Summary
Clinical characteristics.
Dyskeratosis congenita and related telomere biology disorders (DC/TBD) are caused by impaired telomere maintenance resulting in short or very short telomeres. The phenotypic spectrum of telomere biology disorders is broad and includes individuals with classic dyskeratosis congenita (DC) as well as those with very short telomeres and an isolated physical finding. Classic DC is characterized by a triad of dysplastic nails, lacy reticular pigmentation of the upper chest and/or neck, and oral leukoplakia, although this may not be present in all individuals. People with DC/TBD are at increased risk for progressive bone marrow failure (BMF), myelodysplastic syndrome or acute myelogenous leukemia, solid tumors (usually squamous cell carcinoma of the head/neck or anogenital cancer), and pulmonary fibrosis. Other findings can include eye abnormalities (epiphora, blepharitis, sparse eyelashes, ectropion, entropion, trichiasis), taurodontism, liver disease, gastrointestinal telangiectasias, and avascular necrosis of the hips or shoulders. Although most persons with DC/TBD have normal psychomotor development and normal neurologic function, significant developmental delay is present in both forms; additional findings include cerebellar hypoplasia (Hoyeraal Hreidarsson syndrome) and bilateral exudative retinopathy and intracranial calcifications (Revesz syndrome and Coats plus syndrome). Onset and progression of manifestations of DC/TBD vary: at the mild end of the spectrum are those who have only minimal physical findings with normal bone marrow function, and at the severe end are those who have the diagnostic triad and early-onset BMF.
Diagnosis/testing.
A majority of individuals with DC/TBD have abnormally short telomeres for their age, as determined by multicolor flow cytometry fluorescence in situ hybridization (flow-FISH) on lymphocyte subsets. To date, ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, POT1, RPA1, RTEL1, STN1, TERC, TERT, TINF2, WRAP53, and ZCCHC8 are the genes in which pathogenic variants are known to cause DC/TBD and to result in very short telomeres. Pathogenic variants in one of these 16 genes have been identified in approximately 80% of individuals who meet clinical diagnostic criteria for DC/TBD.
Management.
Treatment of manifestations: Treatment is tailored to the individual. Hematopoietic cell transplantation (HCT) is the only curative treatment for BMF and leukemia, but long-term outcome has historically been poor due to treatment toxicity; if a suitable donor is not available, androgen therapy may be considered for BMF. Treatment of other cancers is tailored to the type of cancer. Of note, cancer therapy may pose an increased risk for prolonged cytopenias as well as pulmonary and hepatic toxicity. Treatment of pulmonary fibrosis is primarily supportive, although lung transplantation may be considered.
Surveillance: For BMF: complete blood count (CBC) annually if normal and more often if abnormal; annual bone marrow aspirate and biopsy. For those on androgen therapy: routine monitoring of CBC, liver function, liver ultrasound, and endocrinology evaluation. For cancer risk: monthly self-examination for oral, head, and neck cancer; annual cancer screening by an otolaryngologist and dermatologist; annual gynecologic examination. For pulmonary fibrosis: annual pulmonary function tests starting either at diagnosis or when the individual can perform the test (often age ~8 years); bubble echocardiogram to look for pulmonary arteriovenous malformations if suspected based on clinical symptoms. Routine dental screening every six months and good oral hygiene are recommended.
Agents/circumstances to avoid: Blood donation by family members if HCT is being considered; non-leukodepleted and non-irradiated blood products; the combination of androgens and granulocyte colony-stimulating factor in treatment of BMF (has been associated with splenic rupture); toxic agents implicated in tumorigenesis (e.g., smoking, excessive sun exposure).
Evaluation of relatives at risk: If a relative has signs or symptoms suggestive of DC/TBD or is being evaluated as a potential HCT donor, telomere length testing – or, if the pathogenic variant(s) in the family are known, molecular genetic testing – is warranted.
Genetic counseling.
The mode of inheritance of DC/TBD varies by gene:
- X-linked: DKC1
- Autosomal dominant: NAF1, RPA1, TERC, TINF2, and ZCCHC8
- Autosomal dominant or autosomal recessive: ACD, PARN, RTEL1, and TERT
- Autosomal recessive: CTC1, NHP2, NOP10, POT1, STN1, and WRAP53
Genetic counseling regarding risk to family members depends on accurate diagnosis, determination of the mode of inheritance in each family, and results of molecular genetic testing. Once the DC/TBD-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
GeneReview Scope
Table
Classic dyskeratosis congenita Hoyeraal Hreidarsson syndrome
Diagnosis
Dyskeratosis congenita and related telomere biology disorders (DC/TBD) are caused by impaired telomere maintenance resulting in short or very short telomeres. The phenotypic spectrum of telomere biology disorders is broad and includes individuals with classic dyskeratosis congenita (DC) as well as those with very short telomeres and an isolated physical finding [Savage & Bertuch 2010, Dokal 2011, Ballew & Savage 2013, Bertuch 2016, Niewisch & Savage 2019, Niewisch et al 2022].
The criteria for classic DC were described by Vulliamy et al [2006]; see Suggestive Findings. Note, however, that individuals may develop features of a DC/TBD at variable rates and ages, which can make proper diagnosis challenging.
Suggestive Findings
A telomere biology disorder (TBD), including dyskeratosis congenita (DC), should be suspected in individuals with the following clinical findings.
Physical characteristics. One of the following:
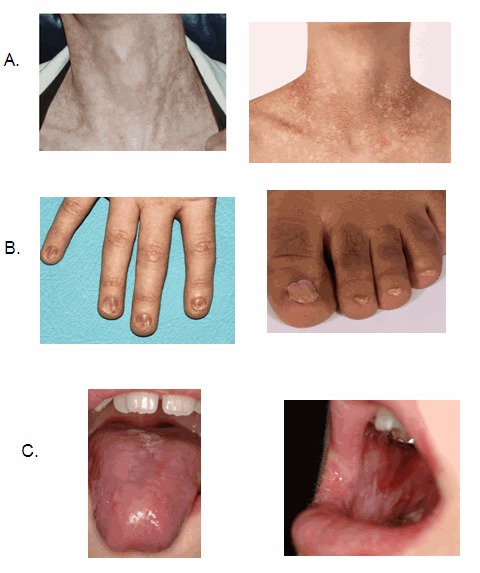
- At least two features of the classic DC clinical triad (Figure 1):
- Dysplastic nails. Findings may be subtle with ridging, flaking, or poor growth, or more diffuse with nearly complete loss of nails.
- Lacy reticular pigmentation of the upper chest and/or neck. May be subtle or diffuse hyper- or hypopigmentation. Note that abnormal pigmentation changes are not restricted to the upper chest and neck.
- Oral leukoplakia (white patches in the mouth)
- One feature of the classic triad or suggestive family history (occurrence of bone marrow failure [BMF], myelodysplastic syndrome [MDS], acute myelogenous leukemia [AML], early-onset head/neck squamous cell cancer [HNSCC], and/or pulmonary fibrosis [PF] in a first- or second-degree relative) in combination with:
- Progressive BMF. May appear at any age and may be a presenting sign. Macrocytosis and elevated hemoglobin F levels may be seen.
- MDS or AML. May be the presenting sign.
- Solid tumors, usually HNSCC or anogenital adenocarcinoma, in persons younger than age 50 years and without other risk factors. Solid tumors may be the first manifestation of DC/TBD in individuals who do not have BMF.
- Any feature of the classic triad plus two or more of the following:
- Epiphora (excessive watering of the eye[s])
- Blepharitis (inflammation of the eyelids, often due to epiphora)
- Abnormal eyelash growth
- Prematurely gray hair
- Alopecia
- Taurodontism (enlarged tooth pulp chambers) or decreased tooth root-to-crown ratio
- Developmental delay
- Short stature
- Microcephaly
- Hypogonadism
- Esophageal stenosis
- Urethral stenosis
- Liver disease
- Osteoporosis
- Avascular necrosis of the hips or shoulders
- Gastrointestinal telangiectasias
- Pulmonary arteriovenous malformations
Note: Individuals with TBDs may have none of the above additional findings or the findings may appear or worsen with age.
Figure 1.
Examples of the dyskeratosis congenita diagnostic triad A. Skin pigmentation
Establishing the Diagnosis
The diagnosis of DC/TBD is established in a proband with characteristic clinical findings and either of the following:
- Shortened telomere length as determined by lymphocyte telomere length testing by automated multicolor flow cytometry fluorescence in situ hybridization (flow-FISH) in the six-cell panel assay. Telomere length less than the first centile for age in lymphocytes is 97% sensitive and 91% specific for DC/TBD. In individuals with complex or atypical DC/TBD, the six-cell panel may be more informative than the two-panel test of total lymphocytes and granulocytes [Alter et al 2012].
- Identification of biallelic pathogenic (or likely pathogenic) variants in one of the six genes known to solely cause autosomal recessive DC/TBD; or a heterozygous pathogenic (or likely pathogenic) variant in one of the five genes known to cause autosomal dominant DC/TBD; or a mono- or biallelic pathogenic (or likely pathogenic) variant in one of the four genes associated with autosomal recessive and dominant DC/TBD; or a hemizygous pathogenic (or likely pathogenic) variant in DKC1 known to cause X-linked DC/TBD (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) The identification of variant(s) of uncertain significance cannot be used to confirm or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of DC/TBD has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
A multigene panel that includes the genes listed in Table 1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD)
Tissue-restricted mosaicism has been observed in a limited number of individuals with DC/TBD. A TERC germline pathogenic variant that was not observed by molecular genetic testing of DNA extracted from peripheral blood cells was detected in DNA extracted from other cells (e.g., skin fibroblasts) of the individual [Jongmans et al 2012]. Similarly, hematopoietic somatic reversion was also reported in an individual with a germline RPA1 variant Sharma et al [2022]. The assumption is that the selective advantage of the revertant hematopoietic cells allows them to populate the bone marrow, resulting in the inability to detect the pathogenic variant in DNA extracted from these cells. Molecular genetic testing of a second tissue source may be considered in individuals who meet the diagnostic criteria for DC/TBD but do not have a pathogenic variant identified on molecular genetic testing of peripheral blood cells.
Clinical Characteristics
Clinical Description
The classic dyskeratosis congenita (DC) triad of abnormal fingernails and toenails, lacy, reticular pigmentation of the neck and upper chest, and oral leukoplakia is diagnostic (Figure 1); however, these features are not present in all individuals with DC and related telomere biology disorders (DC/TBD) and may or may not develop over time after the appearance of other complications listed here [Savage & Bertuch 2010, Dokal 2011, Ward et al 2018, Niewisch et al 2022]. The time of onset for these medical complications varies considerably among individuals even within the same family and thus the manifestations of DC/TBD do not progress in a predictable pattern. The spectrum ranges from individuals who develop bone marrow failure (BMF) first and then years later develop other classic findings such as nail abnormalities, to others who have severe nail problems and abnormalities of skin pigmentation but normal bone marrow function.
Three forms of DC/TBD with more severe manifestations have been identified: Hoyeraal Hreidarsson syndrome, Revesz syndrome, and Coats plus syndrome (see Severe Forms of DC/TBD).
Table 2.
Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD): Frequency of Select Features
Dermatologic. Lacy, reticular pigmentation primarily of the neck and chest may be subtle or diffuse hyper- or hypopigmentation. Changes in skin pigmentation may not be present at time of diagnosis, but may develop or become more pronounced over time.
Dysplastic fingernails and toenails may worsen significantly over time and nails may eventually "disappear."
Additional common skin findings in DC/TBD include epiphora, loss of dermatoglyphics, early graying, palmoplantar hyperkeratosis, eyelash loss, and hair loss from scalp [Ward et al 2018]. Dermatoglyphics may be lost with age.
Hyperhidrosis is noted infrequently in some individuals.
Ears, nose, and throat. Oral leukoplakia is part of the diagnostic triad. It may be a presenting sign found in childhood or may develop over time.
Deafness has been reported but is rare.
Ophthalmic. Epiphora caused by stenosis of the lacrimal drainage system can result in blepharitis.
Abnormal eyelash growth includes sparse eyelashes, ectropion, entropion, and trichiasis, which can lead to corneal abrasions, scarring, or infection if not treated.
Bilateral exudative retinopathy seen in Revesz syndrome or Coats plus syndrome can lead to blindness.
Dental. Decreased root-to-crown ratio is attributed to abnormal tooth development.
Taurodontism (enlarged pulp chambers of the teeth) may be noted on dental x-ray.
Growth and development. Short stature has been reported but height is variable.
Intrauterine growth restriction (IUGR) has been noted in children with the more severe Coats plus syndrome, Hoyeraal Hreidarsson syndrome, or Revesz syndrome.
Developmental delay may be present in some. It can be more pronounced in persons with Hoyeraal Hreidarsson syndrome, Revesz syndrome, or Coats plus syndrome.
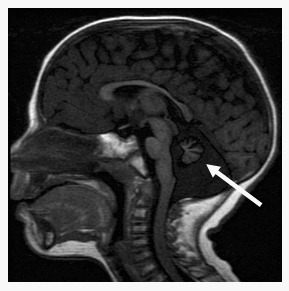
Neurologic. Although most persons with DC/TBD have normal psychomotor development and normal neurologic function, significant developmental delay / intellectual disability is present in Hoyeraal Hreidarsson syndrome and Revesz syndrome. Cerebellar hypoplasia is present in Hoyeraal Hreidarsson syndrome (Figure 2), and intracranial calcifications have been reported in Revesz syndrome and Coats plus syndrome. Microcephaly has also been reported in some persons with DC/TBD, most commonly in Hoyeraal Hreidarsson syndrome and Revesz syndrome.
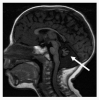
Figure 2.
MRI of cerebellar hypoplasia in an individual with Hoyeraal Hreidarsson syndrome. Arrow indicates the hypoplastic cerebellum.
Psychiatric. Clinically significant psychiatric diagnosis was reported in 27% of 44 individuals with DC/TBD in a recent retrospective study. The psychiatric diagnoses in adults included depression and/or anxiety/panic attacks, autism spectrum disorder, and bipolar disorder [Bhala et al 2019]. However, the true prevalence of psychiatric disorders is unknown.
Endocrine. Hypogonadism has been noted in a small number of severely affected males.
Gastrointestinal. Esophageal stenosis has been reported in several persons with DC/TBD and may worsen over time.
Enteropathy, which may result in poor growth, has been reported, specifically in Hoyeraal Hreidarsson syndrome.
Liver disease, including nodular regenerative hyperplasia, fibrosis, cirrhosis, portal hypertension, and hepatopulmonary syndrome, is increasingly recognized as a potential severe complication of DC/TBD and may lead to liver transplant [Gorgy et al 2015, Kapuria et al 2019, Niewisch et al 2022]. Individuals who have undergone hematopoetic cell transplant (HCT) need close monitoring for liver disease.
Gastrointestinal telangiectasias and bleeding may occur in the context of portal hypertension or independently.
Genitourinary. Urethral stenosis in males may be present at diagnosis or develop over time.
Musculoskeletal. Osteoporosis and osteopenia have been reported. The contribution of prior treatment and comorbid conditions to these complications is not known. Some individuals have reported bone fractures after minor trauma.
Avascular necrosis of the hips and shoulders can result in pain and reduced function. Several individuals have required hip replacement surgery at young ages.
Cardiovascular. Rare reported congenital heart defects include atrial and ventricular septal defects, myocardial fibrosis, and dilated cardiomyopathy.
Hematologic. Bone marrow failure is a common presenting sign, may develop at any age, and may progress over time. Approximately one half of individuals with DC/TBD develop some degree of bone marrow failure by age 40 years.
Individuals with DC/TBD are at increased risk for leukemia (see Cancer).
Immunologic. Immunodeficiency of variable severity has been reported in DC/TBD. It has not been fully characterized, but it appears that some individuals may have reduced numbers of B cells, T cells, and/or NK cells.
Cancer. Persons with DC/TBD are at high risk for leukemia and squamous cell cancer of the head and neck or anogenital region. Elevated risk of cervical squamous cell cancer has also been reported in DC/TBD.
Investigators at the National Cancer Institute analyzed data from 15 years of follow up in a longitudinal cohort study to further quantify cancer risk in individuals with DC/TBD (n=197). The median age of onset for all cancers was 38 years (range 18-63 years), with an O/E (observed deaths to expected deaths) ratio of 4.2 compared to the normal population. The most frequent solid tumors were head and neck squamous cell carcinomas (with an O/E ratio of 74), followed by leukemia, non-Hodgkin lymphoma, and anorectal carcinoma. Of note, the risk of tongue cancer was increased by 216-fold over the normal population. In this study, myelodysplastic syndrome (MDS) was observed at a 578-fold increased risk, and the median age of MDS onset was 31 years (range 4-73 years).
Respiratory. Pulmonary fibrosis may be a presenting sign or may develop over time. It may be more common in individuals who have had HCT. Pulmonary fibrosis is manifest as bibasilar reticular abnormalities, ground glass opacities, or diffuse nodular lesions on high-resolution computed tomography and abnormal pulmonary function studies that include evidence of restriction (reduced vital capacity with an increase in FEV1/FVC ratio) and/or impaired gas exchange (increased P(A-a)O2 with rest or exercise or decreased diffusion capacity of the lung for carbon monoxide).
Pulmonary arteriovenous malformations have recently been reported in individuals with DC/TBD [Khincha et al 2017]. They may be present in individuals with hypoxia in the absence of pulmonary fibrosis and can be diagnosed by bubble echocardiography.
Severe Forms of DC/TBD
Hoyeraal Hreidarsson syndrome, a very severe form of DC/TBD, presents in early childhood [Walne & Dokal 2008]. In addition to features of DC/TBD, cerebellar hypoplasia is required to establish the diagnosis (see Figure 2). The findings in the original cases included cerebellar hypoplasia, developmental delay, immunodeficiency, IUGR, and bone marrow failure, as well as the DC/TBD diagnostic triad [Hoyeraal et al 1970].
Revesz syndrome has many of the features of DC/TBD and presents in early childhood [Revesz et al 1992]. In addition to features of DC/TBD, bilateral exudative retinopathy is required to establish the diagnosis. The individuals in the original reports had intracranial calcifications, IUGR, bone marrow failure, and sparse, fine hair in addition to nail dystrophy and oral leukoplakia.
Coats plus syndrome may include DC/TBD-related mucocutaneous changes, such as dystrophic nails and sparse or graying hair [Anderson et al 2012, Polvi et al 2012]. Additionally, it typically includes bilateral exudative retinopathy, retinal telangiectasias, IUGR, intracranial calcifications, osteopenia with tendency to fracture with poor bone healing, and gastrointestinal vascular ectasias [Linnankivi et al 2006, Briggs et al 2008].
Clinical Findings of X-Linked DC/TBD in Females
Clinical manifestations in females heterozygous for DKC1 pathogenic variants have been reported, including skin pigmentation abnormalities, nail dysplasia, and bone marrow failure [Vulliamy et al 2006, Xu et al 2016, Hirvonen et al 2019].
Phenotype Correlations by Gene
Classic DC with severe bone marrow failure and mucocutaneous triad features is associated with autosomal recessive, X-linked, or TINF2-related autosomal dominant DC/TBD. Significant better overall survival is observed in those with DC/TBD with an underlying autosomal dominant variant (excluding TINF2) versus those with DC associated with other types of variants [Niewisch et al 2022].
Individuals with autosomal recessive or X-linked DC/TBD have been reported to show more neurologic findings than those with autosomal dominant DC/TBD [Bhala et al 2019].
Pulmonary fibrosis without prior HCT is more frequently reported in individuals with autosomal dominant forms of DC/TBD [Armanios et al 2007, Giri et al 2019, Niewisch et al 2022].
Genotype-Phenotype Correlations
Due to the rarity of DC/TBD, no specific genotype-phenotype correlations have been identified.
Penetrance
The penetrance of DC/TBD and DC/TBD-associated medical complications is not well understood. Due to the variability between individuals (even within the same family) and the observation that medical complications may increase with age, penetrance may appear incomplete. Recent studies report somatic TERT promotor variants as possible modifiers to germline pathogenic variants [Maryoung et al 2017, Gutierrez-Rodrigues et al 2019]. However, additional studies are needed to further understand the components leading to variable penetrance of DC/TBD-causing variants.
Nomenclature
Revesz syndrome [Revesz et al 1992], Hoyeraal Hreidarsson syndrome [Hoyeraal et al 1970, Hreidarsson et al 1988], and Coats plus syndrome [Linnankivi et al 2006, Briggs et al 2008], previously thought to be distinct disorders, are now recognized to be part of the phenotypic spectrum of dyskeratosis congenita.
A few case reports of a syndrome of ataxia and pancytopenia are actually describing DC/TBD caused by pathogenic variants in TINF2 [Tsangaris et al 2008].
Prevalence
The prevalence of DC/TBD in the general population is not known; DC/TBD is believed to be rare. As of March 2022, the authors are aware of approximately 800-1,000 affected individuals.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, RPA1, RTEL1, STN1, TERC, TERT, TINF2, WRAP53, or ZCCHC8.
Heterozygous germline pathogenic variants in POT1 are associated with POT1 tumor predisposition (POT1-TDP). POT1-TPD is characterized by an increased lifetime risk for multiple cutaneous melanomas, chronic lymphocytic leukemia, angiosarcoma (particularly cardiac angiosarcomas), and gliomas. The majority of POT1-associated cancers are diagnosed in adulthood.
Differential Diagnosis
Disorders with clinical features that overlap those of dyskeratosis congenita and related telomere biology disorders (DC/TBD) are described below.
Disorders with Nail Dysplasia
Table 3.
Disorders with Nail Dysplasia in the Differential Diagnosis of Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD)
Inherited Bone Marrow Failure Syndromes
Bone marrow failure syndromes are a complex set of related disorders that may have bone marrow failure as the first presenting sign.
Table 4.
Inherited Bone Marrow Failure Syndromes in the Differential Diagnosis of Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD)
Acquired Aplastic Anemia
Characterized by tri-lineage bone marrow cytopenias [Young 2018], acquired aplastic anemia is often progressive and may occur at any age. Telomere length testing helps identify the subset of individuals with later-onset aplastic anemia who have a telomere biology disorder; these individuals may have a few or none of the other clinical findings of DC/TBD. Other known causes of aplastic anemia include an immune process, infection, or drug reaction. In many individuals the cause of acquired aplastic anemia is unknown.
Idiopathic Pulmonary Fibrosis
Idiopathic pulmonary fibrosis (IPF) is the most frequent idiopathic interstitial pneumonia. It results in progressive fibrotic lung disease and has high morbidity and mortality. Persons with DC/TBD may develop IPF and it is conceivable that IPF in a young person could be the first manifestation of DC/TBD; thus, DC/TBD should be considered in young persons with IPF. See Pulmonary Fibrosis Predisposition Overview.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with dyskeratosis congenita (DC) or a related telomere biology disorder (TBD), it is important to note that the clinical spectrum of DC/TCB is broad and signs and symptoms develop at various ages and rates. The evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD)
Treatment of Manifestations
The specific treatment for DC/TBD-related complications must be tailored to the individual. The recommendations in this section were first discussed at a DC clinical research workshop in 2008 and subsequently at a meeting of experts convened to review the publication of the first edition of the Dyskeratosis Congenita and Telomere Biology Disorders: Diagnosis and Management Guidelines [Savage et al 2009, Savage & Cook 2015]. Because of the rarity of DC/TBD, the recommendations are not based on large-scale clinical trials. Affected individuals may have few or many of the complications associated with DC/TBD. Comprehensive coordinated care among specialties is required.
Clinical care guidelines are also available at Team Telomere.
Table 6.
Treatment of Manifestations in Individuals with Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD)
Surveillance
The recommendations in this section were discussed at the first DC clinical research workshop in 2008 and updated in 2014 at a consensus conference that led to publication of the first edition of the Dyskeratosis Congenita and Telomere Biology Disorders: Diagnosis and Management Guidelines [Savage et al 2009, Savage & Cook 2015]. Because of the rarity of DC/TBD, the recommendations are not based on large-scale clinical trials.
Table 7.
Recommended Surveillance for Individuals with Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD)
Agents/Circumstances to Avoid
Blood transfusions
- Transfusions of red cells or platelets should be avoided or minimized for those who are candidates for hematopoietic cell transplantation (HCT).
- To minimize the chances of sensitization, family members must not act as blood donors if HCT is being considered.
- All blood products should be leukodepleted and irradiated.
Radiation. It is prudent to minimize exposure to therapeutic radiation as data on radiation side effects are limited.
Androgens and growth factors. The combination of androgens and granulocyte colony-stimulating factor was associated with splenic peliosis ("blood lakes") and rupture in two individuals; thus, the combination should be avoided [Giri et al 2007].
Cancer prevention. Given the increased susceptibility of individuals with DC/TBD to developing leukemias and other malignancies, individuals with DC/TBD are advised to avoid toxic agents that have been implicated in tumorigenesis (including smoking cigarettes or drinking alcohol), to use sunscreen regularly, and to avoid excessive sun exposure.
Evaluation of Relatives at Risk
For early diagnosis and treatment. It is appropriate to evaluate apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those who would benefit from preventive measures, hematologic surveillance, and prompt initiation of treatment.
Evaluations include:
- Telomere length testing;
- Molecular genetic testing if a molecular diagnosis has been established in the proband.
For hematopoietic cell transplantation (HCT) donor evaluation. Any relative who is a potential HCT donor should undergo telomere length testing or molecular genetic testing (if the pathogenic variant[s] in the family are known).
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Individuals with DC/TBD who become pregnant may develop pancytopenia or existing cytopenias may worsen. They should be followed closely by a perinatologist.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
The mode of inheritance of dyskeratosis congenita and related telomere biology disorders (DC/TBD) depends on the associated gene (see Table 8).
Table 8.
Dyskeratosis Congenita and Related Telomere Biology Disorders (DC/TBD): Mode of Inheritance by Gene
Note: If the causative pathogenic variant(s) have not been identified in an affected family member, telomere length testing of first-degree relatives is recommended (see Management, Evaluation of Relatives at Risk).
X-Linked Inheritance – Risk to Family Members
Parents of a male proband
- The father of an affected male will not have the disorder nor will he be hemizygous for the DKC1 pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected male, the mother of an affected male is an obligate heterozygote. If a woman has more than one affected child and the DKC1 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote, the affected male may have a de novo pathogenic variant (in which case the mother is not a heterozygote), or the mother may have somatic/germline mosaicism.
- Molecular genetic testing of the mother is recommended to confirm her genetic status and to allow reliable recurrence risk assessment.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband has a pathogenic variant, the chance of transmitting it in each pregnancy is 50%:
- Males who inherit the pathogenic variant will be affected.
- Females who inherit the pathogenic variant will be heterozygotes. Clinical manifestations in females heterozygous for DKC1 pathogenic variants have been reported, including skin pigmentation abnormalities, nail dysplasia, and bone marrow failure [Vulliamy et al 2006, Xu et al 2016, Hirvonen et al 2019].
- If the proband represents a simplex case and if the DKC1 pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is presumed to be low but greater than that of the general population because of the possibility of maternal germline mosaicism.
Offspring of a male proband. Affected males transmit the DKC1 pathogenic variant to:
- All their daughters, who will be heterozygotes;
- None of their sons.
Other family members. The maternal aunts and maternal cousins of a male proband may be at risk of having a DKC1 pathogenic variant.
Heterozygote detection. Heterozygote testing for at-risk female relatives requires prior identification of the DKC1 pathogenic variant in the family.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
- Some individuals diagnosed with autosomal dominant DC/TBD have an affected parent.
- A proband with autosomal dominant DC/TBD may have the disorder as the result of a de novo DC/TBD-causing pathogenic variant. With the exception of TINF2 – in which the majority of pathogenic variants appear to occur de novo in the proband [Savage et al 2008, Walne et al 2008, Sasa et al 2012] – the proportion of individuals with DC/TBD caused by a de novo pathogenic variant is unknown.
- If the causative pathogenic variant has been identified in the proband and the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling. If the causative pathogenic variant has not been identified in the proband, telomere length testing of the parents is recommended.
- If the proband has a known pathogenic variant that cannot be detected in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only. Walne et al [2008] reported a family with two affected sibs, in one of whom a TINF2 pathogenic variant was identified (the other was deceased and could not be tested); neither parent had the pathogenic variant, suggesting germline mosaicism in a parent.
- The proband inherited a DC/TBD-causing pathogenic variant from a parent with somatically acquired loss of heterozygosity with preferential loss of the chromosome with the pathogenic variant. This scenario may cause a false negative molecular result when testing leukocyte DNA. Note: To date, this has only been observed in individuals with germline TERC or RPA1 pathogenic variants.
- The family history of some individuals diagnosed with autosomal dominant DC/TBD may appear to be negative because of a milder phenotypic presentation. Therefore, an apparently negative family history cannot be confirmed without molecular genetic testing (to establish that neither parent is heterozygous for the pathogenic variant identified in the proband).
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. The age of onset and manifestations of DC/TBD may vary considerably among heterozygous family members (see Penetrance).
- If the DC/TBD-causing pathogenic variant found in the proband cannot be detected in the DNA of either parent, the recurrence risk to sibs is slightly greater than that of the general population because of:
- The possibility of parental germline mosaicism [Walne et al 2008]; or
- A false negative result in a parent due to preferential loss of the chromosome with the DC/TBD-causing pathogenic variant. Note: Revertant mosaicism (i.e., loss of heterozygosity for the deleterious allele) in peripheral blood cells has only been observed in individuals with germline TERC or RPA1 pathogenic variants.
- If the parents are clinically unaffected but they have not undergone molecular genetic testing or telomere testing, sibs are presumed to be at increased risk for DC/TBD for one of two possible reasons:
- A parent has germline mosaicism; or
- A parent is heterozygous but does not have apparent manifestations of DC/TBD because of phenotypic modification resulting from additional genetic events that confer a protective effect.
Offspring of a proband. Each child of an individual with autosomal dominant DC/TBD has a 50% chance of inheriting the DC/TBD-causing pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, his or her family members may be at risk of having the DC/TBD-causing pathogenic variant and the associated clinical manifestations including leukemia and squamous cell cancer (see Clinical Description).
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for one DC/TBD-causing pathogenic variant.
- If the causative pathogenic variants have been identified in the proband, molecular genetic testing is recommended for the parents of the proband to confirm that both parents are heterozygous for a DC/TBD-causing pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes may or not may not be affected.
- Pathogenic variants in ACD, PARN, RTEL, or TERT can cause both autosomal dominant and autosomal recessive DC/TBD; the effect of heterozygosity for one ACD, PARN, RTEL, or TERT pathogenic variant in individuals from families in which DC/TBD has been inherited in an autosomal recessive manner has not been thoroughly studied, but these individuals may be at elevated risk of DC/TBD-related complications.
- Heterozygotes for a pathogenic variant in CTC1, NHP2, NOP10, PARN, POT1, or WRAP53 are predicted to be asymptomatic for DC/TBD manifestations. Individuals who are heterozygous for a POT1 pathogenic variant should be evaluated for POT1 tumor predisposition.
Sibs of a proband
- If both parents are known to be heterozygous for a DC/TBD-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting two DC/TBD-causing pathogenic variants, a 50% chance of inheriting one pathogenic variant, and a 25% chance of inheriting neither of the familial DC/TBD-causing pathogenic variants.
- The age of onset and manifestations of DC/TBD may vary considerably among family members with biallelic pathogenic variants.
- Heterozygotes may or not may not be affected.
- Pathogenic variants in ACD, PARN, RTEL, or TERT can cause both autosomal dominant and autosomal recessive DC/TBD; the effect of heterozygosity for one ACD, PARN, RTEL, or TERT pathogenic variant in individuals from families in which DC/TBD has been inherited in an autosomal recessive manner has not been thoroughly studied but these individuals may be at elevated risk of DC/TBD-related complications.
- Heterozygotes for a pathogenic variant in CTC1, NHP2, NOP10, PARN, POT1, or WRAP53 are predicted to be asymptomatic for DC/TBD manifestations. Individuals who are heterozygous for a POT1 pathogenic variant should be evaluated for POT1 tumor predisposition.
Offspring of a proband. The offspring of an individual with autosomal recessive DC/TBD are obligate heterozygotes for a DC/TBD-related pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being heterozygous for a DC/TBD-related pathogenic variant.
Heterozygote detection. Heterozygote testing for at-risk relatives requires prior identification of the DC/TBD-related pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis, treatment, and evaluation as a potential hematopoietic cell transplantation donor.
Predictive testing for at-risk asymptomatic family members requires prior identification of the pathogenic variant(s) in the family or, if the causative pathogenic variant(s) in the family are unknown, documentation of short telomere length in an affected relative.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are heterozygous, or are at risk of being heterozygous.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the DC/TBD-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- DC ActionUnited KingdomEmail: info@dcaction.org
- Team Telomere1562 First Avenue #205-4093New York NY 10028-4004Email: info@teamtelomere.org
- National Cancer Institute Inherited Bone Marrow Failure Syndromes (IBMFS) Cohort RegistryPhone: 800-518-8474Email: NCI.IBMFS@westat.com
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Dyskeratosis Congenita and Related Telomere Biology Disorders: Genes and Databases
Table B.
OMIM Entries for Dyskeratosis Congenita and Related Telomere Biology Disorders (View All in OMIM)
Molecular Pathogenesis
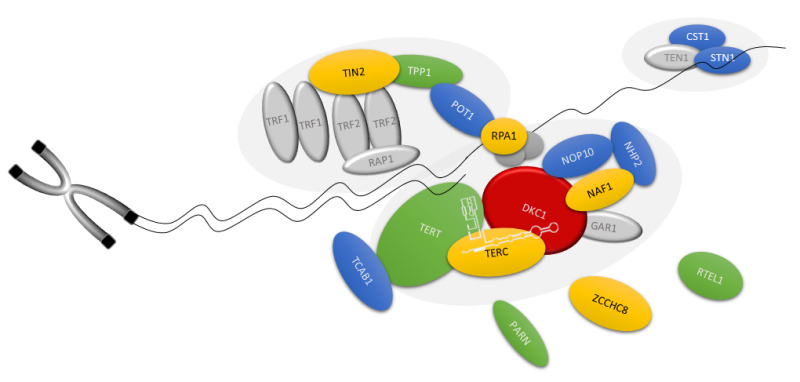
Dyskeratosis congenita and related telomere biology disorders (DC/TBD) are caused by impaired telomere maintenance resulting in short or very short telomeres. The telomere is a complex structure consisting of long nucleotide repeats (TTAGGG)n and a protein complex at chromosome ends that are essential to chromosomal integrity. The TTAGGG nucleotide repeats at the chromosome end fold back to create a T-loop. Many proteins bind to the T-loop and others bind to those proteins to form a stable telomere "cap." Pathogenic variants in 16 different genes (ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, POT1, RPA1, RTEL1, STN1, TERC, TERT, TINF2, WRAP53, and ZCCHC8) encoding critical components of the telomere have been found in individuals with DC/TBD (Figure 3).

Figure 3.
Schematic of the telomere and the proteins affected in dyskeratosis congenita and related telomere biology disorders Colors refer to reported inheritance pattern in the coding genes of depicted proteins:
Mechanism of disease causation. DC/TBD are caused by a loss-of-function mechanism [Bertuch 2016].
Notable variants by gene. See Table 9.
Note: The Telomerase Database, which is maintained by the Arizona State University, is a widely used tool in the telomere research community. Specifically, it includes a collection of variants published in the context of DC/TBD that may be useful in interpreting detected variants in individuals with suspected DC/TBD [Podlevsky et al 2008].
Table 9.
DC/TBD: Notable Pathogenic Variants by Gene
Chapter Notes
Author Notes
Website: marrowfailure.cancer.gov
Acknowledgments
Drs Blanche Alter, Neelam Giri, and Lisa McReynolds, NCI, contributed invaluable advice and insight into patient diagnosis and management.
This work was supported (in part) by the intramural research program of the National Cancer Institute, National Institutes of Health.
Revision History
- 19 January 2023 (sw/sas) Revision: updated number of families with pathogenic variants in WRAP53 detected by sequence analysis (see Table 1)
- 31 March 2022 (ha) Comprehensive update posted live
- 21 November 2019 (aa) Revision: added mode of inheritance and OMIM links for idiopathic pulmonary fibrosis
- 26 May 2016 (ha) Comprehensive update posted live
- 3 January 2013 (cd) Revision: mutations in CTC1 found to cause dyskeratosis congenita
- 13 September 2012 (cd) Revision: sequence analysis of the entire coding regions of NHP2 and NOP10 available clinically
- 10 May 2012 (me) Comprehensive update posted live
- 12 November 2009 (me) Review posted live
- 15 May 2009 (sas) Original submission
Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Dyskeratosis Congenita and Related Telomere Biology Disorders" is in the public domain in the United States of America.
References
Literature Cited
- Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica. 2018;103:30–9. [PMC free article: PMC5777188] [PubMed: 29051281]
- Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97:353–9. [PMC free article: PMC3291588] [PubMed: 22058220]
- Anderson BH, Kasher PR, Mayer J, Szynkiewicz M, Jenkinson EM, Bhaskar SS, Urquhart JE, Daly SB, Dickerson JE, O'Sullivan J, Leibundgut EO, Muter J, Abdel-Salem GM, Babul-Hirji R, Baxter P, Berger A, Bonafé L, Brunstom-Hernandez JE, Buckard JA, Chitayat D, Chong WK, Cordelli DM, Ferreira P, Fluss J, Forrest EH, Franzoni E, Garone C, Hammans SR, Houge G, Hughes I, Jacquemont S, Jeannet PY, Jefferson RJ, Kumar R, Kutschke G, Lundberg S, Lourenço CM, Mehta R, Naidu S, Nischal KK, Nunes L, Ounap K, Philippart M, Prabhakar P, Risen SR, Schiffmann R, Soh C, Stephenson JB, Stewart H, Stone J, Tolmie JL, van der Knaap MS, Vieira JP, Vilain CN, Wakeling EL, Wermenbol V, Whitney A, Lovell SC, Meyer S, Livingston JH, Baerlocher GM, Black GC, Rice GI, Crow YJ. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet. 2012;44:338–42. [PubMed: 22267198]
- Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, Lawson WE, Xie M, Vulto I, Phillips JA III, Lansdorp PM, Greider CW, Loyd JE. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356:1317–26. [PubMed: 17392301]
- Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2013;2013;6:327–37. [PubMed: 23782086]
- Balogh E, Chandler JC, Varga M, Tahoun M, Menyhárd DK, Schay G, Goncalves T, Hamar R, Légrádi R, Szekeres Á, Gribouval O, Kleta R, Stanescu H, Bockenhauer D, Kerti A, Williams H, Kinsler V, Di WL, Curtis D, Kolatsi-Joannou M, Hammid H, Szőcs A, Perczel K, Maka E, Toldi G, Sava F, Arrondel C, Kardos M, Fintha A, Hossain A, D'Arco F, Kaliakatsos M, Koeglmeier J, Mifsud W, Moosajee M, Faro A, Jávorszky E, Rudas G, Saied MH, Marzouk S, Kelen K, Götze J, Reusz G, Tulassay T, Dragon F, Mollet G, Motameny S, Thiele H, Dorval G, Nürnberg P, Perczel A, Szabó AJ, Long DA, Tomita K, Antignac C, Waters AM, Tory K. Pseudouridylation defect due to DKC1 and NOP10 mutations causes nephrotic syndrome with cataracts, hearing impairment, and enterocolitis. Proc Natl Acad Sci U S A. 2020;117:15137–47. [PMC free article: PMC7334496] [PubMed: 32554502]
- Belaya Z, Golounina O, Nikitin A, Tarbaeva N, Pigarova E, Mamedova E, Vorontsova M, Shafieva I, Demina I, Van Hul W. Multiple bilateral hip fractures in a patient with dyskeratosis congenita caused by a novel mutation in the PARN gene. Osteoporos Int. 2021;32:1227–31. [PubMed: 33244623]
- Benyelles M, O'Donohue MF, Kermasson L, Lainey E, Borie R, Lagresle-Peyrou C, Nunes H, Cazelles C, Fourrage C, Ollivier E, Marcais A, Gamez AS, Morice-Picard F, Caillaud D, Pottier N, Ménard C, Ba I, Fernandes A, Crestani B, de Villartay JP, Gleizes PE, Callebaut I, Kannengiesser C, Revy P. NHP2 deficiency impairs rRNA biogenesis and causes pulmonary fibrosis and Høyeraal-Hreidarsson syndrome. Hum Mol Genet. 2020;29:907–22. [PubMed: 31985013]
- Bergstrand S, Böhm S, Malmgren H, Norberg A, Sundin M, Nordgren A, Farnebo M. Biallelic mutations in WRAP53 result in dysfunctional telomeres, Cajal bodies and DNA repair, thereby causing Hoyeraal-Hreidarsson syndrome. Cell Death Dis. 2020;11:238. [PMC free article: PMC7165179] [PubMed: 32303682]
- Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol. 2016;13:696–706. [PMC free article: PMC4993306] [PubMed: 26400640]
- Bhala S, Best AF, Giri N, Alter BP, Pao M, Gropman A, Baker EH, Savage SA. CNS manifestations in patients with telomere biology disorders. Neurol Genet. 2019;5:370. [PMC free article: PMC6878838] [PubMed: 31872047]
- Bonfim C. Special pre- and posttransplant considerations in inherited bone marrow failure and hematopoietic malignancy predisposition syndromes. Hematology Am Soc Hematol Educ Program. 2020;2020:107–14. [PMC free article: PMC7727534] [PubMed: 33275667]
- Brailovski E, Tsui H, Chen YB, Velsher L, Liu J, Buckstein R. Previously unreported WRAP53 gene variants in a patient with dyskeratosis congenita. Ann Hematol. 2022;101:907–9. [PubMed: 34599657]
- Briggs TA, Abdel-Salam GM, Balicki M, Baxter P, Bertini E, Bishop N, Browne BH, Chitayat D, Chong WK, Eid MM, Halliday W, Hughes I, Klusmann-Koy A, Kurian M, Nischal KK, Rice GI, Stephenson JB, Surtees R, Talbot JF, Tehrani NN, Tolmie JL, Toomes C, van der Knaap MS, Crow YJ. Cerebroretinal microangiopathy with calcifications and cysts (CRMCC). Am J Med Genet A. 2008;146A:182–90. [PubMed: 18076099]
- Burris AM, Ballew BJ, Kentosh JB, Turner CE, Norton SA, Giri N, Alter BP, Nellan A, Gamper C, Hartman KR, Savage SA, et al. Hoyeraal-Hreidarsson syndrome due to PARN mutations: fourteen years of follow-up. Pediatr Neurol. 2016;56:62–8.e1. [PMC free article: PMC4789174] [PubMed: 26810774]
- Denny CC, Wilfond BS, Peters JA, Giri N, Alter BP. All in the family: disclosure of "unwanted" information to an adolescent to benefit a relative. Am J Med Genet A. 2008;146A:2719–24. [PMC free article: PMC3143002] [PubMed: 18831063]
- Dodson LM, Baldan A, Nissbeck M, Gunja SMR, Bonnen PE, Aubert G, Birchansky S, Virtanen A, Bertuch AA. From incomplete penetrance with normal telomere length to severe disease and telomere shortening in a family with monoallelic and biallelic PARN pathogenic variants. Hum Mutat. 2019;40:2414–29. [PMC free article: PMC6874886] [PubMed: 31448843]
- Dokal I. Dyskeratosis congenita. Hematology Am Soc Hematol Educ Program. 2011;2011:480–6. [PubMed: 22160078]
- Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: dyskeratosis congenita - update 2015. Eur J Hum Genet. 2015:23. [PMC free article: PMC4667501] [PubMed: 25182133]
- Eiler ME, Frohnmayer D, Frohnmayer L, Larsen K, Owen J, eds. Fanconi Anemia: Guidelines for Diagnosis and Management. 3 ed. Fanconi Anemia Research Fund, Inc. Available online. 2008. Accessed 1-13-23.
- Fioredda F, Iacobelli S, Korthof ET, Knol C, van Biezen A, Bresters D, Veys P, Yoshimi A, Fagioli F, Mats B, Zecca M, Faraci M, Miano M, Arcuri L, Maschan M, O'Brien T, Diaz MA, Sevilla J, Smith O, Peffault de Latour R, de la Fuente J, Or R, Van Lint MT, Tolar J, Aljurf M, Fisher A, Skorobogatova EV, Diaz de Heredia C, Risitano A, Dalle JH, Sedláček P, Ghavamzadeh A, Dufour C. Outcome of haematopoietic stem cell transplantation in dyskeratosis congenita. Br J Haematol. 2018;183:110–18. [PubMed: 29984823]
- Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, Read EJ, Lansdorp PM, Young NS. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet. 2003;362:1628–30. [PubMed: 14630445]
- Gable DL, Gaysinskaya V, Atik CC, Talbot CC Jr, Kang B, Stanley SE, Pugh EW, Amat-Codina N, Schenk KM, Arcasoy MO, Brayton C, Florea L, Armanios M. ZCCHC8, the nuclear exosome targeting component, is mutated in familial pulmonary fibrosis and is required for telomerase RNA maturation. Genes Dev. 2019;33:1381–96. [PMC free article: PMC6771387] [PubMed: 31488579]
- Giri N, Pitel PA, Green D, Alter BP. Splenic peliosis and rupture in patients with dyskeratosis congenita on androgens and granulocyte colony-stimulating factor. Br J Haematol. 2007;138:815–17. [PubMed: 17760812]
- Giri N, Ravichandran S, Wang Y, Gadalla SM, Alter BP, Fontana J, Savage SA. Prognostic significance of pulmonary function tests in dyskeratosis congenita, a telomere biology disorder. ERJ Open Res. 2019;5:00209–2019. [PMC free article: PMC6856494] [PubMed: 31754622]
- Glousker G, Touzot F, Revy P, Tzfati Y, Savage SA. Unraveling the pathogenesis of Hoyeraal-Hreidarsson syndrome, a complex telomere biology disorder. Br J Haematol. 2015;170:457–71. [PMC free article: PMC4526362] [PubMed: 25940403]
- Gorgy AI, Jonassaint NL, Stanley SE, Koteish A, DeZern AE, Walter JE, Sopha SC, Hamilton JP, Hoover-Fong J, Chen AR, Anders RA, Kamel IR, Armanios M. Hepatopulmonary syndrome is a frequent cause of dyspnea in the short telomere disorders. Chest. 2015;148:1019–26. [PMC free article: PMC4594621] [PubMed: 26158642]
- Guo Y, Kartawinata M, Li J, Pickett HA, Teo J, Kilo T, Barbaro PM, Keating B, Chen Y, Tian L, Al-Odaib A, Reddel RR, Christodoulou J, Xu X, Hakonarson H, Bryan TM. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014;124:2767–74. [PMC free article: PMC4215308] [PubMed: 25205116]
- Gutierrez-Rodrigues F, Donaires FS, Pinto A, Vicente A, Dillon LW, Clé DV, Santana BA, Pirooznia M, Ibanez MDPF, Townsley DM, Kajigaya S, Hourigan CS, Cooper JN, Calado RT, Young NS. Pathogenic TERT promoter variants in telomere diseases. Genet Med. 2019;21:1594–602. [PMC free article: PMC6555700] [PubMed: 30523342]
- Higgs C, Crow YJ, Adams DM, Chang E, Hayes D Jr, Herbig U, Huang JN, Himes R, Jajoo K, Johnson FB, Reynolds SD, Yonekawa Y, Armanios M, Boulad F, DiNardo CD, Dufour C, Goldman FD, Khan S, Kratz C, Myers KC, Raghu G, Alter BP, Aubert G, Bhala S, Cowen EW, Dror Y, El-Youssef M, Friedman B, Giri N, Helms Guba L, Khincha PP, Lin TF, Longhurst H, McReynolds LJ, Nelson A, Olson T, Pariser A, Perona R, Sasa G, Schratz K, Simonetto DA, Townsley D, Walsh M, Stevens K, Agarwal S, Bertuch AA, Savage SA, et al. Understanding the evolving phenotype of vascular complications in telomere biology disorders. Angiogenesis. 2019;22:95–102. [PubMed: 30168024]
- Hirvonen EAM, Peuhkuri S, Norberg A, Degerman S, Hannula-Jouppi K, Välimaa H, Kilpivaara O, Wartiovaara-Kautto U. Characterization of an X-chromosome-linked telomere biology disorder in females with DKC1 mutation. Leukemia. 2019;33:275–8. [PubMed: 30185935]
- Hoyeraal HM, Lamvik J, Moe PJ. Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr Scand. 1970;59:185–91. [PubMed: 5442429]
- Hreidarsson S, Kristjansson K, Johannesson G, Johannsson JH. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatr Scand. 1988;77:773–5. [PubMed: 3201986]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jongmans MC, Verwiel ET, Heijdra Y, Vulliamy T, Kamping EJ, Hehir-Kwa JY, Bongers EM, Pfundt R, van Emst L, van Leeuwen FN, van Gassen KL, Geurts van Kessel A, Dokal I, Hoogerbrugge N, Ligtenberg MJ, Kuiper RP. Revertant somatic mosaicism by mitotic recombination in dyskeratosis congenita. Am J Hum Genet. 2012;90:426–33. [PMC free article: PMC3309184] [PubMed: 22341970]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Kapuria D, Ben-Yakov G, Ortolano R, Cho MH, Kalchiem-Dekel O, Takyar V, Lingala S, Gara N, Tana M, Kim YJ, Kleiner DE, Young NS, Townsley DM, Koh C, Heller T. The spectrum of hepatic involvement in patients with telomere disease. Hepatology. 2019;69:2579–85. [PMC free article: PMC7440774] [PubMed: 30791107]
- Khincha PP, Bertuch AA, Agarwal S, Townsley DM, Young NS, Keel S, Shimamura A, Boulad F, Simoneau T, Justino H, Kuo C, Artandi S, McCaslin C, Cox DW, Chaffee S, Collins BF, Giri N, Alter BP, Raghu G, Savage SA. Pulmonary arteriovenous malformations: an uncharacterised phenotype of dyskeratosis congenita and related telomere biology disorders. Eur Respir J. 2017;49:1601640. [PMC free article: PMC5841586] [PubMed: 27824607]
- Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA. Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol. 2014;165:349–57. [PMC free article: PMC3984599] [PubMed: 24666134]
- Kocak H, Ballew BJ, Bisht K, Eggebeen R, Hicks BD, Suman S, O'Neil A, Giri N, Maillard I, Alter BP, Keegan CE, Nandakumar J, Savage SA, et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014;28:2090–102. [PMC free article: PMC4180972] [PubMed: 25233904]
- Linnankivi T, Valanne L, Paetau A, Alafuzoff I, Hakumäki JM, Kivelä T, Lönnqvist T, Mäkitie O, Pääkkönen L, Vainionpää L, Vanninen R, Herva R, Pihko H. Cerebroretinal microangiopathy with calcifications and cysts. Neurology. 2006;67:1437–43. [PubMed: 16943371]
- Maryoung L, Yue Y, Young A, Newton CA, Barba C, van Oers NS, Wang RC, Garcia CK. Somatic mutations in telomerase promoter counterbalance germline loss-of-function mutations. J Clin Invest. 2017;127:982–6. [PMC free article: PMC5330735] [PubMed: 28192371]
- Moon DH, Segal M, Boyraz B, Guinan E, Hofmann I, Cahan P, Tai AK, Agarwal S. Poly(A)-specific ribonuclease (PARN) mediates 3'-end maturation of the telomerase RNA component. Nat Genet. 2015;47:1482–8. [PMC free article: PMC4791094] [PubMed: 26482878]
- Niewisch MR, Giri N, McReynolds LJ, Alsaggaf R, Bhala S, Alter BP, Savage SA. Disease progression and clinical outcomes in telomere biology disorders. Blood. 2022;139:1807–19. [PMC free article: PMC8952184] [PubMed: 34852175]
- Niewisch MR, Savage SA. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2019;12:1037–52. [PMC free article: PMC9400112] [PubMed: 31478401]
- Podlevsky JD, Bley CJ, Omana RV, Qi X, Chen JJ-L. The telomerase database. Nucleic Acids Res. 2008;36:D339–D343. [PMC free article: PMC2238860] [PubMed: 18073191]
- Polvi A, Linnankivi T, Kivelä T, Herva R, Keating JP, Mäkitie O, Pareyson D, Vainionpää L, Lahtinen J, Hovatta I, Pihko H, Lehesjoki AE. Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet. 2012;90:540–9. [PMC free article: PMC3309194] [PubMed: 22387016]
- Revesz T, Fletcher S, al Gazali LI, DeBuse P. Bilateral retinopathy, aplastic anaemia, and central nervous system abnormalities: a new syndrome? J Med Genet. 1992;29:673–5. [PMC free article: PMC1016105] [PubMed: 1404302]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Sasa GS, Ribes-Zamora A, Nelson ND, Bertuch AA. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin Genet. 2012;81:470–8. [PMC free article: PMC3844870] [PubMed: 21477109]
- Savage SA, Bertuch AA. The genetics and clinical manifestations of telomere biology disorders. Genet Med. 2010;12:753–64. [PMC free article: PMC3825100] [PubMed: 21189492]
- Savage SA, Cook EF, eds. Dyskeratosis Congenita and Telomere Biology Disorders: Diagnosis and Management Guidelines. Dyskeratosis Congenita Outreach, Inc. Available online. 2015. Accessed 1-13-23.
- Savage SA, Dokal I, Armanios M, Aubert G, Cowen EW, Domingo DL, Giri N, Greene MH, Orchard PJ, Tolar J, Tsilou E, Van Waes C, Wong JM, Young NS, Alter BP. Dyskeratosis congenita: the first NIH clinical research workshop. Pediatr Blood Cancer. 2009;53:520–3. [PMC free article: PMC2739803] [PubMed: 19415736]
- Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008;82:501–9. [PMC free article: PMC2427222] [PubMed: 18252230]
- Shao Y, Feng S, Huang J, Huo J, You Y, Zheng Y. A unique homozygous WRAP53 Arg298Trp mutation underlies dyskeratosis congenita in a Chinese Han family. BMC Med Genet. 2018;19:40. [PMC free article: PMC5842585] [PubMed: 29514627]
- Sharma R, Sahoo SS, Honda M, Granger SL, Goodings C, Sanchez L, Kunstner A, Busch H, Beier F, Pruett-Miller SM, Valentine MB, Fernandez AG, Chang TC, Geli V, Churikov D, Hirschi S, Pastor VB, Boerries M, Lauten M, Kelaidi C, Cooper MA, Nicholas S, Rosenfeld JA, Polychronopoulou S, Kannengiesser C, Saintome C, Niemeyer CM, Revy P, Wold MS, Spies M, Erlacher M, Coulon S, Wlodarski MW. Gain-of-function mutations in RPA1 cause a syndrome with short telomeres and somatic genetic rescue. Blood. 2022;139:1039–51. [PMC free article: PMC8854676] [PubMed: 34767620]
- Shi L, Webb BD, Birch AH, Elkhoury L, McCarthy J, Cai X, Oishi K, Mehta L, Diaz GA, Edelmann L, Kornreich R. Comprehensive population screening in the Ashkenazi Jewish population for recurrent disease-causing variants. Clin Genet. 2017;91:599–604. [PMC free article: PMC5237408] [PubMed: 27415407]
- Simon AJ, Lev A, Zhang Y, Weiss B, Rylova A, Eyal E, Kol N, Barel O, Cesarkas K, Soudack M, Greenberg-Kushnir N, Rhodes M, Wiest DL, Schiby G, Barshack I, Katz S, Pras E, Poran H, Reznik-Wolf H, Ribakovsky E, Simon C, Hazou W, Sidi Y, Lahad A, Katzir H, Sagie S, Aqeilan HA, Glousker G, Amariglio N, Tzfati Y, Selig S, Rechavi G, Somech R. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. 2016;213:1429–40. [PMC free article: PMC4986528] [PubMed: 27432940]
- Stanley SE, Gable DL, Wagner CL, Carlile TM, Hanumanthu VS, Podlevsky JD, Khalil SE, DeZern AE, Rojas-Duran MF, Applegate CD, Alder JK, Parry EM, Gilbert WV, Armanios M. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8:351ra107. [PMC free article: PMC5351811] [PubMed: 27510903]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Takai H, Jenkinson E, Kabir S, Babul-Hirji R, Najm-Tehrani N, Chitayat DA, Crow YJ, de Lange T. A. POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes Dev. 2016;30:812–26. [PMC free article: PMC4826397] [PubMed: 27013236]
- Townsley DM, Dumitriu B, Young NS. Danazol treatment for telomere diseases. N Engl J Med. 2016;375:1095–6. [PubMed: 27626528]
- Tsangaris E, Adams SL, Yoon G, Chitayat D, Lansdorp P, Dokal I, Dror Y. Ataxia and pancytopenia caused by a mutation in TINF2. Hum Genet. 2008;124:507–13. [PubMed: 18979121]
- Tummala H, Walne A, Collopy L, Cardoso S, de la Fuente J, Lawson S, Powell J, Cooper N, Foster A, Mohammed S, Plagnol V, Vulliamy T, Dokal I. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125:2151–60. [PMC free article: PMC4463202] [PubMed: 25893599]
- van der Vis JJ, van der Smagt JJ, Hennekam FAM, Grutters JC, van Moorsel CHM. Pulmonary fibrosis and a TERT founder mutation with a latency period of 300 years. Chest. 2020;158:612–9. [PubMed: 32315675]
- Velazquez I, Alter BP. Androgens and liver tumors: Fanconi's anemia and non-Fanconi's conditions. Am J Hematol. 2004;77:257–67. [PubMed: 15495253]
- Vulliamy T, Beswick R, Kirwan M, Marrone A, Digweed M, Walne A, Dokal I. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc Natl Acad Sci USA. 2008;105:8073–8. [PMC free article: PMC2430361] [PubMed: 18523010]
- Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006;107:2680–5. [PubMed: 16332973]
- Walne AJ, Dokal I. Dyskeratosis congenita: a historical perspective. Mech Ageing Dev. 2008;129:48–59. [PubMed: 18054794]
- Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–600. [PMC free article: PMC2572788] [PubMed: 18669893]
- Walne AJ, Vulliamy T, Marrone A, Beswick R, Kirwan M, Masunari Y, Al Qurashi FH, Aljurf M, Dokal I. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16:1619–29. [PMC free article: PMC2882227] [PubMed: 17507419]
- Ward SC, Savage SA, Giri N, Alter BP, Rosenberg PS, Pichard DC, Cowen EW. Beyond the triad: Inheritance, mucocutaneous phenotype, and mortality in a cohort of patients with dyskeratosis congenita. J Am Acad Dermatol. 2018;78:804–6. [PMC free article: PMC5857208] [PubMed: 29042228]
- Xu J, Khincha PP, Giri N, Alter BP, Savage SA, Wong JM. Investigation of chromosome X inactivation and clinical phenotypes in female carriers of DKC1 mutations. Am J Hematol. 2016;91:1215–20. [PMC free article: PMC7466628] [PubMed: 27570172]
- Young NS. Aplastic anemia. N Engl J Med. 2018;379:1643–56. [PMC free article: PMC6467577] [PubMed: 30354958]
- Zeng T, Lv G, Chen X, Yang L, Zhou L, Dou Y, Tang X, Yang J, An Y, Zhao X. CD8+ T-cell senescence and skewed lymphocyte subsets in young dyskeratosis congenita patients with PARN and DKC1 mutations. J Clin Lab Anal. 2020;34:e23375. [PMC free article: PMC7521304] [PubMed: 32452087]
- Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, Chen R, Alter BP, Artandi SE. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011;25:11–16. [PMC free article: PMC3012932] [PubMed: 21205863]
Publication Details
Author Information and Affiliations
Clinical Director, Division of Cancer Epidemiology and Genetics
National Cancer Institute
National Institutes of Health
Bethesda, Maryland
Division of Cancer Epidemiology and Genetics
National Cancer Institute
National Institutes of Health
Bethesda, Maryland
Hannover Medical School
Hannover, Germany
Publication History
Initial Posting: November 12, 2009; Last Revision: January 19, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Savage SA, Niewisch MR. Dyskeratosis Congenita and Related Telomere Biology Disorders. 2009 Nov 12 [Updated 2023 Jan 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.