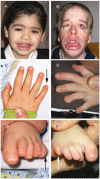
Clinical Characteristics
Clinical Description
The following information has been compiled from data included in two reports by the Coffin-Siris Syndrome International Collaborators [Kosho et al 2014b, Santen et al 2014]. This section focuses on features common to the molecular subtypes; the findings that vary in frequency or severity between genetic etiologies are noted in Genotype-Phenotype Correlations.
Early Characteristics
Prenatal findings are typically unremarkable, with growth within normal limits. Rarely, CNS or cardiac anomalies, IUGR, and microcephaly have been noted.
Infancy. Although many individuals with Coffin-Siris syndrome (CSS) may not be clinically distinguishable at birth, several of the congenital anomalies may be noted:
Hypoplasia of the fifth digits/nails. Most individuals have at a minimum brachydactyly of the fifth digit (seen in 65% of affected infants) and hypoplasia of one or more nails (80%). It should be noted that some individuals with a molecularly confirmed diagnosis of CSS have little or no fifth digit involvement.
Dysmorphic facial features (~30% at birth). Because facial features typically coarsen over time, the characteristic facies may not be apparent until later in
childhood.
Hirsutism often noted
Other findings appearing in infancy that may be the first indication of CSS:
Feeding problems (90%) and slow growth
Hypotonia (75%)
Seizures (50%)
Hearing impairment (45%)
Visual impairment (~40%)
Findings in Childhood
Developmental delays. The developmental/cognitive delay is typically apparent when delayed developmental milestones are noted and/or formal cognitive testing is performed.
On average, children with CSS learn to sit at 12 months, walk at 30 months, and speak their first words at 24 months.
Expressive language is more severely affected than receptive language, with no speech in a significant subset of individuals.
Intellectual disability is present in most and typically moderate to severe (IQ range: 40 to 69); however, IQ as high as 97 has been reported [
Santen et al 2012].
Behavioral abnormalities include hyperactivity (~10%), aggressiveness (~10%), and occasionally autistic features.
Brain/CNS issues
CNS malformations include Dandy-Walker variant, gyral simplification, and agenesis of the corpus callosum.
Seizures and tics. A variety of types of seizures are reported. There is no typical age of onset for seizures or tics.
Hypotonia (75%), noted in infancy, is typically persistent.
Facial features (See .)
Coarse facies (95%)
Thick eyebrows (90%)
Prominent eyelashes (85%)
Flat nasal bridge (50%)
Short nose (50%)
Anteverted nares (50%)
Broad nasal tip (75%)
Wide nasal base (50%)
Thick alae nasi (70%)
Broad philtrum (70%)
Wide mouth (80%)
Thin vermilion of the upper lip (50%)
Thick vermilion of the lower lip (80%)
Musculoskeletal findings
Skin and hair findings
Hypertrichosis (95%) may appear in areas unexpected for an individual’s ethnicity (i.e., back, shoulders).
A low anterior hairline is common (75%).
Sparse scalp hair (60%); hair may appear at an appropriate age but may be very thin.
Feeding difficulties. Children may have oral aversion or difficulty feeding in the absence of any evident intestinal malformations.
Growth issues
Weight and height is below the 50th percentile for most, and below the 5th percentile for 20%.
Bone age typically lags about two to three years behind chronologic age.
Dentition is delayed (40%).
Hearing impairment (45%) is often associated with recurrent upper respiratory tract infections.
Ophthalmologic abnormalities
Ptosis (50%)
Strabismus (50%)
Myopia (20%)
Frequent infections (60%). These are poorly characterized, but often are consistent with upper respiratory viral infections.
Malformations
Cardiac anomalies (35%) including ventricular septal defects, atrial septal defects, tetralogy of Fallot, and patent ductus arteriosus
Renal and genitourinary malformations (~35%) including cryptorchidism most commonly, but also horseshoe kidney, hypospadias, and other abnormalities
Tumor risk. Although pathogenic variants in a subset of the genes causing CSS have been implicated in tumorigenesis (see Cancer and Benign Tumors), data on tumor risk in CSS are lacking. Tumors have been reported in three individuals with CSS:
An individual with a 4.2-Mb deletion that included (among 14 genes)
ARID1B developed papillary thyroid cancer [
Vengoechea et al 2014].
Prognosis
In the absence of long-term studies, information on life span in individuals with Coffin-Siris syndrome is not available. Children have been reported to die of aspiration pneumonia and/or seizures, although this is not common [Schrier et al 2012]. Efforts are in progress by the Coffin-Siris Syndrome International Consortium [Kosho et al 2014a] to better understand prognosis in individuals with CSS.
Phenotype Correlations by Gene
Phenotype correlations by gene have been seen in clinically diagnosed individuals with pathogenic variants in ARID1A, ARID1B, ARID2, DPF2, SMARCA4, SMARCB1, SMARCC2, SMARCE1, SOX4, and SOX11 [Wieczorek et al 2013, Kosho et al 2014b, Santen et al 2014, Tsurusaki et al 2014a, Hempel et al 2016, Vasileiou et al 2018, Gazdagh et al 2019, Machol et al 2019, Zawerton et al 2019].
ARID1B. Individuals with pathogenic ARID1B variants are typically at the milder end of the spectrum of CSS and often have normal growth. Moderately severe feeding problems are noted in two thirds, seizures in one third, and hypoplasia of the corpus callosum in one third. Facial gestalt is consistent with CSS, albeit at times milder.
ARID2. Affected individuals typically do not have birth defects.
SMARCA4. Individuals with a pathogenic variant in SMARCA4 appear to have growth impairment that is mild prenatally and mild to moderate postnatally; sucking/feeding difficulty is almost always observed. While individuals can sometimes have severe developmental delays, significant behavioral challenges tend to be more characteristic. Facial features have demonstrated less coarseness, while hypoplastic fifth fingers or toes and hypoplastic fifth fingernails or toenails are a near-constant finding (and hypoplasia of other fingernails or toenails an occasional finding). Prominence of interphalangeal joints and distal phalanges is also noted in some.
SMARCB1. Individuals with a pathogenic variant in SMARCB1 typically have a more severely affected phenotype and all have growth impairment, usually mild prenatally and moderate to severe postnatally, with sucking/feeding difficulty. Structural CNS abnormalities with hypotonia and seizures are typical findings accompanied by severe developmental delay/intellectual disability; individuals are typically nonverbal. Typical skeletal findings include hypoplastic fifth fingers or toes, hypoplastic other fingernails or toenails, prominent distal phalanges, and scoliosis. Some individuals may walk independently. Gastrointestinal complications and hernia as well as cardiovascular and genitourinary complications are common.
SMARCE1. Individuals with pathogenic SMARCE1 variants tend to have severe intellectual disability, typical facial gestalt, and hypoplastic or absent fifth finger- and toenails associated with hypoplasia of other nails. The hands are characterized by long and slender fingers. Individuals are typically small for gestational age and have postnatal short stature and severe microcephaly, complex congenital heart defects, feeding difficulties, and seizures.
SOX4. Severely affected individuals may show neurologic complications including hypotonia, spastic quadriparesis, and epilepsy.
SOX11. Neurodevelopmental abnormalities tend to be more prevalent than organ-system or physical complications.
Penetrance
Penetrance for Coffin-Siris syndrome appears to be complete.
More females than males with CSS were reported in the literature prior to 2001 [Fleck et al 2001]; however, in cases of molecularly confirmed CSS, male:female ratios are similar [Kosho et al 2014b, Santen et al 2014]. No evidence exists for X-linked dominant, sex-limited, or mitochondrial inheritance.
Prevalence
Fewer than 200 individuals with molecularly confirmed Coffin-Siris syndrome have been reported, indicating that the diagnosis is rare. However, this is likely an underestimate, as not all individuals may have come to medical attention.
In addition, the identification of a pathogenic variant in ARID1B in some members of a large cohort with intellectual disability [Hoyer et al 2012] suggests that the prevalence of pathogenic variants in genes associated with CSS (and possibly of subtle phenotypic features of CSS) may be higher than currently appreciated among those with intellectual disability.
Differential Diagnosis
Nicolaides-Baraitser syndrome
(NCBRS) is characterized by sparse scalp hair, prominence of the interphalangeal joints and distal phalanges due to decreased subcutaneous fat, characteristic coarse facial features, microcephaly, seizures, and developmental delay/intellectual disability. Developmental delay / intellectual disability is severe in nearly half of individuals with NCBRS, moderate in a third, and mild in the remainder. Nearly a third never develop speech. Of note, after heterozygous SMARCA2 pathogenic variants were identified in NCBRS [Van Houdt et al 2012], reevaluation of an individual initially thought to have CSS determined that findings were more consistent with NCBRS [Tsurusaki et al 2012]. Inheritance is autosomal dominant; to date, all affected individuals have had a de novo
SMARCA2 pathogenic variant.
Borjeson-Forssman-Lehmann syndrome (BFLS) (OMIM 301900) is typically characterized by males with severe intellectual disability, epilepsy, hypogonadism, hypometabolism, marked obesity, swelling of subcutaneous tissue of face, narrow palpebral fissure, and large but not deformed ears. Females with pathogenic variants in PHF6, which causes BFLS, demonstrate some phenotypic overlap with individuals with CSS [Wieczorek et al 2013]. The two syndromes, however, are still considered distinctly separate entities [Zweier et al 2013].
Mosaic trisomy 9. An individual with mosaic trisomy 9 had features similar to those of CSS, including facial features (wide, bulbous nose), hirsutism, and hypoplasia of the fifth digits [Kushnick & Adessa 1976].
Brachymorphism-onychodysplasia-dysphalangism (BOD) syndrome (OMIM 113477) is characterized by short stature, tiny dysplastic nails, short fifth fingers, a wide mouth with broad nose, and mild intellectual deficits [Verloes et al 1993, Elliott & Teebi 2000]. This latter characteristic is most likely to distinguish individuals with BOD syndrome from those with CSS, as the cognitive disability in CSS is nearly always moderate to severe. Inheritance appears to be autosomal dominant.
DOORS (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures) syndrome. Features in common with CSS include hypoplastic terminal phalanges and/or nail anomalies, deafness, and neurologic abnormalities. DOORS syndrome is inherited in an autosomal recessive manner and is caused by biallelic pathogenic variants in TBC1D24 (see TBC1D24-Related Disorders).
Fetal alcohol spectrum (FAS). Small nails, prenatal and postnatal growth retardation, dysmorphic facial features, and cognitive disabilities may be seen in FAS.
Fetal hydantoin/phenytoin embryopathy. Small nails with hypoplasia of distal phalanges, dysmorphic facial features, digitalized thumbs, low hairline, short or webbed neck, growth retardation, and cognitive disabilities have been described in this syndrome, caused by prenatal exposure to phenytoin.
Mabry syndrome (hyperphosphatasia with mental retardation syndrome 1; OMIM 239300). Mabry syndrome is characterized by delayed development, seizures, coarse facial features, hypoplastic fifth digits, and elevated serum concentrations of alkaline phosphatase [Gomes & Hunter 1970, Kruse et al 1988, Thompson et al 2010]. It is inherited in an autosomal recessive manner and caused by biallelic pathogenic variants in PIGV [Krawitz et al 2010].
Cornelia de Lange syndrome
(CdLS). Classic CdLS is characterized by distinctive craniofacial features (arched eyebrows, synophrys, upturned nose, small teeth, microcephaly); growth retardation; and limb anomalies, which can at times include fifth finger hypoplasia similar to CSS. Other findings may include cardiac defects, gastrointestinal anomalies, and genitourinary malformations. Pathogenic variants in NIPBL, SMC1A, SMC3, HDAC8, or RAD21 are causative. CdLS is inherited in an autosomal dominant (NIPBL, SMC3, and RAD21) or X-linked (SMC1A and HDAC8) manner.
4q deletion syndrome. This chromosome deletion syndrome results in a characteristic curved, volar, fifth-digit nail, which may resemble a hypoplastic distal phalanx.
BICRA-related disorder (OMIM 619325) is classically characterized by coarse facial features including microcephaly, frontal bossing, epicanthal folds, prominent nasal tips, and low-set ears. These features are also commonly expressed in individuals with CSS. Barish et al [2020] identified 14 individuals with pathogenic variants in BICRA, all of whom have coarse facial features and varying magnitudes of intellectual disability. While phenotypic similarities between CSS and BICRA have been identified, an assessment of a larger cohort of individuals with BICRA pathogenic variants will be needed to determine whether it is clinically similar to or distinct from CSS. BICRA-related disorder is inherited in an autosomal dominant manner.
SMARCD1-related disorder (OMIM 618779). Nixon et al [2019] identified five individuals with pathogenic variants in SMARCD1. These individuals presented with developmental delay, intellectual disability, hypotonia, and feeding difficulties. While similarities between CSS and SMARCD1-related disorder have been identified, an assessment of a larger cohort of individuals with SMARCD1 variants will be needed to determine whether it is clinically similar or distinct from CSS. SMARCD1-related disorder is inherited in an autosomal dominant manner.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Coffin-Siris syndrome (CSS) is inherited in an autosomal dominant manner. Most affected individuals reported to date have had a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
Most probands reported to date have the disorder as the result of a de novo CSS-causing pathogenic variant.
The proportion of cases caused by a de novo pathogenic variant is unknown, but likely approaches 100%, given the paucity of reports of affected parents in the literature.
Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include testing of the parents for the pathogenic variant identified in the proband.
If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, the proband most likely has a
de novo pathogenic variant
. In rare cases, a parent may have germline mosaicism [
Ben-Salem et al 2016].
Evaluation of parents may determine that one is affected but has escaped previous diagnosis because of a milder phenotype. Therefore, an apparently negative family history cannot be fully confirmed until appropriate evaluations have been performed.
Sibs of a proband
The risk to the sibs of the proband depends on the genetic status of the proband’s parents.
In the rare circumstance that a parent of the proband is affected and is heterozygous for a CSS-causing pathogenic variant, the risk to the sibs is 50%.
When the parents are clinically unaffected, the risk to the sibs of a proband appears to be low.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to sibs is approximately 1% because of the theoretic possibility of parental germline mosaicism.
One report of CSS in two sisters and partial expression in their father has been published [
Haspeslagh et al 1984], suggesting parental somatic (and germline) mosaicism. However, there has been no molecular confirmation of the diagnosis, and the affected family members may have a disorder other than CSS.
Offspring of a proband. Each child of an individual with CSS has a 50% chance of inheriting the CSS-related pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: in the rare event of an affected parent, other family members may be at risk.
Prenatal Testing and Preimplantation Genetic Testing
Risk to future pregnancies is presumed to be low as the familial proband most likely has a de novo CSS-causing pathogenic variant. However, prenatal testing or preimplantation genetic testing are options to consider, as the risk may be greater than in the general population because of the possibility of parental germline mosaicism.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.