Summary
Clinical characteristics.
Coffin-Siris syndrome (CSS) is classically characterized by aplasia or hypoplasia of the distal phalanx or nail of the fifth and additional digits, developmental or cognitive delay of varying degree, distinctive facial features, hypotonia, hirsutism/hypertrichosis, and sparse scalp hair. Congenital anomalies can include malformations of the cardiac, gastrointestinal, genitourinary, and/or central nervous systems. Other findings commonly include feeding difficulties, slow growth, ophthalmologic abnormalities, and hearing impairment.
Diagnosis/testing.
Before the molecular basis was known, the diagnosis of CSS was based solely on clinical findings (although consensus clinical diagnostic criteria have not yet been published). The diagnosis of CSS is established in a proband with suggestive findings by identification of a heterozygous pathogenic variant in one of the genes listed in Table 1.
Management.
Treatment of manifestations: Occupational, physical, and/or speech therapies to optimize developmental outcomes. Feeding therapy, nutritional supplementation and/or gastrostomy tube placement as needed to meet nutritional needs. Routine management of ophthalmologic abnormalities and hearing loss.
Surveillance: Yearly evaluation by a developmental pediatrician to assess developmental progress and therapeutic and educational interventions; follow up with a gastroenterologist and feeding specialists as needed to monitor feeding and weight gain. Routine follow up of ophthalmologic and/or audiologic abnormalities.
Genetic counseling.
CSS is inherited in an autosomal dominant manner; however, most affected individuals have the disorder as the result of de novo CSS-causing pathogenic variant. If the CSS-causing pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Formal clinical diagnostic criteria for Coffin-Siris syndrome (CSS) have not been established; however, several key features are useful in making a clinical diagnosis.
Suggestive Findings
Coffin-Siris syndrome (CSS) should be suspected in individuals with the following findings [Fleck et al 2001, Schrier et al 2012, Kosho et al 2014b, Santen et al 2014]:
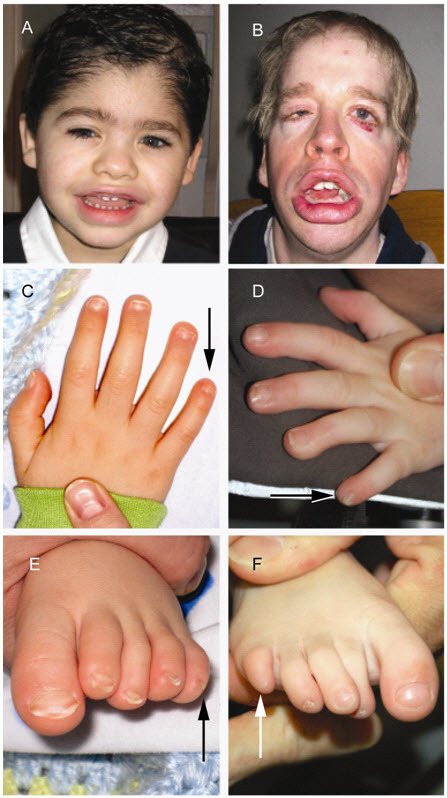
- Fifth-digit nail / distal phalanx hypoplasia/aplasia. Typically, individuals with a clinical diagnosis of CSS have either aplasia or hypoplasia of the distal phalanx or absence of the nail, typically involving the fifth finger, but other digits may also be affected (Figure 1C, D, E, F). Toes can also be affected, where the finding tends to involve multiple digits.
- Developmental or cognitive delay of variable degree
- Facial features [Schrier et al 2012]. Individuals with typical features demonstrate a wide mouth with thick, everted upper and lower lips, broad nasal bridge with broad nasal tip, thick eyebrows, and long eyelashes. Together, these features can give a suggestion of coarseness in individuals with CSS (Figure 1A, B).
- Hypotonia that is central in origin
- Hirsutism/hypertrichosis. Hair growth in atypical areas (e.g., the back) or excessive hair growth on the arms or face
- Sparse scalp hair, especially in infancy, particularly in the temporal regions
Figure 1.
Coffin-Siris syndrome classic features Facial features (i.e., bushy eyebrows, coarse facies, and thick, everted lips) in (A) a clinically diagnosed boy age five years and (B) a clinically diagnosed man age 29 years
Establishing the Diagnosis
The diagnosis of CSS is established in a proband with suggestive findings and a heterozygous pathogenic variant in one of the genes listed in Table 1 identified by molecular genetic testing.
Note: Identification of a heterozygous variant of uncertain significance in one of the genes listed in Table 1 does not establish or rule out the diagnosis of this disorder.
Molecular testing approaches typically include use of a multigene panel or more comprehensive genomic testing.
Note: Serial single-gene testing in the order in which pathogenic variants most commonly occur is another option, but given the number of genes associated with this phenotype, such testing is not commonly done. However, because mutation of ARID1B is found in a relatively large number of affected individuals compared to mutation in the other genes listed in Table 1, sequence analysis of ARID1B, followed by gene targeted deletion/duplication analysis, can be considered as a first step. If this is not diagnostic, use of a multigene panel or more comprehensive genomic testing is often performed next.
- A multigene panel that includes the genes listed in Table 1 and other genes of interest (see Differential Diagnosis) may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel provides the best opportunity to identify the genetic cause of the condition. (3) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
Table 1.
Molecular Genetic Testing Used in Coffin-Siris Syndrome
Karyotype and chromosomal microarray (CMA). Many individuals who present with congenital anomalies and developmental delay have their chromosomes evaluated through karyotype and/or CMA as part of the initial evaluation. If an individual presenting with features of CSS does not have a pathogenic variant identified by the testing described, a karyotype and/or CMA should be considered based on the occurrence of rare rearrangements that have been reported to cause CSS [Backx et al 2011, Halgren et al 2012, Malli et al 2014].
Clinical Characteristics
Clinical Description
The following information has been compiled from data included in two reports by the Coffin-Siris Syndrome International Collaborators [Kosho et al 2014b, Santen et al 2014]. This section focuses on features common to the molecular subtypes; the findings that vary in frequency or severity between genetic etiologies are noted in Genotype-Phenotype Correlations.
Early Characteristics
Prenatal findings are typically unremarkable, with growth within normal limits. Rarely, CNS or cardiac anomalies, IUGR, and microcephaly have been noted.
Infancy. Although many individuals with Coffin-Siris syndrome (CSS) may not be clinically distinguishable at birth, several of the congenital anomalies may be noted:
- Hypoplasia of the fifth digits/nails. Most individuals have at a minimum brachydactyly of the fifth digit (seen in 65% of affected infants) and hypoplasia of one or more nails (80%). It should be noted that some individuals with a molecularly confirmed diagnosis of CSS have little or no fifth digit involvement.
- Dysmorphic facial features (~30% at birth). Because facial features typically coarsen over time, the characteristic facies may not be apparent until later in childhood.
- Hirsutism often noted
- Malformations affecting the CNS and cardiac and genitourinary systems (See Findings in Childhood.)
- Other findings appearing in infancy that may be the first indication of CSS:
- Feeding problems (90%) and slow growth
- Hypotonia (75%)
- Seizures (50%)
- Hearing impairment (45%)
- Visual impairment (~40%)
Findings in Childhood
Developmental delays. The developmental/cognitive delay is typically apparent when delayed developmental milestones are noted and/or formal cognitive testing is performed.
- On average, children with CSS learn to sit at 12 months, walk at 30 months, and speak their first words at 24 months.
- Expressive language is more severely affected than receptive language, with no speech in a significant subset of individuals.
- Intellectual disability is present in most and typically moderate to severe (IQ range: 40 to 69); however, IQ as high as 97 has been reported [Santen et al 2012].
- Behavioral abnormalities include hyperactivity (~10%), aggressiveness (~10%), and occasionally autistic features.
Brain/CNS issues
- CNS malformations include Dandy-Walker variant, gyral simplification, and agenesis of the corpus callosum.
- Seizures and tics. A variety of types of seizures are reported. There is no typical age of onset for seizures or tics.
- Hypotonia (75%), noted in infancy, is typically persistent.
Facial features (See Figure 1.)
- Coarse facies (95%)
- Thick eyebrows (90%)
- Prominent eyelashes (85%)
- Flat nasal bridge (50%)
- Short nose (50%)
- Anteverted nares (50%)
- Broad nasal tip (75%)
- Wide nasal base (50%)
- Thick alae nasi (70%)
- Broad philtrum (70%)
- Wide mouth (80%)
- Thin vermilion of the upper lip (50%)
- Thick vermilion of the lower lip (80%)
Musculoskeletal findings
- Small nails on 5th finger or toe (80%)
- Clinodactyly (40%)
- Delayed bone age (40%)
- Joint laxity (66%)
- Scoliosis (30%)
- Hernias (10%)
Skin and hair findings
- Hypertrichosis (95%) may appear in areas unexpected for an individual’s ethnicity (i.e., back, shoulders).
- A low anterior hairline is common (75%).
- Sparse scalp hair (60%); hair may appear at an appropriate age but may be very thin.
Feeding difficulties. Children may have oral aversion or difficulty feeding in the absence of any evident intestinal malformations.
Growth issues
- Weight and height is below the 50th percentile for most, and below the 5th percentile for 20%.
- Bone age typically lags about two to three years behind chronologic age.
- Dentition is delayed (40%).
Hearing impairment (45%) is often associated with recurrent upper respiratory tract infections.
Ophthalmologic abnormalities
- Ptosis (50%)
- Strabismus (50%)
- Myopia (20%)
Frequent infections (60%). These are poorly characterized, but often are consistent with upper respiratory viral infections.
Malformations
- Cardiac anomalies (35%) including ventricular septal defects, atrial septal defects, tetralogy of Fallot, and patent ductus arteriosus
- Renal and genitourinary malformations (~35%) including cryptorchidism most commonly, but also horseshoe kidney, hypospadias, and other abnormalities
Tumor risk. Although pathogenic variants in a subset of the genes causing CSS have been implicated in tumorigenesis (see Cancer and Benign Tumors), data on tumor risk in CSS are lacking. Tumors have been reported in three individuals with CSS:
- Hepatoblastoma was reported in one of eight individuals with an ARID1A pathogenic variant [Tsurusaki et al 2012, Kosho et al 2014b].
- An individual with a 4.2-Mb deletion that included (among 14 genes) ARID1B developed papillary thyroid cancer [Vengoechea et al 2014].
- Multiple studies have reported an individual who has CSS, schwannomatosis, and a pathogenic variant in SMARCB1 [Carter et al 2012, Schrier et al 2012, Gossai et al 2015].
Prognosis
In the absence of long-term studies, information on life span in individuals with Coffin-Siris syndrome is not available. Children have been reported to die of aspiration pneumonia and/or seizures, although this is not common [Schrier et al 2012]. Efforts are in progress by the Coffin-Siris Syndrome International Consortium [Kosho et al 2014a] to better understand prognosis in individuals with CSS.
Phenotype Correlations by Gene
Phenotype correlations by gene have been seen in clinically diagnosed individuals with pathogenic variants in ARID1A, ARID1B, ARID2, DPF2, SMARCA4, SMARCB1, SMARCC2, SMARCE1, SOX4, and SOX11 [Wieczorek et al 2013, Kosho et al 2014b, Santen et al 2014, Tsurusaki et al 2014a, Hempel et al 2016, Vasileiou et al 2018, Gazdagh et al 2019, Machol et al 2019, Zawerton et al 2019].
ARID1B. Individuals with pathogenic ARID1B variants are typically at the milder end of the spectrum of CSS and often have normal growth. Moderately severe feeding problems are noted in two thirds, seizures in one third, and hypoplasia of the corpus callosum in one third. Facial gestalt is consistent with CSS, albeit at times milder.
ARID2. Affected individuals typically do not have birth defects.
SMARCA4. Individuals with a pathogenic variant in SMARCA4 appear to have growth impairment that is mild prenatally and mild to moderate postnatally; sucking/feeding difficulty is almost always observed. While individuals can sometimes have severe developmental delays, significant behavioral challenges tend to be more characteristic. Facial features have demonstrated less coarseness, while hypoplastic fifth fingers or toes and hypoplastic fifth fingernails or toenails are a near-constant finding (and hypoplasia of other fingernails or toenails an occasional finding). Prominence of interphalangeal joints and distal phalanges is also noted in some.
SMARCB1. Individuals with a pathogenic variant in SMARCB1 typically have a more severely affected phenotype and all have growth impairment, usually mild prenatally and moderate to severe postnatally, with sucking/feeding difficulty. Structural CNS abnormalities with hypotonia and seizures are typical findings accompanied by severe developmental delay/intellectual disability; individuals are typically nonverbal. Typical skeletal findings include hypoplastic fifth fingers or toes, hypoplastic other fingernails or toenails, prominent distal phalanges, and scoliosis. Some individuals may walk independently. Gastrointestinal complications and hernia as well as cardiovascular and genitourinary complications are common.
SMARCE1. Individuals with pathogenic SMARCE1 variants tend to have severe intellectual disability, typical facial gestalt, and hypoplastic or absent fifth finger- and toenails associated with hypoplasia of other nails. The hands are characterized by long and slender fingers. Individuals are typically small for gestational age and have postnatal short stature and severe microcephaly, complex congenital heart defects, feeding difficulties, and seizures.
SOX4. Severely affected individuals may show neurologic complications including hypotonia, spastic quadriparesis, and epilepsy.
SOX11. Neurodevelopmental abnormalities tend to be more prevalent than organ-system or physical complications.
Penetrance
Penetrance for Coffin-Siris syndrome appears to be complete.
More females than males with CSS were reported in the literature prior to 2001 [Fleck et al 2001]; however, in cases of molecularly confirmed CSS, male:female ratios are similar [Kosho et al 2014b, Santen et al 2014]. No evidence exists for X-linked dominant, sex-limited, or mitochondrial inheritance.
Prevalence
Fewer than 200 individuals with molecularly confirmed Coffin-Siris syndrome have been reported, indicating that the diagnosis is rare. However, this is likely an underestimate, as not all individuals may have come to medical attention.
In addition, the identification of a pathogenic variant in ARID1B in some members of a large cohort with intellectual disability [Hoyer et al 2012] suggests that the prevalence of pathogenic variants in genes associated with CSS (and possibly of subtle phenotypic features of CSS) may be higher than currently appreciated among those with intellectual disability.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview have been associated with pathogenic variants in ARID1A, ARID2, DPF2, SMARCC2, SMARCE1, SOX4, or SOX11.
Phenotypes in addition to classic Coffin-Siris syndrome have been associated with pathogenic variants of ARID1B, SMARCA4, and SMARCB1.
- ARID1B. Both pathogenic variants in and microdeletions containing ARID1B have been reported in several individuals with isolated intellectual disability (ID) [Nagamani et al 2009, Hoyer et al 2012, Michelson et al 2012]. Deletions have also been reported in several individuals with features in addition to ID, including agenesis of the corpus callosum (ACC), seizures, hypertrichosis, hearing loss, and myopia [Santen et al 2014]. See ARID1B-Related Disorder.
- SMARCA4. Heterozygous germline pathogenic variants in SMARCA4 have been reported to cause the rhabdoid tumor predisposition [Schneppenheim et al 2010, Hasselblatt et al 2011, Biegel et al 2014].
- SMARCB1. One individual with Kleefstra syndrome has been found to have a heterozygous de novo p.Arg37His variant [Kleefstra et al 2012]. This variant is at the N-terminus of the protein, while those in CSS have been localized at the C-terminal end of the SNF5 domain (see Molecular Pathogenesis).Heterozygous germline pathogenic variants in SMARCB1 have been reported to cause the rhabdoid tumor predisposition syndrome via the classic two-hit model of tumorigenesis [Roberts & Biegel 2009, Biegel et al 2014].Heterozygous germline pathogenic variants in SMARCB1 cause schwannomatosis, an autosomal dominant tumor suppressor syndrome with reduced penetrance, characterized by a predisposition to develop multiple schwannomas and (less frequently) meningiomas [Merker et al 2012].
Of note, several individuals initially ascertained as having CSS were subsequently found to have pathogenic variants in SMARCA2 [Santen et al 2012, Tsurusaki et al 2012] or PHF6 [Wieczorek et al 2013]. Further review of these individuals has suggested that they more consistently fit the diagnoses of Nicolaides-Baraitser and Borjeson-Forssman-Lehmann syndromes, respectively [Kosho et al 2014b, Tsurusaki et al 2014b, Zweier et al 2014]. See Table 1, footnotes 8 and 13, and Differential Diagnosis.
Differential Diagnosis
Nicolaides-Baraitser syndrome (NCBRS) is characterized by sparse scalp hair, prominence of the interphalangeal joints and distal phalanges due to decreased subcutaneous fat, characteristic coarse facial features, microcephaly, seizures, and developmental delay/intellectual disability. Developmental delay / intellectual disability is severe in nearly half of individuals with NCBRS, moderate in a third, and mild in the remainder. Nearly a third never develop speech. Of note, after heterozygous SMARCA2 pathogenic variants were identified in NCBRS [Van Houdt et al 2012], reevaluation of an individual initially thought to have CSS determined that findings were more consistent with NCBRS [Tsurusaki et al 2012]. Inheritance is autosomal dominant; to date, all affected individuals have had a de novo SMARCA2 pathogenic variant.
Borjeson-Forssman-Lehmann syndrome (BFLS) (OMIM 301900) is typically characterized by males with severe intellectual disability, epilepsy, hypogonadism, hypometabolism, marked obesity, swelling of subcutaneous tissue of face, narrow palpebral fissure, and large but not deformed ears. Females with pathogenic variants in PHF6, which causes BFLS, demonstrate some phenotypic overlap with individuals with CSS [Wieczorek et al 2013]. The two syndromes, however, are still considered distinctly separate entities [Zweier et al 2013].
Mosaic trisomy 9. An individual with mosaic trisomy 9 had features similar to those of CSS, including facial features (wide, bulbous nose), hirsutism, and hypoplasia of the fifth digits [Kushnick & Adessa 1976].
Brachymorphism-onychodysplasia-dysphalangism (BOD) syndrome (OMIM 113477) is characterized by short stature, tiny dysplastic nails, short fifth fingers, a wide mouth with broad nose, and mild intellectual deficits [Verloes et al 1993, Elliott & Teebi 2000]. This latter characteristic is most likely to distinguish individuals with BOD syndrome from those with CSS, as the cognitive disability in CSS is nearly always moderate to severe. Inheritance appears to be autosomal dominant.
DOORS (deafness, onychodystrophy, osteodystrophy, mental retardation, and seizures) syndrome. Features in common with CSS include hypoplastic terminal phalanges and/or nail anomalies, deafness, and neurologic abnormalities. DOORS syndrome is inherited in an autosomal recessive manner and is caused by biallelic pathogenic variants in TBC1D24 (see TBC1D24-Related Disorders).
Fetal alcohol spectrum (FAS). Small nails, prenatal and postnatal growth retardation, dysmorphic facial features, and cognitive disabilities may be seen in FAS.
Fetal hydantoin/phenytoin embryopathy. Small nails with hypoplasia of distal phalanges, dysmorphic facial features, digitalized thumbs, low hairline, short or webbed neck, growth retardation, and cognitive disabilities have been described in this syndrome, caused by prenatal exposure to phenytoin.
Mabry syndrome (hyperphosphatasia with mental retardation syndrome 1; OMIM 239300). Mabry syndrome is characterized by delayed development, seizures, coarse facial features, hypoplastic fifth digits, and elevated serum concentrations of alkaline phosphatase [Gomes & Hunter 1970, Kruse et al 1988, Thompson et al 2010]. It is inherited in an autosomal recessive manner and caused by biallelic pathogenic variants in PIGV [Krawitz et al 2010].
Cornelia de Lange syndrome (CdLS). Classic CdLS is characterized by distinctive craniofacial features (arched eyebrows, synophrys, upturned nose, small teeth, microcephaly); growth retardation; and limb anomalies, which can at times include fifth finger hypoplasia similar to CSS. Other findings may include cardiac defects, gastrointestinal anomalies, and genitourinary malformations. Pathogenic variants in NIPBL, SMC1A, SMC3, HDAC8, or RAD21 are causative. CdLS is inherited in an autosomal dominant (NIPBL, SMC3, and RAD21) or X-linked (SMC1A and HDAC8) manner.
4q deletion syndrome. This chromosome deletion syndrome results in a characteristic curved, volar, fifth-digit nail, which may resemble a hypoplastic distal phalanx.
BICRA-related disorder (OMIM 619325) is classically characterized by coarse facial features including microcephaly, frontal bossing, epicanthal folds, prominent nasal tips, and low-set ears. These features are also commonly expressed in individuals with CSS. Barish et al [2020] identified 14 individuals with pathogenic variants in BICRA, all of whom have coarse facial features and varying magnitudes of intellectual disability. While phenotypic similarities between CSS and BICRA have been identified, an assessment of a larger cohort of individuals with BICRA pathogenic variants will be needed to determine whether it is clinically similar to or distinct from CSS. BICRA-related disorder is inherited in an autosomal dominant manner.
SMARCD1-related disorder (OMIM 618779). Nixon et al [2019] identified five individuals with pathogenic variants in SMARCD1. These individuals presented with developmental delay, intellectual disability, hypotonia, and feeding difficulties. While similarities between CSS and SMARCD1-related disorder have been identified, an assessment of a larger cohort of individuals with SMARCD1 variants will be needed to determine whether it is clinically similar or distinct from CSS. SMARCD1-related disorder is inherited in an autosomal dominant manner.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with Coffin-Siris syndrome (CSS), the following evaluations are recommended:
- Consultation with a clinical geneticist and/or genetic counselor
- Neurologic and/or developmental examination to record developmental milestones and identify neurologic symptoms or deficits
- Evaluation for occupational, speech, or physical therapy as needed
- Gastrointestinal evaluation for feeding difficulties or poor growth
- Dietary evaluation by a nutritionist as needed
- Ophthalmologic examination, including a dilated fundus examination and visual acuity
- Audiology evaluation with auditory brain stem response testing and otoacoustic emission testing to assess for hearing loss
- Echocardiogram to evaluate for structural cardiac defects
- Renal ultrasonography to evaluate for structural kidney or genitourinary anomalies
Treatment of Manifestations
The following are appropriate:
- Occupational, physical, and/or speech therapies to optimize developmental outcomes
- Feeding therapy, nutritional supplementation, and/or gastrostomy tube placement as needed to meet nutritional needs
- Spectacles as needed to correct refractive errors and surgery as needed for strabismus and/or ptosis
- Hearing aids as needed
Prevention of Secondary Complications
Therapies and interventions which can prevent secondary complications mirror the recommended treatments for an individual’s particular needs. This may include developmental therapies, appropriate cardiac, gastrointestinal, and neurologic evaluations and treatments, and ophthalmologic and audiologic surveillance.
Surveillance
Surveillance includes the following:
- Yearly evaluation by a developmental pediatrician to assess developmental progress and therapeutic and educational interventions
- Annual follow up with a gastroenterologist and feeding specialists as needed to monitor feeding and weight gain
- Regular follow up of ophthalmologic and/or audiologic abnormalities
Because of the rarity of tumors in CSS, the utility of tumor surveillance has not been determined.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
As no females with CSS have been reported to reproduce, potential complications of pregnancy are unknown.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Coffin-Siris syndrome (CSS) is inherited in an autosomal dominant manner. Most affected individuals reported to date have had a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- Most probands reported to date have the disorder as the result of a de novo CSS-causing pathogenic variant.
- The proportion of cases caused by a de novo pathogenic variant is unknown, but likely approaches 100%, given the paucity of reports of affected parents in the literature.
- Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include testing of the parents for the pathogenic variant identified in the proband.
- If the pathogenic variant found in the proband cannot be detected in leukocyte DNA of either parent, the proband most likely has a de novo pathogenic variant. In rare cases, a parent may have germline mosaicism [Ben-Salem et al 2016].
- Evaluation of parents may determine that one is affected but has escaped previous diagnosis because of a milder phenotype. Therefore, an apparently negative family history cannot be fully confirmed until appropriate evaluations have been performed.
Sibs of a proband
- The risk to the sibs of the proband depends on the genetic status of the proband’s parents.
- In the rare circumstance that a parent of the proband is affected and is heterozygous for a CSS-causing pathogenic variant, the risk to the sibs is 50%.
- When the parents are clinically unaffected, the risk to the sibs of a proband appears to be low.
- If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the empiric recurrence risk to sibs is approximately 1% because of the theoretic possibility of parental germline mosaicism.
- One report of CSS in two sisters and partial expression in their father has been published [Haspeslagh et al 1984], suggesting parental somatic (and germline) mosaicism. However, there has been no molecular confirmation of the diagnosis, and the affected family members may have a disorder other than CSS.
Offspring of a proband. Each child of an individual with CSS has a 50% chance of inheriting the CSS-related pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: in the rare event of an affected parent, other family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to couples who have had an affected child.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown).
Prenatal Testing and Preimplantation Genetic Testing
Risk to future pregnancies is presumed to be low as the familial proband most likely has a de novo CSS-causing pathogenic variant. However, prenatal testing or preimplantation genetic testing are options to consider, as the risk may be greater than in the general population because of the possibility of parental germline mosaicism.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Coffin-Siris Syndrome FoundationPhone: 720-514-9904Email: Foundation@coffinsiris.org
- Genetic and Rare Diseases Information Center (GARD)Phone: 888-205-2311
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- CDC - Child DevelopmentPhone: 800-232-4636
- MedlinePlus
- Clinical Registry of Individuals with Coffin-Siris Syndrome and Other BAF-Related PhenotypesEmail: Samantha.vergano@chkd.org
- CoRDS RegistrySanford ResearchPhone: 605-312-6300
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Coffin-Siris Syndrome: Genes and Databases
Table B.
OMIM Entries for Coffin-Siris Syndrome (View All in OMIM)
Molecular Pathogenesis
Many of the proteins identified in CSS to date encode human homologs of proteins first identified in yeast and drosophila in the BRG1- and BRM-associated factor (BAF) complex, originally called the mammalian switch/sucrose non-fermentable (mSWI/SNF)-like nucleosome remodeling complex. This complex contains a DNA-stimulated ATPase activity capable of destabilizing histone-DNA interactions in an ATP-dependent manner [Ronan et al 2013]. SOX11 is predicted to act downstream of the BAF complex in neurogenesis and conversion of postnatal glia into neurons [Ninkovic et al 2013].
Table 2.
Coffin-Siris Syndrome: Mechanism of Disease Causation
Table 3.
Coffin-Siris Syndrome: Gene-Specific Laboratory Considerations
Table 4.
Coffin-Siris Syndrome: Notable Pathogenic Variants by Gene
Cancer and Benign Tumors
SMARCA4. Heterozygous germline pathogenic variants in SMARCA4 have been reported to cause rhabdoid tumor predisposition; likewise, somatic pathogenic variants in SMARCA4 have been found in atypical teratoid and rhabdoid tumors [Schneppenheim et al 2010, Hasselblatt et al 2011, Biegel et al 2014].
SMARCB1. Heterozygous germline pathogenic variants in SMARCB1 have been reported to cause the rhabdoid tumor predisposition syndrome in which most tumors are associated with biallelic loss-of-function variants, and correspondingly, somatic pathogenic variants in the SMARCB1 have been found in atypical teratoid and rhabdoid tumors [Roberts & Biegel 2009, Biegel et al 2014].
Chapter Notes
Acknowledgments
Dr Schrier Vergano would like to acknowledge Ashley Vasko, BS, and Catherine Nguyen, MS, who assisted with the 2021 revision of this chapter.
Author Notes
All of the authors of this review study the clinical features and molecular basis of the Coffin-Siris syndrome.
Revision History
- 12 August 2021 (aa) Revision: added genes: BICRA, DPF2, SMARCC2, SMARCD1, SOX4
- 8 February 2018 (aa) Revision: ARID2-related intellectual disability added to Differential Diagnosis
- 12 May 2016 (ha) Comprehensive update posted live
- 11 July 2013 (aa) Revision: ARID1B deletion/duplication analysis available on a clinical basis
- 4 April 2013 (me) Review posted live
- 19 July 2012 (md) Original submission
References
Literature Cited
- Backx L, Seuntjens E, Devriendt K, Vermeesch J, Van Esch H. A balanced translocation t(6;14)(q25.3;q13.2) leading to reciprocal fusion transcripts in a patient with intellectual disability and agenesis of corpus callosum. Cytogenet Genome Res. 2011;132:135–43. [PubMed: 21042007]
- Barish S, Barakat TS, Michel BC, Mashtalir N, Phillips JB, Valencia AM, Ugur B, Wegner J, Scott TM, Bostwick B, Murdock DR, Dai H, Perenthaler E, Nikoncuk A, van Slegtenhorst M, Brooks AS, Keren B, Nava C, Mignot C, Douglas J, Rodan L, Nowak C, Ellard S, Stals K, Lynch SA, Faoucher M, Lesca G, Edery P, Engleman KL, Zhou D, Thiffault I, Herriges J, Gass J, Louie RJ, Stolerman E, Washington C, Vetrini F, Otsubo A, Pratt VM, Conboy E, Treat K, Shannon N, Camacho J, Wakeling E, Yuan B, Chen CA, Rosenfeld JA, Westerfield M, Wangler M, Yamamoto S, Kadoch C, Scott DA, Bellen HJ, et al. BICRA, a SWI/SNF complex member, is associated with BAF disorder-related phenotypes in humans and model organisms. Am J Hum Genet. 2020;107:1096–112. [PMC free article: PMC7820627] [PubMed: 33232675]
- Ben-Salem S, Sobreira N, Akawi NA, Al-Shamsi AM, John A, Pramathan T, Valle D, Ali BR, Al-Gazali L. Gonadal mosaicism in ARID1B gene causes intellectual disability and dysmorphic features in three siblings. Am J Med Genet A. 2016;170A:156–61. [PMC free article: PMC5448135] [PubMed: 26395437]
- Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014;166C:350–66. [PMC free article: PMC4516040] [PubMed: 25169151]
- Carter JM, O'hara C, Dundas G, Gilchrist D, Collins MS, Eaton K, Judkins AR, Biegel JA, Folpe AL. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with "neuroblastoma-like" schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol. 2012;36:154–60. [PMC free article: PMC3241826] [PubMed: 22082606]
- Elliott AM, Teebi AS. New autosomal dominant syndrome reminiscent of Coffin-Siris syndrome and brachymorphism-onychodysplasia-dysphalangism syndrome. Clin Dysmorphol. 2000;9:15–9. [PubMed: 10649791]
- Fleck BJ, Pandya A, Vanner L, Kerkering K, Bodurtha J. Coffin-Siris syndrome: review and presentation of new cases from a questionnaire study. Am J Med Genet. 2001;99:1–7. [PubMed: 11170086]
- Gazdagh G, Blyth M, Scurr I, Turnpenny P, Mehta S, Armstrong R, McEntagart M, Newbury-Ecob R, Tobias E, Joss S, et al. Extending the clinical and genetic spectrum of ARID2 related intellectual disability. A case series of 7 patients. Eur J Med Genet. 2019;62:27–34. [PubMed: 29698805]
- Gomes WJ, Hunter JL. Mental retardation, cataracts, and unexplained hyperphosphatasia. Arch Dis Child. 1970;45:726–7. [PMC free article: PMC1647537] [PubMed: 5477707]
- Gossai N, Biegel JA, Messiaen L, Berry SA, Moertel CL. Report of a patient with a constitutional missense mutation in SMARCB1, Coffin-Siris phenotype, and schwannomatosis. Am J Med Genet A. 2015;167A:3186–91. [PubMed: 26364901]
- Halgren C, Kjaergaard S, Bak M, Hansen C, El-Schich Z, Anderson CM, Henriksen KF, Hjalgrim H, Kirchhoff M, Bijlsma EK, Nielsen M, Den Hollander NS, Ruivenkamp CA, Isidor B, Le Caignec C, Zannolli R, Mucciolo M, Renieri A, Mari F, Anderlid BM, Andrieux J, Dieux A, Tommerup N, Bache I. Corpus callosum abnormalities, intellectual disability, speech impairment, and autism in patients with haploinsufficiency of ARID1B. Clin Genet. 2012;82:248–55. [PMC free article: PMC3464360] [PubMed: 21801163]
- Haspeslagh M, Fryns JP, Van Den Berghe H. The Coffin-Siris syndrome: report of a family and further delineation. Clin Genet. 1984;26:374–8. [PubMed: 6499251]
- Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, Giannini C, Judkins AR, Fruhwald MC, Obser T, Schneppenheim R, Siebert R, Paulus W. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35:933–5. [PubMed: 21566516]
- Hempel A, Pagnamenta AT, Blyth M, Mansour S, Mcconnell V, Kou I, Ikegawa S, Tsurusaki Y, Matsumoto N, Lo-Castro A, Plessis G, Albrecht B, Battaglia A, Taylor JC, Howard MF, Keays D, Sohal AS, Kuhl SJ, Kini U, Mcneill A, et al. Deletions and de novo mutations of SOX11 are associated with a neurodevelopmental disorder with features of Coffin-Siris syndrome. J Med Genet. 2016;53:152–62. [PMC free article: PMC4789813] [PubMed: 26543203]
- Hoyer J, Ekici AB, Endele S, Popp B, Zweier C, Wiesener A, Wohlleber E, Dufke A, Rossier E, Petsch C, Zweier M, Gohring I, Zink AM, Rappold G, Schrock E, Wieczorek D, Riess O, Engels H, Rauch A, Reis A. Haploinsufficiency of ARID1B, a member of the SWI/SNF-a chromatin-remodeling complex, is a frequent cause of intellectual disability. Am J Hum Genet. 2012;90:565–72. [PMC free article: PMC3309205] [PubMed: 22405089]
- Kleefstra T, Kramer JM, Neveling K, Willemsen MH, Koemans TS, Vissers LE, Wissink-Lindhout W, Fenckova M, Van Den Akker WM, Kasri NN, Nillesen WM, Prescott T, Clark RD, Devriendt K, Van Reeuwijk J, De Brouwer AP, Gilissen C, Zhou H, Brunner HG, Veltman JA, Schenck A, Van Bokhoven H. Disruption of an EHMT1-associated chromatin-modification module causes intellectual disability. Am J Hum Genet. 2012;91:73–82. [PMC free article: PMC3397275] [PubMed: 22726846]
- Kosho T, Miyake N, Carey JC. Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet. 2014a;166C:241–51. [PubMed: 25169878]
- Kosho T, Okamoto N, et al. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. Am J Med Genet C Semin Med Genet. 2014b;166C:262–75. [PubMed: 25168959]
- Krawitz PM, Schweiger MR, Rodelsperger C, Marcelis C, Kolsch U, Meisel C, Stephani F, Kinoshita T, Murakami Y, Bauer S, Isau M, Fischer A, Dahl A, Kerick M, Hecht J, Kohler S, Jager M, Grunhagen J, De Condor BJ, Doelken S, Brunner HG, Meinecke P, Passarge E, Thompson MD, Cole DE, Horn D, Roscioli T, Mundlos S, Robinson PN. Identity-by-descent filtering of exome sequence data identifies PIGV mutations in hyperphosphatasia mental retardation syndrome. Nat Genet. 2010;42:827–9. [PubMed: 20802478]
- Kruse K, Hanefeld F, Kohlschutter A, Rosskamp R, Gross-Selbeck G. Hyperphosphatasia with mental retardation. J Pediatr. 1988;112:436–9. [PubMed: 3346785]
- Kushnick T, Adessa GM. Partial trisomy 9 with resemblance to Coffin-Siris syndrome. J Med Genet. 1976;13:237–9. [PMC free article: PMC1013400] [PubMed: 933124]
- Machol K, Rousseau J, Ehresmann S, Garcia T, Nguyen TTM, Spillmann RC, Sullivan JA, Shashi V, Jiang YH, Stong N, Fiala E, Willing M, Pfundt R, Kleefstra T, Cho MT, McLaughlin H, Rosello Piera M, Orellana C, Martínez F, Caro-Llopis A, Monfort S, Roscioli T, Nixon CY, Buckley MF, Turner A, Jones WD, van Hasselt PM, Hofstede FC, van Gassen KLI, Brooks AS, van Slegtenhorst MA, Lachlan K, Sebastian J, Madan-Khetarpal S, Sonal D, Sakkubai N, Thevenon J, Faivre L, Maurel A, Petrovski S, Krantz ID, Tarpinian JM, Rosenfeld JA, Lee BH, Campeau PM, et al. Expanding the spectrum of BAF-related disorders: de novo variants in SMARCC2 cause a syndrome with intellectual disability and developmental delay. Am J Hum Genet. 2019;104:164–78. [PMC free article: PMC6323608] [PubMed: 30580808]
- Malli T, Duba HC, Erdel M, Marschon R, Kranewitter W, Deutschbauer S, Kralik J, Diel E, Guenther B, Mueller D, Webersinke G. Disruption of the ARID1B and ADAMTS6 loci due to a t(5;6)(q12.3;q25.3) in a patient with developmental delay. Am J Med Genet A. 2014;164A:3126–31. [PubMed: 25250687]
- Merker VL, Esparza S, Smith MJ, Stemmer-Rachamimov A, Plotkin SR. Clinical features of schwannomatosis: a retrospective analysis of 87 patients. Oncologist. 2012;17:1317–22. [PMC free article: PMC3481897] [PubMed: 22927469]
- Michelson M, Ben-Sasson A, Vinkler C, Leshinsky-Silver E, Netzer I, Frumkin A, Kivity S, Lerman-Sagie T, Lev D. Delineation of the interstitial 6q25 microdeletion syndrome: refinement of the critical causative region. Am J Med Genet A. 2012;158A:1395–9. [PubMed: 22585544]
- Milone R, Gnazzo M, Stefanutti E, Serafin D, Novelli A. A new missense mutation in DPF2 gene related to Coffin Siris syndrome 7: Description of a mild phenotype expanding DPF2-related clinical spectrum and differential diagnosis among similar syndromes epigenetically determined. Brain Dev. 2020;42:192–8. [PubMed: 31706665]
- Nagamani SC, Erez A, Eng C, Ou Z, Chinault C, Workman L, Coldwell J, Stankiewicz P, Patel A, Lupski JR, Cheung SW. Interstitial deletion of 6q25.2-q25.3: a novel microdeletion syndrome associated with microcephaly, developmental delay, dysmorphic features and hearing loss. Eur J Hum Genet. 2009;17:573–81. [PMC free article: PMC2986272] [PubMed: 19034313]
- Ninkovic J, Steiner-Mezzadri A, Jawerka M, Akinci U, Masserdotti G, Petricca S, Fischer J, Von Holst A, Beckers J, Lie CD, Petrik D, Miller E, Tang J, Wu J, Lefebvre V, Demmers J, Eisch A, Metzger D, Crabtree G, Irmler M, Poot R, Gotz M. The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross-regulatory transcriptional network. Cell Stem Cell. 2013;13:403–18. [PMC free article: PMC4098720] [PubMed: 23933087]
- Nixon KCJ, Rousseau J, Stone MH, Sarikahya M, Ehresmann S, Mizuno S, Matsumoto N, Miyake N, Baralle D, McKee S, Izumi K, Ritter AL, Heide S, Héron D, Depienne C, Titheradge H, Kramer JM, Campeau PM. A syndromic neurodevelopmental disorder caused by mutations in SMARCD1, a core SWI/SNF subunit needed for context-dependent neuronal gene regulation in flies. Am J Hum Genet. 2019;104:596–610. [PMC free article: PMC6451697] [PubMed: 30879640]
- Roberts CW, Biegel JA. The role of SMARCB1/INI1 in development of rhabdoid tumor. Cancer Biol Ther. 2009;8:412–6. [PMC free article: PMC2709499] [PubMed: 19305156]
- Ronan JL, Wu W, Crabtree GR. From neural development to cognition: unexpected roles for chromatin. Nat Rev Genet. 2013;14:347–59. [PMC free article: PMC4010428] [PubMed: 23568486]
- Santen GW, Aten E, Sun Y, Almomani R, Gilissen C, Nielsen M, Kant SG, Snoeck IN, Peeters EA, Hilhorst-Hofstee Y, Wessels MW, Den Hollander NS, Ruivenkamp CA, Van Ommen GJ, Breuning MH, Den Dunnen JT, Van Haeringen A, Kriek M. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat Genet. 2012;44:379–80. [PubMed: 22426309]
- Santen GW, Aten E, Vulto-Van Silfhout AT, Pottinger C, Van Bon BW, Van Minderhout IJ, Snowdowne R, Van Der Lans CA, Boogaard M, Linssen MM, Vijfhuizen L, Van Der Wielen MJ, Vollebregt MJ, Coffin-Siris C, Breuning MH, Kriek M, Van Haeringen A, Den Dunnen JT, Hoischen A, Clayton-Smith J, De Vries BB, Hennekam RC, Van Belzen MJ. Coffin-Siris syndrome and the BAF complex: genotype-phenotype study in 63 patients. Hum Mutat. 2013;34:1519–28. [PubMed: 23929686]
- Santen GW, Clayton-Smith J, et al. The ARID1B phenotype: what we have learned so far. Am J Med Genet C Semin Med Genet. 2014;166C:276–89. [PubMed: 25169814]
- Schneppenheim R, Fruhwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Martin Subero JI, Obser T, Oyen F, Vater I, Siebert R. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet. 2010;86:279–84. [PMC free article: PMC2820190] [PubMed: 20137775]
- Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MA, D'avanzo M G, Lynch SA, Musio A, Nyazov DM, Sanchez-Lara PA, Shalev SA, Deardorff MA. The Coffin-Siris syndrome: a proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet A. 2012;158A:1865–76. [PMC free article: PMC3402612] [PubMed: 22711679]
- Thompson MD, Nezarati MM, Gillessen-Kaesbach G, Meinecke P, Mendoza-Londono R, Mornet E, Brun-Heath I, Squarcioni CP, Legeai-Mallet L, Munnich A, Cole DE. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am J Med Genet A. 2010;152A:1661–9. [PubMed: 20578257]
- Tsurusaki Y, Koshimizu E, Ohashi H, Phadke S, Kou I, Shiina M, Suzuki T, Okamoto N, Imamura S, Yamashita M, Watanabe S, Yoshiura K, Kodera H, Miyatake S, Nakashima M, Saitsu H, Ogata K, Ikegawa S, Miyake N, Matsumoto N. De novo SOX11 mutations cause Coffin-Siris syndrome. Nat Commun. 2014a;5:4011. [PubMed: 24886874]
- Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, Kaname T, Naritomi K, Kawame H, Wakui K, Fukushima Y, Homma T, Kato M, Hiraki Y, Yamagata T, Yano S, Mizuno S, Sakazume S, Ishii T, Nagai T, Shiina M, Ogata K, Ohta T, Niikawa N, Miyatake S, Okada I, Mizuguchi T, Doi H, Saitsu H, Miyake N, Matsumoto N. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–8. [PubMed: 22426308]
- Tsurusaki Y, Okamoto N, Ohashi H, Mizuno S, Matsumoto N, Makita Y, Fukuda M, Isidor B, Perrier J, Aggarwal S, Dalal AB, Al-Kindy A, Liebelt J, Mowat D, Nakashima M, Saitsu H, Miyake N, Matsumoto N. Coffin-Siris syndrome is a SWI/SNF complex disorder. Clin Genet. 2014b;85:548–54. [PubMed: 23815551]
- Van Houdt JK, Nowakowska BA, Sousa SB, Van Schaik BD, Seuntjens E, Avonce N, Sifrim A, Abdul-Rahman OA, Van Den Boogaard MJ, Bottani A, Castori M, Cormier-Daire V, Deardorff MA, Filges I, Fryer A, Fryns JP, Gana S, Garavelli L, Gillessen-Kaesbach G, Hall BD, Horn D, Huylebroeck D, Klapecki J, Krajewska-Walasek M, Kuechler A, Lines MA, Maas S, Macdermot KD, Mckee S, Magee A, De Man SA, Moreau Y, Morice-Picard F, Obersztyn E, Pilch J, Rosser E, Shannon N, Stolte-Dijkstra I, Van Dijck P, Vilain C, Vogels A, Wakeling E, Wieczorek D, Wilson L, Zuffardi O, Van Kampen AH, Devriendt K, Hennekam R, Vermeesch JR. Heterozygous missense mutations in SMARCA2 cause Nicolaides-Baraitser syndrome. Nat Genet. 2012;44:445–9. [PubMed: 22366787]
- Vasileiou G, Vergarajauregui S, Endele S, Popp B, Büttner C, Ekici A, Gerard M, Bramswig N, Albrecht B, Clayton-Smith J, Morton J, Tomkins S, Low K, Weber A, Wenzel M, Altmüller J, Li Y. Bernd Wollnik, George Hoganson, Maria-Renée Plona, Megan T. Cho, Christian T. Thiel, Lüdecke H, Strom T, Calpena E, Wilkie A, Wieczorek D, Engel F, Reis A. Mutations in the BAF-complex subunit DPF2 are associated with Coffin-Siris syndrome. Am J Hum Genet. 2018;102:468–79. [PMC free article: PMC5985265] [PubMed: 29429572]
- Vengoechea J, Carpenter L, Zarate YA. Papillary thyroid cancer in a patient with interstitial 6q25 deletion including ARID1B. Am J Med Genet A. 2014;164A:1857–9. [PubMed: 24700687]
- Verloes A, Bonneau D, Guidi O, Berthier M, Oriot D, Van Maldergem L, Koulischer L. Brachymorphism-onychodysplasia-dysphalangism syndrome. J Med Genet. 1993;30:158–61. [PMC free article: PMC1016276] [PubMed: 8445623]
- Wieczorek D, Bogershausen N, Beleggia F, Steiner-Haldenstatt S, Pohl E, Li Y, Milz E, Martin M, Thiele H, Altmuller J, Alanay Y, Kayserili H, Klein-Hitpass L, Bohringer S, Wollstein A, Albrecht B, Boduroglu K, Caliebe A, Chrzanowska K, Cogulu O, Cristofoli F, Czeschik JC, Devriendt K, Dotti MT, Elcioglu N, Gener B, Goecke TO, Krajewska-Walasek M, Guillen-Navarro E, Hayek J, Houge G, Kilic E, Simsek-Kiper PO, Lopez-Gonzalez V, Kuechler A, Lyonnet S, Mari F, Marozza A, Mathieu Dramard M, Mikat B, Morin G, Morice-Picard F, Ozkinay F, Rauch A, Renieri A, Tinschert S, Utine GE, Vilain C, Vivarelli R, Zweier C, Nurnberg P, Rahmann S, Vermeesch J, Ludecke HJ, Zeschnigk M, Wollnik B. A comprehensive molecular study on Coffin-Siris and Nicolaides-Baraitser syndromes identifies a broad molecular and clinical spectrum converging on altered chromatin remodeling. Hum Mol Genet. 2013;22:5121–35. [PubMed: 23906836]
- Zawerton A, Yao B, Yeager JP, Pippucci T, Haseeb A, Smith JD, Wischmann L, Kühl SJ, Dean JCS, Pilz DT, Holder SE, McNeill A, Graziano C, Lefebvre V, et al. De novo SOX4 variants cause a neurodevelopmental disease associated with mild dysmorphism. Am J Hum Genet. 2019;104:246–59. [published correction appears in Am J Hum Genet. 2019;104:777] [PMC free article: PMC6369454] [PubMed: 30661772]
- Zweier C, Kraus C, Brueton L, Cole T, Degenhardt F, Engels H, Gillessen-Kaesbach G, Graul-Neumann L, Horn D, Hoyer J, Just W, Rauch A, Reis A, Wollnik B, Zeschnigk M, Ludecke HJ, Wieczorek D. A new face of Borjeson-Forssman-Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype. J Med Genet. 2013;50:838–47. [PubMed: 24092917]
- Zweier C, Rittinger O, Bader I, Berland S, Cole T, Degenhardt F, Di Donato N, Graul-Neumann L, Hoyer J, Lynch SA, Vlasak I, Wieczorek D. Females with de novo aberrations in PHF6: clinical overlap of Borjeson-Forssman-Lehmann with Coffin-Siris syndrome. Am J Med Genet C Semin Med Genet. 2014;166C:290–301. [PubMed: 25099957]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: April 4, 2013; Last Revision: August 12, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Schrier Vergano S, Santen G, Wieczorek D, et al. Coffin-Siris Syndrome. 2013 Apr 4 [Updated 2021 Aug 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.