Summary
Clinical characteristics.
Donnai-Barrow syndrome (DBS) is characterized by typical craniofacial features (large anterior fontanelle, wide metopic suture, widow's peak, markedly widely spaced eyes, enlarged globes, downslanted palpebral fissures, posteriorly rotated ears, depressed nasal bridge, and short nose. Ocular complications include high myopia, retinal detachment, retinal dystrophy, and progressive vision loss. Additional common features include agenesis of the corpus callosum, sensorineural hearing loss, intellectual disability, and congenital diaphragmatic hernia and/or omphalocele. Both inter- and intrafamilial phenotypic variability are observed.
Diagnosis/testing.
The diagnosis of DBS is established in a proband with: the characteristic clinical features and a distinctive pattern of low-molecular-weight proteinuria; and/or biallelic pathogenic variants in LRP2 identified by molecular genetic testing.
Management.
Treatment of manifestations: Surgical repair of diaphragmatic hernia and/or omphalocele; corrective lenses for myopia; preventive treatments for retinal detachment; hearing aids and/or cochlear implants for hearing loss; antiepileptic drugs for seizures; supplementation as needed for low serum vitamins A and D; education tailored to degree of intellectual, visual, and hearing abilities.
Surveillance: Ophthalmologic surveillance to monitor for retinal detachment; serial audiologic examinations; serial measurement of renal function including blood urea nitrogen and serum creatinine concentrations, urinalysis, and serum vitamin A and D; monitor developmental progress and educational needs.
Genetic counseling.
DBS is inherited in an autosomal recessive manner. In general, the parents of an affected child are obligate heterozygotes with each carrying one pathogenic variant; one instance of uniparental disomy has been reported. When both parents are known to be carriers of a pathogenic variant, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. If the pathogenic variants in the family are known, carrier testing for at-risk relatives and prenatal testing of a pregnancy at increased risk are possible.
Diagnosis
Suggestive Findings
Donnai-Barrow syndrome (DBS) should be suspected in individuals with the following clinical and radiographic features. No single clinical feature is pathognomonic for DBS, nor have diagnostic criteria been formalized.
Clinical features
- Characteristic facial features (see Figure 1):
- Large anterior fontanelle in infants and young children
- Wide metopic suture in infants and young children
- Widow's peak
- Widely spaced eyes, typically marked
- Enlarged globes leading to the appearance of prominent eyes
- Downslanted palpebral fissures
- Posteriorly rotated ears
- Depressed nasal bridge
- Short nose with broad or bifid tip
- Ophthalmologic abnormalities. High myopia (-6 diopters or worse), retinal detachment (30%), retinal dystrophy and optic nerve hypoplasia, progressive visual loss, iris coloboma or iris hypoplasia in some individuals
- Sensorineural hearing loss. Onset in infancy or childhood (100%)
- Developmental delay. Almost always present
- Omphalocele (or umbilical hernia) (~40%)
- Diaphragmatic hernia (or eventration) (~40%)

Figure 1.
Facial characteristics over time in the same male with Donnai-Barrow syndrome A. At age six months; broad forehead, markedly widely spaced eyes, left iris coloboma, and short nose
Radiographic features. Agenesis of corpus callosum (complete or partial)
Establishing the Diagnosis
The diagnosis of Donnai-Barrow syndrome (DBS) is established in a proband with the characteristic clinical features and a distinctive pattern of low-molecular-weight proteinuria and/or biallelic pathogenic variants in LRP2 identified by molecular genetic testing (see Table 1).
Biochemical Testing
Absent or abnormally functioning megalin, the protein encoded by LRP2, prevents normal renal proximal tubule reuptake of megalin ligands, resulting in excess spillage of proteins with low molecular weight in the urine in 100% of individuals with DBS. Among proteins identified by urinary protein electrophoresis* are:
- Retinol-binding protein (RBP)
- Vitamin D-binding protein (VDBP)
* Note: The use of a "dipstick" is not an adequate substitute to protein electrophoresis to identify megalin ligands in the urine of individuals with DBS.
Molecular Genetic Testing
Molecular genetic testing approaches can include gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of DBS has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of DBS, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of LRP2 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis first. If only one or no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.Note: As LRP2 is among the longest genes in the human genome and contains several private benign variants, extreme caution must be used in interpreting sequence changes. The use of an interpretative framework (e.g., the published guidelines from the American College of Medical Genetics and Genomics and the Association for Molecular Pathology) is recommended [Richards et al 2015].
- A multigene panel that includes LRP2 and other genes of interest (see Differential Diagnosis) may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of DBS is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Donnai-Barrow Syndrome
Clinical Characteristics
Clinical Description
The following information is based on case reports reviewed in Patel et al [2007], Pober et al [2009], Shaheen et al [2010], Roane et al [2012], Storm et al [2013], Dachy et al [2015], Anglani et al [2018], and Khan & Ghazi [2018].
Craniofacial features. Widely spaced eyes, depressed nasal bridge, short nose with a broad and occasionally indented tip, broad forehead, and prominent parietal frontal bossing are characteristic findings and are consistently present in individuals with DBS and confirmed biallelic LRP2 pathogenic variants. Downslanted palpebral fissures and low-set posteriorly angulated ears are very frequent findings. While some of these features may change over time, the facial gestalt remains characteristic in the few reported adults.
Ophthalmologic abnormalities. Progressive visual loss resulting in legal blindness is common and seemingly the result of severe myopia and an attendant risk for retinal detachment, although prompt treatment can improve the chances of useful vision. Retinal detachment can be unilateral or bilateral, complete or partial, and can occur in young children. Prophylactic treatment with peripheral barrier photocoagulation to prevent retinal detachment has been successful in several individuals. Progressive retinal dystrophy has been observed in a few individuals; the exact nature and rate of progression of the retinal dystrophy remain to be determined. Optic nerve hypoplasia has been reported in at least one individual. The combination of a small and recessed optic nerve head, surrounded by multiple rings of abnormal pigmentation masking the neuroretinal rim, and the presence of multiple thin vessels emanating from the optic disk was proposed as a characteristic finding on fundoscopy. Iris coloboma, frequently bilateral or affecting the right eye, is common and often present alongside hypoplastic iris stroma. Glaucoma and cataracts have only rarely been reported.
Sensorineural hearing loss. Profound bilateral sensorineural hearing loss is frequently acquired and diagnosed in early childhood. Despite a relative paucity of data, it appears that hearing can deteriorate over time, but not every individual progresses to complete loss. Some individuals have useful hearing using hearing aids and seven reported individuals have successfully received cochlear implants (see Deafness and Hereditary Hearing Loss Overview for definitions).
Renal disease. The majority of individuals with DBS present with asymptomatic low-molecular weight proteinuria (LMWP). In the adult kidney, megalin is highly expressed in proximal tubules where it plays a key role in the reuptake of several proteins by receptor-mediated endocytosis. The characteristic urinary protein profile can be detected by Coomassie staining of a random urine sample. Increased excretion of vitamin D-binding protein (VDBP) and retinol-binding protein (RBP) are consistent with the presence of DBS and may lead to hypovitaminoses in some individuals. Urinary loss of VDBP and RBP, however, are not specific to DBS and may be present in individuals with tubular dysfunction. Urinary cubilin and its ligands are detectable in urine samples of individuals with DBS. Hypercalciuria, nephrocalcinosis, nephrolithiasis, and focal segmental glomerulosclerosis have been reported. Progression to nephropathy and end-stage renal disease is a rare but life-threatening complication, especially in adults, and urinary function should be monitored in individuals with DBS. One of the first reported individuals died of renal failure at age 30 years. No renal phenotype has been convincingly documented in individuals with a heterozygous LRP2 pathogenic variant.
Developmental delay, intellectual disability, and behavioral features. Motor milestones are only slightly delayed and most children become continent. No systematic studies of intellectual functioning exist, but available data suggest that all individuals with DBS have intellectual disabilities of varying degrees ranging from mild to moderate. Formal assessment is difficult because of the severe vision and hearing deficits. Roane at al [2012] described a child age five years engaging in self-injurious behavior that was responsive to behavioral therapy; however, self-injurious behavior is not typically observed in individuals with DBS. Teenagers and young adults attend school with special provisions for hearing and vision problems. Some communicate using sign language, and can perform routine tasks and retain a variable degree of independence. An adult individual reportedly suffered from psychosis since childhood. Mood swings and depressive symptoms in this individual were compatible with her diagnosis of schizoaffective disorder. Overall, interactions with her were described as pleasant. She did not exhibit deterioration of cognitive or functional abilities beyond what was caused by her co-occurring psychiatric disorder but she did not live independently.
Diaphragmatic hernia (or eventration) and/or omphalocele (or umbilical hernia) are each reported in approximately 40% of affected individuals. The two defects infrequently occur together [Gripp et al 1997, Kantarci et al 2007, Patel et al 2007]. The absence of either or both does not exclude the diagnosis.
Seizures. A few individuals have developed tonic-clonic seizures in childhood or adolescence. In one individual, the first episode of seizures developed into status epilepticus leading to her demise. No single antiepileptic drug has been demonstrated to be the treatment of choice specifically for this disorder, and therapy should follow standard guidelines.
Growth. Relatively high birth weight (~4 kg) has been recorded in several infants. Macrocephaly is present in many individuals with DBS and it is often apparent at birth. Height and weight eventually appear to be within the normal range but a few of the older children are tall, with heights on the 90th centile.
Neuroimaging. Agenesis of corpus callosum, either complete or partial, is reported in most individuals with DBS (see Figure 2). The presence of additional brain anomalies, such as periventricular nodular heterotopia and abnormalities of gyral patterns, is increasingly appreciated.
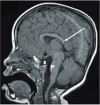
Figure 2.
MRI in a female age three years demonstrates numerous abnormalities including hypogenesis of the corpus callosum (most notably involving the rostrum and splenium) (long white arrow), partially empty sella turcica (S), and small pons (P).
Other. Involvement of other organ systems (e.g., ventriculoseptal defect, bicornuate uterus) has been reported but is rare. Pubertal development occurs at the appropriate times.
Prognosis. Information on long-term follow up and natural history of DBS is limited to a few individuals because many affected pregnancies are interrupted or result in perinatal death secondary to congenital malformations. Major risks are due to complications of retinal detachment leading to visual loss in individuals already compromised by hearing defects, and progression of renal dysfunction. Only two adults older than age 50 years with molecularly confirmed LRP2 pathogenic variants have been reported [Stora et al 2009, Anglani et al 2018]. The most notable health concern is deteriorating renal function, resulting in renal transplantation in one individual and renal rickets in the other.
Genotype-Phenotype Correlations
No genotype-phenotype correlations are known.
Nomenclature
Donnai-Barrow syndrome (DBS) [Donnai & Barrow 1993] and faciooculoacousticorenal (FOAR) syndrome [Holmes & Schepens 1972, Schowalter et al 1997] were reported as two distinct entities, although overlapping clinical features were appreciated [Devriendt et al 1998, Chassaing et al 2003]. It is now known that these syndromes represent the same clinical entity and are caused by pathogenic variants in LRP2 [Kantarci et al 2007].
The following terms should no longer be used when referring to DBS:
- Syndrome of ocular and facial anomalies, telecanthus, and deafness
- Holmes-Schepens syndrome
- Diaphragmatic hernia-exomphalos-hypertelorism syndrome
- Diaphragmatic hernia-hypertelorism-myopia-deafness syndrome
Prevalence
No population-based incidence or prevalence data are available. Only 49 individuals with sufficient clinical and/or molecular data to establish the diagnosis of DBS have been reported in the medical literature. Many of these individuals are members of a few large consanguineous families.
DBS has been reported in different ethnic groups, including northern and central European, Middle Eastern, American of European origin, and African American. No one ethnic group predominates.
Genetically Related (Allelic) Disorders
No phenotypes other than Donnai-Barrow syndrome are currently known to be associated with pathogenic variants in LRP2.
Differential Diagnosis
Donnai-Barrow syndrome (DBS) is associated with congenital diaphragmatic hernia (CDH) (see Congenital Diaphragmatic Hernia Overview).
Table 2.
Disorders to Consider in the Differential Diagnosis of Donnai-Barrow Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Donnai-Barrow syndrome (DBS), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with Donnai-Barrow Syndrome
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Donnai-Barrow Syndrome
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Developmental Delay / Intellectual Disability Management Issues
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the United States, early intervention is a federally funded program available in all states.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed.
Ages 5-21 years
- In the US, an IEP based on the individual's level of function should be developed by the local public school district. Affected children are permitted to remain in the public school district until age 21.
- Discussion about transition plans including financial, vocation/employment, and medical arrangements should begin at age 12 years. Developmental pediatricians can provide assistance with transition to adulthood.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life.
Consideration of private supportive therapies based on the affected individual's needs is recommended. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
In the US:
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Communication issues. Consider evaluation for alternative means of communication (e.g., Augmentative and Alternative Communication [AAC]) for individuals who have expressive language difficulties.
Social/Behavioral Concerns
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavioral management strategies or providing prescription medications, such as medication used to treat attention deficit hyperactivity disorder, when necessary. Repetitive self-injurious behavior has been reported in a single individual [Roane et al 2012] and psychosis in another [Stora et al 2009] (see Clinical Description).
Surveillance
Table 5.
Recommended Surveillance for Individuals with DBS
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Donnai-Barrow syndrome (DBS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are usually obligate heterozygotes (i.e., carriers of one LRP2 pathogenic variant).
- Paternal uniparental isodisomy for chromosome 2 accounted for homozygous LRP2 pathogenic variants in a proband whose father was heterozygous for the variant and whose mother had two normal LRP2 alleles [Kantarci et al 2008] (see Table 6).
- Heterozygotes (carriers) are asymptomatic in that they do not manifest structural birth defects, craniofacial dysmorphology, or kidney dysfunction.
Sibs of a proband
- When both parents are carriers of an LRP2 pathogenic variant, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Given the variability among affected sibs of the occurrence of major structural birth defects (i.e., omphalocele or CDH), the presence of one of the defects in one sib does not predict the presence of either or both in another sib.
- In the case of uniparental isodisomy (see Table 6), the recurrence risk for sibs of the proband is very low.
- Heterozygotes (carriers) are asymptomatic in that they do not manifest structural birth defects, craniofacial dysmorphology, or kidney dysfunction.
Offspring of a proband. No reports of reproduction in individuals with DBS have been published.
Other family members. When a parent of the proband is a carrier of a pathogenic variant, each of his/her sibs is at a 50% risk of being a carrier.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the LRP2 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the LRP2 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Fetal ultrasonography. Prenatal diagnosis using ultrasound examination or fetal MRI scanning for pregnancies at increased risk for DBS can be achieved by detecting anomalies such as partial or complete agenesis of the corpus callosum, diaphragmatic hernia, omphalocele, and the characteristic facial appearance including ocular hypertelorism and prominent eyes. However, failure to detect these structural birth defects in a pregnancy at increased risk for DBS does not eliminate the possibility of an affected fetus.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- American Society for Deaf ChildrenPhone: 800-942-2732 (ASDC)Email: info@deafchildren.org
- CDH InternationalEmail: info@cdhi.org
- Congenital Diaphragmatic Hernia (CDH) InternationalPhone: 919-610-0087Fax: 815-425-9155Email: info@cdhi.org
- National Eye InstitutePhone: 301-496-5248Email: 2020@nei.nih.gov
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Donnai-Barrow Syndrome: Genes and Databases
Table B.
OMIM Entries for Donnai-Barrow Syndrome (View All in OMIM)
Molecular Pathogenesis
Donnai-Barrow syndrome (DBS) is caused by loss-of-function pathogenic variants in LRP2, which encodes the low-density lipoprotein receptor-related protein 2, an endocytic transmembrane glycoprotein also referred to as megalin or LRP-2/gp330. Megalin is widely expressed in specialized absorptive epithelia [Willnow & Christ 2017].
Many features of DBS are recapitulated in animal models of megalin deficiency; for example:
- Forebrain anomalies, including agenesis of corpus callosum and mild holoprosencephaly (not reported in individuals with DBS) [Christ et al 2016]
- Enlarged and exophthalmic eyes (buphthalmos) [Storm et al 2014]
- Glaucoma or glaucomatous changes in some but not every model [Veth et al 2011, Cases et al 2015, Christ et al 2015]
- Defects of cardiovascular development, specifically of the outflow tract and the atrioventricular canal [Baardman et al 2016] (The incidence of heart defects in individuals with DBS may be underestimated [Pober et al 2009].)
Megalin is required for endocytosis [Anzenberger et al 2006, Fisher & Howie 2006]. Dozens of megalin ligands have been identified (reviewed in Christensen & Birn 2002]). Increased excretion of vitamin D-binding protein (VDBP), retinol-binding protein (RBP) [Kantarci et al 2007], cubilin, and type 3 carbonic anhydrase (CAIII) as in Dent's disease [Dachy et al 2015] have been reported in humans with DBS. Megalin is also necessary for the CYP27B1-mediated activation of 25-hydroxyvitamin D to 1,25 dihydroxyvitamin D in the renal proximal tubule, and for its regulation by CYP24A1-mediated inactivation [Nykjaer et al 1999, Chapron et al 2018]. Deficiencies manifested as hypovitaminosis D and osteomalacia have been described in a few individuals [Storm et al 2013, Anglani et al 2018].
Gene structure. LRP2 spans approximately 236 kb of genomic DNA and contains 79 coding exons (NM_004525.2). For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. Twenty-two pathogenic or likely pathogenic variants have been documented in 17 kindreds [Kantarci et al 2007, Kantarci et al 2008, Stora et al 2009, Shaheen et al 2010, Storm et al 2013, Schrauwen et al 2014, Dachy et al 2015, Khalifa et al 2015, Anglani et al 2018, Khan & Ghazi 2018]. Typically, pathogenic and likely pathogenic variants are distributed throughout the gene and are unique to each kindred. The homozygous or compound heterozygous LRP2 variants include small deletions or insertions and conserved splice site, nonsense, and missense variants inherited from each heterozygous carrier parent. As a rare event, one affected individual was homozygous for an LRP2 pathogenic variant resulting from paternal isodisomy of chromosome 2; the father was a heterozygous carrier of c.11469_11472delTTTG [Kantarci et al 2008].
Table 6.
LRP2 Pathogenic Variants Discussed in This GeneReview
Normal gene product. The 600-kd megalin protein comprises 4655 amino acids. Megalin is a member of the low-density lipoprotein receptor gene family, which is expressed predominantly in apical surface of absorptive or secretory epithelia. A variety of tissues including the brain, eye, renal proximal tubule, lung, intestine, uterus, oviduct, male reproductive tract, and embryonic yolk sac express megalin. Megalin binds more than 50 ligands, including lipoproteins, vitamin-binding proteins, hormones, enzymes, and immune- and stress-response-related proteins. It has been proposed that megalin interacts with the sonic hedgehog protein (reviewed in Christ et al [2016]). Megalin and a membrane receptor cubilin (gp280) share many ligands [Fisher & Howie 2006].
Abnormal gene product. The pathogenic variants in LRP2 that cause DBS result in absent or nonfunctional megalin protein. The c.2639+1G>A splice variant was shown to result in complete absence of megalin staining in proximal tubular cells [Storm et al 2013], supporting the loss of function hypothesis. Weak and diffuse cytoplasmic staining was detected in the kidney biopsy of an individual who was compound heterozygous for the c.7564T>C and c.12623C>A LRP2 variants [Dachy et al 2015]. Based on this observation, one or both missense variants are likely to result in a trafficking defect and/or protein instability. In the same tissue sample, the normal apical signal of cubilin at the brush border of the proximal tubule was not affected, suggesting that processing of megalin and cubilin is at least partially independent.
References
Literature Cited
- Anglani F, Terrin L, Brugnara M, Battista M, Cantaluppi V, Ceol M, Bertoldi L, Valle G, Joy MP, Pober BR, Longoni M. Hypercalciuria and nephrolithiasis: expanding the renal phenotype of Donnai-Barrow syndrome. Clin Genet. 2018;94:187–8. [PMC free article: PMC5995642] [PubMed: 29532936]
- Anzenberger U, Bit-Avragim N, Rohr S, Rudolph F, Dehmel B, Willnow TE, Abdelilah-Seyfried S. Elucidation of megalin/LRP2-dependent endocytic transport processes in the larval zebrafish pronephros. J Cell Sci. 2006;119:2127–37. [PubMed: 16638803]
- Baardman ME, Zwier MV, Wisse LJ, Gittenberger-de Groot AC, Kerstjens-Frederikse WS, Hofstra RMW, Jurdzinski A, Hierck BP, Jongbloed MR, Berger RM, Plösch T, DeRuiter MC. Common arterial trunk and ventricular non-compaction in Lrp2 knockout mice indicate a crucial role of LRP2 in cardiac development. Dis Model Mech. 2016;9:413–25. [PMC free article: PMC4852499] [PubMed: 26822476]
- Cases O, Joseph A, Obry A, Santin MD, Ben-Yacoub S, Pâques M, Amsellem-Levera S, Bribian A, Simonutti M, Augustin S, Debeir T, Sahel JA, Christ A, de Castro F, Lehéricy S, Cosette P, Kozyraki R. Foxg1-Cre mediated Lrp2 inactivation in the developing mouse neural retina, ciliary and retinal pigment epithelia models congenital high myopia. PLoS One. 2015;10:e0129518. [PMC free article: PMC4480972] [PubMed: 26107939]
- Chapron BD, Chapron A, Phillips B, Okoli MC, Shen DD, Kelly EJ, Himmelfarb J, Thummel KE. Reevaluating the role of megalin in renal vitamin D homeostasis using a human cell-derived microphysiological system. ALTEX. 2018;35:504–15. [PMC free article: PMC6896899] [PubMed: 29999169]
- Chassaing N, Lacombe D, Carles D, Calvas P, Saura R, Bieth E. Donnai-Barrow syndrome: four additional patients. Am J Med Genet A. 2003;121A:258–62. [PubMed: 12923867]
- Christ A, Christa A, Klippert J, Eule JC, Bachmann S, Wallace VA, Hammes A, Willnow TE. LRP2 acts as SHH clearance receptor to protect the retinal margin from mitogenic stimuli. Dev Cell. 2015;35:36–48. [PubMed: 26439398]
- Christ A, Katja Herzog K, Willnow TE. LRP2, an auxiliary receptor that controls sonic hedgehog signaling in development and disease. Dev Dyn. 2016;245:569–79. [PubMed: 26872844]
- Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol. 2002;3:256–66. [PubMed: 11994745]
- Dachy A, Paquot F, Debray G, Bovy C, Christensen EI, Collard L, Jouret F. In-depth phenotyping of a Donnai–Barrow patient helps clarify proximal tubule dysfunction. Pediatr Nephrol. 2015;30:1027–31. [PubMed: 25822460]
- Devriendt K, Standaert L, Van Hole C, Devlieger H, Fryns JP. Proteinuria in a patient with the diaphragmatic hernia-hypertelorism-myopia-deafness syndrome: further evidence that the facio-oculo-acoustico-renal syndrome represents the same entity. J Med Genet. 1998;35:70–1. [PMC free article: PMC1051192] [PubMed: 9475100]
- Donnai D, Barrow M. Diaphragmatic hernia, exomphalos, absent corpus callosum, hypertelorism, myopia, and sensorineural deafness: a newly recognized autosomal recessive disorder? Am J Med Genet. 1993;47:679–82. [PubMed: 8266995]
- Fisher CE, Howie SEM. The role of megalin (LRP-2/Gp330) during development. Dev Biol. 2006;296:279–97. [PubMed: 16828734]
- Gripp KW, Donnai D, Clericuzio CL, McDonald-McGinn DM, Guttenberg M, Zackai EH. Diaphragmatic hernia-exomphalos-hypertelorism syndrome: a new case and further evidence of autosomal recessive inheritance. Am J Med Genet. 1997;68:441–4. [PubMed: 9021018]
- Holmes LB, Schepens CL. Syndrome of ocular and facial anomalies, telecanthus, and deafness. J Pediatr. 1972;81:552–5. [PubMed: 4626128]
- Kantarci S, Al-Gazali L, Hill RS, Donnai D, Black GCM, Bieth E, Chassaing N, Lacombe D, Devriendt K, Teebi A, Loscertales M, Robson C, Liu T, MacLaughlin DT, Noonan KM, Russell MK, Walsh CA, Donahoe PK, Pober BR. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39:957–9. [PMC free article: PMC2891728] [PubMed: 17632512]
- Kantarci S, Ragge NK, Thomas NS, Robinson DO, Noonan KM, Russell MK, Donnai D, Raymond FL, Walsh CA, Donahoe PK, Pober BR. Donnai-Barrow syndrome (DBS/FOAR) in a child with a homozygous LRP2 mutation due to complete chromosome 2 paternal isodisomy. Am J Med Genet A. 2008;146A:1842–7. [PMC free article: PMC2891749] [PubMed: 18553518]
- Khalifa O, Al-Sahlawi Z, Imtiaz F, Ramzan K, Allam R, Al-Mostafa A, Abdel-Fattah M, et al. Variable expression pattern in Donnai-Barrow syndrome: report of two novel LRP2 mutations and review of the literature. Eur J Med Genet. 2015;58:293–9. [PubMed: 25682901]
- Khan AO, Ghazi NG. The distinct optic disk and peripapillary appearance in Donnai-Barrow syndrome. Ophthalmic Genet. 2018;39:321–4. [PubMed: 29388841]
- Nykjaer A, Dragun D, Walther D, Vorum H, Jacobsen C, Herz J, Flemming Melsen F, Christensen EI, Willnow TE. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell. 1999;96:507–15. [PubMed: 10052453]
- Patel N, Hejkal T, Katz A, Margalit E. Ocular manifestations of Donnai-Barrow syndrome. J Child Neurol. 2007;22:462–4. [PubMed: 17621530]
- Pober BR, Longoni M, Noonan KM. A review of Donnai-Barrow and facio-oculo-acoustico-renal (DB/FOAR) syndrome: clinical features and differential diagnosis. Birth Defects Res A Clin Mol Teratol. 2009;85:76–81. [PMC free article: PMC2882234] [PubMed: 19089858]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Roane H, Bouxsein K, Fulton C. Assessment and treatment of self-injurious behavior associated with Donnai-Barrow syndrome. J Dev Phys Disabil. 2012;24:327–35. [PMC free article: PMC4306227] [PubMed: 25632217]
- Schowalter DB, Pagon RA, Kalina RE, McDonald R. Facio-oculo-acoustico-renal (FOAR) syndrome: case report and review. Am J Med Genet. 1997;69:45–9. [PubMed: 9066882]
- Schrauwen I, Sommen M, Claes C, Pinner J, Flaherty M, Collins F, Van Camp G. Broadening the phenotype of LRP2 mutations: A new mutation in LRP2 causes a predominantly ocular phenotype suggestive of Stickler syndrome. Clin Genet. 2014;86:282–6. [PubMed: 23992033]
- Shaheen IS, Finlay E, Prescott K, Russell M, Longoni M, Joss S. Focal segmental glomerulosclerosis in a female patient with Donnai-Barrow syndrome. Clin Dysmorphol. 2010;19:35–7. [PubMed: 19952924]
- Stora S, Conte M, Chouery E, Richa S, Jalkh N, Gillart AC, Laure de Joannis A, Mégarbané A. A. 56-year-old female patient with facio-oculo-acoustico-renal syndrome (FOAR) syndrome. Report on the natural history and of a novel mutation. Eur J Med Genet. 2009;52:341–3. [PubMed: 19577669]
- Storm T, Heegaard S, Christensen EI, Nielsen R. Megalin deficiency causes high myopia, retinal pigment epithelium-macromelanosomes and abnormal development of the ciliary body in mice. Cell Tissue Res. 2014;358:99–107. [PMC free article: PMC4186978] [PubMed: 24980834]
- Storm T, Tranebjærg L, Frykholm C, Birn H, Verroust PJ, Nevéus T, Sundelin B, Hertz JM, Holmström G, Ericson K, Christensen EI, Nielsen R. Renal phenotypic investigations of megalin-deficient patients: novel insights into tubular proteinuria and albumin filtration. Nephrol Dial Transplant. 2013;28:585–91. [PubMed: 23048173]
- Veth KN, Willer JR, Collery RF, Gray MP, Willer GB, Wagner DS, Mullins MC, Udvadia AJ, Smith RS, John SW, Gregg RG, Link BA. Mutations in zebrafish Lrp2 result in adult-onset ocular pathogenesis that models myopia and other risk factors for glaucoma. PLoS Genet. 2011;7:e1001310. [PMC free article: PMC3040661] [PubMed: 21379331]
- Willnow TE, Christ A. Endocytic receptor LRP2/megalin - of holoprosencephaly and renal Fanconi syndrome. Pflugers Arch. 2017;469:907–16. [PubMed: 28497274]
Chapter Notes
Acknowledgments
We thank Dr Patricia K Donahoe for her leadership and continuous support. We also thank the many physicians who referred patients with DBS/FOAR to us. Finally, we gratefully thank all families who have participated in our study, through which we were able to find the gene responsible for DBS/FOAR.
Author History
Dian Donnai, MD (2008-present)
Sibel Kantarci, PhD, FACMG (2008-present)
Mauro Longoni, MD (2018-present)
Kristin M Noonan, MD; Medical College of Wisconsin (2008-2018)
Barbara R Pober, MD (2008-present)
Revision History
- 21 November 2018 (sw) Comprehensive update posted live
- 28 June 2011 (me) Comprehensive update posted live
- 28 August 2008 (me) Review posted live
- 28 April 2008 (brp) Original submission
Publication Details
Author Information and Affiliations
Assistant, Massachusetts General Hospital
Boston, Massachusetts
Quest Diagnostics Nichols Institute
Cytogenetics and Genomics
San Juan Capistrano, California
University of Manchester
St Mary's Hospital
Manchester, United Kingdom
Harvard Medical School
Boston, Massachusetts
Publication History
Initial Posting: August 28, 2008; Last Update: November 21, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Longoni M, Kantarci S, Donnai D, et al. Donnai-Barrow Syndrome. 2008 Aug 28 [Updated 2018 Nov 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

