Summary
Clinical characteristics.
Bohring-Opitz syndrome (BOS) is characterized by distinctive facial features and posture, growth failure, variable but usually severe intellectual disability, and variable anomalies. The facial features may include microcephaly or trigonocephaly / prominent (but not fused) metopic ridge, hypotonic facies with full cheeks, synophrys, glabellar and eyelid nevus flammeus (simplex), prominent globes, widely set eyes, palate anomalies, and micrognathia. The BOS posture, which is most striking in early childhood and often becomes less apparent with age, is characterized by flexion at the elbows with ulnar deviation and flexion of the wrists and metacarpophalangeal joints. Feeding difficulties in early childhood, including cyclic vomiting, have a significant impact on overall health; feeding tends to improve with age. Seizures are common and typically responsive to standard epileptic medications. Minor cardiac anomalies and transient bradycardia and apnea may be present. Affected individuals may experience recurrent infections, which also tend to improve with age. Isolated case reports suggest that individuals with BOS are at greater risk for Wilms tumor than the general population, but large-scale epidemiologic studies have not been conducted.
Diagnosis/testing.
The diagnosis of Bohring-Opitz syndrome (BOS) is established in a proband with suggestive clinical features and/or the identification of a constitutional heterozygous pathogenic variant in ASXL1 by molecular genetic testing.
Management.
Treatment of manifestations. Cyclic vomiting may be managed by identification and avoidance of triggers, daily maintenance medication, and early abortive treatment; G-tubes or GJ-tubes may decrease aspiration and improve nutrition. Due to the prevalence of obstructive sleep apnea, polysomnography should be considered. Referral to a craniofacial team should be considered for those with palatal abnormalities, micrognathia, or obstructive sleep apnea. Tracheostomy may be considered for those with recurrent aspiration who develop secondary lung disease, or in those with severe sleep apnea that is not adequately treated with noninvasive pressure support (e.g., CPAP, BiPAP) or surgical intervention (e.g., mandibular distraction). Standard management is indicated for seizures, congenital heart defects, intellectual disability, myopia, urinary tract infections, urinary retention, and renal stones.
Prevention of primary manifestations. Adequate treatment of severe emesis can decrease hospitalizations, infectious exposures, and ascending aspiration.
Surveillance: Renal ultrasound every three months from birth to age eight to screen for the development of Wilms tumor; frequent monitoring of growth and development; close monitoring of feeding intolerance with a gastroenterology specialist; regular follow up for vision optimization.
Agents/circumstances to avoid. Triggers for vomiting should be avoided and managed with prophylactic antiemetics prior to the exposure.
Genetic counseling.
Bohring-Opitz syndrome (BOS) is typically the result of a de novo pathogenic variant in ASXL1. When BOS results from a de novo variant, the risk to the sibs of a proband is small. No individuals with BOS have been reported to reproduce. Although the vast majority of BOS occurs as the result of a de novo variant in ASXL1, molecular genetic testing can be used to evaluate a pregnancy at theoretically increased risk as a result of constitutional and/or germline mosaicism for an ASXL1 pathogenic variant in a clinically unaffected parent.
Diagnosis
Prior to the identification of the molecular cause of Bohring-Opitz syndrome (BOS), Hastings et al [2011] had proposed clinical diagnostic criteria for the condition. Ultimately, only five individuals used to develop these clinical diagnostic criteria were molecularly confirmed to have BOS. Therefore, the specificity of these diagnostic criteria is unclear.
Suggestive Findings
Bohring-Opitz syndrome should be suspected in individuals with the following clinical features [Bohring et al 1999, Bohring et al 2006, Hastings et al 2011, Magini et al 2012, Russell et al 2015].
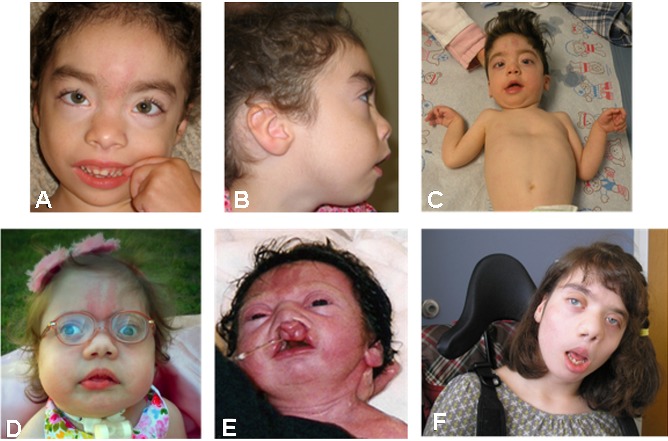
Craniofacial appearance (Figure 1)

Figure 1.
Facial features of individuals with BOS at varying ages A-B. 2 years
- Microcephaly or trigonocephaly / prominent (but not necessarily fused) metopic ridge
- Glabellar and eyelid nevus flammeus (simplex) that fades with age
- Prominent globes
- Cleft lip
- Palatal anomalies: cleft palate, high arched palate, or prominent palatine ridges
- Micrognathia and/or retrognathia
Growth and feeding
- Intrauterine growth restriction
- Severe feeding difficulties with chronic emesis that typically improves with age
- Poor postnatal weight gain and linear growth, often exacerbated by severe feeding intolerance
Neurologic
- Developmental delay or intellectual disability in the severe-to-profound range with minimal or complete lack of expressive language
- Seizures
Respiratory. Recurrent infections (commonly respiratory) in early childhood that improve with age
Sleep
- Sleep disturbance
- Obstructive sleep apnea
Ophthalmologic
- High myopia presenting in infancy that may worsen over the first years of life
- Variable optic nerve and retinal anomalies
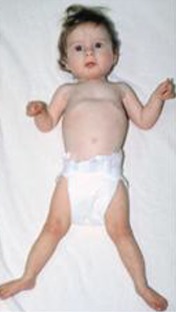
BOS posture (Figure 2)
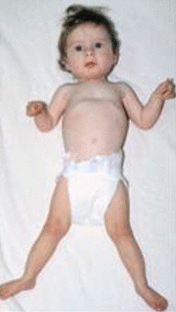
Figure 2.
Typical BOS posture with flexion at the elbows, ulnar deviation, flexion of the wrists and metacarpophalangeal joints, and hypertonic extremities with central hypotonia From Hastings et al [2011]. Republished with permission.
- Flexion at the elbows with ulnar deviation and flexion of the wrists and metacarpophalangeal joints; most noticeable in early childhood and usually less obvious with age
- Truncal hypotonia with hypertonia of the extremities
Establishing the Diagnosis
The diagnosis of Bohring-Opitz syndrome is established in a proband with suggestive clinical features (see Suggestive Findings) and/or by identification of a constitutional heterozygous pathogenic variant in ASXL1 on molecular genetic testing (see Table 1).
Molecular genetic testing approaches can include single-gene testing, use of a multigene panel, or more comprehensive genomic testing:
- Single-gene testing. Sequence analysis of ASXL1 is performed first and followed by gene-targeted deletion/duplication analysis if no pathogenic variant is found.
- A multigene panel that includes ASXL1 and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered if single-gene testing (and/or use of a multigene panel that includes ASXL1) fails to confirm a diagnosis in an individual with features of Bohring-Opitz syndrome. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Table 1.
Molecular Genetic Testing Used in Bohring-Opitz Syndrome
Clinical Characteristics
Clinical Description
Bohring-Opitz syndrome (BOS) is a rare condition characterized by distinctive facial features and posture, variable but usually severe intellectual disability, growth failure, and variable anomalies. Feeding difficulties have a significant impact on overall health in early childhood; feeding tends to improve with age. This section summarizes clinical data from numerous case reports and case series; see Russell et al [2015] and references therein, Dangiolo et al [2015], and Suggested Reading. Additional references are cited where appropriate.
Craniofacial. Individuals with BOS have a characteristic facial appearance (see Suggestive Findings), although significant variability is observed. The most striking and consistent features are a prominent glabellar nevus flammeus (simplex) that fades with age, synophrys that becomes more prominent with age, hypotonic facies with full cheeks, and prominent or proptotic eyes. The majority of affected individuals also have hypertrichosis with rapidly growing hair and nails.
Other features widely, but variably, reported include cleft lip with or without cleft palate, high arched palate, widely spaced eyes (hypertelorism), depressed and wide nasal bridge, anteverted nares, and low-set ears with increased posterior angulation.
Growth. Mild intrauterine growth restriction has been noted, but many infants are products of healthy pregnancies with average or low-average birth weights. Poor growth is typically noted in the first year of life and is often clinically attributed to chronic emesis and feeding intolerance. Adequate nutrition does play a role in early growth, but even those without feeding intolerance typically display poor long-term growth.
Feeding. Most children have had feeding issues beginning in infancy and generally improving or resolving in early childhood. Historically, feeding issues have been presumed to be secondary to severe gastroesophageal reflux, but initial case reports did not produce diagnostic evidence of gastroesophageal reflux or demonstrate improvement on traditional antireflux therapies. Recent publications have suggested a neurogenic etiology, including cyclic vomiting with possible poor gastric motility, as the underlying cause of the chronic emesis [Russell et al 2015]. Given the frequency of emesis, there is a high risk for aspiration and dehydration, typically resulting in multiple hospitalizations.
Development and behavior. All affected individuals reported in the literature have severe-to-profound intellectual disability. Few are able to speak, but many have been able to express basic needs using augmentative and alternative communication (AAC) devices as well as gestures with associated basic vocalizations. Individuals with BOS often have a happy and pleasant demeanor [Russell et al 2015]. Typically, they are able to recognize caregivers and have a social, interactive nature. Most are unable to walk independently, but some have had success using walkers and braces in late childhood. Given the recent increase in rate of diagnosis due to genomic testing, it is expected that affected individuals with a milder phenotype may be reported in the future.
Neurologic. Seizures are common in individuals with BOS and are typically responsive to standard antiepileptic medications. Affected individuals have also been described to have truncal hypotonia with hypertonia of the extremities. A wide range of primary brain anomalies have been reported. Corpus callosum defects are the most common and range from hypoplastic to absent. Dandy-Walker malformation, delayed myelination, and enlarged ventricles have also been described.
Cardiovascular. Idiopathic and transient bradycardia as well as apnea were widely reported in the initial literature that predated the identification of the genetic cause of BOS. Cardiovascular deaths associated with bradycardia and apnea account for four (33%) of the 12 deaths published in the literature (although none of those individuals had a molecular confirmation of BOS). Other minor cardiac anomalies including septal defects and cardiac hypertrophy have also been described in a small number of affected individuals.
Respiratory. In addition to apnea and bradycardia, respiratory infections are common in infancy and account for about 42% of deaths (5/12). When chronic emesis is treated or improves with age, the rate of respiratory infections decreases [Russell et al 2015]. Tracheostomies have been necessary for some (see Management).
Sleep. Obstructive sleep apnea and sleep disturbances, including difficulty falling asleep and staying asleep, are frequently reported. Affected individuals with micrognathia may also exhibit tongue-based airway obstruction.
Ophthalmologic. Myopia, often severe, is common in individuals with BOS. Most affected individuals require corrective lenses in infancy. Retinal and optic nerve abnormalities including colobomas, retinal and optic nerve atrophy, and abnormal coloration of the retinas are also frequently reported.
Immunologic. Recurrent infections may be frequent in early life, although immunodeficiency has not been reported in the literature. The frequency of infections typically declines with age.
Urologic. Urinary retention and recurrent urinary tract infections have been reported in individuals with BOS [Russell et al 2015]. There also appears to be an increased risk for renal stones [Author, personal observation].
Skeletal. The typical BOS posture (see Suggestive Findings) is most notable in early childhood and usually becomes less obvious with age. Congenital contractures, dislocations, and pectus excavatum have also been reported.
Malignancy. Isolated case reports suggest that children with BOS are at greater risk for Wilms tumor than the general population; large-scale epidemiologic studies have not been conducted due to the limited number of individuals diagnosed with BOS. Of three individuals with a clinical diagnosis of BOS and renal neoplasia, two had documented pathogenic variants in ASXL1 and bilateral Wilms tumor; Wilms tumor was diagnosed in one at age two years and in the other at age six years [Russell et al 2015]. Another individual with BOS had nephroblastomatosis on autopsy at age five months. This infant later underwent molecular genetic testing of ASXL1; no pathogenic variant was identified [Brunner et al 2000, Hoischen et al 2011]. Overall, Wilms tumor appears to affect about 7% of individuals with BOS. This risk estimate is based on a very small number of reported cases and thus is likely to change over time as larger cohorts of children and adults with BOS are investigated.
The only other neoplastic process reported was medulloblastoma in a child age five years with clinical features of BOS in whom a pathogenic ASXL1 variant was not identified [Hastings et al 2010, Hoischen et al 2011].
Other
- Annular pancreas has been described in some affected individuals.
- Gallstones have been reported in several affected individuals [Author, personal observation].
- Historically, this condition had been associated with high infant mortality (27% based on data published before 2015), but the current survival rate is likely to be much better due to advances in pediatric care and more aggressive interventions [Russell et al 2015].
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been reported to date, but the total number of individuals in whom ASXL1 pathogenic variants have been identified is limited.
Nomenclature
BOS is occasionally known as Oberklaid-Danks syndrome after F Oberklaid and DM Danks, who described one of the early cases of BOS [Oberklaid & Danks 1975]. BOS is the more commonly used term.
Prevalence
The prevalence of BOS is unknown; out of 46 clinically diagnosed individuals reported in the literature, 20 had the diagnosis molecularly confirmed.
Note: Not all clinically diagnosed individuals reported in the literature have undergone molecular genetic testing of ASXL1.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in ASXL1.
Sporadic malignancies (including myelodysplastic syndrome) occurring in the absence of any other findings of Bohring-Opitz syndrome frequently harbor somatic variants in ASXL1 that are not present in the germline. In these circumstances predisposition to these tumors is not heritable. For more details see Cancer and Benign Tumors.
Differential Diagnosis
Table 2.
Disorders to Consider in the Differential Diagnosis of Bohring-Opitz Syndrome (BOS)
Management
Evaluations Following Initial Diagnosis
To establish the spectrum of manifestations and medical needs in an individual diagnosed with Bohring-Opitz syndrome (BOS), the following evaluations are recommended if they have not already been completed.
Table 3.
Recommended Evaluations Following Initial Diagnosis of Bohring-Opitz Syndrome
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with BOS
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years. In the US, an IEP based on the individual's level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility.
- Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction. Assuming that the individual is safe to eat by mouth, feeding therapy – typically from an occupational or speech therapist – is recommended for affected individuals who have difficulty feeding due to poor oral motor control.
Communication issues. Consider evaluation for alternative means of communication (e.g., Augmentative and Alternative Communication [AAC] for individuals who have expressive language difficulties.
Prevention of Primary Manifestations
Adequate treatment of severe emesis can decrease hospitalizations, infectious exposures, and aspiration.
Surveillance
The following are appropriate:
- Renal ultrasound every three months from birth to age eight years to screen for the development of Wilms tumor [Russell et al 2015]
- Frequent monitoring of growth and development with interventions as needed (see Treatment of Manifestations)
- Close management of feeding intolerance with a gastroenterology specialist
- Regular follow up with an ophthalmologist for vision optimization
Agents/Circumstances to Avoid
Triggers for vomiting should be avoided and managed with prophylactic antiemetics prior to the exposure (see Treatment of Manifestations).
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Bohring-Opitz syndrome (BOS) is inherited in an autosomal dominant manner.
Note: A previously published sib pair identified as having BOS (which could have implied either autosomal recessive inheritance or germline mosaicism in a parent) have subsequently been identified as having another genetic condition [Greenhalgh et al 2003, Bruel et al 2017].
Risk to Family Members
Parents of a proband
- Most probands with BOS reported to date have the disorder as a result of a de novo ASXL1 pathogenic variant.
- Recommendations for the evaluation of parents of a proband with an apparent de novo ASXL1 pathogenic variant include molecular genetic testing.
Sibs of a proband
- Most affected individuals reported to date have had a de novo ASXL1 pathogenic variant, suggesting a low risk to sibs.
- If the ASXL1 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
Offspring of a proband. Individuals with BOS are not known to reproduce.
Other family members. Given that most probands with BOS reported to date have the disorder as a result of a de novo ASXL1 pathogenic variant, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes, allelic variants, and diseases will improve in the future, consideration should be given to banking DNA of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Once the ASXL1 pathogenic variant has been identified in an affected family member, prenatal testing and preimplantation genetic testing are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- ASXL Rare Research Endowment FoundationEmail: info@arrefoundation.org
- Bohring-Opitz SyndromeA worldwide exchange of information and awareness.
- Bohring-Opitz Syndrome FoundationEmail: info@bos-foundation.org
- Bohring-Opitz Syndrome & ASXL Related Disorders RegistryEmail: ASXL-CHROMATIN-REGISTRY@mednet.ucla.edu
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Bohring-Opitz Syndrome: Genes and Databases
Table B.
OMIM Entries for Bohring-Opitz Syndrome (View All in OMIM)
Molecular Pathogenesis
ASXL1 is one of the human orthologs of the additional sex combs gene (Asx), which is an atypical polycomb group (PcG) protein that in Drosophila regulates gene expression by binding to chromatin and by regulating ubiquitination of specific histones (e.g., histone H2A) through activation of specific deubiquitinases. In humans, ASXL1, ASXL2, and ASXL3 (Table 2) code for putative PcG proteins (for ASXL1 see www.uniprot.org/uniprot/Q8IXJ9) that regulate transcription through various mechanisms, including recruitment of histone H3 in specific cell types and deubiquitination of specific histone H2A. See Russell & Graham [2013] and references therein for insights into conditions associated with members of the ASXL family.
Gene structure. NM_015338 is the longest ASXL1 transcript variant; it is composed of 13 exons. Alternatively spliced transcripts have been reported. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. In one report, de novo heterozygous inactivating pathogenic variants were detected in 50% of individuals who had features consistent with a diagnosis of BOS [Dangiolo et al 2015]. See Table A, LSDB and HGMD.
NOTE: ASXL1 variants, including BOS-associated variants, have been identified in the elderly or in cohorts of individuals with cancer; such variants can be found in reference population databases. However, these variants are thought to result from hematopoiectic somatic mosaicism as opposed to BOS-associated germline variants [Carlston et al 2017 and references therein]. Failure to consider somatic mosaicism may lead to the misclassification of potentially pathogenic variants. See Cancer and Benign Tumors.
Normal gene product. The transcript NM_015338 encodes the protein isoform NP_056153 (isoform 1) with 1,541 amino acid residues known as putative polycomb group protein ASXL1.
Abnormal gene product. The de novo frameshift and nonsense ASXL1 pathogenic variants suggest a loss-of-function mechanism as a cause of BOS [Hoischen et al 2011, Dangiolo et al 2015].
Cancer and Benign Tumors
Sporadic malignancies (including myelodysplastic syndrome) occurring in the absence of any other findings of BOS frequently harbor somatic variants in ASXL1 that are not present in the germline. In these circumstances predisposition to these tumors is not heritable.
Somatic ASXL1 pathogenic variants are known to be associated with a worse prognosis in persons with myeloid leukemia [Abdel-Wahab et al 2012]. Mice with somatic pathogenic Asxl variants are at high risk for myelodysplastic conditions [Abdel-Wahab et al 2013], but constitutive Asxl1 loss does not produce long-term cell line dysfunction [Fisher et al 2010a, Fisher et al 2010b]. Leukemias have not been reported in individuals with ASXL1-related BOS.
References
Literature Cited
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, Perna F, Zhao X, Taylor JE, Park CY, Carroll M, Melnick A, Nimer SD, Jaffe JD, Aifantis I, Bernstein BE, Levine RL. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012;22:180–93. [PMC free article: PMC3422511] [PubMed: 22897849]
- Abdel-Wahab O, Gao J, Adli M, Dey A, Trimarchi T, Chung YR, Kuscu C, Hricik T, Ndiaye-Lobry D, Lafave LM, Koche R, Shih AH, Guryanova OA, Kim E, Li S, Pandey S, Shin JY, Telis L, Liu J, Bhatt PK, Monette S, Zhao X, Mason CE, Park CY, Bernstein BE, Aifantis I, Levine RL. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J Exp Med. 2013;210:2641–59. [PMC free article: PMC3832937] [PubMed: 24218140]
- Angius A, Uva P, Buers I, Oppo M, Puddu A, Onano S, Persico I, Loi A, Marcia L, Höhne W, Cuccuru G, Fotia G, Deiana M, Marongiu M, Atalay HT, Inan S, El Assy O, Smit LM, Okur I, Boduroglu K, Utine GE, Kılıç E, Zampino G, Crisponi G, Crisponi L, Rutsch F. Bi-allelic mutations in KLHL7 cause a Crisponi/CISS1-like phenotype associated with early-onset retinitis pigmentosa. Am J Hum Genet. 2016;99:236–45. [PMC free article: PMC5005468] [PubMed: 27392078]
- Balasubramanian M, Willoughby J, Fry AE, Weber A, Firth HV, Deshpande C, Berg JN, Chandler K, Metcalfe KA, Lam W, Pilz D, Tomkins S. Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo, heterozygous, loss-of-function mutations in ASXL3 and review of published literature. J Med Genet. 2017;54:537–43. [PubMed: 28100473]
- Bohring A, Oudesluijs GG, Grange DK, Zampino G, Thierry P. New cases of Bohring-Opitz syndrome, update, and critical review of the literature. Am J Med Genet Part A. 2006;140:1257–63. [PubMed: 16691589]
- Bohring A, Silengo M, Lerone M, Superneau DW, Spaich C, Braddock SR, Poss A, Opitz JM. Severe end of Opitz trigonocephaly (C) syndrome or new syndrome? Am J Med Genet. 1999;85:438–46. [PubMed: 10405439]
- Bruel AL, Bigoni S, Kennedy J, Whiteford M, Buxton C, Parmeggiani G, Wherlock M, Woodward G, Greenslade M, Williams M, St-Onge J, Ferlini A, Garani G, Ballardini E, van Bon B, Acuna-Hidalgo R, Bohring A, Deleuze J, Boland A, Meyer V, Olaso R, Ginglinger E, Study D, Rivière J, Brunner HG, Hoischen A, Newbury-Ecob R, Faivre L, Thauvin-Robinet C, Thevenon J. Expanding the clinical spectrum of recessive truncating mutations of KLHL7 to a Bohring-Opitz-like phenotype. J Med Genet. 2017;54:830–5. [PubMed: 29074562]
- Brunner HG, Van Tintelen JP, De Boer RJ. Bohring syndrome. Am J Med Genet. 2000;92:366–8. [PubMed: 10861670]
- Carlston CM, O'Donnell-Luria AH, Underhill HR, Cummings BB, Weisburd B, Minikel EV, Birnbaum DP, Tvrdik T, MacArthur DG, Mao R, et al. Pathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz syndrome. Hum Mutat. 2017;38:517–23. [PMC free article: PMC5487276] [PubMed: 28229513]
- Dangiolo SB, Wilson A, Jobanputra V, Anyane-Yeboa K. Bohring-Opitz syndrome (BOS) with a new ASXL1 pathogenic variant: review of the most prevalent molecular and phenotypic features of the syndrome. Am J Med Genet Part A. 2015;167A:3161–6. [PubMed: 26364555]
- Fisher CL, Lee I, Bloyer S, Bozza S, Chevalier J, Dahl A, Bodner C, Helgason CD, Hess JL, Humphries RK, Brock HW. Additional sex combs-like 1 belongs to the enhancer of trithorax and polycomb group and genetically interacts with Cbx2 in mice. Dev Biol. 2010a;337:9–15. [PMC free article: PMC2807749] [PubMed: 19833123]
- Fisher CL, Pineault N, Brookes C, Helgason CD, Ohta H, Bodner C, Hess JL, Humphries RK, Brock HW. Loss-of-function additional sex combs like 1 mutations disrupt hematopoiesis but do not cause severe myelodysplasia or leukemia. Blood. 2010b;115:38–46. [PMC free article: PMC2803690] [PubMed: 19861679]
- Greenhalgh KL, Newbury-Ecob RA, Lunt PW, Dolling CL, Hargreaves H, Smithson SF. Siblings with Bohring-Opitz syndrome. Clin Dysmorphol. 2003;12:15–9. [PubMed: 12514360]
- Hastings R, Cobben JM, Gillessen-Kaesbach G, Goodship J, Hove H, Kjaergaard S, Kemp H, Kingston H, Lunt P, Mansour S, McGowan R, Metcalfe K, Murdoch-Davis C, Ray M, Rio M, Smithson S, Tolmie J, Turnpenny P, van Bon B, Wieczorek D, Newbury-Ecob R. Bohring-Opitz (Oberklaid-Danks) syndrome: clinical study, review of the literature, and discussion of possible pathogenesis. Eur J Hum Genet. 2011;19:513–9. [PMC free article: PMC3083618] [PubMed: 21368916]
- Hastings RW, Newbury-Ecob R, Lunt PW. A case of probable Bohring-Opitz syndrome with medulloblastoma. Clin Dysmorphol. 2010;19:202–5. [PubMed: 20717007]
- Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, de Vries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R, Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M, Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA, Brunner HG, de Vries BBBA. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011;43:729–31. [PubMed: 21706002]
- Kaiser FJ, Ansari M, Braunholz D, Concepción Gil-Rodríguez M, Decroos C, Wilde JJ, Fincher CT, Kaur M, Bando M, Amor DJ, Atwal PS, Bahlo M, Bowman CM, Bradley JJ, Brunner HG, Clark D, Del Campo M, Di Donato N, Diakumis P, Dubbs H, Dyment DA, Eckhold J, Ernst S, Ferreira JC, Francey LJ, Gehlken U, Guillén-Navarro E, Gyftodimou Y, Hall BD, Hennekam R, Hudgins L, Hullings M, Hunter JM, Yntema H, Innes AM, Kline AD, Krumina Z, Lee H, Leppig K, Lynch SA, Mallozzi MB, Mannini L, McKee S, Mehta SG, Micule I, Mohammed S, Moran E, Mortier GR, Moser JA, Noon SE, Nozaki N, Nunes L, Pappas JG, Penney LS, Pérez-Aytés A, Petersen MB, Puisac B, Revencu N, Roeder E, Saitta S, Scheuerle AE, Schindeler KL, Siu VM, Stark Z, Strom SP, Thiese H, Vater I, Willems P, Williamson K, Wilson LC, Hakonarson H, Quintero-Rivera F, Wierzba J, Musio A, Gillessen-Kaesbach G, Ramos FJ, Jackson LG, Shirahige K, Pié J, Christianson DW, Krantz ID, Fitzpatrick DR, Deardorff MA, et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014;23:2888–900. [PMC free article: PMC4014191] [PubMed: 24403048]
- Kaname T, Yanagi K, Chinen Y, Makita Y, Okamoto N, Maehara H, Owan I, Kanaya F, Kubota Y, Oike Y, Yamamoto T, Kurosawa K, Fukushima Y, Bohring A, Opitz JM, Yoshiura K, Niikawa N, Naritomi K. Mutations in CD96, a member of the immunoglobulin superfamily, cause a form of the C (Opitz trigonocephaly) syndrome. Am J Hum Genet. 2007;81:835–41. [PMC free article: PMC2227933] [PubMed: 17847009]
- Kuechler A, Czeschik JC, Graf E, Grasshoff U, Hüffmeier U, Busa T, Beck-Woedl S, Faivre L, Rivière JB, Bader I, Koch J, Reis A, Hehr U, Rittinger O, Sperl W, Haack TB, Wieland T, Engels H, Prokisch H, Strom TM, Lüdecke HJ, Wieczorek D. Bainbridge-Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition. Eur J Hum Genet. 2017;25:183–91. [PMC free article: PMC5255962] [PubMed: 27901041]
- Magini P, Della Monica M, Uzielli ML, Mongelli P, Scarselli G, Gambineri E, Scarano G, Seri M. Two novel patients with Bohring-Opitz syndrome caused by de novo ASXL1 mutations. Am J Med Genet Part A. 2012;158A:917–21. [PubMed: 22419483]
- Oberklaid F, Danks D. The Opitz trigonocephaly syndrome. Am J Dis Child. 1975;129:1348–9. [PubMed: 1190170]
- Parenti I, Gervasini C, Pozojevic J, Wendt KS, Watrin E, Azzollini J, Braunholz D, Buiting K, Cereda A, Engels H, Garavelli L, Glazar R, Graffmann B, Larizza L, Lüdecke HJ, Mariani M, Masciadri M, Pié J, Ramos FJ, Russo S, Selicorni A, Stefanova M, Strom TM, Werner R, Wierzba J, Zampino G, Gillessen-Kaesbach G, Wieczorek D, Kaiser FJ. Expanding the clinical spectrum of the 'HDAC8-phenotype' - implications for molecular diagnostics, counseling and risk prediction. Clin Genet. 2016;89:564–73. [PubMed: 26671848]
- Peña-Padilla C, Marshall CR, Walker S, Scherer SW, Tavares-Macías G, Razo-Jiménez G, Bobadilla-Morales L, Acosta-Fernández E, Corona-Rivera A, Mendoza-Londono R, Corona-Rivera JR. Compound heterozygous mutations in the IFT140 gene cause Opitz trigonocephaly C syndrome in a patient with typical features of a ciliopathy. Clin Genet. 2017;91:640–6. [PubMed: 27874174]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Russell B, Graham JM. Expanding our knowledge of conditions associated with the ASXL gene family. Genome Med. 2013;2013;5:16. [PMC free article: PMC3706972] [PubMed: 23672984]
- Russell B, Johnston JJ, Biesecker LG, Kramer N, Pickart A, Rhead W, Tan W-H, Brownstein CA, Kate Clarkson L, Dobson A, Rosenberg AZ, Vergano SAS, Helm BM, Harrison RE, Graham Jr JM. Clinical management of patients with ASXL1 mutations and Bohring–Opitz syndrome, emphasizing the need for Wilms tumor surveillance. Am J Med Genet Part A. 2015;167A:2122–31. [PMC free article: PMC4760347] [PubMed: 25921057]
- Shashi V, Pena LD, Kim K, Burton B, Hempel M, Schoch K, Walkiewicz M, McLaughlin HM, Cho M, Stong N, Hickey SE, Shuss CM, Freemark MS, Bellet JS, Keels MA, Bonner MJ, El-Dairi M, Butler M, Kranz PG, Stumpel CT, Klinkenberg S, Oberndorff K, Alawi M, Santer R, Petrovski S, Kuismin O, Korpi-Heikkilä S, Pietilainen O, Aarno P, Kurki MI, Hoischen A, Need AC, Goldstein DB, Kortüm F, et al. De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet. 2016;99:991–9. [PMC free article: PMC5065681] [PubMed: 27693232]
Chapter Notes
Author Notes
Dr Bianca Russell is a clinical geneticist at Cincinnati Children's Hospital who has been studying and caring for patients with Bohring-Opitz Syndrome since meeting two patients with the condition in 2012. She met these patients while under the mentorship of Dr John Graham at Cedars Sinai Medical Center. Dr Graham has been a leader in the field of clinical dysmorphology for decades and an outstanding mentor. Dr Wen-Hann Tan is a clinical geneticist at Boston Children's Hospital who cares for several patients with BOS and was an integral part of their 2015 publication on clinical management of BOS.
Dr Russell and Dr Tan are continuing their work with the BOS community through a registry for disorders caused by pathogenic variants in the ASXL family of genes.
Acknowledgments
The authors would like to thank the BOS families and community for their commitment to understanding BOS and providing outstanding care for their children. They continue to be a true inspiration to the authors of this review.
Revision History
- 15 February 2018 (ma) Review posted live
- 13 March 2017 (br) Initial submission
Publication Details
Author Information and Affiliations
Cincinnati, Ohio
Boston, Massachusetts
Harbor-UCLA Medical Center
Emeritus Professor of Pediatrics, David Geffen School of Medicine at UCLA
Los Angeles, California
Publication History
Initial Posting: February 15, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Russell B, Tan WH, Graham JM Jr. Bohring-Opitz Syndrome. 2018 Feb 15. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.