Bestrophinopathies
Ian M MacDonald, MDCM, MSc, Thomas Lee, MD, MSc, and Jessica Lawrence, MSc.
Author Information and AffiliationsInitial Posting: September 30, 2003; Last Update: July 16, 2020.
Estimated reading time: 24 minutes
Summary
Clinical characteristics.
Bestrophinopathies, the spectrum of ophthalmic disorders caused by pathogenic variants in BEST1, are typically characterized by retinal degeneration. The four recognized phenotypes are the three autosomal dominant disorders: Best vitelliform macular dystrophy (BVMD), BEST1 adult-onset vitelliform macular dystrophy (AVMD), and autosomal dominant vitreoretinochoroidopathy (ADVIRC); and autosomal recessive bestrophinopathy (ARB). Onset is usually in the first decade (except AVMD in which onset is age 30 to 50 years). Slow visual deterioration is the usual course. Choroidal neovascularization can occur in rare cases. ADVIRC is also associated with panophthalmic involvement including nanophthalmos, microcornea, hyperopia, and narrow anterior chamber angle with angle closure glaucoma.
Diagnosis/testing.
The diagnosis of autosomal dominant bestrophinopathy is established in a proband with suggestive findings and a heterozygous BEST1 pathogenic (or likely pathogenic) variant identified by molecular genetic testing. The diagnosis of autosomal recessive bestrophinopathy is established in a proband with suggestive findings and biallelic BEST1 pathogenic variants.
Management.
Treatment of manifestations: For individuals with significant visual impairment, referral to a low vision clinic; attention to special education needs for children with visual impairment; and occupational counseling. Regarding advanced BVMD fundus lesions, no clinical trials have compared conservative treatment vs laser photocoagulation for choroidal neovascularization (CNV) and hemorrhage. Also, there are no ongoing clinical trials regarding the effectiveness of treatment with anti-VEGF (vascular endothelial growth factor) agents.
Surveillance: Annual ophthalmologic examination (including best corrected visual acuity, visual fields, and spectral domain optical coherence tomography) to monitor progression of fundus lesions and to evaluate for coincidental development of CNV; in childhood, perform annual ophthalmologic examinations to help prevent the development of amblyopia.
Agents/circumstances to avoid: Cessation of smoking to help prevent neovascularization of the retina.
Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from prompt ophthalmologic evaluation and routine follow up.
Genetic counseling.
BVMD, AVMD, and ADVIRC are inherited in an autosomal dominant (AD) manner. By definition, autosomal recessive bestrophinopathy (ARB) is inherited in an autosomal recessive (AR) manner.
AD bestrophinopathy. Each child of an affected individual has a 50% chance of inheriting the BEST1 pathogenic variant.
AR bestrophinopathy. If both parents are known to be heterozygous for a BEST1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Once the BEST1 pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for a bestrophinopathy are possible.
GeneReview Scope
View in own window
| Bestrophinopathies: Included Phenotypes |
|---|
Best vitelliform macular dystrophy (BVMD) Adult-onset vitelliform macular dystrophy (AVMD) Autosomal dominant vitreoretinochoroidopathy (ADVIRC) Autosomal recessive bestrophinopathy (ARB)
|
Diagnosis
No consensus diagnostic criteria for bestrophinopathies have been published.
Suggestive Findings
A bestrophinopathy should be suspected in individuals with following the ophthalmologic findings and electrophysiologic studies by phenotype, and family history.
Ophthalmologic
findings by phenotype
Best vitelliform macular dystrophy (BVMD)
Onset age three to 15 years
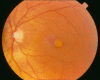
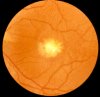
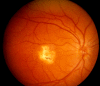
Fundus examination. A typical yellow yolk-like macular lesion may be present, usually bilateral, but in some cases unilateral. Multiple lesions and lesions outside the macula occur in at least 25% of individuals. See , , .
BEST1 adult-onset vitelliform macular dystrophy (AVMD)
Onset age 30-50 years
Fundus examination. Subretinal, small, circular, yellow vitelliform lesion; vitelliform lesion can become atrophic over time.
Autosomal dominant vitreoretinochoroidopathy (ADVIRC)
Onset in the first decade of life
Fundus examination. Peripheral retinal pigmentation, white retinal opacities
Other ocular findings. Nanophthalmos, hyperopia, microcornea, narrow-angle glaucoma
Autosomal recessive bestrophinopathy (ARB)
Best vitelliform macular dystrophy: Vitelliform stage (Stage 2)
Best vitelliform macular dystrophy: Pseudohypopyon (Stage 3)
Best vitelliform macular dystrophy: Central scarring (Stage 4b)
Electrophysiologic studies
Electrooculogram (EOG) measures indirectly the standing potential of the eye. A normal light peak / dark trough ratio (Arden ratio) is >1.8. (As the Arden ratio decreases with age after the fourth decade, this value is not absolute.)
BVMD. Usually abnormal with a reduced light peak / dark trough ratio (Arden ratio) <1.5, most often between 1.0 and 1.3.
Note: Occasionally individuals with molecularly confirmed Best vitelliform macular dystrophy have a normal EOG [
Testa et al 2008].
AVMD. Normal or only slightly reduced
ADVIRC. Abnormal
ARB. Abnormal
Full-field electroretinogram (ERG)
Spectral-domain optical coherence tomography (OCT)
BVMD and AVMD. Splitting and elevation of outer retina and retinal pigment epithelial layer with dome-like hyporeflective or hyperreflective material and subretinal fluid
ADVIRC. Retinal atrophy usually present; possible cystoid macular edema
ARB. Subretinal deposits with subretinal and/or intraretinal fluid
Fundus autofluorescence (AF)
BVMD and AVMD. Hyperautofluorescence at early stages progressing to hypofluorescence in late atrophic stages
ADVIRC. Typically normal centrally with blocked fluorescence in the periphery
ARB. Diffuse, discrete small areas of hyper- and hypoautofluorescence
Family history is typically consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations); however, autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity) has been reported in some families. Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of bestrophinopathy is established in a proband with suggestive findings and a heterozygous BEST1 pathogenic (or likely pathogenic) variant or biallelic BEST1 pathogenic (or likely pathogenic) variants identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a variant(s) of uncertain significance does not establish or rule out the diagnosis of bestrophinopathy.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive
genomic testing (exome sequencing and genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of a bestrophinopathy has not been considered may be more likely to be diagnosed using genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of BEST1 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Targeted analysis for the c.383G>C pathogenic variant can be performed first in individuals of Swedish ancestry ("pedigree S1" in Petrukhin et al [1998]). See Table 3).
A multigene panel that includes BEST1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Bestrophinopathies
View in own window
| Gene 1 | Method | Proportion of Probands with a Pathogenic Variant 2 Detectable by Method |
|---|
|
BEST1
| Sequence analysis 3 | >99% 4 |
| Gene-targeted deletion/duplication analysis 5 | Rare 4 |
- 1.
- 2.
- 3.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include a range of techniques such as quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
Clinical Characteristics
Clinical Description
Bestrophinopathies, the spectrum of ophthalmic disorders caused by pathogenic variants in BEST1, are typically characterized by retinal degeneration but may also be pan ophthalmic. The four recognized phenotypes are the three autosomal dominant disorders: Best vitelliform macular dystrophy (BVMD), BEST1 adult-onset vitelliform macular dystrophy (AVMD), and autosomal dominant vitreoretinochoroidopathy (ADVIRC); and autosomal recessive bestrophinopathy (ARB).
Table 2.
Bestrophinopathies: Frequency of Phenotypes
View in own window
| Phenotype | Ophthalmologic Findings | Comment |
|---|
| Most Common | Common | Infrequent |
|---|
| BVMD | "Egg yolk"-like vitelliform lesion |
|
|
|
| AVMD | Bilateral, small, circular vitelliform lesion | Atrophic scar | Choroidal neovascularization |
|
| ADVIRC | Peripheral chorioretinal atrophy w/white retinal opacities |
| Breakdown of blood-retinal barrier w/retinal neovascularization | |
| ARB |
|
| Choroidal neovascularization |
|
Autosomal Dominant Vitreoretinochoroidopathy (ADVIRC)
ADVIRC is characterized by circumscribed hyperpigmentation in the peripheral retina. A sharp demarcation line exists in the midperiphery between normal and abnormal retina. White pre-retinal opacities occur with the areas of hyperpigmentation. Cystoid macular degeneration is common along with pre-retinal neovascularization. Vitreous cells and vitreous fibrillar condensation can obscure vision. Affected individuals maintain good visual acuity until later in the disease course, when the entire retina becomes involved.
There is an association with nanophthalmos, microcornea, hyperopia, and narrow anterior chamber angle with angle closure glaucoma [Yardley et al 2004].
Autosomal Recessive Bestrophinopathy (ARB)
Typical age of onset in ARB is in the first decade but can be as late as the fifth decade. ARB is a more severe retinopathy than BVMD. Visual acuity can range from normal to less than 20/200 depending on the macular involvement of the disease.
The multifocal subretinal yellow deposits seen in the macula and peripheral retina are associated with subretinal fibrosis and intraretinal and subretinal fluid.
Affected individuals are hyperopic with shallow anterior chambers, making them prone to angle closure glaucoma.
Six children from three different families had biallelic pathogenic variants in BEST1 [Bitner et al 2011, Iannaccone et al 2011, Zhao et al 2012] (see Genotype-Phenotype Correlations). Four of the six had multiple vitelliform lesions. Heterozygotes (i.e., carriers) did not develop disease.
Genotype-Phenotype Correlations
For most BEST1 variants, genotype-phenotype correlations have not been demonstrated. However, a few missense variants have been associated with a milder phenotype:
Other observations:
Six children from three different families had biallelic pathogenic variants in
BEST1 [
Bitner et al 2011,
Iannaccone et al 2011,
Zhao et al 2012]. Three of the six were compound heterozygotes for the variants p.Leu41Pro and p.Ile201Thr; two of the six were homozygous for the c.1415delT variant; and one of the six was a compound heterozygote for p.Arg141Ser and p.Arg141His. Heterozygotes (i.e., carriers) did not develop disease.
Penetrance
BVMD shows high but reduced (>70%) penetrance, especially when electrooculogram is used as evidence of clinical expression.
Individuals heterozygous for a BEST1 variant associated with ARB are generally clinically unaffected [Zhao et al 2012].
Nomenclature
Other terms used to refer to BVMD include Best disease, early-onset vitelliform macular dystrophy, juvenile-onset vitelliform macular dystrophy, and polymorphic vitelline macular degeneration.
Prevalence
BVMD is a rare disorder. The prevalence has been estimated at 1:5,500 in a North American population [Dalvin et al 2017].
The BEST1 founder variant c.383G>C (p.Trp93Cys) has been identified in individuals who can trace their ancestry to a large Swedish kindred ("pedigree S1" in Petrukhin et al [1998]).
Differential Diagnosis
Best vitelliform macular dystrophy (BVMD) is the second most common hereditary macular dystrophy. The most common heritable juvenile-onset macular dystrophy is Stargardt disease.
Although the cause of adult-onset vitelliform macular dystrophy (AVMD) in most individuals is unknown, this phenotype is observed in the spectrum of bestrophinopathies and with heterozygous pathogenic variants in PRPH2 (encoding peripherin-2) (OMIM 179605), IMPG1 (OMIM 616151) and IMPG2 (OMIM 616152).
Choroidal neovascularization is an acquired disorder.
Management
No clinical practice guidelines for bestrophinopathies have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a bestrophinopathy, the evaluations summarized below (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Ophthalmologic examination, including best-corrected visual acuity, fundus examination, fundus photographs, and spectral-domain optical coherence tomography (SD-OCT) to determine the stage of disease
Consultation with a low vision specialist/clinic as needed
Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature, mode of inheritance, and implications of bestrophinopathies in order to facilitate medical and personal decision making
Treatment of Manifestations
Low vision aids benefit those individuals with significantly reduced visual acuity.
In the United States (US), educational issues for children with visual impairment can be addressed in the following:
Individualized education plan (IEP) services:
An IEP provides specially designed instruction and related services to children who qualify.
IEP services will be reviewed annually to determine whether any changes are needed.
As required by special education law, children should be in the least restrictive environment feasible at school and included in general education as much as possible and when appropriate.
Vision consultants should be a part of the child's IEP team to support access to academic material.
A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
Occupational counseling, often available through state agencies, patient advocacy groups, and health plans, should be offered.
Best vitelliform macular dystrophy (BVMD) stage 4c fundus lesions (see Clinical Description) or choroidal neovascularization and hemorrhage can be managed by direct laser photocoagulation. Marano et al [2000] suggested a conservative approach in the treatment of choroidal neovascularization based on two individuals with BVMD whose visual acuity improved. No clinical trials comparing the efficacy of laser photocoagulation to conservative treatment have been conducted.
Anti-VEGF (vascular endothelial growth factor) agents are the standard treatment for individuals with subfoveal choroidal neovascularization (CNV). Leu et al [2007] injected intravitreal bevacizumab in a boy age 13 years with BVMD and CNV, hastening visual recovery and regression of the CNV. Intravitreal ranibizumab has also been used with success [Querques et al 2009]. There are no reports on the use of aflibercept. Long-term follow up of these affected individuals is unknown. Currently there are no clinical trials to demonstrate the effectiveness of anti-VEGF agents on CNV in BVMD.
Andrade et al [2003] performed photodynamic therapy (PDT) using verteporfin for CNV on one person with BVMD. The CNV regressed and the subretinal hemorrhage resolved. The authors suggested that PDT may be an option for treatment of CNV in BVMD.
Surveillance
Ophthalmologic examination (including best-corrected visual acuity, visual fields, and SD-OCT) should be performed annually to monitor the progression of the fundus lesions and to evaluate for coincident development of choroidal neovascularization (CNV).
In children, annual examinations are important in preventing the development of amblyopia, especially if there is a significant difference in the best-corrected visual acuity of one eye. A trial of conventional patching therapy of the better-seeing eye may be able to determine if amblyopia is present.
Affected individuals should be advised to see their ophthalmologist in the event of decreased vision or metamorphopsia (straight lines appearing wavy), which could be signs of CNV. In some cases, affected individuals can be advised to use an Amsler grid for self-evaluation.
Agents/Circumstances to Avoid
Cessation of smoking helps prevent neovascularization of the retina [Clemons et al 2005].
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from prompt ophthalmologic evaluation and routine follow up.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Best vitelliform macular dystrophy (BVMD), BEST1 adult-onset vitelliform macular dystrophy (AVMD), and autosomal dominant vitreoretinochoroidopathy (ADVIRC) are inherited in an autosomal dominant manner.
By definition, autosomal recessive bestrophinopathy (ARB) is inherited in an autosomal recessive manner.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Most individuals diagnosed with BVMD, AVMD, or ADVIRC have an affected parent.
Molecular genetic testing is recommended for the parents of a proband to confirm their genetic status and to allow reliable recurrence risk counseling. Note: Fundoscopic examination may be unreliable for the evaluation of the parents because the fundus may appear normal in affected individuals.
If the pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
The proband has a
de novo pathogenic variant. Note: A pathogenic variant is reported as "
de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed
de novo" [
Richards et al 2015].
The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Though theoretically possible, no instances of a proband inheriting a pathogenic variant from a parent with germline mosaicism have been reported. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism.
The family history of some individuals diagnosed with BVMD, AVMD, or ADVIRC may appear to be negative because of failure to recognize the disorder in family members and/or reduced penetrance in a heterozygous parent. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs depends on the genetic status of the proband's parents:
If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs is 50%. Note: The age of onset, clinical manifestations, and degree of functional impairment is highly variable among heterozygous family members.
If the
BEST1 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents have not been tested for the BEST1 pathogenic variant but are clinically unaffected based on electrooculogram (EOG), the risk to sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still at increased risk for BVMD, AVMD, or ADVIRC because of the possibility of reduced penetrance in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with an autosomal dominant bestrophinopathy has a 50% chance of inheriting the BEST1 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is heterozygous for the BEST1 pathogenic variant, the parent's family members are also at risk of having the pathogenic variant.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
The parents of an affected child are typically heterozygotes (i.e., carriers of one BEST1 pathogenic variant).
Accurate recurrence risk counseling relies on carrier testing of both parents to determine if each is heterozygous for a BEST1 pathogenic variant. If carrier testing detects the variant in only one parent:
Individuals who are heterozygous for a
BEST1 variant associated with ARB are generally clinically unaffected [
Zhao et al 2012]. However, mildly diminished responses on multifocal ERG and/or mild symptoms of maculopathy have been reported in several individuals with heterozygous ARB-related
BEST1 pathogenic variants [
MacDonald et al 2012,
Wivestad Jansson et al 2016].
Sibs of a proband
If both parents are known to be heterozygous for a BEST1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Individuals who are heterozygous for a
BEST1 variant associated with ARB are generally clinically unaffected [
Zhao et al 2012]. However, mildly diminished responses on multifocal ERG and/or mild symptoms of maculopathy have been reported in several individuals with heterozygous ARB-related
BEST1 pathogenic variants [
MacDonald et al 2012,
Wivestad Jansson et al 2016].
Offspring of a proband. The offspring of an individual with ARB are obligate heterozygotes for a pathogenic variant in BEST1.
Other family members. Each sib of the proband's parents has a 50% chance of being heterozygous for a BEST1 pathogenic variant.
Heterozygote Detection
Heterozygote testing for at-risk relatives requires prior identification of the BEST1 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the BEST1 pathogenic variant(s) have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible. Note: Because intrafamilial clinical variability is observed in the bestrophinopathies, expression and age of onset cannot be predicted on the basis of prenatal molecular genetic test results.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Macular Degeneration Foundation
PO Box 531313
Henderson NV 89053
Phone: 888-633-3937 (toll-free); 702-450-2908
Fax: 702-450-3396
Email: liz@eyesight.org
National Library of Medicine Genetics Home Reference
NCBI Genes and Disease
Association for Macular Diseases
210 East 64th Street
8th Floor
New York NY 10065
Phone: 212-605-3719
Fax: 212-605-3795
Email: association@retinal-research.org
Foundation Fighting Blindness
Phone: 800-683-5555
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Bestrophinopathies: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
BEST1 encodes bestrophin, a chloride ion channel that is sensitive to intracellular calcium [Petrukhin et al 1998, Sun et al 2002]. Bestrophin is highly expressed by the retinal pigment epithelium (RPE) and localizes to the basolateral plasma membrane [Marmorstein et al 2000]. Bestrophin functions either as a chloride channel or as a regulator of voltage-gated calcium channels in the RPE [Hartzell et al 2008, Yu et al 2008].
Expression of disease-associated missense variants causes reduced or abolished membrane current. Bestrophin undergoes dephosphorylation by a protein phosphatase, suggesting that bestrophin participates in a signal transduction pathway that may be related to the modulation of the light peak on electrooculogram [Marmorstein et al 2002].
Mechanism of disease causation. Loss of function. Pathogenic variants in BEST1 alter ion transport by the RPE, resulting in the accumulation of fluid between the RPE and the photoreceptors [Qu et al 2006, Yu et al 2007, Hartzell et al 2008].
Table 3.
Notable BEST1 Pathogenic Variants
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Chapter Notes
Author History
Jessica Lawrence, MSc (2020-present)
Thomas Lee, MD (2003-present)
Ian M MacDonald, MD, CM (2003-present)
Dean Y Mah, MSc, MD; University of Alberta (2003-2009)
Revision History
16 July 2020 (bp) Comprehensive update posted live
12 December 2013 (me) Comprehensive update posted live
7 April 2009 (me) Comprehensive update posted live
8 December 2005 (me) Comprehensive update posted live
27 October 2003 (imd) Revision: sequence analysis clinically available
30 September 2003 (me) Review posted live
14 July 2003 (imd) Original submission
References
Literature Cited
Andrade RE, Farah ME, Costa RA. Photodynamic therapy with verteporfin for subfoveal choroidal neovascularization in best disease.
Am J Ophthalmol. 2003;136:1179–81. [
PubMed: 14644242]
Apushkin MA, Fishman GA, Taylor CM, Stone EM. Novel de novo mutation in a patient with Best macular dystrophy.
Arch Ophthalmol. 2006;124:887–9. [
PubMed: 16769844]
Atchaneeyasakul LO, Jinda W, Sakolsatayadorn N, Trinavarat A, Ruangvoravate N, Thanasombatskul N, Thongnoppakhun W, Limwongse C. Mutation analysis of the VMD2 gene in Thai families with Best macular dystrophy.
Ophthalmic Genet. 2008;29:139–44. [
PubMed: 18766995]
Bitner H, Mizrahi-Meissonnier L, Griefner G, Erdinest I, Sharon D, Banin E. A homozygous frameshift mutation in BEST1 causes the classical form of Best disease in an autosomal recessive mode.
Invest Ophthalmol Vis Sci. 2011;52:5332–8. [
PubMed: 21467170]
Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene.
Prog Retin Eye Res. 2009;28:187–205. [
PubMed: 19375515]
Boon CJ, van den Born LI, Visser L, Keunen JE, Bergen AA, Booij JC, Riemslag FC, Florijn RJ, van Schooneveld MJ. Autosomal recessive bestrophinopathy: differential diagnosis and treatment options.
Ophthalmology. 2013;120:809–20. [
PubMed: 23290749]
Clemons TE, Milton RC, Klein R, Seddon JM, Ferris FL 3rd, et al. Risk factors for the incidence of advanced age-related macular degeneration in the Age-Related Eye Disease Study (AREDS) AREDS report no. 19.
Ophthalmology. 2005;112:533–9. [
PMC free article: PMC1513667] [
PubMed: 15808240]
Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O'Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa.
Am J Hum Genet. 2009;85:581–92. [
PMC free article: PMC2775838] [
PubMed: 19853238]
Ellingford JM, Campbell C, Barton S, Bhaskar S, Gupta S, Taylor RL, Sergouniotis PI, Horn B, Lamb JA, Michaelides M, Webster AR, Newman WG, Panda B, Ramsden SC, Black GC. Validation of copy number variation analysis for next-generation sequencing diagnostics. Version 2.
Eur J Hum Genet. 2017;25:719–24. [
PMC free article: PMC5427176] [
PubMed: 28378820]
Eksandh L, Bakall B, Bauer B, Wadelius C, Andréasson S. Best's vitelliform macular dystrophy caused by a new mutation (Val89Ala) in the VMD2 gene.
Ophthalmic Genet. 2001;22:107–15. [
PubMed: 11449320]
Glybina IV, Frank RN. Localization of multifocal electroretinogram abnormalities to the lesion site: findings in a family with Best disease.
Arch Ophthalmol. 2006;124:1593–600. [
PubMed: 17102007]
Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies.
Physiol Rev. 2008;88:639–72. [
PubMed: 18391176]
Iannaccone A, Kerr NC, Kinnick TR, Calzada JI, Stone EM. Autosomal recessive best vitelliform macular dystrophy: report of a family and management of early-onset neovascular complications.
Arch Ophthalmol. 2011;129:211–7. [
PubMed: 21320969]
Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland.
Nature. 2017;549:519–22. [
PubMed: 28959963]
Khan KN, Islam F, Moore AT, Michaelides M. The fundus phenotype associated with the p.ala243val best1 mutation.
Retina. 2018;38:606–13. [
PubMed: 28225368]
Kinnick TR, Mullins RF, Dev S, Leys M, Mackey DA, Kay CN, Lam BL, Fishman GA, Traboulsi E, Iezzi R, Stone EM. Autosomal recessive vitelliform macular dystrophy in a large cohort of vitelliform macular dystrophy patients.
Retina. 2011;31:581–95. [
PubMed: 21273940]
Leu J, Schrage NF, Degenring RF. Choroidal neovascularisation secondary to Best's disease in a 13-year-old boy treated by intravitreal bevacizumab.
Graefes Arch Clin Exp Ophthalmol. 2007;245:1723–5. [
PubMed: 17605026]
Li Y, Wang G, Dong B, Sun X, Turner MJ, Kamaya S, Zhang K. A novel mutation of the VMD2 gene in a Chinese family with best vitelliform macular dystrophy.
Ann Acad Med Singapore. 2006;35:408–10. [
PubMed: 16865191]
MacDonald IM, Gudiseva HV, Villanueva A, Greve M, Caruso R, Ayyagari R. Phenotype and genotype of patients with autosomal recessive bestrophinopathy.
Ophthalmic Genet. 2012;33:123–9. [
PubMed: 21809908]
Marano F, Deutman AF, Leys A, Aandekerk AL. Hereditary retinal dystrophies and choroidal neovascularization.
Graefes Arch Clin Exp Ophthalmol. 2000;238:760–4. [
PubMed: 11045344]
Marchant D, Yu K, Bigot K, Roche O, Germain A, Bonneau D, Drouin-Garraud V, Schorderet DF, Munier F, Schmidt D, Le Neindre P, Marsac C, Menasche M, Dufier JL, Fischmeister R, Hartzell C, Abitbol M. New VMD2 gene mutations identified in patients affected by Best vitelliform macular dystrophy.
J Med Genet. 2007;44:e70. [
PMC free article: PMC2598027] [
PubMed: 17287362]
Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium.
Proc Natl Acad Sci U S A. 2000;97:12758–63. [
PMC free article: PMC18837] [
PubMed: 11050159]
Marmorstein LY, McLaughlin PJ, Stanton JB, Yan L, Crabb JW, Marmorstein AD. Bestrophin interacts physically and functionally with protein phosphatase 2A.
J Biol Chem. 2002;277:30591–7. [
PubMed: 12058047]
Mullins RF, Oh KT, Heffron E, Hageman GS, Stone EM. Late development of vitelliform lesions and flecks in a patient with best disease: clinicopathologic correlation.
Arch Ophthalmol. 2005;123:1588–94. [
PubMed: 16286623]
Palmowski AM, Allgayer R, Heinemann-Vernaleken B, Scherer V, Ruprecht KW. Detection of retinal dysfunction in vitelliform macular dystrophy using the multifocal ERG (MF-ERG).
Doc Ophthalmol. 2003;106:145–52. [
PubMed: 12678279]
Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, Vujic M, Bergen AA, McGarty-Dugan V, Figueroa D, Austin CP, Metzker ML, Caskey CT, Wadelius C. Identification of the gene responsible for Best macular dystrophy.
Nat Genet. 1998;19:241–7. [
PubMed: 9662395]
Pianta MJ, Aleman TS, Cideciyan AV, Sunness JS, Li Y, Campochiaro BA, Campochiaro PA, Zack DJ, Stone EM, Jacobson SG. In vivo micropathology of Best macular dystrophy with optical coherence tomography.
Exp Eye Res. 2003;76:203–11. [
PubMed: 12565808]
Qu Z, Chien LT, Cui Y, Hartzell HC. The anion-selective pore of the bestrophins, a family of chloride channels associated with retinal degeneration.
J Neurosci. 2006;26:5411–9. [
PMC free article: PMC6675304] [
PubMed: 16707793]
Querques G, Regenbogen M, Quijano C, Delphin N, Soubrane G, Souied EH. High-definition optical coherence tomography features in vitelliform macular dystrophy.
Am J Ophthalmol. 2008;146:501–7. [
PubMed: 18619572]
Querques G, Zerbib J, Santacroce R, Margaglione M, Delphin N, Querques L, Rozet JM, Kaplan J, Souied EH. The spectrum of subclinical Best vitelliform macular dystrophy in subjects with mutations in BEST1 gene.
Invest Ophthalmol Vis Sci. 2011;52:4678–84. [
PubMed: 21436265]
Querques G, Zerbib J, Santacroce R, Margaglione M, Delphin N, Rozet JM, Kaplan J, Martinelli D, Delle Noci N, Soubrane G, Souied EH. Functional and clinical data of Best vitelliform macular dystrophy patients with mutations in the BEST1 gene.
Mol Vis. 2009;15:2960–72. [
PMC free article: PMC2802291] [
PubMed: 20057903]
Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation.
Nat Genet. 2016;48:126–33. [
PMC free article: PMC4731925] [
PubMed: 26656846]
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology.
Genet Med. 2015;17:405–24. [
PMC free article: PMC4544753] [
PubMed: 25741868]
Rudolph G, Kalpadakis P. Topographic mapping of retinal function with the SLO-mfERG under simultaneous control of fixation in Best's disease.
Ophthalmologica. 2003;217:154–9. [
PubMed: 12592056]
Scholl HP, Schuster AM, Vonthein R, Zrenner E. Mapping of retinal function in Best macular dystrophy using multifocal electroretinography.
Vision Res. 2002;42:1053–61. [
PubMed: 11934455]
Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting.
Hum Genet. 2020;139:1197–207. [
PMC free article: PMC7497289] [
PubMed: 32596782]
Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels.
Proc Natl Acad Sci U S A. 2002;99:4008–13. [
PMC free article: PMC122639] [
PubMed: 11904445]
Testa F, Rossi S, Passerini I, Sodi A, Di Iorio V, Interlandi E, Della Corte M, Menchini U, Rinaldi E, Torricelli F, Simonelli F. A normal electro-oculography in a family affected by Best disease with a novel spontaneous mutation of the BEST1 gene.
Br J Ophthalmol. 2008;92:1467–70. [
PubMed: 18703557]
Van Schil K, Naessens S, Van de Sompele S, Carron M, Aslanidis A, Van Cauwenbergh C, Kathrin Mayer A, Van Heetvelde M, Bauwens M, Verdin H, Coppieters F, Greenberg ME, Yang MG, Karlstetter M, Langmann T, De Preter K, Kohl S, Cherry TJ, Leroy BP, De Baere E, et al. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations.
Genet Med. 2018;20:202–13. [
PMC free article: PMC5787040] [
PubMed: 28749477]
Wivestad Jansson R, Berland S, Bredrup C, Austeng D, Andréasson S, Wittström E. Biallelic mutations in the BEST1 gene: additional families with autosomal recessive bestrophinopathy.
Ophthalmic Genet. 2016;37:183–93. [
PubMed: 26333019]
Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM, Perveen R, Reddy MA, Bhattacharya SS, Traboulsi E, Baralle D, De Laey JJ, Puech B, Kestelyn P, Moore AT, Manson FD, Black GC. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC).
Invest Ophthalmol Vis Sci. 2004;45:3683–9. [
PubMed: 15452077]
Yu K, Qu Z, Cui Y, Hartzell HC. Chloride channel activity of bestrophin mutants associated with mild or late-onset macular degeneration.
Invest Ophthalmol Vis Sci. 2007;48:4694–705. [
PubMed: 17898294]
Yu K, Xiao Q, Cui G, Lee A, Hartzell HC. The Best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains.
J Neurosci. 2008;28:5660–70. [
PMC free article: PMC2587081] [
PubMed: 18509027]
Zhao L, Grob S, Corey R, Krupa M, Luo J, Du H, Lee C, Hughes G, Lee J, Quach J, Zhu J, Shaw PX, Kozak I, Zhang K. A novel compound heterozygous mutation in the BEST1 gene causes autosomal recessive Best vitelliform macular dystrophy.
Eye (Lond). 2012;26:866–71. [
PMC free article: PMC3376281] [
PubMed: 22422030]