Summary
Clinical characteristics.
GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) is the most common genetic cause of congenital (present at birth) severe-to-profound non-progressive sensorineural hearing loss in many world populations. In countries where available, newborn hearing screening (NBHS) typically identifies severe-to-profound hearing loss.
GJB2-AR NSHL can also be mild to moderate and is usually not progressive; however, it can progress. Congenital mild-to-moderate GJB2-AR NSHL is not detected by NBHS.
GJB2-AR NSHL has no related systemic findings.
Diagnosis/testing.
The diagnosis of GJB2-AR NSHL is established in a proband with suggestive findings and biallelic GJB2 pathogenic variants identified by molecular genetic testing. Of note: About 1% of individuals with GJB2-AR NSHL are compound heterozygotes for one GJB2 pathogenic variant and one of several different deletions that include sequences upstream of GJB2 (comprising either GJB6 and portions of CRYL1 or just portions of CRYL1) that delete cis-regulatory regions of GJB2, thereby abolishing GJB2 expression. Occasionally, the deletion also includes GJB2.
Management.
Treatment of manifestations: It is recommended that NBHS be completed by age one month, the genetic diagnosis be established by age three months, and early intervention begun by age six months. (This recommendation is also known as "the 1-3-6 benchmark.") In the United States, the states that recommend the 1-3-6 benchmark should actually strive for a "1-2-3" timeline.
Because children with severe-to-profound hearing loss who are candidates for cochlear implantation can attain levels of social functioning and education indistinguishable from those of normal-hearing peers, cochlear implantation should be performed as soon as possible.
Children with mild-to-moderate hearing loss can be treated with hearing aids customized to the child's age and severity of hearing loss.
Surveillance: Most children with severe-to-profound GJB2-AR NSHL who are cochlear implant recipients initially require frequent follow-up visits with their cochlear implant team (otolaryngologist, audiologist, and speech-language pathologist) for assessment of speech recognition and equipment check. Once the cochlear implant recipient and their family become comfortable with the cochlear implant, many of these tasks can be performed by the family at home.
All children with mild-to-moderate GJB2-AR NSHL require follow-up audiograms annually to detect any progression of hearing loss. Children using hearing aids typically have an annual evaluation by an otolaryngologist, audiologist, and hearing aid specialist to examine the ears, obtain an audiogram, and check hearing aid function.
Agents/circumstances to avoid: Individuals with mild-to-moderate hearing loss should avoid environmental exposures known to cause hearing loss. Most important for persons with GJB2-related mild-to-moderate hearing loss is avoidance of repeated overexposure to loud noises (i.e., >75 decibels), particularly secondary to earbud use. Note that the headphone safety feature built into many smartphones can be set to a maximum limit of decibels.
Evaluation of relatives at risk: With limited exceptions, it is appropriate to clarify the genetic status of sibs of a proband with GJB2-AR NSHL; early identification of infants and children with hearing loss allows appropriate support and management to be provided to the child and family.
Genetic counseling.
GJB2-AR NSHL is inherited in an autosomal recessive manner. The parents of a child with GJB2-AR NSHL are typically heterozygous for a GJB2 pathogenic variant. In populations with a high carrier rate, it is possible that either one or both parents of a child with GJB2-AR NSHL also have GJB2-AR NSHL. If both parents are known to be heterozygous for a pathogenic variant, each sib of an individual with GJB2-AR NSHL has at conception a 25% chance of inheriting biallelic pathogenic variants and having hearing loss, a 50% chance of inheriting one pathogenic variant, and a 25% chance of inheriting neither of the familial GJB2 pathogenic variants. Once the GJB2 pathogenic variants have been identified in a family member with GJB2-AR NSHL, prenatal and preimplantation genetic testing for GJB2-AR NSHL are possible.
Diagnosis
Suggestive Findings
The diagnosis of GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) should be considered in two scenarios: an abnormal newborn hearing screening (NBHS) result and a symptomatic individual.
Scenario 1: Abnormal Newborn Hearing Screening (NBHS) Result
Universal NBHS using physiologic screening (either otoacoustic emissions [OAE], which measure the response of cochlear outer hair cells to auditory stimuli, or automated auditory brain stem response [ABR], which measures the physiologic response of cochlear inner hair cells, auditory nerve, brain stem, and brain to auditory stimuli) is required by law or rule in all 50 states in the United States and is performed on >98% of children in the US typically within days after birth (see 2020 Summary of National CDC EHDI Data).
Note: (1) NBHS is designed to detect moderate-to-profound hearing loss and may miss neonates with mild hearing loss, an important consideration, since data from the Centers for Disease Control and Prevention (CDC) show that more than 25% of bilateral deafness in newborns is slight or mild (<40 dB) (see 2020 Type and Severity Summary of Identified Cases of Hearing Loss). An even larger percentage of newborns may be affected by this degree of hearing loss, as very mild hearing loss can go undetected by NBHS (depending on the hearing thresholds and testing methods used). Although implementing physiologic screening at lower hearing thresholds would detect more mild hearing loss, it would also increase the rate of false positive test results and burden confirmatory diagnostic audiology evaluations. (2) The exact hearing thresholds used in NBHS are difficult to define precisely because manufacturers of equipment used in auditory screening tests choose the stimulus levels for their equipment, and these cannot be adjusted by the operator [Shearer et al 2019].
The following evaluations need to begin immediately on receipt of an abnormal NBHS result:
- The first step is to perform confirmatory audiometric testing, typically diagnostic ABR testing.
- The next step is a medical evaluation by an otolaryngologist, who is often the first point of contact for children with newly diagnosed hearing loss and will perform examinations including: (1) otomicroscopic evaluation for other causes of hearing loss such as conductive hearing loss resulting from otitis media (fluid in the middle ear) and outer and middle ear abnormalities; and (2) evaluate for features of a syndrome that may be associated with hearing loss.
- The otolaryngologist will consider family history, gestational history, physical examination, audiometric testing, and molecular genetic testing to determine the underlying diagnosis (see Establishing the Diagnosis).
Scenario 2: Symptomatic Individual
GJB2-AR NSHL should be suspected in a three- to five-year-old child with the following clinical findings and family history.
Clinical findings
- Mild-to-moderate typically non-progressive sensorineural hearing loss as measured by ABR testing or pure tone audiometry
- No related systemic findings identified by medical history and physical examination
Hearing is measured in decibels (dB). The threshold or 0 dB mark for each frequency refers to the level at which young adults with normal hearing perceive a tone burst 50% of the time. Hearing is considered normal if an individual's hearing thresholds are within 15 dB of normal thresholds. Severity of hearing loss is graded as shown in Table 1.
Table 1.
Severity of Hearing Loss in Decibels (dB)
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis. Note: Pseudodominant inheritance (i.e., an autosomal recessive condition present in individuals in two or more generations) has been reported in some families (see Genetic Counseling).
Establishing the Diagnosis
The diagnosis of GJB2-AR NSHL is established in a proband with suggestive findings who has EITHER of the following identified by molecular genetic testing (see Table 2):
- Biallelic GJB2 pathogenic (or likely pathogenic) variants (99%);
OR
- Compound heterozygosity for one GJB2 pathogenic (or likely pathogenic) variant and one of the following findings:
- A deletion that is either (1) an intragenic deletion or (2) a whole-gene deletion (see Molecular Genetics, GJB2-specific laboratory technical considerations);
- A noncoding pathogenic (or likely pathogenic) variant upstream or downstream of GJB2; or
- Mosaic uniparental disomy of GJB2.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. For expert specification of the ACMG/AMP variant interpretation for genetic hearing loss, see Oza et al [2018] (full text). Reference to "pathogenic variants" in this section is understood to include likely pathogenic variants. (2) Identification of biallelic GJB2 variants of uncertain significance (or of one known GJB2 pathogenic variant and one GJB2 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Note: Single-gene testing (sequence analysis of GJB2, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
Option 1
A hearing loss multigene panel that includes all genes implicated in nonsyndromic hearing loss and disorders that mimic nonsyndromic hearing loss including GJB2 and other genes of interest (see Differential Diagnosis and Genetic Hearing Loss Overview) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Analytic methods used for this panel must include the detection of deletions of GJB2, either intragenic or whole gene, and deletions that include sequences upstream of GJB2 (comprising either GJB6 and portions of CRYL1 or just portions of CRYL1) that delete cis-regulatory regions of GJB2, thereby abolishing GJB2 expression). See also Molecular Genetics, GJB2-specific laboratory technical considerations.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in GJB2-Related Autosomal Recessive Nonsyndromic Hearing Loss
Clinical Characteristics
Clinical Description
GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) is the most common genetic cause of congenital (present at birth) severe-to-profound non-progressive sensorineural hearing loss in many world populations. GJB2-AR NSHL can also be mild to moderate and is usually not progressive; however, there are exceptions.
Severe-to-profound GJB2-AR NSHL is typically congenital and diagnosed on newborn hearing screening (NBHS); however, there are occasional exceptions.
In two examples, a newborn passed his NBHS auditory brain stem response (ABR)-based test, but was diagnosed as deaf at age 15 months when an ABR test obtained at the request of his parents showed no responses through 90 dB. Another child had a normal free-field audiogram performed by an experienced audiologist at age five months, but at age nine months had ABR-confirmed severe hearing loss [Green et al 2000].
Severe-to-profound GJB2-AR NSHL is most frequently associated with truncating GJB2 variants (see Genotype-Phenotype Correlations).
Mild-to-moderate GJB2-AR NSHL may escape detection on the NBHS, especially when the hearing loss is mild. Most mild-to-moderate GJB2-AR NSHL is not progressive; however, an important exception is the unique trajectory of biallelic p.Val37Ile pathogenic variants, which has been well documented by Chen et al [2022] (see Genotype-Phenotype Correlations).
Mild-to-moderate GJB2-AR NSHL is most often associated with either biallelic missense variants or a missense variant in trans with a truncating variant (see Genotype-Phenotype Correlations).
Intrafamilial variability in the degree of GJB2-AR NSHL. The majority (67%) of sib pairs will have similar levels of hearing loss (within 20 dB); however, in a significant percentage (33%), there will be a prominent difference in hearing levels (>30 dB) between sibs [Fujioka et al 2020].
All GJB2-AR NSHL. Vestibular function is normal. Affected infants and young children do not experience balance problems and learn to sit and walk at age-appropriate times.
Except for the hearing impairment, individuals with GJB2-AR NSHL are healthy; life span is normal.
Genotype-Phenotype Correlations
Class of GJB2 Pathogenic Variants
Truncating vs nontruncating variants. The strong genotype-phenotype correlations with GJB2-AR NSHL can be recognized by classifying GJB2 variants as truncating (T) or nontruncating (NT) and then defining three genotype classes: biallelic truncating (T/T), biallelic nontruncating (NT/NT), and compound heterozygous (T/NT) [Snoeckx et al 2005]. Hearing loss across these classes is nonrandomly distributed, with loss in the T/T class being more severe than in the T/NT class, which is more severe than in the NT/NT class. See Figure 3 in Snoeckx et al [2005] (full text), which provides audiograms for various genotypes, showing the average hearing thresholds (50th centile) and the expected range (10th centile and 90th centile) for common genotypes.
- In the T/T class, hearing loss is most typically severe to profound (84%-92% of persons), although moderate (10%-12%) and mild (0%-3%) hearing losses are reported.
- In the T/NT class, 34%-47% of persons have severe-to-profound hearing loss.
- In the NT/NT class, only one person in five has severe-to-profound hearing loss, with more than half of persons having mild hearing loss.
Large deletions involving GJB2
- When a large GJB2 deletion occurs in the compound heterozygous state with either a GJB2 truncating variant or a missense variant, hearing loss is severe to profound.
- Homozygosity for a large GJB2 deletion, which is very rare, is associated with severe to profound sensorineural hearing loss [Pandya et al 2020].
Specific GJB2 Variants
p.Val37Ile. Chen et al [2022] reported that more than 40% of newborns either homozygous for p.Val37Ile or compound heterozygous for p.Val37Ile and a truncating GJB2 variant have hearing thresholds lower than 20 dB (i.e., normal hearing levels) on click-evoked ABR-based testing and pass their distortion-product otoacoustic emissions (DPOAE)-based NBHS. However, hearing thresholds decline with age so that ~60% of 40- to 60-year-olds and ~80% of 60- to 85-year-olds with this genotype have moderate hearing loss.
Nomenclature
GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) may also be referred by its genetic locus, DFNB1 (DFN for DeaFNess, B to indicate autosomal recessive inheritance, and 1 to indicate the relative order of hearing loss-related gene mapping). However, locus-based naming can be problematic when more than one gene is mapped to a given locus, as is the case with GJB2 and GJB6, which both map to the DFNB1 locus (13q12.11). For clarity, gene-based naming is used in this GeneReview rather than locus-based naming.
Prevalence
The prevalence of GJB2-AR NSHL and the carrier rate for pathogenic GJB2 variants are population dependent (see Figure 1). Some of this variability may reflect ascertainment bias. For example, although Adadey et al [2022] present the most comprehensive data of the landscape of genetic hearing loss to date, they note that in Africa the causes of genetic hearing loss are still to be investigated in a large majority of African countries. Based on currently available data, the highest carrier rate (8.3%) is reported in the East Asian population, in which the most common pathogenic variant is p.Val37Ile.

Genetically Related (Allelic) Disorders
Fewer than 30 GJB2 variants are associated with autosomal dominant inheritance [Azaiez et al 2018].
In most of these case reports, the severity of the hearing loss is variable and is associated with a spectrum of skin manifestations such as keratitis-ichthyosis-deafness syndrome (OMIM 148210), hystrix-like ichthyosis with deafness (OMIM 602540), Vohwinkel syndrome (OMIM 124500), Bart-Pumphrey syndrome (OMIM 149200), and palmoplantar keratoderma with deafness (OMIM 148350) [Iossa et al 2011].
Two variants at amino acid residue 75 – p.Arg75Gln and p.Arg75Trp – have been reported to cause autosomal dominant hearing loss. Both of these missense changes affect the same amino acid and are associated with hearing loss that is fully penetrant and a skin phenotype (palmoplantar keratoderma) that is not fully penetrant [Birkenhäger et al 2010]. This phenotypic variability occurs even within the same family [Manzoli et al 2013].
Differential Diagnosis
As of this writing, more than 70 genes have been associated with autosomal recessive nonsyndromic hearing loss. For a list of selected genes associated with distinctive clinical features, see Genetic Hearing Loss Overview, Table 3.
For a current, comprehensive list of all identified autosomal recessive nonsyndromic hearing loss genes, see Hereditary Hearing Loss Homepage.
Management
No clinical practice guidelines specific to GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) have been published.
Management ideally occurs in the context of a multidisciplinary clinic with specialists in otolaryngology, audiology, and genetic counseling. See Genetic Hearing Loss Overview, Management.
Evaluations Following Initial Diagnosis
To establish the extent of involvement and needs in an individual diagnosed with GJB2-AR NSHL, the following evaluations by an otolaryngologist are recommended:
- Comprehensive medical history focused on gestational and perinatal events, newborn hearing screen (NBHS) results, and cytomegalic virus (CMV) test results (if performed)
- Assessment of hearing using age-appropriate tests such as auditory brain stem response (ABR) testing, auditory steady-state response (ASSR) testing, and pure-tone audiometry to determine severity of hearing loss (see Table 1) and laterality (expected to be symmetric)
- Physical examination including an evaluation for syndromic features (which should be absent) and a thorough examination of the head and neck, including an otomicroscopic evaluation of the ear to evaluate for otitis media
- Ophthalmologic examination to identify refractive errors (unrelated to GJB2-AR NSHL) that require spectacle correction (glasses) to improve visual acuity
- Consultation with a medical geneticist, certified genetic counselor, or certified advanced genetic nurse to inform affected individuals and their families about the nature, mode of inheritance, and implications of GJB2-AR NSHL in order to facilitate medical and personal decision making
- Assessment of need for family support and resources including community or online resources such as Parent to Parent, social work involvement for parental support, and home nursing referral
Treatment of Manifestations
Management of children with GJB2-AR NSHL involves discussion of the following issues with the family.
Uncorrected hearing loss (regardless of etiology) has consistent sequelae. Auditory deprivation during the first two years of life is associated with poor reading performance, poor communication skills, and poor speech production, deficiencies that cannot be completely remediated even with educational intervention. Early auditory intervention is effective – whether through cochlear implantation or amplification [Smith et al 2005].
To optimize intervention for children with hearing loss, the Joint Committee on Infant Hearing [2019] recommends universal NBHS be completed by age one month, diagnosis established by age three months, and early intervention begun by age six months (also known as "the 1-3-6 benchmark"). In the United States, the states that recommend the 1-3-6 benchmark should actually strive for a "1-2-3" timeline.
Children with severe-to-profound hearing loss who are candidates for cochlear implantation can attain levels of social functioning and education indistinguishable from those of normal-hearing peers [Loy et al 2010, Langereis & Vermeulen 2015]. Cochlear implantation performed as soon as possible is associated with significantly better long-term auditory performance and speech intelligibility than later cochlear implantation [Wu et al 2015, Eshraghi et al 2020]. Nonetheless, there is no upper age limit for cochlear implantation; however, at older ages habilitation outcomes are less predictable.
Children with mild-to-moderate hearing loss can be treated with hearing aids customized to the child's age and severity of hearing loss.
Surveillance
To monitor the individual's response to habituation and to identify any changes in severity of hearing loss, the following evaluations are recommended.
Most children with severe-to-profound GJB2-AR NSHL with a cochlear implant will initially have frequent evaluations recommended by their cochlear implant team (otolaryngologist, audiologist, and speech and language pathologist).
At follow-up appointments, the following may be performed: speech recognition testing, equipment checks (including device adjustment and troubleshooting), and provision of replacement or upgraded equipment [Cullington et al 2016].
As the cochlear implant recipient becomes comfortable with the cochlear implant, many of the above tasks can be performed by the family at home, obviating the need for routinely scheduled appointments unless the need for a clinic visit arises.
Children with mild-to-moderate GJB2-AR NSHL
- An annual evaluation by an otolaryngologist, audiologist, and hearing aid specialist is recommended to examine the ears, obtain an audiogram, and check hearing aid function.
- This annual follow up is essential, as progression of hearing loss can occur. For most GJB2 genotypes associated with mild-to-moderate hearing loss, hearing is stable; however, the variant p.Val37Ile is associated with progressive hearing loss. In one study, one of eight persons homozygous for the p.Val37Ile pathogenic variant underwent cochlear implantation due to progression of mild-to-moderate hearing loss; six of eight used hearing aids; and one of eight was not treated [Lee et al 2021].
Agents/Circumstances to Avoid
Individuals with hearing loss should avoid environmental exposures known to cause hearing loss.
Most important for persons with GJB2-related mild-to-moderate hearing loss is avoidance of repeated overexposure to loud noises, particularly secondary to earbud use. The headphone safety feature built into most smartphones can be set a maximum limit of 75 dB.
Headphone/earbud safety features can be found in the phone settings menu:
- In iPhones, under Settings > Sounds & Haptics > Headphone Safety
- In Android phones, under Settings > Sounds & Vibrations > Volume > Media volume limit
Also see these general resources on noise reduction:
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of sibs of a proband with GJB2-AR NSHL if:
- A newborn sib has an abnormal result on universal NBHS;
- A newborn sib has a normal result on NBHS and the GJB2 pathogenic variants identified in the proband are associated with mild hearing loss (as NBHS may miss newborns with mild hearing loss); or
- A sib did not undergo NBHS and/or NBHS results are unknown.
Early identification of infants and children with hearing loss allows appropriate support and management to be provided to the child and family.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this condition.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
By definition, GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) is inherited in an autosomal recessive manner.
Note: Pseudodominant inheritance (i.e., an autosomal recessive condition present in individuals in two or more generations) is reported in some families [Pampanos et al 2000, Tekin et al 2003] and is more likely to occur in populations with a high carrier rate (see Figure 1).
Risk to Family Members
Parents of a proband
- The parents of a child with GJB2-AR NSHL are typically heterozygous for a GJB2 pathogenic variant.
- In populations with a high carrier rate (e.g., the East Asian population, in which the carrier frequency for p.Val37Ile is 8.3% [see Figure 1]), it is possible that either one or both parents of a child with GJB2-AR NSHL also have GJB2-AR NSHL.
- Molecular genetic testing of the parents is recommended to confirm that both parents are heterozygous for a GJB2 pathogenic variant and to allow reliable counseling regarding the likelihood of recurrence.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A deletion of a portion of GJB2 in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity [Abe et al 2018];
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) of a GJB2 pathogenic variant associated with autosomal recessive nonsyndromic hearing loss are asymptomatic.
Sibs of a proband
- If both parents are known to be heterozygous for a GJB2 pathogenic variant, each sib of an individual with GJB2-AR NSHL has at conception a 25% chance of inheriting biallelic GJB2 pathogenic variants and having hearing loss, a 50% chance of inheriting one GJB2 pathogenic variant, and a 25% chance of inheriting neither of the familial GJB2 pathogenic variants.
- Strong genotype-phenotype correlations are observed in GJB2-AR NSHL.
- The majority of sibs (67%) who inherit biallelic GJB2 pathogenic variants have a hearing threshold similar (within 20 dB) to that of the proband; a prominent difference (>30 dB) in hearing thresholds is seen in 33% of sib pairs [Snoeckx et al 2005, Fujioka et al 2020].
- Heterozygotes (carriers) of a GJB2 pathogenic variant associated with autosomal recessive nonsyndromic hearing loss are asymptomatic.
Offspring of a proband
- Unless an affected individual's reproductive partner also has GJB2-AR NSHL or is a carrier, offspring will be obligate heterozygotes (carriers) for a pathogenic variant in GJB2.
- In populations with a high carrier rate (see Figure 1), the reproductive partner of the proband may have two GJB2 pathogenic variants or be heterozygous. Thus, the likelihood of GJB2-AR NSHL recurrence in offspring is most accurately determined after GJB2 molecular genetic testing of the proband's reproductive partner.
Other family members. Each sib of the proband's parents has a 50% chance of being a carrier of a GJB2 pathogenic variant.
Carrier Detection
Carrier testing for relatives of an individual with GJB2-AR NSHL requires prior identification of the GJB2 pathogenic variants in the family.
Related Genetic Counseling Issues
See Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
The following points are noteworthy:
- Clear communication between individuals with hearing loss, families, and health care providers is key. Deaf and hard-of-hearing persons may use a variety of communication methods, including spoken language, sign language, lip reading, and written notes. For deaf individuals and families who use sign language, a certified sign language interpreter must be used. Communication aids such as visual aids and verbal cues when changing topics can be helpful.
- It is important to ascertain and address the questions and concerns of the family/individual. Deaf and hard-of-hearing persons may be interested in obtaining information about the cause of their own deafness, including information on medical, educational, and social services. Others may seek information about the chance of deaf/hard-of-hearing children and information for family planning decisions.
- The use of neutral or balanced terminology can enhance the provision of services; for example: use of the term "chance" instead of "risk"; "deaf" or "hearing" instead of "affected" or "unaffected"; and "deaf" or "hard-of-hearing" instead of "hearing impaired." Members of the Deaf community may view deafness as a distinguishing characteristic and not as a handicap, impairment, or medical condition requiring a "treatment" or "cure," or to be "prevented." Terms such as "handicap" should be avoided.
Family planning
- The optimal time for determination of genetic status and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of the probability of hearing loss in offspring and reproductive options) to young adults with hearing loss.
- Because GJB2-AR NSHL is associated with extensive allelic heterogeneity, reliable carrier testing for the reproductive partners of individuals with GJB2-AR NSHL (and individuals who are carriers of GJB2-AR NSHL) requires GJB2 sequence analysis. Note: Screening for the large deletions shown in Figure 2 is not universally recommended because of the very low frequency of these deletions.
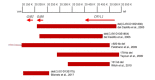
Figure 2.
Schematic of large (>10 kilobase) pathogenic deletions associated with GJB2-related autosomal recessive nonsyndromic hearing loss Modified from Abe et al [2018]
Prenatal Testing and Preimplantation Genetic Testing
Once the GJB2 pathogenic variants have been identified in a family member with GJB2-AR NSHL, prenatal and preimplantation genetic testing for GJB2-AR NSHL are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Medical Home Portal
- Alexander Graham Bell Association for the Deaf and Hard of HearingPhone: 866-337-5220 (toll-free); 202-337-5221 (TTY)Fax: 202-337-8314Email: info@agbell.org
- American Society for Deaf ChildrenPhone: 800-942-2732 (ASDC)Email: info@deafchildren.org
- BabyHearing.orgThis site, developed with support from the National Institute on Deafness and Other Communication Disorders, provides information about newborn hearing screening and hearing loss.
- MedlinePlus
- National Association of the DeafPhone: 301-587-1788 (Purple/ZVRS); 301-328-1443 (Sorenson); 301-338-6380 (Convo)Fax: 301-587-1791Email: nad.info@nad.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
GJB2-Related Autosomal Recessive Nonsyndromic Hearing Loss: Genes and Databases
Table B.
OMIM Entries for GJB2-Related Autosomal Recessive Nonsyndromic Hearing Loss (View All in OMIM)
Molecular Pathogenesis
GJB2 encodes gap junction beta-2 protein, also called connexin 26. Connexins aggregate in groups of six around a central 2.3-nm pore to form a connexon. Connexin 26 forms functional gap junctions with itself, connexin 32, connexin 46, and connexin 50.
Connexons from adjoining cells covalently bond, forming a channel between cells. Gap junctions permit direct intercellular exchange of ions and molecules through their central aqueous pores and permit synchronization of activity in excitable tissues and the exchange of metabolites and signal molecules in nonexcitable tissues.
Mechanism of disease causation. Loss of function
GJB2-specific laboratory technical considerations
- Large deletions have been described, such as sequences upstream of GJB2 (comprising either GJB6 and portions of CRYL1 or just portions of CRYL1) that delete cis-regulatory regions of GJB2, thereby abolishing GJB2 expression (see Figure 2) [Abe et al 2018].
- Uniparental disomy (UPD). Mosaicism for the GJB2 c.235delC variant has been reported in an individual with maternal UPD [Lin et al 2021].
Table 3.
GJB2 Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Author Notes
Richard JH Smith, MD, is a pediatric otolaryngologist and human geneticist at the University of Iowa. He directs the Molecular Otolaryngology and Renal Research Laboratories (MORL), which offer comprehensive genetic testing for hearing loss. He is interested in collaborating with clinicians treating families affected by genetic hearing loss in whom no causative variant has been identified through molecular genetic testing of the genes known to be involved in this group of disorders. He also is actively involved in research regarding individuals with GJB2-related autosomal recessive nonsyndromic hearing loss (GJB2-AR NSHL) and would be happy to communicate with persons who have any questions regarding diagnosis of GJB2-AR NSHL or other considerations. For questions about hearing loss and the diagnosis of GJB2-AR NSHL, email ude.awoiu@lrom. MORL website: morl.lab.uiowa.edu
Hela Azaiez, MS, PhD, is a human geneticist specializing in the molecular genetics and genomics of hearing loss at the Department of Otolaryngology at the University of Iowa. She is actively engaged in basic and translational research, with a focus on the genetic etiology of hearing loss. Her research aims to decode the molecular mechanisms of hearing and deafness, intending to translate this knowledge into improved clinical diagnostics and enhanced patient care. She also maintains a keen interest in collaborating with clinicians who treat families affected by genetic hearing loss, particularly when no causative variant has been identified through molecular genetic testing of known genes associated with hearing loss.
Acknowledgments
Supported in part by grant RO1-DC02842 from the NIDCD (RJHS).
Author History
Hela Azaiez, MS, PhD (2023-present)
Kevin Booth, PhD (2023-present)
Mary-Kayt N Jones, BA; University of Iowa (2016-2023)
Daryl A Scott, MD, PhD; University of Iowa (1998-2001)
Val C Sheffield, MD, PhD; University of Iowa (1998-2001)
Richard JH Smith, MD (1998-present)
Guy Van Camp, PhD; University of Antwerp (1998-2016)
Revision History
- 20 July 2023 (bp) Comprehensive update posted live
- 18 August 2016 (bp) Comprehensive update posted live
- 2 January 2014 (me) Comprehensive update posted live
- 14 July 2011 (me) Comprehensive update posted live
- 11 July 2008 (me) Comprehensive update posted live
- 21 December 2005 (me) Comprehensive update posted live
- 27 October 2003 (me) Comprehensive update posted live
- 24 April 2001 (me) Comprehensive update posted live
- 4 April 1998 (rjs) Original submission
References
Published Guidelines / Consensus Statements
- The Joint Committee on Infant Hearing. Year 2019 position statement: principles and guidelines for early hearing detection and intervention programs. Available online. Accessed 7-10-2023.
Literature Cited
- Abe S, Nishio SY, Yokota Y, Moteki H, Kumakawa K, Usami SI. Diagnostic pitfalls for GJB2-related hearing loss: a novel deletion detected by array-CGH analysis in a Japanese patient with congenital profound hearing loss. Clin Case Rep. 2018;6:2111–6. [PMC free article: PMC6230644] [PubMed: 30455902]
- Adadey SM, Wonkam-Tingang E, Aboagye ET, Quaye O, Awandare GA, Wonkam A. Hearing loss in Africa: current genetic profile. Hum Genet. 2022;141:505–17. [PMC free article: PMC9034983] [PubMed: 34609590]
- Azaiez H, Booth KT, Ephraim SS, Crone B, Black-Ziegelbein EA, Marini RJ, Shearer AE, Sloan-Heggen CM, Kolbe D, Casavant T, Schnieders MJ, Nishimura C, Braun T, Smith RJH. Genomic landscape and mutational signatures of deafness-associated genes. Am J Hum Genet. 2018;103:484–97. [PMC free article: PMC6174355] [PubMed: 30245029]
- Birkenhäger R, Lüblinghoff N, Prera E, Schild C, Aschendorff A, Arndt S. Autosomal dominant prelingual hearing loss with palmoplantar keratoderma syndrome: variability in clinical expression from mutations of R75W and R75Q in the GJB2 gene. Am J Med Genet A. 2010;152A:1798–802. [PubMed: 20583176]
- Chen Y, Wang Z, Jiang Y, Lin Y, Wang X, Wang Z, Tang Z, Wang Y, Wang J, Gao Y, Shi W, Huang Z, Li Y, Shi J, Wang X, Yu Q, Ma Y, Zhou J, Yang T, Wu H. Biallelic p.V37I variant in GJB2 is associated with increasing incidence of hearing loss with age. Genet Med. 2022;24:915–23. [PubMed: 35016843]
- Cullington H, Kitterick P, DeBold L, Weal M, Clarke N, Newberry E, Aubert L. Personalised long-term follow-up of cochlear implant patients using remote care, compared with those on the standard care pathway: study protocol for a feasibility randomised controlled trial. BMJ Open. 2016;6:e011342. [PMC free article: PMC4874122] [PubMed: 27178980]
- Eshraghi AA, Polineni SP, Davies C, Shahal D, Mittal J, Al-Zaghal Z, Sinha R, Jindal U, Mittal R. Genotype-phenotype correlation for predicting cochlear implant outcome: current challenges and opportunities. Front Genet. 2020;11:678. [PMC free article: PMC7381205] [PubMed: 32765579]
- Fujioka M, Hosoya M, Nara K, Morimoto N, Sakamoto H, Otsu M, Nakano A, Arimoto Y, Masuda S, Sugiuchi T, Masuda S, Morita N, Ogawa K, Kaga K, Matsunaga T. Differences in hearing levels between siblings with hearing loss caused by GJB2 mutations. Auris Nasus Larynx. 2020;47:938–42. [PubMed: 32553771]
- Green GE, Smith RJ, Bent JP, Cohn ES. Genetic testing to identify deaf newborns. JAMA. 2000;284:1245. [PubMed: 10979110]
- Iossa S, Marciano E, Franzé A. GJB2 gene mutations in syndromic skin diseases with sensorineural hearing loss. Curr Genomics. 2011;12:475–785. [PMC free article: PMC3219843] [PubMed: 22547955]
- Joint Committee on Infant Hearing. Year 2019 position statement: principles and guidelines for early hearing detection and intervention programs. J Early Hear Detect Interv. 2019;4:1–44.
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Langereis M, Vermeulen A. School performance and wellbeing of children with CI in different communicative–educational environments. Int J Pediatr Otorhinolaryngol. 2015;79:834–9. [PubMed: 25840945]
- Lee SY, Han SC, Han JH, Kim MY, Oh DY, Kim NJ, Song JJ, Koo JW, Lee JH, Oh SH, Choi BY. Natural course of residual hearing with reference to GJB2 and SLC26A4 genotypes: clinical implications for hearing rehabilitation. Ear Hear. 2021;42:644–53. [PubMed: 33928925]
- Lin YH, Wu PC, Tsai CY, Lin YH, Lo MY, Hsu SJ, Lin PH, Erdenechuluun J, Wu HP, Hsu CJ, Wu CC, Chen PL. Hearing impairment with monoallelic GJB2 variants: a GJB2 cause or non-GJB2 cause? J Mol Diagn. 2021;23:1279–91. [PubMed: 34325055]
- Loy B, Warner-Czyz AD, Tong L, Tobey EA, Roland PS. The children speak: an examination of the quality of life of pediatric cochlear implant users. Otolaryngol Head Neck Surg. 2010;142:247–53. [PMC free article: PMC2852181] [PubMed: 20115983]
- Manzoli GN, Abe-Sandes K, Bittles AH, da Silva DS. Fernandes Lda C, Paulon RM, de Castro IC, Padovani CM, Acosta AX. Non-syndromic hearing impairment in a multi-ethnic population of Northeastern Brazil. Int J Pediatr Otorhinolaryngol. 2013;77:1077–82. [PubMed: 23684175]
- Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti LA, Murry JB, Hasadsri L, Nara K, Kenna M, Booth KT, Azaiez H, Griffith A, Avraham KB, Kremer H, Rehm HL, Amr SS, Abou Tayoun AN, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–613. [PMC free article: PMC6188673] [PubMed: 30311386]
- Pampanos A, Neou P, Iliades T, Apostolopoulos N, Voyiatzis N, Grigoriadou M, Katsichti L, Skevas A, Petersen MB. Pseudodominant inheritance of DFNB1 deafness due to the common 35delG mutation. Clin Genet. 2000;57:232–4. [PubMed: 10782932]
- Pandya A, O'Brien A, Kovasala M, Bademci G, Tekin M, Arnos KS. Analyses of del(GJB6-D13S1830) and del(GJB6-D13S1834) deletions in a large cohort with hearing loss: Caveats to interpretation of molecular test results in multiplex families. Mol Genet Genomic Med. 2020;8:e1171. [PMC free article: PMC7196463] [PubMed: 32067424]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Shearer AE, Shen J, Amr S, Morton CC, Smith RJ, et al. A proposal for comprehensive newborn hearing screening to improve identification of deaf and hard-of-hearing children. Genet Med. 2019;21:2614–30. [PMC free article: PMC6831511] [PubMed: 31171844]
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJ. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441–50. [PMC free article: PMC4796320] [PubMed: 26969326]
- Smith RJ, Bale JF Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879–90. [PubMed: 15752533]
- Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, Mueller-Malesinska M, Pollak A, Ploski R, Murgia A, Orzan E, Castorina P, Ambrosetti U, Nowakowska-Szyrwinska E, Bal J, Wiszniewski W, Janecke AR, Nekahm-Heis D, Seeman P, Bendova O, Kenna MA, Frangulov A, Rehm HL, Tekin M, Incesulu A, Dahl HH, du Sart D, Jenkins L, Lucas D, Bitner-Glindzicz M, Avraham KB, Brownstein Z, Del Castillo I, Moreno F, Blin N, Pfister M, Sziklai I, Toth T, Kelley PM, Cohn ES, Van Maldergem L, Hilbert P, Roux AF, Mondain M, Hoefsloot LH, Cremers CW, Lopponen T, Lopponen H, Parving A, Gronskov K, Schrijver I, Roberson J, Gualandi F, Martini A, Lina-Granade G, Pallares-Ruiz N, Correia C, Fialho G, Cryns K, Hilgert N, Van de Heyning P, Nishimura CJ, Smith RJ, Van Camp G. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77:945–57. [PMC free article: PMC1285178] [PubMed: 16380907]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tekin M, Duman T, Boğoçlu G, Incesulu A, Cin S, Akar N. Moderate hearing loss and pseudodominant inheritance due to L90P/35delG mutations in the GJB2 (connexin 26) gene. Genet Couns. 2003;14:379–86. [PubMed: 14738110]
- Wu CM, Ko HC, Tsou YT, Lin YH, Lin JL, Chen CK, Chen PL, Wu CC. Long-term cochlear implant outcomes in children with GJB2 and SLC26A4 mutations. PLoS One. 2015;10:e0138575. [PMC free article: PMC4580418] [PubMed: 26397989]
Publication Details
Author Information and Affiliations
University of Iowa Hospitals and Clinics
Iowa City, Iowa
University of Iowa Hospitals and Clinics
Iowa City, Iowa
Publication History
Initial Posting: September 28, 1998; Last Update: July 20, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Smith RJH, Azaiez H, Booth K. GJB2-Related Autosomal Recessive Nonsyndromic Hearing Loss. 1998 Sep 28 [Updated 2023 Jul 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

