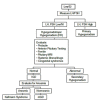
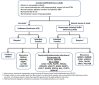
Molecular Pathogenesis
Over the last three decades, a combination of clinical investigational strategies and contemporary genetic approaches has revealed more than 25 causal/contributory genes for the nonsyndromic forms of IGD, with varied modes of inheritance. Broadly, two groups of genetic pathways are linked to IGD: (i) Neurodevelopmental genes govern the origin of the GnRH neurons and typically cause the Kallmann syndrome (KS) form of IGD. (ii) A group of neuroendocrine genes that control the secretion or action of GnRH cause the normosmic isolated GnRH deficiency (nIGD) form IGD. A small subset of genes may cause both KS and nIGD forms of IGD and this suggests that they govern both GnRH migration and GnRH secretion/action. (see Table 2a and Table 2b).
In addition to Mendelian modes of inheritance, a complex genetic architecture for IGD (occurring in 10%-15% of cases) has now been documented wherein pathogenic variants in two or more IGD-related genes are present in a single individual. These pathogenic variants are typically heterozygous and by themselves are not sufficient to cause IGD but require the presence of additional pathogenic variants in a second gene to cause IGD [Sykiotis et al 2010b]. Almost all of the known IGD-related genes have been associated with oligogenic inheritance. These oligogenic pathogenic variants presumably act in a synergistic manner, potentially accounting for some of the variable expressivity and reduced penetrance that is characteristic of IGD.
ANOS1 (KAL1)
Gene structure.
ANOS1 (KAL1) comprises 14 exons and has no alternative spice variants.
Pathogenic variants. Reported pathogenic variants in ANOS1 include deletion of the entire gene, deletion of one or more exons, deletion of several nucleotides, pathogenic missense variants, pathogenic nonsense variants, and splice variants. For more information, see Table A.
Normal gene product. The protein encoded by ANOS1, anosmin 1, has 680 amino acids with functional similarities to molecules involved in neural development [Rugarli et al 1993]. The N-terminus domains share homologies with a consensus sequence of the whey acid protein family and a motif found in protease inhibitors. The C terminus contains a series of fibronectin type III repeats similar to those found in neural cell adhesion molecules.
Abnormal gene product. Impaired function of anosmin results in a migratory defect of the olfactory and GnRH neurons from the olfactory placode during development [Cariboni et al 2004]. The obstructed migration of these neurons accounts for the tell-tale signs of KS, IGD, and anosmia, and leads to olfactory bulb malformation detectable by MRI in the majority of individuals.
CHD7
Gene structure.
CHD7 comprises 38 exons.
Pathogenic variants. Pathogenic variants of CHD7 resulting in IGD are predominantly missense variants that are either hypomorphic alleles or dominant alleles; in contrast, loss-of-function (i.e., truncating) pathogenic variants in CHD7 lead to a more extensive phenotypic presentation as seen in individuals with CHARGE syndrome [Balasubramanian et al 2014].
Normal gene product. The normal gene product is chromodomain helicase DNA-binding protein 7. It belongs to a family of proteins that are thought to alter nucleosome structures and mediate chromatin interactions.
Abnormal gene product.
CHD7 pathogenic variants reported in individuals with KS or nIGD result in truncated proteins or amino acid substitutions of conserved residues when compared with CHD7 orthologs [Kim et al 2008].
FGFR1
Gene structure.
FGFR1 comprises 18 exons with a known splice variant at the end of exon 10.
Pathogenic variants. Pathogenic variants in FGFR1 include pathogenic deletions and missense, nonsense, and splice variants. For more information, see Table A.
Normal gene product.
FGFR1 encodes a membrane receptor with three extracellular immunoglobulin-like domains and an intracellular tyrosine kinase domain [Lee et al 1989]. Ligand binding results in receptor dimerization and recruitment of intracellular signaling proteins.
Abnormal gene product. Abnormal FGFR1 gene products result in impaired receptor signaling. The gene dose effect of anosmin and its interaction with FGFR1 in guiding GnRH neuronal migration have been proposed as explanations for the greater predominance of the IGD phenotype in males than females [Dodé et al 2003].
GNRHR
Gene structure.
GNRHR comprises three coding exons and one alternative splice variant.
Pathogenic variants.
GNRHR pathogenic variants, typically missense variants, cause normosmic IGD in an autosomal recessive inheritance pattern. Other pathogenic variants in GNRHR include nonsense and frameshift variants that occasionally cause autosomal recessive IGD. Heterozygous pathogenic variants (missense, frameshift, and nonsense) are also seen in persons with IGD with diverse clinical phenotypes, suggesting an oligogenic inheritance pattern [Gianetti et al 2012].
Normal gene product.
GNRHR encodes for the gonadotropin-releasing hormone receptor, GNRHR, a G protein-coupled transmembrane receptor for the decapeptide, GNRH.
Abnormal gene product. Abnormal GNRHR gene products result in diminished or absent GNRH signaling through the receptor resulting in hypogonadotropism.
IL17RD
Gene structure.
IL17RD comprises 17 coding exons and seven alternative splice variants.
Pathogenic variants.
IL17RD missense variants in both homozygous and heterozygous state cause the KS form of IGD, including oligogenic inheritance.
Normal gene product. This gene encodes a membrane protein that belongs to the interleukin-17 receptor (IL-17R) protein family and is the component of the interleukin-17 receptor signaling complex. The gene product affects fibroblast growth factor signaling, inhibiting or stimulating growth through MAPK/ERK signaling.
Abnormal gene product. Abnormal IL17RD gene product results in increased FGF8 signaling in vitro, resulting in apoptosis of olfactory progenitor cells.
PROKR2
Gene structure.
PROKR2 comprises two exons.
Pathogenic variants. Pathogenic variants of PROKR2 described include missense and nonsense variants.
Normal gene product. The normal gene product encodes the prokineticin receptor 2, a G protein-coupled transmembrane receptor for PROK2.
Abnormal gene product. The PROKR2 pathogenic variants identified in individuals with KS/nIGD result in diminished receptor function and impaired signaling [Cole et al 2008, Monnier et al 2009, Martin et al 2011]. Functional studies of selected PROKR2 pathogenic variants have failed to demonstrate a dominant negative effect. Knockout mice lack olfactory bulbs and have severe atrophy of the reproductive system related to the absence of gonadotropin-releasing hormone (Gnrh)-synthesizing neurons in the hypothalamus [Matsumoto et al 2006, Martin et al 2011].
SOX10
Gene structure.
SOX10 comprises four coding exons and four alternative splice variants.
Pathogenic variants.
SOX10 nonsense, frameshift, or missense variants cause the KS form of IGD in an autosomal dominant pattern with variable penetrance.
Normal gene product.
SOX10 encodes a transcription factor, SRY (sex determining region Y)-box 10 which is involved in the regulation of neural crest and peripheral nervous system development.
Abnormal gene product. Abnormal SOX10 gene products resulting in transcriptional activity and Sox10-deficient mice show impaired development of olfactory ensheathing cells and impaired migration of GnRH neurons.
TACR3
Gene structure.
TACR3 comprises five coding exons and no alternative splice variants.
Pathogenic variants.
TACR3 nonsense, frameshift, or missense variants cause normosmic IGD in an autosomal recessive inheritance pattern. Occasionally, heterozygous pathogenic variants are also seen in persons with IGD, suggesting an oligogenic inheritance pattern.
Normal gene product.
TACR3 encodes for TACR3, a G protein-coupled transmembrane receptor for neurokinin B.
Abnormal gene product. Abnormal TACR3 gene products result in diminished or absent neurokinin B signaling through the receptor, resulting in secondary GnRH deficiency and consequent hypogonadotropism.
For information about genes in Table 2b, click here (pdf).