Clinical Description
CYLD cutaneous syndrome (CCS) encompasses the clinical phenotypes described in individuals with germline pathogenic CYLD variants. Historically descriptive names including Brooke-Spiegler syndrome (BSS), familial cylindromatosis (FC), and multiple familial trichoepithelioma (MFT) were assigned on the basis of the predominant tumor type and location; these conditions are now recognized to constitute a clinical spectrum. Individuals with the clinical phenotypes of BSS, FC, and MFT can all present in a single family, and the lack of prognostication offered by these historical labels favors the use of CYLD cutaneous syndrome as a diagnostic term for those with this single gene disorder.
Table 2.
Features of CYLD Cutaneous Syndrome
View in own window
| Feature 1 | # of Persons w/Feature | Comment |
|---|
Phenotype of predominantly
cylindromas/spiradenomas | 14/26 (∼54%) | |
| Phenotype of predominantly trichoepitheliomas | 8/26 (∼30%) | |
Phenotype of mixed cylindroma/spiradenoma/
trichoepithelioma | 4/26 (∼15%) | |
Severe phenotype
necessitating
complete scalp excision | 6/26 (∼23%) | In a further study of a Hungarian pedigree, 2 5/21 individuals w/a germline pathogenic CYLD variant had this severe phenotype. |
| Salivary gland tumors | ~5% | |
| Pulmonary cylindromas | 3 persons 3 | May result in respiratory compromise if lesion affects the large airways |
Except where otherwise noted, the table summarizes a single study by Rajan et al [2009a], which analyzed the clinical features of 26 individuals with germline pathogenic CYLD variants.
- 1.
While by definition most individuals with CYLD cutaneous syndrome will be affected by one of more of the characteristic tumors, precise detail about frequency of the clinical features is lacking in most published studies.
- 2.
- 3.
To date, more than 100 pedigrees have been identified with a germline pathogenic variant in CYLD, most of whom have family-specific pathogenic variants with few mutational hot spots identified [Rajan et al 2009a, Grossmann et al 2013, Nagy et al 2015]. The following description of the phenotypic features associated with this condition is based on these reports.
CYLD cutaneous syndrome typically manifests in the second or third decade with the appearance of multiple skin tumors including cylindromas, spiradenomas, and trichoepitheliomas. The first tumor typically presents at puberty, but tumors have been reported to present in children as young as age eight years. Tumors progressively accumulate through adulthood. A female preponderance was reported in early studies of small pedigrees; however, assessment of larger pedigrees supports equal penetrance in both males and females, with increased expressivity in females. The presence of tumors at sites of secondary sexual hair development, the timing of onset, and the greater severity in females (vs males) with cylindromas suggest a role of hormonal factors in tumor pathogenesis [Rajan et al 2009b].
Natural history studies in CYLD cutaneous syndrome are limited, but a recent study that followed three affected individuals with a combined total of 32 skin tumors visible on serial CT scans (6.7-23.5 mm across) showed progressive growth in 30 out of 32 lesions over a period of one year [Brown et al 2018b]. The progressive growth of these benign tumors supports the case for considering early excision of skin lesions rather than waiting until a more extensive procedure is required (see Table 5). Affected individuals frequently need repeated procedures due to the development of new tumors and, in some cases, the recurrence of incompletely excised tumors.
Individuals with CYLD cutaneous syndrome may present with more than one tumor type discussed below.
Tumors typically arise on the scalp and face but can also arise on the torso and sun-protected sites, such as the genital and axillary skin.
Tumors are often painful and may cause sexual dysfunction when they arise on genital skin.
Tumors arising within the ear (a favored site for tumor formation) can occlude the external auditory canal and result in conductive hearing loss.
Although the tumors are usually benign, malignant transformation is recognized, and affected individuals should be guided to report tumors which are rapidly growing, bleeding, ulcerating, or different in appearance from their usual tumors.
Cylindromas
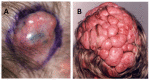
Cylindromas are well-circumscribed, smooth, pale pink nodular tumors, often with arborizing vessels visible. The tumors are slow growing and vary in size from a few millimeters to >5 mm (see ). The finding of mixed cylindroma and spiradenoma histology within a single tumor is common in CYLD cutaneous syndrome, and the two terms have been used to describe variants of the same tumor [Rajan et al 2011a].
A. Cylindroma demonstrating a well-circumscribed pink nodular lesion with arborizing blood vessels visible on the surface B. Confluent scalp cylindromas in a severely affected individual with CYLD cutaneous syndrome
Confluent scalp tumors. In severe cases, tumors may cover most of the scalp, referred to as confluent scalp tumors (see ).
Pulmonary cylindromas. Single or multiple pulmonary cylindromas that originate from the skin have been described in three individuals with CYLD cutaneous syndrome [Vernon et al 1988, Brown et al 2018b]. In these cases, there is no history of a primary malignant cutaneous cylindroma in the skin, lymph node disease is absent, and pulmonary histology is benign, leading them to be categorized as "benign" metastases.
One individual presented at age 64 with breathlessness on exertion and was found to have multiple pulmonary tumors. This individual required serial monitoring with pulmonary imaging, and received endocopic laser ablation to maintain large airway patency.
A second individual had four asymptomatic pulmonary tumors discovered incidentally on a chest radiograph at age 80 years. The tumors were histologically confirmed on autopsy.
Whole-exome and genome sequencing have shown pulmonary cylindromas to harbor an additional pathogenic variant in
AKT1 and a UV mutation signature confirming cutaneous origin [
Davies et al 2019].
The true prevalence of pulmonary cylindromas in individuals with CYLD cutaneous syndrome is not known, as prospective radiologic imaging studies of large numbers of affected individuals have yet to be performed.
Currently, there are no routine surveillance imaging guidelines that can be recommended; however clinicians who care for individuals with CYLD cutaneous syndrome need to be aware that these tumors do arise and may need to be monitored to determine rate of growth, and that surgical interventions may be necessary to ablate pulmonary tumors that threaten large airway patency.
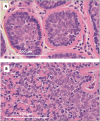
Histopathology. The histologic appearance of well-circumscribed cylindrical nests of basaloid cells in the dermis led to the term "cylindroma." Each cluster consists of darker, basophilic cells at the periphery, and larger pale cells centrally and is surrounded by a thick, hyaline membrane consisting of extracellular matrix proteins (including collagen IV and VII and laminin-332) in the basement membrane of the skin (see ).
A. Histopathology of cylindroma B. Histopathology of spiradenoma
Spiradenomas
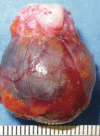
Spiradenomas are nodular tumors that are often blue/black in color. They tend to be painful and can grow up to 10 cm in diameter (see ).
Spiradenoma: a well-circumscribed nodular lesion (excision specimen) with characteristic blue/black appearance Rajan & Ashworth [2015]; republished with permission of author.
Histopathology. Spiradenomas are relatively disorganized histologically when compared to cylindromas and consist of sheets of epithelial cells associated with a lymphocytic infiltrate (mixed T and B cells). Some affected individuals present with histologic features of both cylindroma and spiradenoma within a single tumor specimen, giving rise to the term spiradenocylindroma. In addition, there is evidence that histologically organized cylindroma and histologically disorganized spiradenoma represent extremes of a spectrum of histophenotype of the same tumor [Rajan et al 2011a] (see ).
Trichoepitheliomas
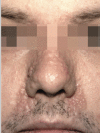
Trichoepitheliomas are skin-colored papules or firm nodules, mainly found on the central face (see ). They are often symmetrically distributed and usually no more than 2-5 mm across.
Trichoepithelioma: small skin-colored papules with a predilection for the central face Rajan & Ashworth [2015]; republished with permission of author.
Histopathology. Trichepitheliomas demonstrate clusters of basaloid germinative cells with keratinizing cystic spaces and superficial follicular differentiation surrounded by a fibrocytic stroma. Intra-stromal clefts and mesenchymal papillary buds may be seen.
Other Findings
Salivary lesions. Affected individuals are also at risk of developing tumors of the salivary glands, typically membranous basal cell adenoma (MBCA) [Jungehülsing et al 1999] usually after age 40 years.
Malignant transformation has been (rarely) reported in preexisting spiradenomas, cylindromas, spiradenocylindromas, and MBCA [Hyman et al 1988, Kazakov 2016]. Malignant tumors tend to be aggressive carcinomas with frequent local infiltrative growth or metastases.
Death from metastatic disease has been described in affected individuals in the early fifth decade [
Kazakov et al 2009].
Malignant histopathology. Four diverse histologic patterns of malignant cylindroma or spiradenoma have been described [
Kazakov et al 2009], including:
Salivary gland type basal cell adenocarcinoma-like pattern, low-grade
Salivary gland type basal cell adenocarcinoma-like pattern, high-grade
Invasive adenocarcinoma, not otherwise specified
Sarcomatoid (metaplastic) carcinoma
Other skin lesions and malignancies
Affected individuals may also develop small milia cysts on the skin of the face [
Bajwa et al 2018].
Vulval cysts consisting histologically of multiple epidermal inclusion cysts and milia were reported in one affected female [
Dubois et al 2017].
Basal cell carcinoma (BCC) is the most common human cancer, and coexistence in individuals with
CYLD cutaneous syndrome may be coincidental or related to overlap in the appearance of each under the microscope. However, in a recent study of a large South American family with
CYLD cutaneous syndrome, BCC was a reported in 25% of affected family members from this kindred [
Arruda et al 2020].
Segmental disease as a result of mosaic pathogenic CYLD variants should be considered when individuals develop unilateral clusters of any cylindromas, spiradenomas, or trichoepitheliomas [Arefi et al 2019]. Findings may include skin lesions arranged in a linear fashion [Furuichi et al 2012] following embryologic skin development lines (lines of Blaschko). The presentation of unilateral clusters may reflect either a pathogenic CYLD variant in the skin alone (late postzygotic mosaicism) or a pathogenic variant in both the blood and the skin (early postzygotic mosaicism) (see ) [Arefi et al 2019].
Mosaic presentations of CYLD cutaneous syndrome Arefi et al [2019]; published under Creative Commons license.
Nomenclature
Historically, descriptive names including Brooke-Spiegler syndrome (BSS), familial cylindromatosis (FC), and multiple familial trichoepithelioma (MFT) were assigned on the basis of the predominant tumor type and location. These conditions are now recognized to constitute a clinical spectrum and individuals with the clinical phenotypes of BSS, FC, and MFT can all present in a single family. The lack of prognostication offered by these historical labels favors the use of CYLD cutaneous syndrome as a diagnostic term for individuals with this single-gene disorder.
Outdated terms previously used in the literature to refer to CYLD cutaneous syndrome include the following:
Ancell-Spiegler cylindromas
Dermal eccrine cylindroma
Turban tumor syndrome (now denoted as confluent scalp tumors)