Summary
NOTE: THIS PUBLICATION HAS BEEN RETIRED. THIS ARCHIVAL VERSION IS FOR HISTORICAL REFERENCE ONLY, AND THE INFORMATION MAY BE OUT OF DATE.
Clinical characteristics.
Charcot-Marie-Tooth neuropathy type 1 (CMT1) is a demyelinating peripheral neuropathy characterized by distal muscle weakness and atrophy, sensory loss, and slow nerve conduction velocity. It is usually slowly progressive and often associated with pes cavus foot deformity and bilateral foot drop. Affected individuals usually become symptomatic between age five and 25 years. Fewer than 5% of individuals become wheelchair dependent. Life span is not shortened.
Diagnosis/testing.
CMT1A (70%-80% of all CMT1) involves duplication of PMP22. CMT1B (6%-10% of all CMT1) is associated with single-nucleotide variants in MPZ. CMT1C (1%-2% of all CMT1) is associated with pathogenic variants in LITAF, and CMT1D (<2% of all CMT1) is associated with pathogenic variants in EGR2. CMT1E (<5% of all CMT1) is associated with single-nucleotide variants in PMP22. CMT2E/1F (<5% of all CMT1) is associated with pathogenic variants in NEFL.
Management.
Treatment of manifestations: Treatment by a multidisciplinary team including a neurologist, physiatrist, orthopedic surgeon, physical and occupational therapists; special shoes and/or ankle/foot orthoses to correct foot drop and aid walking; surgery as needed for severe pes cavus; forearm crutches, canes, wheelchairs as needed for mobility; exercise as tolerated.
Prevention of secondary complications: Daily heel cord stretching to prevent Achilles' tendon shortening.
Surveillance: Regular foot examination for pressure sores.
Agents/circumstances to avoid: Obesity (makes ambulation more difficult); medications (e.g., vincristine, isoniazid, nitrofurantoin) known to cause nerve damage.
Pregnancy management: Affected pregnant women may experience worsening symptoms during or after gestation; a higher occurrence of presentation anomalies, use of forceps, and operative delivery; and/or an increased incidence of post-partum bleeding.
Genetic counseling.
CMT1 is inherited in an autosomal dominant manner. About two thirds of probands with CMT1A have inherited the PMP22 duplication; about one third have CMT1A as the result of a de novo pathogenic variant. Similar data are not available for the other subtypes of CMT1. The offspring of an individual with any of the subtypes of CMT1 have a 50% chance of inheriting the altered gene. Prenatal testing is possible if the pathogenic variant has been identified in the family. Requests for prenatal testing for typically adult-onset diseases that do not affect intellect or life span are uncommon.
GeneReview Scope
View in own window
| Charcot-Marie-Tooth Neuropathy Type 1: Included Disorders |
|---|
CMT1A CMT1B CMT1C CMT1D CMT1E CMT2E/1F
|
Diagnosis
Clinical Diagnosis
Charcot-Marie-Tooth neuropathy type (CMT1) is diagnosed in individuals with the following:
A progressive peripheral motor and sensory neuropathy
Slow nerve conduction velocity (NCV). NCVs are typically 10-30 meters per second, with a range of 5-38 m/s (normal: >40-45 m/s).
Palpably enlarged nerves, especially the ulnar nerve at the olecranon groove and the greater auricular nerve running along the lateral aspect of the neck
Molecular Genetic Testing
Genes. The CMT1 subtypes and the genes associated with them are summarized in Table 1. The complicated genetic diversity of hereditary neuropathies with emphasis on CMT syndrome has been addressed by Baets et al [2014], Pareyson et al [2014] and Bird [Charcot-Marie-Tooth Hereditary Neuropathy Overview.] Many genetic testing strategies have been proposed including that of Saporta et al [2011a].
Clinical testing
Table 1.
Summary of Molecular Genetic Testing Used in Charcot-Marie-Tooth Neuropathy Type 1 (CMT1)
View in own window
| CMT1 Subtype | Gene 1 | Proportion of CMT1 Attributed to Pathogenic Variants in This Gene | Test Method |
|---|
| CMT1A |
PMP22
| 70%-80% | Targeted analysis for pathogenic variants 2 |
| CMT1B | MPZ 3 | 5%-10% | Sequence analysis 4 |
| Deletion/duplication analysis 5 |
| CMT1C | LITAF 3
(previously known as SIMPLE) | 1%-2% | Sequence analysis 4 |
| Deletion/duplication analysis 5 |
| CMT1D | EGR2 3 | <2% | Sequence analysis 4 |
| Deletion/duplication analysis 5 |
| CMT1E | PMP22 3 | <5% | Sequence analysis 4 |
| Deletion/duplication analysis 5 |
| CMT2E/1F | NEFL 3 | <5% | Sequence analysis 4 |
| Deletion/duplication analysis 5 |
| Unknown 6 | NA | NA |
- 1.
- 2.
Detects a1.5-Mb duplication at 17p11.2 that includes PMP22 resulting in the presence of three copies of PMP22 in all individuals with CMT1A. The test method is a deletion/duplication analysis targeted specifically at the PMP22 duplication; a variety of test methods can be used (see footnote 5) in addition to FISH.
- 3.
Each of these subtypes is identified based on detection of a pathogenic variant in the associated gene; hence, the variant detection rate is 100%.
- 4.
- 5.
- 6.
Testing Strategy
To confirm/establish the diagnosis in a proband with slow nerve conduction velocities, one genetic testing strategy is serial single gene molecular genetic testing based on the order in which pathogenic variants most commonly occur.
- 1.
Because CMT1A (caused by the 1.5-Mb duplication at 17p11.2 including PMP22) is by far the most common type of CMT1, it is appropriate to test a proband with very slow nerve conduction velocities for this duplication first [Klein & Dyck 2005].
- 2.
If no PMP22 duplication is identified, the next step is molecular genetic testing of MPZ and GJB1 (a cause of X-linked CMT). Note: If the family history shows male to male transmission, testing of GJB1, mutation of which causes Charcot-Marie-Tooth Neuropathy X Type 1, is not appropriate.
- 3.
If no PMP22 duplication, MPZ pathogenic variant, or GJB1 pathogenic variant is identified, consider sequence analysis of LITAF, EGR2, PMP22 (single nucleotide variants) and NEFL [Saporta et al 2011a].
Note: This testing strategy is different from that for axonal neuropathies and autosomal recessive neuropathies.
An alternative genetic testing strategy is use of a multigene panel that includes PMP22, MPZ, GJB1, and other genes of interest (see Table 1 and Differential Diagnosis). Note: The genes included and the methods used in multigene panels vary by laboratory and over time. Success of this approach is demonstrated by Klein et al [2014], who were able to identify the genetic cause of CMT in five of 15 kindreds (using exome sequencing) who had escaped earlier detection by single-gene analysis. For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Clinical Characteristics
Clinical Description
Classic CMT1 Phenotype
Individuals with CMT1 usually become symptomatic between age five and 25 years [Marques et al 2005, Houlden & Reilly 2006, van Paassen et al 2014]; age of onset ranges from infancy (resulting in delayed walking) to the fourth and subsequent decades. Clinical severity is variable, ranging from extremely mild disease that goes unrecognized by the affected individual and physician to considerable weakness and disability.
The typical presenting symptom of CMT1 is weakness of the feet and ankles [Ferrarin et al 2012]. The initial physical findings are depressed or absent tendon reflexes with weakness of foot dorsiflexion at the ankle. The typical affected adult has bilateral foot drop, symmetric atrophy of muscles below the knee (stork leg appearance), atrophy of intrinsic hand muscles, and absent tendon reflexes in both upper and lower extremities.
Onset in the first year of life often suggests an autosomal recessive cause of CMT but autosomal dominant types of CMT caused by duplication of PMP22 (CMT1A) and pathogenic missense variants in PMP22 (CMT1E), MPZ (CMT1B), and NEFL have been reported in this age group [Baets et al 2011].
Proximal muscles usually remain strong.
Mild to moderate sensory deficits of position, vibration, and pain/temperature commonly occur in the feet, but many affected individuals are unaware of this finding. Pain, especially in the feet, is reported by 20%-30% of individuals [Carter et al 1998, Gemignani et al 2004, Carvalho et al 2005]. The pain is often musculoskeletal in origin but may be neuropathic in some cases [Pazzaglia et al 2010].
Poretti et al [2013] have shown that the vestibular impairment may contribute to the poor balance often present in CMT1.
In a study of 61 subjects with CMT1, Boentert et al [2014] found that 37% had obstructive sleep apnea and 40% had restless leg syndrome. If these findings are confirmed they would represent an important newly recognized aspect of the CMT1 phenotype.
Episodic pressure palsies have been reported [Kleopa et al 2004].
In CMT1A, prolonged distal motor latencies may already be present in the first months of life, and slow motor nerve conduction velocities (NCVs) have been found in some individuals by age two years [Krajewski et al 2000]. However, the full clinical picture may not occur until the second decade of life or later [García et al 1998]. In a study of 57 individuals with CMT1A, three had floppy infant syndrome, two had marked proximal and distal weakness (one requiring a wheelchair), one had severe scoliosis, five had calf muscle hypertrophy, and three had hand deformity [Marques et al 2005].
Some individuals with CMT1B have onset in the first decade of life; others have a much later onset. The age of onset trend tends to run true in families [Hattori et al 2003].
CMT1 is slowly progressive over many years. Affected individuals experience long plateau periods without obvious deterioration [Teunissen et al 2003]. NCVs slow progressively over the first two to six years of life and are relatively stable throughout adulthood. Early onset of symptoms and severity of disease show some correlation with slower NCVs, but this is only a general trend. Muscle weakness correlates with progressive decrease in the compound muscle action potential (CMAP) and suggests that developing axonal pathology is of considerable clinical relevance [Hattori et al 2003, Pareyson et al 2006].
In a study of persons with CMT1A over a five-year period, Verhamme et al [2009a] found increasing disability at least partially related to “a process of normal aging.” In a study of a large family with CMT1A over two decades, Berciano et al [2010] found that deterioration varied from mild to marked. It remains unclear why such a wide range of severity is observed in persons with CMT1A with the same pathogenic variant (PMP22 dup).
In CMT1A, Kim et al [2012] found that severity of weakness and sensory loss correlated with CMAPs and SNAPs (sensory nerve action potential), but not with conduction velocities.
The disease does not decrease life span.
Other findings in individuals with CMT1. A few men with CMT1 have reported impotence [Bird et al 1994].
Pes cavus foot deformity is common (>50%) and hip dysplasia may be under-recognized [Walker et al 1994, McGann & Gurd 2002].
Pulmonary insufficiency and sleep apnea are sometimes seen [Dematteis et al 2001].
Deafness has been occasionally reported in the CMT1 phenotype. Impaired auditory perception and processing has been reported as common (>60%) both in children with CMT1 and in those with CMT2 [Rance et al 2012]. Hearing loss has been associated with single-nucleotide variants in PMP22 (CMT1E) [Kovach et al 1999, Sambuughin et al 2003, Postelmans & Stokroos 2006] and MPZ (CMT1B) [Starr et al 2003, Seeman et al 2004].
Vestibular abnormalities have been reported both in persons with CMT1A and in those with CMTX [Poretti et al 2013].
Lower-limb muscle atrophy and fatty infiltration can be demonstrated by MRI and followed longitudinally [Gallardo et al 2006].
Chanson et al [2013] reported MRI findings of decreased white matter volume in both CMT1A and hereditary neuropathy with liability to pressure palsies (HNPP). This was confirmed in one pathologic examination of a brain from a subject with CMT1A.
Colomban et al [2014] reported earlier onset of symptoms (8.6 vs 14 years) and higher deterioration of quality of life in affected women compared to affected men.
People with CMT1A can have symptoms that mimic those of HNPP [Mathis et al 2014].
Quality of life from the affected individual’s perspective has been studied by Johnson et al [2014]. Foot and ankle weakness, impaired balance, pain, and fatigue were viewed as important disabling symptoms and tended to be more prevalent in affected women. Ramdharry et al [2012b] also reported a high prevalence of fatigue as a symptom in persons with CMT.
Pregnancy. See Pregnancy Management.
CMT1 Subtypes
The CMT1 subtypes, identified solely by molecular findings, are often clinically indistinguishable.
CMT1A. NCVs vary. Mean median motor NCVs were 21±5.7 m/s in one study [Hattori et al 2003] and 16.5 m/s (range: 5-26.5 m/s) in another [Carvalho et al 2005]. In a third study, the range was 12.6-35 m/s [Marques et al 2005]. CMAP is decreased [Hattori et al 2003].
CMT1B. The NCV shows a bimodal curve, with some families having slow median motor NCV (mean: 16.5 m/s) and others having normal or near-normal NCV (mean: 44.3 m/s). The individuals in this latter "normal" NCV group tend to have lower CMAP, later age of onset, and more frequent hearing loss and pupillary abnormalities. These findings suggest the existence of two types of CMT1B: primarily demyelinating and primarily axonal. The two types probably reflect functional differences (early onset gain of function versus late onset loss of function of the MPZ protein) caused by different pathogenic variants in MPZ (see Genotype-Phenotype Correlations) [Hattori et al 2003, Shy et al 2004, Grandis et al 2008].
CMT1C. This subtype appears to be clinically identical to CMT1A [Bennett et al 2004, Saifi et al 2005, Latour et al 2006]. NCVs range from 7.5 to 27 m/s with occasional temporal dispersion [Bennett et al 2004] and conduction block with variable age of onset including early childhood [Gerding et al 2009]. Using ultrasound, Luigetti et al [2015] found enlarged peripheral nerves in individuals with CMT1C.
CMT1D. A few families with CMT1D have been identified [Warner et al 1998, Nelis et al 1999b, Numakura et al 2003, Shiga et al 2012].
CMT1E. An amino acid substitution in PMP22 in exon 3 (p.Ala67Pro) is associated with deafness in a family with CMT1 previously reported by Kousseff et al [1982], Kovach et al [1999], Kovach et al [2002].
The amino acid substitution p.Trp28Arg was associated with profound deafness in one family [Boerkoel et al 2002].
The amino acid substitution p.Ser22Phe in PMP22 is associated with pressure palsies as well as the CMT1 phenotype in a Cypriot family [Kleopa et al 2004].
In addition to the above, the following findings in affected families demonstrate further heterogeneity in the CMT1 phenotype:
Neuropathology
CMT1A. Microscopically, the enlarged nerves show hypertrophy and onion bulb formation thought to result from repeated demyelination and remyelination of Schwann cell wrappings around individual axons [Carvalho et al 2005, Schröder 2006].
CMT1B. Individuals with slow NCVs tend to have demyelinating features on nerve biopsy, whereas those with normal NCVs have more axonal pathology with axonal sprouting [Hattori et al 2003]. Onion bulb formation has been seen [Bai et al 2006]. Excessive myelin folding and thickness were reported in a family with a c.336delA null variant in MPZ [De Angelis et al 2004].
Genotype-Phenotype Correlations
CMT1A. A relative gene dosage effect exists regarding genotype-phenotype correlation:
Severe neuropathy has been reported in persons with CMT1A and a second neuropathy-causing disease such as CMT1C [Meggouh et al 2005], CMTX1, myotonic dystrophy type 1 (DM1) or adrenomyeloneuropathy (see X-Linked Adrenoleukodystrophy) [Hodapp et al 2006].
CMT1B
MPZ pathogenic variants with normal or near-normal NCVs include:
p.Ser44Phe,
p.Ser59Thr,
p.Asp75Val,
p.His81Arg,
p.Tyr82His,
p.Thr124Met,
p.Lys130Arg, and
p.Gly167Arg [
Marrosu et al 1998,
De Jonghe et al 1999,
Young et al 2001,
Hattori et al 2003,
Bienfait et al 2006a,
Finsterer et al 2006].
Brozková et al [2010] discuss the care which must be taken in sorting out the pathogenicity of ten different DNA variants in
MPZ.
CMT1C
CMT1D
CMT1E
Individuals with
PMP22 single-nucleotide variants tend to have more severe clinical disability than persons with a single 17p11.2
duplication, presumably because of a
dominant-negative or loss of protein-function effect [
Fabrizi et al 2001b].
Abe et al [2010] reported a child with severe CMT and compound heterozygosity for complete
deletion of
PMP22 on one
allele and deletion of
PMP22 exon 5 on the other allele.
CMT1F. Two different pathogenic variants in codon 22 of NEFL (p.Pro22Thr and p.Pro22Arg) have been reported with demyelinating autosomal dominant CMT1F [Shin et al 2008]. The p.Pro22Ser pathogenic variant in NEFL is associated with autosomal recessive CMT2E.
Penetrance
Penetrance of CMT1 is usually nearly 100%, but the wide range in age of onset and severity may result in under-recognition of individuals with mild or late-onset disease.
Nomenclature
CMT1A/CMT1E. CMT1A refers to CMT1 caused by duplication of PMP22; CMT1E refers to CMT1 caused by single-nucleotide variants in PMP22.
CMT2E/1F. Some individuals with pathogenic variants in NEFL, which typically cause CMT2E, may have slow NCVs, resulting in a diagnosis of CMT1F. To accommodate these two phenotypes associated with mutation of NEFL, the designation CMT2E/1F has been used.
Dejerine-Sottas syndrome (DSS). The severe phenotype associated with onset in early childhood has in the past been called Dejerine-Sottas syndrome (DSS). However, DSS is a confusing term because it no longer refers to a specific phenotype caused by pathogenic variants in a specific gene. Pathogenic variants in at least three genes (PMP22, MPZ, and EGR2) have been associated with a severe early-onset phenotype:
Heterozygosity for
de novo autosomal dominant single-nucleotide variants in both
PMP22 and
MPZ and homozygosity for
PMP22 pathogenic variants have been found in individuals with severe childhood-onset disease.
Three
missense variants at codon 72 of
PMP22 are associated with this
phenotype, suggesting that codon 72 pathogenic variants lead to a severe phenotype [
Nelis et al 1999a].
Autosomal recessive forms of CMT may cause the DSS
phenotype.
Prevalence
The overall prevalence of hereditary neuropathies is estimated at approximately 30:100,000 population. The prevalence of CMT1 is 15:100,000-20:100,000. The prevalence of CMT1A is approximately 10:100,000. These numbers hold true in a great variety of regions including China [Song et al 2006, Szigeti et al 2006].
CMT1A represents about 70% of CMT1 [Reilly & Shy 2009] and CMT1B represents about 6%-10% of CMT1 [Mandich et al 2009].
In a large study of German individuals with a CMT1 phenotype (776), Gess et al [2013] found the following percentages: CMT1A (51%), CMTX1 (9%), and CMT1B (5%). Among those with a CMT1 phenotype, 66% had a genetic diagnosis.
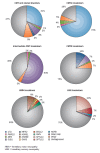
shows the frequency of various genetic causes of CMT [Rossor et al 2013], indicating that the PMP22 duplication on chromosome 17p is responsible for approximately 31% of all CMT cases and approximately 70% of those with the CMT1 phenotype.
Genetic diagnoses in CMT and related disorders From Rossor et al [2013]; reprinted with permission
In a Chinese population Liu et al [2013] found pathogenic variants in MPZ in 3% of individuals with CMT1 and in 6% of those with CMT2.
Differential Diagnosis
Acquired causes of neuropathy and other inherited neuropathies need to be considered (see CMT Overview). The differential diagnosis includes other genetic neuropathies, especially CMTX, CMT2, CMT4, and HNPP, all of which show considerable phenotypic overlap [Bienfait et al 2006b].
FBLN5.
Auer-Grumbach et al [2011] found pathogenic variants in FBLN5 in families with features of CMT1; FBLN5 pathogenic variants were additionally associated with age-related macular degeneration and cutis laxa. Šafka Brozková et al [2013] have found the same pathogenic missense variant in FBLN5 in a Czech family with CMT1 and a different background haplotype compared with the Austrian family reported by Auer-Grumbach.
GJB3.
López-Bigas et al [2001] have described an autosomal dominant neuropathy associated with hearing impairment caused by a pathogenic variant in GJB3. Although the sural nerve pathology showed demylination compatible with CMT1, the nerve conduction velocities (NCVs) were not markedly slow and may suggest an axonal neuropathy.
Familial slow NCV (OMIM 608236). Verhoeven et al [2003] have described a family with no symptoms or signs, but with slow NCVs associated with a pathogenic variant in ARHGEF10, encoding the protein rho guanine nucleotide exchange factor 10.
In the autosomal dominant intermediate forms of CMT, individuals have a relatively typical CMT phenotype with NCVs that overlap those observed in CMT1 (demyelinating neuropathy) and CMT2 (axonal neuropathy) [Villanova et al 1998]. Motor NCVs in these families usually range between 25 and 50 m/s. Five types are recognized to date:
It is usually not possible to differentiate between intermediate forms of CMT and most CMT2 subtypes based on clinical findings [Nicholson & Myers 2006] unless cataract and/or neutropenia (occasional findings in DI-CMTB) are present.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Charcot-Marie-Tooth neuropathy type 1 (CMT1), the following evaluations are recommended:
Physical examination to determine extent of weakness and atrophy, pes cavus, gait stability, and sensory loss
NCV to help distinguish demyelinating, axonal, and mixed forms of neuropathy
Detailed family history
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Individuals with CMT1 are often evaluated and managed by a multidisciplinary team that includes neurologists, physiatrists, orthopedic surgeons, and physical and occupational therapists [Carter 1997, Grandis & Shy 2005].
Treatment is symptomatic and may include the following:
Special shoes, including those with good ankle support; affected individuals often require ankle/foot orthoses (AFOs) to correct foot drop and aid walking [
Ramdharry et al 2012a].
Forearm crutches or canes for gait stability for some individuals; fewer than 5% of individuals need wheelchairs.
Exercise within the individual's capability; many remain physically active. Exercise is
not detrimental to persons with CMT [
Piscosquito et al 2014].
Accurate identification, as far as possible, of the cause of pain:
Career and employment counseling to address persistent weakness of hands and/or feet
Interventions designed to improve leg cramps, tremor, agility, endurance, and ankle flexibility, thereby improving quality of life; see
Burns et al [2010] study of children with CMT1A.
Prevention of Primary Manifestations
No treatment reverses or slows the natural progression of CMT.
Prevention of Secondary Complications
Daily heel cord stretching exercises to prevent Achilles' tendon shortening are desirable.
Surveillance
Individuals should be evaluated regularly by a team comprising physiatrists, neurologists, and physical and occupational therapists to determine neurologic status and functional disability.
Agents/Circumstances to Avoid
Obesity is to be avoided because it makes walking more difficult.
Medications that are toxic or potentially toxic to persons with CMT comprise a spectrum of risk ranging from definite high risk to negligible risk. See the Charcot-Marie-Tooth Association website (pdf) for an up-to-date list.
Pregnancy Management
Rudnik-Schöneborn et al [1993] evaluated 45 pregnancies in 21 women with CMT1. Worsening of the CMT1 symptoms during or after gestation was reported in about half of pregnancies. A follow-up study of 63 pregnancies in 33 women with CMT showed no serious complications but 20% of women reported a worsening of symptoms during pregnancy [Awater et al 2012]. In a study of affected pregnant women in Norway, deliveries involved a higher occurrence of presentation anomalies, use of forceps, and operative delivery; the women also experienced increased post-partum bleeding [Hoff et al 2005].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Charcot-Marie-Tooth neuropathy type 1 (CMT1) is inherited an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Note: Although most individuals diagnosed with CMT1 have an affected parent, the family history may appear to be negative because of failure to recognize the disorder in family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. If the parent is the individual in whom the pathogenic variant first occurred, s/he may have somatic mosaicism for the pathogenic variant and may be mildly/minimally affected.
Sibs of a proband
The risk to the sibs depends on the genetic status of the
proband's parents.
If a parent has the
PMP22,
MPZ,
LITAF, or
EGR2 pathogenic variant, the risk to sibs is 50%.
When the parents are clinically unaffected, the risk to the sibs of a
proband appears to be low.
Offspring of a proband. Every child of an individual with CMT1 has a 50% chance of inheriting the PMP22, MPZ, LITAF, or EGR2 pathogenic variant.
Other family members of a proband
The risk to other family members depends on the status of the
proband's parents.
Prenatal Testing
If the PMP22, MPZ, LITAF, or EGR2 pathogenic variant has been identified in an affected family member, prenatal testing for pregnancies at increased risk may be available from a clinical laboratory that offers either testing of the gene or custom prenatal testing.
Requests for prenatal testing for typically adult-onset conditions which (like CMT1) do not affect intellect or life span are not common. Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. Although most centers would consider decisions about prenatal testing to be the choice of the parents, discussion of these issues is appropriate.
Preimplantation genetic testing (PGT) may be an option for some families in which the PMP22, MPZ, LITAF, or EGR2 pathogenic variant has been identified. Successful use of PGT for CMT1A has been reported [Lee et al 2013].
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Association CMT France
France
Phone: 820 077 540; 2 47 27 96 41
Charcot-Marie-Tooth Association (CMTA)
PO Box 105
Glenolden PA 19036
Phone: 800-606-2682 (toll-free); 610-499-9264
Fax: 610-499-9267
Email: info@cmtausa.org
European Charcot-Marie-Tooth Consortium
Department of Molecular Genetics
University of Antwerp
Antwerp Antwerpen B-2610
Belgium
Fax: 03 2651002
Email: gisele.smeyers@ua.ac.be
Hereditary Neuropathy Foundation, Inc.
432 Park Avenue South
4th Floor
New York NY 10016
Phone: 855-435-7268 (toll-free); 212-722-8396
Fax: 917-591-2758
Email: info@hnf-cure.org
My46 Trait Profile
National Library of Medicine Genetics Home Reference
NCBI Genes and Disease
TREAT-NMD
Institute of Genetic Medicine
University of Newcastle upon Tyne
International Centre for Life
Newcastle upon Tyne NE1 3BZ
United Kingdom
Phone: 44 (0)191 241 8617
Fax: 44 (0)191 241 8770
Email: info@treat-nmd.eu
Association Francaise contre les Myopathies (AFM)
1 Rue de l'International
BP59
Evry cedex 91002
France
Phone: +33 01 69 47 28 28
Email: dmc@afm.genethon.fr
European Neuromuscular Centre (ENMC)
Lt Gen van Heutszlaan 6
3743 JN Baarn
Netherlands
Phone: 31 35 5480481
Fax: 31 35 5480499
Email: enmc@enmc.org
Muscular Dystrophy Association - USA (MDA)
222 South Riverside Plaza
Suite 1500
Chicago IL 60606
Phone: 800-572-1717
Email: mda@mdausa.org
Muscular Dystrophy UK
61A Great Suffolk Street
London SE1 0BU
United Kingdom
Phone: 0800 652 6352 (toll-free); 020 7803 4800
Email: info@musculardystrophyuk.org
RDCRN Patient Contact Registry: Inherited Neuropathies Consortium
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Charcot-Marie-Tooth Neuropathy Type 1: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
118200 | CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1B; CMT1B |
|
118220 | CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1A; CMT1A |
|
118300 | CHARCOT-MARIE-TOOTH DISEASE AND DEAFNESS |
|
129010 | EARLY GROWTH RESPONSE 2; EGR2 |
|
159440 | MYELIN PROTEIN ZERO; MPZ |
|
162280 | NEUROFILAMENT PROTEIN, LIGHT POLYPEPTIDE; NEFL |
|
601097 | PERIPHERAL MYELIN PROTEIN 22; PMP22 |
|
601098 | CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1C; CMT1C |
|
603795 | LIPOPOLYSACCHARIDE-INDUCED TUMOR NECROSIS FACTOR-ALPHA FACTOR; LITAF |
|
607678 | CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1D; CMT1D |
|
607734 | CHARCOT-MARIE-TOOTH DISEASE, DEMYELINATING, TYPE 1F; CMT1F |
PMP22 (CMT1A, CMT1E)
Gene structure.
PMP22 transcript variant 1 (NM_000304.2) has 1828 nucleotides and five exons, four of which encode amino acids [Patel et al 1992]. It is similar to a growth arrest-specific gene in mouse and rat. For a detailed summary of gene and protein information for the following genes, see Table A, Gene.
Pathogenic variants
Table 2.
Selected PMP22 Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.47T>C | p.Leu16Pro |
NM_000304.2
NP_000295.1
|
| c.65C>T | p.Ser22Phe |
| c.82T>C | p.Trp28Arg |
| c.117G>C | p.Trp39Cys |
| c.199G>C | p.Ala67Pro |
| c.353C>T 2 | p.Thr118Met |
| c.469C>T 3 | p.Arg157Trp |
| c.281dupG 3 | p.Arg95GlnfsTer128
(Gly94fsTer222) |
| (1.5-Mb duplication at 17p11.2) | -- |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
- 2.
- 3.
Normal gene product. Peripheral myelin protein 22 is a 160-amino acid protein that is present in compact myelin and has four transmembrane domains.
Abnormal gene product. Duplication of PMP22 is associated with increased mRNA message for PMP22 in peripheral nerve and by an unknown mechanism that results in abnormal myelination [Gabriel et al 1997].
Most pathogenic missense variants are localized in the transmembrane domains of peripheral myelin protein 22, indicating the functional importance of these domains. Individuals with PMP22 single-nucleotide variants tend to have more severe clinical disability than those with a single 17p11.2 duplication, presumably because of a dominant-negative or loss-of-protein function effect [Sereda & Nave 2006].
A mouse containing eight copies of human PMP22 shows a phenotype similar to but more severe than that seen in individuals with CMT1A, while mice containing 16 and 30 additional copies of mouse PMP22 show severe hypomyelination [Nelis et al 1999a]. This supports the hypothesis that more copies of PMP22 result in a more severe phenotype [Giambonini-Brugnoli et al 2005].
Perea et al [2001] have generated a transgenic mouse model in which mouse PMP22 over-expression can be regulated, possibly providing a system for evaluation of potential therapeutic approaches.
MPZ (CMT1B)
Gene structure.
MPZ spans approximately seven kilobases and contains six exons. The reference sequence was updated in 2010 to encode the 248 amino acid protein NM_000530.6, which should be considered when referring to MPZ pathogenic variants (for detailed information, see entry for MPZ at Inherited Peripheral Neuropathies Mutation Database).
Pathogenic variants. Nearly 100 pathogenic variants in MPZ have been reported [De Jonghe et al 1997, Nelis et al 1999a, Kochański et al 2004, Lee et al 2004, Shy 2006]. More than 70% of the pathogenic variants are localized in exons 2 and 3 of MPZ, which code for the extracellular domain, indicating the functional importance of this domain. Intronic variants affecting MPZ splicing have been reported [Sabet et al 2006]. (For more information, see Table A.) A duplication of the entire MPZ gene was detected in a Norwegian family with an autosomal dominant, early onset (first decade), severe, demyelinating CMT syndrome [Høyer et al 2011].
Table 3.
Selected MPZ Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.89T>C 2 | p.Ile30Thr |
NM_000530.6
NP_000521.2
|
| c.131C>T 2 | p.Ser44Phe |
| c.164G>T 2 | p.Ser55Ile |
| c.181G>A | p.Asp61Asn |
| c.175T>A 2 | p.Ser59Thr |
| c.224A>T 2 | p.Asp75Val |
| c.241C>T | p.His81Tyr |
| c.[241C>T;337G>T] 2, 3 | p.[His81Tyr;Val113Phe] |
| c.244T>C | p.Tyr82His |
| c.296T>C 4 | p.Ile99Thr |
| c.306delA 2, 5 | p.Asp104ThrfsTer14 |
| c.347A>G | p.Asn116Ser |
| c.337G>T 2 | p.Val113Phe |
| c.371C>T 2 | p.Thr124Met |
| c.389A>G 2 | p.Lys130Arg |
| c.393C>A 6 | p.Asn131Lys |
| c.487G>A | p.Gly163Arg |
| c.499G>A 2 | p.Gly167Arg |
| c.588dupT | p.Met197TyrTer38
(Met207TyrfsTer38) |
| c.670G>T 2 | p.Asp224Tyr |
| c.645+1G>T 2 | NA |
| c.649C>T 2 | p.Pro217Ser |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
- 2.
- 3.
Nomenclature for variants in cis configuration
- 4.
- 5.
- 6.
Normal gene product. P0 myelin protein is a major structural component of peripheral myelin, representing about 50% of peripheral myelin protein by weight and about 7% of Schwann cell message [Wells et al 1993]. It is a homophilic adhesion molecule of the immunoglobulin family that plays an important role in myelin compaction. It has a single transmembrane domain, a large extracellular domain, and a smaller intracellular domain. It is also expressed in glomerular epithelial cells of the kidney [Plaisier et al 2005].
Abnormal gene product. Different pathogenic variants affect all portions of the protein and may alter myelin adhesion or produce an unfolded protein response [Wrabetz et al 2006]. Either demyelinating or axonal phenotypes can result. Grandis et al [2008] found that pathogenic variants associated with late-onset disease cause a partial loss of function in transfected cells, whereas pathogenic variants associated with early-onset disease cause abnormal gain of function. Abnormal MPZ is retained in the endoplasmic reticulum of Schwann cells causing a transitory canonic unfolded protein response [Pennuto et al 2008, Saporta et al 2012].
LITAF (CMT1C)
Gene structure.
LITAF has three coding exons. For a detailed summary of gene and protein information for the following genes, see Table A, Gene.
Benign variants. A benign variant was reported by Bennett et al [2004].
Pathogenic variants. Missense variants have been reported in LITAF [Street et al 2003, Bennett et al 2004, Saifi et al 2005, Latour et al 2006] (Table 4). (For more information, see Table A.) The pathogenicity of some DNA changes is difficult to determine [Kochański 2006].
Table 4.
Selected LITAF Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.332C>G | p.Ala111Gly |
NM_004862.3
NP_004853.2
|
| c.334G>A 1 | p.Gly112Ser |
| c.344C>A | p.Thr115Asn |
| c.346T>G | p.Trp116Gly |
| c.385G>A | p.Ala129Thr |
| c.403C>T | p.Pro135Ser |
| c.403C>A | p.Pro135Thr |
| c.404C>G | p.Pro135Arg |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Normal gene product. The protein product of LITAF has two names: lipopolysaccaride-induced tumor necrosis factor-α factor (LITAF) and small integral membrane protein of the lysosome/late endosome (SIMPLE) [Saifi et al 2005]. The gene may play a role in the lysosomal sorting of plasma membrane proteins [Shirk et al 2005].
Abnormal gene product. Mutation may alter the ability of the Schwann cell to degrade proteins.
EGR2 (CMT1D)
Gene structure.
EGR2 spans 4.3 kb and contains two coding exons. For a detailed summary of gene and protein information for the following genes, see Table A, Gene.
Pathogenic variants. Selected autosomal dominant pathogenic variants are listed in Table 5 [Timmerman et al 1999, Pareyson et al 2000]. (For more information, see Table A.) The pathogenicity of some DNA changes is difficult to determine [Kochański 2006].
Table 5.
Selected EGR2 Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.1075C>T 1 | p.Arg359Trp |
NM_000399.3
NP_000390.2
|
| c.1076G>A 1 | p.Arg359Gln |
| c.1142G>A 1 | p.Arg381His |
| c.1144A>C or c.1146T>A | p.Ser382Arg |
| c.1147G>T 1 | p.Asp383Tyr |
| c.1160C>A 1 | p.Thr387Asn |
| c.1225C>T | p.Arg409Trp |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Normal gene product. Early growth response-2 protein is a zinc finger transcription factor. It is the ortholog of the murine Krox-2 protein. EGR2 induces expression of several proteins involved in myelin sheath formation and maintenance.
Abnormal gene product.
Krox-2
null mice show a block in Schwann cell differentiation.
NEFL (CMT2E/1F)
Gene structure. Both the mouse and human NEFL have four coding exons; the 5' UTRs are highly conserved. For a detailed summary of gene and protein information for the following genes, see Table A, Gene.
Pathogenic variants. See Table 6. (For more information, see Table A.)
Table 6.
Selected NEFL Pathogenic Variants
View in own window
| DNA Nucleotide Change | Predicted Protein Change
(Alias 1) | Reference Sequences |
|---|
| c.23C>G | p.Pro8Arg |
NM_006158.3
NP_006149.2
|
| c.64C>T | p.Pro22Ser |
| c.64C>A 2 | p.Pro22Thr |
| c.65C>G 2 | p.Pro22Arg |
| c.1001A>C | p.Gln334Pro
(Gln333Pro) |
| c.293A>G | p.Asn98Ser
(Asn97Ser) |
| c.446C>T | p.Ala149Val
(Ala148Val) |
Variants listed in the table have been provided by the author. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
- 2.
Normal gene product. The protein encoded by NEFL contains 543 amino acids with a head, rod, and tail domain. Neurofilaments form the cytoskeletal component of myelinated axons.
Abnormal gene product. Knockout mice lacking neurofilments have diminished axon caliber and delayed regeneration of myelinated axons following crush injury. A mouse with a single-nucleotide variant in NEFL has massive degeneration of spinal motor neurons and abnormal neurofilament accumulation with severe neurogenic skeletal muscle atrophy.
References
Published Guidelines / Consensus Statements
Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available
online. 2013. Accessed 4-30-18.
National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset conditions. Available
online. 2017. Accessed 4-30-18.
Literature Cited
Abe A, Nakamura K, Kato M, Numakura C, Honma T, Seiwa C, Shirahata E, Itoh A, Kishikawa Y, Hayasaka K. Compound heterozygous PMP22 deletion mutations causing severe Charcot-Marie-Tooth disease type 1.
J Hum Genet. 2010;55:771–3. [
PubMed: 20739940]
Auer-Grumbach M, Weger M, Fink-Puches R, Papic L, Fröhlich E, Auer-Grumbach P, El Shabrawi-Caelen L, Schabhüttl M, Windpassinger C, Senderek J, Budka H, Trajanoski S, Janecke AR, Haas A, Metze D, Pieber TR, Guelly C. Fibulin-5 mutations link inherited neuropathies, age-related macular degeneration and hyperelastic skin.
Brain. 2011;134:1839–52. [
PMC free article: PMC3272386] [
PubMed: 21576112]
Awater C, Zerres K, Rudnik-Schöneborn S. Pregnancy course and outcome in women with hereditary neuromuscular disorders: comparison of obstetric risks in 178 patients.
Eur J Obstet Gynecol Reprod Biol. 2012;162:153–9. [
PubMed: 22459654]
Baets J, Deconinck T, De Vriendt E, Zimoń M, Yperzeele L, Van Hoorenbeeck K, Peeters K, Spiegel R, Parman Y, Ceulemans B, Van Bogaert P, Pou-Serradell A, Bernert G, Dinopoulos A, Auer-Grumbach M, Sallinen SL, Fabrizi GM, Pauly F, Van den Bergh P, Bilir B, Battaloglu E, Madrid RE, Kabzińska D, Kochanski A, Topaloglu H, Miller G, Jordanova A, Timmerman V, De Jonghe P. Genetic spectrum of hereditary neuropathies with onset in the first year of life.
Brain. 2011;134:2664–76. [
PMC free article: PMC3170533] [
PubMed: 21840889]
Baets J, De Jonghe P, Timmerman V. Recent advances in Charcot-Marie-Tooth disease.
Curr Opin Neurol. 2014;27:532–40. [
PubMed: 25110935]
Bai Y, Ianokova E, Pu Q, Ghandour K, Levinson R, Martin JJ, Ceuterick-de Groote C, Mazanec R, Seeman P, Shy ME, Li J. Effect of an R69C mutation in the myelin protein zero gene on myelination and ion channel subtypes.
Arch Neurol. 2006;63:1787–94. [
PubMed: 17172621]
Bennett CL, Shirk AJ, Huynh HM, Street VA, Nelis E, Van Maldergem L, De Jonghe P, Jordanova A, Guergueltcheva V, Tournev I, Van Den Bergh P, Seeman P, Mazanec R, Prochazka T, Kremensky I, Haberlova J, Weiss MD, Timmerman V, Bird TD, Chance PF. SIMPLE mutation in demyelinating neuropathy and distribution in sciatic nerve.
Ann Neurol. 2004;55:713–20. [
PubMed: 15122712]
Berciano J, Gallardo E, García A, Ramón C, Infante J, Combarros O. Clinical progression in Charcot-Marie-Tooth disease type 1A duplication: clinico-electrophysiological and MRI longitudinal study of a family.
J Neurol. 2010;257:1633–41. [
PubMed: 20443018]
Bienfait HM, Baas F, Gabreëls-Festen AA, Koelman JH, Langerhorst CT, de Visser M. Two amino-acid substitutions in the myelin protein zero gene of a case of Charcot-Marie-Tooth disease associated with light-near dissociation.
Neuromuscul Disord. 2002;12:281–5. [
PubMed: 11801400]
Bienfait HM, Faber CG, Baas F, Gabreëls-Festen AA, Koelman JH, Hoogendijk JE, Verschuuren JJ, Wokke JH, de Visser M. Late onset axonal Charcot-Marie-Tooth phenotype caused by a novel myelin protein zero mutation.
J Neurol Neurosurg Psychiatry. 2006a;77:534–7. [
PMC free article: PMC2077493] [
PubMed: 16543539]
Bienfait HM, Verhamme C, van Schaik IN, Koelman JH, de Visser BW, de Haan RJ, Baas F, van Engelen BG, de Visser M. Comparison of CMT1A and CMT2: similarities and differences.
J Neurol. 2006b;253:1572–80. [
PubMed: 16941080]
Bird TD, Lipe HP, Crabtree LD. Impotence associated with the Charcot-Marie-Tooth syndrome.
Eur Neurol. 1994;34:155–7. [
PubMed: 8033941]
Boentert M, Knop K, Schuhmacher C, Gess B, Okegwo A, Young P. Sleep disorders in Charcot-Marie-Tooth disease type 1.
J Neurol Neurosurg Psychiatry. 2014;85:319–25. [
PubMed: 23704315]
Boerkoel CF, Takashima H, Garcia CA, Olney RK, Johnson J, Berry K, Russo P, Kennedy S, Teebi AS, Scavina M, Williams LL, Mancias P, Butler IJ, Krajewski K, Shy M, Lupski JR. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation.
Ann Neurol. 2002;51:190–201. [
PubMed: 11835375]
Boffeli TJ, Tabatt JA. Minimally invasive early operative treatment of progressive foot and ankle deformity associated with Charcot-Marie-Tooth disease.
J Foot Ankle Surg. 2015;54:701–8. [
PubMed: 25131389]
Brozková D, Mazanec R, Haberlová J, Sakmaryová I, Seeman P. Clinical and in silico evidence for and against pathogenicity of 11 new mutations in the MPZ gene.
Clin Genet. 2010;78:81–7. [
PubMed: 20456450]
Bueno KC, Gouvêa SP, Lourenço CM, Marques W Jr. Mutational analysis of LITAF, EGR2 and NEFL genes in a Brazilian population of Charcot-Marie-Tooth neuropathy type I. Poster session. Potomac, MD: Meeting of the Peripheral Nerve Society. 2011.
Burns J, Ramchandren S, Ryan MM, Shy M, Ouvrier RA. Determinants of reduced health-related quality of life in pediatric inherited neuropathies.
Neurology. 2010;75:726–31. [
PMC free article: PMC2931653] [
PubMed: 20733147]
Carter GT. Rehabilitation management in neuromuscular disease.
J Neuro Rehab. 1997;11:69–80. [
PubMed: 19730017]
Carter GT, Jensen MP, Galer BS, Kraft GH, Crabtree LD, Beardsley RM, Abresch RT, Bird TD. Neuropathic pain in Charcot-Marie-Tooth disease.
Arch Phys Med Rehabil. 1998;79:1560–4. [
PubMed: 9862301]
Carvalho AA, Vital A, Ferrer X, Latour P, Lagueny A, Brechenmacher C, Vital C. Charcot-Marie-Tooth disease type 1A: clinicopathological correlations in 24 patients.
J Peripher Nerv Syst. 2005;10:85–92. [
PubMed: 15703022]
Chanson JB, Echaniz-Laguna A, Blanc F, Lacour A, Ballonzoli L, Kremer S, Namer IJ, Lannes B, Tranchant C, Vermersch P, de Seze J. Central nervous system abnormalities in patients with PMP22 gene mutations: a prospective study.
J Neurol Neurosurg Psychiatry. 2013;84:392–7. [
PubMed: 23243264]
Chapon F, Latour P, Diraison P, Schaeffer S, Vandenberghe A. Axonal phenotype of Charcot-Marie-Tooth disease associated with a mutation in the myelin protein zero gene.
J Neurol Neurosurg Psychiatry. 1999;66:779–82. [
PMC free article: PMC1736388] [
PubMed: 10329755]
Chung KW, Sunwoo IN, Kim SM, Park KD, Kim WK, Kim TS, Koo H, Cho M, Lee J, Choi BO. Two missense mutations of EGR2 R359W and GJB1 V136A in a Charcot-Marie-Tooth disease family.
Neurogenetics. 2005;6:159–63. [
PubMed: 15947997]
Ciotti P, Luigetti M, Geroldi A, Capponi S, Pezzini I, Gulli R, Pazzaglia C, Padua L, Massa R, Mandich P, Bellone E. A novel LITAF/SIMPLE mutation within a family with a demyelinating form of Charcot-Marie-Tooth disease.
J Neurol Sci. 2014;343:183–6. [
PubMed: 24880540]
Colomban C, Micallef J, Lefebvre MN, Dubourg O, Gonnaud PM, Stojkovic T, Jouve E, Blin O, Pouget J, Attarian S. Clinical spectrum and gender differences in a large cohort of Charcot-Marie-Tooth type 1A patients.
J Neurol Sci. 2014;336:155–60. [
PubMed: 24246498]
De Angelis MV, Di Muzio A, Capasso M, Angiari C, Cavallaro T, Fabrizi GM, Rizzuto N, Uncini A. Segmental conduction abnormalities and myelin thickenings in Val102/fs null mutation of MPZ gene.
Neurology. 2004;63:2180–3. [
PubMed: 15596778]
De Jonghe P, Timmerman V, Ceuterick C, Nelis E, De Vriendt E, Löfgren A, Vercruyssen A, Verellen C, Van Maldergem L, Martin JJ, Van Broeckhoven C. The Thr124Met mutation in the peripheral myelin protein zero (MPZ) gene is associated with a clinically distinct Charcot-Marie-Tooth phenotype.
Brain. 1999;122:281–90. [
PubMed: 10071056]
De Jonghe P, Timmerman V, Nelis E, Martin JJ, Van Broeckhoven C. Charcot-Marie-Tooth disease and related peripheral neuropathies.
J Peripher Nerv Syst. 1997;2:370–87. [
PubMed: 10975746]
Dematteis M, Pépin JL, Jeanmart M, Deschaux C, Labarre-Vila A, Lévy P. Charcot-Marie-Tooth disease and sleep apnoea syndrome: a family study.
Lancet. 2001;357:267–72. [
PubMed: 11214130]
de Vries SD, Verhamme C, van Ruissen F, van Paassen BW, Arts WF, Kerkhoff H, van Engelen BG, Lammens M, de Visser M, Baas F, van der Kooi AJ. The phenotype of the Gly94fsX222 PMP22 insertion.
J Peripher Nerv Syst. 2011;16:113–8. [
PubMed: 21692910]
Dillmann U, Heide G, Dietz B, Teshmar E, Schimrigk K. Hereditary motor and sensory neuropathy with spastic paraplegia and optic atrophy: report on a family.
J Neurol. 1997;244:562–5. [
PubMed: 9352453]
Donaghy M, Sisodiya SM, Kennett R, McDonald B, Haites N, Bell C. Steroid responsive polyneuropathy in a family with a novel myelin protein zero mutation.
J Neurol Neurosurg Psychiatry. 2000;69:799–805. [
PMC free article: PMC1737181] [
PubMed: 11080236]
Dyck PJ, Swanson CJ, Low PA, Bartleson JD, Lambert EH. Prednisone-responsive hereditary motor and sensory neuropathy.
Mayo Clin Proc. 1982;57:239–46. [
PubMed: 7070119]
Fabrizi GM, Cavallaro T, Angiari C, Cabrini I, Taioli F, Malerba G, Bertolasi L, Rizzuto N. Charcot-Marie-Tooth disease type 2E, a disorder of the cytoskeleton.
Brain. 2007;130:394–403. [
PubMed: 17052987]
Fabrizi GM, Ferrarini M, Cavallaro T, Jarre L, Polo A, Rizzuto N. A somatic and germline mosaic mutation in MPZ/P(0) mimics recessive inheritance of CMT1B.
Neurology. 2001a;57:101–5. [
PubMed: 11445635]
Fabrizi GM, Pellegrini M, Angiari C, Cavallaro T, Morini A, Taioli F, Cabrini I, Orrico D, Rizzuto N. Gene dosage sensitivity of a novel mutation in the intracellular domain of P0 associated with Charcot-Marie-Tooth disease type 1B.
Neuromuscul Disord. 2006;16:183–7. [
PubMed: 16488608]
Fabrizi GM, Simonati A, Taioli F, Cavallaro T, Ferrarini M, Rigatelli F, Pini A, Mostacciuolo ML, Rizzuto N. PMP22 related congenital hypomyelination neuropathy.
J Neurol Neurosurg Psychiatry. 2001b;70:123–6. [
PMC free article: PMC1763468] [
PubMed: 11118262]
Ferrarin M, Bovi G, Rabuffetti M, Mazzoleni P, Montesano A, Pagliano E, Marchi A, Magro A, Marchesi C, Pareyson D, Moroni I. Gait pattern classification in children with Charcot-Marie-Tooth disease type 1A.
Gait Posture. 2012;35:131–7. [
PMC free article: PMC3909942] [
PubMed: 21944474]
Finsterer J, Miltenberger G, Rauschka H, Janecke A. Novel C59T leader peptide mutation in the MPZ gene associated with late-onset, axonal, sensorimotor polyneuropathy.
Eur J Neurol. 2006;13:1149–52. [
PubMed: 16987171]
Fledrich R, Stassart RM, Klink A, Rasch LM, Prukop T, Haag L, Czesnik D, Kungl T, Abdelaal TA, Keric N, Stadelmann C, Brück W, Nave KA, Sereda MW. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A.
Nat Med. 2014;20:1055–61. [
PubMed: 25150498]
Floroskufi P, Panas M, Karadima G, Vassilopoulos D. New mutation of the MPZ gene in a family with the Dejerine-Sottas disease phenotype.
Muscle Nerve. 2007;35:667–9. [
PubMed: 17143884]
Gabriel JM, Erne B, Pareyson D, Sghirlanzoni A, Taroni F, Steck AJ. Gene dosage effects in hereditary peripheral neuropathy. Expression of peripheral myelin protein 22 in Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies nerve biopsies.
Neurology. 1997;49:1635–40. [
PubMed: 9409359]
Gallardo E, García A, Combarros O, Berciano J. Charcot-Marie-Tooth disease type 1A duplication: spectrum of clinical and magnetic resonance imaging features in leg and foot muscles.
Brain. 2006;129:426–37. [
PubMed: 16317020]
García A, Combarros O, Calleja J, Berciano J. Charcot-Marie-Tooth disease type 1A with 17p duplication in infancy and early childhood: a longitudinal clinical and electrophysiologic study.
Neurology. 1998;50:1061–7. [
PubMed: 9566395]
Gemignani F, Melli G, Alfieri S, Inglese C, Marbini A. Sensory manifestations in Charcot-Marie-Tooth disease.
J Peripher Nerv Syst. 2004;9:7–14. [
PubMed: 14871449]
Gerding WM, Koetting J, Epplen JT, Neusch C. Hereditary motor and sensory neuropathy caused by a novel mutation in LITAF.
Neuromuscul Disord. 2009;19:701–3. [
PubMed: 19541485]
Gess B, Schirmacher A, Boentert M, Young P. Charcot-Marie-Tooth disease: frequency of genetic subtypes in a German neuromuscular center population.
Neuromuscul Disord. 2013;23:647–51. [
PubMed: 23743332]
Giambonini-Brugnoli G, Buchstaller J, Sommer L, Suter U, Mantei N. Distinct disease mechanisms in peripheral neuropathies due to altered peripheral myelin protein 22 gene dosage or a Pmp22 point mutation.
Neurobiol Dis. 2005;18:656–68. [
PubMed: 15755691]
Ginsberg L, Malik O, Kenton AR, Sharp D, Muddle JR, Davis MB, Winer JB, Orrell RW, King RH. Coexistent hereditary and inflammatory neuropathy.
Brain. 2004;127:193–202. [
PubMed: 14607795]
Grandis M, Shy ME. Current Therapy for Charcot-Marie-Tooth Disease.
Curr Treat Options Neurol. 2005;7:23–31. [
PubMed: 15610704]
Grandis M, Vigo T, Passalacqua M, Jain M, Scazzola S, La Padula V, Brucal M, Benvenuto F, Nobbio L, Cadoni A, Mancardi GL, Kamholz J, Shy ME, Schenone A. Different cellular and molecular mechanisms for early and late-onset myelin protein zero mutations.
Hum Mol Genet. 2008;17:1877–89. [
PubMed: 18337304]
Guyton GP, Mann RA. The pathogenesis and surgical management of foot deformity in Charcot-Marie-Tooth disease.
Foot Ankle Clin. 2000;5:317–26. [
PubMed: 11232233]
Hattori N, Yamamoto M, Yoshihara T, Koike H, Nakagawa M, Yoshikawa H, Ohnishi A, Hayasaka K, Onodera O, Baba M, Yasuda H, Saito T, Nakashima K, Kira J, Kaji R, Oka N, Sobue G., Study Group for Hereditary Neuropathy in Japan. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32): a clinicopathological study of 205 Japanese patients.
Brain. 2003;126:134–51. [
PubMed: 12477701]
Hodapp JA, Carter GT, Lipe HP, Michelson SJ, Kraft GH, Bird TD. Double trouble in hereditary neuropathy: concomitant mutations in the PMP-22 gene and another gene produce novel phenotypes.
Arch Neurol. 2006;63:112–7. [
PubMed: 16401743]
Hoff JM, Gilhus NE, Daltveit AK. Pregnancies and deliveries in patients with Charcot-Marie-Tooth disease.
Neurology. 2005;64:459–62. [
PubMed: 15699375]
Houlden H, Reilly MM. Molecular genetics of autosomal-dominant demyelinating Charcot-Marie-Tooth disease.
Neuromolecular Med. 2006;8:43–62. [
PubMed: 16775366]
Høyer H, Braathen GJ, Eek AK, Skjelbred CF, Russell MB. Charcot-Marie-Tooth caused by a copy number variation in myelin protein zero.
Eur J Med Genet. 2011;54:e580–3. [
PubMed: 21787890]
Jen J, Baloh RH, Ishiyama A, Baloh RW. Dejerine-Sottas syndrome and vestibular loss due to a point mutation in the PMP22 gene.
J Neurol Sci. 2005;237:21–4. [
PubMed: 15992829]
Johnson NE, Heatwole CR, Dilek N, Sowden J, Kirk CA, Shereff D, Shy ME, Herrmann DN., Inherited Neuropathies Consortium. Quality-of-life in Charcot-Marie-Tooth disease: The patient's perspective.
Neuromuscul Disord. 2014;24:1018–23. [
PMC free article: PMC4253871] [
PubMed: 25092060]
Jordanova A, De Jonghe P, Boerkoel CF, Takashima H, De Vriendt E, Ceuterick C, Martin JJ, Butler IJ, Mancias P, Papasozomenos SCh, Terespolsky D, Potocki L, Brown CW, Shy M, Rita DA, Tournev I, Kremensky I, Lupski JR, Timmerman V. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot-Marie-Tooth disease.
Brain. 2003;126:590–7. [
PubMed: 12566280]
Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D'Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, Timmerman V. Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy.
Nat Genet. 2006;38:197–202. [
PubMed: 16429158]
Kim YH, Chung HK, Park KD, Choi KG, Kim SM, Sunwoo IN, Choi YC, Lim JG, Lee KW, Kim KK, Lee DK, Joo IS, Kwon KH, Gwon SB, Park JH, Kim DS, Kim SH, Kim WK, Suh BC, Kim SB, Kim NH, Sohn EH, Kim OJ, Kim HS, Cho JH, Kang SY, Park CI, Oh J, Shin JH, Chung KW, Choi BO. Comparison between clinical disabilities and electrophysiological values in Charcot-Marie-Tooth 1A patients with PMP22 duplication.
J Clin Neurol. 2012;8:139–45. [
PMC free article: PMC3391619] [
PubMed: 22787498]
Kleffner I, Schirmacher A, Gess B, Boentert M, Young P. Four novel mutations of the myelin protein zero gene presenting as a mild and late-onset polyneuropathy.
J Neurol. 2010;2010;257:1864–8. [
PubMed: 20556410]
Klein CJ, Dyck PJ. Genetic testing in inherited peripheral neuropathies.
J Peripher Nerv Syst. 2005;10:77–84. [
PubMed: 15703021]
Klein CJ, Middha S, Duan X, Wu Y, Litchy WJ, Gu W, Dyck PJ, Gavrilova RH, Smith DI, Kocher JP, Dyck PJ. Application of whole exome sequencing in undiagnosed inherited polyneuropathies.
J Neurol Neurosurg Psychiatry. 2014;85:1265–72. [
PubMed: 24604904]
Kleopa KA, Georgiou DM, Nicolaou P, Koutsou P, Papathanasiou E, Kyriakides T, Christodoulou K. A novel PMP22 mutation Ser22Phe in a family with hereditary neuropathy with liability to pressure palsies and CMT1A phenotypes.
Neurogenetics. 2004;5:171–5. [
PubMed: 15205993]
Kochański A. How to assess the pathogenicity of mutations in Charcot-Marie-Tooth disease and other diseases?
J Appl Genet. 2006;47:255–60. [
PubMed: 16877806]
Kochański A, Kabzińska D, Drac H, Ryniewicz B, Rowińska-Marcińska K, Hausmanowa-Petrusewicz I. Early onset Charcot-Marie-Tooth type 1B disease caused by a novel Leu190fs mutation in the myelin protein zero gene.
Eur J Paediatr Neurol. 2004;8:221–4. [
PubMed: 15261887]
Kousseff BG, Hadro TA, Treiber DL, Wollner T, Morris C. Charcot-Marie-Tooth disease with sensorineural hearing loss--an autosomal dominant trait.
Birth Defects Orig Artic Ser. 1982;18:223–8. [
PubMed: 7139106]
Kovach MJ, Campbell KC, Herman K, Waggoner B, Gelber D, Hughes LF, Kimonis VE. Anticipation in a unique family with Charcot-Marie-Tooth syndrome and deafness: delineation of the clinical features and review of the literature.
Am J Med Genet. 2002;108:295–303. [
PubMed: 11920834]
Kovach MJ, Lin JP, Boyadjiev S, Campbell K, Mazzeo L, Herman K, Rimer LA, Frank W, Llewellyn B, Jabs EW, Gelber D, Kimonis VE. A unique point mutation in the PMP22 gene is associated with Charcot-Marie-Tooth disease and deafness.
Am J Hum Genet. 1999;64:1580–93. [
PMC free article: PMC1377901] [
PubMed: 10330345]
Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, Kamholz J, Shy ME. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A.
Brain. 2000;123:1516–27. [
PubMed: 10869062]
Latour P, Gonnaud PM, Ollagnon E, Chan V, Perelman S, Stojkovic T, Stoll C, Vial C, Ziegler F, Vandenberghe A, Maire I. SIMPLE mutation analysis in dominant demyelinating Charcot-Marie-Tooth disease: three novel mutations.
J Peripher Nerv Syst. 2006;11:148–55. [
PubMed: 16787513]
Lee YC, Lee TC, Lin KP, Lin MW, Chang MH, Soong BW. Clinical characterization and genetic analysis of a possible novel type of dominant Charcot-Marie-Tooth disease.
Neuromuscul Disord. 2010;20:534–9. [
PubMed: 20627571]
Lee YC, Soong BW, Lin KP, Lee HY, Wu ZA, Kao KP. Myelin protein zero gene mutations in Taiwanese patients with Charcot-Marie-Tooth disease type 1.
J Neurol Sci. 2004;219:95–100. [
PubMed: 15050444]
Lewis RA, McDermott MP, Herrmann DN, Hoke A, Clawson LL, Siskind C, Feely SM, Miller LJ, Barohn RJ, Smith P, Luebbe E, Wu X, Shy ME., Muscle Study Group. High-dosage ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A: results of a randomized, double-masked, controlled trial.
JAMA Neurol. 2013;70:981–7. [
PMC free article: PMC3752369] [
PubMed: 23797954]
Liu L, Li X, Zi X, Huang S, Zhan Y, Jiang M, Guo J, Xia K, Tang B, Zhang R. Two novel MPZ mutations in Chinese CMT patients.
J Peripher Nerv Syst. 2013;18:256–60. [
PubMed: 24028194]
López-Bigas N, Olivé M, Rabionet R, Ben-David O, Martínez-Matos JA, Bravo O, Banchs I, Volpini V, Gasparini P, Avraham KB, Ferrer I, Arbonés ML, Estivill X. Connexin 31 (GJB3) is expressed in the peripheral and auditory nerves and causes neuropathy and hearing impairment.
Hum Mol Genet. 2001;10:947–52. [
PubMed: 11309368]
Luigetti M, Fabrizi GM, Taioli F, Del Grande A, Lo Monaco M. A novel LITAF/SIMPLE variant within a family with minimal demyelinating Charcot-Marie-Tooth disease.
Neurol Sci. 2014;35:2005–7. [
PubMed: 24844793]
Luigetti M, Sabatelli M, Bellone E, Fabrizi GM, Padua L, Granata G. Nerve ultrasound in patients with CMT1C: Description of three cases.
Muscle Nerve. 2015;51:781–2. [
PubMed: 25286909]
Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA, Chakravarti A, Patel PI. DNA duplication associated with Charcot-Marie-Tooth disease type 1A.
Cell. 1991;66:219–32. [
PubMed: 1677316]
Maeda MH, Mitsui J, Soong BW, Takahashi Y, Ishiura H, Hayashi S, Shirota Y, Ichikawa Y, Matsumoto H, Arai M, Okamoto T, Miyama S, Shimizu J, Inazawa J, Goto J, Tsuji S. Increased gene dosage of Myelin Protein Zero causes Charcot-Marie-Tooth disease.
Ann Neurol. 2012;71:84–92. [
PubMed: 22275255]
Mandich P, Fossa P, Capponi S, Geroldi A, Acquaviva M, Gulli R, Ciotti P, Manganelli F, Grandis M, Bellone E. Clinical features and molecular modelling of novel MPZ mutations in demyelinating and axonal neuropathies.
Eur J Hum Genet. 2009;17:1129–34. [
PMC free article: PMC2986589] [
PubMed: 19293842]
Marques W Jr, Freitas MR, Nascimento OJ, Oliveira AB, Calia L, Melo A, Lucena R, Rocha V, Barreira AA. 17p duplicated Charcot-Marie-Tooth 1A: characteristics of a new population.
J Neurol. 2005;252:972–9. [
PubMed: 15765265]
Marrosu MG, Vaccargiu S, Marrosu G, Vannelli A, Cianchetti C, Muntoni F. Charcot-Marie-Tooth disease type 2 associated with mutation of the myelin protein zero gene.
Neurology. 1998;50:1397–401. [
PubMed: 9595994]
Mathis S, Corcia P, Tazir M, Camu W, Magdelaine C, Latour P, Biberon J, Guennoc AM, Richard L, Magy L, Funalot B, Vallat JM. Peripheral myelin protein 22 gene duplication with atypical presentations: a new example of the wide spectrum of Charcot-Marie-Tooth 1A disease.
Neuromuscul Disord. 2014;24:524–8. [
PubMed: 24792522]
McGann R, Gurd A. The association between Charcot-Marie-Tooth disease and developmental dysplasia of the hip.
Orthopedics. 2002;25:337–9. [
PubMed: 11918042]
Meggouh F, de Visser M, Arts WF, De Coo RI, van Schaik IN, Baas F. Early onset neuropathy in a compound form of Charcot-Marie-Tooth disease.
Ann Neurol. 2005;57:589–91. [
PubMed: 15786462]
Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy.
Ann Neurol. 2007;61:61–72. [
PubMed: 17262851]
Mikesová E, Hühne K, Rautenstrauss B, Mazanec R, Baránková L, Vyhnálek M, Horácek O, Seeman P. Novel EGR2 mutation R359Q is associated with CMT type 1 and progressive scoliosis.
Neuromuscul Disord. 2005;15:764–7. [
PubMed: 16198564]
Nelis E, Haites N, Van Broeckhoven C. Mutations in the peripheral myelin genes and associated genes in inherited peripheral neuropathies.
Hum Mutat. 1999a;13:11–28. [
PubMed: 9888385]
Nelis E, Timmerman V, De Jonghe P, Van Broeckhoven C, Rautenstrauss B. Molecular genetics and biology of inherited peripheral neuropathies: a fast-moving field.
Neurogenetics. 1999b;2:137–48. [
PubMed: 10541586]
Nicholson G, Myers S. Intermediate forms of Charcot-Marie-Tooth neuropathy: a review.
Neuromolecular Med. 2006;8:123–30. [
PubMed: 16775371]
Numakura C, Lin C, Oka N, Akiguchi I, Hayasaka K. Hemizygous mutation of the peripheral myelin protein 22 gene associated with Charcot-Marie-Tooth disease type 1.
Ann Neurol. 2000;47:101–3. [
PubMed: 10632107]
Numakura C, Shirahata E, Yamashita S, Kanai M, Kijima K, Matsuki T, Hayasaka K. Screening of the early growth response 2 gene in Japanese patients with Charcot-Marie-Tooth disease type 1.
J Neurol Sci. 2003;210:61–4. [
PubMed: 12736090]
Pareyson D, Reilly MM, Schenone A, Fabrizi GM, Cavallaro T, Santoro L, Vita G, Quattrone A, Padua L, Gemignani F, Visioli F, Laurà M, Radice D, Calabrese D, Hughes RA, Solari A. CMT-TRIAAL; CMT-TRAUK groups. Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial.
Lancet Neurol. 2011;10:320–8. [
PMC free article: PMC3154498] [
PubMed: 21393063]
Pareyson D, Saveri P, Piscosquito G. Charcot-Marie-Tooth disease and related hereditary neuropathies: from gene function to associated phenotypes.
Curr Mol Med. 2014;14:1009–33. [
PubMed: 25323870]
Pareyson D, Scaioli V, Laurà M. Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease.
Neuromolecular Med. 2006;8:3–22. [
PubMed: 16775364]
Pareyson D, Taroni F, Botti S, Morbin M, Baratta S, Lauria G, Ciano C, Sghirlanzoni A. Cranial nerve involvement in CMT disease type 1 due to early growth response 2 gene mutation.
Neurology. 2000;54:1696–8. [
PubMed: 10762521]
Parman Y, Planté-Bordeneuve V, Guiochon-Mantel A, Eraksoy M, Said G. Recessive inheritance of a new point mutation of the PMP22 gene in Dejerine-Sottas disease.
Ann Neurol. 1999;45:518–22. [
PubMed: 10211478]
Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, Robaglia-Schlupp A, Pellissier JF, Fontés M. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease.
Nat Med. 2004;10:396–401. [
PubMed: 15034573]
Patel PI, Pleasure D. Whither hope for pharmacological treatment of Charcot-Marie-Tooth disease type 1A?
JAMA Neurol. 2013;70:969–71. [
PubMed: 23797977]
Patel PI, Roa BB, Welcher AA, Schoener-Scott R, Trask BJ, Pentao L, Snipes GJ, Garcia CA, Francke U, Shooter EM, Lupski JR, Suter U. The gene for the peripheral myelin protein PMP-22 is a candidate for Charcot-Marie-Tooth disease type 1A.
Nat Genet. 1992;1:159–65. [
PubMed: 1303228]
Patzkó A, Bai Y, Saporta MA, Katona I, Wu X, Vizzuso D, Feltri ML, Wang S, Dillon LM, Kamholz J, Kirschner D, Sarkar FH, Wrabetz L, Shy ME. Curcumin derivatives promote Schwann cell differentiation and improve neuropathy in R98C CMT1B mice.
Brain. 2012;135:3551–66. [
PMC free article: PMC3577101] [
PubMed: 23250879]
Pazzaglia C, Vollono C, Ferraro D, Virdis D, Lupi V, Le Pera D, Tonali P, Padua L, Valeriani M. Mechanisms of neuropathic pain in patients with Charcot-Marie-Tooth 1 A: a laser-evoked potential study.
Pain. 2010;149:379–85. [
PubMed: 20334975]
Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice.
Neuron. 2008;57:393–405. [
PMC free article: PMC2267889] [
PubMed: 18255032]
Perea J, Robertson A, Tolmachova T, Muddle J, King RH, Ponsford S, Thomas PK, Huxley C. Induced myelination and demyelination in a conditional mouse model of Charcot-Marie-Tooth disease type 1A.
Hum Mol Genet. 2001;10:1007–18. [
PubMed: 11331611]
Piscosquito G, Reilly MM, Schenone A, Fabrizi GM, Cavallaro T, Santoro L, Vita G, Quattrone A, Padua L, Gemignani F, Visioli F, Laurà M, Calabrese D, Hughes RA, Radice D, Solari A, Pareyson D. CMT-TRIAAL & CMT-TRAUK Group. Is overwork weakness relevant in Charcot-Marie-Tooth disease?
J Neurol Neurosurg Psychiatry. 2014;85:1354–8. [
PubMed: 24659795]
Plaisier E, Mougenot B, Verpont MC, Jouanneau C, Archelos JJ, Martini R, Kerjaschki D, Ronco P. Glomerular permeability is altered by loss of P0, a myelin protein expressed in glomerular epithelial cells.
J Am Soc Nephrol. 2005;16:3350–6. [
PubMed: 16162811]
Planté-Bordeneuve V, Guiochon-Mantel A, Lacroix C, Lapresle J, Said G. The Roussy-Lévy family: from the original description to the gene.
Ann Neurol. 1999;46:770–3. [
PubMed: 10553995]
Poretti A, Palla A, Tarnutzer AA, Petersen JA, Weber KP, Straumann D, Jung HH. Vestibular impairment in patients with Charcot-Marie-Tooth disease.
Neurology. 2013;80:2099–105. [
PubMed: 23658384]
Postelmans JT, Stokroos RJ. Cochlear implantation in a patient with deafness induced by Charcot-Marie-Tooth disease (hereditary motor and sensory neuropathies).
J Laryngol Otol. 2006;120:508–10. [
PubMed: 16772060]
Potocki L, Bi W, Treadwell-Deering D, Carvalho CM, Eifert A, Friedman EM, Glaze D, Krull K, Lee JA, Lewis RA, Mendoza-Londono R, Robbins-Furman P, Shaw C, Shi X, Weissenberger G, Withers M, Yatsenko SA, Zackai EH, Stankiewicz P, Lupski JR. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype.
Am J Hum Genet. 2007;80:633–49. [
PMC free article: PMC1852712] [
PubMed: 17357070]
Raeymaekers P, Timmerman V, Nelis E, De Jonghe P, Hoogendijk JE, Baas F, Barker DF, Martin JJ, De Visser M, Bolhuis PA. Duplication in chromosome 17p11.2 in Charcot-Marie-Tooth neuropathy type 1a (CMT 1a). The HMSN Collaborative Research Group.
Neuromuscul Disord. 1991;1:93–7. [
PubMed: 1822787]
Ramdharry GM, Day BL, Reilly MM, Marsden JF. Foot drop splints improve proximal as well as distal leg control during gait in Charcot-Marie-Tooth disease.
Muscle Nerve. 2012a;46:512–9. [
PubMed: 22987691]
Ramdharry GM, Thornhill A, Mein G, Reilly MM, Marsden JF. Exploring the experience of fatigue in people with Charcot-Marie-Tooth disease.
Neuromuscul Disord. 2012b;22 Suppl 3:S208–13. [
PubMed: 23182641]
Rance G, Ryan MM, Bayliss K, Gill K, O'Sullivan C, Whitechurch M. Auditory function in children with Charcot-Marie-Tooth disease.
Brain. 2012;135:1412–22. [
PubMed: 22522939]
Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies.
J Neurol Neurosurg Psychiatry. 2009;80:1304–14. [
PubMed: 19917815]
Rose KJ, Raymond J, Refshauge K, North KN, Burns J. Serial night casting increases ankle dorsiflexion range in children and young adults with Charcot-Marie-Tooth disease: a randomised trial.
J Physiother. 2010;56:113–9. [
PubMed: 20482478]
Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease.
Nat Rev Neurol. 2013;9:562–71. [
PubMed: 24018473]
Rudnik-Schöneborn S, Röhrig D, Nicholson G, Zerres K. Pregnancy and delivery in Charcot-Marie-Tooth disease type 1.
Neurology. 1993;43:2011–6. [
PubMed: 8413959]
Sabet A, Li J, Ghandour K, Pu Q, Wu X, Kamholz J, Shy ME, Cambi F. Skin biopsies demonstrate MPZ splicing abnormalities in Charcot-Marie-Tooth neuropathy 1B.
Neurology. 2006;67:1141–6. [
PubMed: 17030746]
Šafka Brozková D, Lassuthová P, Neupauerová J, Krutová M, Haberlová J, Stejskal D, Seeman P. Czech family confirms the link between FBLN5 and Charcot-Marie-Tooth type 1 neuropathy.
Brain. 2013;136:e232. [
PubMed: 23328402]
Sagliocco L, Orlandi G, Calabrese R, Pellegrinetti A, Baglini O, Castelli F, Baldinotti F, Sartucci F. Electrodiagnostic evidence of phrenic nerve demyelination in Charcot-Marie-Tooth disease 1A.
Am J Phys Med Rehabil. 2003;82:754–9. [
PubMed: 14508405]
Sahenk Z, Nagaraja HN, McCracken BS, King WM, Freimer ML, Cedarbaum JM, Mendell JR. NT-3 promotes nerve regeneration and sensory improvement in CMT1A mouse models and in patients.
Neurology. 2005;65:681–9. [
PubMed: 16157899]
Sahenk Z, Galloway G, Clark KR, Malik V, Rodino-Klapac LR, Kaspar BK, Chen L, Braganza C, Montgomery C, Mendell JR. AAV1.NT-3 gene therapy for Charcot-Marie-Tooth neuropathy.
Mol Ther. 2014;22:511–21. [
PMC free article: PMC3944324] [
PubMed: 24162799]
Saifi GM, Szigeti K, Wiszniewski W, Shy ME, Krajewski K, Hausmanowa-Petrusewicz I, Kochanski A, Reeser S, Mancias P, Butler I, Lupski JR. SIMPLE mutations in Charcot-Marie-Tooth disease and the potential role of its protein product in protein degradation.
Hum Mutat. 2005;25:372–83. [
PubMed: 15776429]
Sambuughin N, de Bantel A, McWilliams S, Sivakumar K. Deafness and CMT disease associated with a novel four amino acid deletion in the PMP22 gene.
Neurology. 2003;60:506–8. [
PubMed: 12578939]
Saporta MA, Katona I, Zhang X, Roper H, McClelland L, Macdonald F, Brueton L, Blake J, Suter U, Reilly MM, Shy ME, Li J. Neuropathy in a human without the PMP22 gene.
Arch Neurol. 2011b;68:814–21. [
PMC free article: PMC3711535] [
PubMed: 21670407]
Saporta MA, Shy BR, Patzko A, Bai Y, Pennuto M, Ferri C, Tinelli E, Saveri P, Kirschner D, Crowther M, Southwood C, Wu X, Gow A, Feltri ML, Wrabetz L, Shy ME. MPZ R98C arrests Schwann cell development in a mouse model of early-onset Charcot-Marie-Tooth disease type 1B.
Brain. 2012;135:2032–47. [
PMC free article: PMC3381724] [
PubMed: 22689911]
Schröder JM. Neuropathology of Charcot-Marie-Tooth and related disorders.
Neuromolecular Med. 2006;8:23–42. [
PubMed: 16775365]
Seeman P, Mazanec R, Huehne K, Suslíková P, Keller O, Rautenstrauss B. Hearing loss as the first feature of late-onset axonal CMT disease due to a novel P0 mutation.
Neurology. 2004;63:733–5. [
PubMed: 15326256]
Sereda MW, Nave KA. Animal models of Charcot-Marie-Tooth disease type 1A.
Neuromolecular Med. 2006;8:205–16. [
PubMed: 16775377]
Shiga K, Noto Y, Mizuta I, Hashiguchi A, Takashima H, Nakagawa M. A novel EGR2 mutation within a family with a mild demyelinating form of Charcot-Marie-Tooth disease.
J Peripher Nerv Syst. 2012;17:206–9. [
PubMed: 22734907]
Shin JS, Chung KW, Cho SY, Yun J, Hwang SJ, Kang SH, Cho EM, Kim SM, Choi BO. NEFL Pro22Arg mutation in Charcot-Marie-Tooth disease type 1.
J Hum Genet. 2008;53:936–40. [
PubMed: 18758688]
Shirk AJ, Anderson SK, Hashemi SH, Chance PF, Bennett CL. SIMPLE interacts with NEDD4 and TSG101: evidence for a role in lysosomal sorting and implications for Charcot-Marie-Tooth disease.
J Neurosci Res. 2005;82:43–50. [
PubMed: 16118794]
Shy ME. Peripheral neuropathies caused by mutations in the myelin protein zero.
J Neurol Sci. 2006;242:55–66. [
PubMed: 16414078]
Shy ME, Jáni A, Krajewski K, Grandis M, Lewis RA, Li J, Shy RR, Balsamo J, Lilien J, Garbern JY, Kamholz J. Phenotypic clustering in MPZ mutations.
Brain. 2004;127:371–84. [
PubMed: 14711881]
Shy ME, Scavina MT, Clark A, Krajewski KM, Li J, Kamholz J, Kolodny E, Szigeti K, Fischer RA, Saifi GM, Scherer SS, Lupski JR. T118M PMP22 mutation causes partial loss of function and HNPP-like neuropathy.
Ann Neurol. 2006;59:358–64. [
PubMed: 16437560]
Soong BW, Huang YH, Tsai PC, Huang CC, Pan HC, Lu YC, Chien HJ, Liu TT, Chang MH, Lin KP, Tu PH, Kao LS, Lee YC. Exome sequencing identifies GNB4 mutations as a cause of dominant intermediate Charcot-Marie-Tooth disease.
Am J Hum Genet. 2013;92:422–30. [
PMC free article: PMC3591844] [
PubMed: 23434117]
Song S, Zhang Y, Chen B, Zhang Y, Wang M, Wang Y, Yan M, Zou J, Huang Y, Zhong N. Mutation frequency for Charcot-Marie-Tooth disease type 1 in the Chinese population is similar to that in the global ethnic patients.
Genet Med. 2006;8:532–5. [
PubMed: 16912585]
Speevak MD, Farrell SA. Charcot-Marie-Tooth 1B caused by expansion of a familial myelin protein zero (MPZ) gene duplication.
Eur J Med Genet. 2013;56:566–9. [
PubMed: 23811036]
Starr A, Michalewski HJ, Zeng FG, Fujikawa-Brooks S, Linthicum F, Kim CS, Winnier D, Keats B. Pathology and physiology of auditory neuropathy with a novel mutation in the MPZ gene (Tyr145->Ser).
Brain. 2003;126:1604–19. [
PubMed: 12805115]
Steck AJ, Erne B, Pareyson D, Sghirlanzoni A, Taroni F, Schaeren-Wiemers N. Normal expression of myelin protein zero with frame-shift mutation correlates with mild phenotype.
J Peripher Nerv Syst. 2006;11:61–6. [
PubMed: 16519783]
Street VA, Bennett CL, Goldy JD, Shirk AJ, Kleopa KA, Tempel BL, Lipe HP, Scherer SS, Bird TD, Chance PF. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C.
Neurology. 2003;60:22–6. [
PubMed: 12525712]
Street VA, Meekins G, Lipe HP, Seltzer WK, Carter GT, Kraft GH, Bird TD. Charcot-Marie-Tooth neuropathy: clinical phenotypes of four novel mutations in the MPZ and Cx 32 genes.
Neuromuscul Disord. 2002;12:643–50. [
PubMed: 12207932]
Szigeti K, Garcia CA, Lupski JR. Charcot-Marie-Tooth disease and related hereditary polyneuropathies: molecular diagnostics determine aspects of medical management.
Genet Med. 2006;8:86–92. [
PubMed: 16481890]
Szigeti K, Wiszniewski W, Saifi GM, Sherman DL, Sule N, Adesina AM, Mancias P, Papasozomenos SCh, Miller G, Keppen L, Daentl D, Brophy PJ, Lupski JR. Functional, histopathologic and natural history study of neuropathy associated with EGR2 mutations.
Neurogenetics. 2007;8:257–62. [
PubMed: 17717711]
Taioli F, Cabrini I, Cavallaro T, Acler M, Fabrizi GM. Inherited demyelinating neuropathies with micromutations of peripheral myelin protein 22 gene.
Brain. 2011;134:608–17. [
PubMed: 21252112]
Taioli F, Bertolasi L, Ajena D, Ferrarini M, Cabrini I, Crestanello A, Fabrizi GM. Parental mosaicism of a novel PMP22 mutation with a minimal neuropathic phenotype.
J Peripher Nerv Syst. 2012;17:414–7. [
PubMed: 23279344]
Teunissen LL, Notermans NC, Franssen H, Van Engelen BG, Baas F, Wokke JH. Disease course of Charcot-Marie-Tooth disease type 2: a 5-year follow-up study.
Arch Neurol. 2003;60:823–8. [
PubMed: 12810486]
Timmerman V, De Jonghe P, Ceuterick C, De Vriendt E, Löfgren A, Nelis E, Warner LE, Lupski JR, Martin JJ, Van Broeckhoven C. Novel missense mutation in the early growth response 2 gene associated with Dejerine-Sottas syndrome phenotype.
Neurology. 1999;52:1827–32. [
PubMed: 10371530]
van Paassen BW, van der Kooi AJ, van Spaendonck-Zwarts KY, Verhamme C, Baas F, de Visser M. PMP22 related neuropathies: Charcot-Marie-Tooth disease type 1A and Hereditary Neuropathy with liability to Pressure Palsies.
Orphanet J Rare Dis. 2014;9:38. [
PMC free article: PMC3994927] [
PubMed: 24646194]
Verhamme C, van Schaik IN, Koelman JH, de Haan RJ, de Visser M. The natural history of Charcot-Marie-Tooth type 1A in adults: a 5-year follow-up study.
Brain. 2009a;132:3252–62. [
PubMed: 19843647]
Verhamme C, de Haan RJ, Vermeulen M, Baas F, de Visser M, van Schaik IN. Oral high dose ascorbic acid treatment for one year in young CMT1A patients: a randomised, double-blind, placebo-controlled phase II trial.
BMC Med. 2009b;7:70. [
PMC free article: PMC2784478] [
PubMed: 19909499]
Verhoeven K, De Jonghe P, Van de Putte T, Nelis E, Zwijsen A, Verpoorten N, De Vriendt E, Jacobs A, Van Gerwen V, Francis A, Ceuterick C, Huylebroeck D, Timmerman V. Slowed conduction and thin myelination of peripheral nerves associated with mutant rho Guanine-nucleotide exchange factor 10.
Am J Hum Genet. 2003;73:926–32. [
PMC free article: PMC1180612] [
PubMed: 14508709]
Verhoeven K, Villanova M, Rossi A, Malandrini A, De Jonghe P, Timmerman V. Localization of the gene for the intermediate form of Charcot-Marie-Tooth to chromosome 10q24.1-q25.1.
Am J Hum Genet. 2001;69:889–94. [
PMC free article: PMC1226075] [
PubMed: 11533914]
Villanova M, Timmerman V, De Jonghe P, Malandrini A, Rizzuto N, Van Broeckhoven C, Guazzi G, Rossi A. Charcot-Marie-Tooth disease: an intermediate form.
Neuromuscul Disord. 1998;8:392–3. [
PubMed: 9713856]
Walker JL, Nelson KR, Heavilon JA, Stevens DB, Lubicky JP, Ogden JA, VandenBrink KA. Hip abnormalities in children with Charcot-Marie-Tooth disease.
J Pediatr Orthop. 1994;14:54–9. [
PubMed: 8113373]
Ward CM, Dolan LA, Bennett DL, Morcuende JA, Cooper RR. Long-term results of reconstruction for treatment of a flexible cavovarus foot in Charcot-Marie-Tooth disease.
J Bone Joint Surg Am. 2008;90:2631–42. [
PMC free article: PMC2663331] [
PubMed: 19047708]
Warner LE, Mancias P, Butler IJ, McDonald CM, Keppen L, Koob KG, Lupski JR. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies.
Nat Genet. 1998;18:382–4. [
PubMed: 9537424]
Warner LE, Svaren J, Milbrandt J, Lupski JR. Functional consequences of mutations in the early growth response 2 gene (EGR2) correlate with severity of human myelinopathies.
Hum Mol Genet. 1999;8:1245–51. [
PubMed: 10369870]
Wells CA, Saavedra RA, Inouye H, Kirschner DA. Myelin P0-glycoprotein: predicted structure and interactions of extracellular domain.
J Neurochem. 1993;61:1987–95. [
PubMed: 7504078]
Wrabetz L, D'Antonio M, Pennuto M, Dati G, Tinelli E, Fratta P, Previtali S, Imperiale D, Zielasek J, Toyka K, Avila RL, Kirschner DA, Messing A, Feltri ML, Quattrini A. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice.
J Neurosci. 2006;26:2358–68. [
PMC free article: PMC6674823] [
PubMed: 16495463]
Yonekawa T, Komaki H, Saito Y, Takashima H, Sasaki M. Congenital hypomyelinating neuropathy attributable to a de novo p.Asp61Asn mutation of the myelin protein zero gene.
Pediatr Neurol. 2013;48:59–62. [
PubMed: 23290023]
Young P, Grote K, Kuhlenbäumer G, Debus O, Kurlemann H, Halfter H, Funke H, Ringelstein EB, Stögbauer F. Mutation analysis in Chariot-Marie Tooth disease type 1: point mutations in the MPZ gene and the GJB1 gene cause comparable phenotypic heterogeneity.
J Neurol. 2001;248:410–5. [
PubMed: 11437164]
Young T, Shuey N, Partridge J, Bremner FD, Nicholl DJ. Compound Charcot-Marie-Tooth disease: a kindred with severe hereditary neuropathy, pupil abnormalities and a novel MPZ mutation.
J Neurol Neurosurg Psychiatry. 2013;84:234–6. [
PubMed: 23197742]
Zschüntzsch J, Dibaj P, Pilgram S, Kötting J, Gerding WM, Neusch C. Severe demyelinating hypertrophic polyneuropathy caused by a de novo frameshift mutation within the intracellular domain of myelin protein zero (MPZ/P0).
J Neurol Sci. 2009;281:113–5. [
PubMed: 19344920]
Suggested Reading
Lupski JR, Garcia CA. Charcot-Marie-Tooth peripheral neuropathies and related disorders. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 227. New York, NY: McGraw-Hill.
Chapter Notes
Revision History
26 March 2015 (ks) Revision: MPZ pathogenic variants renamed to correspond with RefSeq NM_000530.6
18 December 2014 (me) Comprehensive update posted live
20 February 2014 (tb) Revision: Lee et al 2013 added to Preimplantation genetic diagnosis
7 November 2013 (tb) Revision: additions to Prevalence; figure added [
Rossor et al 2013]
11 July 2013 (tb) Revision: additions to Prevalence and Natural History
18 October 2012 (me) Comprehensive update posted live
18 August 2011 (tb) Revision: Høyer et al 2011; see Testing, Genotype-Phenotype Correlations, Molecular Genetics
16 June 2011 (tb) Revision: additions to Differential Diagnosis – FBLN5
1 March 2011 (cd) Revision: edits to Testing Strategy
14 September 2010 (me) Comprehensive update posted live
30 March 2007 (me) Comprehensive update posted to live Web site
30 December 2005 (cd) Revision:
prenatal diagnosis and mutation scanning clinically available for CMT1C
26 April 2005 (me) Comprehensive update posted live
9 September 2004 (tb,cd) Revision: addition of
LITAF;
sequence analysis clinically available
10 May 2004 (tb) Author revisions
29 December 2003 (tb) Author revisions
22 April 2003 (tb) Author revisions
27 March 2003 (me) Comprehensive update posted live
10 May 2002 (tb) Author revisions
20 December 2001 (tb) Author revisions
12 September 2001 (tb) Author revisions
24 July 2001 (tb) Author revisions
27 June 2001 (tb) Author revisions
1 June 2001 (tb) Author revisions
16 January 2001 (tb) Author revisions
25 August 2000 (ca) Comprehensive update posted live
15 June 2000 (tb) Author revisions
15 May 2000 (tb) Author revisions
14 January 2000 (tb) Author revisions
31 August 1999 (tb) Author revisions
18 June 1999 (tb) Author revisions
8 April 1999 (tb) Author revisions
5 March 1999 (tb) Author revisions
12 October 1998 (tb) Author revisions
31 August 1998 (pb) Review posted live
April 1996 (tb) Original submission