Clinical Description
Cerebrotendinous xanthomatosis (CTX) is a lipid storage disease characterized by infantile-onset diarrhea, childhood-onset cataract, adolescent- to young adult-onset tendon xanthomas, and adult-onset progressive neurologic dysfunction (dementia, psychiatric disturbances, pyramidal and/or cerebellar signs, dystonia, atypical parkinsonism, peripheral neuropathy, and seizures). Intrafamilial variability is considerable. A suspicion index for diagnosis has been reported based on clinical and laboratory findings [Mignarri et al 2014].
Table 3.
Cerebrotendinous Xanthomatosis: Frequency of Select Features
View in own window
| Feature | % of Persons w/Feature |
|---|
| Infantile-onset diarrhea | 40% |
| Childhood-onset cataract | 89% |
| Adolescent- to young adult-onset tendon xanthomas | 78% |
| Cardiovascular findings | 25% |
| Osteopenia | 67% |
| Adult-onset progressive neurologic dysfunction | Intellectual disability | 60% |
| Psychiatric disturbances | 44% |
| Ataxia | 36% |
| Spastic paraparesis | 64% |
| Parkinsonism | 9% |
| Peripheral neuropathy | 70% |
| Seizures | 33% |
Gastrointestinal and hepatic findings. Chronic diarrhea from infancy, even in the neonatal period, may be the earliest clinical manifestation of CTX [Cruysberg 2002, Gong et al 2017]. Gallstones have been reported on occasion. Neonatal cholestasis has been identified as a presenting manifestation of CTX [Zhang et al 2021]. Cases with fatal cholestasis [von Bahr et al 2005] and infantile hepatitis in infancy [Clayton et al 2002] have been also reported.
Eye. In approximately 75% of affected individuals, cataracts are the first finding, often appearing in the first decade of life. In 25% of individuals, cataracts are first observed after age 40 years. Cataracts may be visually significant opacities requiring lensectomy or visually insignificant cortical opacities. The appearance can include irregular cortical opacities, anterior polar cataracts, and dense posterior subcapsular cataracts [Cruysberg et al 1995]. Among large study groups of individuals with juvenile-onset cataracts, CTX was diagnosed in 1.8% in the United States [Freedman et al 2019] and 1.55% in Turkey [Atilla et al 2021].
Other findings include palpebral xanthelasmas, optic nerve atrophy and proptosis, paleness of the optic disk, premature retinal senescence with retinal vessel sclerosis, cholesterol-like deposits along vascular arcades, and myelinated nerve fibers [Dotti et al 2001].
Khan et al [2013] reported the unique finding of fleck lenticular opacities in three children with CTX; these affected children also had capsular opacities (posterior only or posterior and anterior) that caused visual symptoms.
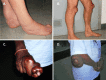
Xanthomas appear in the second or third decade. In addition to the classic xanthomas of the Achilles tendon, xanthomas also occur on the extensor tendons of the elbow and hand, the patellar tendon (see ), and the neck tendons. Xanthomas have been reported in the lung, bones, and central nervous system [Brienza et al 2015].
Cardiovascular system. Premature atherosclerosis and coronary artery disease have been reported [Valdivielso et al 2004, Androdias et al 2012], as has lipomatous hypertrophy of the atrial septum [Dotti et al 1998, Frih-Ayed et al 2005].
Skeleton. Bone involvement is characterized by granulomatous lesions in the lumbar vertebrae and femur, osteoporosis and increased risk of bone fractures, and impaired adsorption of radiocalcium, which improves with chenodeoxycholic acid treatment [Martini et al 2013]. Osteoporosis is evident by total body densitometry in untreated individuals. Individuals may have marked thoracic kyphosis.
Premature aging. Early-onset cataract, osteopenia with bone fractures and loss of teeth, atherosclerosis, and neurologic impairment with dementia and/or parkinsonism (associated with the characteristic facies) suggest a generalized premature aging process [Dotti et al 1991].
Neurologic Signs
Intellectual disability or dementia following slow deterioration in intellectual abilities occurs in the third decade in more than 50% of individuals [Verrips et al 2000a]. Some individuals show cognitive impairment from early infancy, whereas the majority have normal or only slightly impaired intellectual function until puberty. In the spinal form, mainly characterized by myelopathy and spastic paraparesis, intellect is almost always normal.
Neuropsychiatric symptoms including behavioral changes, hallucinations, agitation, aggression, depression, and suicide attempts may be prominent [Fraidakis 2013].
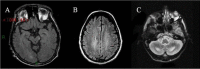
Pyramidal signs (i.e., spasticity) and/or cerebellar signs are almost invariably present between ages 20 and 30 years. The clinical findings are related to the primary involvement of corticospinal tracts, subcortical white matter, dentate nuclei, and cerebellum cortex involvement that is evident on MRI [Dotti et al 1994, Inglese et al 2003, Mignarri et al 2017, Rosini et al 2017, Catarino et al 2018, Makary et al 2018].
Some individuals present with a spinal form, in which progressive spastic paraparesis is the main clinical concern [Nicholls et al 2015, Catarino et al 2018].
Extrapyramidal manifestations can be considered a late disease manifestation, with parkinsonism the most frequently reported, followed by dystonia, myoclonus, and postural tremor. In a recent review of 79 individuals with CTX, the mean age at onset of a movement disorder was 40±12 years (median 40, range 13-62 years). Movement disorders were found to be mixed in 23% of individuals and were usually part of a complex clinical picture, rather than a prominent finding. Still, in 18% of individuals, a movement disorder was the presenting manifestation [Stelten et al 2019].
Seizures are reported in approximately 50% of individuals with CTX [Pedroso et al 2012].
Peripheral neuropathy is evident on electrophysiologic studies [Ginanneschi et al 2013, Zhang et al 2020], which reveal decreased nerve conduction velocities and abnormalities in somatosensory, motor, brain stem, and visual evoked potentials. Clinical manifestations related to peripheral nerve involvement are distal muscle atrophy and pes cavus. Sensory abnormalities are rarely described.
Heterozygotes
Heterozygotes are generally asymptomatic; however, clinical findings have been reported in heterozygotes ranging from an increased incidence of cardiovascular disorders to gallstones [Author, personal observation].