Isolated sulfite oxidase deficiency (ISOD), a rare inborn error of metabolism of sulfur-containing amino acids, comprises a spectrum ranging from classic early-onset (severe) disease to late-onset (mild) disease [Johnson & Duran 2001].
Classic ISOD
Classic ISOD typically manifests within few hours to days of life with intractable seizures, feeding difficulties, and rapidly progressive encephalopathy [Sass et al 2010, Salih et al 2013, Bosley et al 2014]. The clinical features resemble that of neonatal hypoxic ischemic encephalopathy.
Almost all affected infants are born after uncomplicated pregnancy. Of note, sustained abdominal trembling resembling vibration of mobile phones (interpreted as fetal seizures) has been reported in the third trimester in one instance [Chen et al 2014]. Delivery is usually uncomplicated, although Apgar scores may be depressed.
Multiple types of seizures including tonic-clonic and multifocal myoclonic seizures not responding to treatment are the main feature [Sass et al 2010, Bindu et al 2011, Salih et al 2013, Bosley et al 2014]. Rarely, infantile spasms and hypsarrhythmia, exaggerated startle response, or hyperekplexia also have been reported [Bosley et al 2014, Holder et al 2014]. Electroencephalography (EEG) shows low-amplitude or disorganized background [Sass et al 2010, Holder et al 2014], multifocal epileptiform discharges [Holder et al 2014], or a burst suppression pattern [Relinque et al 2015].
Neurologic examination reveals abnormalities in tone including opisthotonus, spastic quadriplegia, and pyramidal signs.
All affected children develop profound psychomotor retardation: neurologic development is halted at the level of brain stem reflexes and children lack any response to environmental stimulation except for exaggerated startle and intermittent seizures [Bosley et al 2014].
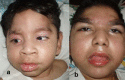
Although head circumference is normal at birth, severe progressive postnatal microcephaly develops [Bosley et al 2014]. Progressive facial dysmorphism is an additional feature.
Systemic manifestations are rare and can include such findings as severe asthma and pyloric stenosis [Bindu et al 2011, Bosley et al 2014].
Prognosis is poor and children usually die during the first few months of life.
A significant proportion of infants who survive the newborn period develop ectopia lentis (lens dislocation or subluxation) [Johnson & Duran 2001, Bosley et al 2014]. The natural history of ectopia lentis is not known: it may not be present (or at least not obvious) during the first few months and may only become evident (and detectable by ophthalmologic examination) during the first year. Subtle lens dislocation may be obvious only after dilation of the pupil and slit lamp examination (over time) [Lueder & Steiner 1995].
Late-Onset ISOD
Late-onset ISOD differs from the classic form in that onset is between ages six and 18 months and some suggestive findings (e.g., intractable seizures, ectopia lentis) may be absent.
The main clinical features are developmental delay/regression, movement disorder characterized by dystonia and choreoathetosis, ataxia, and (rarely) acute hemiplegia as a result of metabolic stroke. The clinical course may be progressive or episodic. In the episodic form encephalopathy, dystonia, choreoathetosis, and/or ataxia are intermittent [Van der Knaap &Valk 2005, Del Rizzo et al 2013, Rocha et al 2014].
Ectopia lentis (lens dislocation or subluxation) may or may not be present at the time of presentation.