmune disorders (in individual cases)
Neoplasia (including benign tumors)
- Lymphoma (Hodgkin and non-Hodgkin lymphoma)
- Carcinoma (thyroid, breast, skin, tongue, liver)
- Multiple neoplastic lesions (thyroid/breast adenomas, gliomas)
Other and/or infrequent findings
- Urticaria and other skin rashes
- Vasculitis
- Panniculitis
- Arthritis and arthralgia
- Recurrent oral ulcers
- Humoral immunodeficiency
- Pulmonary infiltrates
- Premature ovarian insufficiency
- Hydrops fetalis
- Organic brain syndrome (mental status changes, seizures, headaches)
The natural history of ALPS is not well understood. While non-malignant lymphoproliferative manifestations often regress or improve over time, autoimmunity appears to show no permanent remission with advancing age. Moreover, the risk for development of lymphoma likely is lifelong. Thus, in the absence of curative treatment, the overall prognosis for ALPS remains guarded, necessitating long-term clinical studies to better understand its natural history. Two publications have provided significant new insights into the features, complications, natural history, and prognosis of ALPS. These studies are subsequently referred to in this GeneReview as the "French cohort" and the "NIH cohort" [Neven et al 2011, Price et al 2014].
ALPS-FAS
ALPS-FAS is the most common and best-characterized type of ALPS. The following are the main consequences of perturbed lymphocyte homeostasis in ALPS-FAS.
Chronic non-malignant lymphoproliferation. Expansion of antigen-specific lymphocyte populations that are not eliminated through apoptosis leads to expansion of the lymphoid compartment, resulting in lymphadenopathy, splenomegaly, hypersplenism, and, less frequently, hepatomegaly. In most individuals with ALPS-FAS, this finding typically manifests in the first years of life. In some individuals, splenomegaly is the predominant or only manifestation of lymphoproliferation [Bleesing 2003, Rieux-Laucat et al 2003].
The median age of onset was three years in the French cohort and 2.7 years in the NIH cohort. Lymphadenopathy was present in 85% in the French cohort and 97% in the NIH cohort, while splenomegaly was present in 94% in the French cohort (with 73% showing hypersplenism) and 95% in the NIH cohort [Neven et al 2011, Price et al 2014].
In many individuals, lymphadenopathy tends to decrease early in the second decade, whereas splenomegaly often does not. Furthermore, long-term follow up in several individuals has shown that diminution of lymphadenopathy is not accompanied by significant changes in the overall expansion of lymphocyte subsets in peripheral blood [Bleesing et al 2001]. The lymphoproliferation waxes and wanes for reasons that are not entirely clear. Intercurrent viral and bacterial infections can influence lymphadenopathy, perhaps reflecting activation of other (intact) apoptosis pathways.
The overall prognosis of lymphoproliferation is relatively good; few individuals require long-term treatment with immunosuppressive agents to control lymphoproliferation [Bleesing 2003, Rieux-Laucat et al 2003, Neven et al 2011, Price et al 2014].
Laboratory findings of lymphoproliferation show expansion of most lymphocyte subsets including the pathognomonic α/β-DNT cells as well as other T- and B-cell subsets.
Autoimmunity, a common feature of ALPS, is often not present at the time of diagnosis or at the time of the most extensive lymphoproliferation. The reason for the delay in onset is unclear but may be related to age-dependent acquisition of secondary pathogenic factors that interact with defective Fas-mediated apoptosis. In many individuals with ALPS autoantibodies can be detected years before the appearance of clinical manifestations of autoimmune disease [Bleesing 2003, Rieux-Laucat et al 2003].
The French cohort and NIH cohort revealed that, in general, affected individuals with later disease onset often present with autoimmune disease, while younger individuals typically present with lymphoproliferative disease, followed by autoimmune disease, with a two- to three-year delay between lymphoproliferative disease onset and autoimmune disease onset. However, many affected individuals in both age groups presented with autoimmune disease as their first manifestation of ALPS [Neven et al 2011, Price et al 2014].
Although autoimmune manifestations can also wax and wane, current knowledge suggests that autoimmune disease poses a lifelong burden. In the NIH cohort, 37% of affected individuals were described as having a severe autoimmune disease phenotype (as determined by the presence of grade 3 or 4 cytopenias) within two years of disease onset [Price et al 2014].
Autoimmunity most often involves combinations of Coombs-positive hemolytic anemia and immune thrombocytopenia (together referred to as Evans syndrome); autoimmune neutropenia is less common. The observation of primary lymphopenia, contrasting with the typical presence of lymphocytosis, suggests the possibility of autoimmune lymphopenia (as seen in other autoimmune diseases).
The presence of Evans syndrome without significant lymphoproliferation can be consistent with ALPS, especially if α/β-DNT cells are present [Seif et al 2010].
Autoimmune cytopenias may be difficult to distinguish from the effects of concomitant hypersplenism; examination of blood smears for evidence of hemolysis and measurement of autoantibodies and the degree of reticulocytosis may help in establishing the distinction.
Additional autoimmune features can be found, often in patterns that appear to be family specific, suggesting the influence of other (background) genetic information [Rieux-Laucat et al 1999, Vaishnaw et al 1999, Kanegane et al 2003].
Laboratory findings include among others: autoantibodies detected by direct and indirect antiglobulin tests (Coombs' test), antiplatelet antibodies, antineutrophil antibodies, antinuclear antibodies (ANA), and antiphospholipid antibodies.
Lymphoma. Individuals with ALPS-FAS are at an increased risk for both Hodgkin and non-Hodgkin lymphoma, underscoring the role of Fas as a tumor-suppressor gene. Based on calculations in one study, the increased risk is 14-fold and 51-fold for non-Hodgkin lymphoma (NHL) and Hodgkin lymphoma (HL), respectively [Straus et al 2001].
More recently, updated risk calculations were provided through the French cohort and the NIH cohort. The French cohort provided a 15% cumulative risk of lymphoma before age 30 years. This represented seven cases of lymphoma (3 cases of HL and 4 cases of NHL) out of a total of 90 affected individuals [Neven et al 2011].
In the NIH cohort, 18 cases of lymphoma out of a total of 150 affected individuals were identified with a median age of detection of 18 years and a male-to-female ratio of 3.5 to 1. Sixteen (89%) of 18 cases were of B-cell origin. It was determined that 17/18 cases occurred in individuals with pathogenic variants affecting the death domain of FAS. Using published expected cases of HL and NHL in the general population, the 16 cases of B-cell lymphoma conferred a standardized incidence ratio of 149 for HL and 61 for NHL. These numbers are significantly different from those previously published by the NIH group [Straus et al 2001, Price et al 2014].
Lymphoma typically originates in B cells, but has been found in T cells as well, although much less frequently (2/18 cases in the NIH cohort) [Price et al 2014]. Lymphoma is not related to Epstein-Barr virus (EBV) infection (based on absence of EBV in tumor biopsies).
Current experience suggests that lymphomas can occur at any age in ALPS-FAS and do respond to conventional chemotherapeutic treatment. Individuals with other forms of ALPS may also be at an increased risk for lymphoma; however, further data are needed to provide a detailed risk assessment. Because of the frequent concomitant presence of benign (i.e., "typical") lymphadenopathy and splenomegaly, distinguishing a "good" node from a "bad" node is a diagnostic challenge. Important clues are B-type symptoms including fever, night sweats, itching, and weight loss. In addition, PET-based imaging may be helpful in distinguishing "good" from "bad" nodes on the basis of presumed higher metabolic activity of malignant lymphoid tissue [Rao et al 2006].
A number of studies have looked at associations between Fas and neoplasms, including somatic pathogenic variants in solid tumors, leukemias, and lymphomas. For further discussion, see Müschen et al [2002], Houston & O'Connell [2004], Poppema et al [2004], and Peter et al [2005].
ALPS-FAS Caused by Biallelic Pathogenic Variants
Chronic non-malignant lymphoproliferation. Individuals with homozygous or compound heterozygous FAS pathogenic variants often present with severe lymphoproliferation at or shortly after birth [Rieux-Laucat et al 1995, Le Deist et al 1996, Kasahara et al 1998, van der Burg et al 2000].
Autoimmunity. In several individuals reported, the delay between onset of autoimmunity and lymphoproliferation was minimal, while in others this was not the case. The rarity of and poor prognosis in ALPS-FAS resulting from biallelic pathogenic variants make it difficult to draw firm conclusions regarding autoimmunity in this type of ALPS [Rieux-Laucat et al 1995, Le Deist et al 1996, Kasahara et al 1998, van der Burg et al 2000].
Lymphoma. Because of the severity of ALPS-FAS caused by biallelic pathogenic variants, affected individuals typically succumb to lymphoproliferation and/or autoimmunity at an early age.
ALPS-sFAS
Somatic FAS pathogenic variants in selected cell populations (notably the α/β-DNT cells) have been identified in individuals with ALPS-sFAS. Individuals with somatic FAS pathogenic variants now constitute the second largest group of ALPS. Most of the clinical and laboratory features of ALPS-FAS are recapitulated in individuals with somatic FAS pathogenic variants including age of presentation, although lower incidence of splenectomy and lower lymphocyte counts have been reported in ALPS-sFAS and no cases of lymphoma have yet been published.
The population of α/β-DNT cells is expanded; however, as noted initially [Holzelova et al 2004, Rössler et al 2005, Magerus-Chatinet et al 2011], Fas-mediated apoptosis in vitro is typically not defective, although defective Fas-mediated apoptosis has been noted in some recently published cases [Dowdell et al 2010].
Pathogenesis of ALPS
The phenotype of ALPS results from defective apoptosis of lymphocytes mediated through the Fas/Fas ligand (FasL) pathway. This pathway normally limits the size of the lymphocyte compartment by eliminating/removing autoreactive lymphocytes; therefore, defects in this pathway lead to expansion of antigen-specific lymphocyte populations. Although Fas also appears to play a role in suppression of malignant transformation of lymphocytes, it remains to be firmly established whether this involves the Fas/FasL pathway in a similar way. It should be noted that the pathogenesis of ALPS remains an ongoing topic of research.
Somatic FAS pathogenic variants are of particular interest in understanding the pathogenesis of ALPS, for example, with regard to the observed delay between lymphoproliferation and autoimmunity: the somatic pathogenic variant is mostly confined to the α/β-DNT cells and typically not found (at least not in large proportion) in other lymphocyte subsets such as B cells. Perhaps this observation will help to characterize the impact of the FAS pathogenic variant relative to other potential pathogenic factors.
Genotype-Phenotype Correlations
ALPS-FAS. The clinical lymphoproliferative and autoimmune phenotype of ALPS is associated with pathogenic variants which affect any domain of Fas. Lymphomas, in contrast, seem thus far to be associated mostly with pathogenic variants affecting the intracellular domains of Fas, although independent confirmation is required [Straus et al 2001, Price et al 2014].
In the majority of affected individuals, heterozygous FAS pathogenic variants are associated with ALPS-FAS by the mechanism of dominant-negative interference; however, with certain pathogenic variants affecting extracellular domain, the proposed mechanism is haploinsufficiency. In the latter case, the ALPS clinical phenotype may be less severe, linked to less defective in vitro apoptosis [Kuehn et al 2011]. (For further discussion see Molecular Pathogenesis.)
ALPS-FASLG and ALPS-CASP10. Because of their rarity, genotype-phenotype correlations are not clearly established for FASLG and CASP10 pathogenic variants.
Penetrance
ALPS-FAS. A distinction needs to be made between the penetrance of the cellular phenotype (defective Fas-mediated apoptosis) and the penetrance of the clinical phenotype (i.e., ALPS).
Family studies to date suggest that penetrance for the defective Fas-mediated apoptosis cellular phenotype approximates 100% (i.e., every individual heterozygous for an inherited [germline] pathogenic variant has defective apoptosis) whereas the penetrance for the clinical phenotype is reduced because a significant proportion of relatives heterozygous for the pathogenic variant have no clinical findings of ALPS. In addition, other relatives have laboratory findings of ALPS (e.g., expansion of lymphocyte subsets and/or autoantibodies) without clinical evidence of either lymphoproliferation or autoimmunity [Infante et al 1998, Jackson et al 1999, Bleesing et al 2001].
The factors that determine the penetrance of clinical ALPS are not entirely understood. Penetrance appears to be determined by the location and type of pathogenic variant [Rieux-Laucat et al 1999, Le Deist 2004]. In initial studies, the highest penetrance (70%-90%) for the clinical phenotype occurred with missense variants affecting the intracellular domains (ICD), followed by variants leading to truncation of the ICDs. For pathogenic variants affecting the extracellular domains (ECD) the highest penetrance was estimated at approximately 30% [Jackson et al 1999].
In the French cohort ECD pathogenic variants had a penetrance of 52% (higher than previous data) and ICD pathogenic variants had a 63% penetrance (lower than previously reported). The penetrance of missense variants affecting the death domain (part of the ICD) was 73% [Jackson et al 1999, Neven et al 2011].
The reduced penetrance for ALPS in some families suggests that one or more additional pathogenic factors interact with defective Fas-mediated apoptosis. However, the high penetrance for the clinical phenotype in certain families associated with specific types of FAS pathogenic variants (e.g., missense variants affecting the death domain) casts doubt on that assumption, suggesting that under certain conditions a single defect in Fas-mediated apoptosis is sufficient to cause ALPS [Infante et al 1998, Jackson et al 1999, Le Deist 2004].
An observation that may shed more light on the issue of penetrance, particularly as it relates to pathogenic variants affecting intracellular vs extracellular domains (as well as on pathogenesis and natural history of ALPS): in a small subset of affected individuals, clinical disease appeared to develop as a consequence of both an inherited heterozygous (germline) FAS pathogenic variant and a somatic genetic event in the second FAS allele [Magerus-Chatinet et al 2011]. Analysis of α/β-DNT cells revealed that the second genetic event involved either a somatic missense or nonsense variant in the second FAS allele or loss of heterozygosity by telomeric uniparental disomy of chromosome 10. These observations were recently confirmed in a family with ALPS in which affected individuals had a heterozygous germline FAS start codon variant with somatic loss of heterozygosity [Hauck et al 2013].
Disease penetrance differs between males and females. In the French cohort, the likelihood of a male with a heterozygous germline FAS pathogenic variant developing ALPS was about 75%, compared to 51% for females. In the NIH cohort the likelihood of developing ALPS for males and females was 69% and 46%, respectively. The ratio of affected males to affected females was 2.2 (French cohort) and 1.6 (NIH cohort). Lastly, in the French cohort, the ratio of affected males to affected females increased from 2.2. to 2.9 if autoimmune disease was present and to 4.2 if autoimmune disease included autoimmune cytopenias [Neven et al 2011, Price et al 2014].
Anticipation
Anticipation has not been documented in ALPS.
Nomenclature
Table 2.
Revised Classification of ALPS
ALPS has also been referred to as Canale-Smith syndrome.
Prevalence
Nearly 500 patients with ALPS in more than 300 families have been reported worldwide with no racial or ethnic predilection. However, the true prevalence of ALPS is still unknown as many individuals are undiagnosed or misdiagnosed [Shah et al 2014].
Genetically Related (Allelic) Disorders
FAS, FASLG, and CASP10. No phenotype other than ALPS is currently known to be associated with germline or somatic pathogenic variants in these genes.
Differential Diagnosis
ALPS-like disorders. Pathogenic variants in genes inside (other than FAS, FASLG, and CASP10) and outside Fas/FasL pathway have been reported in association with clinical findings similar to ALPS. However, they either do not fulfill the current diagnostic criteria of ALPS [Oliveira et al 2010] or have other distinct phenotypes, and therefore are listed as ALPS-like disorders. These include:
- Dianzani autoimmune lymphoproliferative disease (DALD) (OMIM 605233). Individuals with DALD present with autoimmunity, lymphoproliferation, splenomegaly, and defective Fas function without expansion of DNT cells. A genetic basis for this disorder is suspected, but no associated genes have been identified to date [Oliveira et al 2010, Boggio et al 2014].
- Ras-associated autoimmune leukoproliferative disorder (RALD) (OMIM 614470). RALD results from somatic gain-of-function variants in NRAS and KRAS, which are present only in blood cells. Similar to ALPS, RALD is a primary immunodeficiency disorder of defective apoptosis. Abnormal apoptosis in RALD results from a defect in a secondary apoptosis pathway, rather than the FAS-mediated apoptosis pathway in ALPS. RALD is characterized by mild peripheral lymphadenopathy, (hepato)splenomegaly, and autoimmunity. Recurrent respiratory tract infections are reported in some affected individuals. Because of the rarity of this condition, the risk for lymphoma is not known [Chun et al 2002, Oliveira et al 2007, Niemela et al 2011, Takagi et al 2011, Lanzarotti et al 2014].
- Caspase-8 deficiency state (CEDS) (OMIM 607271). CEDS is a rare, autosomal recessive immunodeficiency syndrome resulting in lymphadenopathy, splenomegaly, marginal elevation of double-negative T cells, and defective FAS-mediated apoptosis, in addition to frequent bacterial and viral infections secondary to defective activation of T and B lymphocytes and NK cells. Autoimmunity has not been reported to date in individuals with CEDS. The risk of lymphoma in people with CASP8 pathogenic variants is not known, nor has the full spectrum of the disease been elucidated given the rarity of individuals with known pathogenic variants [Chun et al 2002, Oliveira 2013].
- Fas-associated via death domain (FADD) deficiency (OMIM 613759). FADD deficiency is a rare, autosomal recessive primary immunodeficiency syndrome characterized by severe bacterial and viral infections, congenital heart defects, and recurrent episodes of fever, liver dysfunction, and seizures. Biochemical markers are consistent with ALPS, but the affected individuals described to date do not have the characteristic clinical features of ALPS, including lymphadenopathy and splenomegaly [Bolze et al 2010, Oliveira 2013, Savic et al 2015]. FADD deficiency is caused by biallelic pathogenic variants in FADD.
- Common variable immunodeficiency 9 (PRKCD deficiency) (OMIM 615559). PRKCD deficiency is a rare, autosomal recessive primary immunodeficiency characterized by recurrent infections, lymphadenopathy, (hepato)splenomegaly, autoimmunity, and NK cell dysfunction. The full spectrum of the disease has not been elucidated given the small number of individuals with known pathogenic variants in PRKCD [Kuehn et al 2013, Salzer et al 2013].
- CTLA4 haploinsufficiency with autoimmune infiltration (CHAI) (OMIM 616100). CHAI is an autosomal dominant immune dysregulation syndrome with incomplete penetrance that is characterized by recurrent infections, autoimmune thrombocytopenias, CD4+ T-cell lymphopenia, B cell abnormalities, hypogammaglobulinemia, and abnormal lymphocytic infiltration of nonlymphoid organs such as brain, lungs and gastrointestinal tract. Additional features include diffuse lymphadenopathy, hepatosplenomegaly, and EBV-associated Hodgkin lymphoma [Kuehn et al 2014, Schubert et al 2014]. CHAI is caused by heterozygous pathogenic loss-of-function variants in CTLA4.
Within the differential diagnosis for ALPS are other immunodeficiency disorders characterized or complicated by lymphoproliferation, autoimmune disease, and lymphoma. These include the following:
- Common variable immune deficiency (CVID) has an estimated incidence of one in 50,000 and occurs equally in males and females. CVID is characterized by humoral immune deficiency with onset after age 24 months and usually in young adulthood, resulting in increased susceptibility to infections and diminished responses to protein and polysaccharide vaccines. Although multiple genes have been associated with CVID (see OMIM PS607594), the genetic etiology of CVID in most affected individuals is currently unknown. From a clinical and immunologic standpoint, CVID can be roughly classified into two groups, depending on the presence or absence of mature B-cells in peripheral blood. Individuals with CVID with B-cells (but absent or decreased memory B-cells) are at an increased risk for autoimmune disease that often targets blood cells and for chronic lymphoproliferation including lymphadenopathy, splenomegaly, and lymphoma [Warnatz et al 2002, Piqueras et al 2003]. CVID with present B cells should be regarded in the differential diagnosis of ALPS, while the variant characterized by low or absent B cells and generally low serum concentrations of immunoglobulins should not.The overlap between ALPS and CVID is also illustrated by the report of two individuals with CVID who were found to have heterozygous pathogenic variants in CASP8 [Chun et al 2002, Rensing-Ehl et al 2010] and multiple individuals from unrelated families with CVID and enteropathy or autoimmunity who were found to have heterozygous pathogenic variants in CTLA4 [Schubert et al 2014]. Biomarkers have also confirmed the overlap between ALPS and CVID [Roberts et al 2013].
- Hyper IgM (HIGM) syndrome. Several non-X-linked forms of hyper IgM syndrome have now been identified. In varying degrees, they share features with the X-linked form (see X-Linked Hyper IgM Syndrome), caused by pathogenic variants in CD40LG. Shared features include recurrent bacterial infections such as otitis media, sinusitis, and pneumonias. Autoimmune hematologic disorders including neutropenia, thrombocytopenia, and hemolytic anemia are also found. Other complications may include lymphomas and other malignancies as well as gastrointestinal complications. Serum concentration of IgM is elevated while other immunoglobulin levels are normal; specific antibody responses are defective. In contrast to HIGM1, T-cell function in ALPS is typically within normal limits, reflected in an absence of opportunistic infections.HIGM2 (OMIM 605258) is caused by pathogenic variants in AICDA, encoding single-stranded DNA cytosine deaminase. Inheritance is usually autosomal recessive, but in rare cases autosomal dominant [Revy et al 2000, Lee et al 2005]. Recurrent bacterial, respiratory, and gastrointestinal infections are typical; opportunistic infections are rare. Lymphoid hyperplasia, seen in ALPS, has been reported in HIGM2 [Revy et al 2000, Lee et al 2005]. AICDA pathogenic variants typically affect only B-cell differentiation.HIGM3, HIGM4, and HIGM5 are other forms of non-X-linked hyper IgM syndrome (OMIM PS308230). Their inclusion in the differential diagnosis of ALPS is less clear on the basis of known clinical presentation and inheritance pattern [Ferrari et al 2001, Imai et al 2003].
- X-linked lymphoproliferative disease (XLP) caused by hemizygous pathogenic variants in SH2D1A is associated with an inappropriate immune response to Epstein-Barr virus (EBV) infection resulting in unusually severe and often fatal infectious mononucleosis, dysgammaglobulinemia, and/or lymphoproliferative disorders (typically of B-cell origin). Clinical manifestations of XLP vary, even among affected family members. The most common presentation is a near-fatal or fatal EBV infection associated with an unregulated and exaggerated immune response with widespread proliferation of cytotoxic T cells, EBV-infected B cells, and macrophages. Mortality is greater than 90%. In approximately one third of males with XLP, hypogammaglobulinemia of one or more immunoglobulin subclasses is diagnosed prior to EBV infection or in rare survivors of EBV infection. The prognosis for males with this phenotype is more favorable if they are managed with regular intravenous immune globulin (IVIG) administration. Lymphomas or other lymphoproliferative diseases occur in approximately one third of males with XLP, some of whom have hypogammaglobulinemia or have survived an initial EBV infection. The lymphomas seen in individuals with XLP are typically high-grade B-cell lymphomas (non-Hodgkin type), are often extranodal, and particularly involve the intestine. Demonstration of defective T-cell receptor restimulation apoptosis in persons with XLP suggests that altered lymphocyte homeostasis affects disease pathogenesis as well [Snow et al 2008]. Allogeneic bone marrow transplantation (BMT) is the only curative therapy for XLP. Average life expectancy without curative BMT has been estimated at less than ten years. Note: XLP caused by pathogenic variants in XIAP has not been associated with lymphoproliferation or lymphoma to date. See Lymphoproliferative Disease, X-Linked.
- WAS-related disorders, which include Wiskott-Aldrich syndrome, X-linked thrombocytopenia (XLT), and X-linked congenital neutropenia (XLN), are a spectrum of disorders of hematopoietic cells, with predominant defects of platelets and lymphocytes caused by mutation of WAS. WAS-related disorders usually present in infancy. Affected males have thrombocytopenia with intermittent mucosal bleeding, bloody diarrhea, and intermittent or chronic petechiae and purpura; eczema; and recurrent bacterial and viral infections, particularly recurrent ear infections. At least 40% of those who survive the early complications develop one or more autoimmune conditions including hemolytic anemia, immune thrombocytopenic purpura, immune-mediated neutropenia, arthritis, vasculitis of small and large vessels, and immune-mediated damage to the kidneys and liver. Individuals with a WAS-related disorder, particularly those who have been exposed to EBV, are at increased risk of developing lymphomas, which often occur in unusual, extranodal locations such as the brain, lung, or gastrointestinal tract. Males with XLT have thrombocytopenia with small platelets; other complications of Wiskott-Aldrich syndrome, including eczema and immune dysfunction, are mild or absent. See WAS-Related Disorders.
- STAT3 gain-of-function variants causing autoimmune disease can result in clinical features such as lymphadenopathy, elevated α/β-DNT cells, and autoimmune cytopenia, resembling ALPS. Other findings include recurrent infections, short stature, and multiorgan autoimmunity. In contrast, loss-of-function variants in STAT3 are associated with immunodeficiency and hyper-IgE recurrent infection syndrome [Milner et al 2015].
- Lymphoma without other manifestations of ALPS has been observed in families with ALPS-FAS. Thus, both B-cell and T-cell lymphoma should be considered in the differential diagnosis of ALPS [van der Werff ten Bosch et al 1999, Poppema et al 2004].
Management
Evaluations Following Initial Diagnosis
To determine the presence and extent of disease and needs in an individual diagnosed with autoimmune lymphoproliferative syndrome (ALPS), the following evaluations are recommended:
- Complete blood counts and flow cytometric immunophenotyping of lymphocytes, especially with regard to α/β-DNT cells, in combination with physical examination and imaging studies to assess lymphadenopathy and hepatosplenomegaly
- If significant lymphadenopathy is present, more extensive diagnostic procedures to detect lymphoma, especially if constitutional symptoms (e.g., fever, night sweats, weight loss) are present
- Measurement of autoantibodies to assess for autoimmunity
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
In the absence of curative treatment, current management is focused on the following:
- Monitoring for and treatment of lymphoproliferation, hypersplensim, and lymphomas
- Management of cytopenias and other autoimmune diseases [Madkaikar et al 2011, Rao & Oliveira 2011, Teachey 2012]
Monitoring for and treatment of lymphoproliferation and hypersplenism
- Manifestations of lymphoproliferation require close clinical observation, as well as serial CT and PET scans every two to three years.
- Biopsy is indicated whenever there is a clinical suspicion of lymphoma.
- Corticosteroids and immunosuppressive drugs do not decrease lymphadenopathy long term in individuals with ALPS, and are generally reserved for severe complications of lymphoproliferation (e.g., airway obstruction, significant hypersplenism associated with splenomegaly) and/or autoimmune manifestations.
- Experience with sirolimus suggests that it is the preferred agent in treating lymphoproliferation in a more sustained manner, including for maintenance of remission after initial treatment followed by a period of discontinued use [Teachey et al 2009, Bride et al 2016]. However, sirolimus is not without side effects.
- In severe cases, more potent (lympho-depleting) agents may be required to sufficiently control lymphoproliferative manifestations. Agents include cyclophosphamide, antithymocyte globulin (ATG), and select monoclonal antibodies such as alemtuzumab (Campath®).
Treatment of lymphoma is according to conventional protocols. The presence of defective Fas-mediated apoptosis does not appear to hinder the response to chemotherapeutic agents or radiation.
Treatment of cytopenias and other autoimmune diseases
- Autoimmune cytopenias are typically treated by immune suppression with corticosteroids as well as corticosteroid-sparing agents, if prolonged treatment of autoimmune cytopenias is required and/or in cases of refractory cytopenias.
- Recent data from a multi-institutional study suggest that sirolimus should be considered as a first-line – corticosteroid-sparing – agent [Bride et al 2016]. Sirolimus requires monitoring for drug levels and toxic side effects.
- Mycophenolate mofetil (MMF) can be used in cases when a less immunosuppressive drug (e.g., compared with sirolimus) seems sufficient, as steroids are tapered. In addition, if drug level and toxic side effects cannot be adequately monitored, MMF could be used as a first-line agent.
- Individuals with severe autoimmune hemolytic anemia may benefit from IVIG in combination with corticosteroids.
- Rituximab has been used successfully in the treatment of refractory cytopenias in ALPS. However, because of its immune toxicity, its use is generally avoided until other immunosuppression therapies have failed [Rao et al 2009, Rao & Oliveira 2011].
- Splenectomy is reserved as an option of last resort in the treatment of life-threatening refractory cytopenias and/or severe hypersplenia because of the high risk of recurrence of cytopenias and sepsis post-splenectomy in those with ALPS [Rao & Oliveira 2011, Price et al 2014]. Given the known risks associated with splenectomy, patients requiring this last-resort approach should be considered for curative bone marrow transplantation.
- Individuals with isolated chronic neutropenia may improve on low-dose G-CSF [Rao & Oliveira 2011]. In addition, use of a thrombopoietin (TPO) receptor agonist should be considered for isolated immune thrombocytopenia (ITP).
Prevention of Primary Manifestations
Bone marrow (hematopoietic stem cell) transplantation (BMT/HSCT) is currently the only curative treatment for ALPS. Because of the risks associated with BMT, it has so far been performed mostly in individuals with ALPS with severe clinical phenotypes, such as those with homozygous or compound heterozygous pathogenic variants in FAS, those with severe and/or refractory autoimmune cytopenias, those with lymphoma, and those who have developed complications from (often long-term) immunosuppressive therapy. It is likely, however, that individuals with undiagnosed forms of ALPS, including ALPS-FAS, have been transplanted.
Successful (reported) BMT in several individuals indicates that defective Fas-mediated apoptosis does not pose a barrier to this treatment option [Benkerrou et al 1997, Sleight et al 1998, Dowdell et al 2010].
Prevention of Secondary Complications
Vaccinations pre-splenectomy (with consideration of post-splenectomy boost vaccinations) and penicillin prophylaxis are strongly recommended for individuals who undergo splenectomy.
Surveillance
Clinical assessment, imaging, and laboratory studies outlined in Evaluations Following Initial Diagnosis can be used in surveillance for manifestations of lymphoproliferation and autoimmunity.
Specialized imaging studies such as combined PET and CT scanning in combination with clinical and laboratory surveillance may be helpful in detection of malignant transformation, keeping in mind that PET/CT scanning is often abnormal in ALPS, such that distinguishing between "typical" ALPS findings and a new malignancy (lymphoma) can be difficult [Rao & Oliveira 2011]. Because of this, careful consideration of imaging modalities that expose the patient to radiation is warranted.
Agents/Circumstances to Avoid
Splenectomy to control autoimmune cytopenias and/or massive splenomagaly is discouraged because it typically does not lead to permanent remission of autoimmunity and may be associated with an increased risk for infections. The two recent cohort studies reveal clear-cut consequences of splenectomy. In the French cohort, nine (30%) of 30 affected individuals who underwent splenectomy suffered 17 cases of severe invasive bacterial infection with four deaths after splenectomy; in the NIH cohort, 27 (41%) of 66 affected individuals suffered one or more episodes of sepsis with seven deaths. Of note: Antimicrobial prophylaxis and appropriate vaccinations did not prevent the majority of episodes of sepsis, although poor compliance was found to be a risk factor in the French cohort [Neven et al 2011, Price et al 2014].
The use of over-the-counter medications such as aspirin and other nonsteroidal anti-inflammatory drugs should be discussed with a physician as some of these medications can interfere with platelet function.
Evaluation of Relatives at Risk
It is appropriate to perform molecular genetic testing on relatives at risk for ALPS-FAS, ALPS-FASLG, or ALPS-CASP10 if the pathogenic variant has been identified in the proband.
Relatives who have the family-specific pathogenic variant should:
- Be advised of their increased risk for ALPS if the type and location of the ALPS-related pathogenic variant is predicted to have a high penetrance for clinical ALPS;
- Undergo ALPS-specific evaluations at initial diagnosis (e.g., enumeration of α/β-DNT cells, detection of autoantibodies, IL-10/soluble FasL measurement) (see Evaluations Following Initial Diagnosis);
- Be advised that ALPS-specific evaluations or other assessments may need to be repeated at regular intervals, particularly if the family member is young and/or if new health-related issues consistent with ALPS or ALPS-related complications (e.g., lymphoma) become apparent (see Surveillance).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
In addition to the risks and benefits to a woman with ALPS associated with treatment with corticosteroids, mycophenylate mofitil, or sirolimus during pregnancy, the potential teratogenic risks of these exposures to the fetus must also be weighed.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Inheritance of ALPS-CASP10, most cases of ALPS-FAS, and some cases of ALPS-FASLG is autosomal dominant. Inheritance of most cases of ALPS-FASLG and severe ALPS associated with biallelic FAS pathogenic variants is autosomal recessive.
ALPS-FAS can also be the result of somatic mosaicism. Somatic pathogenic variants have not been reported in ALPS-FASLG or ALPS-CASP10 to date.
Autosomal Dominant ALPS – Risk to Family Members
Parents of a proband
- Most individuals diagnosed with ALPS-FAS have a parent who has a FAS pathogenic variant. Individuals who are heterozygous for a FAS pathogenic variant all have defective Fas-mediated apoptosis but may have no clinical findings of ALPS (see Penetrance).
- An insufficient number of cases of ALPS-FASLG and ALPS-CASP10 are available to determine the likelihood that the FASLG or CASP10 pathogenic variant was inherited from a parent.
- A proband with ALPS-FAS, ALPS-FASLG, or ALPS-CASP10 may have the disorder as the result of a de novo germline pathogenic variant. The proportion of cases caused by a de novo FAS germline pathogenic variant is small (the proportion of cases caused by a de novo FASLG or CASP10 is unknown).
- Recommendations for the evaluation of parents of a proband with a possible de novo pathogenic variant (i.e., neither parent is known to be affected with ALPS) include molecular genetic testing for the pathogenic variant identified in the proband.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, possible explanations include a de novo somatic or germline pathogenic variant in the proband or germline mosaicism in a parent.
- Although most individuals diagnosed with ALPS have a parent with a FAS pathogenic variant, the family history may appear to be negative because of reduced penetrance of the clinical symptoms of ALPS (as opposed to the nearly complete penetrance of the cellular phenotype in individuals with a FAS pathogenic variant), failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, molecular genetic testing is the most accurate means of determining the genetic status of at-risk individuals.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents.
- If a parent of the proband has a FAS, FASLG, or CASP10 pathogenic variant, each sib has a 50% chance of inheriting the variant. The risk of developing ALPS-related complications, however, depends on the nature of the variant, as well as the presence of other as-yet incompletely understood genetic or environmental factors.
- If the FAS, FASLG, or CASP10 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to sibs is approximately 1% because of the theoretic possibility of parental germline mosaicism.
Offspring of a proband
- Each child of an individual with ALPS-FAS, ALPS-FASLG, or ALPS-CASP10 has a 50% chance of inheriting the FAS, FASLG, or CASP10 pathogenic variant.
- The risk to a child who has inherited the pathogenic variant of developing ALPS-related complications depends on the nature of the pathogenic variant as well as the presence of other as-yet incompletely understood genetic or environmental factors (see Penetrance).
Other family members. The risk to other family members depends on the genetic status of the proband's parents: if a parent has a FAS, FASLG, or CASP10 pathogenic variant, his or her family members may have inherited the same variant and are potentially at some increased risk of developing ALPS-related complications.
Autosomal Recessive ALPS – Risk to Family Members
Parents of a proband
- The parents of a child with ALPS-FAS or ALPS-FASLG resulting from biallelic pathogenic variants are likely to be heterozygotes, in which case each parent would have one FAS or FASLG pathogenic variant.
- Heterozygotes may present with ALPS-related findings or may be clinically asymptomatic.
Sibs of a proband
- At conception, each sib of a child with ALPS-FAS or ALPS-FASLG resulting from biallelic pathogenic variants has:
- An overall 75% chance of having one or two FAS/FASLG pathogenic variants;
- A 25% chance of inheriting two FAS/FASLG pathogenic variants, which would most likely result in a severe ALPS phenotype;
- A 50% chance of inheriting a single FAS/FASLG pathogenic variant, which could result in clinical manifestations of ALPS;
- A 25% chance of inheriting one normal FAS/FASLG allele from each parent and having no clinical manifestations of ALPS.
- Heterozygotes may present with ALPS-related symptoms or may be clinically asymptomatic.
Offspring of a proband. Individuals with ALPS resulting from biallelic pathogenic variants are more likely to die at an early age and thus are not as likely to reproduce.
Other family members. If a parent of the proband has an ALPS-related pathogenic variant, his/her sibs are at a 50% risk of having the variant.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Testing of at-risk asymptomatic family members for FAS, FASLG, or CASP10 pathogenic variants is possible once the variant(s) are identified in the proband. Although the factors that determine the penetrance of clinical ALPS are not entirely understood, penetrance appears to be determined by the location and type of variant. Results of testing of at-risk asymptomatic family members can reduce morbidity and mortality through early diagnosis and treatment and may be helpful in predicting phenotype.
Molecular genetic testing of asymptomatic individuals should in general be undertaken following thorough genetic counseling and assessment of family-specific risks.
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
- Prior to pregnancy, affected women should be advised about the teratogenic risks associated with medications used to treat ALPS.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Once the FAS, FASLG, or CASP10 pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for ALPS are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Institute of Allergy and Infectious Diseases (NIAID)NIAID Office of Communications and Government Relations6610 Rockledge DriveMSC 6612Bethesda MD 20892-6612Phone: 866-284-4107 (toll-free); 301-496-5717; 800-877-8339 (toll-free TDD)Fax: 301-402-3573Email: ocpostoffice@niaid.nih.gov
- American Autoimmune Related Diseases Association, Inc. (AARDA)22100 Gratiot AvenueEast Detroit MI 48021Phone: 800-598-4668 (toll-free); 586-776-3900Fax: 586-776-3903Email: aarda@aarda.org
- ImmUnity CanadaCanadaPhone: 250-381-7134; 877 -607-2476Email: info@immunitycanada.org
- Jeffrey Modell Foundation/National Primary Immunodeficiency Resource CenterEmail: info@jmfworld.org
- European Society for Immunodeficiencies (ESID) RegistryEmail: esid-registry@uniklinik-freiburg.de
- United States Immunodeficiency Network (USIDNET) RegistryEmail: contact@usidnet.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Autoimmune Lymphoproliferative Syndrome: Genes and Databases
Table B.
OMIM Entries for Autoimmune Lymphoproliferative Syndrome (View All in OMIM)
Molecular Pathogenesis
Autoimmune lymphoproliferative syndrome (ALPS) can be considered a prototypic disorder of defective lymphocyte homeostasis [Sneller et al 1992, Fisher et al 1995, Rieux-Laucat et al 1995]. Although it appears that the full clinical spectrum of ALPS may depend on the interplay of several pathogenic factors, defective activation-induced cell death (also known as apoptosis or cellular suicide) through the Fas/FasL pathway is central in the etiology of ALPS [Lenardo et al 1999]. (Note: Bleesing [2002], Figure 1 diagrams this process; login or purchase required.)
CASP10
Gene structure. CASP10 comprises 11 exons and spans approximately 48 kb [Hadano et al 2001]. For a detailed summary of gene and protein information, see Table A, Gene. There are two isoforms of CASP10 transcripts. The CASP10L isoform encodes an additional 43 amino acids at the end of the prodomain, but its C terminus is the same as the short CASP10 isoform. The two isoforms are expressed equally.
Pathogenic variants. To date, four CASP10 pathogenic variants have been reported in individuals with ALPS [Wang et al 1999, Zhu et al 2006, Cerutti et al 2007] (see Table 3). Two missense variants, p.Leu285Phe and p.Ile406Leu, were identified in one and two kindreds, respectively, with ALPS-CASP10 characterized by abnormal lymphocyte and dendritic cell homeostasis and immune regulatory defects [Wang et al 1999, Zhu et al 2006].
Cerutti et al [2007] described two patients who demonstrated coinheritance of both a FAS and a CASP10 pathogenic variant. FAS expression and CASP10 activity were decreased in both patients. Additionally, a common variant of CASP10 (p.Thr446Cys) originally thought to be benign [Wang et al 1999] has been associated with ALPS [Zhu et al 2006].
Table 3.
Selected CASP10 Pathogenic Variants
Normal gene product. The physiologic function of caspase-10 is poorly understood. Gene transfection assays verified its function as a death-inducing caspase [Chaudhary et al 1997, Pan et al 1997, Schneider et al 1997, Vincenz & Dixit 1997]. Moreover, Wang et al [2001] showed that caspase-10 can function independently of caspase-8 in initiating Fas and tumor necrosis factor-related apoptosis.
Abnormal gene product. Pathogenic variants result in decreased caspase activity and dominantly interfere with death receptor-induced apoptosis, particularly with that stimulated by FasL and TRAIL.
FAS
Gene structure. FAS comprises nine exons. Exons 1 and 2 encode a signal sequence that, upon trafficking of the Fas protein to the cell surface, is cleaved off. Exons 3, 4, and 5 encode three extracellular cysteine-rich domains (CRD). Exon 6 encodes the transmembrane domain. The intracellular domains of Fas are encoded by exons 7-9, with exon 9 representing the death domain that interacts with the intracellular, apoptosis-inducing signal transduction pathway [Jackson et al 1999]. Genomic DNA of FAS spans approximately 25 kb. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. To date, more than 130 pathogenic variants have been reported, including missense and nonsense variants, splicing defects, small deletions/insertions, gross deletions, and complex deletion/duplications. Many of these pathogenic variants are loss-of-function alleles.
The discovery of individuals with ALPS with somatic pathogenic variants in FAS may offer new insights as the presence of pathogenic variants in some, but not all, lymphocyte subsets could allow dissection of the molecular mechanisms of ALPS in a manner that cannot be achieved in individuals with germline variants in FAS.
Normal gene product. FAS encodes a 16-amino acid signal sequence, followed by a mature protein of 319 amino acids with a single transmembrane domain and a molecular mass of approximately 36 kd.
The protein encoded by FAS is a member of the TNF-receptor superfamily and contains a death domain; it has been shown to play a central role in the physiologic regulation of programmed cell death. The interaction of Fas with its ligand allows the formation of a death-inducing signaling complex that includes Fas-associated death domain protein (FADD), caspase-8, and caspase-10. The autoproteolytic processing of the inductor caspases in the complex triggers a downstream effector caspase cascade, leading to apoptosis. Fas has also been shown to activate NF-kappaβ, MAPK3/ERK1, and MAPK8/JNK, leading to the transduction of proliferating signals in normal diploid fibroblast and T cells.
Abnormal gene product. Dominant-negative interference by abnormal Fas chains has been demonstrated for heterozygous pathogenic variants in the death domain, which lead to defective Fas-mediated apoptosis in many cases of ALPS-FAS [Jackson et al 1999, Martin et al 1999]. Fas and FasL form homotrimers; therefore, normal Fas trimers (consisting of 3 normal proteins) occur in only one out of eight trimers, assuming equal amounts of mutated and wild type Fas protein [Fisher et al 1995, Jackson et al 1999].
Extracellular heterozygous pathogenic variants affecting the FasL-binding domain (CRD2 and CRD3) are also associated with dominant-negative interference because Fas proteins self-associate into trimers prior to FasL interaction [Siegel et al 2000]. For other extracellular heterozygous pathogenic variants, including variants that affect the domain of the protein that regulates self-association into trimers, defective apoptosis can be explained by interference of truncated and/or soluble fragments of mutated Fas, or by haploinsufficiency, in which the total amount of Fas generated is below a threshold needed for physiologic induction of apoptosis [Roesler et al 2005, Kuehn et al 2011].
In individuals with homozygous or compound heterozygous pathogenic variants, defective Fas-mediated apoptosis can be explained by loss of function [van der Burg et al 2000]. In contrast to those with heterozygous pathogenic variants, these individuals display absent or reduced surface expression of Fas on lymphocytes.
FASLG
Gene structure. FASLG (FASL) spans approximately 8 kb and comprises four exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. See Table 4. To date, fewer than ten pathogenic alleles have been reported in FASLG in association with an ALPS phenotype.
Table 4.
Selected FASLG Pathogenic Variants
Normal gene product. The FASLG cDNA encodes a protein of 281 amino acids. Fas ligand (FasL) is a type II transmembrane protein that belongs to the tumor necrosis factor family. It is expressed in activated splenocytes and thymocytes, consistent with its involvement in T cell-mediated cytotoxicity and in several non-lymphoid tissues (e.g., testis, liver, lung, ovary, heart), where its function is unclear.
Abnormal gene product. The individual with the homozygous pathogenic missense variant (p.Ala247Glu) showed decreased Fas-mediated cell death and Fas-dependent cytotoxicity [Del-Rey et al 2006]. The heterozygous p.Arg156Gly pathogenic variant affects the extracellular Fas-binding region of FASL. The variant produces a dominant interfering FasL protein that binds to wild type FasL, preventing Fas-mediated apoptosis [Bi et al 2007].
References
Literature Cited
- Benkerrou M, Le Deist F, de Villartay JP, Caillat-Zucman S, Rieux-Laucat F, Jabado N, Cavazzana-Calvo M, Fischer A. Correction of Fas (CD95) deficiency by haploidentical bone marrow transplantation. Eur J Immunol. 1997;27:2043–7. [PubMed: 9295043]
- Bi LL, Pan G, Atkinson TP, Zheng L, Dale JK, Makris C, Reddy V, McDonald JM, Siegel RM, Puck JM, Lenardo MJ, Straus SE. Dominant inhibition of Fas ligand-mediated apoptosis due to a heterozygous mutation associated with autoimmune lymphoproliferative syndrome type Ib. BMC Med Genet. 2007;8:41. [PMC free article: PMC1931585] [PubMed: 17605793]
- Bleesing JJ. Autoimmune lymphoproliferative syndrome (ALPS). Curr Pharm Des. 2003;9:265–78. [PubMed: 12570831]
- Bleesing JJ. Sorting out the causes of ALPS. J Pediatr. 2005;147:571–4. [PubMed: 16291343]
- Bleesing JJ, Brown MR, Straus SE, Dale JK, Siegel RM, Johnson M, Lenardo MJ, Puck JM, Fleisher TA. Immunophenotypic profiles in families with autoimmune lymphoproliferative syndrome. Blood. 2001;98:2466–73. [PubMed: 11588044]
- Bleesing JJH. Autoimmune lymphoproliferative syndrome: a genetic disorder of abnormal lymphocyte apoptosis. Immunol Allergy Clin North Am. 2002;22:339–55.
- Boggio E, Clemente N, Mondino A, Cappellano G, Orilieri E, Gigliotti CL, Toth E, Ramenghi U, Dianzani U, Chiocchetti A. IL-17 protects T cells from apoptosis and contributes to development of ALPS-like phenotypes. Blood. 2014;123:1178–86. [PubMed: 24363402]
- Bolze A, Byun M, McDonald D, Morgan NV, Abhyankar A, Premkumar L, Puel A, Bacon CM, Rieux-Laucat F, Pang K, Britland A, Abel L, Cant A, Maher ER, Riedl SJ, Hambleton S, Casanova JL. Whole-exome-sequencing-based discovery of human FADD deficiency. Am J Hum Genet. 2010;87:873–81. [PMC free article: PMC2997374] [PubMed: 21109225]
- Bowen RA, Dowdell KC, Dale JK, Drake SK, Fleisher TA, Hortin GL, Remaley AT, Nexo E, Rao VK. Elevated vitamin B12 levels in autoimmune lymphoproliferative syndrome attributable to elevated haptocorrin in lymphocytes. Clin Biochem. 2012;45:490–2. [PMC free article: PMC3307947] [PubMed: 22306884]
- Bride KL, Vincent T, Smith-Whitley K, Lambert MP, Bleesing JJ, Seif AE, Manno CS, Casper J, Grupp SA, Teachey DT. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial. Blood. 2016;127:17–28. [PMC free article: PMC4705607] [PubMed: 26504182]
- Caminha I, Fleisher TA, Hornung RL, Dale JK, Niemela JE, Price S, Davis J, Perkins K, Dowdell KC, Brown MR, Rao VK, Oliveira JB. Using biomarkers to predict the presence of FAS mutations in patients with features of the autoimmune lymphoproliferative syndrome. J Allergy Clin Immunol. 2010;125:946–9.e6. [PMC free article: PMC3412519] [PubMed: 20227752]
- Carter LB, Procter JL, Dale JK, Straus SE, Cantilena CC. Description of serologic features in autoimmune lymphoproliferative syndrome. Transfusion. 2000;40:943–8. [PubMed: 10960521]
- Cerutti E, Campagnoli MF, Ferretti M, Garelli E, Crescenzio N, Rosolen A, Chiocchetti A, Lenardo MJ, Ramenghi U, Dianzani U. Co-inherited mutations of Fas and caspase-10 in development of the autoimmune lymphoproliferative syndrome. BMC Immunol. 2007;8:28. [PMC free article: PMC2211507] [PubMed: 17999750]
- Chaudhary PM, Eby M, Jasmin A, Bookwalter A, Murray J, Hood L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity. 1997;7:821–30. [PubMed: 9430227]
- Chun HJ, Zheng L, Ahmad M, Wang J, Speirs CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, Skoda-Smith S, Atkinson TP, Straus SE, Lenardo MJ. Pleiotropic defects in lymphocyte activation caused by caspase-8 mutations lead to human immunodeficiency. Nature. 2002;419:395–9. [PubMed: 12353035]
- Del-Rey M, Ruiz-Contreras J, Bosque A, Calleja S, Gomez-Rial J, Roldan E, Morales P, Serrano A, Anel A, Paz-Artal E, Allende LM. A homozygous Fas ligand gene mutation in a patient causes a new type of autoimmune lymphoproliferative syndrome. Blood. 2006;108:1306–12. [PubMed: 16627752]
- Dowdell KC, Niemela JE, Price S, Davis J, Hornung RL, Oliveira JB, Puck JM, Jaffe ES, Pittaluga S, Cohen JI, Fleisher TA, Rao VK. Somatic FAS mutations are common in patients with genetically undefined autoimmune lymphoproliferative syndrome. Blood. 2010;115:5164–9. [PMC free article: PMC2892951] [PubMed: 20360470]
- Ferrari S, Giliani S, Insalaco A, Al-Ghonaium A, Soresina AR, Loubser M, Avanzini MA, Marconi M, Badolato R, Ugazio AG, Levy Y, Catalan N, Durandy A, Tbakhi A, Notarangelo LD, Plebani A. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc Natl Acad Sci U S A. 2001;98:12614–9. [PMC free article: PMC60102] [PubMed: 11675497]
- Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, Strober W, Lenardo MJ, Puck JM. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46. [PubMed: 7540117]
- Hadano S, Yanagisawa Y, Skaug J, Fichter K, Nasir J, Martindale D, Koop BF, Scherer SW, Nicholson DW, Rouleau GA, Ikeda J, Hayden MR. Cloning and characterization of three novel genes, ALS2CR1, ALS2CR2, and ALS2CR3, in the juvenile amyotrophic lateral sclerosis (ALS2) critical region at chromosome 2q33-q34: candidate genes for ALS2. Genomics. 2001;71:200–13. [PubMed: 11161814]
- Hauck F, Magerus-Chatinet A, Vicca S, Rensing-Ehl A, Roesen-Wolff A, Roesler J, Rieux-Laucat F. Somatic loss of heterozygosity, but not haploinsufficiency alone, leads to full-blown autoimmune lymphoproliferative syndrome in 1 of 12 family members with FAS start codon mutation. Clin Immunol. 2013;147:61–8. [PubMed: 23524443]
- Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, Blanche S, Bartunkova J, Vilmer E, Fischer A, Le Deist F, Rieux-Laucat F. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–18. [PubMed: 15459302]
- Houston A, O'Connell J. The Fas signalling pathway and its role in the pathogenesis of cancer. Curr Opin Pharmacol. 2004;4:321–6. [PubMed: 15251123]
- Imai K, Catalan N, Plebani A, Maródi L, Sanal O, Kumaki S, Nagendran V, Wood P, Glastre C, Sarrot-Reynauld F, Hermine O, Forveille M, Revy P, Fischer A, Durandy A. Hyper-IgM syndrome type 4 with a B lymphocyte-intrinsic selective deficiency in Ig class-switch recombination. J Clin Invest. 2003;112:136–42. [PMC free article: PMC162294] [PubMed: 12840068]
- Infante AJ, Britton HA, DeNapoli T, Middelton LA, Lenardo MJ, Jackson CE, Wang J, Fleisher T, Straus SE, Puck JM. The clinical spectrum in a large kindred with autoimmune lymphoproliferative syndrome caused by a Fas mutation that impairs lymphocyte apoptosis. J Pediatr. 1998;133:629–33. [PubMed: 9821419]
- Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, Dale JK, Fleisher TA, Middelton LA, Sneller MC, Lenardo MJ, Straus SE, Puck JM. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64:1002–14. [PMC free article: PMC1377824] [PubMed: 10090885]
- Kanegane H, Vilela MM, Wang Y, Futatani T, Matsukura H, Miyawaki T. Autoimmune lymphoproliferative syndrome presenting with glomerulonephritis. Pediatr Nephrol. 2003;18:454–6. [PubMed: 12736807]
- Kasahara Y, Wada T, Niida Y, Yachie A, Seki H, Ishida Y, Sakai T, Koizumi F, Koizumi S, Miyawaki T, Taniguchi N. Novel Fas (CD95/APO-1) mutations in infants with a lymphoproliferative disorder. Int Immunol. 1998;10:195–202. [PubMed: 9533447]
- Kuehn HS, Caminha I, Niemela JE, Rao VK, Davis J, Fleisher TA, Oliveira JB. FAS haploinsufficiency is a common disease mechanism in the human autoimmune lymphoproliferative syndrome. J Immunol. 2011;186:6035–43. [PMC free article: PMC3725553] [PubMed: 21490157]
- Kuehn HS, Niemela JE, Rangel-Santos A, Zhang M, Pittaluga S, Stoddard JL, Hussey AA, Evbuomwan MO, Priel DA, Kuhns DB, Park CL, Fleisher TA, Uzel G, Oliveira JB. Loss-of-function of the protein kinase C δ (PKCδ) causes a B-cell lymphoproliferative syndrome in humans. Blood. 2013;121:3117–25. [PMC free article: PMC3630827] [PubMed: 23430113]
- Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, Frucht DM, Dumitriu B, Scheinberg P, Folio LR, Frein CA, Price S, Koh C, Heller T, Seroogy CM, Huttenlocher A, Rao VK, Su HC, Kleiner D, Notarangelo LD, Rampertaap Y, Olivier KN, McElwee J, Hughes J, Pittaluga S, Oliveira JB, Meffre E, Fleisher TA, Holland SM, Lenardo MJ, Tangye SG, Uzel G. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345:1623–7. [PMC free article: PMC4371526] [PubMed: 25213377]
- Lanzarotti N, Bruneau J, Trinquand A, Stolzenberg MC, Neven B, Fregeac J, Levy E, Jeremiah N, Suarez F, Mahlaoui N, Fischer A, Magerus-Chatinet A, Cavé H, Rieux-Laucat F. RAS-associated lymphoproliferative disease evolves into severe juvenile myelo-monocytic leukemia. Blood. 2014;123:1960–3. [PubMed: 24652966]
- Le Deist F. Autoimmune lymphoproliferative syndrome. Orphanet Encyclopedia. 2004.
- Le Deist F, Emile JF, Rieux-Laucat F, Benkerrou M, Roberts I, Brousse N, Fischer A. Clinical, immunological, and pathological consequences of Fas-deficient conditions. Lancet. 1996;348:719–23. [PubMed: 8806292]
- Lee WI, Torgerson TR, Schumacher MJ, Yel L, Zhu Q, Ochs HD. Molecular analysis of a large cohort of patients with the hyper immunoglobulin M (IgM) syndrome. Blood. 2005;105:1881–90. [PubMed: 15358621]
- Lenardo M, Chan KM, Hornung F, McFarland H, Siegel R, Wang J, Zheng L. Mature T lymphocyte apoptosis--immune regulation in a dynamic and unpredictable antigenic environment. Annu Rev Immunol. 1999;17:221–53. [PubMed: 10358758]
- Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler-Stevenson M, Strober W, Sneller MC, Puck JM, Lenardo MJ, Elenitoba-Johnson KS, Lin AY, Raffeld M, Jaffe ES. Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol. 1998;153:1541–50. [PMC free article: PMC1853411] [PubMed: 9811346]
- Madkaikar M, Mhatre S, Gupta M, Ghosh K. Advances in autoimmune lymphoproliferative syndromes. Eur J Haematol. 2011;87:1–9. [PubMed: 21447005]
- Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, Schaffner C, Cluet-Dennetiere S, Haerynck F, Michel G, Bole-Feysot C, Zarhrate M, Radford-Weiss I, Romana SP, Picard C, Fischer A, Rieux-Laucat F. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 2011;121:106–12. [PMC free article: PMC3007148] [PubMed: 21183795]
- Magerus-Chatinet A, Stolzenberg MC, Lanzarotti N, Neven B, Daussy C, Picard C, Neveux N, Desai M, Rao M, Ghosh K, Madkaikar M, Fischer A, Rieux-Laucat F. Autoimmune lymphoproliferative syndrome caused by a homozygous null FAS ligand (FASLG) mutation. J Allergy Clin Immunol. 2013;131:486–90. [PMC free article: PMC3824280] [PubMed: 22857792]
- Magerus-Chatinet A, Stolzenberg MC, Loffredo MS, Neven B, Schaffner C, Ducrot N, Arkwright PD, Bader-Meunier B, Barbot J, Blanche S, Casanova JL, Debré M, Ferster A, Fieschi C, Florkin B, Galambrun C, Hermine O, Lambotte O, Solary E, Thomas C, Le Deist F, Picard C, Fischer A, Rieux-Laucat F. FAS-L, IL-10, and double-negative CD4- CD8- TCR alpha/beta+ T cells are reliable markers of autoimmune lymphoproliferative syndrome (ALPS) associated with FAS loss of function. Blood. 2009;113:3027–30. [PubMed: 19176318]
- Maric I, Pittaluga S, Dale JK, Niemela JE, Delsol G, Diment J, Rosai J, Raffeld M, Puck JM, Straus SE, Jaffe ES. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol. 2005;29:903–11. [PubMed: 15958855]
- Martin DA, Zheng L, Siegel RM, Huang B, Fisher GH, Wang J, Jackson CE, Puck JM, Dale J, Straus SE, Peter ME, Krammer PH, Fesik S, Lenardo MJ. Defective CD95/APO-1/Fas signal complex formation in the human autoimmune lymphoproliferative syndrome, type Ia. Proc Natl Acad Sci U S A. 1999;96:4552–7. [PMC free article: PMC16370] [PubMed: 10200300]
- Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, Lyons JJ, Engelhardt KR, Zhang Y, Topcagic N, Roberson ED, Matthews H, Verbsky JW, Dasu T, Vargas-Hernandez A, Varghese N, McClain KL, Karam LB, Nahmod K, Makedonas G, Mace EM, Sorte HS, Perminow G, Rao VK, O'Connell MP, Price S, Su HC, Butrick M, McElwee J, Hughes JD, Willet J, Swan D, Xu Y, Santibanez-Koref M, Slowik V, Dinwiddie DL, Ciaccio CE, Saunders CJ, Septer S, Kingsmore SF, White AJ, Cant AJ, Hambleton S, Cooper MA. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. 2015;125:591–9. [PMC free article: PMC4304103] [PubMed: 25359994]
- Müschen M, Rajewsky K, Krönke M, Küppers R. The origin of CD95-gene mutations in B-cell lymphoma. Trends Immunol. 2002;23:75–80. [PubMed: 11929130]
- Nabhani S, Hönscheid A, Oommen PT, Fleckenstein B, Schaper J, Kuhlen M, Laws HJ, Borkhardt A, Fischer U. A novel homozygous Fas ligand mutation leads to early protein truncation, abrogation of death receptor and reverse signaling and a severe form of the autoimmune lymphoproliferative syndrome. Clin Immunol. 2014;155:231–7. [PubMed: 25451160]
- Neven B, Bruneau J, Stolzenberg MC, Meyts I, Magerus-Chatinet A, Moens L, Lanzarotti N, Weller S, Amiranoff D, Florkin B, Bader-Meunier B, Leverger G, Ferster A, Chantrain C, Blanche S, Picard C, Molina TJ, Brousse N, Durandy A, Rizzi M, Bossuyt X, Fischer A, Rieux-Laucat F. Defective anti-polysaccharide response and splenic marginal zone disorganization in ALPS patients. Blood. 2014;124:1597–609. [PubMed: 24970930]
- Neven B, Magerus-Chatinet A, Florkin B, Gobert D, Lambotte O, De Somer L, Lanzarotti N, Stolzenberg MC, Bader-Meunier B, Aladjidi N, Chantrain C, Bertrand Y, Jeziorski E, Leverger G, Michel G, Suarez F, Oksenhendler E, Hermine O, Blanche S, Picard C, Fischer A, Rieux-Laucat F. A survey of 90 patients with autoimmune lymphoproliferative syndrome related to TNFRSF6 mutation. Blood. 2011;118:4798–807. [PubMed: 21885602]
- Niemela JE, Lu L, Fleisher TA, Davis J, Caminha I, Natter M, Beer LA, Dowdell KC, Pittaluga S, Raffeld M, Rao VK, Oliveira JB. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117:2883–6. [PMC free article: PMC3062298] [PubMed: 21079152]
- Oliveira JB. The expanding spectrum of the autoimmune lymphoproliferative syndromes. Curr Opin Pediatr. 2013;25:722–9. [PMC free article: PMC4435794] [PubMed: 24240292]
- Oliveira JB, Bidère N, Niemela JE, Zheng L, Sakai K, Nix CP, Danner RL, Barb J, Munson PJ, Puck JM, Dale J, Straus SE, Fleisher TA, Lenardo MJ. NRAS mutation causes a human autoimmune lymphoproliferative syndrome. Proc Natl Acad Sci U S A. 2007;104:8953–8. [PMC free article: PMC1885609] [PubMed: 17517660]
- Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, Rieux-Laucat F, Siegel RM, Su HC, Teachey DT, Rao VK. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40. [PMC free article: PMC2953894] [PubMed: 20538792]
- Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. [PubMed: 9242610]
- Peter ME, Legembre P, Barnhart BC. Does CD95 have tumor promoting activities? Biochim Biophys Acta. 2005;1755:25–36. [PubMed: 15907590]
- Piqueras B, Lavenu-Bombled C, Galicier L, Bergeron-van der Cruyssen F, Mouthon L, Chevret S, Debré P, Schmitt C, Oksenhendler E. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23:385–400. [PubMed: 14601647]
- Poppema S, Maggio E, van den Berg A. Development of lymphoma in Autoimmune Lymphoproliferative Syndrome (ALPS) and its relationship to Fas gene mutations. Leuk Lymphoma. 2004;45:423–31. [PubMed: 15160902]
- Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, Perkins K, Hornung RL, Folio L, Rosenberg PS, Puck JM, Hsu AP, Lo B, Pittaluga S, Jaffe ES, Fleisher TA, Rao VK, Lenardo MJ. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123:1989–99. [PMC free article: PMC3968385] [PubMed: 24398331]
- Rao VK, Carrasquillo JA, Dale JK, Bacharach SL, Whatley M, Dugan F, Tretler J, Fleisher T, Puck JM, Wilson W, Jaffe ES, Avila N, Chen CC, Straus SE. Fluorodeoxyglucose positron emission tomography (FDG-PET) for monitoring lymphadenopathy in the autoimmune lymphoproliferative syndrome (ALPS). Am J Hematol. 2006;81:81–5. [PubMed: 16432855]
- Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011;118:5741–51. [PMC free article: PMC3228494] [PubMed: 21885601]
- Rao VK, Price S, Perkins K, Aldridge P, Tretler J, Davis J, Dale JK, Gill F, Hartman KR, Stork LC, Gnarra DJ, Krishnamurti L, Newburger PE, Puck J, Fleisher T. Use of rituximab for refractory cytopenias associated with autoimmune lymphoproliferative syndrome (ALPS). Pediatr Blood Cancer. 2009;52:847–52. [PMC free article: PMC2774763] [PubMed: 19214977]
- Rensing-Ehl A, Janda A, Lorenz MR, Gladstone BP, Fuchs I, Abinun M, Albert M, Butler K, Cant A, Cseh AM, Ebinger M, Goldacker S, Hambleton S, Hebart H, Houet L, Kentouche K, Kühnle I, Lehmberg K, Mejstrikova E, Niemeyer C, Minkov M, Neth O, Dückers G, Owens S, Rösler J, Schilling FH, Schuster V, Seidel MG, Smisek P, Sukova M, Svec P, Wiesel T, Gathmann B, Schwarz K, Vach W, Ehl S, Speckmann C. Sequential decisions on FAS sequencing guided by biomarkers in patients with lymphoproliferation and autoimmune cytopenia. Haematologica. 2013;98:1948–55. [PMC free article: PMC3856970] [PubMed: 23850805]
- Rensing-Ehl A, Warnatz K, Fuchs S, Schlesier M, Salzer U, Draeger R, Bondzio I, Joos Y, Janda A, Gomes M, Abinun M, Hambleton S, Cant A, Shackley F, Flood T, Waruiru C, Beutel K, Siepermann K, Dueckers G, Niehues T, Wiesel T, Schuster V, Seidel MG, Minkov M, Sirkiä K, Kopp MV, Korhonen M, Schwarz K, Ehl S, Speckmann C. Clinical and immunological overlap between autoimmune lymphoproliferative syndrome and common variable immunodeficiency. Clin Immunol. 2010;137:357–65. [PubMed: 20832369]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, Tezcan I, Ersoy F, Kayserili H, Ugazio AG, Brousse N, Muramatsu M, Notarangelo LD, Kinoshita K, Honjo T, Fischer A, Durandy A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell. 2000;102:565–75. [PubMed: 11007475]
- Rieux-Laucat F, Blachère S, Danielan S, De Villartay JP, Oleastro M, Solary E, Bader-Meunier B, Arkwright P, Pondaré C, Bernaudin F, Chapel H, Nielsen S, Berrah M, Fischer A, Le Deist F. Lymphoproliferative syndrome with autoimmunity: A possible genetic basis for dominant expression of the clinical manifestations. Blood. 1999;94:2575–82. [PubMed: 10515860]
- Rieux-Laucat F, Fischer A, Deist FL. Cell-death signaling and human disease. Curr Opin Immunol. 2003;15:325–31. [PubMed: 12787759]
- Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–9. [PubMed: 7539157]
- Roberts CA, Ayers L, Bateman EA, Sadler R, Magerus-Chatinet A, Rieux-Laucat F, Misbah SA, Ferry BL. Investigation of common variable immunodeficiency patients and healthy individuals using autoimmune lymphoproliferative syndrome biomarkers. Hum Immunol. 2013;74:1531–5. [PubMed: 23993982]
- Roesler J, Izquierdo JM, Ryser M, Rösen-Wolff A, Gahr M, Valcarcel J, Lenardo MJ, Zheng L. Haploinsufficiency, rather than the effect of an excessive production of soluble CD95 (CD95{Delta}TM), is the basis for ALPS Ia in a family with duplicated 3' splice site AG in CD95 intron 5 on one allele. Blood. 2005;106:1652–9. [PubMed: 15870181]
- Rössler J, Enders A, Lahr G, Heitger A, Winkler K, Fuchs H, Kopp M, Niemeyer C, Ehl S. Identical phenotype in patients with somatic and germline CD95 mutations requires a new diagnostic approach to autoimmune lymphoproliferative syndrome. J Pediatr. 2005;147:691–4. [PubMed: 16291365]
- Ruiz-García R, Mora S, Lozano-Sánchez G, Martínez-Lostao L, Paz-Artal E, Ruiz-Contreras J, Anel A, González-Granado LI, Moreno-Pérez D, Allende LM. Decreased activation-induced cell death by EBV-transformed B-cells from a patient with autoimmune lymphoproliferative syndrome caused by a novel FASLG mutation. Pediatr Res. 2015;78:603–8. [PubMed: 26334989]
- Salzer E, Santos-Valente E, Klaver S, Ban SA, Emminger W, Prengemann NK, Garncarz W, Müllauer L, Kain R, Boztug H, Heitger A, Arbeiter K, Eitelberger F, Seidel MG, Holter W, Pollak A, Pickl WF, Förster-Waldl E, Boztug K. B-cell deficiency and severe autoimmunity caused by deficiency of protein kinase C δ. Blood. 2013;121:3112–6. [PMC free article: PMC3630826] [PubMed: 23319571]
- Savic S, Parry D, Carter C, Johnson C, Logan C, Gutierrez BM, Thomas JE, Bacon CM, Cant A, Hambleton S. A new case of Fas-associated death domain protein deficiency and update on treatment outcomes. J Allergy Clin Immunol. 2015;136:502–5.e4. [PubMed: 25794656]
- Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, Holler N, Tschopp J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity. 1997;7:831–6. [PubMed: 9430228]
- Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schäffer AA, Grüning BA, Unger S, Frede N, Baumann U, Witte T, Schmidt RE, Dueckers G, Niehues T, Seneviratne S, Kanariou M, Speckmann C, Ehl S, Rensing-Ehl A, Warnatz K, Rakhmanov M, Thimme R, Hasselblatt P, Emmerich F, Cathomen T, Backofen R, Fisch P, Seidl M, May A, Schmitt-Graeff A, Ikemizu S, Salzer U, Franke A, Sakaguchi S, Walker LS, Sansom DM, Grimbacher B. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med. 2014;20:1410–6. [PMC free article: PMC4668597] [PubMed: 25329329]
- Seif AE, Manno CS, Sheen C, Grupp SA, Teachey DT. Identifying autoimmune lymphoproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood. 2010;115:2142–5. [PubMed: 20068224]
- Shah S, Wu E, Rao VK, Tarrant TK. Autoimmune lymphoproliferative syndrome: an update and review of the literature. Curr Allergy Asthma Rep. 2014;14:462–4. [PMC free article: PMC4148697] [PubMed: 25086580]
- Siegel RM, Frederiksen JK, Zacharias DA, Chan FK, Johnson M, Lynch D, Tsien RY, Lenardo MJ. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–7. [PubMed: 10875918]
- Sleight BJ, Prasad VS, DeLaat C, Steele P, Ballard E, Arceci RJ, Sidman CL. Correction of autoimmune lymphoproliferative syndrome by bone marrow transplantation. Bone Marrow Transplant. 1998;22:375–80. [PubMed: 9722073]
- Sneller MC, Straus SE, Jaffe ES, Jaffe JS, Fleisher TA, Stetler-Stevenson M, Strober W. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90:334–41. [PMC free article: PMC443107] [PubMed: 1386609]
- Snow AL, Oliveira JB, Zheng L, Dale JK, Fleisher TA, Lenardo MJ. Critical role for BIM in T cell receptor restimulation-induced death. Biol Direct. 2008;3:34. [PMC free article: PMC2529272] [PubMed: 18715501]
- Sobh A, Crestani E, Cangemi B, Kane J, Chou J, Pai SY, Notarangelo LD, Al-Herz W, Geha RS, Massaad MJ. Autoimmune lymphoproliferative syndrome caused by a homozygous FasL mutation that disrupts FasL assembly. J Allergy Clin Immunol. 2016;137:324–7. [PubMed: 26456038]
- Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, Peters AM, Sneller MC, Hallahan CW, Wang J, Fischer RE, Jackson CM, Lin AY, Bäumler C, Siegert E, Marx A, Vaishnaw AK, Grodzicky T, Fleisher TA, Lenardo MJ. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. [PubMed: 11418480]
- Takagi M, Shinoda K, Piao J, Mitsuiki N, Takagi M, Matsuda K, Muramatsu H, Doisaki S, Nagasawa M, Morio T, Kasahara Y, Koike K, Kojima S, Takao A, Mizutani S. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–90. [PubMed: 21063026]
- Teachey DT. New advances in the diagnosis and treatment of autoimmune lymphoproliferative syndrome. Curr Opin Pediatr. 2012;24:1–8. [PMC free article: PMC3673763] [PubMed: 22157362]
- Teachey DT, Greiner R, Seif A, Attiyeh E, Bleesing J, Choi J, Manno C, Rappaport E, Schwabe D, Sheen C, Sullivan KE, Zhuang H, Wechsler DS, Grupp SA. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145:101–6. [PMC free article: PMC2819393] [PubMed: 19208097]
- Vaishnaw AK, Toubi E, Ohsako S, Drappa J, Buys S, Estrada J, Sitarz A, Zemel L, Chu JL, Elkon KB. The spectrum of apoptotic defects and clinical manifestations, including systemic lupus erythematosus, in humans with CD95 (Fas/APO-1) mutations. Arthritis Rheum. 1999;42:1833–42. [PubMed: 10513797]
- van der Burg M, de Groot R, Comans-Bitter WM, den Hollander JC, Hooijkaas H, Neijens HJ, Berger RM, Oranje AP, Langerak AW, van Dongen JJ. Autoimmune lymphoproliferative syndrome (ALPS) in a child from consanguineous parents: a dominant or recessive disease? Pediatr Res. 2000;47:336–43. [PubMed: 10709732]
- van der Werff ten Bosch J, Delabie J, Böhler T, Verschuere J, Thielemans K. Revision of the diagnosis of T-zone lymphoma in the father of a patient with autoimmune lymphoproliferative syndrome type II. Br J Haematol. 1999;106:1045–8. [PubMed: 10520011]
- Vincenz C, Dixit VM. Fas-associated death domain protein interleukin-1beta-converting enzyme 2 (FLICE2), an ICE/Ced-3 homologue, is proximally involved in CD95- and p55-mediated death signaling. J Biol Chem. 1997;272:6578–83. [PubMed: 9045686]
- Wang J, Chun HJ, Wong W, Spencer DM, Lenardo MJ. Caspase-10 is an initiator caspase in death receptor signaling. Proc Natl Acad Sci U S A. 2001;98:13884–8. [PMC free article: PMC61136] [PubMed: 11717445]
- Wang J, Zheng L, Lobito A, Chan FK, Dale J, Sneller M, Yao X, Puck JM, Straus SE, Lenardo MJ. Inherited human Caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II. Cell. 1999;98:47–58. [PubMed: 10412980]
- Warnatz K, Denz A, Dräger R, Braun M, Groth C, Wolff-Vorbeck G, Eibel H, Schlesier M, Peter HH. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–51. [PubMed: 11861266]
- Wu J, Wilson J, He J, Xiang L, Schur PH, Mountz JD. Fas ligand mutation in a patient with systemic lupus erythematosus and lymphoproliferative disease. J Clin Invest. 1996;1996;98:1107–13. [PMC free article: PMC507531] [PubMed: 8787672]
- Zhu S, Hsu AP, Vacek MM, Zheng L, Schäffer AA, Dale JK, Davis J, Fischer RE, Straus SE, Boruchov D, Saulsbury FT, Lenardo MJ, Puck JM. Genetic alterations in caspase-10 may be causative or protective in autoimmune lymphoproliferative syndrome. Hum Genet. 2006;119:284–94. [PubMed: 16446975]
Chapter Notes
Author History
Jack JH Bleesing, MD, PhD (2006-present)
Judith Johnson, MS, CGC; Cincinnati Children's Hospital (2006-2017)
Chinmayee Nagaraj, MS, CGC (2017-present)
Kejian Zhang, MD, MBA (2006-present)
Revision History
- 24 August 2017 (ha) Comprehensive update posted live
- 11 September 2014 (me) Comprehensive update posted live
- 8 September 2011 (me) Comprehensive update posted live
- 7 April 2009 (me) Comprehensive update posted live
- 9 July 2007 (cd) Revision: sequence analysis and prenatal diagnosis available clinically for CASP10
- 14 September 2006 (me) Review posted live
- 2 December 2005 (jj) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: September 14, 2006; Last Update: August 24, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Bleesing JJH, Nagaraj CB, Zhang K. Autoimmune Lymphoproliferative Syndrome. 2006 Sep 14 [Updated 2017 Aug 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
