Clinical Characteristics
Clinical Description
Classic Joubert syndrome (JS) is characterized by the three primary findings of: a distinctive cerebellar and brain stem malformation called the molar tooth sign (MTS), hypotonia, and developmental delays. Often these findings are accompanied by episodic tachypnea or apnea and/or atypical eye movements. In general, the breathing abnormalities improve with age, truncal ataxia develops over time, and acquisition of gross motor milestones is delayed. Cognitive abilities are variable, ranging from severe intellectual disability to normal. Additional findings can include retinal dystrophy, renal disease, ocular colobomas, occipital encephalocele, hepatic fibrosis, polydactyly, oral hamartomas, and endocrine abnormalities. Table 2 associates phenotypic features with genes; Table 3 associates genes with phenotypic features. Both intra- and interfamilial phenotypic variation are seen in JS.
Many of the clinical features of JS are evident in infancy [Joubert et al 1969, Boltshauser & Isler 1977]. The findings of nystagmus, oculomotor apraxia, and abnormal breathing patterns can be observed in all clinical subtypes. Most children with JS develop truncal ataxia and, in combination with hypotonia, exhibit delayed acquisition of gross motor milestones.
Nystagmus. Many children with Joubert syndrome demonstrate horizontal nystagmus at birth that improves with age. Torsional and pendular rotatory nystagmus have also been observed.
Oculomotor apraxia is often identified in childhood rather than in infancy, perhaps because of under-recognition of the finding [Steinlin et al 1997]. Many children with oculomotor apraxia demonstrate head thrusting as a compensatory mechanism for their inability to initiate saccades [Hodgkins et al 2004, Khan et al 2008, Weiss et al 2009]. Horizontal head titubation (i.e., a "no-no" head tremor) has been described in infants and young children younger than age two years [Poretti et al 2014]. Visual acuity and functional vision may improve with age as a result of visual maturation, in spite of significantly aberrant eye movements at birth [M Parisi and A Weiss, personal observation].
Respiratory findings. Many children with JS exhibit apnea, tachypnea, or both, sometimes alternating, particularly in the neonatal period [Saraiva & Baraitser 1992, Steinlin et al 1997, Maria et al 1999a, Valente et al 2008]. Although some infants have died of apnea, episodic apnea generally improves with age and may completely disappear [Maria et al 1999b]. Children with JS are at increased risk for sleep apnea, including central (particularly in infancy and childhood) and obstructive (particularly in later childhood/adolescence related to tongue hypertrophy, hypotonia, and obesity) [Parisi 2009]. A survey of self-reported sleep behaviors in individuals with JS using a validated sleep questionnaire suggested sleep-related breathing disorders in six of the 14 individuals surveyed [Kamdar et al 2011]. Some individuals with Leber congenital amaurosis resulting from biallelic pathogenic variants in CEP290 have also been found to have abnormalities in motile respiratory cilia that may predispose to respiratory symptoms including chronic rhinitis, recurrent sinusitis, and bronchitis [Papon et al 2010].
Central nervous system findings
Abnormal EEG and/or seizures are present in some affected individuals; the exact incidence is unknown [
Saraiva & Baraitser 1992]. One study identified greater cognitive impairment in individuals with JS and an abnormal EEG [
Summers et al 2017].
Behavioral findings including inattention, hyperactivity, and atypical behaviors such as temper tantrums are present in some children and adolescents [
Deonna & Ziegler 1993,
Hodgkins et al 2004,
Farmer et al 2006]. Emotional and behavior issues were reported in almost 40% in one survey of 54 individuals with JS [
Bulgheroni et al 2016]. In another survey of 76 individuals, behavior issues were more likely to manifest as internalizing (anxiety, depression) than externalizing (aggression, oppositional defiance) [
Summers et al 2017].
JS Clinical Subtypes
See Table 2 and Table 3.
Table 2.
Joubert Syndrome: Clinical Subtypes
View in own window
| Name of Clinical Subtype | Mandatory Features in Addition to Primary Criteria 1 | Strongly Associated Features 2 | Other Names | Genes
(bold = major gene) |
|---|
| Classic or pure Joubert syndrome | | | JS; JS type A | Many genes |
| Joubert syndrome w/retinal disease (JS-Ret) | Retinal dystrophy (including LCA) | | JS type B |
AHI1
CEP290
CEP41
INPP5E
MKS1
TMEM107
TMEM138
TMEM216
|
| Joubert syndrome w/renal disease (JS-Ren) | NPHP (includes cystic kidney disease) | | |
AHI1
CC2D2A
CEP290
NPHP1
OFD1
RPGRIP1L
TMEM138
TMEM216
TMEM237
ZNF423
|
| Joubert syndrome w/oculorenal disease (JS-OR) | Retinal dystrophy (incl LCA); NPHP | CHF (occasional) | JS type B; CORS; Senior-Løken syndrome; Dekaban-Arima syndrome |
AHI1
CC2D2A
CEP290
NPHP1
POC1B
RPGRIP1L
TMEM216
TMEM231
TMEM237
|
| Joubert syndrome w/hepatic disease (JS-H) | CHF | Colobomas; NPHP | COACH syndrome; Gentile syndrome |
CC2D2A
CEP290
INPP5E
RPGRIP1L
TMEM67
|
| Joubert syndrome w/oral-facial-digital features (JS-OFD) | Tongue hamartomas; oral frenulae; polydactyly 3 | Cleft lip/palate | Varadi-Papp syndrome; OFD VI; OFD IV; Mohr-Majewski syndrome |
B9D2
C2CD3
CPLANE1
CEP120
KIF7
OFD1
TCTN2
TCTN3
TMEM107
TMEM216
|
| Joubert syndrome w/acro-callosal features (JS-AC) | Agenesis of corpus callosum; polydacyly 3 | Hydrocephalus | Acrocallosal syndrome |
KIF7
|
| Joubert syndrome w/Jeune asphyxiating thoracic dystrophy features (JS-JATD) | Skeletal dysplasia (short ribs, small thorax, short limbs, renal cystic disease) | Polydactyly 3; cone-shaped epiphyses; CHF | Jeune asphyxiating thoracic dystrophy; Mainzer-Saldino syndrome |
CEP120
CSPP1
IFT172
KIAA0586
|
Adapted from Brancati et al [2010]. This classification scheme should not be interpreted as definitive, given the extreme clinical heterogeneity of the manifestations and the variable age of onset of many of these features.
AC = acro-callosal; CHF = congenital hepatic fibrosis ; COACH = cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, and hepatic fibrosis; CORS = cerebello-oculo-renal syndromes; H = hepatic; JATD = Jeune asphyxiating thoracic dystrophy; LCA = Leber congenital amaurosis; NPHP = nephronophthisis; OFD = oral-facial-digital syndrome; OR = oculorenal; Ren = renal; Ret = retinal
- 1.
Primary criteria = molar tooth sign (MTS), hypotonia, developmental delay (DD)
- 2.
Other features including encephalocele, postaxial polydactyly, other structural brain malformations (including polymicrogyria), congenital heart defects, Hirschsprung disease, and situs defects can be seen in these subtypes but are not major features.
- 3.
Polydactyly is often postaxial, especially of hands, and preaxial, especially of feet. Distinctive for OFD VI syndrome: mesaxial or central polydactyly with a Y-shaped metacarpal.
Joubert syndrome with retinal disease (JS-Ret) is characterized by a pigmentary retinopathy that may be indistinguishable from classic retinitis pigmentosa; it can occasionally be severe with neonatal onset of congenital blindness and an attenuated or extinguished electroretinogram that resembles Leber congenital amaurosis (LCA) [Tusa & Hove 1999]. However, the retinal disease may not be progressive and is not always present in infancy or early childhood [Steinlin et al 1997]. One survey of 235 families with JSRD identified retinal dystrophy in 30% [Doherty 2009].
Ocular colobomas are most often described as chorioretinal [
Saraiva & Baraitser 1992,
Parisi 2009] and may be associated with hepatic fibrosis, as in the COACH syndrome variant [
Doherty et al 2010]. One survey described colobomas in 19% of families with JSRD [
Doherty 2009]. A retinal change described as the "morning glory disc anomaly" has been described in an extended Austrian family from the Tyrolean region with biallelic
TMEM237 pathogenic variants [
Janecke et al 2004,
Huang et al 2011].
Other. Variably present:
Joubert syndrome with renal disease (JS-Ren) has been described traditionally in two forms (nephronophthisis and cystic dysplasia); however, these now appear to be part of a continuum with the specific renal manifestation varying by stage of renal disease. Juvenile nephronophthisis, a form of chronic tubulointerstitial nephropathy, often presents in the first or second decade of life with polydipsia, polyuria, urine-concentrating defects, growth restriction, and/or anemia. Progression to end-stage kidney disease occurs on average by age 13 years [Hildebrandt et al 1998]. Renal changes visible on ultrasound examination occur late in the course and consist of small, scarred kidneys with increased echogenicity and occasional cysts at the corticomedullary junction, findings consistent with cystic dysplasia (i.e., multiple variably sized cysts in immature kidneys with fetal lobulations) [Saraiva & Baraitser 1992, Steinlin et al 1997, Satran et al 1999].
In addition to the nephronophthisis and cystic dysplasia spectrum, a second type of renal disease that resembles autosomal recessive polycystic kidney disease (ARPKD) has been reported.
Three individuals with JS caused by biallelic
TMEM67 pathogenic variants were reported to have renal disease more typical of ARPKD, with enlarged, diffusely microcystic kidneys and early-onset severe hypertension as well as congenital hepatic fibrosis; in addition, they exhibited chronic anemia characteristic of nephronophthisis [
Gunay-Aygun et al 2009].
In the Hutterite population, approximately 70% of probands with JS caused by biallelic
TMEM237 pathogenic variants have cystic renal disease and abnormal renal function, with hypertension reported in some [
Boycott et al 2007,
Huang et al 2011].
Renal disease has been reported in 23% [Doherty 2009] and 30% [Saraiva & Baraitser 1992] of persons with JS. These prevalence values may increase as a cohort ages, as renal disease can develop during childhood and adolescence [Steinlin et al 1997].
Joubert syndrome with oculorenal disease (JS-OR). Retinal disease and renal impairment often occur together in the same individual, and many of JS-related genes are associated with both renal cystic disease and retinal dystrophy, a combination sometimes known as Senior-Løken syndrome [Parisi 2009, Brancati et al 2010] (Table 2). In the past JS-OR was also known as Dekaban Arima syndrome (retinopathy, cystic dysplastic kidneys), which can be evident prenatally or at birth.
Joubert syndrome with hepatic disease (JS-H). Hepatic fibrosis is usually progressive but rarely symptomatic at birth. Congenital hepatic fibrosis is a developmental disorder of the portobiliary system characterized histologically by defective remodeling of the ductal plate (ductal plate malformation), abnormal branching of the intrahepatic portal veins, and progressive fibrosis of the portal tracts. Clinical findings include enlarged, abnormally shaped liver, relatively well-preserved hepatocellular function, and portal hypertension resulting in splenomegaly, hypersplenism, and gastroesophageal varices.
Hepatic fibrosis was observed in 18% of individuals with JS in one cohort [Doherty 2009].
When present in JS, hepatic fibrosis is often associated with chorioretinal colobomas and sometimes with renal disease. The combination of colobomas, cognitive impairment ("oligophrenia"), ataxia, cerebellar vermis hypoplasia, and hepatic fibrosis has been termed COACH syndrome [Satran et al 1999, Gleeson et al 2004, Doherty et al 2010].
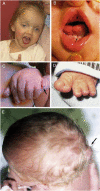
Joubert syndrome with oral-facial-digital features (JS-OFD). Oral findings can include midline upper-lip cleft, midline groove of tongue, hamatomas of the alveolar ridge (), cleft palate, oral frenulae, and tongue lobulations or hamartomas. Craniofacial features often include wide-spaced eyes or telecanthus, hypoplastic alae nasi, and micrognathia.
Clinical features in JSRD A. Facial features in a girl with JS/COACH syndrome at age 27 months showing broad forehead, arched eyebrows, strabismus, eyelid ptosis (on right eye), and open mouth configuration indicating reduced facial tone
Polydactyly is described in 8%-19% of probands [Doherty 2009, Brancati et al 2010]. Polydactyly can be unilateral or bilateral and is often postaxial (), although preaxial polydactyly of the toes is also frequently reported () [Saraiva & Baraitser 1992].
Mesaxial polydactyly, in which the extra digit occurs between the central digits and is often accompanied by a Y-shaped metacarpal, has been described in some individuals with JS, many of whom have other features of oral-facial-digital syndrome type VI/Varadi-Papp syndrome [Gleeson et al 2004]. OFD VI has now been defined as a form of JS, requiring the MTS as well as one or more of the following features: tongue hamartoma/oral frenula/upper-lip notch, mesaxial polydactyly, and hypothalamic hamartoma [Poretti et al 2012].
Joubert syndrome with acrocallosal features (JS-AC). Agenesis of the corpus callosum is common in JS [Valente et al 2005]. In one survey of 20 individuals with JS, 80% had some degree of callosal dysgenesis [Senocak et al 2010]. Callosal abnormalities are relatively frequent in those with biallelic KIF7 pathogenic variants [Bachmann-Gagescu et al 2015a], suggesting overlap with acrocallosal syndrome (see Genetically Related Disorders) in which polydactyly and hydrocephalus are also seen [Putoux et al 2011].
Joubert syndrome with Jeune asphyxiating thoracic dystrophy (JS-JATD). Features of JATD (see Genetically Related Disorders) and the related short-rib thoracic dysplasia condition, Mainzer-Saldino syndrome, have been reported in several children with a JS, reflecting the shared ciliary origin of these conditions [Lehman et al 2010, Halbritter et al 2013, Shaheen et al 2015b].
Other Findings in JS Not Specific to a Given Subtype
Scoliosis has been described, most likely related to early hypotonia.
Endocrine abnormalities have been described; they include pituitary hormone dysfunction ranging from isolated growth hormone deficiency or thyroid hormone deficiency to more extensive panhypopituitarism or micropenis in males [Delous et al 2007, Wolf et al 2007, Parisi 2009, Sanders et al 2015].
Obesity may be increased in JS, suggesting an association with the ciliary disorder Bardet-Biedl syndrome; the identification of biallelic pathogenic variants in INPP5E in both JS and MORM syndrome (mental retardation, obesity, retinal dystrophy, and micropenis) reinforces this association [Bielas et al 2009, Jacoby et al 2009].
Typical facial features including long face with bitemporal narrowing, high-arched eyebrows, ptosis, prominent nasal bridge with anteverted nostrils, triangular-shaped mouth, prognathism, and low-set ears are sometimes described [Maria et al 1999a] (); however, these features can be difficult to discern in infancy and are thus far nonspecific. Nonetheless, many observers report a "Joubert syndrome facies" [Braddock et al 2007]. The craniofacial features in those with biallelic KIF7 pathogenic variants often include macrocephaly, frontal bossing, hypertelorism, high palate, and micrognathia [Dafinger et al 2011, Putoux et al 2011].
Heart defects have been described in a number of individuals with JS, in some cases assocated with features of oral-facial-digital syndrome, and have included septal defects, aortic valve anomalies, and coarctation of the aorta [Bachmann-Gagescu et al 2015a].
Laterality defects including situs inversus are seen in some individuals [Parisi 2009].
Hirschsprung disease has been described in a few individuals [Brancati et al 2010].
Conductive hearing loss may result from middle ear infections [Kroes et al 2010]. Sensorineural hearing loss has been described.
Tongue hypertrophy. Many have rhythmic tongue movements that may lead to tongue hypertrophy.
Other CNS malformations
Abnormal collections of cerebrospinal fluid in the fourth ventricle or the posterior fossa that resemble Dandy-Walker malformation; in approximately 10% of individuals in one survey [
Maria et al 2001] and in ~42% in another [
Poretti et al 2017]
Abnormal brain stem and hypothalamic hamartomas, particularly in those with oral-facial-digital syndrome type VI-related findings [
Poretti et al 2011]
Abnormal nuclei and tracts of the pons, cerebellum, and medulla based on neuropathologic evaluation [
Doherty 2009]; absence of decussation of the corticospinal and superior cerebellar tracts based on diffusion tensor imaging [
Poretti et al 2007]; and abnormal activation patterns during motor tasks based on functional MRI studies [
Parisi et al 2004b]
Phenotype Correlations by Gene
Table 3 includes preliminary information on genotype-phenotype correlations.
Table 3.
Genes Associated with JS by Phenotypic Features
View in own window
| Gene | Phenotypic Feature (in addition to the molar tooth sign) | Allelic/Related Disorder 3 |
|---|
| Retinal dystrophy | Coloboma 1 | Renal | Oculorenal 2 | Hepatic 1 | Oral | Polydactyly | Other |
|---|
|
AHI1
| ++ 4 | (+) | + 5 | + | (+) | | | Polymicrogyria 6 | |
|
CPLANE1
| | | | | (+) | + 7 | + 7 | Founder effects in French Canadian 8 & Dutch populations 9 | OFD VI |
|
CC2D2A
| + | + | + | + | + 10 | | | Encephalocele, ventriculomegaly, seizures 11; milder phenotype in French Canadian population 12 | Meckel syndrome 13 |
|
CEP290
| ++ | + | ++ | ++ 14 | + | | | Encephalocele; cardiac; situs inversus; other 15 | LCA, Meckel syndrome, BBS |
|
CSPP1
| (+) | | (+) | (+) | (+) | | | SNHL; corpus callosum hypoplasia; encephalocele; founder variant in Hutterite population 16 | Meckel syndrome, JATD 17 |
|
INPP5E
| + | | + | + | (+) | | + | | MORM syndrome 18 |
|
KIAA0586
| (+) | + | | | | (+) | (+) | Broad range of phenotypes: severe HLS (& cleft palate) to JATD w/short ribs & narrow thorax to pure JS 19 | HLS, JATD |
|
MKS1
| (+) | | | | | | | Kidney/liver findings & PD described in 1 individual 20 | Meckel syndrome |
|
NPHP1
| + | | ++ | + | | | | "Mild molar tooth" sometimes described 21 | Juvenile NPHP type 1, Cogan syndrome |
|
RPGRIP1L
| (+) | (+) | ++ | + | (+) | | (+) | Encephalocele | Meckel syndrome, retinal disease 22 |
| TCTN2 21 | | | | | | | | Clubfoot 23 | Meckel syndrome 24 |
|
TMEM67
| | + 25 | + | | ++ 26, 27 | | (+) | Encephalocele | Meckel syndrome 28 |
| TMEM216 29 | (+) | (+) | ++ | + | (+) | + | + | Cardiac findings; encephalocele | Meckel syndrome |
(+) = feature is uncommon but has been described; + = feature is present in some cases; ++ = Major feature; HLS = hydrolethalus syndrome; NPHP = nephronophthisis; LCA = Leber congenital amaurosis; BBS = Bardet-Biedl syndrome; MORM = mental retardation, truncal obesity, retinal dystrophy, micropenis [Jacoby et al 2009]; OFD = oral-facial-digital syndrome; PD = polydactyly; JATD = jeune asphyxiating thoracic dystrophy; SNHL = sensorineural hearing loss
- 1.
May include COACH syndrome: cerebellar vermis hypoplasia, oligophrenia, ataxia, coloboma, and hepatic fibrosis
- 2.
This refers to retinal disease plus kidney disease; terms used in the past include: Senior-Løken syndrome (retinopathy and juvenile-onset nephronophthisis); Dekaban-Arima syndrome (retinopathy, cystic dysplastic kidneys).
- 3.
- 4.
The most common clinical association in those with biallelic AHI1 pathogenic variants is retinal dystrophy, present in approximately 80% [Valente et al 2008]. Early-onset congenital blindness [Valente et al 2006a] and liver involvement [Vilboux et al 2017] have been described.
- 5.
- 6.
- 7.
The phenotype most closely resembles pure or classic Joubert syndrome, with several individuals exhibiting preaxial, postaxial, and/or mesaxial polydactyly and a few with retinal involvement [Srour et al 2015] or liver involvement [Vilboux et al 2017]. None of the affected individuals (ranging in age from 1.5 to 52 years) has evidence of renal impairment or liver disease [Srour et al 2012a, Srour et al 2012b, Srour et al 2015]. Pathogenic variants in this gene also cause OFD VI, with features of preaxial and/or mesaxial polydactyly and hypothalamic hamartoma typical [Lopez et al 2014, Romani et al 2015].
- 8.
Pathogenic variants in this gene are the cause of JS in the original family described by Joubert et al [1969]. Several pathogenic variants recur in the French Canadian population found in the lower St. Lawrence region of Quebec province [Srour et al 2012b, Srour et al 2015].
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
Up to 50% of individuals with both retinal and renal involvement harbor biallelic pathogenic variants in CEP290 [Valente et al 2008].
- 15.
The phenotypic spectrum is very broad, including congenital blindness, ocular colobomas, renal disease, encephaloceles, septal heart disease, and situs abnormalities.
- 16.
- 17.
Pathogenic variants in this gene have been described in phenotypes ranging from classic JS with occasional retinopathy and sensorineural hearing loss [Akizu et al 2014] to the JS-JATD phenotype with features of Jeune skeletal dysplasia [Tuz et al 2014] to a lethal MKS-like phenotype [Shaheen et al 2014]. Thin corpus callosum, occipital encephalocele, and heterotopias have also been described [Akizu et al 2014, Tuz et al 2014].
- 18.
- 19.
Pathogenic variants in this gene cause a wide spectrum of ciliopathy phenotypes, from "pure" JS with relatively mild manifestations and impairment [Bachmann-Gagescu et al 2015b, Roosing et al 2015] to features of Jeune asphyxiating thoracic dystrophy (small chest, short ribs, short stature) [Alby et al 2015, Malicdan et al 2015] to severe features of hydrolethalus syndrome with hydrocephalus and fetal or perimatal demise [Alby et al 2015]. This broad range of phenotypes is not explained by the nature of the pathogenic variants, as many afftected individuals have homozygous or compound heterozygous truncating variants due to frameshifts, aberrant splice, or nonsense variants.
- 20.
Individuals with JS caused by MKS1 pathogenic variants have at least one variant with partial function (e.g., a missense variant that retains some function), in contrast to more severe variants described in those with MKS [Romani et al 2014, Slaats et al 2016]. Most of the affected individuals have a relatively mild phenotype, characterized by classic JS with or without retinal dystrophy. Only one reported individual (out of a group of 9 with pathogenic variants in this gene) had additional features of renal echogenicity, liver fibrosis, and postaxial polydactyly [Slaats et al 2016].
- 21.
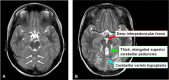
Some individuals with biallelic pathogenic variants in NPHP1 and JS have a distinctive appearance of the molar tooth sign: elongated but thin superior cerebellar peduncles and milder vermis hypoplasia [Parisi et al 2004a].
- 22.
RPGRIP1L pathogenic variants also cause Meckel syndrome. Of note, more severe loss-of-function pathogenic variants predict a more severe (and in many cases, lethal) Meckel phenotype [Delous et al 2007, Wolf et al 2007].
- 23.
A limited number of individuals with pathogenic variants in this gene has been described; thus, the phenotypic spectrum is unknown [Sang et al 2011].
- 24.
- 25.
Biallelic pathogenic variants in TMEM67 were present in 53% of those with ocular coloboma regardless of liver status [Doherty et al 2010].
- 26.
- 27.
- 28.
More severe loss-of-function variants in TMEM67 have been identified in individuals with lethal forms of Meckel syndrome [Smith et al 2006], in comparison with variants with partial function causing JS with hepatic disease or nephronophthisis and liver fibrosis in the absence of the molar tooth sign and other neurologic symptoms [Otto et al 2009, Doherty et al 2010].
- 29.
Nomenclature
The term "Joubert syndrome and related disorders" (JSRD) has been used in the past to describe conditions that share the molar tooth sign and the clinical features of classic Joubert syndrome and also have other organ system involvement. In an evolving nomenclature designed to reduce reliance on confusing and inconsistently used eponyms, at least eight clinical subtypes of JS that share the three primary findings have been proposed (Table 2) [Brancati et al 2010]. More recently, "Joubert syndrome" has become the accepted term to describe all forms of JS.
In the past, some of the following disorders were described as distinct syndromes, but more recent studies indicate that many individuals with these disorders demonstrate the molar tooth sign [Satran et al 1999, Gleeson et al 2004]. Examples of such autosomal recessive disorders include the following:
Dekaban-Arima syndrome (retinopathy, cystic dysplastic kidneys) [
Dekaban 1969]
Varadi-Papp syndrome (oral-facial-digital syndrome VI [OFD VI]) includes cerebellar vermis hypoplasia, oral frenulae, tongue hamartomas, and midline cleft lip as well as the distinctive feature of central polydactyly with a Y-shaped metacarpal [
Münke et al 1990]. Renal and cardiac involvement have been described.
Prevalence
The prevalence of Joubert syndrome (JS) has not been determined. Many authors use a range between 1:80,000 and 1:100,000, but this may represent an underestimate [Kroes et al 2007, Parisi et al 2007, Brancati et al 2010].
There is a relatively high prevalence of JS in the French Canadian population, with several founder variants noted. The family first described by Joubert et al [1969] has been traced to a founder who immigrated to Quebec from France in the 1600s [Badhwar et al 2000]. However, in this family and others, it appears that there are multiple CPLANE1 pathogenic variant-containing haplotypes in the French Canadian population. In fact, in 35 French Canadian families, pathogenic variants were identified in 33 (94%) in the following genes (number of affected families given in parentheses): CPLANE1 (14), CC2D2A (9), NPHP1 (3), TMEM231 (2); and CEP290, TMEM67, TCTN1, OFD1, B9D1, C2CD3, and CEP104 (1 family each). Many French Canadian individuals are compound heterozygous for different pathogenic variants in either CPLANE1, CC2D2A, TMEM231, or NPHP1 [Srour et al 2012a, Srour et al 2012b, Srour et al 2015].
A different founder variant in CPLANE1, p.Arg2904Ter, occurs in the Dutch population [Kroes et al 2016].
A TMEM216 founder variant, p.Arg73Leu, has a carrier rate of 1:92-1:100 in the Ashkenazi Jewish population [Edvardson et al 2010, Valente et al 2010].
In a Canadian Hutterite population, ten related individuals with a MKS-like phenotype including encephaloceles and cystic kidneys were homozygous for a nonsense pathogenic variant (c.52C>T; p.Arg18Ter) in TMEM237, reflecting a carrier frequency of 6% in this population [Huang et al 2011]. Two different Schmiedeleut Hutterite families had the same homozygous pathogenic frameshift variant, c.363_364delTA, in CSPP1 [Shaheen et al 2014], representing a separate founder variant.
In a survey of Japanese families with JS, 6/27 had pathogenic variants in CEP290, with c.6012-12T>A found on nine out of 12 disease alleles; 7/27 families had pathogenic variants in TMEM67 but no founder alleles were identified [Suzuki et al 2016].
Pathogenic variants in genes that cause Joubert syndrome (JS) have also been identified in disorders with clinical findings that overlap with JS; thus, in many instances it has become difficult to determine if a previously recognized disorder is truly distinct from JS (i.e., is an allelic disorder) or is part of the spectrum of JS (see Table 3). Brief descriptions of some of those disorders follow.
Acrocallosal syndrome (ACLS) (OMIM 200990), an autosomal recessive disorder, is characterized by macrocephaly, intellectual disability, agenesis of the corpus callosum and occasional posterior fossa abnormalities, ocular hypertelorism, polyaxial polydactyly of the hands, and preaxial polydactyly of the feet. It has been postulated that ACLS is allelic to hydrolethalus syndrome. Identification of several families with both disorders and KIF7 pathogenic variants confirms the proposed association; of note, several of the probands had evidence of the molar tooth sign (MTS) on cranial imaging, suggesting that ACLS and JS may represent overlapping ciliopathies [Putoux et al 2011].
Bardet-Biedl syndrome
(BBS), usually inherited in an autosomal recessive manner, is characterized by cone-rod retinal dystrophy, truncal obesity, postaxial polydactyly, cognitive impairment, hypogonadotropic hypogonadism in males, genital malformations in females, and renal disease that may include structural malformations, renal hypoplasia, hydronephrosis, cystic kidneys, and glomerulonephritis. Progressive retinal impairment often causes blindness; renal failure may cause significant morbidity. Some affected individuals have hepatic fibrosis. Although many individuals are ataxic with poor coordination, cerebellar involvement or structural malformations are not typical [Baskin et al 2002]. Pathogenic variants in at least 19 genes, all of which play a role in the primary cilium, have been described. Pathogenic variants in CEP290, MKS1, and NPHP1 have been shown to cause both BBS and JS [Leitch et al 2008, Zaghloul & Katsanis 2009, Knopp et al 2015].
Cogan syndrome (OMIM 257550), an autosomal recessive familial form of congenital oculomotor apraxia, is characterized by defective horizontal voluntary eye movements with jerkiness. Oculomotor apraxia is also a common manifestation of JS. Detailed neuroimaging via fiber tracking suggests that there may be subtle differences in some of the pathways in Cogan syndrome versus JS [Merlini et al 2010].
Some individuals with Cogan syndrome also have cerebellar vermis hypoplasia with evidence of the molar tooth sign [Whitsel et al 1995, Sargent et al 1997], and occasionally develop nephronophthisis. The approximately 290-kb NPHP1 homozygous deletion or compound heterozygosity for the approximately 290-kb deletion and an NPHP1 sequence variant have been identified in some individuals with Cogan syndrome [Saunier et al 1997, Betz et al 2000].
Hydrolethalus syndrome (HLS) (OMIM PS236680), a lethal autosomal recessive disorder, is associated with midline brain anomalies (usually hydrocephaly or anencephaly with a keyhole foramen magnum), migrognathia, postaxial polydactyly of the hands, and preaxial polydactyly of the feet. In the Finnish population, pathogenic variants in HYLS1 have been identified [Mee et al 2005]. Pathogenic variants in KIF7 were identified in a consanguineous Algerian pedigree in which four affected fetuses had features consistent with HLS, but also a midbrain-hindbrain malformation similar to the MTS [Putoux et al 2011]. Pathogenic variants in KIAA0586 have also been described in fetuses with HLS as well as in individuals with JS and a variety of ciliopathy phenotypes [Alby et al 2015].
Jeune asphyxiating thoracic dystrophy (JATD) is an autosomal recessive skeletal dysplasia characterized by a long, narrow thorax, short stature, short limbs, polydactyly, and renal cystic disease, with skeletal findings that may include cone-shaped epiphyses in hands and feet, irregular metaphyses, shortened ilium, and trident-shaped acetabulum. It is often lethal in infancy secondary to respiratory insufficiency. More than 12 ciliary genes and/or loci have been identified (including several intraflagellar transport proteins). Heterozygous pathogenic variants in TTC21B have been identified in three families with JATD with one proband demonstrating compound heterozygosity for a null allele and a hypomorphic allele [Davis et al 2011]. Pathogenic variants in CSPP1 [Tuz et al 2014] and KIAA0586 [Malicdan et al 2015] have been identified in individuals with JS and manifestations of JATD.
Leber congenital amaurosis
(LCA), a severe dystrophy of the retina, typically becomes evident in the first year of life. Visual function is usually poor and often accompanied by nystagmus, sluggish or near-absent pupillary responses, photophobia, high hyperopia, and keratoconus. Visual acuity is rarely better than 20/400. A characteristic finding is Franceschetti's oculodigital sign, comprising eye poking, pressing, and rubbing. The appearance of the fundus is extremely variable. While the retina may initially appear normal, a pigmentary retinopathy reminiscent of retinitis pigmentosa is frequently observed later in childhood. The electroretinogram is characteristically "nondetectable" or severely subnormal. Pathogenic variants in at least 17 genes cause LCA, and pathogenic variants in CEP290 account for about 20% of LCA, with one homozygous intronic pathogenic variant accounting for at least 20% of isolated congenital blindness in European cohorts [den Hollander et al 2006].
Mainzer-Saldino syndrome (MZSDS) is an autosomal recessive disorder described by the three diagnostic criteria of retinal dystrophy, renal disease (typically nephronophthisis), and phalangeal cone-shaped epiphyses. Variable findings include cerebellar hypoplasia, a narrow thorax, hepatic fibrosis, and dolichocephaly, with significant overlap with features of JATD and pathogenic variants in IFT140 described in both conditions [Mainzer et al 1970, Perrault et al 2012]. Pathogenic variants in IFT172, another component of the intraflagellar transport apparatus, have been described in those with JATD, MZSDS, and JS [Halbritter et al 2013].
A term that has been used to encompasse Ellis-van Creveld syndrome (EVC), short-rib polydactyly syndrome (SRPS), JATD, and MZSDS is short-rib thoracic dysplasia (SRTD) (OMIM PS208500) with or without polydactyly; these conditions are autosomal recessive skeletal ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a "trident" appearance of the acetabular roof. There is clearly a great deal of overlap between these skeletal dysplasias and some forms of JS.
Meckel syndrome (OMIM PS249000), an autosomal recessive disorder, is characterized by the triad of cystic renal disease, posterior fossa abnormalities (usually occipital encephalocele), and the hepatic ductal plate malformation leading to hepatic fibrosis and bile duct proliferation. Polydactyly is relatively common. Cerebellar vermis hypoplasia has been described in some individuals. Meckel syndrome is usually lethal in the prenatal or perinatal period [Kyttälä et al 2006, Smith et al 2006]. Pathogenic variants in at least 21 genes have been identified in Meckel syndrome [Knopp et al 2015]. Pathogenic variants in at least 18 of these genes, CEP290, TMEM67, RPGRIP1L, CC2D2A, CEP41, MKS1, B9D1, B9D2, TMEM138, TMEM231, TCTN2, TCTN3, TMEM237, CPLANE1, CSPP1, CEP120, TMEM107, and TMEM216, have also been identified in individuals with JS [Parisi 2009, Valente et al 2010, Thomas et al 2012, Romani et al 2014, Bachmann-Gagescu et al 2015a, Knopp et al 2015, Shaheen et al 2015a, Roosing et al 2016a, Slaats et al 2016]. In many cases, pathogenic variants that predict a more severe effect on protein function such as transcription termination or null variants are associated with the lethal Meckel syndrome phenotype, while milder pathogenic variants such as missense variants are associated with JS [Romani et al 2014, Slaats et al 2016]. In some families the identical pathogenic variants can be found in a fetus with Meckel syndrome and a child with a JS, highlighting that these disorders can represent a spectrum [Valente et al 2010].
MORM (mental retardation, truncal obesity, retinal dystrophy, micropenis) syndrome (OMIM 610156), an autosomal recessive disorder, appears to be related to Bardet-Biedl syndrome and is caused by pathogenic variants in INPP5E [Bielas et al 2009, Jacoby et al 2009]. Individuals with this condition have normal growth parameters and life span with a congenital non-progressive retinal dystrophy and static mild-to-moderate cognitive impairment; in contrast to Bardet-Biedl syndrome, there is no polydactyly, apparent hypogonadism, or obvious renal disease [Hampshire et al 2006].
Nephronophthisis, an autosomal recessive kidney disease characterized by renal tubular atrophy and progressive interstitial fibrosis with later development of medullary cysts, is caused by pathogenic variants in at least 19 genes [Hildebrandt et al 2009, Hurd & Hildebrandt 2011, Wolf 2015]. The age of onset of end-stage kidney disease can be variable, thereby defining subtypes such as infantile, juvenile, and adolescent. A homozygous, approximately 290-kb deletion of NPHP1 is identified in approximately 25% of individuals with juvenile nephronophthisis [Hoefele et al 2005, Saunier et al 2005, Hildebrandt et al 2009] and is causative in a small subset of individuals with JS. Note: The most common form, juvenile nephronophthisis, can also be a renal manifestation in JS. Conversely, it is estimated that 10% of individuals with nephronophthisis have extrarenal findings, which can include the molar tooth sign in some cases [Saunier et al 2005].
Oral-facial-digital syndrome describes a heterogeneous group of disorders characterized by facial features, oral abnormalities (often lobulated tongue and oral frenula), and digital anomalies such as polydactyly. Based on other associated clinical features, at least 13 clinical subtypes have been described. These features also overlap considerably with Meckel syndrome, short-rib polydactyly syndrome, and JS. Of the genes identified thus far for OFD, all have all had ciliary roles, and several overlap with JS.
Oral-facial-digital syndrome type I
(OFD1) is associated with dysfunction of primary cilia and is characterized by the following abnormalities:
Oral (lobed tongue, hamartomas or lipomas of the tongue, cleft of the hard or soft palate, accessory gingival frenulae, hypodontia and other dental abnormalities)
Facial (ocular hypertelorism or telecanthus, hypoplasia of the alae nasi, median cleft or pseudocleft of the upper lip, micrognathia)
Digital (brachydactyly, syndactyly of varying degrees, and clinodactyly of the fifth finger; duplicated hallux [great toe]; preaxial or postaxial polydactyly of the hands)
Brain (intracerebral cysts, corpus callosum agenesis, cerebellar agenesis with or without Dandy-Walker malformation)
Kidney (polycystic kidney disease)
Up to 50% of individuals with OFD1 have some degree of intellectual disability that is usually mild. Almost all affected individuals are female. However, males with OFD1 have been described, mostly as malformed fetuses delivered by women with OFD1.
Of note, the phenotypic spectrum was broadened with recognition that the clinical features described in four individuals (hydrops fetalis, jaundice, brisk deep tendon reflexes, seizures, and trilobate left lung) [Terespolsky et al 1995, Brzustowicz et al 1999] were associated with pathogenic variants in OFD1 [Budny et al 2006]. Pathogenic variants in OFD1 have also been described in rare males with JS and features of OFD [Coene et al 2009, Field et al 2012].
Oral-facial-digital syndrome type IV (OFD IV, Mohr-Majewski syndrome) (OMIM 258860) is characterized by hallucal and postaxial polysyndactyly, tibial dysplasia, and variable short ribs, cystic kidneys, and brain anomalies. Pathogenic truncating variants in TCTN3 were identified in several pedigrees with a severe lethal OFD IV phenotype and bowing of long bones, cystic kidneys, occipital encephalocele, and bile duct proliferation of the liver but without short ribs; several of these fetuses also displayed vermis agenesis suggestive of the molar tooth sign [Thomas et al 2012]. Of note, this phenotype overlaps with Meckel syndrome and with JS.
Oral-facial-digital syndrome type VI (OFD VI, Varadi-Papp syndrome) (OMIM 277170). Individuals with OFD VI often have mesaxial polydactyly, in which the extra digit occurs between the central digits and is often accompanied by a Y-shaped metacarpal, as well as cerebellar vermis hypoplasia, oral frenulae, tongue lobulations or hamartomas (), and craniofacial features that include wide-spaced eyes and midline lip groove. Renal and cardiac involvement have been described [Münke et al 1990]. Problems with mastication, swallowing, and respiration may result. OFD VI has been defined as a form of JS, requiring the MTS as well as one or more of the following features: tongue hamartoma/oral frenula/upper lip notch, mesaxial polydactyly, and hypothalamic hamartoma [Poretti et al 2012]. One group identified pathogenic variants in CPLANE1 in 9/11 families with OFD VI [Lopez et al 2014]. Features of preaxial and/or mesaxial polydactyly and hypothalamic hamartoma were more likely related to CPLANE1 pathogenic variants, whereas tongue hamartomas and lingual frenula were not associated with pathogenic variants in this gene [Lopez et al 2014, Romani et al 2015]. Another group identified CPLANE1 pathogenic variants in only two of 17 individuals with OFD VI; pathogenic variants in TMEM216, TMEM107, and OFD1 have also been reported in OFD VI [Romani et al 2015, Lambacher et al 2016].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Joubert syndrome (JS) is inherited predominantly in an autosomal recessive manner.
OFD1-related JS is inherited in an X-linked manner (click here (pdf) for discussion of X-linked inheritance).
Risk to Family Members (Autosomal Recessive Inheritance)
Parents of a proband
The parents of an affected child are obligate heterozygotes (i.e., carriers of one pathogenic variant in a JS-related gene).
Heterozygotes are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband
The offspring of a proband are obligate heterozygotes (carriers) for a pathogenic variant in a JS-related gene.
Although no individuals with JS are reported to have reproduced, the broad spectrum of cognitive impairment now known in this condition may increase the likelihood that reports of individuals who have had offspring will be forthcoming.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a pathogenic variant in a JS-related gene.
For information about risk to family members ‒ X-linked inheritance (OFD1-related) click here (pdf).
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the JS-related pathogenic variant(s) in the family.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the JS-related pathogenic variant(s) have been identified in an affected family member, prenatal and preimplantation genetic testing for JS are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Prenatal imaging. First-trimester diagnosis of JS for pregnancies at 25% risk has been reported using ultrasound examination to identify structural brain abnormalities such as encephalocele [van Zalen-Sprock et al 1996, Wang et al 1999]. More typically, prenatal diagnosis in at-risk fetuses has been accomplished by prenatal ultrasound examination of the posterior fossa and/or kidneys (for cysts and enlarged and/or hyperechoic kidneys) and digits (for polydactyly) as early as the second trimester [Ní Scanaill et al 1999, Aslan et al 2002, Doherty et al 2005]. In reality, prenatal sonographic findings in fetuses with JS are relatively nonspecific and include increased nuchal translucency, enlarged cisterna magna, cerebellar vermis aplasia/hypoplasia, occipital encephalocele, and ventriculomegaly, making definitive diagnosis of JS difficult in the absence of a family history. Moreover, the cerebellar vermis is a relatively late-developing structure, and may not cover the fourth ventricle until 18 weeks' gestation, making visualization of the molar tooth sign (MTS) difficult earlier in gestation [Bromley et al 1994]. The use of 2D ultrasound and 3D sonographic reconstruction with surface rendering has allowed visualization of the MTS as early as 22 weeks in several fetuses without a prior family history of JS [Quarello et al 2014].
Accurate prenatal diagnosis of JS in an at-risk fetus has been achieved by serial prenatal ultrasound imaging starting at 11 to 12 weeks' gestation, with detailed evaluation of cerebellar and other fetal anatomy through 20 weeks' gestation, followed by fetal MRI imaging at 20 to 22 weeks' gestation [Doherty et al 2005]. In a series of 12 pregnancies at 25% risk of having a fetus with JS, one center was able to correctly diagnose JS in the three affected fetuses based on fetal MRI findings at the pontomesencephalic junction (including the MTS) as early as 22 weeks' gestation [Saleem & Zaki 2010]. In the earliest reported diagnoses to date, MTS was identified in two separate at-risk pregnancies at 17 to 18 weeks' gestation via fetal MRI [Saleem et al 2011]. Although prenatal imaging, including fetal MRI, is useful in the diagnosis of posterior fossa anomalies, its sensitivity and specificity for the diagnosis of JS is unknown, and its use has not been systematically evaluated.
For a couple who has already had a child with JS, the presence of findings that suggest a prenatal diagnosis of Joubert syndrome and related disorders (e.g., encephalocele, renal cystic changes, polydactyly, or posterior fossa anomalies on fetal imaging) is highly significant; however, the absence of these signs does not preclude a diagnosis of Joubert syndrome and related disorders because of the unknown sensitivity of imaging and because of intrafamilial variability.