Summary
The purpose of this GeneReview is to:
- 1.
Define HCM;
- 2.
Identify the categories of HCM;
- 3.
Provide the evaluation strategy for a proband with HCM to establish (when possible) the specific genetic cause;
- 4.
Provide a basic view of genetic risk assessment of at-risk asymptomatic relatives of a proband with HCM to inform cardiac surveillance and to allow early detection and treatment of HCM to improve long-term outcome.
1. Hypertrophic Cardiomyopathy: Definition
Hypertrophic cardiomyopathy (HCM) is typically defined by the presence of unexplained left ventricular hypertrophy (LVH) with a maximum wall thickness ≥15 mm in adults or a z score >3 in children [Gersh et al 2011, Elliott et al 2014]. If there is a family history of HCM, or if genetic testing confirms that a relative has inherited the family's pathogenic sarcomere variant, a maximum LV wall thickness ≥13 mm supports diagnosis. Such LVH occurs in a non-dilated ventricle in the absence of other cardiac or systemic disease capable of producing the observed magnitude of increased LV wall thickness, such as pressure overload or storage/infiltrative disorders.
The diagnosis of HCM is most often established with noninvasive cardiac imaging, including echocardiography and/or cardiac magnetic resonance imaging (cardiac MRI).
While asymmetric septal hypertrophy is the most common pattern of hypertrophy, the degree and location of hypertrophy vary. LVH can be concentric, or confined to other walls or the LV apex.
Findings on transthoracic echocardiography may also include:
Systolic anterior motion (SAM) of the mitral valve with associated left ventricular outflow tract obstruction and mitral regurgitation;
Mid-ventricular obstruction as a result of systolic cavity obliteration;
Diastolic dysfunction, including restrictive physiology. Of note, impaired LV relaxation can be detected in individuals with a pathogenic variant in a gene that encodes a component of the sarcomere (see
Nonsyndromic HCM) who have normal LV wall thickness [
Nagueh et al 2001,
Ho et al 2002], suggesting that diastolic dysfunction is an early phenotype of HCM rather than a secondary consequence of LVH.
Although LVH and a clinical diagnosis of HCM often become apparent during adolescence around the onset of puberty or during young adulthood, onset can be earlier (in infancy and childhood) or later in life [Niimura et al 2002]. Common symptoms include shortness of breath (particularly with exertion), chest pain, palpitations, orthostasis, presyncope, and syncope.
Variability and progression. The clinical manifestations of HCM are highly variable, ranging from asymptomatic LVH to arrhythmias (atrial fibrillation as well as malignant ventricular arrhythmias) to refractory heart failure. Moreover, manifestations can vary even within the same family.
Approximately one third of persons with HCM have detectable intracavitary obstruction at rest, and another third of persons with HCM can develop outflow tract obstruction with provocation (e.g., reduction of preload or afterload) [Maron et al 2003, Elliott et al 2014]. The degree of obstruction does not strictly correlate with the severity of symptoms or risk for sudden cardiac death. Observational studies have reported that persons with HCM with outflow tract obstruction may be at higher risk for symptom progression and death than those without outflow tract obstruction [Maron et al 2003, Sorajja et al 2009]; high gradients may also be well tolerated over long periods of time.
Individuals with HCM are at an increased risk for atrial fibrillation (AF), which can have significant morbidity due to increased risk of thromboembolism and symptomatic deterioration. The prevalence of AF increases with age and duration of disease. The overall prevalence of AF in individuals with HCM is ~20%, but prevalence is ~60% by age 60 years for individuals diagnosed with HCM by age 40 years [Elliott et al 2014, Ho et al 2018]. In individuals with HCM and AF the prevalence of thromboembolic complications has been estimated at 27% [Elliott et al 2014].
Approximately 5%-10% of individuals with HCM progress to end-stage disease with impaired systolic function and, in some cases, left ventricular dilatation and regression of LVH. The annual mortality rate in individuals with end-stage disease is estimated at 11% [Harris et al 2006] and cardiac transplantation may be required.
Sudden cardiac death (SCD), most likely related to ventricular tachycardia / ventricular fibrillation, is an important but relatively rare consequence of HCM.
Life span. Compared to the US general population, the mortality rate in individuals with HCM is approximately threefold higher, but the mortality rate in younger individuals with HCM, ages 20-29, is as much as fourfold higher than expected. Sudden death accounts for 16% of deaths [Ho et al 2018].
2. Hypertrophic Cardiomyopathy: Categories
The differential diagnosis for HCM includes increased left ventricular wall thickness due to acquired, syndromic (with other systemic involvement), and nonsyndromic (without other systemic involvement) disorders.
Acquired (Secondary) Left Ventricular Hypertrophy
Secondary left ventricular hypertrophy (LVH) can be pathologic, occurring in response to pressure overload (e.g., systemic hypertension, aortic stenosis). This type of adverse remodeling can lead to diastolic abnormalities and heart failure. Physiologic hypertrophy (athlete's heart) may result from rigorous athletic training. Such training may result in increased left ventricular wall thickness accompanied by increased LV cavity size. This type of remodeling is thought to be adaptive and not associated with adverse consequences. Both pathologic and physiologic forms of secondary hypertrophy can regress if the underlying stimulus is removed (e.g., by adequate treatment of high blood pressure or a period of detraining for an athlete).
Syndromic HCM (with Other Systemic Involvement)
In GeneReviews, "syndromic" refers to a disorder characterized by a constellation of phenotypic features that either (a) specifically suggests the diagnosis (which can be confirmed by molecular genetic testing) or (b) allows diagnosis of the disorder in the absence of confirmatory molecular genetic findings. A selected list of syndromic HCM is provided in Table 1. Syndromic HCM will not be discussed further in this overview.
Table 1.
Syndromic Hypertrophic Cardiomyopathy – A Selected List
View in own window
AD = autosomal dominant; AR = autosomal recessive; MOI = mode of inheritance; XL = X-linked
- 1.
Disorders are in alphabetic order.
- 2.
The RASopathies are a group of syndromes that have overlapping clinical features resulting from a common pathogenetic mechanism [Tidyman & Rauen 2009].
Nonsyndromic HCM (HCM without Other Systemic Involvement)
Individuals with HCM who do not have acquired (secondary) HCM or syndromic HCM (Table 1) have nonsyndromic hypertrophic cardiomyopathy (defined in this GeneReview as HCM with no other systemic involvement). For the remainder of this GeneReview, genes causing HCM without other systemic involvement will be referred to as HCM genes. See Table 2 for a current list of known HCM genes; the strength of the evidence associating each gene with HCM varies significantly [Ingles et al 2019]. The genes with the strongest clinical validity encode different components of the sarcomere [Ingles et al 2019].
Pathogenic variants in one of the genes encoding a component of the sarcomere are found in approximately 50%-60% of probands (adults and children) with a family history of HCM, and approximately 20%-30% of probands without a family history of HCM [Alfares et al 2015]. Approximately 3%-5% of affected individuals have more than one sarcomere gene variant (either biallelic variants in 1 gene or heterozygous variants in >1 gene) although fewer than 1% will have more than one pathogenic or likely pathogenic variant [Alfares et al 2015, Burns et al 2017].
The Clinical Genome Resource (ClinGen) HCM Gene Curation Expert Panel has classified HCM genes using the ClinGen framework for the strength of their relationship with monogenic, nonsyndromic HCM. A summary of the data curated for each gene can be accessed via the links provided in the ClinGen Classification column (see also Ingles et al [2019]).
Table 2.
Hypertrophic Cardiomyopathy Genes
View in own window
AD = autosomal dominant; AR = autosomal recessive; ARVC = arrhythmogenic right ventricular cardiomyopathy; DCM = dilated cardiomyopathy; LGMD2G = limb-girdle muscular dystrophy type 2G; LGMD2J = limb-girdle muscular dystrophy type 2J; MOI = mode of inheritance
- 1.
Genes are ordered first by validity classification, then frequency of causation of HCM, and then alphabetically.
- 2.
Prevalence data list for genes included in Alfares et al [2015]. "Rare" denotes genes not included in this paper.
- 3.
Allelic disorders = other phenotypes caused by pathogenic variants in the same gene
- 4.
PLN and ACTN2 were curated for intrinsic cardiomyopathy given their association with a spectrum of cardiac phenotypes, including isolated LVH and HCM.
3. Establishing (when Possible) the Specific Genetic Cause of Hypertrophic Cardiomyopathy
Genetic testing is recommended (1) in individuals fulfilling diagnostic criteria for HCM to enable cascade screening of relatives and (2) to confirm the diagnosis in individuals with clinical evidence that is suggestive of HCM [Elliott et al 2014].
The purposes of establishing a molecular diagnosis of HCM are to (1) identify syndromic HCM (see Table 1) that could have different treatment and/or management and (2) inform risk assessment of relatives of a proband (see Genetic Risk Assessment and Cardiac Surveillance). Elements of the personal and family history (i.e., family history of HCM, septal morphology, younger age at diagnosis) have been associated with a higher likelihood of a positive genetic test result [Ingles et al 2013, Bos et al 2014].
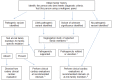
A general approach to identifying the specific genetic cause in individuals with hypertrophic cardiomyopathy (HCM) is summarized in . Genetic testing for HCM is best viewed as a family test rather than a test of an individual since results are most accurately interpreted after integrating genetic and medical test (echocardiogram, EKG) results from multiple family members. In this manner, a cohesive understanding of the family's phenotype can be used to determine if variants segregate with the disorder within the family (the suspected pathogenic variant should be present in affected family members, but not in unaffected family members).
Familial hypertrophic cardiomyopathy: algorithm for genetic testing and clinical cardiac screening Notes:
Initial evaluation should always include the following.
Family history. Effort should be made to obtain a three-generation family history with attention to other relatives with a history of any of the following: heart failure, HCM, cardiac transplantation, unexplained or sudden death (particularly in relatives age <40 years), cardiac conduction system disease and/or arrhythmia, or unexplained stroke or other thromboembolic disease. Note: The family member who will undergo molecular genetic testing should have an unequivocal diagnosis of HCM and is, ideally, the most severely affected person in the family.
Note: (1) Confirming the absence of other affected relatives can be complicated by various medical issues (e.g., failure to undergo appropriate cardiac screening, reduced penetrance, early death from other causes before onset of HCM) and/or social issues (e.g., isolation from family members, undisclosed adoption, and alternate paternity or maternity). Thus, it may not be possible to distinguish whether the proband with HCM is truly a simplex case (i.e., a single occurrence in the family). (2) Clinical evaluation of family members can provide valuable information about the family history (e.g., diagnosis of a family member with previously unrecognized HCM). (3) Family history should be reviewed and updated periodically.
Genetic counseling is recommended for all individuals with HCM, whether or not genetic testing will be used [Elliott et al 2014].
Molecular genetic testing for HCM relies on use of multigene panels, which are comprehensive (i.e., comprising genes known to be associated with HCM and genes associated with a variety of genetic cardiomyopathies). See Table 2 for a current list of known HCM genes.
An HCM multigene panel that includes the genes with established clinical validity listed in Table 2 is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 2).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
When an HCM multigene panel is not diagnostic, exome (or genome) sequencing is another possible testing method, though the anticipated incremental yield is low [Cirino et al 2017b]. If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Health care providers ordering genetic testing should be familiar with the genetics of HCM. Given the complexity of interpreting genetic test results and their implications for surveillance and management, health care providers should consider referral to a cardiovascular genetics center or a genetic counselor specializing in cardiac genetics (see NSGC – Find a Genetic Counselor). Basic guidance on the clinical implications of different variant classification categories can be found in Table 3.
Table 3.
Variant Classification Categories and Clinical Implications
View in own window
| Variant Classification |
|---|
| Benign or No Variant Found | Likely Benign | Of Uncertain Significance | Likely Pathogenic | Pathogenic |
|---|
|
Meaning
|
NEGATIVE
|
UNCERTAIN
|
POSITIVE
|
| No important variants detected. Genetic disease cannot be excluded. | Variant detected is likely harmless.
Genetic disease cannot be excluded. | Ambiguous result;
insufficient data re variant pathogenicity.
Segregation studies may provide additional data. | Likely cause of HCM;
segregation studies may provide additional evidence for causality. | Responsible for causing HCM |
Utility for
proband
| None | None | Unknown | Suggests HCM diagnosis; may inform mgmt or lead to additonal diagnostic studies. | Establishes HCM diagnosis.
May inform mgmt. |
Utility for
family
| No option for predictive genetic testing;
rely on longitudinal phenotypic eval. | No option for predictive genetic testing;
rely on longitudinal phenotypic eval. | Predictive genetic not recommended;
rely on longitudinal phenotypic eval.
Consider segregation testing in affected relatives. | Predictive genetic testing should be approached carefully & may be combined w/phenotypic eval & surveillance. | Can be used for predictive genetic testing. |
4. Genetic Risk Assessment and Cardiac Surveillance of At-Risk Relatives for Detection of Early Treatable Manifestations of Hypertrophic Cardiomyopathy
Practice guidelines recommend construction of a three- (or more) generation family history in all persons with HCM to help identify at-risk family members [Hershberger et al 2018]. At-risk family members should seek clinical evaluation according to the guidelines listed in Table 4 and be offered genetic testing if a pathogenic variant has been identified in an affected family member. Consideration should be given to the timing of testing in unaffected children and the potential benefits and harms to the child should be weighed [Elliott et al 2014].
If the pathogenicity of the variant identified in the affected family member is uncertain (i.e., likely pathogenic or of unknown significance), testing other affected family members as part of a segregation analysis can help in variant interpretation; observation that the variant occurs with HCM in other affected family members (and does not occur in those with a normal phenotype) provides further support for pathogenicity.
The number of relatives tested is an important consideration, as the a priori chance that the variant is present in first-degree relatives is 50%. In contrast, the absence of the variant in a single affected individual provides strong evidence that the variant is not pathogenic.
It is appropriate to combine genetic testing with clinical evaluation in at-risk relatives to provide more comprehensive information about the disease and variant transmission in the family.
When pathogenicity of a variant is refuted by segregation analysis, this information should be relayed back to the genetic testing laboratory.
If the variant identified in the tested family member is of uncertain significance, testing of unaffected at-risk family members for the variant is not helpful, as this information will not aid in interpretation of the variant and will not reliably modify the a priori risk to that relative of developing HCM.
If no variant is identified in the tested family member, no further genetic testing can be pursued (at this time) to clarify the genetic status of at-risk family members.
Genetic Risk Assessment
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Hypertrophic cardiomyopathy (HCM) is typically inherited in an autosomal dominant manner; pathogenic variants in genes associated with autosomal recessive inheritance have been reported in rare families.
In rare instances, more than one pathogenic variant in a gene encoding a sarcomere protein has been identified in a single individual (i.e., double heterozygosity) [Alfares et al 2015, Burns et al 2017]. Therefore, determining the pattern of inheritance for each variant in an individual is critical for accurate risk assessment of other family members.
Autosomal Dominant Inheritance – Risk to Family Members
Parents of a proband
Some individuals diagnosed as having HCM have an affected parent.
A proband with HCM may also have the disorder as the result of a de novo pathogenic variant. The proportion of cases caused by de novo pathogenic variants is unknown.
Recommendations for the evaluation of parents of a proband with an apparent de novo pathogenic variant include molecular genetic testing for the pathogenic variant identified in the proband, echocardiogram, EKG, and physical examination by a cardiologist familiar with HCM.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the pathogenic variant most likely occurred
de novo in the proband. Another possible explanation is that the proband inherited a pathogenic variant from a parent with germline mosaicism. A case of germline mosaicism has been reported [
Forissier et al 2000] but the incidence is unknown.
Note: Although some individuals diagnosed with HCM have an affected parent, the family history may appear to be negative because of failure to recognize the disorder in asymptomatic or mildly symptomatic family members, early death of the parent before the onset of symptoms, or late onset of the disease in the affected parent. Therefore, an apparently negative family history cannot be confirmed unless appropriate clinical evaluation and/or molecular genetic testing has been performed on the parents of the proband.
Sibs of a proband. The risk to sibs depends on the genetic status of the proband's parents:
If a parent of the proband has the pathogenic variant, the risk to the sibs of inheriting the allele is 50%. However, clinical severity and age of onset cannot be predicted in sibs who inherit a familial HCM-related pathogenic variant.
If the pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be ~1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
Offspring of a proband. Each child of an individual with HCM has a 50% chance of inheriting the pathogenic variant and therefore being at risk for developing HCM. However, penetrance may be reduced and clinical severity and age of onset cannot be predicted in offspring who inherit a familial HCM-related pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected and/or has an HCM-related pathogenic variant, the parent's family members may be at risk.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being a carrier, and a 25% chance of not being a carrier.
Typically, risk of disease in heterozygotes (carriers) is not increased over that of the general population; however,
ALPK3 carriers may be at risk of developing cardiomyopathy in adulthood [
Almomani et al 2016].
Offspring of a proband. The offspring of an individual with autosomal recessive HCM are obligate heterozygotes (carriers) for an HCM-related pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an HCM-related pathogenic variant.
Cardiac Surveillance
It is appropriate to clarify the clinical and genetic status of asymptomatic family members at risk for HCM prior to the onset of manifestations to identify those with asymptomatic HCM and allow early diagnosis for those yet to develop disease.
Family members of an affected individual who has a known pathogenic variant in an HCM-related gene. If a definitive pathogenic variant is identified in the affected individual, predictive molecular genetic testing can be performed in at-risk relatives to clarify their genetic risk. Those identified as heterozygous for the pathogenic variant present in the affected family member (and thus at high risk for developing HCM) should undergo clinical cardiovascular screening by physical examination, EKG, and echocardiography performed in accordance with published recommendations [Ommen et al 2020] (see Table 4a).
Table 4a.
Guidelines for Clinical Screening of Asymptomatic Family Members of a Proband With a Known HCM-Related Pathogenic Variant
View in own window
| Genetic Status | Age of Asymptomatic Relative 1 | Risk for Developing HCM | When To Initiate Screening | Repeat EKG & 2D Echo 2 |
|---|
| Heterozygous for the familial HCM-related pathogenic variant | Children & adolescents | High 3 | At the time HCM is diagnosed in another family member | Every 1-2 yrs |
| Adults | Every 3-5 yrs |
| Not heterozygous for the familial HCM-related pathogenic variant | Children & adolescents | Not at ↑ risk | May be discharged from cardiac surveillance | NA |
| Adults |
Echo = echocardiography; EKG = electrocardiogram; NA = not applicable
- 1.
Includes first-degree relatives. May include more distant relatives based on clinical judgment.
- 2.
Screening interval may be modified based on symptom development and/or family history
- 3.
Lorenzini et al [2020] found ~50% penetrance over 15 years of follow up in at-risk relatives who were heterozygous for the familial HCM-related pathogenic variant.
Family members of an affected individual in whom the specific genetic cause of HCM has not been identified. Clinical cardiovascular screening by physical examination, EKG, and echocardiography should be performed in accordance with published recommendations [Ommen et al 2020] in all asymptomatic first-degree relatives (adults and children) of an individual with HCM in whom a pathogenic variant has not been identified (see Table 4b).
Table 4b.
Guidelines for Clinical Screening of Asymptomatic Family Members of a Proband in Whom the Specific Genetic Cause of HCM Has Not Been Identified
View in own window
| Age of Asymptomatic Relative 1 | Age of Onset in Affected Family Member(s) | When To Initiate Screening | Repeat EKG & 2D Echo 2 |
|---|
| Children & adolescents | Early onset (i.e., onset in infancy &/or childhood) | At the time HCM is diagnosed in another family member | Every 1-2 years |
| Onset during or after puberty | At the time HCM is diagnosed in another family member but no later than puberty | Every 2-3 years |
| Adults | Any age | At the time HCM is diagnosed in another family member | Every 3 to 5 years |
Echo = echocardiography; EKG = electrocardiogram
- 1.
Includes first-degree relatives. May include more distant relatives based on clinical judgment.
- 2.
Screening interval may be modified based on symptom development and/or family history.
Because penetrance of diagnostic features (i.e., LVH) is age dependent, a single unremarkable evaluation does not exclude the possibility of future development of HCM. Diagnostic clinical manifestations are often not present in infancy / early childhood; they commonly develop during adolescence and early adulthood, but may also develop late in life. Therefore, longitudinal follow up is required at a frequency based on the individual's age and family history and physician discretion. Screening should be performed in response to any symptoms that develop or any change in clinical status.
Note: As understanding of human DNA variation evolves, it is possible that the classification of a variant will change, potentially affecting the recommendations made to a person/family. It is recommended that families of individuals with HCM follow up with a cardiovascular genetics clinic and/or genetic counselor on a regular basis as genetic testing methods improve.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Children's Cardiomyopathy Foundation
Hypertrophic Cardiomyopathy Association (HCMA)
Phone: 973-983-7429
Email: support@4hcm.org
American Heart Association
Phone: 800-242-8721
Cardiomyopathy UK
United Kingdom
Phone: 0800 018 1024 (UK only)
Email: contact@cardiomyopathy.org
Chapter Notes
Revision History
8 July 2021 (aa/ha) Revision: in
Table 2 – added ClinGen gene validity hyperlinks, updated allelic disorders information, removed
OBSCN11 February 2021 (aa/ha) Revision: added 2020 AHA/ACC Guidelines and revised screening guidelines provided (
Table 4a,
Table 4b) for consistency with 2020 AHA/ACC Guidelines
6 June 2019 (ha) Comprehensive update posted live
16 January 2014 (me) Comprehensive update posted live
5 August 2008 (me) Review posted live
11 June 2007 (ac) Original submission
References
Published Guidelines / Consensus Statements
Colan SD, Lipshultz SE, Lowe AM, Sleeper LA, Messere J, Cox GF, Lurie PR, Orav EJ, Towbin JA. Epidemiology and case-specific outcomes in hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Available
online. 2007. Accessed 8-16-23.
Elliott PM ,Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy. Eur Heart J. 2014;35:2733-79. [
PubMed]
Garratt CJ, Elliott P, Behr E, Camm AJ, Cowan C, Cruickshank S, Grace A, Griffith MJ, Jolly A, Lambiase P, McKeown P, O'Callagan P, Stuart G, Watkins H. Heart Rhythm UK position statement on clinical indications for implantable cardioverter defibrillators in adult patients with familial sudden cardiac death syndrome. Available
online. 2010. Accessed 8-16-23.
Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines. Available
online. 2011. Accessed 8-16-23.
Literature Cited
Alfares
AA, Kelly
MA, McDermott
G, Funke
BH, Lebo
MS, Baxter
SB, Shen
J, McLaughlin
HM, Clark
EH, Babb
LJ, Cox
SW, DePalma
SR, Ho
CY, Seidman
JG, Seidman
CE, Rehm
HL. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity.
Genet Med.
2015;17:880-8.
[
PubMed: 25611685]
Almomani
R, Verhagen
JMA, Herkert
JC, Brosens
E, van Spaedonck-Zwarts
KY, Asimaki
A, van der Zwaag
PA. Frohn-Mulder IME, Bertollo-Avella AM, Boven LG, van Slegtenhorst MA, van der Smagt JJ, van IJcken WFJ, Timmer B, van Stuijvenberg M, Verdijk RM, Saffitz JE, du Plessis FA, Michels M, Hofstra RM, Sinke RJ, van Tintelen JP, Wessels MW, Jongbloed JD, van de Laar IM. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy.
J Am Coll Cardiol.
2016;67: 515-25.
[
PubMed: 26846950]
Bagnall
RD, Weintraub
RG, Ingles
J, Duflou
J, Yeates
L, Lam
L, Davis
AM, Thompson
T, Connell
V, Wallace
J, Naylor
C, Crawford
J, Love
DR, Hallam
L, White
J, Lawrence
C, Lynch
M, Morgan
N, James
P, du Sart
D, Puranik
R, Langlois
N, Vohra
J, Winship
I, Atherton
J, McGaughran
J, Skinner
JR, Semsarian
C. N Engl J Med.
2016;374: 2441-52.
[
PubMed: 27332903]
Bos
JM, Will
ML, Gersh
BJ, Kruisselbrink
TM, Ommen
SR, Ackerman
MJ. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy.
Mayo Clin Proc
2014;89:727-37.
[
PMC free article: PMC4234122] [
PubMed: 24793961]
Burns
C, Bagnall
RD, Lam
L, Semsarian
C, Ingles
J. Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing.
Circ Cardiovasc Genet.
2017;10:e001666.
[
PubMed: 28790153]
Cirino
AL, Harris
S, Lakdawala
NK, Michels
M, Olivotto
I, Day
SM, Abrams
DJ, Charron
P, Caleshu
C, Semsarian
C, Ingles
J, Rakowski
H, Judge
DP, Ho
CY. Role of genetic testing in inherited cardiovascular disease: a review.
JAMA Cardiol.
2017a;2:1153-60.
[
PubMed: 28793145]
Cirino
AL, Lakdawala
NK, McDonough
B, Conner
L, Adler
D, Weinfeld
M, O'GaraP, Rehm HL, Machini K, Lebo M, Blout C, Green RC, MacRae CA, Seidman CE, Ho CY. A comparison of whole genome sequencing to multigene panel testing in hypertrophic cardiomyopathy patients.
Circ Cardiovasc Genet.
2017b;10:e001768.
[
PMC free article: PMC5683423] [
PubMed: 29030401]
Eckart
RE, Shry
EA, Burke
AP, McNear
JA, Appel
DA, Castillo-Rojas
LM, Avedissian
L, Pearse
LA, Potter
RN, Tremaine
L, Gentlesk
PJ, Huffer
L, Reich
SS, Stevenson
WG. Sudden death in young adults: autopsy-based series of a population undergoing active surveillance.
J Am Coll Cardiol.
2011;58:1254-61.
[
PubMed: 21903060]
Elliott
PM, Anastasakis
A, Borger
MA, Borggrefe
M, Cecchi
F, Charron
P, Hagege
AA, Lafont
A, Limongelli
G, Mahrholdt
H, McKenna
WJ, Mogensen
J, Nihoyannopoulos
P, Nistri
S, Pieper
PG, Pieske
B, Rapezzi
C, Rutten
FH, Tillmanns
C, Watkins
H. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy.
Eur Heart J.
2014;35:2733-79.
[
PubMed: 25173338]
Finocchiaro
G, Papadakis
M, Tanzarella
G, Dhutia
H, Miles
C, Tome
M, Behr
ER, Sharma
S, Sheppard
MN. Sudden death can be the first manifestation of hypertrophic cardiomyopathy: data from a United Kingdom pathology registry.
JACC Clin Electrophysiol.
2019;5:252-4.
[
PubMed: 30784699]
Forissier
JF, Richard
P, Briault
A, Ledeuil
C, Dubourg
O, Charbonnier
B, Carrier
L, Moraine
C, Boone
G, Komajda
M, Schwartz
K, Hainque
B. First description of germline mosaicism in familial hypertrophic cardiomyopathy.
J Med Genet.
2000;37:132-4.
[
PMC free article: PMC1734529] [
PubMed: 10662815]
Gersh
BJ, Maron
BJ, Bonow
RO, Dearani
JA, Fifer
MA, Link
MS, Naidu
SS, Nishimura
RA, Ommen
SR, Rakowski
H, Seidman
CE, Towbin
JA, Udelson
JE, Yancy
CW. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force of Practice Guidelines.
Circulation.
2011;124:2761-96.
[
PubMed: 22068435]
Harmon
KG, Drezner
JA, Maleszewski
JJ, Lopez-Anderson
M, Owns
D, Prutkin
JM, Asif
IM, Klossner
D, Ackerman
MJ. Pathogenesis of sudden cardiac death in national collegiate athletic associated athletes.
Circ Arrhythm Electrophysiol.
2014;7:198-204.
[
PubMed: 24585715]
Harris
KM, Spirito
P, Maron
MS, Zenovich
AG, Formisano
F, Lesser
JR, Mackey-Bojack
S, Manning
WJ, Udelson
JE, Maron
BJ. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy.
Circulation.
2006;114:216-25.
[
PubMed: 16831987]
Hershberger
RE, Givertz
MM, Ho
CY, Judge
DP, Kantor
PF, McBride
KL, Morales
A, Taylor
MRG, Vatta
M, Ware
S. Genetic evaluation of cardiomyopathy- a Heart Failure Society of America practice guideline.
J Card Fail.
2018:24:281-302.
[
PMC free article: PMC9903357] [
PubMed: 29567486]
Ho
CY, Day
SM, Ashley
EA, Michels
M, Pereira
AC, Jacoby
D, Cirino
AL, Fox
JC, Lakdawala
NK, Ware
JS, Caleshu
CA, Helms
AS, Colan
SD, Girolami
F, Cecchi
F, Seidman
CE, Sajeev
G, Signorovitch
J, Green
EM, Olivotto
I, et al.
Genotype and lifetime burden of disease in hypertrophic cardiomyopathy. Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe).
Circulation
2018;138:1387-98.
[
PMC free article: PMC6170149] [
PubMed: 30297972]
Ho
CY, Sweitzer
NK, McDonough
B, Maron
BJ, Casey
SA, Seidman
JG, Seidman
CE, Solomon
SD. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy.
Circulation.
2002;105:2992–7.
[
PubMed: 12081993]
Ingles
J, Goldstein
J, Thaxton
C, Caleshu
C, Corty
EW, Crowley
SB, Dougherty
K, Harrison
SM, McGlaughon
J, Milko
LV, Morales
A, Seifert
BA, Strande
N, Thomson
K, Peter van Tintelen
J, Wallace
K, Walsh
R, Wells
Q, Whiffin
N, Witkowski
L, Semsarian
C, Ware
JS, Hershberger
RE, Funke
B. Evaluating the clinical validity of hypertrophic cardiomyopathy genes.
Circ Genom Precis Med.
2019;12:e002460.
[
PMC free article: PMC6410971] [
PubMed: 30681346]
Ingles
J, Sarina
T, Yeates
L, Hunt
L, Macciocca
I, McCormack
L, Winship
I, McGaughran
J, Atherton
J, Semsarian
C. Clinical predictors of genetic testing outcomes in hypertrophic cardiomyopathy.
Genet Med.
2013;15:972-7.
[
PubMed: 23598715]
Lorenzini
M, Norrish
G, Field
E, Ochoa
JP, Cicerchia
M, Akhtar
MM, Syrris
P, Lopes
LR, Kaski
JP, Elliott
PM. Penetrance of Hypertrophic Cardiomyopathy in Sarcomere Protein Mutation Carriers.
J Am Coll Cardiol.
2020;76:550-9.
[
PMC free article: PMC7397507] [
PubMed: 32731933]
Maron
BJ. Sudden death in young athletes.
N Engl J Med.
2003;349:1064-75.
[
PubMed: 12968091]
Maron
BJ, Olivotto
I, Spirito
P, Casey
SA, Bellone
P, Gohman
TE, Graham
KJ, Burton
DA, Cecchi
F. Epidemiology of hypertrophic cardiomyopathy-related death: revisited in a large non-referral-based patient population.
Circulation.
2000;102:858-64.
[
PubMed: 10952953]
Maron
MS, Olivotto
I, Betocchi
S, Casey
SA, Lesser
JR, Losi
MA, Cecchi
F, Maron
BJ. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy.
N Engl J Med.
2003;348:295-303.
[
PubMed: 12540642]
Nagueh
SF, Bachinski
LL, Meyer
D, Hill
R, Zoghbi
WA, Tam
JW, Quinones
MA, Roberts
R, Marian
AJ. Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy.
Circulation.
2001;104:128-30.
[
PMC free article: PMC2900859] [
PubMed: 11447072]
Niimura
H, Patton
KK, McKenna
WJ, Soults
J, Maron
BJ, Seidman
JG, Seidman
CE. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly.
Circulation.
2002;105:446-51.
[
PubMed: 11815426]
Ommen
SR, Mital
S, Burke
MA, Day
SM, Deswal
A, Elliott
P, Evanovich
LL, Hung
J, Joglar
JA, Kantor
P, Kimmelstiel
C, Kittleson
M, Link
MS, Maron
MS, Martinez
MW, Miyake
CY, Schaff
HV, Semsarian
C, Sorajja
P. 2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy: Executive Summary: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines.
Circulation.
2020;142:e533-e557.
[
PubMed: 33215938]
Rahbari
R, Wuster
A, Lindsay
SJ, Hardwick
RJ, Alexandrov
LB, Turki
SA, Dominiczak
A, Morris
A, Porteous
D, Smith
B, Stratton
MR, Hurles
ME, et al.
Timing, rates and spectra of human germline mutation.
Nat Genet.
2016;48:126-33.
[
PMC free article: PMC4731925] [
PubMed: 26656846]
Sorajja
P, Nishimura
RA, Gersh
BJ, Dearani
JA, Hodge
DO, Wiste
HJ, Ommen
SR. Outcome of mildly symptomatic or asymptomatic obstructive hypertrophic cardiomyopathy: a long-term follow-up study.
J Am Coll Cardiol.
2009;54:234-41.
[
PubMed: 19589436]