Summary
Clinical characteristics.
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is a disorder of the urea cycle and ornithine degradation pathway. Clinical manifestations and age of onset vary among individuals even in the same family.
Neonatal onset (~8% of affected individuals). Manifestations of hyperammonemia usually begin 24-48 hours after feeding begins and can include lethargy, somnolence, refusal to feed, vomiting, tachypnea with respiratory alkalosis, and/or seizures.
Infantile, childhood, and adult onset (~92%). Affected individuals may present with:
- Chronic neurocognitive deficits (including developmental delay, ataxia, spasticity, learning disabilities, cognitive deficits, and/or unexplained seizures);
- Acute encephalopathy secondary to hyperammonemic crisis precipitated by a variety of factors; and
- Chronic liver dysfunction (unexplained elevation of liver transaminases with or without mild coagulopathy, with or without mild hyperammonemia and protein intolerance).
Neurologic findings and cognitive abilities can continue to deteriorate despite early metabolic control that prevents hyperammonemia.
Diagnosis/testing.
The biochemical diagnosis of HHH syndrome is established in a proband with the classic metabolic triad of episodic or postprandial hyperammonemia, persistent hyperornithinemia, and urinary excretion of homocitrulline. The molecular diagnosis of HHH syndrome is established in a symptomatic individual with or without suggestive metabolic/biochemical findings by identification of biallelic pathogenic variants in SLC25A15.
Management.
Treatment of manifestations: Acute and long-term management is best performed in conjunction with a metabolic specialist. Of primary importance is the use of established protocols to rapidly control hyperammonemic episodes by discontinuation of protein intake, intravenous infusion of glucose and, as needed, infusion of supplemental arginine and the ammonia removal drugs sodium benzoate and sodium phenylacetate. Hemodialysis is performed if hyperammonemia persists and/or the neurologic status deteriorates.
Prevention of primary manifestations: Individuals with HHH syndrome should be maintained on an age-appropriate protein-restricted diet, citrulline supplementation, and sodium phenylbutyrate to maintain plasma concentrations of ammonia, glutamine, arginine, and essential amino acids within normal range. Note: Liver transplantation is not indicated when metabolic control can be achieved with this regimen as liver transplantation may correct the hyperammonemia but will not correct tissue-specific metabolic abnormalities that also contribute to the neuropathology.
Surveillance: Routine assessment of height, weight, and head circumference from the time of diagnosis to adolescence. Routine assessment of plasma ammonia concentration, plasma and urine amino acid concentrations, urine organic acids, and urine orotic acid based on age and history of adherence and metabolic control. Routine developmental and educational assessment to assure optional interventions. Attention to subtle changes in mood, behavior, and eating and/or the onset of vomiting, which may suggest that plasma concentrations of glutamine and ammonia are increasing. Periodic neurologic evaluation to monitor for neurologic deterioration even when metabolic control is optimal.
Agents/circumstances to avoid: Excess dietary protein intake; nonprescribed protein supplements such as those used during exercise regimens; prolonged fasting during an illness or weight loss; oral and intravenous steroids; and valproic acid, which exacerbates hyperammonemia in urea cycle disorders.
Evaluation of relatives at risk: Once the pathogenic variants in a family are known, use molecular genetic testing to clarify the genetic status of at-risk relatives to allow early diagnosis and treatment, perhaps even before symptoms occur.
Pregnancy management: In general, pregnant women should continue dietary protein restriction and supplementation with citrulline and ammonia-scavenging medications based on their clinical course before pregnancy. Due to increased protein and energy requirements in pregnancy and, oftentimes, difficulty with patient adherence, weekly to every two-week monitoring of plasma amino acids and ammonia is recommended, especially in the first and third trimester, and close monitoring immediately after delivery.
Genetic counseling.
HHH syndrome is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SLC25A15 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible. However, the identification of familial SLC25A15 pathogenic variants cannot predict clinical outcome because of significant intrafamilial phenotypic variability.
Diagnosis
Suggestive Findings
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome should be suspected in symptomatic individuals with the following age-related clinical, laboratory, and neuroimaging findings.
Clinical Findings
Neonatal presentation (~8% of individuals). Manifestations of hyperammonemia usually begin 24-48 hours after the start of feeding and can include lethargy, somnolence, refusal to feed, vomiting, tachypnea with respiratory alkalosis, and/or seizures.
Infantile, childhood, and adolescent/adult presentation (~92% of individuals) may exhibit any of the following:
- Chronic neurocognitive deficits including developmental and speech delay, ataxia, spasticity, learning disabilities, cognitive deficits, and/or unexplained seizures
- Acute encephalopathy secondary to hyperammonemic crisis, which can be precipitated by infection, fasting, or injury (or occur for no apparent reason) and can manifest as lethargy, decreased appetite, nausea, vomiting, increased respiratory rate, and seizures
- Chronic liver dysfunction characterized by unexplained elevation of liver enzymes (AST and ALT) with or without mild coagulopathy and with or without mild hyperammonemia.
- Mild encephalopathy manifesting as disorientation, irritability, and episodic confusion with mild hyperammonemia, which is difficult to detect as it may resolve spontaneously without treatment or quickly normalize with IV solutions that include glucose [Qadri et al 2016].
Laboratory Findings
Episodic or postprandial mild to moderate hyperammonemia. Plasma ammonia concentrations at the time of diagnosis are summarized in Table 1. Note that neonates have a higher median plasma ammonia level than older affected individuals.
Note: (1) In HHH syndrome the degree of hyperammonemia is usually significantly less than in other urea cycle disorders such as OTC, ASS, or CPS-I deficiency (see Urea Cycle Disorders). (2) Once an affected individual is placed on a protein-restricted diet and treated with sodium phenylbutyrate (see Management), plasma ammonia concentrations return to normal.
Table 1.
Plasma Ammonia Concentrations Observed in HHH Syndrome by Age of Diagnosis
Hyperornithinemia (increased plasma concentration of ornithine). At the time of initial diagnosis, plasma concentration of ornithine can range from 200 to 1915 μmol/L (normal: 30-110 μmol/L).
Note: While plasma concentration of ornithine decreases significantly with a protein-restricted diet, it very rarely normalizes.
Homocitrullinuria (urinary excretion of homocitrulline) is a key feature of HHH syndrome; however, exceptions exist: some infants with neonatal-onset HHH syndrome do not excrete homocitrulline in significant amounts and individuals with HHH syndrome who self-restrict protein intake may excrete minimal or no homocitrulline in the urine [Valle & Simell 2001]. In controls, homocitrulline is not detected in the urine.
Note: Homocitrulline may be found in infant formulas due to the carbamylation of lysine during manufacture and, thus, may cause a false positive result.
Of note, in neonates, the classic metabolic triad of hyperammonemia, hyperornithinemia, and homocitrullinuria may be absent or subtle; alternatively, it may be obscured by the abnormal plasma amino acid profile and aminoaciduria characteristic of hepatic dysfunction and prematurity [Wild et al 2019].
Neuroimaging findings include evidence of cortical or subtentorial atrophy, abnormal white matter signal, demyelination, stroke-like lesions, and/or calcifications/lesions of the basal ganglia [Al-Hassnan et al 2008, Martinelli et al 2015, Guan et al 2017]. For example:
- At initial presentation brain MRI of a previously undiagnosed male age 36 years demonstrated multiple foci of subcortical white matter gliosis and moderate atrophy of the frontoparietal opercula and cerebellar hemispheres [Filosto et al 2013].
- The initial brain MRI findings (pre-diagnosis of HHH syndrome) in a male age 48 years were normal; seven months after extended treatment for hyperammonemic coma requiring dialysis, brain MRI showed evidence of severe hyperammonemic encephalopathy: brain gliosis, widespread hemorrhagic necrosis in the tips of the temporal lobes, and widened horns of the lateral ventricles [Silfverberg et al 2018].
Establishing the Diagnosis
Biochemical Diagnosis
The biochemical diagnosis of hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is established in a proband with the classic metabolic triad of episodic or postprandial hyperammonemia, persistent hyperornithinemia, and urinary excretion of homocitrulline. Note: An incomplete metabolic triad may be observed because of the following: (a) individuals whose protein intake was restricted during early childhood may never have experienced hyperammonemia; (b) affected individuals who come to medical attention because of learning disabilities or school difficulties may only have isolated persistent hyperornithinemia at the time of evaluation; or (c) a low-protein diet can be associated with little to no homocitrulline in the urine.
Molecular Diagnosis
The molecular diagnosis of HHH syndrome is established in a symptomatic individual with or without suggestive metabolic/biochemical findings by identification of biallelic pathogenic (or likely pathogenic) variants in SLC25A15 (Table 2).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic SLC25A15 variants of uncertain significance (or of one known SLC25A15 pathogenic variant and one SLC25A15 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches that depend on the clinical and biochemical findings can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (typically exome sequencing and exome array).
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive laboratory findings of HHH syndrome described in Suggestive Findings are likely to be diagnosed using gene-targeted testing, whereas symptomatic individuals with nonspecific but suggestive clinical, laboratory, and neuroimaging findings in whom the diagnosis of HHH syndrome has not been considered are more likely to be diagnosed using either a multigene panel or comprehensive genomic testing.
Single-gene testing. Sequence analysis of SLC25A15 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no SLC25A15 variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications (see Table 2).
Targeted analysis for the following pathogenic variants (see Table 8) may be considered.
- French-Canadian founder variant
- Accounts for 28% of individuals with HHH syndrome [Debray et al 2008, Martinelli et al 2015]
- Japanese and Middle Eastern founder variant
- Accounts for 16% of individuals with HHH syndrome [Debray et al 2008, Martinelli et al 2015]
A hyperammonemia or urea cycle disorder multigene panel that includes SLC25A15 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 2).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive genomic testing. When the diagnosis of HHH syndrome has not been considered because an individual has nonspecific clinical and laboratory findings, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is an option. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 2.
Molecular Genetic Testing Used in Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome
Clinical Characteristics
Clinical Description
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is characterized by variable clinical presentation and age of onset ranging from the neonatal period to adulthood. Those with neonatal onset are normal for the first 24-48 hours, followed by onset of symptoms related to hyperammonemia (poor feeding, vomiting, lethargy, low temperature, rapid breathing). Those with later onset may present with chronic neurocognitive deficits and/or unexplained seizures, spasticity, acute encephalopathy secondary to hyperammonemic crisis, or chronic liver dysfunction. Neurologic findings and cognitive abilities can continue to deteriorate despite early metabolic control that prevents hyperammonemia.
Unless otherwise indicated, the data used in this chapter are from a total of 122 individuals with HHH syndrome: Martinelli et al 2015 (n=111), six subsequent case reports [Qadri et al 2016, Guan et al 2017, Ono et al 2018, Silfverberg et al 2018, Ho et al 2019, Wild et al 2019] and three unpublished affected individuals [Author, personal observation].
Table 3.
Selected Clinical Findings in Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH) Syndrome
The overall survival rate in individuals with HHH syndrome is 94% (109/116). Of the 21% (13/62) of individuals with HHH syndrome who manifested in the neonatal period, the mortality rate is 15% (2/13). Treatment with a protein-restricted diet resolves hepatic dysfunction (elevated transaminases and coagulopathy). Since hyperammonemia in HHH syndrome responds quickly to treatment, early diagnosis leads to an overall improved long-term outcome regardless of the age of onset.
The long-term neurodevelopmental outcome usually (but not always) correlates with the severity and duration of the hyperammonemic insult.
Pyramidal tract findings and intellectual disability, which range from mild to severe, are generally evident by childhood since almost 70% of HHH syndrome manifests in infancy and childhood [Martinelli et al 2015]. While treatment with a protein-restricted diet prevents postprandial and acute hyperammonemia, outcomes vary. For example, individuals with HHH syndrome with limited clinical manifestations throughout life have been reported [Filosto et al 2013; Silfverberg et al 2018; L Merritt, MD & E Font-Montgomery, MD, personal communication (see Outcomes and Presentation; pdf). Conversely, three of four adults with HHH syndrome who maintained normal levels of plasma ammonia for 11 to 38 years exhibited progressive neurologic and cognitive deterioration with serious outcomes [Kim et al 2012].
Neonatal diagnosis (birth – 1 month). About 8% (9/116) of affected individuals were diagnosed during the neonatal period, usually following an uncomplicated pregnancy and delivery. The clinical course is indistinguishable from that of other neonatal-onset urea cycle disorders: the infant is asymptomatic for the first 24-48 hours and, thereafter, has episodes of poor feeding, vomiting, lethargy, low temperature, and/or rapid breathing related to hyperammonemia (see Table 1). If left untreated seizures, coma, and even death may ensue. Hepatic dysfunction and coagulopathy in the neonatal period are common [Martinelli et al 2015, Wild et al 2019].
Given the small number of case studies published to date, little is known about the long-term outcome of individuals with neonatal onset of HHH syndrome. One child died from hyperammonemic encephalopathy at birth and another at age two months. Survivors ranged in age from one to 23 years – one demonstrated normal development at age six years, four had progressive pyramidal signs, and one with significant neuromotor impairment underwent liver transplantation at age seven years. In eight survivors with neonatal onset, cognitive abilities in four ranged from normal to mild deficiency, three exhibited severe cognitive impairment, and one was a recently reported premature infant [Martinelli et al 2015, Wild et al 2019].
Click here (pdf) for more details about outcomes in the neonatal-onset cases described above.
Infantile (>1 month – 1 year) age at diagnosis. Approximately 10% (12/116) of individuals with HHH syndrome present in this timeframe. Highly variable manifestations in infancy can include hypotonia, lethargy, failure to thrive, seizures, psychomotor delay, hepatomegaly, hepatic dysfunction (coagulopathy and elevated transaminases), hyperammonemia, feeding difficulties, and recurrent vomiting. Some children come to medical attention only after experiencing environmental stressors, most commonly infection.
Unique case studies include an affected individual who presented with fulminant hepatic failure and a recent example of the clinical progression of symptoms in an undiagnosed infant.
Click here (pdf) for more details about the presentation of infantile-onset HHHS described above.
Childhood (>1 year to 12 years) presentation accounts for almost half (56/116) of all HHH syndrome. Children in this group come to medical attention either for findings related to mild hyperammonemia with or without liver dysfunction or for evaluation of developmental and speech delay, dysarthria, intellectual disability, learning disabilities, hyperactivity, recurring vomiting, academic difficulties, spasticity, ataxia, and/or unexplained seizure activity. Environmental triggers (i.e., surgery, infection, medication) may also induce manifestations in previously healthy children.
A salient characteristic of affected individuals diagnosed in childhood who have the same SLC25A15 pathogenic variants is marked phenotypic variability.
Click here (pdf) for more details about outcomes in childhood-onset HHHS.
Liver dysfunction, a predominant feature at time of diagnosis, generally manifests as mild coagulopathy and elevated liver enzymes (AST and ALT) with or without hyperammonemia. In a few reports acute liver failure prompted consideration of liver transplantation [Fecarotta et al 2006, Mhanni et al 2008]. However, the liver dysfunction that may occur during the initial clinical presentation does not appear to cause long-term complications. Once the hyperammonemia is treated in a standard manner (see Treatment of Manifestations), the liver dysfunction subsides [Martinelli et al 2015, Ono et al 2018].
Despite early detection and adequate metabolic control (i.e., absence of hyperammonemia), some individuals with HHH syndrome continue to worsen neurologically with pyramidal tract involvement and cognitive decline [Camacho et al 2006, Debray et al 2008, Tessa et al 2009, Martinelli et al 2015]. Subclinical hyperammonemia is thought to be a major factor in neurocognitive decline, but not in the cause or progression of pyramidal syndrome. In some individuals with early-childhood onset, gait abnormalities and spasticity are the predominant findings.
The Urea Cycle Disorders Consortium reported developmental quotients (DQ) in four preschool children (age 4-5 years) with HHH syndrome: two were in the normal range and two were <71. Two also exhibited anxiety and acting out behaviors [Waisbren et al 2016].
Adolescence/adulthood (>12 years) accounts for about one third (39/116) of persons with HHH syndrome. After infancy, these individuals quickly learn to self-restrict their protein intake to avoid the malaise and vomiting that accompanies protein-rich meals. The milder form of the disease and self-adherence to a low-protein diet allow these individuals to lead a relatively symptom-free life and remain undetected until they inadvertently overwhelm their compromised ability to detoxify harmful elevations of plasma ammonia. Ammonia overload may result from catabolic events (i.e., surgery, infection, prolonged fasting, pregnancy, internal bleeding), deviation from a protein-restricted diet, or certain medications (e.g., valproate, steroids).
Individuals diagnosed in adolescence/adulthood may present with recurrent encephalopathy secondary to hyperammonemia (lethargy, disorientation, episodic confusion, unexplained seizures), intellectual disabilities, recurrent vomiting, chronic behavioral problems, cerebellar signs (dizziness, loss of balance, poor coordination, abnormal gait/posture), and pyramidal tract dysfunction (inability to perform fine movements, positive Babinski reflex, muscle weakness, ataxia, hyperreflexia, and spasticity).
Click here (pdf) for more details about outcomes in adolescent/adult-onset HHHS.
Cognitive development in persons with HHH syndrome ranges from normal (34%) to severe impairment (34%), with the majority having normal to mild neurocognitive deficit (59%). In some reports, affected individuals have significant neurologic findings such as spasticity and ataxia without cognitive impairment [Martinelli et al 2015]. Of note, pyramidal signs of the lower extremities (hyperreflexia, clonus, tip-toe gait, and/or spastic ataxia) may develop years after the initial diagnosis [Salvi et al 2001b, Debray et al 2008, Tessa et al 2009].
The Urea Cycle Disorder Consortium reported findings of two successive neurocognitive evaluations given to one adult with HHH syndrome: full scale IQ was 100 and 84 at ages 21 years and 26 years, respectively; performance was significantly diminished across all neuropsychological tests. No cognitive or behavioral issues were noted [Waisbren et al 2016].
Additional clinical biochemical abnormalities in HHHS can include the following:
- Mildly elevated plasma glutamine concentration (1.5- to 2-fold the upper limits of control values)
- Plasma lysine can range from normal to moderately decreased
- Increased urinary excretion of:
- Orotic acid (2.5- to 12-fold the upper limit of control values)
- Organic acids. An increase in the urinary excretion of components of the Krebs cycle (succinate, citrate, fumaric, α-ketoglutaric), methylcitrate, and lactate has been documented in a few reports [Korman et al 2004, Fecarotta et al 2006].
- Statistically significant elevation of AFP and pronounced liver ultrasound abnormalities at follow up [Ranucci et al 2019]
Genotype-Phenotype Correlations
The SLC25A15 genotype does not correlate with the clinical or biochemical phenotype of HHH syndrome [Fiermonte et al 2003, Camacho et al 2006, Tessa et al 2009].
Nomenclature
The name "hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome" was coined by Vivian Shih, MD in 1969 for the disorder in which a "block in the ornithine metabolic pathway" has biochemical findings "not concordant with those in patients with proven hepatic ornithine transcarbamylase deficiency" [Shih et al 1969].
In 1999 after ORNT1 (now known as SLC25A15), the gene encoding ORNT1, was identified, the term ORNT1 deficiency was introduced and used interchangeably with HHH syndrome [Camacho et al 1999].
HHH syndrome may also be referred to as "ornithine transporter deficiency" or "ornithine translocase deficiency."
Prevalence
Since the description of the first individual with HHH syndrome by Shih et al [1969], approximately 122 individuals with HHH syndrome have been reported [Martinelli et al 2015, Qadri et al 2016, Guan et al 2017, Ono et al 2018, Silfverberg et al 2018, Ho et al 2019, Wild et al 2019].
Summar et al [2013], using two large longitudinal registries in the US (NIH-sponsored Urea Cycle Disorders (UCD) Consortium and The National UCD Foundation) and one in Europe (European Registry and Network for Intoxication-Type Metabolic Diseases), calculated an incidence for HHH syndrome of 1% and 3% of all UCDs in the US and Europe, respectively. In the US, the frequency of HHH syndrome is less than 1:2,000,000 live births [Summar et al 2013].
The incidence of HHH syndrome is highest in individuals of French-Canadian ancestry because of the SLC25A15 founder variant c.562_564delTTC (p.Phe188del) in this population (see Table 8) [Camacho et al 1999, Debray et al 2008]. A study of this founder variant in an isolated northern Saskatchewan population of mixed French-Canadian and Aboriginal descent suggested that the incidence of HHH syndrome in this population is 1:1,550 live births [Sokoro et al 2010].
Another common pathogenic variant, c.535C>T (p.Arg179Ter) (see Table 8), seen in 14% of individuals with HHH syndrome, is frequent in persons with HHH syndrome of Japanese and Middle Eastern descent [Tsujino et al 2000, Salvi et al 2001a].
Genetically Related (Allelic) Disorders
No clinical or biochemical phenotypes other than those discussed in this GeneReview are known to be associated with biallelic pathogenic variants in SLC25A15.
Differential Diagnosis
Hyperammonemia
Most commonly, neonates with hyperammonemia and neonatal-onset HHH syndrome are initially suspected of having sepsis. See Häberle et al [2019] for a comprehensive algorithm for the differential diagnosis of neonatal hyperammonemia based on plasma and urine metabolites.
Like other urea cycle disorders (UCDs), HHH syndrome should be included in the differential diagnosis of any individual with hyperammonemia, including women who experience hyperammonemia during or following pregnancy. The onset and severity of findings in HHH syndrome are more variable and less severe when compared to UCDs like ornithine transcarbamylase (OTC) deficiency or carbamyl phosphate synthase (CPS-I) deficiency (OMIM 237300). UCDs usually present with isolated elevation in plasma ammonia concentration and metabolic alkalosis. Plasma amino acid analysis and acylcarnitine profile, urine amino acid analysis, urine organic acid analysis, and urine orotic acid measurements allow diagnosis of the specific UCD (see Urea Cycle Disorders) or HHH syndrome. Neonates with a UCD may have hypoglycemia.
A complete chemistry panel (CMP), CBC and differential, lactate determination, arterial blood gases, serum creatine kinase (CK), and urinalysis (to check for ketonuria) should always be included in the evaluation of any person with an elevated plasma ammonia concentration to evaluate for conditions including the following:
- Organic acidemias (present with acidosis)
- Lysinuric protein intolerance (associated with low plasma ornithine, lysine, and arginine)
- Fatty-acid oxidation defects (associated with nonketotic hypoglycemia and liver dysfunction). See MCAD Deficiency.
- Pyruvate carboxylase deficiency (presents with lactic acidosis and hypoglycemia)
- Mitochondrial disease. Given that the neurologic nonacute presentation for HHH syndrome may be indistinguishable from primary mitochondrial disease, urine organic acid analysis should also be ordered. In some cases of HHH syndrome, urinary excretion of Krebs cycle components (succinate, fumarate, citrate, and α-ketoglutarate) and lactate have been reported [Korman et al 2004]. This pattern of excretion of organic acids, which is commonly seen in children and adults with defects in mitochondrial complex I or III, may create the impression that persons with HHH syndrome have a primary rather than a secondary mitochondrial defect.
Hyperornithinemia
The only other condition that causes chronic elevations in plasma ornithine concentration is deficiency of ornithine amino transferase (OAT) (OMIM 258870), a mitochondrial matrix enzyme involved in the ornithine degradation pathway. However, OAT deficiency never presents with the neurologic and clinical biochemical features of HHH syndrome (e.g., elevation in plasma ammonia concentration and glutamine, urinary excretion of homocitrulline and/or orotic acid). OAT deficiency presents mostly with ophthalmologic findings known as hyperornithinemia with gyrate atrophy of the choroid and retina that manifest as chorioretinal degeneration with loss of peripheral vision, night blindness, and often posterior subcapsular cataracts [Kim et al 2013].
Homocitrullinuria
Homocitrulline is a by-product of canned milk production that arises from the reaction of cyanate and the terminal ε-amino group of lysine. In canned formulas, cyanate is produced from heat-induced urea breakdown. When homocitrulline is consumed in the diet from sources such as these, it is absorbed in the small intestine via a transport system similar to that of cationic amino acids and excreted in the urine. In contrast, homocitrullinuria detected in neonates given IV glucose only (and no dietary source of protein) indicates the presence of a metabolic disorder.
Some individuals with lysinuric protein intolerance (LPI) have been shown to excrete homocitrulline [Habib et al 2013]. Although these individuals may also have hyperammonemia, their clinical biochemical profile demonstrates low concentrations of plasma ornithine, lysine, and arginine and persistent urinary excretion of lysine, ornithine, and arginine.
Neurologic Findings
In those individuals with early-childhood onset HHH syndrome in whom gait abnormalities and spasticity predominate, the differential diagnosis should also include early-onset inherited spastic paraplegia (see Hereditary Spastic Paraplegia Overview).
Arginase I deficiency is the only other urea cycle disorder (UCD) in which a prominent manifestation is progressive pyramidal tract involvement leading to spastic paraparesis. In general, spastic paraparesis in arginase I deficiency manifests earlier in childhood.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH) Syndrome
Treatment of Manifestations
It is critical that the acute and long-term management of individuals with HHH syndrome be performed in conjunction with a metabolic specialist. Of primary importance is the rapid control of acute hyperammonemic episodes that may result from changes in diet (e.g., protein intake), infection, dehydration, fasting, injury, or unknown cause. Long-term management focuses on prevention of postprandial hyperammonemia and acute hyperammonemic episodes and efforts to lower the plasma ornithine concentration.
Acute hyperammonemic episodes are treated like hyperammonemic episodes in other UCDs: assessment of plasma ammonia concentration, complete chemistry panel, arterial blood gases, CBC and differential (to evaluate for an infectious process), urinalysis, urine and plasma amino acids, and urine orotic acid. Urine organic acids should also be included to determine possible secondary mitochondrial dysfunction. Viral studies are indicated to evaluate infection as a possible cause of catabolism.
Plasma ammonia concentrations ≥80 μmol/L (~2x control value) should be treated immediately.
Discontinue all oral intake until the affected individual is stabilized to stop all protein intake (a cornerstone of acute treatment) and to reduce the risk of vomiting from hyperammonemia and/or nitrogen-scavenging medications. Of note, use of the recently approved ammonia-scavenging medication glycerol phenylbutyrate (RAVICTI®) should be stopped as it relies on pancreatic lipases and, thus, only works when administered with food [Berry et al 2017].
Provide intravenous fluid with dextrose and intralipids to reverse catabolism while optimizing caloric intake:
- Initial intravenous infusion should be 10% dextrose (with 1/4 normal saline and 20 mEq/L KCl) at twice maintenance; ammonia and glucose/Na/K/Cl/CO2 concentrations should be monitored every two hours or when neurologic status changes.
- It is important to maintain an approximate glucose infusion rate (GIR) between 10 to 15 mg/kg/min in order to prevent catabolism (protein sparring effect) and maintain adequate blood glucose levels between 100 and 150 mg/dL. The concentration of glucose may need to be increased (12.5% to 15%) to prevent fluid overload, particularly when secondary brain edema is present. As the affected individual's metabolic condition stabilizes, it is important to keep a stable GIR and not decrease or stop infusion rate in response to hyperglycemia (>150 mg/dL). An insulin drip should be used to maintain blood glucose at ~150-160 mg/dL during the critical phase of the hyperammonemic event and may be quickly withdrawn or reduced in rate if hypoglycemia develops.
- The use of IV intralipids (2-3 g/kg/day) is an additional source of energy used in individuals with prolonged episodes of hyperammonemia common during periods of infection.
- An individual with HHH syndrome is more likely to respond to the initial IV infusion of dextrose and to normalize the individual's plasma ammonia concentration when compared to an individual with a urea cycle disorder such as OTC deficiency or ASS deficiency. If clinical status does not improve, infusion of supplemental arginine and ammonia removal drugs is added to the regimen.
Of note, as clinical manifestations often do not correlate with rising plasma ammonia concentrations, treatment decisions should consider both plasma ammonia concentration and neurologic status.
Follow published protocols for treatment of acute hyperammonemic episodes similar to those instituted for OTC deficiency (see National Organization for Rare Diseases Physician Guide to the Urea Cycle Disorders) [Brusilow & Horwich 2001]. See also Häberle et al [2019] and Matsumoto et al [2019] for protocols specific to HHH syndrome. These protocols consist of arginine supplementation and use of intravenous bolus and maintenance infusions of the ammonia removal drugs sodium benzoate and sodium phenylacetate.
The New England Consortium of Metabolic Programs complete set of treatment protocols for neonatal hyperammonemia and OTC deficiency focusing on the use of Ammonul® (Ucyclyd Pharma) are freely available. Alfadhel et al [2016] provide an excellent online step-by-step resource for management of hyperammonemia.
It is important to note that unless the affected individual is transferred to a specialized metabolic center, treatment with scavenging agents to remove excess nitrogen and ammonia from the blood will depend on availability of medications.
An initial priming dose of arginine, benzoate, and phenylacetate is given (see Table 5).
Table 5.
Initial Priming Dose of Arginine, Benzoate, and Phenylacetate by Age Group
If ammonia levels stabilize, the same arginine, benzoate, and phenylacetate solution is infused over 24 hrs.
Note: All preparations of arginine and ammonia removal drugs should be double- or triple-checked given the potential for drug intoxication (if high doses are given) or for continued CNS ammonia toxicity (if low doses are given). If sodium benzoate or sodium phenylacetate solutions are not available, infusion of arginine should be started.
- Sodium phenylbutyrate (sodium phenylacetate prodrug) may be given either by mouth or by nasogastric tube (Buphenyl® powder) during acute episodes of hyperammonemia in hospitals that do not carry IV Ammonul® (sodium benzoate/phenylacetate). Moreover, in individuals who use glycerol phenylbutyrate for maintenance at home, sodium phenylbutyrate should continue after formula and food are started until discharge to avoid double medication errors.
- Dose of sodium phenylbutyrate:
- ≤25 kg: 550-600 mg/kg/day
- >25 kg: 9.9-13.0 g/m2/day
Table 6 summarizes mechanisms of drug actions in the treatment of hyperammonemia.
Table 6.
Mechanisms of Drug Action in Treatment of Hyperammonemia
Concurrent with the above, the trigger for the hyperammonemic event (most commonly infection or internal bleeding) should be identified and treated to prevent ongoing protein catabolism and allow for the metabolic stabilization of the affected individual.
Hemodialysis. If the individual fails to respond to the above treatment of hyperammonemia, if the plasma ammonia concentration increases, and/or if the neurologic status deteriorates, dialysis should be started promptly to remove ammonia from the circulation. Infusion of arginine, benzoate, and phenylacetate should continue during dialysis. The preferred method to remove increased levels of ammonia in neonates, infants, and children is continuous renal replacement therapy (CRRT), especially when there is hemodynamic instability [Häberle et al 2019, Matsumoto et al 2019].
Need for dialysis (CRRT) falls into one of the following three categories based on age and plasma ammonia level [Alfadhel et al 2016, Häberle et al 2019]:
- Neonates and children
- Unlikely: <200 µmol/L
- Possible: ≥200-300 µmol/L if no response within four hours
- Definitely: >400-500 µmol/L
- Adolescents and adults. Definitely if ammonia >200µmol/L
Dialysis may be prolonged if the catabolic state (e.g., infection) persists.
Nutrition. Twenty-four to 36 hours after initial admission oral intake should start, including daily doses of only essential amino acids, carnitine, vitamins, and lipids to help avoid a catabolic state, which will prolong hyperammonemia. Note: Nonessential amino acids (i.e., glutamine, proline, and glycine) should be avoided since they increase the nitrogen load to an already compromised urea cycle.
Long-term management includes maintenance on an age-appropriate protein-restricted diet that controls hyperammonemia, but also provides sufficient protein for normal growth and development, especially for infants and children.
The recommended amount of total protein, calories, and fluids consumed per day varies with age. The ratio of natural protein to essential amino acids is approximately 60:40. The Ross Manual [Acosta & Yannicelli 2001] can serve as a guide for the recommended daily nutrient intake for infants, children, and adults. For example, individuals age 12 years or older usually need approximately 0.7-1.0 g/kg/day of total protein.
- Dietary supplementation with Cyclinex®-1 formula (infants and toddlers) or Cyclinex®-2 formula (children, adolescents, and adults) that provides only essential amino acids and other nutritional supplements has been helpful for some individuals. Children older than age 10-12 years and adults with a urea cycle disorder use VITAFLO® essential amino acid supplement.
- Caloric supplements such as ProPhree® (4.5 calories per gram) are available for neonates, infants, and toddlers.
- UCD-Trio® provides isoleucine, leucine, and valine for toddlers and children whose branched-chain amino acid levels are consistently low, often due to ammonia-scavenging drugs.
- Citrulline supplementation at 0.17 g/kg/day or 3.8 g/m2/day is preferred to arginine because citrulline provides better metabolic control and avoids secondary creatine deficiency [Martinelli et al 2015]. Citrulline accepts an aspartate (via the arginosuccinate synthase reaction) and therefore eliminates two amino groups per cycle. Molema et al [2019] reported that in individuals with OTC deficiency, CPS1 deficiency, or HHH syndrome, the relatively greater bioavailability of citrulline leads to higher plasma arginine levels than no supplementation or supplementation with arginine.
- Sodium phenylbutyrate (Buphenyl®) is given in three divided doses either as 450-600 mg/kg/day for individuals ≤25 kg or as 9.9-13.0 g/m2/day for individuals ≥25 kg when glycerol phenylbuyrate (RAVICTI®) is not available. Note that sodium phenylbutyrate initially is imported into the mitochondria where it undergoes β-oxidation to produce phenylacetate.
- Effective January 2019, the FDA approved the use of glycerol phenylbutyrate (RAVICTI®, Horizon Pharma) for individuals of all ages (including neonates) for treatment of hyperammonemic episodes due to urea cycle disorders [Author, personal communication].
- Lysine supplementation is indicated when plasma lysine concentrations are low. Low plasma lysine concentrations have been associated with delayed growth and development.
- Creatine supplementation has also been recommended in HHH syndrome and urea cycle disorders such as OTC and ASS deficiencies [Boenzi et al 2012]. In HHH syndrome, the synthesis of creatine from arginine is decreased due to ornithine inhibition of the first step, which depends on glycine transaminidase.
- Carnitine supplementation may be necessary due to carnitine deficiency secondary to dietary restriction (e.g., meat) and/or conjugation of carnitine with ammonia removal drug metabolites (phenylacetate).
- Plasma concentrations of ammonia, glutamine, arginine, ornithine, and essential amino acids (in particular lysine, isoleucine, leucine, and valine) should be maintained within the normal range.Note: (1) Although elevated plasma ornithine concentrations may decrease significantly if dietary management is followed, complete normalization of plasma ornithine concentration is rarely observed. (2) Even in the absence of hyperammonemic episodes, affected individuals may continue to develop neurologic complications such as spasticity or learning disabilities. Maintaining as low a level of plasma ornithine as possible by restricting protein intake could help prevent some of the progressive neurologic complications seen in these individuals [Valle & Simell 2001].
Three individuals with HHH syndrome who had acute fulminant hepatic failure and coagulopathy rapidly stabilized after protein restriction and arginine or citrulline supplementation [Fecarotta et al 2006, Mhanni et al 2008, Lee et al 2014].
Liver transplantation is not indicated when metabolic control can be achieved with diet, supplemental citrulline, and ammonia-scavenging medications. Because SLC25A15 and the ornithine degradation pathway are expressed in all tissues (e.g., brain, kidney) and most cell types (e.g., astrocytes, fibroblasts), liver transplantation may correct the hyperammonemia, but it will not correct tissue-specific metabolic abnormalities that also contribute to the neuropathology.
Two instances of liver transplantation for HHH syndrome were reported:
- A child age six years with a history of continued biochemical abnormalities, protein intolerance, developmental delay, and abnormal posture [Guan et al 2017];
- A child age seven years with poor metabolic control who had presented at age four weeks with hyperammonemic coma (plasma ammonia 2,300 µmol/L) [Verloo et al 2007].
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician and social worker is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine if any changes are needed.
- As required by special education law, children should be in the least restricted environment feasible at school and included in general education as much as possible and when appropriate.
- Vision and hearing consultants should be a part of the child’s IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual’s needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Surveillance
All surveillance of individuals with HHH syndrome (Table 7) should be a combined effort of the general pediatrician or adult practitioner and a metabolic team (metabolic geneticist, metabolic dietician, and social worker).
Table 7.
Recommended Surveillance for Individuals with Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH) syndrome
Agents/Circumstances to Avoid
Avoid the following:
- Excess dietary protein intake
- Nonprescribed protein supplements such as those used to increase size of skeletal muscle during exercise regimens
- Prolonged fasting during an illness or weight loss
- Use of oral and intravenous steroids
- Valproic acid, which induces and exacerbates hyperammonemia in urea cycle disorders
- Exposure to communicable diseases
Evaluation of Relatives at Risk
Testing of at-risk sibs is warranted to allow for early diagnosis and treatment.
If the SLC25A15 pathogenic variants in the family are known and if prenatal testing has not been performed on an at-risk sib, test for the familial SLC25A15 pathogenic variants in the newborn period while restricting the diet (≤1.2 g/kg/day protein using breastfeeding and a zero-protein formula) and monitoring plasma ammonia, plasma amino acids, and urine orotic acid.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Ho et al [2019] reported pregnancy management and outcomes of four women with HHH syndrome: a patient of their own who had two pregnancies; and three women reported in the literature. They provide a systematic protocol for severe metabolic decompensation during pregnancy.
Management of the affected mother before and during pregnancy to achieve metabolic control to enable normal fetal growth and development. In general, pregnant women should continue dietary protein restriction and supplementation with citrulline and ammonia-scavenging medications (after an appropriate benefit/risk calculation) based on their clinical course before pregnancy. Protein restriction during pregnancy is challenging given the complications that commonly arise during pregnancy (i.e., nausea, vomiting, anorexia) plus the natural aversion to protein exhibited by individuals with HHH syndrome. Due to increased protein and energy requirements in pregnancy and, oftentimes, difficulty with patient adherence, weekly to every two-week monitoring of plasma amino acids and ammonia is recommended, especially in the first and third trimester, and close monitoring immediately after delivery. Plasma amino acid levels can help guide quick adjustments to diet in order to achieve normal plasma amino acid profiles that prevent catabolism and hyperammonemia while allowing for normal fetal growth and development. Protein intake during pregnancy varied from 0.8 g/kg/day to 1.3 g/kg/day (normal dietary reference intakes: 1.1 g/kg/day to 1.2-1.52 g/kg/day) [Ho et al 2019].
Delivery by cesarean section is recommended in order to optimize metabolic control.
Häberle et al [2019] provide a table detailing protein intake and extra energy requirements in pregnancy and lactation.
Management of metabolic complications during pregnancy in women with HHH syndrome. Complications related to HHH syndrome arose during pregnancy and delivery and in the immediate postpartum period in three of the four women reported by Ho et al [2019]. Mild hyperammonemic episodes and seizures during pregnancy were treated by adjustments to dietary protein intake (decrease) and anti-seizure medication (carbamazepine), respectively [Kim et al 2012, Ho et al 2019].
- One woman experienced mild hyperammonemia at the end of the first trimester of her first pregnancy. In her second pregnancy, she experienced metabolic decompensation (ammonia 295 µmol/L) prior to the second trimester and responded well to emergency treatment with Ammonul® and arginine [Ho et al 2019].
- A second woman experienced mild hyperammonemia at the end of the first trimester of her first pregnancy, and developed petit mal seizures in the second trimester and elevations in ammonia (21-103 µmol/L) post partum. In her second pregnancy, she experienced hyperammonemia ten hours post-C-section delivery that responded to treatment with oral sodium benzoate and IV arginine. During her third pregnancy, she had a seizure.
- A third woman experienced mild elevation of plasma ammonia (43-126 µmol/L) during labor.
Normal pregnancy and delivery were documented in two women with HHH syndrome.
- One, diagnosed with HHH syndrome at age 13 years in the course of evaluation of an affected sib, was clinically asymptomatic before pregnancy. During the pregnancy, she was maintained on citrulline and an appropriate protein-restricted diet [Rebecca Mardach, MD, Kaiser Permanente, personal communication].
- The other, a woman age 22 years with no neurologic findings, had learned to self-restrict protein intake after severe protein intolerance in infancy [Ho et al 2019].
Fetal outcomes. Ho et al [2019] summarized the outcome of five previously reported infants born to three women with HHH who were primarily on dietary management or older ammonia-scavenging medications (lactulose) during pregnancy. Three of the five pregnancies resulted in healthy newborns.
- One female had intrauterine growth restriction (IUGR); development at age two years was normal.
- One of the neonates who had normal development at birth experienced transient respiratory distress requiring mechanical ventilation.
There are no well-controlled epidemiologic studies of the fetal effects of sodium benzoate, phenylacetate, or phenylbutyrate during human pregnancy, although there are several case reports.
- Redonnet-Vernhet et al [2000] reported a woman with symptomatic ornithine transcarbamylase (OTC) deficiency who was treated with sodium benzoate during the first 11 weeks of gestation and was subsequently transitioned to sodium phenylbutyrate for the remainder of pregnancy. She delivered a healthy female who continued to do well at age two years.
- Lamb et al [2013] reported another woman with symptomatic OTC who was treated throughout pregnancy with sodium benzoate (4 g/4x/day), sodium phenylbutyrate (2 g/4x/day) and arginine (300 mg/4x/day) who delivered a healthy, unaffected male who was doing well at age six weeks.
- Ho et al [2019] are the first to document the use of sodium phenylbutyrate throughout two sequential pregnancies in a woman with HHH syndrome:
- In the first pregnancy sodium phenylbutyrate (5.5 g/4x/day) was used as maintenance therapy. This resulted in the delivery of a healthy female who was noted to have typical growth and development at age five years.
- In the second pregnancy, emergency treatment with Ammonul® (sodium phenylacetate/sodium benzoate) to manage hyperammonemic crisis (ammonia 295 µmol/L) was used in addition to maintenance therapy of sodium phenylbutyrate (5 g/4x/day).Although the mother responded well to emergency treatment, the baby experienced IUGR and remained in the NICU due to prematurity and low birth weight. At age two years the child exhibited speech delay and autism.How severe metabolic decompensation, elevated plasma ornithine, and/or side effects of sodium phenylbutyrate, phenylacetate, and/or benzoate may have contributed to the speech delay and/or autism is not known.
- Ho et al [2019] prefer and recommend the use of sodium benzoate if deemed medically necessary during pregnancy, but did not advise switching maintenance medications during pregnancy
Theoretic concerns. Sodium benzoate has been reported to lead to malformations and neurotoxicity/nephrotoxicity in zebrafish larvae [Tsay et al 2007]. As a known differentiating agent, sodium phenylbutyrate also functions as a histone deacetylase (HDAC) inhibitor with potential teratogenicity given its ability to alter gene expression in fetal mice [Di Renzo et al 2007]. Theoretically, the use of benzoate/phenylacetate and in particular sodium phenylbutyrate should be avoided during pregnancy, especially during the first trimester. The use of these medications should be carefully evaluated for each individual (benefit/risk ratio) in consultation with a metabolic genetics specialist.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one SLC25A15 pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a SLC25A15 pathogenic variant and allow reliable recurrence risk assessment. Although a de novo pathogenic variant has not been reported in HHH syndrome to date, de novo variants are known to occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017].
- Heterozygotes (carriers) are clinically asymptomatic and are not at risk of developing HHH syndrome.
Sibs of a proband
- If both parents are known to be heterozygous for a SLC25A15 pathogenic variant, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Marked phenotypic variability is observed among sibs who have the same SLC25A15 pathogenic variants [Camacho et al 2006, Debray et al 2008, Tessa et al 2009, Martinelli et al 2015].
- Heterozygotes (carriers) are clinically asymptomatic and are not at risk of developing HHH syndrome [Valle & Simell 2001].
Offspring of a proband. The offspring of an individual with HHH syndrome are obligate heterozygotes (carriers) for a pathogenic variant in SLC25A15.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier.
Carrier Detection
Carrier testing for at-risk family members requires prior identification of the SLC25A15 pathogenic variants in the family.
Carrier testing using clinical biochemical parameters is unreliable; heterozygotes (e.g., parents and carrier sibs) do not exhibit biochemical abnormalities in plasma or urine; therefore, molecular genetic testing is the only reliable method of carrier detection.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC25A15 pathogenic variants have been identified in an affected family member, molecular genetic testing for a pregnancy at increased risk for HHH syndrome and preimplantation genetic testing are possible. However, the identification of familial SLC25A15 pathogenic variants cannot predict clinical outcome as significant intrafamilial phenotypic variability is observed in HHH syndrome.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Connecting Families - Urea Cycle Disorders (UCD) FoundationPhone: 918-490-3055
- National Library of Medicine Genetics Home Reference
- National Urea Cycle Disorders FoundationPhone: 626-578-0833
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Urea Cycle Disorders ConsortiumPhone: 202-306-6489Email: jseminar@childrensnational.org
- European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD)
- National Urea Cycle Disorders Foundation International Patient RegistryEmail: coordinator@ucdparegistry.org
- Urea Cycle Disorders Consortium RegistryChildren's National Medical Center
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome: Genes and Databases
Table B.
OMIM Entries for Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome (View All in OMIM)
Molecular Pathogenesis
Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is a disorder of the urea cycle (UC) and ornithine degradation pathway. The urea cycle (Figure 1) eliminates 80% of toxic nitrogenous waste by converting nitrogen (in the form of ammonia) to urea that is excreted by the kidneys. In humans, protein catabolism generates ammonia, which is a neurotoxin.
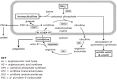
HHH syndrome is caused by pathogenic variants in SLC25A15, which encodes the mitochondrial ornithine transporter, ORNT1. Ornithine, derived from the hydrolysis of arginine in the last step of the UC in the cytosol, must enter the mitochondrial matrix. ORNT1, which translocates cytosolic ornithine into the mitochondrial matrix, is a member of a family of mitochondrial carrier (MCF) proteins that are responsible for the selective transport of solutes into and out of the mitochondrial matrix. Defective ORNT1 results in:
- Absence of intramitochondrial ornithine. The UC is disrupted in the liver and hyperammonemia ensues. The toxic effects of ammonia and glutamine (product of ammonia and glutamate) include brain edema and, if not treated, coma. Moreover, effects on astrocytes, which play a key role in long-term memory, may explain "memory problems" that are common in individuals with HHH syndrome [Brooks 2018, Matsumoto et al 2019].
- Disruption of the ornithine amino transferase reaction, the pathway for ornithine catabolism. Ornithine accumulates, leading to hyperornithinemia.
- Underused carbamoyl phosphate either reacting with lysine to form homocitrulline or entering the pyrimidine pathway to form orotic acid. Consequently, their accumulation leads to increased urinary excretion of homocitrulline (homocitrullinuria) and orotic acid.
- Increased cytosolic ornithine leading to increased polyamine synthesis. Polyamines in excess increase intramitochondrial calcium (promotes apoptosis), mitochondrial autophagy, and cell proliferation [Valle & Simell 2001, Minois et al 2011].
- Inhibition of AGAT, which reduces endogenous creatine synthesis in the cerebrum and cerebellum, increasing the brain’s susceptibility to local ammonia-induced damage [Valle & Simell 2001, Boenzi et al 2012].
Two other mitochondrial ornithine transporters, ORNT2 (SLC25A2) and ORNT3 (SLC25A29), exist; ORNT2 has been shown to co-transport citrulline and, thus, may compensate for a defective ORNT1 in the urea cycle. Hence, gene redundancy may in part be responsible for the later onset and less severe clinical presentation in individuals with HHH syndrome [Camacho & Rioseco-Camacho 2009, Porcelli et al 2014, Monné et al 2015].
Mechanism of disease causation. HHH syndrome is caused by loss of ORNT1 function.
SLC25A15-specific laboratory considerations. Five non-processed pseudogenes derived from SLC25A15 (13q14.11, NC-000013.11) have been identified on three different chromosomes, in addition to chromosome 13:
- Chr Yq11.13 (NG_024787, SLC25A15 pseudogene 1)
- Chr 21p11.2 (NG_025052, SLC25A15 pseudogene 4)
- Chr 22q11.1 (NG_027407, SLC25A15 pseudogene 5)
- Chr 13q12.3 (NG_024903, SLC25A15 pseudogene 3)
- Chr 13q12.11 (NG_032358, SLC25A15 pseudogene 2)
When performing SLC25A15 mRNA analysis, note that SLC25A2 (ORNT2) is intronless and arose by retrotransposition. SLC25A15 and SLC25A2 are 87% and 88% identical at the nucleotide and protein level, respectively [Camacho et al 2003].
Table 8.
Notable SLC25A15 Pathogenic Variants
Chapter Notes
Acknowledgments
We would like to acknowledge Drs William Nyhan, Rebecca Mardach, Yong Qu, Kamer Tezcan, Alicia Chan, Esperanza Font-Montgomery, and J Lawrence Merritt for referring or sharing clinical information of patients with HHH syndrome.
Dr José A Camacho's work was previously supported by a Robert Wood Johnson Foundation-Dr Harold Amos Faculty Development Award.
Revision History
- 13 February 2020 (bp) Comprehensive update posted live
- 31 May 2012 (me) Review posted live
- 17 June 2011 (jac) Original submission
References
Literature Cited
- Acosta PB, Yannicelli S, eds. Nutrition Support Protocols: The Ross Metabolic Formula System. 4 ed. Columbus, OH: Abbott Nutrition; 2001.
- Alfadhel M, Mutairi FA, Makhseed N, Jasmi FA, Al-Thihli K, Al-Jishi E, AlSayed M, Al-Hassnan ZN, Al-Murshedi F, Häberle J, Ben-Omran T. Guidelines for acute management of hyperammonemia in the Middle East region. Ther Clin Risk Manag. 2016;12:479–87. [PMC free article: PMC4820220] [PubMed: 27099506]
- Al-Hassnan ZN, Rashed MS, Al-Dirbashi OY, Patay Z, Rahbeeni Z, Abu-Amero KK. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome with stroke-like imaging presentation: clinical, biochemical and molecular analysis. J Neurol Sci. 2008;264:187–94. [PubMed: 17825324]
- Berry SA, Longo N, Diaz GA, McCandless SE, Smith WE, Harding CO, Zori R, Ficicioglu C, Lichter-Konecki U, Robinson B, Vockley J. Safety and efficacy of glycerol phenylbutyrate for management of urea cycle disorders in patients aged 2 months to 2 years. Mol Genet Metab. 2017;122:46–53. [PubMed: 28916119]
- Boenzi S, Pastore A, Martinelli D, Goffredo BM, Boiani A, Rizzo C, Dionisi-Vici C. Creatine metabolism in urea cycle defects. J Inherit Metab Dis. 2012;35:647–653. [PubMed: 22644604]
- Brooks GA. The science and translation of lactate shuttle theory. Cell Metab. 2018;27:757–85. [PubMed: 29617642]
- Brusilow SW, Horwich AL. Urea cycle enzymes. In Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Basis of Inherited Disease. 8 ed, vol 2. New York, NY: McGraw Hill; 2001:1909-64.
- Bürk K, Sival DA. Scales for the clinical evaluation of cerebellar disorders. Handb Clin Neurol. 2018;154:329–39. [PubMed: 29903450]
- Camacho JA, Rioseco-Camacho N. The human and mouse SLC25A29 mitochondrial transporters rescue the deficient ornithine metabolism in fibroblasts of patients with the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr Res. 2009;66:35–41. [PubMed: 19287344]
- Camacho JA, Mardach R, Rioseco-Camacho N, Ruiz-Pesini E, Derbeneva O, Andrade D, Zaldivar F, Qu Y, Cederbaum SD. Clinical and functional characterization of a human ORNT1 mutation (T32R) in the hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome. Pediatr Res. 2006;60:423–9. [PubMed: 16940241]
- Camacho JA, Obie C, Biery B, Goodman BK, Hu CA, Almashanu S, Steel G, Casey R, Lambert M, Mitchell GA, Valle D. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome is caused by mutations in a gene encoding a mitochondrial ornithine transporter. Nat Genet. 1999;22:151–8. [PubMed: 10369256]
- Camacho JA, Rioseco-Camacho N, Andrade D, Porter J, Kong J. Cloning and characterization of human ORNT2: a second mitochondrial ornithine transporter that can rescue a defective ORNT1 in patients with the hyperornithinemia-hyperammonemia-homocitrullinuria syndrome, a urea cycle disorder. Mol Genet Metab. 2003;79:257–71. [PubMed: 12948741]
- Debray FG, Lambert M, Lemieux B, Soucy JF, Drouin R, Fenyves D, Dube J, Maranda B, Laframboise R, Mitchell GA. Phenotypic variability among patients with hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome homozygous for the delF188 mutation in SLC25A15. J Med Genet. 2008;45:759–64. [PubMed: 18978333]
- Di Renzo F, Broccia ML, Giavini E, Menegola E. Relationship between embryonic histonic hyperacetylation and axial skeletal defects in mouse exposed to the three HDAC inhibitors apicidin, MS-275, and sodium butyrate. Toxicol Sci. 2007;98:582–8. [PubMed: 17517827]
- Fecarotta S, Parenti G, Vajro P, Zuppaldi A, Della Casa R, Carbone MT, Correra A, Torre G, Riva S, Dionisi-Vici C, Santorelli FM, Andria G. HHH syndrome (hyperornithinaemia, hyperammonaemia, homocitrullinuria), with fulminant hepatitis-like presentation. J Inherit Metab Dis. 2006;2006;29:186–9. [PubMed: 16601889]
- Fiermonte G, Dolce V, David L, Santorelli FM, Dionisi-Vici C, Palmieri F, Walker JE. The mitochondrial ornithine transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution of two human isoforms. J Biol Chem. 2003;278:32778–83. [PubMed: 12807890]
- Filosto M, Alberici A, Tessa A, Padovani A, Santorelli FM. Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome in adulthood: a rare recognizable condition. Neurol Sci. 2013;34:1699–701. [PubMed: 23247599]
- Guan HZ, Ding Y, Li DX, Dong H, Song JQ, Jin Y, Zhu ZJ, Sun LY, Yang YL. Clinical diagnosis and treatment of three cases with hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Zhonghua Er Ke Za Zhi. 2017;55:428–33. [PubMed: 28592010]
- Häberle J, Burlina A, Chakrapani A, Dixon M, Karall D, Lindner M, Mandel H, Martinelli D, Pintos-Morell G, Santer R, Skouma A, Servais A, Tal G, Rubio V, Huemer M, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders: First revision. J Inherit Metab Dis. 2019;42:1192–230. [PubMed: 30982989]
- Habib A, Yunus Z, Azize NA, Ch'ng GS, Ong P, Chen BC, Hsu HT, Wong KJ, Pitt J, Ngu LH. Hyperexcretion of homocitrulline in a Malaysian patient with lysinuric protein intolerance. Eur J Pediatr. 2013;172:1277–81. [PubMed: 23358709]
- Ho B, MacKenzie J, Walia J, Geraghty M, Smith G, Nedvidek J, Guerin A. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome in pregnancy: Considerations for management and review of the literature. JIMD Rep. 2019;46:28–34. [PMC free article: PMC6498866] [PubMed: 31240152]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Kim SJ, Lim DH, Kim JH, Kang SW. Gyrate atrophy of the choroid and retina diagnosed by ornithine-delta-aminotransferase gene analysis: a case report. Korean J Ophthalmol. 2013;27:388–91. [PMC free article: PMC3782588] [PubMed: 24082780]
- Kim SZ, Song WJ, Nyhan WL, Ficicioglu C, Mandell R, Shih VE. Long-term follow-up of four patients affected by HHH syndrome. Clin Chim Acta. 2012;413:1151–5. [PubMed: 22465082]
- Korman SH, Kanazawa N, Abu-Libdeh B, Gutman A, Tsujino S. Hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome with evidence of mitochondrial dysfunction due to a novel SLC25A15 (ORNT1) gene mutation in a Palestinian family. J Neurol Sci. 2004;218:53–8. [PubMed: 14759633]
- Kumar K, Agarwal A, Agarwal A, Dhawan A, Chandani N, Raj P. Ocular manifestations in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome: a rare association. Retin Cases Brief Rep. 2015;9:134–7. [PubMed: 25411929]
- Lamb S, Aye CY, Murphy E, Mackillop L. Multidisciplinary management of ornithine transcarbamylase (OTC) deficiency in pregnancy: essential to prevent hyperammonemic complications. BMJ Case Rep. 2013;2013. [PMC free article: PMC3604418] [PubMed: 23283608]
- Lee HH, Poon KH, Lai CK, Au KM, Siu TS, Lai JP, Mak CM, Yuen YP, Lam CW, Chan AY. Hyperornithinaemia-hyperammonaemia-homocitrullinuria syndrome: a treatable genetic liver disease warranting urgent diagnosis. Hong Kong Med J. 2014;20:63–6. [PubMed: 24473688]
- Martinelli D, Diodato D, Ponzi E, Monné M, Boenzi S, Bertini E, Fiermonte G, Dionisi-Vici C. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J Rare Dis. 2015;10:29. [PMC free article: PMC4358699] [PubMed: 25874378]
- Matsumoto S, Häberle J, Kido J, Mitsubuchi H, Endo F, Nakamura K. Urea cycle disorders-update. J Hum Genet. 2019;64:833–47. [PubMed: 31110235]
- Mhanni AA, Chan A, Collison M, Seifert B, Lehotay DC, Sokoro A, Huynh HQ, Greenberg CR. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome (HHH) presenting with acute fulminant hepatic failure. J Pediatr Gastroenterol Nutr. 2008;46:312–5. [PubMed: 18376250]
- Minois N, Carmona-Gutierrez D, Madeo F. Polyamines in aging and disease. Aging (Albany NY). 2011;3:716–32. [PMC free article: PMC3184975] [PubMed: 21869457]
- Molema F, Gleich F, Burgard P, van der Ploeg AT, Summar ML, Chapman KA, Baric I, Lund AM, Kolker S, Williams M. Evaluation of dietary treatment and amino acid supplementation in organic acidurias and urea-cycle disorders: On the basis of information from a European multicenter registry. J Inherit Metab Dis. 2019;42:1162–75. [PubMed: 30734935]
- Monné M, Miniero DV, Daddabbo L, Palmieri L, Porcelli V, Palmieri F. Mitochondrial transporters for ornithine and related amino acids: a review. Amino Acids. 2015;47:1763–77. [PubMed: 26002808]
- Ono H, Tamada T, Shigematsu Y. Lactate/pyruvate in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Pediatr Int. 2018;2018;60:762–4. [PubMed: 30058227]
- Porcelli V, Fiermonte G, Longo A, Palmieri F. The human gene SLC25A29, of solute carrier family 25, encodes a mitochondrial transporter of basic amino acids. J Biol Chem. 2014;289:13374–84. [PMC free article: PMC4036346] [PubMed: 24652292]
- Qadri SK, Ting TW, Lim JS, Jamuar SS. Milder form of urea cycle defect revisited: report and review of hyperornithinaemia-hyperammonaemia-homocitrullinuria (HHH) syndrome diagnosed in a teenage girl presenting with recurrent encephalopathy. Ann Acad Med Singapore. 2016;45:563–6. [PubMed: 28062886]
- Ranucci G, Rigoldi M, Cotugno G, Bernabei SM, Liguori A, Gasperini S, Goffredo BM, Martinelli D, Monti L, Francalanci P, Candusso M, Parini R, Dionisi-Vici C. Chronic liver involvement in urea cycle disorders. J Inherit Metab Dis. 2019;42:1118–27. [PubMed: 31260111]
- Redonnet-Vernhet I, Rouanet F, Pedespan JM, Hocke C, Parrot F. A successful pregnancy in a heterozygote for OTC deficiency treated with sodium phenylbutyrate. Neurology. 2000;54:1008. [PubMed: 10691008]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Salvi S, Dionisi-Vici C, Bertini E, Verardo M, Santorelli FM. Seven novel mutations in the ORNT1 gene (SLC25A15) in patients with hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome. Hum Mutat. 2001a;18:460. [PubMed: 11668643]
- Salvi S, Santorelli FM, Bertini E, Boldrini R, Meli C, Donati A, Burlina AB, Rizzo C, Di Capua M, Fariello G, Dionisi-Vici C. Clinical and molecular findings in hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Neurology. 2001b;57:911–4. [PubMed: 11552031]
- Shih VE, Efron ML, Moser HW. Hyperornithinemia, hyperammonemia, and homocitrullinuria. A new disorder of amino acid metabolism associated with myoclonic seizures and mental retardation. Am J Dis Child. 1969;117:83–92. [PubMed: 5782534]
- Silfverberg T, Sahlander F, Enlund M, Oscarson M, Hardstedt M. Late onset hyperornithinemia-hyperammonemia-homocitrullinuria syndrome - how web searching by the family solved unexplained unconsciousness: a case report. J Med Case Rep. 2018;12:274. [PMC free article: PMC6151189] [PubMed: 30243302]
- Sokoro AA, Lepage J, Antonishyn N, McDonald R, Rockman-Greenberg C, Irvine J, Lehotay DC. Diagnosis and high incidence of hyperornithinemia-hyperammonemia-homocitrullinemia (HHH) syndrome in northern Saskatchewan. J Inherit Metab Dis. 2010;33:S275–81. [PubMed: 20574716]
- Summar ML, Koelker S, Freedenberg D, Le Mons C, Häberle J, Lee HS, Kirmse B. The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179–80. [PMC free article: PMC4364413] [PubMed: 23972786]
- Tessa A, Fiermonte G, Dionisi-Vici C, Paradies E, Baumgartner MR, Chien YH, Loguercio C, de Baulny HO, Nassogne MC, Schiff M, Deodato F, Parenti G, Rutledge SL, Vilaseca MA, Melone MA, Scarano G, Aldamiz-Echevarría L, Besley G, Walter J, Martinez-Hernandez E, Hernandez JM, Pierri CL, Palmieri F, Santorelli FM. Identification of novel mutations in the SLC25A15 gene in hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome: a clinical, molecular, and functional study. Hum Mutat. 2009;30:741–8. [PubMed: 19242930]
- Tsay HJ, Wang YH, Chen WL, Huang MY, Chen YH. Treatment with sodium benzoate leads to malformation of zebrafish larvae. Neurotoxicol Teratol. 2007;29:562–9. [PubMed: 17644306]
- Tsujino S, Kanazawa N, Ohashi T, Eto Y, Saito T, Kira J, Yamada T. Three novel mutations (G27E, insAAC, R179X) in the ORNT1 gene of Japanese patients with hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome. Ann Neurol. 2000;47:625–31. [PubMed: 10805333]
- Valle D, Simell O. The hyperornithinemias. In Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Basis of Inherited Disease. 8 ed, vol 2. New York, NY: McGraw Hill; 2001:1857-96.
- Verloo P, de Meirleir L, Van Hove J, Van Biervliet S, Van Winckel M. Liver transplantation cures biochemical defects in a boy with hyperammonemia-hyperornithinemia-homocitrullinuria (HHH syndrome). J Inherit Metab Dis. 2007;30:85.
- Waisbren SE, Gropman AL, Batshaw ML. Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J Inherit Metab Dis. 2016;39:573–84. [PMC free article: PMC4921309] [PubMed: 27215558]
- Wild KT, Ganetzky RD, Yudkoff M, Ierardi-Curto L. Hyperornithinemia, hyperammonemia, and homocitrullinuria syndrome causing severe neonatal hyperammonemia. JIMD Rep. 2019;44:103–7. [PMC free article: PMC6323011] [PubMed: 30187369]
Publication Details
Author Information and Affiliations
Loma Linda, California
Publication History
Initial Posting: May 31, 2012; Last Update: February 13, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Camacho J, Rioseco-Camacho N. Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome. 2012 May 31 [Updated 2020 Feb 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
